

Visual Abstract

Keywords: autism, dendritic spine, ERK, MAPK, MECP2, spine clustering
Abstract
The inflexible repetitive behaviors and “insistence on sameness” seen in autism imply a defect in neural processes controlling the balance between stability and plasticity of synaptic connections in the brain. It has been proposed that abnormalities in the Ras-ERK/MAPK pathway, a key plasticity-related cell signaling pathway known to drive consolidation of clustered synaptic connections, underlie altered learning phenotypes in autism. However, a link between altered Ras-ERK signaling and clustered dendritic spine plasticity has yet to be explored in an autism animal model in vivo. The formation and stabilization of dendritic spine clusters is abnormally increased in the MECP2-duplication syndrome mouse model of syndromic autism, suggesting that ERK signaling may be increased. Here, we show that the Ras-ERK pathway is indeed hyperactive following motor training in MECP2-duplication mouse motor cortex. Pharmacological inhibition of ERK signaling normalizes the excessive clustered spine stabilization and enhanced motor learning behavior in MECP2-duplication mice. We conclude that hyperactive ERK signaling may contribute to abnormal clustered dendritic spine consolidation and motor learning in this model of syndromic autism.
Significance Statement
It has been proposed that autism-associated genetic mutations lead to altered learning phenotypes by perturbing cell signaling pathways that regulate synaptic plasticity in the brain. The Ras-ERK/MAPK signaling pathway, which promotes stabilization of dendritic spine clusters, has been particularly implicated in autism spectrum disorder (ASD). Here, we show that Ras-ERK signaling is increased in motor cortex following rotarod training in the MECP2-duplication syndrome mouse model of autism, and that the abnormal motor learning and excessive stabilization of clustered dendritic spines previously observed in MECP2-duplication mice can be rescued by pharmacological inhibition of Ras-ERK signaling. This provides additional support to hypotheses that autistic phenotypes arise from disrupted Ras-ERK signaling and synaptic plasticity and suggest potential future paths for therapeutic intervention.
Introduction
It has been proposed that phenotypes of autism spectrum disorder (ASD) arise from an abnormal imbalance between the stability and plasticity of synaptic connections in the brain (Ramocki and Zoghbi, 2008). Syndromic autism refers to a subgroup of ASD caused by genetic abnormalities that cause autism at high penetrance (Sztainberg and Zoghbi, 2016). Modeling these genetic abnormalities in mice has led to the development of autism animal models with improved validity (Sztainberg and Zoghbi, 2016). Abnormal synaptic plasticity is a common feature in animal models of autism (Bourgeron, 2015). Some autism mouse models exhibit impaired synaptic plasticity whereas others show enhanced synaptic plasticity (Bourgeron, 2015). Understanding the molecular mechanisms underlying these plasticity changes could open new avenues for therapeutic intervention. Animal models for the methyl-CpG-binding-protein 2 (MeCP2) disorders Rett syndrome (MeCP2 loss-of-function) and MECP2-duplication syndrome (MeCP2 gain-of-function) in particular have led to a wealth of findings on the molecular biological and neural circuit underpinnings of ASD (Collins et al., 2004; Jiang et al., 2013; Lyst and Bird, 2015; Lu et al., 2016; Sztainberg and Zoghbi, 2016) and identified potential avenues for treatment (Na et al., 2014; Hao et al., 2015; Sztainberg et al., 2015; Achilly et al., 2021).
Enhanced repetitive motor learning on the rotarod is observed in several autism mouse models with prominent behavioral inflexibility, including MECP2-duplication (Collins et al., 2004; Sztainberg et al., 2015), neuroligin-3 (Rothwell et al., 2014), 15q duplication (Nakatani et al., 2009), PTEN (Kwon et al., 2006), and CNTNAP2 mice (Peñagarikano et al., 2011), providing a robust model behavior for studying the abnormal consolidation of repetitive motor routines. It was previously reported that an aberrant increase in the formation and stabilization of dendritic spine clusters in the animal model of MECP2-duplication syndrome correlated with an enhancement in motor performance on the rotarod task (Ash et al., 2021). The increased clustered-spine stability in the ∼9- to 10-μm proximity range that was observed is strikingly similar to the range of a known BDNF-TrkB-Ras-ERK-dependent form of clustered-spine plasticity (Harvey and Svoboda, 2007; Harvey et al., 2008; Makino and Malinow, 2011; Niculescu et al., 2018). Ras-ERK/MAPK signaling in hippocampal pyramidal neurons has been shown in vitro to be involved in the cooperative potentiation of neighboring dendritic spines (Harvey et al., 2008; Patterson et al., 2010; Kwon and Sabatini, 2011). When a spine is activated, Ras enters its GTP-bound active state and diffuses 9–10 μm down the dendrite to invade neighboring spines. There it initiates the ERK phosphorylation cascade orchestrating a number of transcriptional and translational changes associated with synaptic consolidation (Ye and Carew, 2010).
Mutations in Ras-MAPK pathway genes are linked to several forms of autism (Stornetta and Zhu, 2011; Wen et al., 2016; Vithayathil et al., 2018), and both patients and animal models of autism demonstrate abnormal Ras-MAPK signaling (Ebert and Greenberg, 2013; Cheng et al., 2017; Rosina et al., 2019). Ras-MAPK genes have also been shown to be dysregulated in MECP2-duplication mice (Chahrour et al., 2008). We therefore hypothesized that hyperactive ERK signaling could contribute to the abnormal plasticity phenotypes observed in our animals.
Here, we show that Ras-ERK signaling is upregulated in the motor cortex of MECP2-duplication animals after motor training, and both excessive synaptic clustering and enhanced motor learning can be reversed by pharmacological normalization of ERK signaling. These data link a structural dendritic spine phenotype to a specific autism-associated cell-signaling pathway in a mouse model of autism, in vivo.
Materials and Methods
Animals
FVB-background MECP2-duplication (Tg1) mice (Collins et al., 2004), were crossed to C57 thy1-GFP-M homozygotes obtained from The Jackson Laboratory, to generate F1C57;FVB MECP2-duplication;thy1-GFP-M mice and thy1-GFP-M littermate controls. Males and females were used in experiments. Animals were housed in a 12/12 h light/dark cycle (lights on from 7 A.M. to 7 P.M.). All experiments with animals were conducted in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals and were approved by the institution’s Institutional Animal Care and Use Committee.
Blinding
In data reported in Figure 1, mice from each genotype were randomly assigned to the drug or vehicle condition. Material to be injected was placed into individual tubes for each animal, then mice were injected without knowledge of the test tube’s contents. Mice were trained, motor cortex was harvested, and Westerns were run and analyzed blind to condition. In Figure 2 data, imaging, rotarod training, drug/vehicle injection, and data analysis were performed blinded to experimental condition.
Figure 1.
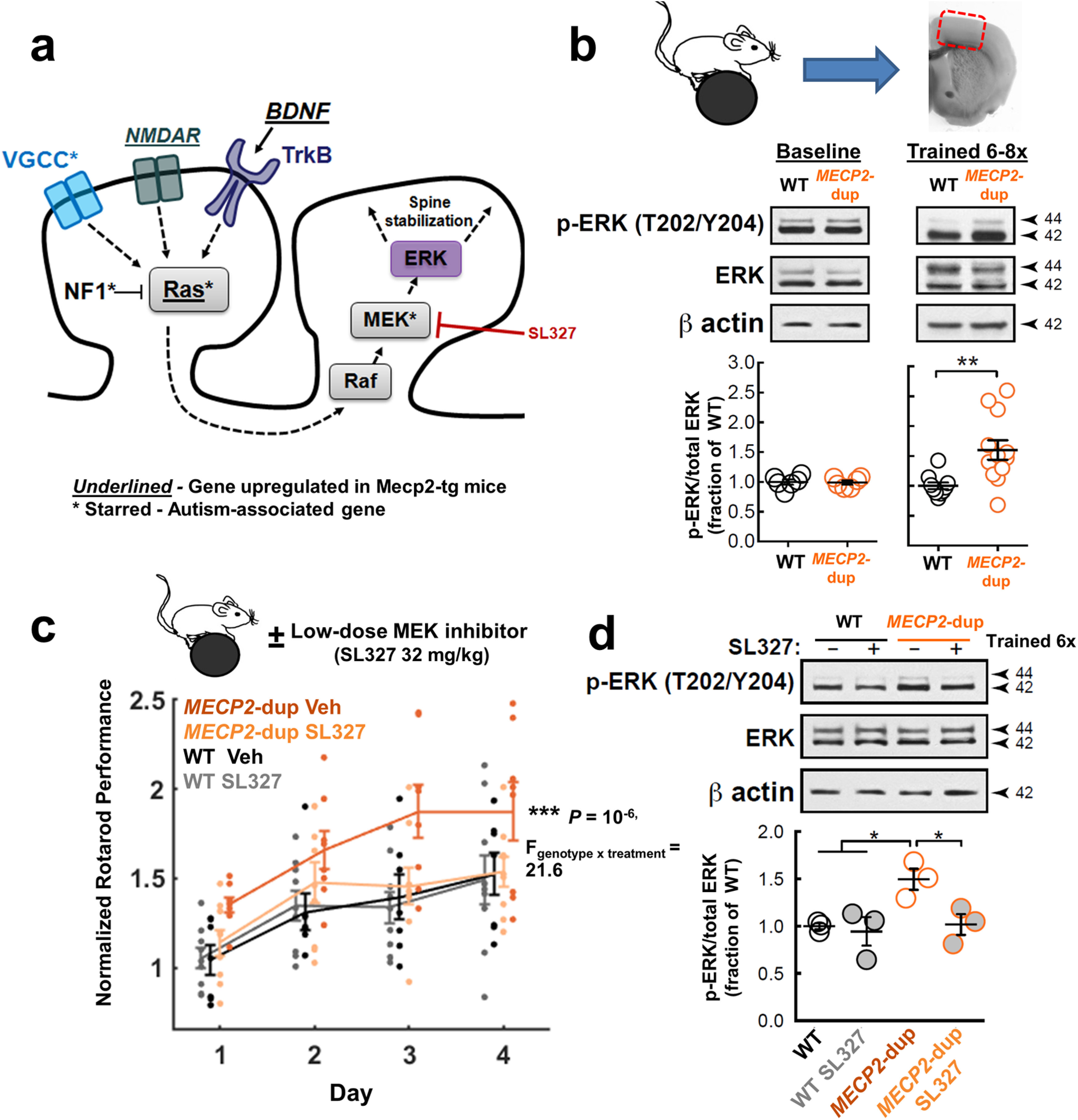
Normalization of enhanced motor learning by pharmacological inhibition of elevated Ras-ERK signaling in MECP2-duplication mice. a, A simplified schema of the Ras-MAPK signaling pathway and how it is hypothesized to contribute to clustered-spine stabilization. Genes transcriptionally upregulated in MECP2-overexpressing mice are underlined. Known autism-associated genes are denoted with an asterisk. b, Rotarod training (six to eight trials) induces enhanced Ras-MAPK signaling (ERK phosphorylation) in MECP2-duplication mice relative to WT littermates, despite equivalent baseline levels before training. Representative Western blottings and densitometric quantification of ERK activation (p-ERK T202/Y204/total ERK immunoreactivity; 42- and 44-kDa bands summed in quantification) at baseline (n = 6 mice/genotype; unpaired two-sided t test: p = 0.9, t(10) = 0.17) and after training (n = 10 WT and 12 MECP2-duplication mice; unpaired two-sided t test: **p = 0.004, t(20) = 3.2). Data are presented as fraction of the WT mean for illustration purposes. Statistical tests were performed on raw per-animal p-ERK immunoreactivity/total-ERK immunoreactivity values. Time spent on the rotarod did not explain differences between genotypes (see text). c, The MEK inhibitor SL327 normalizes rotarod performance in MECP2-duplication mice. SL327 or vehicle was injected intraperitoneally 30 min before training on each training day. Mean ± SEM of the peak performance on each day plotted for vehicle-treated MECP2-duplication (dark orange, n = 8 mice), vehicle-treated WT (black, n = 7), SL327-treated MECP2-duplication (pale orange, n = 9), and SL327-treated WT (gray, n = 9) mice. For illustration purposes, rotarod performance for each animal was normalized to the mean first-day performance of the WT littermates in that animal’s cohort before averaging across animals, to account for systematic variability in performance across cohorts because of animal weight and age; ***p = 10−6, genotype × drug interaction, Fgenotype × treatment(1,1,29) = 21.6; Fgenotype(1,29) = 6.4, p = 0.01; Ftreatment(1,29) = 4.9, p = 0.027; Ftrial(15,29) = 15.8, p = 10−6; mixed effects repeated-measures ANOVA. Statistical analysis was performed on raw per-trial rotarod performance values. Litter was included as an interacting variable to control for across-litter variability in performance. d, Consistent with a role for elevated ERK signaling in the mutant’s enhanced motor learning phenotype, 32 mg/kg SL327 treatment blocked the training-dependent increase in M1 ERK phosphorylation in MECP2-duplication mice. Representative Western blottings and densitometric quantification of ERK activation in vehicle-treated versus SL327-treated WT and MECP2-duplication mice (n = 3 mice/genotype/treatment group; *p < 0.05, Fgenotype(1,8) = 6.7, Ftreatment(1,8) = 5.9, Finteraction(1,1,8) = 3.7, two-way ANOVA with Tukey’s post hoc correction for multiple comparisons. Normalization of data points as in b. Mice in the vehicle-treated condition are also included in b. Error bars represent SEM. Circles show data points from individual animals. Example full length Western blottings are shown in Extended Data Figure 1-1a,b. For blinding procedure, see Materials and Methods.
Figure 2.

Normalization of excessive clustered spine stabilization in MECP2-duplication mice by pharmacological Ras-ERK inhibition. a, Sample images of dendritic segments imaged before rotarod training (left), following 4 d of training (middle), and 4 d after the end of training (right) demonstrating decreased clustered spine stabilization in SL327-treated (bottom) versus vehicle-treated (top) MECP2-duplication mice. b, Total spines formed (left bars) and spines stabilized (right bars) per 100 μm in vehicle-treated (black, n = 4 animals, 57 spines formed, 30 dendritic segments) and SL327-treated (gray, n = 7 animals, 49 spines formed, 49 dendritic segments) littermate control mice. c, Clustered and isolated new-spines stabilized per 100 μm in vehicle-treated (black) and SL327-treated (gray) littermate control mice. d, Total spines formed (left bars) and spines stabilized (right bars) per 100 μm in vehicle-treated (dark orange, n = 7 animals, 184 spines formed, 54 dendritic segments) and SL327-treated (light orange, n = 8 animals, 143 spines formed, 50 dendritic segments) MECP2-duplication mice. e, Clustered and isolated spines stabilized per 100 μm in vehicle-treated (dark orange) and SL327-treated (light orange) MECP2-duplication mice. Note that there are significantly fewer stabilized clustered spines in SL327 treated animals; *p < 0.05, **p < 0.01, specific p values reported in the figure, Mann–Whitney U test.
Example full-length Western blottings, related to Figure 1. A, Example full-length Western blottings relevant to Figure 1B, for p-ERK (T202/204), total ERK, and β-actin. B, Example full-length Western blottings relevant to Figure 1D, showing immunoblots to p-ERK (T202/204), total ERK, and β-actin. Note SL-327 suppresses the level of p-ERK in mutant animals. Download Figure 1-1, TIF file (1.7MB, tif) .
In vivo two-photon imaging
Experiments were performed as in Ash et al. (2021). At least two weeks before the first imaging session (∼12- to 14-week-old mice), a 3-mm-wide opening was drilled over motor cortex, centered at 1.6 mm lateral to bregma based on (Tennant et al., 2011), and a glass coverslip was placed over the exposed brain surface to allow chronic imaging of neuronal morphology (Mostany and Portera-Cailliau, 2008, 2011). Dendritic spines were imaged using a Zeiss in vivo two-photon microscope with Zeiss 20 × 1.0 NA water-immersion objective lens. High-quality craniotomies had a characteristic bright-field appearance with well-defined vasculature and pale gray matter. Under two-photon scanning fluorescent dendrites were reliably clear and visible with low laser power (<20 mW on the pia) and photomultiplier tube voltage.
Only high-quality preparations (low background noise across all time points, <5-pixel (0.25 μm) motion artifact, and dendrites well isolated from other fluorescent structures) were used in the blinded analysis. Green fluorescent protein (GFP)-labeled neurons were first imaged at low resolution (620 × 620 μm FOV, 0.6 μm/pixel in XY, 2.5-μm Z-step size) down to 600–700 μm to capture all of the cell’s dendritic processes and assay cell subtype by morphology, primary apical bifurcation depth, and soma depth (Holtmaat et al., 2006). The apical dendrites from complex-tufted neurons, the corticospinal neurons projecting to the spinal cord and thalamus in M1, were selected based on their large highly ramified dendritic trees, deep primary apical bifurcation, and thick dendrites, and re-imaged at high resolution (310 × 310 to 420 × 420 μm FOV, 0.1 μm/pixel, 1 μm Z-step size) to adequately capture individual dendritic spines. Laser power was maintained under 20 mW (average ∼10 mW) during image stack acquisition.
Analysis of structural plasticity
Raw z stacks were denoised by a custom polynomial interpolation method (Jiang et al., 2013). Spine formation, elimination, and stabilization were quantified with a custom MATLAB user interface and ImageJ (MicroBrightField). Terminal dendrite segments which were well-visualized at all time points were chosen for analysis. Sections of dendrite occluded by other fluorescent structures or blood vessels were excluded from the analysis. Because of in vivo two-photon microscopy’s relatively poor resolving power in the z-axis, only structures protruding laterally along the X-Y plane were included in the analysis, following the standard in this field (Holtmaat et al., 2009). For a protrusion to be selected for analysis it had to project out of the dendritic shaft by at least 4 pixels (∼0.4 μm), which corresponds approximately to 2 SDs of the noise blur on either side of the dendritic shaft. Spines were initially identified at one time point, by moving up and down through individual slices in each Z stack, and the same region of dendrite was examined at other time points to identify the first (formation) and last (elimination) time that the spine was present. Custom MATLAB routines analyzed the stability/survival of each formed spine. Filopodia, which are rare at the analyzed age, were identified morphologically, based on their long length (usually >4 μm), curved shape, and lack of a distinct head (Zuo et al., 2005) and excluded from the analysis as in Yang et al. (2009). Each spine was classified as either clustered or isolated by calculating the distance to its nearest-neighbor stabilized learning-associated spine as in Fu et al. (2012) and Ash et al. (2021).
Motor training
The Ugo Basile mouse rotarod was used for motor training. At least 2 h after imaging sessions, in the late afternoon, mice were placed on the rotarod, and the rotarod gradually accelerated from 5 to 80 rpm over 3 min. Up to five littermate mice with intermixed genotypes were trained in parallel. Single-trial rotarod performance was quantified as the time right before falling or holding on to the dowel rod for two complete rotations without regaining footing. A 5- to 10-min rest period occurred between each trial. Four trials were performed per day. For immunoblot experiments measuring M1 ERK phosphorylation with rotarod training, training-naive mice were trained six to eight times consecutively (Fig. 1b,d). For the pharmacology experiments demonstrating the effect of the specific MEK inhibitor SL327 on rotarod performance, the peak performance of each animal on each day, normalized to the vehicle-treated wild-type (WT) mean to account for across litter variability in performance, was used in the analysis (median performance yielded similar statistically significant results).
Pharmacological Ras-MAPK inhibition
The centrally-acting selective MEK inhibitor SL327 (Atkins et al., 1998; Axonmedchem #1122) was injected intraperitoneally at 32 mg/kg (in 16 mg/ml DMSO) 30 min before rotarod training (Bureau et al., 2010). This dose was selected to be low as it is known to minimally affect motor performance in WT animals (Bureau et al., 2010).
Immunoblots
Deeply anesthetized (isoflurane) four- to five-month-old MECP2-duplication mice and littermate controls were killed 30 min after training and their brains were rapidly dissected on a glass plate over ice. The motor cortex from each hemisphere was isolated from the remaining cortical tissue and lysed in ice-cold homogenization buffer (200 mm HEPES, 50 mm NaCl, 10% glycerol, 1% Triton X-100, 1 mm EDTA, 50 mm NaF, 2 mm Na3VO4, 25 mm β-glycerophosphate, and 1× EDTA-free complete ULTRA protease inhibitor cocktail tablets; Roche). Insoluble material was removed by centrifugation at 14,000 × g for 10 min at 4°C. Protein concentration of the resulting supernatant was determined by Bradford assay (Bio-Rad, reagent 500-0006) and lysates were then diluted in 2× Laemmli Buffer. A total of 30 μg protein/sample was resolved by SDS-PAGE (12.5% acrylamide) and gel contents were transferred to nitrocellulose membranes. Membranes were blocked 30 min in 5% milk, 0.2% Tween 20 tris-buffered saline (TBST). To assess ERK phosphorylation, membranes were first probed overnight at 4°C with phospho-specific rabbit anti-phospho-p44/42 MAPK/Erk1/2 (Cell Signaling Technology, #4370, 1:1000). Blots were then incubated with HRP-conjugated goat anti-rabbit secondary antibody (Jackson ImmunoResearch, 111-035-144, 1:5000) for 1 h at room temperature followed by incubation in Super-Signal West Femto kit substrate (Thermo Scientific, 34096) per the manufacturer’s instructions. Film was exposed to the Super-Signal-treated membranes and then developed. Membranes were then stripped (1.5% glycine and 2.9% NaCl, pH2.8), blocked with 5% milk TBST, then re-probed overnight with rabbit anti-MAPK/ERK1/2 (Cell Signaling Technologies, #9202, 1:1000) to assess total ERK protein levels. β-Actin levels were measured as an additional loading control (mouse anti-β-actin, Millipore, MAB1501, 1:5000). The goat anti-mouse secondary antibody was diluted 1:10,000 for the β-actin blots. Band density was quantified in ImageJ (NIH).
Image presentation
Dendritic spine images are displayed as “best” projection mosaics (Holtmaat et al., 2009). Extraneous fluorescence is masked and images are slightly smoothed for illustration purposes only.
Statistical tests
Statistical significance between unpaired normal samples was assessed by Mann–Whitney U test or two-way ANOVA, except where noted, using MATLAB. Enhanced rotarod performance in MECP2-duplication mice in Figure 1 was tested by repeated-measures ANOVA. Effect of SL327 on rotarod performance in MECP2-duplication mice compared with littermate controls was evaluated by determining the genotype × treatment interaction term using a mixed effects repeated-measures ANOVA in R statistics software. Statistical analysis was performed on raw per-trial rotarod performance values. Litter was included as a term to control for across-litter variability in performance; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. All results are reported as mean ± SEM, unless otherwise noted.
Results
ERK signaling has been shown to promote cooperative plasticity between dendritic spines located at close proximity (9–10 μm) to each other along the dendrite (Harvey and Svoboda, 2007; Harvey et al., 2008). This led us naturally to hypothesize that it mediates the enhanced clustered dendritic spine stabilization previously observed in MECP2-duplication animals (Ash et al., 2021). The experiments described below were targeted a-priori to examine this primary hypothesis.
Enhanced motor learning in MECP2-duplication mice is normalized by Ras-ERK inhibition
A simplified sketch of ERK signaling in clustered-spine plasticity is illustrated in Figure 1a, adapted from (Harvey et al., 2008; Ye and Carew, 2010). Several genes in the ERK pathway are associated with autism (Stornetta and Zhu, 2011; Fig. 1a, asterisks) and MECP2-duplication mice overexpress several genes in this pathway (Chahrour et al., 2008; notably, Bdnf, Nmdar1, and Ras; Fig. 1a, underlined). To directly test whether ERK signaling is increased in motor cortex of MECP2-duplication mice, we performed Western blot analyses on M1 protein extracts isolated from mutant mice and WT littermates at baseline and immediately following rotarod training (Fig. 1b; Extended Data Fig. 1-1a, six to eight consecutive trials in previously-untrained animals). We found that marked hyperphosphorylation of the MAP kinase ERK1/2 (T202/Y204) in MECP2-duplication mouse area M1 occurred with rotarod training (p = 0.004, n = 10 WT M1 extracts from 10 mice, 12 MECP2-duplication M1 extracts from 12 mice, two-tailed unpaired t test; Fig. 1b). In contrast the phosphorylation state of ERK was not significantly altered in mutant M1 lysates without training (p = 0.87, n = 6 mice per genotype, two-tailed unpaired t test; Fig. 1b), as reported previously for untrained Mecp2-null and MECP2-duplication animals (Pitcher et al., 2015). Note that the increase in ERK phosphorylation in area M1 after motor training in MECP2-animals could not be explained solely by increased time spent on the rotarod, as the level of phosphorylation did not correlate with the latency to fall from the rotarod (r = −0.17, p = 0.53). This suggests the induction of ERK phosphorylation by training is exuberant in the motor cortex of MECP2-duplication animals.
Training-induced ERK hyperphosphorylation (Fig. 1b) coupled with the increased clustered-spine stabilization previously observed in MECP2-duplication animals (Ash et al., 2021) implicated elevated ERK signaling in the enhanced motor learning phenotype of the MECP2-duplication syndrome mouse model (Collins et al., 2004; Harvey et al., 2008). To test this hypothesis, in a new round of experiments we administered a low dose (32 mg/kg, i.p.) of the specific centrally-acting MEK inhibitor SL327 or DMSO vehicle 30 min before rotarod training on each day (Bureau et al., 2010), and trained the animals for four consecutive trials daily for four days in a row (Collins et al., 2004; Ash et al., 2021). This dose was chosen to ensure effects on locomotor function in WT are minimal (Antoine et al., 2013; but see Bureau et al., 2010). Accordingly, the performance of WT mice was not affected, whereas SL327 reversed the enhanced motor learning in mutants (p = 10−6, genotype × drug interaction, Fgenotype × treatment = 21.6, n = 7–9 mice per group, mixed effects repeated-measures ANOVA; Fig. 1c). Furthermore, M1 ERK phosphorylation was normalized in SL327-treated mutants versus vehicle-treated mutants (Fig. 1d; Extended Data Fig. 1-1b, p < 0.05, n = 3 mice/group, two-way ANOVA with Tukey’s post hoc correction for multiple comparisons), confirming the effectiveness of the inhibitor in reducing ERK phosphorylation levels.
Enhanced clustered spine stabilization in MECP2-duplication mice is normalized by Ras-ERK inhibition
Lastly, we measured the effect of MEK inhibition on clustered spine stabilization in MECP2-duplication mice and littermate controls crossed to the thy1-GFP M line, which sparsely expresses GFP in layer 5 pyramidal neurons (Feng et al., 2000). As in Ash et al. (2021), chronic cranial windows were implanted over primary motor cortex (M1) in MECP2-duplication mice and littermate controls, and apical dendrites from GFP-expressing L5 pyramidal neurons were imaged with in vivo two-photon microscopy (Holtmaat et al., 2009). Spine analysis was performed on terminal dendritic branches of the apical tuft of these neurons. We first identified baseline spines, then animals were trained on the rotarod task (four trials per day) for 4 d (Fig. 2A). Animals were randomized to receive SL327 32 mg/kg or vehicle (DMSO) injected intraperitoneally 30 min before training across the four rotarod training days. On the fifth day, dendrites were imaged again to identify new spines formed. Following 4 d of rest, dendrites were once again imaged to identify the new spines that stabilized. The follow-up imaging time point was chosen in line with prior studies showing that the vast majority of newly formed dendritic spines which persist for at least 4 d form an electron-microscopy-verified synapse (Knott et al., 2006). Each stabilized spine was classified as clustered or isolated based on its proximity to another newly-formed spine on the same dendrite (clustered: <9 μm interspine distance; isolated: ≥9-μm interspine distance, following Ash et al., 2021).
As observed previously (Ash et al., 2021), clustered spine stabilization was increased in vehicle-treated MECP2-duplication mice compared with controls (WT 1.1 ± 0.2 stabilized per 100 μm, n =4 animals, 30 dendritic segments; MECP2-duplication: 2.2 ± 0.4 stabilized per 100 μm, n = 7 animals, 54 dendritic segments, p = 0.04, Mann–Whitney U test; two-way ANOVA effect of genotype: p = 0.03, F(1,22) = 5.5). Overall spine formation/stabilization as well as clustered and isolated spine stabilization were not significantly affected by MEK inhibition in control mice (all p values > 0.09, Mann–Whitney U test, n = 4 vehicle-treated WT mice, 7 SL327-treated WT mice; Fig. 2b,c), as expected given the low dose of SL327 (Antoine et al., 2013). In contrast, in MECP2-duplication animals overall spine stabilization was significantly reduced by SL327 (effect on stabilization: p = 0.029, Mann–Whitney U test; two-way ANOVA effect of SL327: p = 0.02, F(1,22) = 6.2; interaction: p = 0.4; n = 7 vehicle-treated MECP2-duplication mice, n = 8 SL327-treated MECP2-duplication mice; Fig. 2d), back to levels similar to that of WT controls (SL327-treated mutant spine stabilization rate: 1.0 ± 0.2 per 100 μm; vehicle-treated WT spine stabilization rate: 1.0 ± 0.3 per 100 μm, vehicle-treated mutant spine stabilization rate: 1.7 ± 0.2). Separating stabilized spines into clustered and non-clustered subgroups revealed that this SL327-induced decrease in spine stabilization was mediated entirely by a reduction in the stabilization of clustered spines (p = 0.009, Mann–Whitney U test, Cohen’s d = 1.65; two-way ANOVA effect of SL327: p = 0.01, F(1,22) = 7.7, effect of SL327; effect of genotype: p = 0.01, F(1,22) = 7.7; Interaction: p = 0.06, F(1,22) = 3.7; n = 7 vehicle-treated MECP2-duplication mice, n = 8 SL327-treated MECP2-duplication mice; Fig. 2e). SL327 had a negligible effect on the stabilization of isolated new-spines in mutants (p = 0.61, Mann–Whitney U test, Cohen’s d = 0.3) and also did not affect significantly overall spine formation (effect on mutant formation: p = 0.19, Mann–Whitney U test; two-way ANOVA effect of SL327: p = 0.09; effect of genotype: p = 0.0006, F(1,22) = 15.9; interaction: p = 0.9; Fig. 2d). Interestingly, there was a trend toward decreased stabilization of isolated spines in SL327-treated WT mice not seen in mutants (genotype × SL327 interaction: p = 0.03, F(1,22) = 5.2), although this trend did not survive pairwise comparison (p > 0.1, Mann–Whitney U test). These observations suggest that elevated ERK signaling contributes to the structural stabilization of training-associated clustered synapses in the MECP2-duplication mouse.
Discussion
In summary, we found that the ERK pathway, a major regulator of clustered spine stabilization (Harvey et al., 2008; Frank et al., 2018), is hyperactive following training in MECP2-duplication mouse motor cortex, and both increased spine-cluster stabilization and enhanced motor learning in MECP2-duplication mice can be reversed by ERK-specific pharmacologic inhibition.
Our results build on a growing evidence base implicating Ras-ERK/MAPK signaling in ASD (Vithayathil et al., 2018). Mutations in Ras-MAPK pathway genes including NF1, Ras, MEK, and RSK, together referred to as Rasopathies, cause syndromic autism (Stornetta and Zhu, 2011). Pathway analyses of large scale genome sequencing data confirm that Ras-MAPK pathway mutations are enriched in idiopathic autism (Wen et al., 2016; Mitra et al., 2017), and upregulations in Ras-MAPK pathway activity correlate with symptom severity in autism patients (Rosina et al., 2019). Multiple animal models of autism demonstrate abnormalities in Ras-MAPK signaling including Rett syndrome (Mellios et al., 2018), BTBR strain (Seese et al., 2014; Cheng et al., 2017), 16p11del (Pucilowska et al., 2015), fragile X syndrome (Soong et al., 2008), neurofibromatosis type 1 (Costa et al., 2002), Noonan syndrome (Lee et al., 2014), Costello syndrome (Schreiber et al., 2017), cardio-cutaneo-facial syndrome (Anastasaki et al., 2009), and SynGAP1 syndrome (Rumbaugh et al., 2006). Autism-associated phenotypes have been shown to be rescued by inhibition of Ras-ERK signaling in many of these animal models (Wang et al., 2012; Pucilowska et al., 2018; Moreau et al., 2020; Mullins, 2020). Our results provide the first evidence that Ras-ERK signaling is upregulated following training in the mouse model for MECP2-duplication syndrome, and that its abnormally enhanced motor learning phenotype can be rescued by inhibiting ERK signaling (Fig. 1).
Essentially all autism models that have been formally tested exhibit changes in synaptic plasticity and learning (Bourgeron, 2015), including MECP2-duplication mice (Collins et al., 2004; Na et al., 2012; Jiang et al., 2013; Ash et al., 2018). In prior work, it was shown that clustered spine consolidation is abnormally enhanced in MECP2-duplication animals (Ash et al., 2021). Here, we show that MECP2-duplication clustered spine consolidation returned to WT-like levels after normalizing ERK signaling with the specific pharmacological inhibitor SL327 (Fig. 2). Furthermore, this occurred at a dose that did not alter the rate of new spine formation or the rate of new isolated spine consolidation after training, so the observed effect appears to be specific to co-operative spine consolidation. These observations forge a link between increased ERK signaling, increased clustered spine consolidation, and enhanced learning phenotype in the MECP2-duplication mouse model of syndromic autism.
Our results align with converging evidence on the role of ERK signaling in dendritic spine clustering and autism pathophysiology. Multiple links in the Ras-ERK pathway including BDNF, TrkB, Ras, and ERK are required for clustered spine consolidation ex vivo (Harvey et al., 2008; Niculescu et al., 2018), and CCR5-mutant animals engineered to have hyperactive Ras-ERK signaling exhibit increased clustered spine plasticity (Frank et al., 2018). Our contribution adds to this work, showing that the excessive clustered spine stabilization previously observed in the MECP2-duplication syndrome mouse model (Ash et al., 2021) can be normalized in vivo with a pharmacological inhibitor of ERK signaling.
One remaining puzzle is the fact that excessive clustered spine stabilization in MECP2-duplication animals occurs both with and without training (Ash et al., 2021), whereas excessive ERK phosphorylation occurred with training but not without training (Fig. 1b; Pitcher et al., 2015). It is possible that a subtle elevation in Ras-MAPK signaling (ERK phosphorylation) may be present without training, but not be enough to be detected by Western blot analysis (White and Wolf-Yadlin, 2016), as any difference at baseline would likely be small given our data (Fig. 1b). Other nonexclusive possibilities include that downstream effectors of p-ERK are nonlinearly affected by Ras-ERK activity in MECP2-duplication mice, and/or they can still be activated through ERK-independent mechanisms without training. Finally, Ras-ERK inhibition may modulate motor learning and spine consolidation through mechanisms other than Ras’s role in dendritic spines. Ras-ERK modulates learning and plasticity in the brain through multiple presynaptic and postsynaptic mechanisms in both excitatory and inhibitory neurons (Ryu and Lee, 2016). It is not inconceivable, for example, that MEK inhibition could downregulate spontaneous activity or alter modulatory neurotransmitter release (Ding et al., 2011; Borges et al., 2017), which would in turn indirectly normalize enhanced learning and clustered spine consolidation.
Importantly, we note that we have not conclusively shown that clustered spine stabilization drives enhanced learning, although others previously showed a correlation between spine clustering and learning across animals (Frank et al., 2018; Ash et al., 2021). The fact that the MEK inhibitor normalized motor performance in MECP2-duplication mice even on the first day of training (Fig. 1c), at which point clustered spine stabilization is unlikely to play a significant role, suggests that excessive Ras-MAPK signaling boosts learning in mutants through multiple mechanisms. Verifying a causal link between clustered spine consolidation and learning is an important goal of future work. Finally, we note that in our preparation 32 mg/kg SL327 did not significantly affect WT rotarod performance or M1 ERK phosphorylation, which aligns with (Antoine et al., 2013) but disagrees somewhat with (Bureau et al., 2010), who reported that 30 mg/kg SL327 was sufficient to decrease rotarod performance in WT mice and decrease ERK phosphorylation in the striatum. It is possible that subtle differences in the rotarod training parameters (four trials per day for 4 d, accelerating from 5 to 80 rpm in 3 min in our paradigm, vs 10 trials per day for 4 d, from 4 to 40 rpm in 5 min in their paradigm) and probing for ERK in different brain areas could explain these discrepancies, but this requires follow-up experimental assessment to determine under which conditions SL327 does or does not affect WT learning and memory.
Although enhanced motor learning is not observed in human patients (Ramocki et al., 2010), it is possible that the structural abnormality we describe in mice is also operating in the humans, but manifests with a different developmental trajectory of motor deterioration because of species-specific differences in motor system development and function (Collins et al., 2004; Ramocki et al., 2009). We note again here that several autism mouse models including MECP2-duplication, neuroligin-3, 15q duplication, PTEN, CNTNAP2, and CCR5 have enhanced motor learning and plasticity (Etherton et al., 2011; Piochon et al., 2014; Rothwell et al., 2014; Frank et al., 2018), while several other mouse models including Rett syndrome, fragile X, and Angelman (15q deletion), have impaired motor learning and plasticity (Li et al., 2002; Asaka et al., 2006; Van Woerden et al., 2007; Kondo et al., 2008; Padmashri et al., 2013). In particular, opposite to our findings in the MECP2-duplication mice, it was recently reported that fragile X mice have impaired motor learning and decreased clustered spine stabilization (Bhattacharya et al., 2012; Padmashri et al., 2013; Reiner and Dunaevsky, 2015). Building on other studies positing an axis of synaptic pathophysiology in syndromic autism (Auerbach et al., 2011), these findings together suggest that certain forms of syndromic autism defined by enhanced plasticity and prominent behavioral inflexibility could be particularly amenable to Ras-MAPK modulating agents.
Our results provide in vivo evidence that ERK signaling is involved in learning-associated synaptic stabilization and synaptic clustering and suggest that a training-dependent increase in M1 ERK signaling facilitates procedural motor memory consolidation in MECP2-duplication animals. Our findings provide additional support to hypotheses that learning phenotypes in autism arise from disrupted Ras-ERK signaling and synaptic plasticity (Bourgeron, 2015; Vithayathil et al., 2018). In the future, it will be fruitful to study the regulation of upstream and downstream mediators of ERK signaling in MECP2-duplication mice and explore how excessive ERK signaling contributes to excessive clustered spine stabilization in this mouse model (Alonso et al., 2004; Im et al., 2010; Ye and Carew, 2010; Stornetta and Zhu, 2011; Ebert and Greenberg, 2013; Niculescu et al., 2018). In the future, modulation of structural plasticity through Ras-MAPK inhibitors could provide a potential therapeutic avenue for behavioral inflexibility and other phenotypes in the MECP2-duplication syndrome (Sandweiss et al., 2020).
Acknowledgments
Acknowledgements: We thank G. Allen, S. Torsky, E. Sztainberg, B. Suter, J. Meyer, A. Palagina, J. Patterson, S. Shen, D. Yu, and K. Tolias for technical and theoretical advice on experiments and comments on this manuscript. We also thank H. Lu for the images of mouse coronal brain slices.
Synthesis
Reviewing Editor: Christian Hansel, University of Chicago
Decisions are customarily a result of the Reviewing Editor and the peer reviewers coming together and discussing their recommendations until a consensus is reached. When revisions are invited, a fact-based synthesis statement explaining their decision and outlining what is needed to prepare a revision will be listed below. The following reviewer(s) agreed to reveal their identity: Peyman Golshani, Stéphane Baudouin.
The manuscript shows convincing evidence of a link between dysregulation of ERK signaling, synaptic structural plasticity, and motor deficits in mice with duplication of MeCP2. This work adds to existing scientific evidence showing, in preclinical models, and involvement of Ras-ERK signaling in the pathophysiology of neurodevelopmental disorders, and autism spectrum disorders in particular. These results also replicate results previously obtain in the MeCP2 duplication mouse model (Ash et al 2020) providing important confirmation of published results.
The authors find hyperphosphorylation of ERK 1/2 in the MECP2-duplication mouse only after rotarod training. The MECP2 duplication mice have enhanced motor learning and treatment with MEK inhibitor SL327 blocked this enhancement and blocked clustered stabilization of L5 apical dendritic spines in motor cortex. Overall this is an important paper with exciting findings that have potential relevance to understanding aberrant plasticity in MECP2 duplication and Rett’s. The experiments are well-designed, the analysis is robust and the effect sizes large.
Detailed critique: Abstract/Introduction: I think it would be important for the readers that the authors clarify the relationship between the MeCP2 duplication mouse model and ASD, and in particular address the following points: 1- In the abstract, the authors use the term syndromic autism. This term refers to a very specific notion in ASD, relating to two categories of ASD. The authors should clarify this notion in the introduction 2- The MeCP2 duplication mouse model is not well-studied compared to the MeCP2 deletion model. I would therefore encourage the authors to provide additional background to better describe the model, particularly to clarify the existence of the two models and briefly highlight their differences. Such an introduction could also serve to better present the link between MeCP2, Rett syndrome, and autism spectrum disorders. 3- The authors relate the phenotype of the MeCP2 duplication mouse model to ASD and, in the current manuscript, this behavior is enhanced learning. I do not think that enhanced learning is a characteristic feature of ASD (see my related comment in the discussion). The authors should provide a more in-depth justification in relating their results to ASD and allude to it in the introduction to avoid misleading the readers. Comments related to Figure 1: - The results are in 1B, 1C, and 1D are presented as a fraction of WT and there are multiple data points for WT. I find the nomenclature unclear; it would be better to more clearly state what the ratio is exactly. It would also be important to state which data were used for the statistical analysis. - For 1C specifically, it would be good if the authors could present the results in more detail, as they are presented in Ash et al 2020 for example. - The authors showed an absence of the effect of SL327 on the level of phosphorylation of ERK in WT animals. The argument used by the authors to justify such absence of an effect is that the dose used, 32mg/kg, is too low to affect wild-type animals, referencing Antoine et al. 2013. Bureau et al 2010, also cited in the article, showed that treatment with 30mg/kg SL327 decreases ERK phosphorylation in WT. It would be good if the authors fully acknowledge the existence of these results and discuss the discrepancies. A possible explanation for the discrepancy is the use of different brain regions (striatum versus M1). Also, ERK phosphorylation is increased in wild-type animals following rotarod training (Bureau et al 2020), unlike M1. The authors could try to determine, experimentally, if the difference in brain regions is the reason for the discrepancy. - Similarly, Bureau et al 2020 showed that treatment with 30mg/kg of SL327 impaired motor learning in wild-type mice. This effect is not replicated by the authors but also not mentioned. As above, it would be important to better acknowledge existing results and discuss potential reasons for the discrepancy. - It is unclear to me if SL327 is injected every day before rotarod training, please clarify. Note that I do not think that an absence of replication necessarily invalids the present results. Comments related to Figure 2: - The results are presented in a very synthetic form, using bar graph representations. For reasons of clarity in the results but also consistency in the presentation between figures, it would be better to represent individual data points. - As noted by the authors, results in Figure 1C showed a potential trend for an effect of SL327 on the stability of isolated spines. The number of animals used in this study is rather small compared to Ash et al 2020, which is directly comparable, and it remains possible that such a low number affects the results of the statistical analysis. Such a result could prove important when stated that 30mg/kg of SL327 does not affect wild-type animals. If possible, it would be good to add data points to more firmly conclude on an absence of effect of SL327 in wild-type mice. Discussion: The authors state that “These observations forge a link between increased ERK signaling, increased clustered spine consolidation, and enhanced learning phenotype in the MECP2-duplication mouse model of syndromic autism”. The authors also state their “findings provide additional support to hypotheses that learning phenotypes in autism arise from disrupted Ras-ERK signaling and synaptic plasticity”. Enhanced learning is not a feature particularly associated with ASD and so it would be interesting to develop on the nature of the link between enhanced learning and phenotypes associated with ASD. In particular, it would be interesting to know to which category or subtype of ASD could benefit from treatment with ERK inhibitors.
Minor criticisms: 1. The authors should include the individual data points on top of the bar graphs for Figure 2B-E. 2. Do the authors have any information on the involvement of different spine types in the MECP2 duplication phenotype?
Author Response
Dear Dr. Hansel,
We are glad that our work was well-received by the reviewers, and we were highly motivated to address
their issues and suggestions. Please forgive the delayed revision response, which was necessary due to
the first author’s clinical duties. The reviewers make several constructive and addressable issues related
to readability and data presentation. We are sincerely appreciative of these comments, and in
addressing them we feel that we have made the manuscript a more rigorous and readable scientific
work.
Here we reproduce the reviewers’ comments and our revisions point-by-point:
The manuscript shows convincing evidence of a link between dysregulation of ERK signaling, synaptic
structural plasticity, and motor deficits in mice with duplication of MeCP2. This work adds to existing
scientific evidence showing, in preclinical models, and involvement of Ras-ERK signaling in the
pathophysiology of neurodevelopmental disorders, and autism spectrum disorders in particular. These
results also replicate results previously obtain in the MeCP2 duplication mouse model (Ash et al 2020)
providing important confirmation of published results.
The authors find hyperphosphorylation of ERK 1/2 in the MECP2-duplication mouse only after rotarod
training. The MECP2 duplication mice have enhanced motor learning and treatment with MEK inhibitor
SL327 blocked this enhancement and blocked clustered stabilization of L5 apical dendritic spines in motor
cortex. Overall this is an important paper with exciting findings that have potential relevance to
understanding aberrant plasticity in MECP2 duplication and Rett’s. The experiments are well-designed,
the analysis is robust and the effect sizes large.
Detailed critique:
Abstract/Introduction:
I think it would be important for the readers that the authors clarify the relationship between the MeCP2
duplication mouse model and ASD, and in particular address the following points:
-In the abstract, the authors use the term syndromic autism. This term refers to a very specific
notion in ASD, relating to two categories of ASD. The authors should clarify this notion in the
introduction
--The MeCP2 duplication mouse model is not well-studied compared to the MeCP2 deletion
model. I would therefore encourage the authors to provide additional background to better
describe the model, particularly to clarify the existence of the two models and briefly highlight
their differences. Such an introduction could also serve to better present the link between
MeCP2, Rett syndrome, and autism spectrum disorders.
--The authors relate the phenotype of the MeCP2 duplication mouse model to ASD and, in the
current manuscript, this behavior is enhanced learning. I do not think that enhanced learning is a
characteristic feature of ASD (see my related comment in the discussion). The authors should
provide a more in-depth justification in relating their results to ASD and allude to it in the
introduction to avoid misleading the readers. 2
We are very thankful to the reviewer for these constructive suggestions to improve the
readability and rigor of the abstract and introduction. We have modified these sections based
on the feedback and are happy with the new version.
Comments related to Figure 1:
The results are in 1B, 1C, and 1D are presented as a fraction of WT and there are multiple data
points for WT. I find the nomenclature unclear; it would be better to more clearly state what the
ratio is exactly. It would also be important to state which data were used for the statistical
analysis.
Each data point in Fig. 1b and 1d depicts a single animal's phospho-ERK T202/Y204
immunoreactivity / total ERK immunoreactivity, divided by the WT mean for illustration
purposes. Statistical tests were performed on raw per-animal p-ERK immunoreactivity / total-ERK immunoreactivity values. In Fig. 1c, rotarod performance for each animal was normalized
to the mean first-day performance of the WT littermates in that animal’s cohort, to account
for systematic variability in performance across cohorts due to animal weight and age. This
was done for illustration purposes only, and statistical analysis was performed on raw per-trial rotarod performance values (with litter as an interacting random variable).
We have updated the figure legend in Fig. 1 as suggested by the reviewer to assist in
readability and interpretability of the results.
For 1C specifically, it would be good if the authors could present the results in more detail, as
they are presented in Ash et al 2020 for example.
We appreciate this suggestion. We added individual data points in Fig. 1c, which better
visualizes the robustness of the results.
- The authors showed an absence of the effect of SL327 on the level of phosphorylation of ERK in
WT animals. The argument used by the authors to justify such absence of an effect is that the
dose used, 32mg/kg, is too low to affect wild-type animals, referencing Antoine et al. 2013.
Bureau et al 2010, also cited in the article, showed that treatment with 30mg/kg SL327
decreases ERK phosphorylation in WT. It would be good if the authors fully acknowledge the
existence of these results and discuss the discrepancies. A possible explanation for the
discrepancy is the use of different brain regions (striatum versus M1). Also, ERK phosphorylation
is increased in wild-type animals following rotarod training (Bureau et al 2020), unlike M1. The
authors could try to determine, experimentally, if the difference in brain regions is the reason for
the discrepancy. Similarly, Bureau et al 2020 showed that treatment with 30mg/kg of SL327
impaired motor learning in wild-type mice. This effect is not replicated by the authors but also
not mentioned. As above, it would be important to better acknowledge existing results and
discuss potential reasons for the discrepancy... Note that I do not think that an absence of
replication necessarily invalids the present results.
We are thankful for these suggestions and agree with the reviewer’s contentions. We have
updated the results and discussion to point out these discrepancies between our results and
that of Bureau et al 2010, and describing key methodological differences between the studies.
It is unclear to me if SL327 is injected every day before rotarod training, please clarify.
We apologize that this was not clear. Yes, SL327 was injected 30 minutes prior to training on
all 4 training days. We updated the results and figure legend to make this more clear.3
Comments related to Figure 2:
- The results are presented in a very synthetic form, using bar graph representations. For reasons of
clarity in the results but also consistency in the presentation between figures, it would be better to
represent individual data points.
We agree that this helps visualize the robustness of the results. We updated the plots in Fig. 2 to
include individual data points and are happy with the result.
- As noted by the authors, results in Figure 2C showed a potential trend for an effect of SL327 on the
stability of isolated spines. The number of animals used in this study is rather small compared to Ash et al
2020, which is directly comparable, and it remains possible that such a low number affects the results of
the statistical analysis. Such a result could prove important when stated that 30mg/kg of SL327 does not
affect wild-type animals. If possible, it would be good to add data points to more firmly conclude on an
absence of effect of SL327 in wild-type mice.
We agree that this is potentially an exciting finding worthy of follow-up and are glad that the reviewer
noticed it, and we also recognize that the results are not adequately powered to make a strong claim.
We are working to optimize both our method of plasticity induction and readout, as well as our
method for measuring and modulating Ras-ERK activity, as part of future work to explore these and
related questions, but this is part of a larger project and we feel is beyond the scope of the current
manuscript.
Discussion:
The authors state that “These observations forge a link between increased ERK signaling, increased
clustered spine consolidation, and enhanced learning phenotype in the MECP2-duplication mouse model
of syndromic autism”. The authors also state their “findings provide additional support to hypotheses
that learning phenotypes in autism arise from disrupted Ras-ERK signaling and synaptic plasticity”.
Enhanced learning is not a feature particularly associated with ASD and so it would be interesting to
develop on the nature of the link between enhanced learning and phenotypes associated with ASD. In
particular, it would be interesting to know to which category or subtype of ASD could benefit from
treatment with ERK inhibitors.
We have thought a good amount about these issues and are glad that the reviewer called attention to
them. We decided to include a slightly expanded discussion on these themes while recognizing that
they are somewhat speculative.
Minor criticisms:
1- The authors should include the individual data points on top of the bar graphs for Figure 2B-E.
Done
2- Do the authors have any information on the involvement of different spine types in the MECP2
duplication phenotype?
We first note here that in Jiang et al J Neuro 2013 subtle differences in the densities and
morphologies of different spine types were observed in somatosensory cortex of MECP2-
duplication mice. In pilot studies we attempted to separate spines by subtypes (mushroom,
stubby, thin/filopodia). We found that the filopodia are never stabilized, so we excluded them 4
from the analysis (as stated in the methods). In our hands we did not see prominent
differences in spine formation and stabilization between mushroom and stubby spines, so we
pooled these spine types to simplify the spine counting step (which is a very time-consuming
part of the analysis)
References
- Achilly NP, Wang W, Zoghbi HY (2021) Presymptomatic training mitigates functional deficits in a mouse model of Rett syndrome. Nature 592:596–595. 10.1038/s41586-021-03369-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso M, Medina JH, Pozzo-Miller L (2004) ERK1/2 activation is necessary for BDNF to increase dendritic spine density in hippocampal CA1 pyramidal neurons. Learn Mem 11:172–178. 10.1101/lm.67804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasaki C, Estep AL, Marais R, Rauen KA, Patton EE (2009) Kinase-activating and kinase-impaired cardio-facio-cutaneous syndrome alleles have activity during zebrafish development and are sensitive to small molecule inhibitors. Hum Mol Genet 18:2543–2554. 10.1093/hmg/ddp186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoine MW, Hübner CA, Arezzo JC, Hébert JM (2013) A causative link between inner ear defects and long-term striatal dysfunction. Science 341:1120–1123. 10.1126/science.1240405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asaka Y, Jugloff DGM, Zhang L, Eubanks JH, Fitzsimonds RM (2006) Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol Dis 21:217–227. 10.1016/j.nbd.2005.07.005 [DOI] [PubMed] [Google Scholar]
- Ash RT, Fahey PG, Park J, Zoghbi HY, Smirnakis SM (2018) Increased axonal bouton stability during learning in the mouse model of MECP2 duplication syndrome. eNeuro 5:ENEURO.0056-17.2018. 10.1523/ENEURO.0056-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash RT, Park J, Suter B, Smirnakis SM, Zoghbi HY (2021) Excessive formation and stabilization of dendritic spine clusters in the mecp2-duplication syndrome mouse model of autism. eNeuro 8:ENEURO.0282-20.2020. 10.1523/ENEURO.0282-20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD (1998) The MAPK cascade is required for mammalian associative learning. Nat Neurosci 1:602–609. 10.1038/2836 [DOI] [PubMed] [Google Scholar]
- Auerbach BD, Osterweil EK, Bear MF (2011) Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 480:63–68. 10.1038/nature10658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A, Kaphzan H, Alvarez-Dieppa AC, Murphy JP, Pierre P, Klann E (2012) Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron 76:325–337. 10.1016/j.neuron.2012.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges G, Miguelez C, Neto F, Mico JA, Ugedo L, Berrocoso E (2017) Activation of extracellular signal-regulated kinases (ERK 1/2) in the locus coeruleus contributes to pain-related anxiety in arthritic male rats. Int J Neuropsychopharmacol 20:463–475. 10.1093/ijnp/pyx005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeron T (2015) From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci 16:551–563. 10.1038/nrn3992 [DOI] [PubMed] [Google Scholar]
- Bureau G, Carrier M, Lebel M, Cyr M (2010) Intrastriatal inhibition of extracellular signal-regulated kinases impaired the consolidation phase of motor skill learning. Neurobiol Learn Mem 94:107–115. 10.1016/j.nlm.2010.04.008 [DOI] [PubMed] [Google Scholar]
- Chahrour M, Sung YJ, Shaw C, Zhou X, Wong STC, Qin J, Zoghbi HY (2008) MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320:1224–1229. 10.1126/science.1153252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng N, Alshammari F, Hughes E, Khanbabaei M, Rho JM (2017) Dendritic overgrowth and elevated ERK signaling during neonatal development in a mouse model of autism. PLoS One 12:e0179409. 10.1371/journal.pone.0179409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AL, Levenson JM, Vilaythong AP, Richman R, Armstrong DL, Noebels JL, Sweatt JD, Zoghbi HY (2004) Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet 13:2679–2689. 10.1093/hmg/ddh282 [DOI] [PubMed] [Google Scholar]
- Costa RM, Federov NB, Kogan JH, Murphy GG, Stern J, Ohno M, Kucherlapati R, Jacks T, Silva AJ (2002) Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature 415:526–530. 10.1038/nature711 [DOI] [PubMed] [Google Scholar]
- Ding Y, Won L, Britt JP, Lim SAO, McGehee DS, Kang UJ (2011) Enhanced striatal cholinergic neuronal activity mediates L-DOPA-induced dyskinesia in parkinsonian mice. Proc Natl Acad Sci USA 108:840–845. 10.1073/pnas.1006511108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, Greenberg ME (2013) Activity-dependent neuronal signalling and autism spectrum disorder. Nature 493:327–337. 10.1038/nature11860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etherton M, Földy C, Sharma M, Tabuchi K, Liu X, Shamloo M, Malenka RC, Südhof TC (2011) Autism-linked neuroligin-3 R451C mutation differentially alters hippocampal and cortical synaptic function. Proc Natl Acad Sci USA 108:13764–13769. 10.1073/pnas.1111093108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR (2000) Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28:41–51. 10.1016/s0896-6273(00)00084-2 [DOI] [PubMed] [Google Scholar]
- Frank AC, Huang S, Zhou M, Gdalyahu A, Kastellakis G, Silva TK, Lu E, Wen X, Poirazi P, Trachtenberg JT, Silva AJ (2018) Hotspots of dendritic spine turnover facilitate clustered spine addition and learning and memory. Nat Commun 9:422. 10.1038/s41467-017-02751-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu M, Yu X, Lu J, Zuo Y (2012) Repetitive motor learning induces coordinated formation of clustered dendritic spines in vivo. Nature 483:92–96. 10.1038/nature10844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao S, Tang B, Wu Z, Ure K, Sun Y, Tao H, Gao Y, Patel AJ, Curry DJ, Samaco RC, Zoghbi HY, Tang J (2015) Forniceal deep brain stimulation rescues hippocampal memory in Rett syndrome mice. Nature 526:430–434. 10.1038/nature15694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CD, Svoboda K (2007) Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature 450:1195–1200. 10.1038/nature06416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CD, Yasuda R, Zhong H, Svoboda K (2008) The spread of Ras activity triggered by activation of a single dendritic spine. Science 321:136–140. 10.1126/science.1159675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Wilbrecht L, Knott GW, Welker E, Svoboda K (2006) Experience-dependent and cell-type-specific spine growth in the neocortex. Nature 441:979–983. 10.1038/nature04783 [DOI] [PubMed] [Google Scholar]
- Holtmaat A, Bonhoeffer T, Chow DK, Chuckowree J, De Paola V, Hofer SB, Hübener M, Keck T, Knott G, Lee WCA, Mostany R, Mrsic-Flogel TD, Nedivi E, Portera-Cailliau C, Svoboda K, Trachtenberg JT, Wilbrecht L (2009) Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc 4:1128–1144. 10.1038/nprot.2009.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im HI, Hollander JA, Bali P, Kenny PJ (2010) MeCP2 controls BDNF expression and cocaine intake through homeostatic interactions with microRNA-212. Nat Neurosci 13:1120–1127. 10.1038/nn.2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Ash RT, Baker SA, Suter B, Ferguson A, Park J, Rudy J, Torsky SP, Chao HT, Zoghbi HY, Smirnakis SM (2013) Dendritic arborization and spine dynamics are abnormal in the mouse model of MECP2 duplication syndrome. J Neurosci 33:19518–19533. 10.1523/JNEUROSCI.1745-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott GW, Holtmaat A, Wilbrecht L, Welker E, Svoboda K (2006) Spine growth precedes synapse formation in the adult neocortex in vivo. Nat Neurosci 9:1117–1124. 10.1038/nn1747 [DOI] [PubMed] [Google Scholar]
- Kondo M, Gray LJ, Pelka GJ, Christodoulou J, Tam PPL, Hannan AJ (2008) Environmental enrichment ameliorates a motor coordination deficit in a mouse model of Rett syndrome - Mecp2 gene dosage effects and BDNF expression. Eur J Neurosci 27:3342–3350. 10.1111/j.1460-9568.2008.06305.x [DOI] [PubMed] [Google Scholar]
- Kwon HB, Sabatini BL (2011) Glutamate induces de novo growth of functional spines in developing cortex. Nature 474:100–104. 10.1038/nature09986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF (2006) Pten regulates neuronal arborization and social interaction in mice. Neuron 50:377–388. 10.1016/j.neuron.2006.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Ehninger D, Zhou M, Oh JY, Kang M, Kwak C, Ryu HH, Butz D, Araki T, Cai Y, Balaji J, Sano Y, Nam C, Kim HK, Kaang BK, Burger C, Neel BG, Silva AJ (2014) Mechanism and treatment for learning and memory deficits in mouse models of Noonan syndrome. Nat Neurosci 17:1736–1743. 10.1038/nn.3863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Pelletier MR, Perez Velazquez JL, Carlen PL (2002) Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol Cell Neurosci 19:138–151. 10.1006/mcne.2001.1085 [DOI] [PubMed] [Google Scholar]
- Lu H, Ash RT, He L, Kee SE, Wang W, Yu D, Hao S, Meng X, Ure K, Ito-Ishida A, Tang B, Sun Y, Ji D, Tang J, Arenkiel BR, Smirnakis SM, Zoghbi HY (2016) Loss and gain of MeCP2 cause similar hippocampal circuit dysfunction that is rescued by deep brain stimulation in a Rett syndrome mouse model. Neuron 91:739–747. 10.1016/j.neuron.2016.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyst MJ, Bird A (2015) Rett syndrome: a complex disorder with simple roots. Nat Rev Genet 16:261–274. 10.1038/nrg3897 [DOI] [PubMed] [Google Scholar]
- Makino H, Malinow R (2011) Compartmentalized versus global synaptic plasticity on dendrites controlled by experience. Neuron 72:1001–1011. 10.1016/j.neuron.2011.09.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellios N, Feldman DA, Sheridan SD, Ip JPK, Kwok S, Amoah SK, Rosen B, Rodriguez BA, Crawford B, Swaminathan R, Chou S, Li Y, Ziats M, Ernst C, Jaenisch R, Haggarty SJ, Sur M (2018) MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol Psychiatry 23:1051–1065. 10.1038/mp.2017.86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra I, Lavillaureix A, Yeh E, Traglia M, Tsang K, Bearden CE, Rauen KA, Weiss LA (2017) Reverse pathway genetic approach identifies epistasis in autism spectrum disorders. PLoS Genet 13:e1006516. 10.1371/journal.pgen.1006516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau MM, Pietropaolo S, Ezan J, Robert BJA, Miraux S, Maître M, Cho Y, Crusio WE, Montcouquiol M, Sans N (2020) Scribble controls social behaviors through the regulation of the ERK/Mnk1 pathway. bioRxiv 2020.09.10.289397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostany R, Portera-Cailliau C (2008) A craniotomy surgery procedure for chronic brain imaging. J Vis Exp (12):680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostany R, Portera-Cailliau C (2011) Absence of large-scale dendritic plasticity of layer 5 pyramidal neurons in peri-infarct cortex. J Neurosci 31:1734–1738. 10.1523/JNEUROSCI.4386-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins CA (2020) Persistent behavior in a mouse model of autism: role of L-type calcium channels and striatum. Diss Abstr Int Sect B Sci Eng 77:816–822. [Google Scholar]
- Na ES, Nelson ED, Adachi M, Autry AE, Mahgoub MA, Kavalali ET, Monteggia LM (2012) A mouse model for MeCP2 duplication syndrome: MeCP2 overexpression impairs learning and memory and synaptic transmission. J Neurosci 32:3109–3117. 10.1523/JNEUROSCI.6000-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na ES, Morris MJ, Nelson ED, Monteggia LM (2014) GABAA a receptor antagonism ameliorates behavioral and synaptic impairments associated with MeCP2 overexpression. Neuropsychopharmacology 39:1946–1954. 10.1038/npp.2014.43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani J, Tamada K, Hatanaka F, Ise S, Ohta H, Inoue K, Tomonaga S, Watanabe Y, Chung YJ, Banerjee R, Iwamoto K, Kato T, Okazawa M, Yamauchi K, Tanda K, Takao K, Miyakawa T, Bradley A, Takumi T (2009) Abnormal behavior in a chromosome- engineered mouse model for human 15q11-13 duplication seen in autism. Cell 137:1235–1246. 10.1016/j.cell.2009.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niculescu D, Michaelsen-Preusse K, Güner Ü, van Dorland R, Wierenga CJ, Lohmann C (2018) A BDNF-mediated push-pull plasticity mechanism for synaptic clustering. Cell Rep 24:2063–2074. 10.1016/j.celrep.2018.07.073 [DOI] [PubMed] [Google Scholar]
- Padmashri R, Reiner BC, Suresh A, Spartz E, Dunaevsky A (2013) Altered structural and functional synaptic plasticity with motor skill learning in a mouse model of fragile X syndrome. J Neurosci 33:19715–19723. 10.1523/JNEUROSCI.2514-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson MA, Szatmari EM, Yasuda R (2010) AMPA receptors are exocytosed in stimulated spines and adjacent dendrites in a Ras-ERK-dependent manner during long-term potentiation. Proc Natl Acad Sci USA 107:15951–15956. 10.1073/pnas.0913875107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peñagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, Sonnenblick LI, Gruver R, Almajano J, Bragin A, Golshani P, Trachtenberg JT, Peles E, Geschwind DH (2011) Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 147:235–246. 10.1016/j.cell.2011.08.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piochon C, Kloth AD, Grasselli G, Titley HK, Nakayama H, Hashimoto K, Wan V, Simmons DH, Eissa T, Nakatani J, Cherskov A, Miyazaki T, Watanabe M, Takumi T, Kano M, Wang SSH, Hansel C (2014) Cerebellar plasticity and motor learning deficits in a copy-number variation mouse model of autism. Nat Commun 5:5586. 10.1038/ncomms6586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher MR, Herrera JA, Buffington SA, Kochukov MY, Merritt JK, Fisher AR, Schanen NC, Costa-Mattioli M, Neul JL (2015) Rett syndrome like phenotypes in the R255X Mecp2 mutant mouse are rescued by MECP2 transgene. Hum Mol Genet 24:2662–2672. 10.1093/hmg/ddv030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucilowska J, Vithayathil J, Tavares EJ, Kelly C, Colleen Karlo J, Landreth GE (2015) The 16p11.2 deletion mouse model of autism exhibits altered cortical progenitor proliferation and brain cytoarchitecture linked to the ERK MAPK pathway. J Neurosci 35:3190–3200. 10.1523/JNEUROSCI.4864-13.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucilowska J, Vithayathil J, Pagani M, Kelly C, Karlo JC, Robol C, Morella I, Gozzi A, Brambilla R, Landreth GE (2018) Pharmacological inhibition of ERK signaling rescues pathophysiology and behavioral phenotype associated with 16p11.2 chromosomal deletion in mice. J Neurosci 38:6640–6652. 10.1523/JNEUROSCI.0515-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramocki MB, Zoghbi HY (2008) Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature 455:912–918. 10.1038/nature07457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramocki MB, Peters SU, Tavyev YJ, Zhang F, Carvalho CMB, Schaaf CP, Richman R, Fang P, Glaze DG, Lupski JR, Zoghbi HY (2009) Autism and other neuropsychiatric symptoms are prevalent in individuals with MECP2 duplication syndrome. Ann Neurol 66:771–782. 10.1002/ana.21715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramocki MB, Tavyev YJ, Peters SU (2010) The MECP2 duplication syndrome. Am J Med Genet A 152A:1079–1088. 10.1002/ajmg.a.33184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner BC, Dunaevsky A (2015) Deficit in motor training-induced clustering, but not stabilization, of new dendritic spines in fmr1 knock-out mice. PLoS One 10:e0126572. 10.1371/journal.pone.0126572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosina E, Battan B, Siracusano M, Di Criscio L, Hollis F, Pacini L, Curatolo P, Bagni C (2019) Disruption of mTOR and MAPK pathways correlates with severity in idiopathic autism. Transl Psychiatry 9:50. 10.1038/s41398-018-0335-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell PE, Fuccillo MV, Maxeiner S, Hayton SJ, Gokce O, Lim BK, Fowler SC, Malenka RC, Südhof TC (2014) Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell 158:198–212. 10.1016/j.cell.2014.04.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbaugh G, Adams JP, Kim JH, Huganir RL (2006) SynGAP regulates synaptic strength and mitogen-activated protein kinases in cultured neurons. Proc Natl Acad Sci USA 103:4344–4351. 10.1073/pnas.0600084103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu HH, Lee YS (2016) Cell type-specific roles of RAS-MAPK signaling in learning and memory: implications in neurodevelopmental disorders. Neurobiol Learn Mem 135:13–21. [DOI] [PubMed] [Google Scholar]
- Sandweiss AJ, Brandt VL, Zoghbi HY (2020) Advances in understanding of Rett syndrome and MECP2 duplication syndrome: prospects for future therapies. Lancet Neurol 19:689–698. 10.1016/S1474-4422(20)30217-9 [DOI] [PubMed] [Google Scholar]
- Schreiber J, Grimbergen LA, Overwater I, Van Der Vaart T, Stedehouder J, Schuhmacher AJ, Guerra C, Kushner SA, Jaarsma D, Elgersma Y (2017) Mechanisms underlying cognitive deficits in a mouse model for Costello syndrome are distinct from other RASopathy mouse models. Sci Rep 7:1256. 10.1038/s41598-017-01218-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seese RR, Maske AR, Lynch G, Gall CM (2014) Long-term memory deficits are associated with elevated synaptic ERK1/2 activation and reversed by mGluR5 antagonism in an animal model of autism. Neuropsychopharmacology 39:1664–1673. 10.1038/npp.2014.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soong HK, Markham JA, Weiler IJ, Greenough WT (2008) Aberrant early-phase ERK inactivation impedes neuronal function in fragile X syndrome. Proc Natl Acad Sci USA 105:4429–4434. 10.1073/pnas.0800257105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stornetta RL, Zhu JJ (2011) Ras and Rap signaling in synaptic plasticity and mental disorders. Neuroscientist 17:54–78. 10.1177/1073858410365562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztainberg Y, Zoghbi HY (2016) Lessons learned from studying syndromic autism spectrum disorders. Nat Neurosci 19:1408–1417. 10.1038/nn.4420 [DOI] [PubMed] [Google Scholar]
- Sztainberg Y, Chen HM, Swann JW, Hao S, Tang B, Wu Z, Tang J, Wan YW, Liu Z, Rigo F, Zoghbi HY (2015) Reversal of phenotypes in MECP2 duplication mice using genetic rescue or antisense oligonucleotides. Nature 528:123–126. 10.1038/nature16159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennant KA, Adkins DL, Donlan NA, Asay AL, Thomas N, Kleim JA, Jones TA (2011) The organization of the forelimb representation of the C57BL/6 mouse motor cortex as defined by intracortical microstimulation and cytoarchitecture. Cereb Cortex 21:865–876. 10.1093/cercor/bhq159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Woerden GM, Harris KD, Hojjati MR, Gustin RM, Qiu S, De Avila Freire R, Jiang YH, Elgersma Y, Weeber EJ (2007) Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat Neurosci 10:280–282. 10.1038/nn1845 [DOI] [PubMed] [Google Scholar]
- Vithayathil J, Pucilowska J, Landreth (2018) GE ERK/MAPK signaling and autism spectrum disorders. Prog Brain Res 241:63–112. [DOI] [PubMed] [Google Scholar]
- Wang X, Snape M, Klann E, Stone JG, Singh A, Petersen RB, Castellani RJ, Casadesus G, Smith MA, Zhu X (2012) Activation of the extracellular signal-regulated kinase pathway contributes to the behavioral deficit of fragile x-syndrome. J Neurochem 121:672–679. 10.1111/j.1471-4159.2012.07722.x [DOI] [PubMed] [Google Scholar]
- Wen Y, Alshikho MJ, Herbert MR (2016) Pathway network analyses for autism reveal multisystem involvement, major overlaps with other diseases and convergence upon MAPK and calcium signaling. PLoS One 11:e0153329. 10.1371/journal.pone.0153329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FM, Wolf-Yadlin A (2016) Methods for the analysis of protein phosphorylation-mediated cellular signaling networks. Annu Rev Anal Chem (Palo Alto Calif) 9:295–315. 10.1146/annurev-anchem-071015-041542 [DOI] [PubMed] [Google Scholar]
- Yang G, Pan F, Gan WB (2009) Stably maintained dendritic spines are associated with lifelong memories. Nature 462:920–924. 10.1038/nature08577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Carew TJ (2010) Small G protein signaling in neuronal plasticity and memory formation: the specific role of Ras family proteins. Neuron 68:340–361. 10.1016/j.neuron.2010.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Lin A, Chang P, Gan WB (2005) Development of long-term dendritic spine stability in diverse regions of cerebral cortex. Neuron 46:181–189. 10.1016/j.neuron.2005.04.001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Example full-length Western blottings, related to Figure 1. A, Example full-length Western blottings relevant to Figure 1B, for p-ERK (T202/204), total ERK, and β-actin. B, Example full-length Western blottings relevant to Figure 1D, showing immunoblots to p-ERK (T202/204), total ERK, and β-actin. Note SL-327 suppresses the level of p-ERK in mutant animals. Download Figure 1-1, TIF file (1.7MB, tif) .