

Abstract
Objective
The kainic acid (KA)‐induced status epilepticus (SE) model in rats is a well‐defined model of epileptogenesis. This model closely recapitulates many of the clinical and pathological characteristics of human temporal lobe epilepsy (TLE) that arise following SE or another neurological insult. Spontaneous recurrent seizures (SRS) in TLE can present after a latent period following a neurological insult (traumatic brain injury, SE event, viral infection, etc.). Moreover, this model is suitable for preclinical studies to evaluate the long‐term process of epileptogenesis and screen putative disease‐modifying/antiepileptogenic agents. The burden of human TLE is highly variable, similar to the post‐KA SE rat model. In this regard, this model may have broad translational relevance. This report thus details the pharmacological characterization and methodological refinement of a moderate‐throughput drug screening program using the post‐KA‐induced SE model of epileptogenesis in male Sprague Dawley rats to identify potential agents that may prevent or modify the burden of SRS. Specifically, we sought to demonstrate whether our protocol could prevent the development of SRS or lead to a reduced frequency/severity of SRS.
Methods
Rats were administered either everolimus (2–3 mg/kg po) beginning 1, 2, or 24 h after SE onset, or phenobarbital (60 mg/kg ip) beginning 1 h after SE onset. All treatments were administered once/day for 5–7 days. Rats in all studies (n = 12/treatment dose/study) were then monitored intermittently by video‐electroencephalography (2 weeks on, 2 weeks off, 2 weeks on epochs) to determine latency to onset of SRS and disease burden.
Results
Although no adverse side effects were observed in our studies, no treatment significantly modified disease or prevented the presentation of SRS by 6 weeks after SE onset.
Significance
Neither phenobarbital nor everolimus administered at several time points after SE onset prevented the development of SRS. Nonetheless, we demonstrate a practical and moderate‐throughput screen for potential antiepileptogenic agents in a rat model of TLE.
Keywords: antiepileptogenesis, EEG, everolimus, kainic acid, phenobarbital
Key Points.
Disease‐modifying therapies are needed to prevent or attenuate the burden of epilepsy in at‐risk individuals
We report a moderate‐throughput screening protocol to identify disease‐modifying agents in a rat post‐kainic acid status epilepticus model
Everolimus was administered at multiple time points post‐status epilepticus with no effect on spontaneous seizures up to 6 weeks later
Repeated administration of phenobarbital also did not prevent the development of spontaneous recurrent seizures up to 6 weeks post‐SE
Although we did not identify any effect of either agent, our approach provides a moderate‐throughput screen for antiepileptogenesis in rats
1. INTRODUCTION
Despite over 30 antiseizure drugs (ASDs) available for the treatment of symptomatic seizures, no therapy is yet approved to prevent epilepsy in at‐risk individuals,1, 2 underscoring the need for disease‐modifying and/or antiepileptogenic agents. As recommended by recent National Institute of Neurological Disorders and Stroke (NINDS) Advisory Council Working Group reviews of the long‐standing Epilepsy Therapy Screening Program (ETSP),3, 4 the program refocused its screening workflow not only to evaluate compounds that may treat the symptomatic seizures of drug‐resistant epilepsy, but also to identify agents that may potentially modify or altogether prevent the development of epilepsy. Under the NINDS ETSP’s prime contract (HHSN271201600048C), the University of Utah has subcontracted the University of Washington (UW) to evaluate the potential of putative disease‐modifying agents in a post‐status epilepticus (SE) model of temporal lobe epilepsy (TLE) in rats in an unbiased and blinded manner. One of the strengths of this subcontracted approach is that investigational compounds are subjected to evaluation in a blinded manner by a large scientific team of experienced epilepsy investigators at the NINDS, University of Utah, and UW. Furthermore, this strategy employs an in‐life testing protocol that replicates treatment paradigms in patients at risk for acquired epilepsy.
Spontaneous recurrent seizures (SRS) in clinical TLE are often focal impaired awareness seizures that generalize to tonic–clonic seizures and develop after a latent period following a neurological insult, including SE.5 The rat systemic kainic acid (KA)‐induced SE model of TLE is a technically feasible and well‐characterized preclinical model that is defined by SRS onset within 0–2 weeks following a latent period.6, 7, 8 Post‐KA SE rats exhibit marked reactive gliosis,9 neuroinflammation,10 and behavioral deficits11 days to weeks after SE insult. As a result of the pathophysiological and phenotypic similarities to clinical TLE, as well as the sensitivity of SRS to available ASDs in this model,5, 12, 13 the KA‐induced SE paradigm in rats is well suited for moderate‐throughput drug discovery to screen the disease‐modifying potential of investigational agents on the development and severity of SRS.6, 14
This study aimed to establish and validate an in‐life disease modification screen using the repeated administration of the ASD phenobarbital (PB) or the immunomodulator everolimus (EVL) to rats immediately following the induction of SE. The primary study objective was to rigorously evaluate the extent to which pharmacological intervention after SE induction would modify the severity and/or frequency of SRS up to 6 weeks later. This study describes the approach used to assess the antiepileptogenic potential of two investigational agents and several interventional time points in an effort to iteratively refine a disease modification screening protocol for the NINDS ETSP. Of note, this study was conducted in a blinded manner, with the University of Utah sequentially submitting each compound to investigators at UW as an unidentified compound for independent evaluation at the UW facility. PB was selected because previous studies have suggested the potential to prevent the development of traumatic brain injury‐induced epilepsy in at‐risk patients.15, 16, 17 EVL was selected because it is approved as a disease‐modifying treatment for tuberous sclerosis complex (TSC),18, 19 a condition characterized by chronic seizures and epilepsy.20, 21 EVL is also a rapamycin derivative (rapalog) that inhibits mammalian target of rapamycin (mTOR) activation.22 It has been hypothesized that mTOR inhibition may exert antiepileptogenic effects in epilepsy more broadly.23, 24 Furthermore, repeated administration of rapamycin commencing 24 h after KA‐SE has been previously shown to attenuate the burden of SRS and neuropathology in rats.25 Using a refined and blinded protocol, we observed that neither PB nor escalating doses of EVL administered at discrete time points significantly prevented the development of SRS in our hands. Despite the lack of disease‐modifying effect with either agent using multiple study protocols, we herein establish proof of concept and feasibility of the NINDS‐supported protocol conducted in the post‐KA SE rat model of acquired epilepsy to potentially identify disease‐modifying agents in a moderate‐throughput, rigorous, and unbiased manner.
2. MATERIALS AND METHODS
An abbreviated methods section is included herein to facilitate study comprehension. However, detailed methods of more standardized procedures (i.e., surgical electroencephalographic [EEG] implants, drug formulation, statistics, etc.) are included in the Supplemental Files, as well as described in detail in previously published studies.5, 7, 26, 27
2.1. Animals
All animal experimentation was approved by the UW institutional animal care and use committee. Male CD IGS Sprague Dawley rats (150–200 g; Charles River Laboratories) were housed 5/cage for 1 week to acclimate to the vivarium. After EEG implantation (described in Supplemental Methods), rats were housed individually in custom plexiglass cages with corncob bedding, in a temperature‐controlled vivarium on a 14:10 light/dark cycle. Animals were permitted ad libitum access to irradiated chow (Picolab 5053), filtered water, and enrichment (Nylabones and cardboard tubes). Rats were given a minimum of 24 h to acclimate to the EEG recording suite prior to all experimentation. Rats were euthanized by CO2 asphyxiation at completion of all in‐life studies, in a manner consistent with American Veterinary Medical Association guidelines.28
To accommodate a target video‐EEG (vEEG) monitoring group size of n = 12 rats/treatment group (vehicle [VEH] or investigational compound) and potential for SE‐induced mortality, a total of 34 animals were surgically implanted 1 week prior to KA‐SE for each study (Figure S1). KA administration for SE induction is described in detail in the Supplemental Methods and previously published studies.5, 7, 26, 27 Each study was divided into two cohorts of randomly assigned rats (n = 17/cohort). This allowed for potential KA‐SE mortality of up to 12%–18% (final cohort size of n = 14–15 rats); rats that survived the SE insult were then candidates for drug intervention at the relevant time point (Figure S1). Any rat that lost more than 20% of pre‐SE body weight within 7 days of SE insult and during the investigational drug or VEH administration period was removed from the study.
2.2. Investigational compound and formulation VEHs
The investigational compound, PB (Sigma‐Aldrich catalogue #P1636), was formulated for repeated administration in .9% saline (Day 1) or 40% hydroxy‐propyl beta‐cyclodextrin (Day 2–5; Sigma‐Aldrich catalogue #H107), consistent with previously published methods.29, 30 EVL (MedChem Express #HY‐10218) was initially dissolved in 100% EtOH as a 10‐mg/ml stock solution and stored at −20°C. No solubility or formulation issues were noted with any preparation. For each treatment day, EVL stock was then freshly diluted into a 1‐mg/ml dosing solution with a VEH of 5% PEG‐400 (Sigma‐Aldrich catalogue #06855)/5% Tween‐80 (Sigma‐Aldrich catalogue #P‐1754) in .9% saline. Because there were different formulation VEHs for PB and EVL, it was necessary to have an independent VEH‐treated KA‐SE control group for each investigational compound.
Each investigational compound was administered for 5 (PB and EVL 3 mg/kg) or 7 (EVL 2 mg/kg) consecutive days after SE onset (detailed study protocol is described in Figure 1). For each study, animals were randomized to their treatment group based on time of SE onset. Investigators were blinded to treatment group (n = 12/treatment group). PB (60 mg/kg) or its VEH was administered by the intraperitoneal route on Day 1, and then by the subcutaneous route on Days 2–5 post‐SE to replicate the previously published administration protocol of Sutula and colleagues.31 PB or VEH administration was started 1 h after onset of behavioral SE, with SE confirmed by sustained electrographic spiking. The dose and time of administration of PB were based on prior studies demonstrating no disease‐modifying effect of PB when administered more than 30 min after SE onset.32 EVL (2 mg/kg) or its VEH was administered by the oral route for 7 days beginning 24 h after onset of behavioral and electrographic SE (Figure 1). EVL (3 mg/kg) or its VEH was administered orally for 5 days beginning 2 h after SE onset (Figure 1). The doses of EVL and time points of administration were selected based on prior studies suggesting that rapamycin, an agent with limited brain bioavailability, could modify disease severity in a post‐traumatic brain injury model of epileptogenesis in mice.33 No other pharmacological intervention was administered during the post‐KA SE period or up to 42 days postinsult.
FIGURE 1.
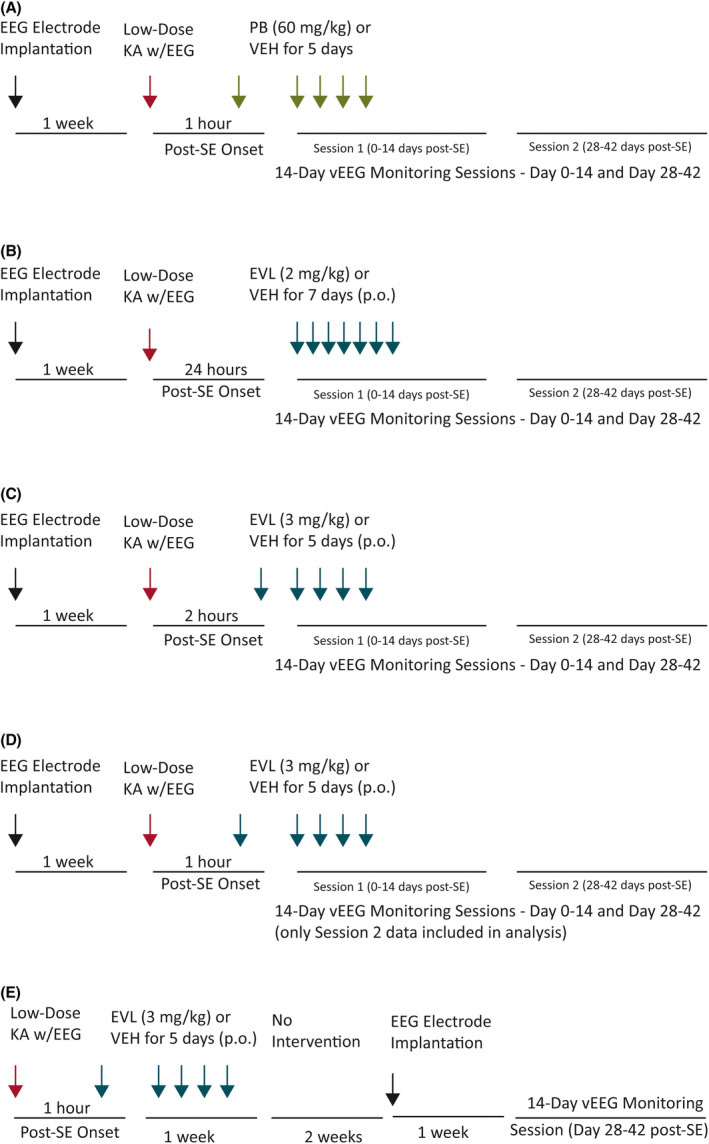
(A–C) Original experimental protocol for the evaluation of the disease‐modifying potential of (A) phenobarbital (PB; 60 mg/kg ip), (B) everolimus (EVL; 2 mg/kg po), or (C) EVL (3 mg/kg po) in male Sprague Dawley rats. (D, E) Upon further refinement, the original protocol (D) was compared in a head‐to‐head study with an (E) expedited protocol to determine whether EVL (3 mg/kg po) would confer any disease‐modifying effects against spontaneous recurrent seizure (SRS) severity or onset up to 6 weeks later. The original protocol design (A–D) included rats that were surgically implanted under ketamine/xylazine anesthesia 7–10 days prior to repeated low‐dose administration of kainic acid (KA) to induce status epilepticus (SE) confirmed by video‐electroencephalography (vEEG) and sustained Racine Stage 4/5 seizures for at least 30 min. Animals were randomly enrolled into either drug or respective vehicle (VEH) treatment groups and administered the first injection at 1 h (A, D, E), 2 h (B), or 24 h (C) after SE induction. Rats were then intermittently monitored by continuous vEEG recording from 0 to 2 weeks and from 4 to 6 weeks after SE onset to determine latency to onset of SRS. In the expedited protocol (E), rats were implanted with EEG electrodes 7 days after KA‐SE and then only monitored from 4 to 6 weeks after SE onset. In all studies, each investigational compound or VEH was administered by an experimenter blinded to treatment condition. Rats that survived the SE insult and investigational drug intervention period were monitored for the presence of spontaneous behavioral and electrographic seizures (or spontaneous behavioral seizures with electrographic correlate) up to 42 days after SE insult. vEEG‐observed events were scored off‐line by a trained investigator and confirmed by a secondary investigator, both of whom were blinded to experimental condition in all studies
3. RESULTS
3.1. Refinement of testing protocol
The primary goal of this study was to rigorously assess the feasibility of a moderate‐throughput disease‐modifying or antiepileptogenic agent screening protocol in a well‐characterized rat model of TLE. Thus, several time points of drug administration after the onset of sustained SE were iteratively evaluated (Figure 1) to ascertain the ability of PB (repeated injections beginning at a single time point) or EVL (repeated injections beginning at multiple time points after SE) to modify the onset and/or severity of SRS up to 6 weeks later. Using a sequential testing strategy over the course of 2 years, we attempted to continuously refine our approach. PB was selected as a positive control, and thus only one time point was evaluated, with administration commencing 1 h after SE onset. EVL was selected as a positive control, and thus administration commenced at three time points: 1, 2, and 24 h after SE onset (Figure 1). In an effort to further refine our screening protocol, we then developed an expedited protocol and assessed the efficacy of EVL in a head‐to‐head comparison study. The “original” testing protocol planned for 24/7 vEEG monitoring for 0–14 days and 28–42 days post‐SE; the “expedited” testing protocol planned for 24/7 vEEG monitoring only from 28–42 days post‐SE (Figure 1E). This expedited protocol resulted in significant cost savings (or efficiency), as we were able to focus our resources on those animals that survived the initial SE; that is, stereotaxic EEG implant surgery was not conducted prior to SE induction, but 3 weeks after SE induction (see Figure 2 for details of different protocols).
FIGURE 2.

Recording session summary of the number of seizures and seizure burden during each 2‐week recording epoch in the original protocol (two 2‐week recording sessions). For seizure burden, data were analyzed separately for each 2‐week monitoring session by Mann–Whitney U test, but presented on a single graph for illustration purposes. (A, B) There was no effect of phenobarbital (PB) on (A) number of seizures during each recording session (0–2 weeks and 4–6 weeks after status epilepticus [SE]) or (B) average seizure burden. (C, D) There was no effect of everolimus (EVL; 2 mg/kg) on (C) the number of seizures during each recording session (0–2 weeks and 4–6 weeks post‐SE) or (D) average seizure burden. (E, F) There was no effect of EVL (3 mg/kg) on (E) the number of seizures during each recording session (0–2 weeks and 4–6 weeks post‐SE) or (F) average seizure burden. VEH, vehicle
3.2. Therapeutic intervention immediately after KA‐induced SE did not significantly affect body weight change
KA‐induced SE is typically associated with some body weight loss as a result of the unremitting seizure activity. Body weight was not significantly affected by a time × treatment interaction during the recovery period 4–7 days after SE insult in any of the original testing protocol studies of this investigation. Specifically, repeated PB treatment did not affect body weight change versus that of VEH‐treated post‐KA SE rats (Figure S1A; F 5, 86 = 143.0, p < .0001). Additionally, EVL at either 2 mg/kg po (Figure S1B; F 7, 140 = .2995, p < .0001) or 3 mg/kg po (Figure S1C; F 7, 126 = 3.320, p = .0028) did not significantly affect body weight change versus matched VEH‐treated rats. In the blinded head‐to‐head comparison of the original versus expedited protocol assessment of EVL (3 mg/kg po), EVL treatment did significantly affect body weight following SE (Figure S2). Notably, EVL administration after EEG implantation was not associated with a significant time × treatment interaction (original protocol, Figure S2A; F 4, 104 = 1.587, p > .18). However, with EVL administration prior to EEG implantation (expedited protocol, Figure S2B; F 4, 108 = 4.241, p = .0032), there was a significant effect of EVL treatment. Specifically, EVL lead to a reduction in body weight at Day 5 post‐SE. Thus, treatment with PB and EVL for the 4–7 days following KA‐induced SE was generally well tolerated, and none of the investigational drug treatments led to any notable worsening or improvement in body weight change versus their respective VEH treatments when administered after the EEG probe implantation (original protocol). However, administration of 3 mg/kg EVL prior to EEG implantation (expedited protocol) was associated with reduced body weight by Day 5 post‐SE.
3.3. Therapeutic intervention did not significantly reduce the number of seizures or seizure burden
Following the KA‐induced SE insult, all rats in the study developed SRS within 6 weeks. Neither PB nor EVL (in any study) significantly reduced the number of seizures that occurred during each of the 2‐week monitoring sessions, and there was no effect of treatment on the seizure burden (i.e., modified Racine stage seizure severity × frequency of Racine stage seizures) during these monitoring sessions. Specifically, there was a significant main effect of time on the total number of observed seizures for all studies, but no main effect of treatment or treatment × time interaction (Figure 2). Both PB‐ and VEH‐treated rats experienced a time‐dependent increase in the total number of SRS (Figure 2A; F 1, 22 = 7.968, p = .0099). There was no significant difference in the seizure burden (i.e., seizure stage × frequency of events) between PB‐ and VEH‐treated rats at either time point (Figure 2B; 0–2 h: U = 53.5, p = .204; 4–6 h: U = 70, p = .9205). EVL‐treated (2 mg/kg) and VEH‐treated rats also experienced a greater number of SRS across time after SE insult (Figure 2C; F 1, 17 = 6.626, p = .0197). There was no significant difference in the seizure burden between EVL‐treated (2 mg/kg) and VEH‐treated rats at either time point (Figure 2D; 0–2 h: U = 53.5, p = .204; 4–6 h: U = 70, p = .9205). Finally, both EVL‐treated (3 mg/kg) and VEH‐treated rats experienced a greater number of SRS (Figure 2E; F 1, 22 = 13.02, p = .0016) with time post‐SE. There was no significant difference in the seizure burden between EVL‐treated (3 mg/kg) and VEH‐treated rats at either time point (Figure 2F; 0–2 h: U = 65, p = .4783; 4–6 h: U = 53, p = .2818). Thus, neither PB nor EVL administration, whether started at 1, 2, or 24 h post‐SE for 5‐ or 7‐day duration, conferred significant effect on either number of seizures or seizure burden in this rat TLE model.
Finally, we attempted to further refine our screening protocol in rats and thus performed a head‐to‐head study of EVL (3 mg/kg) administered in the original versus expedited protocols (Figure 3). That protocol refinement demonstrated that there was no significant effect of protocol design on the number of seizures (F 1, 39 = .08438, p = .77) in either treatment group. There was no effect of treatment on seizure burden in the 4–6‐week post‐SE monitoring period in either protocol design (expedited: U = 45.5, p = .5233; original: U = 59, p = .9603). The expedited protocol would thus be able to detect seizure events to a similar extent to that of the original protocol, at a fraction of the resource demands.
FIGURE 3.

Recording session summary of the number of seizures and seizure burden during the 4–6 week recording epoch in the original (two 2‐week recording sessions) versus expedited protocol design. There was no effect of everolimus (EVL) on (A) number of seizures during the 4–6 weeks post‐status epilepticus (SE) recording session or (B) average seizure burden. Sz, seizure; VEH, vehicle
3.4. Therapeutic interventions investigated did not significantly alter cumulative seizure burden up to 6 weeks post‐SE
This screening protocol was not designed to precisely determine the timing of SRS onset. Nonetheless, it is helpful to ascertain the extent to which an intervention can alter disease trajectory over time. Cumulative seizure burden is an indirect measure of disease trajectory over time and includes the cumulative seizure severity × seizure frequency across all the observation days.34 PB administration did not significantly reduce the cumulative seizure burden versus VEH‐treated post‐SE rats (Figure 4A). Similarly, EVL (2 mg/kg) treatment beginning 24 h after SE onset did not alter the cumulative seizure burden versus VEH‐treated control rats (Figure 4C). Lastly, a higher dose of EVL (3 mg/kg) administered earlier after SE onset (2 h; Figure 1C) did not significantly alter the cumulative seizure burden versus VEH‐treated control rats (Figure 4E). In the head‐to‐head study of the expedited protocol, administration of EVL did not alter cumulative seizure burden in either study design (Figure 4G,I). Thus, cumulative seizure burden was not significantly affected by any treatment versus the VEH‐treated KA‐SE rats within the same study group in this particular experimental design.
FIGURE 4.
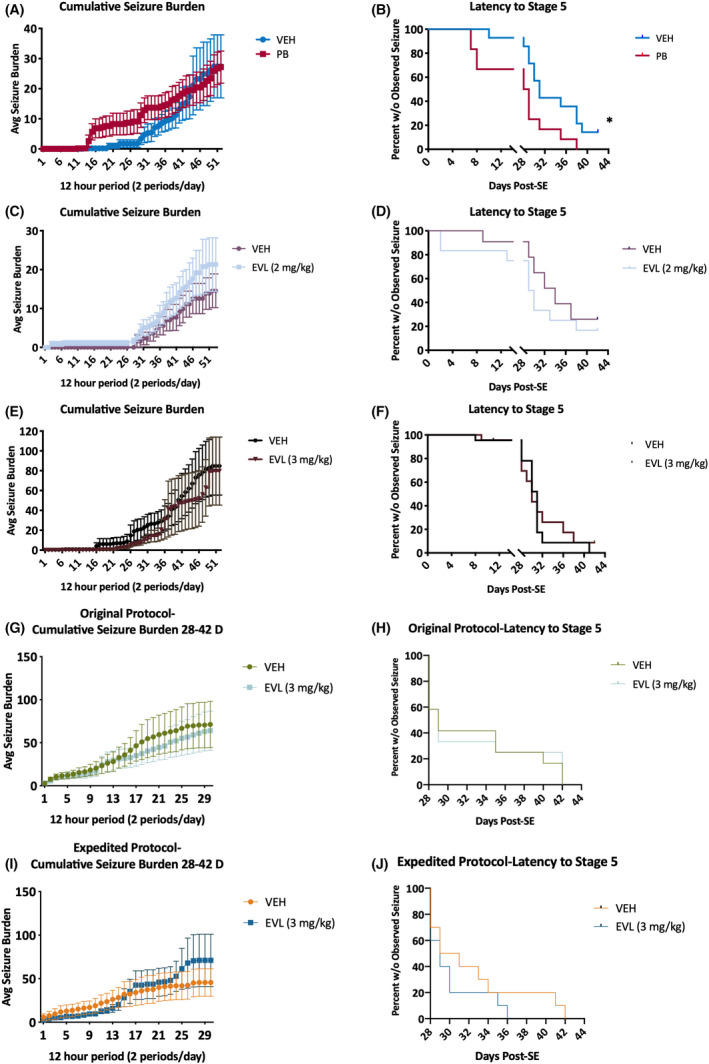
Cumulative seizure burden is the summation of all observed events during each of the two 2‐week‐long recording sessions from 0 to 2 and from 4 to 6 weeks post‐status epilepticus (SE; original protocol: A, C, E). (A, C, E) There was no significant effect of any investigational treatment: (A) phenobarbital (PB), (C) everolimus (EVL; 2 mg/kg), or (E) EVL (3 mg/kg). (B, D, F) The latency to Stage 5 seizures was not significantly improved by any treatment condition for all rats up to 42 days post‐SE in the original protocol. (A) Administration of PB led to a significant reduction in the time until first observed Racine Stage 5 seizure during the two 2‐week‐long monitoring sessions (χ2 = 5.691, *p = .0171). Although it is possible that the intermittent monitoring may have missed seizures that occurred during the 2–4 weeks post‐SE using our original protocol, this analysis was conducted to further characterize disease progression in this novel drug screening model. (G–J) In the head‐to‐head evaluation of EVL efficacy in the original versus expedited protocol, there was also no effect of EVL administration on cumulative seizure burden (G, I) nor on latency to Stage 5 seizures (H, J). This head‐to‐head study demonstrates that the expedited protocol is valid for moderate‐throughput disease modification studies and can demonstrate disease‐modifying potential (or lack thereof) to an equivalent degree as the original, longer duration monitoring study of the original protocol. VEH, vehicle
3.5. Latency to Stage 5 seizures was significantly reduced by PB treatment
The latency to the first observed spontaneously occurring Racine Stage 5 seizure during the vEEG recording sessions was assessed to determine whether any intervention could effectively delay the onset of severe generalized seizures, an alternative metric of disease burden or disease course. Specifically, a compound that delays the time to onset of Stage 5 seizures could suggest a disease‐modifying effect. Although it is possible that the intermittent sampling occurring 0–2 and 4–6 weeks after SE onset (as used in the original study design) may have missed some seizures in the 2–4 weeks post‐SE period, we did include this evaluation metric in an attempt to better characterize disease progression in our drug screening paradigm. Additionally, the expedited study design was similarly limited to only a 4–6 weeks post‐SE session, but used to screen for potential agents that may effectively modify SRS severity or frequency. We herein demonstrate that PB, when administered repeatedly beginning 1 h after SE onset and then four additional times at 24‐h intervals for a total of 5 treatment days, led to a significant reduction in the time until first observed Racine Stage 5 seizures (Figure 4B; χ2 = 5.691, p = .0171); that is, PB‐treated rats presented with Stage 5 seizures earlier than VEH‐treated rats. No dose or time point of EVL administration (Figure 4) led to any significant deviations in the latency to Stage 5 seizures versus the respective VEH‐treated control cohort (Figure 4D, χ2 = 2.280, p = .131; Figure 4F, χ2 = .1155, p = .734). This also includes the head‐to‐head evaluation of EVL in the original versus expedited protocol (original protocol: Figure 4H, χ2 = .0009090, p > .97; expedited protocol: Figure 4J, χ2 = .5971, p > .44). Thus, EVL treatment did not significantly increase or decrease the time to onset of Stage 5 seizures.
4. DISCUSSION
The NINDS ETSP prioritized the identification of agents to treat symptomatic seizures of drug‐resistant epilepsy and of agents that may potentially prevent the development of epileptogenesis or attenuate disease burden in people with epilpesy.3, 4 Our present study established the feasibility and suitability of our screen using two agents administered by four different treatment designs to formally, rigorously, and blindly evaluate the suitability of this paradigm in a well‐established rat model of SE‐induced TLE.7 Furthermore, this study reveals our efforts to refine and optimize a moderate‐throughput screening protocol for disease‐modifying or antiepileptogenic agents. Although no treatment administered in this study significantly altered the burden of epilepsy, our findings indicate the suitability of this approach to screen for potentially efficacious agents in a rigorous and blinded manner.
Preclinical models of epileptogenesis are essential to identify disease‐modifying or antiepileptogenic agents that may transform the management of epilepsy.35, 36 Prevention of epilepsy in at‐risk individuals has long been considered the “holy grail” of epilepsy therapy development.1, 2 Clinical studies have attempted to prevent the development of epilepsy in posttraumatic brain injury patients using conventional ASDs, including PB; however, no such study has yet demonstrated a meaningful effect.17, 37, 38 Clinical studies of antiepileptogenesis are highly time‐ and resource‐intensive, making practical and well‐characterized preclinical models of epileptogenesis critical to screen potentially promising agents. For these reasons, the post‐KA SE rat model provides a number of benefits to screen for disease‐modifying or antiepileptogenic agents. First, SRS of the post‐KA SE rat model of TLE arise after a well‐defined and reliable latent period.7 Second, the model reproduces the interindividual heterogeneity of SRS onset, severity, and frequency,6 which may lead to a greater likelihood of successfully translating preclinical findings to clinical use. Third, the post‐KA SE rat model of TLE is characterized by SRS that are resistant to a number of ASDs.5, 12, 13 Finally, the post‐KA SE rat model exhibits a number of pathophysiological features consistent with clinical TLE, including neuroinflammation,9 neurodegeneration,25 and behavioral deficits.11 Although chemoconvulsant‐induced SE is not a predominate way by which people typically develop epilepsy,39 neurological insult, including SE, traumatic brain injury, and stroke, are well known to cause clinical acquired epilepsy, accounting for some 15% of new epilepsy cases.40, 41 Regardless of whether the post‐KA SE rat model of TLE is the best model to identify all agents to prevent epilepsy in the clinical setting, it does carry a number of practical benefits that make it well suited to drug screening applications. Ultimately, however, the demonstration of clinical potential should be derived from consistent evidence of efficacy in a number of diverse preclinical models, using a rigorous approach, to confirm that the compound is potentially beneficial. Clinical validation of this model will then only be possible upon identification of a compound in this model that is then found to be effective in people.36, 42 As such, this rat model is an approach to the screening of candidate agents within the NINDS ETSP and would inform a comprehensive assessment plan for any potentially disease‐modifying or antiepileptogenic agent.
Our present study evaluated the potential of two mechanistically different agents, PB and EVL, to prevent or modify the development of SRS when administered at three different time points after SE insult. Prior studies have demonstrated a disease‐modifying effect of PB and the close EVL analogue, rapamycin, in this model when administered at various time points relative to KA administration.31, 43 Our study also included the administration of EVL 1–24 h after SE onset. Because of the variety of causes of SE, SE often cannot be treated until well after onset.44, 45 When SE is treated in the clinic, interventions are chosen to immediately stop the seizure. Our study thus intervened after the neurological SE insult so as to define whether any agent could prevent the development of epilepsy without directly preventing SE. Whether this design will identify promising disease‐modifying or antiepileptogenic agents is subject to further scrutiny. In any case, our approach, analytical plan, and interpretation may identify such an agent with the utmost rigor.
EVL and PB are both US Food and Drug Administration‐approved for the management of epilepsy syndromes; EVL is approved for the treatment of TSC, whereas PB is an ASD approved for monotherapy in diverse seizure types. Limited preclinical studies have assessed the antiepileptogenic potential of both agents. More than 1 month of once‐daily PB administration (70 mg/kg ip) beginning 34 min after KA‐SE insult in PND35 rats did not subsequently prevent the development of SRS and cognitive deficits.32 Our present study aligns with these earlier findings using a markedly reduced experimental timeline (5 days of PB treatment). Our study confirms that chronic or subchronic PB is not a valid means to prevent SRS in post‐KA SE rats. Although Sutula and colleagues demonstrated that PB could prevent the SE‐induced damage and modify disease severity when administered during active KA‐induced SE, that study administered PB “immediately after” KA administration.31 Thus, PB likely attenuated the severity of the SE insult itself, which may have contributed to the observed disease‐modifying effects.31 Use of a 1‐h delay may suggest that a critical window of PB intervention (i.e., less than 30 min) exists to effectively prevent the development of SRS, likely through the modification of the insult itself.32 Whether rats treated with PB within 1 h of SE onset would have extensive neuropathology was beyond the scope of this model refinement study and thus remains unknown.
Similar to PB, EVL did not demonstrate a dose‐ or time‐related antiepileptogenic effect in this model, including a head‐to‐head evaluation of the original versus expedited protocol designed to improve screening throughput. To our knowledge, only one other study has specifically evaluated the preclinical efficacy of EVL using the two‐hit mouse model of KA‐SE‐induced TLE.43 That study, however, was limited in that EVL (1 mg/kg ip) was administered between the first and second KA bouts, and significantly reduced the latency to Racine Stage 5 seizures and attenuated SE severity.43 Thus, any previously observed effects of EVL were limited by the initial attenuation of the SE insult. Additionally, there have been conflicting reports regarding the extent to which mTOR inhibition with rapamycin is disease‐modifying in various models of acquired epilepsy,25, 46, 47, 48 including mouse models of TSC.49 Thus, our study suggests, through unbiased assessment, that EVL at the doses and time points of treatment tested did not prevent the development of SRS in the post‐KA SE rat model. Our study design provides a robust and unbiased assessment of disease‐modifying potential after SE insult; any effects can be directly attributed to the compound itself, rather than insult modification.
Our present investigations to refine our drug screening protocol were limited by a number of factors. First, we did not conduct pharmacokinetic assessments of brain concentrations of any compound, nor did we assess the extent to which any of the pharmacological targets (e.g., γ‐aminobutyric acid type A receptors or mTOR inhibition) were engaged. In any case, it is entirely possible that our team can perform such bioanalysis. Second, the original design did not include continuous 24/7 vEEG monitoring throughout the 42‐day period, but instead broke the observations into two 2‐week sessions separated by a 2‐week break. The rationale behind this approach was one of logistics. An alternating 2 week on/2 week off/2 week on monitoring protocol can accommodate 24 chronically implanted rats in a single 12‐unit recording suite in a standard housing room (10 × 10 ft) to conduct long‐term studies in a moderate‐throughput capacity over roughly 10 weeks. Our original protocol minimized resource burden and increased throughput to conduct long‐duration studies. By expediting our study design through this iterative study, we establish a new moderate‐throughput drug screening protocol for disease‐modifying or antiepileptogenic agents. Future studies will thus have a reduced recording burden as a result of these presently reported studies; novel agents only need to be monitored for a single 2‐week recording period from 4 to 6 weeks after SE insult. If a future compound is found to modify disease trajectory using that expedited protocol, subsequent studies with longer duration monitoring can be performed.
In addition to methodological limitations, our present study had a number of limitations related to the model selection itself. First, our study was limited by the goal to not intervene too early after SE insult so as to clearly dissociate insult‐modifying from disease‐modifying agents.35 By delaying intervention for 1 h post‐SE, we can dissociate any potential disease‐modifying effects due to the blockade or reduction in the SE severity itself, in contrast to SE pretreatment studies.31 Additionally, whether the KA‐SE rat model of TLE evokes too severe of an insult to lead to false negative results is certainly a potential limitation of our model selection. Ultimately, however, we aim to prioritize resource utilization to identify agents in a paradigm with a high bar for efficacy. It would be hoped that such a stringent criterion would lead to the identification of agents that could have broad applicability and utility for many epilepsy syndromes. If an agent is found to work in this post‐KA SE rat model, it is reasonable to expect success in other, less severe epilepsy models, and ultimately in humans at risk for developing epilepsy. Thus, our design is not without limitations, but we herein offer the epilepsy research community a rigorous, unbiased, and rational screen to identify possible disease‐modifying or antiepileptogenic agents. Whether our approach will identify any clinical candidates remains to be defined. In any case, this platform supported by the NINDS ETSP offers a critical resource to address an unmet need.3
CONFLICT OF INTEREST
H.S.W. has served on the scientific advisory board of Otsuka Pharmaceuticals, has served as an advisor to Biogen Pharmaceuticals and Acadia Pharma, and is a member of the UCB speakers bureau. H.S.W. is scientific cofounder of NeuroAdjuvants, Salt Lake City, Utah. C.M. is a former employee of NeuroAdjuvants and is a consultant for Sea Pharmaceuticals. K.S.W. serves on the scientific advisory board of Mend Neuroscience and Blackfynn, and is a consultant to Xenon Pharmaceuticals. None of the other authors has any conflict of interest to disclose. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
AUTHOR CONTRIBUTIONS
Conception and experimental design: Melissa Barker‐Haliski, Cameron Metcalf, Karen S. Wilcox, H. Steve White. Acquisition of experimental data: Kevin Knox, Dannielle Zierath, Zachery Koneval, Melissa Barker‐Haliski. Performed data analysis: all authors. Wrote or contributed to the writing of the manuscript: all authors.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful for the scientific input of Drs. Brian Klein and John Kehne of the NINDS ETSP. The authors acknowledge the technical support for KA‐SE induction of Ms. Stephanie Mizuno. The authors also gratefully recognize the efforts of Dr. Gaëlle Batot for effectively managing the subcontract from the University of Utah. This project has been funded with federal funds from the NINDS, National Institutes of Health, Department of Health and Human Services, under contract no. HHSN271201600048C (principal investigator K.S.W.) through a subcontract from the University of Utah to UW.
Barker‐Haliski M, Knox K, Zierath D, Koneval Z, Metcalf C, Wilcox KS, et al. Development of an antiepileptogenesis drug screening platform: Effects of everolimus and phenobarbital. Epilepsia. 2021;62:1677–1688. 10.1111/epi.16955
REFERENCES
- 1.Pitkänen A, Nehlig A, Brooks‐Kayal AR, Dudek FE, Friedman D, Galanopoulou AS, et al. Issues related to development of antiepileptogenic therapies. Epilepsia. 2013;54(Suppl 4):35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.White HS, Loscher W. Searching for the ideal antiepileptogenic agent in experimental models: single treatment versus combinatorial treatment strategies. Neurotherapeutics. 2014;11:373–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.National Advisory Neurological Disorders and Stroke (NANDS) Council . A report of the NINDS Epilepsy Therapy Screening Program Working Group of the National Advisory Neurological Disorders and Stroke (NANDS) Council. 2020. https://www.ninds.nih.gov/sites/default/files/etsp_wg_report_final_redacted_version_508c.pdf. Accessed 29 May 2021.
- 4.Kehne JH, Klein BD, Raeissi S, Sharma S. The National Institute of Neurological Disorders and Stroke (NINDS) Epilepsy Therapy Screening Program (ETSP). Neurochem Res. 2017;42(7):1894–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomson KE, Metcalf CS, Newell TG, Huff J, Edwards SF, West PJ, et al. Evaluation of subchronic administration of antiseizure drugs in spontaneously seizing rats. Epilepsia. 2020;61:1301–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mizuno S, Koneval Z, Zierath DK, Knox KM, White HS, Barker‐Haliski M. Diurnal burden of spontaneous seizures in early epileptogenesis in the post‐kainic acid rat model of epilepsy. Epilepsia Open. 2021. 10.1002/epi4.12485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hellier JL, Patrylo PR, Buckmaster PS, Dudek FE. Recurrent spontaneous motor seizures after repeated low‐dose systemic treatment with kainate: assessment of a rat model of temporal lobe epilepsy. Epilepsy Res. 1998;31:73–84. [DOI] [PubMed] [Google Scholar]
- 8.Ben‐Ari Y, Cossart R. Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci. 2000;23:580–7. [DOI] [PubMed] [Google Scholar]
- 9.Vargas JR, Takahashi DK, Thomson KE, Wilcox KS. The expression of kainate receptor subunits in hippocampal astrocytes after experimentally induced status epilepticus. J Neuropathol Exp Neurol. 2013;72:919–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bertoglio D, Amhaoul H, Van Eetveldt A, Houbrechts R, Van De Vijver S, Ali I, et al. Kainic acid‐induced post‐status epilepticus models of temporal lobe epilepsy with diverging seizure phenotype and neuropathology. Front Neurol. 2017;8:588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stafstrom CE, Chronopoulos A, Thurber S, Thompson JL, Holmes GL. Age‐dependent cognitive and behavioral deficits after kainic acid seizures. Epilepsia. 1993;34:420–32. [DOI] [PubMed] [Google Scholar]
- 12.Grabenstatter HL, Dudek FE. Effect of carbamazepine on spontaneous recurrent seizures recorded from the dentate gyrus in rats with kainate‐induced epilepsy. Epilepsia. 2019;60:636–47. [DOI] [PubMed] [Google Scholar]
- 13.Grabenstatter HL, Ferraro DJ, Williams PA, Chapman PL, Dudek FE. Use of chronic epilepsy models in antiepileptic drug discovery: the effect of topiramate on spontaneous motor seizures in rats with kainate‐induced epilepsy. Epilepsia. 2005;46:8–14. [DOI] [PubMed] [Google Scholar]
- 14.Leite JP, Garcia‐Cairasco N, Cavalheiro EA. New insights from the use of pilocarpine and kainate models. Epilepsy Res. 2002;50:93–103. [DOI] [PubMed] [Google Scholar]
- 15.Murri L, Parenti G, Bonuccelli U, Lenzi B, Del Tacca M. Phenobarbital prophylaxis of post traumatic epilepsy. Ital J Neurol Sci. 1980;1:225–30. [PubMed] [Google Scholar]
- 16.Murri L, Arrigo A, Bonuccelli U, Rossi G, Parenti G. Phenobarbital in the prophylaxis of late posttraumatic seizures. Ital J Neurol Sci. 1992;13:755–60. [DOI] [PubMed] [Google Scholar]
- 17.Temkin NR. Preventing and treating posttraumatic seizures: the human experience. Epilepsia. 2009;50(Suppl 2):10–3. [DOI] [PubMed] [Google Scholar]
- 18.van der Poest Clement E, Jansen FE, Braun KPJ, Peters JM. Update on drug management of refractory epilepsy in tuberous sclerosis complex. Paediatr Drugs. 2020;22:73–84. [DOI] [PubMed] [Google Scholar]
- 19.Lechuga L, Franz DN. Everolimus as adjunctive therapy for tuberous sclerosis complex‐associated partial‐onset seizures. Expert Rev Neurother. 2019;19:913–25. [DOI] [PubMed] [Google Scholar]
- 20.Curatolo P, Moavero R, van Scheppingen J, Aronica E. mTOR dysregulation and tuberous sclerosis‐related epilepsy. Expert Rev Neurother. 2018;18:185–201. [DOI] [PubMed] [Google Scholar]
- 21.Henske EP, Jóźwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Rev Dis Primers. 2016;2:16035. [DOI] [PubMed] [Google Scholar]
- 22.Sasongko TH, Ismail NF, Zabidi‐Hussin Z. Rapamycin and rapalogs for tuberous sclerosis complex. Cochrane Database Syst Rev. 2016;7:CD011272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crino PB. Mechanistic target of rapamycin (mTOR) signaling in status epilepticus. Epilepsy Behav. 2019;101:106550. [DOI] [PubMed] [Google Scholar]
- 24.Wong M. Mammalian target of rapamycin (mTOR) inhibition as a potential antiepileptogenic therapy: from tuberous sclerosis to common acquired epilepsies. Epilepsia. 2010;51:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeng LH, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci. 2009;29:6964–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abraham J, Fox PD, Condello C, Bartolini A, Koh S. Minocycline attenuates microglia activation and blocks the long‐term epileptogenic effects of early‐life seizures. Neurobiol Dis. 2012;46:425–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomson KE, White HS. A novel open‐source drug‐delivery system that allows for first‐of‐kind simulation of nonadherence to pharmacological interventions in animal disease models. J Neurosci Methods. 2014;238:105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leary SL, Underwood MW, Anthony R, Corey D, Grandin T, Greenacre C, et al. AVMA Guidelines for the Euthanasia of Animals: 2013 Edition. J Am Vet Me A. 2013. [Google Scholar]
- 29.Prasad A, Williamson JM, Bertram EH. Phenobarbital and MK‐801, but not phenytoin, improve the long‐term outcome of status epilepticus. Ann Neurol. 2002;51:175–81. https://www.avma.org/sites/default/files/resources/euthanasia‐highres.pdf [DOI] [PubMed] [Google Scholar]
- 30.Loscher W. The pharmacokinetics of antiepileptic drugs in rats: consequences for maintaining effective drug levels during prolonged drug administration in rat models of epilepsy. Epilepsia. 2007;48:1245–58. [DOI] [PubMed] [Google Scholar]
- 31.Sutula T, Cavazos J, Golarai G. Alteration of long‐lasting structural and functional effects of kainic acid in the hippocampus by brief treatment with phenobarbital. J Neurosci. 1992;12:4173–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bolanos AR, Sarkisian M, Yang Y, Hori A, Helmers SL, Mikati M, et al. Comparison of valproate and phenobarbital treatment after status epilepticus in rats. Neurology. 1998;51:41–8. [DOI] [PubMed] [Google Scholar]
- 33.Guo D, Zeng L, Brody DL, Wong M. Rapamycin attenuates the development of posttraumatic epilepsy in a mouse model of traumatic brain injury. PLoS One. 2013;8:e64078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patel DC, Wallis G, Dahle EJ, McElroy PB, Thomson KE, Tesi RJ, et al. Hippocampal TNFalpha signaling contributes to seizure generation in an infection‐induced mouse model of limbic epilepsy. eNeuro. 2017;4:ENEURO.0105‐17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barker‐Haliski ML, Friedman D, French JA, White HS. Disease modification in epilepsy: from animal models to clinical applications. Drugs. 2015;75:749–67. [DOI] [PubMed] [Google Scholar]
- 36.Barker‐Haliski M, Friedman D, White H, French J. How clinical development can, and should, inform translational science. Neuron. 2014;84:582–93. [DOI] [PubMed] [Google Scholar]
- 37.Temkin NR. Antiepileptogenesis and seizure prevention trials with antiepileptic drugs: meta‐analysis of controlled trials. Epilepsia. 2001;42:515–24. [DOI] [PubMed] [Google Scholar]
- 38.Temkin NR, Jarell AD, Anderson GD. Antiepileptogenic agents: how close are we? Drugs. 2001;61:1045–55. [DOI] [PubMed] [Google Scholar]
- 39.Lefebvre KA, Robertson A. Domoic acid and human exposure risks: a review. Toxicon. 2010;56:218–30. [DOI] [PubMed] [Google Scholar]
- 40.Annegers JF, Hauser WA, Coan SP, Rocca WA. A population‐based study of seizures after traumatic brain injuries. N Engl J Med. 1998;338:20–4. [DOI] [PubMed] [Google Scholar]
- 41.Hauser WA, Annegers JF, Kurland LT. Prevalence of epilepsy in Rochester, Minnesota: 1940–1980. Epilepsia. 1991;32:429–45. [DOI] [PubMed] [Google Scholar]
- 42.Barker‐Haliski M, Steve White H. Validated animal models for antiseizure drug (ASD) discovery: advantages and potential pitfalls in ASD screening. Neuropharmacology. 2020;167:107750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang M‐T, Lin Y‐C, Ho W‐H, Liu C‐L, Lee W‐T. Everolimus is better than rapamycin in attenuating neuroinflammation in kainic acid‐induced seizures. J Neuroinflammation. 2017;14:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cook AM, Castle A, Green A, Lesch C, Morrison C, Rhoney D, et al. Practice variations in the management of status epilepticus. Neurocrit Care. 2012;17:24–30. [DOI] [PubMed] [Google Scholar]
- 45.Brophy GM, Bell R, Claassen J, Alldredge B, Bleck TP, Glauser T, et al. Guidelines for the evaluation and management of status epilepticus. Neurocrit Care. 2012;17:3–23. [DOI] [PubMed] [Google Scholar]
- 46.Sliwa A, Plucinska G, Bednarczyk J, Lukasiuk K. Post‐treatment with rapamycin does not prevent epileptogenesis in the amygdala stimulation model of temporal lobe epilepsy. Neurosci Lett. 2012;509:105–9. [DOI] [PubMed] [Google Scholar]
- 47.Huang X, McMahon J, Huang Y. Rapamycin attenuates aggressive behavior in a rat model of pilocarpine‐induced epilepsy. Neuroscience. 2012;215:90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Drion CM, Borm LE, Kooijman L, Aronica E, Wadman WJ, Hartog AF, et al. Effects of rapamycin and curcumin treatment on the development of epilepsy after electrically induced status epilepticus in rats. Epilepsia. 2016;57:688–97. [DOI] [PubMed] [Google Scholar]
- 49.Theilmann W, Gericke B, Schidlitzki A, Muneeb Anjum SM, Borsdorf S, Harries T, et al. Novel brain permeant mTORC1/2 inhibitors are as efficacious as rapamycin or everolimus in mouse models of acquired partial epilepsy and tuberous sclerosis complex. Neuropharmacology. 2020;180:108297. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material