

Abstract
Food allergy (FA), a growing public health burden in the United States, and familial aggregation studies support strong roles for both genes and environment in FA risk. Deepening our understanding of the molecular and cellular mechanisms driving FAs is paramount to improving its prevention, diagnosis and clinical management. In this review, we document lessons learned from the genetics of FA that have aided our understanding of these mechanisms. While current genetic association studies suffer from low power, heterogeneity in definition of FA, and difficulty in our ability to truly disentangle FA from food sensitization and general atopy genetics, they reveal a set of genetic loci, genes and variants that continue to implicate the importance of barrier and immune function genes across the atopic march, and FA in particular. The largest reported effects on FA are from MALT1 (OR=10.99), FLG (average OR~2.9) and HLA (average OR~2.03). The biggest challenge in the field of FA genetics is to elucidate the specific mechanism of action on FA risk and pathogenesis for these loci, and integrative approaches including genetics with transcriptomics, proteomics and metabolomics will be critical next steps to translating these genetic insights into practice.
Keywords: Food allergy, Peanut allergy, Genetics, Genomics, Barrier genes, Immune genes, Atopic march, Dual Allergen Hypothesis, FLG, HLA
INTRODUCTION
Food allergy (FA) is a growing public health burden in the United States and globally 1. Upon exposure to an allergenic food protein, the immune system mounts a response by producing antigen specific IgE, a state termed as food sensitization (FS). On re-exposure the allergenic food binds to specific antibodies and triggers the release of mediators, such as histamine, leading symptomatic FA 2. The Dual Allergen Exposure hypothesis which states that low-dose cutaneous sensitization to food may lead to FS and early consumption of food may induce oral tolerance 3, is one proposed hypothesis for increases noted in FA prevalence 3. Additionally, the mechanism of FA development as illustrated through the atopic march suggests a role for both barrier and immune related genes given the hypothesis that children predisposed to atopic dermatitis (AD) or FA in early childhood have an increased risk of developing other allergic diseases such as asthma and allergic rhinitis (AR), later in life 4, 5. The pathophysiology of FA development is therefore, a complex interplay of environmental factors, including allergen exposure, within a strong interaction framework with genetic risk factors 3, 6.
While there have been a wide array of genetic studies on FA spanning different approaches (Box 1) as detaiedl in Table E1, there are several challenges with these that need to be articulated before evaluating the evidence for FA loci for their robustness, and interpretation of their effects on FA risk. Firstly, very few studies adequately measure environmental exposure either in-utero or early life. Therefore, they are unable to capture gene *environment (GxE) interactions. In a disease where the interaction between genes and environment may be quite large, this could result in a major loss in power to fully identify FA genetic determinants. Studies to date are further limited in power with small sample sizes; the largest study is ~11,000 samples (Table E1). Where potential exists for meta-analysis approaches to overcome the weaknesses of smaller studies, the existing studies are highly variable in definition of outcome ranging from health questionnaires, skin prick testing (SPT), quantitative biomarkers (e.g., specific IgE, sIgE) to double blinded placebo controlled oral food challenges (DBPCFCs). In addition to variability in the definition of FA cases, there is also variability in the definition of controls (e.g., atopic vs. non-atopic controls vs. unphenotyped population-based controls, Table E1). Overall, there is difficulty in teasing apart FA from FS genetics across these studies, and the range of phenotype definition introduces heterogeneity in disease outcome for any combined meta-analysis approach across them. Despite these limitations, there have been numerous genetic loci, many with robust replication, suggested for FA (Table 1) documenting a role for both barrier and immune related genes. Finally, there is considerable overlap between FA genetic loci and genome-wide association study (GWAS) identified loci for AD 7, asthma 8, and AR 9 (Figure 1). The considerable overlap between FA and AD supports the mechanisms for FA suggested by the atopic march 10, 11.
Box 1: Approaches to dissecting the genetics of FA.
Familial aggregation studies are the often relied-on first-line of evidence in documenting a genetic component to disease interrogating whether clustering within families is greater than general population prevalence.
Heritability is the quantification of the overall phenotypic variation/risk that is attributable to genetic factors.
Association testing tests for the correlation between disease and genetic markers (most often SNPs) typically in a case–control design setting wherein allele frequencies at a measured single nucleotide polymorphism (SNP) are compared between case and control samples from the population.
Candidate gene association studies (CGAS) are targeted studies of association between selected genes of interest and a phenotype. The approach begins with selection of a putative candidate gene based on its relevance in the mechanism of the disease or trait being investigated and are most often performed in the framework of a case-control study.
Genome-wide association study (GWAS) rely on the idea that common genetic variation across the genome is correlated in blocks because of linkage disequilibrium (LD), and therefore measuring ~2.5 million genetic variants on a high throughput assay can capture most associations with common variants.
Gene*environment interaction (GxE) studies interrogate how environmental exposures and genetic predisposition work together to modify disease risk and/or outcome. An interaction is indicated when the presence of one factor (e.g. lifestyle and diet) affects the influence of the second factor (e.g. genetic) on disease risk.
(WGS) are the two next-generation sequencing (NGS) techniques to study the common and rare genetic variations in the genome. WES allows variations in the protein-coding region of any gene to be identified whereas WGS offers the ability to interrogate the entire DNA sequence of the genome.
Meta-Analysis is a routine approach to combining smaller GWAS studies using summary statistics/results to overcome the smaller samples sizes of individual studies.
Fine-Mapping is a set of statistical and laboratory approaches to determine the causal genetic variant for associated genetic loci.
Table 1:
Genetic loci and genes associated with FA identified by GWAS, CGAS and GxE interaction studies organized by functional categories. Genes/loci that are bolded are those that are replicated within the study, or have evidence across multiple studies. For genetic loci mapping to multiple genes, the index gene for each category is marked with an asterisk (*).
| Functional Categories | GWAS | CGAS | G*E |
|---|---|---|---|
| Skin Barrier Integrity | FLG-AS179, SERPINB7|SERPINB279 | FLG23–26, 28, SPINK35, 36 | FLG105 |
| Vascular and endothelial cell factors | ANGPT480, CHCHD3|EXOC480, CTNNA380, SKAP180 | ||
| Innate immunity | HLA-DPB178, 82, HLA-DRB176, 78, 82, IL2678, MALT131 | CD1445, 47, HLA-B100, HLA-DPB196, HLA-DRB196, 98, 100, IL1051, 52, IL2|IL21*|KIAA110928, 29, TLR10/1/6*|FAM114A129, TSLP*|WDR3628 | MALT131 |
| Adaptive immunity | C11orf30|LRRC32*|LOC10192881379, 80, HLA-DPB178, 82, HLA-DQA176, 78, 82, HLA-DQA276, 82, HLA-DQB176, 78, 79, 81,82, HLA-DRA76, 82 HLA-DRB176, 78, 82, HLA-DRB582, IL2678, IL4*|KIF3A79, MALT131 | C11ORF30|LRRC32*28, 73, C-REL67, HLA-B100, HLA-DPB196, HLA-DQB130, 96, 99, 100, HLA-DRB196, 98, 100, IL1051, 52, IL2|IL21*|KIAA110928, 29, KIF3A|IL13* 28, 59, STAT6 28, 40, TSLP*|WDR3628 | MALT131 |
| Immune modulation and regulation | IL2678, IL4*|KIF3A79, SERPINB7|SERPINB279 | C-REL67, IL1051, 52, IL28B54, IL2|IL21*|KIAA110928, 29, KIF3A|IL13* 28, 59, STAT6 28, 40 | DBP/GC106 |
| Other | ADGB77, ARHGAP2480, ATP10A78, BCAS178, DLX2|ITGA680, FXR178, GNPDA1|NDFIP131, GYG1P2|RNU6-67P77, IER5L78, LINC00540|BASP1P131, LINC00298|LINC0029931, LINC01260|KCNK15-AS131, LINC01568|LOC10192803531, LINGO278, LMX1A78, LOC10192716680, LOC10192794777, MDN178, MMP12|MMP1380, NAV278, PADI631, PAFAH1B178, PAX278, PLAGL178, PLPP7|PRRC2B31, PYROXD178, RCC2|ARHGEF10L31, RGS2178, RIMS278, RNF13078, SALL378, SLC2A978, SORBS278, SV2C78, TES78, TRIM231, ZNF65277 | CCDC8028, CLEC16A|DEXI28, GLB128, NAT260, NLRP366, OR10A3|NLRP1028, OVOL128, TMEM232|ISLC25A4628, TNFRSF6B|ZGPAT28, ZNF36528, ZNF65228 | |
Fig 1: Overlap between food allergy associated genes (from CGAS) and loci (from GWAS) and asthma, AD and AR GWAS loci.
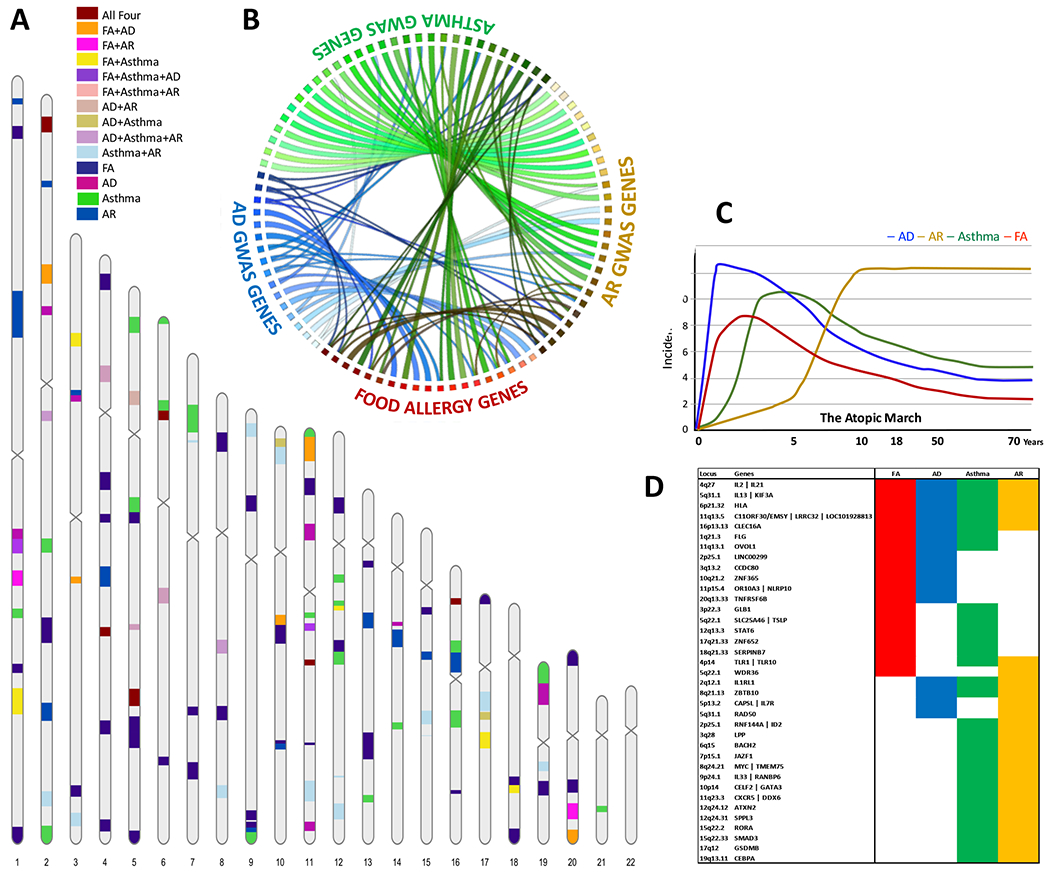
[A] Karyogram shows the genetic loci associated FA from Table 1 and the largest GWAS reported loci for AD 7, asthma 8, and AR 9. The colors represent the overlap in chromosomal position between the four phenotypes of the Atopic March. [B] Circos plot showing connections between overlapping loci for the four phenotypes. FA loci show the highest overlap with AD loci as compared to asthma or AR GWAS loci. [C] The proposed model for atopic march showing the development of FA, Asthma and AR correlates AD severity in early life, adapted from Davidson et al 11. [D] Shared genes among atopic march phenotypes presented in panel B.
FAMILIAL AGGREGATION AND HERITABILITY OF FOOD ALLERGY
The first documented report of familial aggregation of FA was in 1996 in a survey of 622 families with probands (i.e. an index case with disease) of self-reported, suspected and known peanut allergy (PA) in the United Kingdom 12. PA prevalence was found to increase through successive generations: 0.1% in grandparents, 1.6% in parents and 7% of siblings of the proband 12; a pattern often noted in diseases with a genetic predisposition as closer relatives share more genetically.
Twins are a special category of sibling pairs where there is greater shared (early) environment in twins vs. siblings. Additionally, the genetics for monozygotic (MZ) vs. dizygotic (DZ) twins differs with 100% vs 50% shared genetics, respectively; the comparison of concordance (i.e. both individuals within the twin pair have FA) between MZ and DZ twins provides insight into the role of genes vs. environment. In 2000, a small twin study including 58 twin pairs from the United States showed much higher concordance of PA among MZ twins (64.3%) than DZ twins (6.8%), with heritability estimates at 81.6% 13. A study in a Chinese population including 826 twin pairs evaluated sensitization to 9 foods (milk, egg, wheat, peanut, soybean, sesame, walnut, shellfish, and fish), confirmed higher concordance in sensitization for any food in MZ (52.1%) than DZ (39.2%) twins and report a heritability of FS of 56% 14 Higher prevalence of PA in siblings of allergic children than the siblings of non-allergic/non-sensitized children (8.5% vs 1.3%) has also been noted in 514 families from Canada 15. Siblings of peanut allergic children had a 7-fold greater risk for PA, and interestingly birth order also appears to have an effect on risk, with younger siblings bearing the highest risk 15. Elevated risk to siblings of peanut allergic children were also noted in the Chicago Family Cohort Food Allergy study 16 including 581 nuclear families, with evidence for heritability in food-specific IgE (15%-35%).
The largest and most robust familial aggregation study including 5,276 infants was performed in the HealthNuts study cohort in Australia 17 . They used oral food challenge (OFC) to egg, peanut or sesame allergy to define FA, and reported that the risk for FA in the children increased with increasing numbers of atopic family member. However, they found that the risk for FA in infants with two allergic family members compared with no allergic family members was lower in children with both parents born in Australia (OR=2.2,95% CI=1.5-3.3) than those with both born in Asia (OR=4.7, 95% CI=2.3-9.5). The reverse was noted for the risk for FA in infants with one allergic family member compared with no allergic family members; higher in children with both parents born in Australia (OR=1.9, 95% CI=1.3-2.7) than Asia (OR=1.4, 95% CI=0.7-2.9). The overall lower rates of reported FA among parents born in East Asia, but higher rates among their infants supports interactions with environmental factors. They also found that maternal history of eczema and asthma, and sibling history of AR were predictors of egg allergy (EA) in infants whereas maternal and paternal history of asthma with AR were predictors PA in infants 17 Most recently, the Chicago Family Cohort Food Allergy study 18 applied a set of clinical criteria to data gathered from the questionnaires, immunological markers of specific IgE measurements and SPTs to 9 food allergens (egg white, sesame, peanut, soy, cow’s milk, shrimp, walnut, cod fish and wheat) and was able to dissect FA from FS. They reported that among siblings of children with FA, 53% siblings were food sensitized but did not have clinical FA; 33.4% had neither FS nor clinical FA and only 13.6% were both sensitized and had clinical FA; an overall higher prevalence in siblings were noted for FS vs FA.
These studies depict a wide range in the heritability estimates for FA and FS ranging from 15% to 82% 13, 14, 16, and the risk to siblings in FA studies ranges from 1.3-fold to 12-fold 12, 15, 16, 18. The Chicago Family Cohort Food Allergy study even noted that the observation of 1 in 8 siblings at risk of developing FA represented only a minimally higher risk than general population (1 in 12) and concluded that FA in an index child is not an indication to test siblings 18. While this wide spectrum of heritability estimates for FA and FS may seem contradictory at first, these are in fact not unexpected observations. FA is a disease with a strong environmental component; allergen exposure, its timing and route are key components to the development of disease 6 in addition to genetic susceptibility. The less-than-100% concordance in FA between MZ twins, but the higher concordance in MZ twins compared to DZ twins offers the insight that there are genetic factors at play (MZ twins share their full genome while DZ share only half), but there are certainly environmental factors at play as well (given that in <<100% of MZ twin pairs, both twins have FA). The wide range in heritability represents the relative differences between genetic and environmental variability in the specific sample being studied. Take for example a study sample where every child is exposed vs. one where no child is exposed. Even if the genetics were identical, the dramatic difference in exposure rates will result in differing disease prevalence, and consequently wildly differing heritability estimates. This complex interplay of genes and environment in FA is further compounded by the assessment of allergy and/or sensitization in these studies which ranges from brief questionnaire data, to SPT and/or specific IgE measures and OFCs. Nonetheless, the overall collection of studies would support the role of genetics and its interaction with environment in FA, and there has been success in identifying genetic determinants of FA as illustrated below.
CANDIDATE GENE ASSOCIATION STUDIES IN FOOD ALLERGY
Candidate gene association studies (CGAS) for FA have primarily focused on genes known to be associated with asthma, AD and GWAS-identified FA genetic loci. A comprehensive overview of the study designs of the CGASs performed for FS, FA, and food induced anaphylaxis (FIA) is illustrated in Table E1, and reiterates the stated challenges with small sample sizes and definition of disease outcome. Of the 27 CGAS-identified genes/loci (Table 1), filaggrin (FLG) and the set of human leukocyte antigen (HLA) genes have some of the most robust evidence. Given the striking overlap in HLA genes from CGAS and GWAS, and the complexity of HLA associations at the allelic level, these genes are discussed in the separate section of this review.
Filaggrin (FLG) located in the epidermal differentiation complex (EDC) region on chromosome 1q21.3 encodes profilaggrin and plays a key role in epithelial barrier function in allergic skin disease 19 with well-established associations with AD20 , asthma 20 and AR 21 and increased levels of certain food specific IgEs, indicating FS 21. Cutaneous exposure to foods through an impaired skin barrier is hypothesized as a route of FS, and the timing and balance of cutaneous exposure relative to oral exposure may result in FA 22, making FLG a perfect biological candidate. Much attention has been devoted to a set of loss-of-function (LOF)/null mutations including R501X, 2282del4, R2447X, and S3247X mutations. The first evidence for FLA LOF mutations in PA was published almost a decade ago 23 in PA cases confirmed with OFC compared to population-based control subjects from the UK, Netherlands and Ireland. Strong effect sizes (OR=5.3, 95%CI=2.8-10.2) were noted for a null genotype, and findings were replicated in a Canadian replication cohort albeit with weaker effects (OR=1.9, 95%CI=1.4-2.6). The Australian HealthNuts cohort 24 confirmed the strong effects FLG has with FS (OR=3.0, 95%CI=1.0-8.7) and FA (OR=2.9, 95%CI=1.0-8.6) even after adjusting for any association with eczema. Importantly no associations were observed with EA or PA within the egg sensitized or peanut sensitized infants, respectively 24, suggesting no role for allergy beyond sensitization. The relationship between FLG LOF mutations and PA was investigated in a Canadian cohort 25, and significant associations were observed (OR=1.96, 95%CI=1.49-2.58) that were confirmed to be independent of a history of asthma. In a longitudinal study exploring the time-order relationships between FLG LOF mutations and FA and FS 26 in children from the Isle of Wight (IOW) birth cohort 27, sustained effects were noted at younger (OR=31.46, 95%CI=2.86-100) and older ages for FA (OR=4.25, 95%CI=1.55-11.61). They also reported that the effect of FLG LOF mutations on FA was an indirect effect through eczema and FS in the early ages 26. There has been limited investigation into the role of FLG in FA beyond European ancestry populations; with only a single report confirming its role in Japanese subjects 28 with significant associations noted for 6 FLG LOF variants and FA (OR =1.63; 95%CI=1.28-2.07). Two negative reports for FLG have been noted for cow’s milk allergy (CMA) 29, 30. Taken together, FLG is perhaps the strongest candidate gene in FA aside from HLA genes. While there does seem to be some conflict in whether FLG LOF mutations increase risk of FS through impaired skin barrier function caused by filaggrin deficiency with 26 or without a further role in progression to FA 24, 31, the cumulative evidence strongly supports the role of skin barrier function in FA 26. Furthermore, the observation that the associations remain significant even after the adjustment for eczema would suggest an independent effect on FS 24 beyond AD. Importantly, FLG associations seem to not have specificity for type of FA, but a more generalized effect for FS and FA. There is also a consistency of effect, with same FLG LOF mutations increasing risk to phenotypes across the atopic march. The risk effects for FLG LOF mutations are strongest for AD (OR=11.56, 95%CI=5.73-24.74, 32), followed by FA (OR=5.3, 95%CI=2.8-10.2, 23) and asthma (OR=3.55, 95%CI=1.87-7.02, 33); no associations have been reported for AR.
SPINK5 encodes lymphoepithelial Kazal-type-related inhibitor (LEKTI), that is involved in regulation of desquamation (shedding of outermost layer of epidermis) 34. The p.Glu420Lys polymorphism was found to increase risk for FA in a Japanese AD cohort 35 (OR=NR), and associations of multiple SNPs have also been noted for FA in the Australian HealthNuts cohort 36 including a multi-ethnic study sample of Caucasians, Asians (South-East Asians) and Mixed Asian-Caucasian (OR=1.83-2.95, 95%CI = 1.11-1.95 to 3.03-5.83). Similar to FLG, these associations have strong effects (max OR=2.95), and independence of eczema status 36 confirming the importance of an impaired skin barrier in FA.
STAT6 plays a central role in the production of IL-4 and IL-13 cytokines and in their signal transduction pathway for IgE class switching; an important component in allergic responses 37–39. The first reported association was with nut allergy in a UK study 40 (OR=3.9, 95%CI=1.9-8.3) with documentation of a role for not just risk of nut (peanut and/or tree nut) allergy, but also severity of the allergy 40. While negative reports do exist for the same STAT6 genetic variants in a Japanese study, it was very small (66 controls/14 cases) and hampered by low power 41. Evidence for the role of STAT6 as a candidate gene extends beyond nut allergy. In a subgroup of the GENEVA cohort 42, including Dutch children with FA defined by any positive DBPCFC for PA and CMA, STAT6 was found to be associated with PA (OR=NR), higher sIgE levels to peanuts and cow’s milk, more severe symptoms for both peanut and cow’s milk, and greater eliciting doses during the DBPCFC. Theses multiple studies document the role of STAT6 in FA independent of allergenic food (i.e. across multiple allergens), and equally important, they suggest a role in severity of the allergy beyond just risk.
CD14, a known asthma locus 43, encodes a pattern recognition receptor CD14 that binds to lipopolysaccharides, activating antigen presenting cells (APCs) with subsequent release of pro-inflammatory cytokines 44 A promoter polymorphism (c.−159C>T) previously implicated for asthma was shown to be associated with FA in white subjects (OR=1.7, 95%CI=1.1-2.8) 45 but not in a Japanese population 46. Associations with CD14 variants have also been shown 47 within a sibling-pair design, with predominantly European ancestry subjects for PA (OR=1.97, 95%CI=1.02-3.79) and peanut-specific IgE (psIgE) 47. Interestingly, CD14 is a gene with known GxE interactions with endotoxin exposure, and observed population differences given environmental exposures in high-and low-endotoxin environments 48 which may help explain in part differential observations in FA between populations.
IL-10 and TGF-β1 are anti-inflammatory cytokines49, 50 known to suppress allergen specific IgE production. Interestingly, all CGAS on these anti-inflammatory cytokines have been in non-European ancestry populations. The association between FA and polymorphisms of IL10 and TGF-β1 genes were first investigated in Japanese children 51, and the IL10 c.−1082A>G SNP was found to convey an increased risk for the development of FA (OR=2.4, 95%CI=1.0-6.0) 51. In a study looking at IgE-mediated CMA in Brazilian children 52, once again associations were only observed for IL10 with higher risk in a persistent sub-group (OR=1.74, 95%CI=1.14-2.81). In a small-scale study in a Taiwanese population (N<100), while no associations were detected for allergy, the authors report significant associations with egg white specific IgE 53.
A study in Australian children has reported the association between IL-28B gene and IgE mediated FA 54 IL-28B gene encodes cytokine IFN-λ3, a member of type III interferon family that has immunomodulatory functions. Despite the very limited sample size (30 cases with physician diagnosed FA and 35 non-allergic controls of Caucasian ancestry) the authors were able to identify a nominal association for FA (OR=3, 95%CI=1.8-5.2), however there has been no replication beyond this one study, and therefore evidence for IL-28B is less robust. IL-13 mediates IgE class switching necessary for the development of Th2 mediated allergic immune response 55 and has been extensively studied in the context of asthma 56, AD 57 and AR 58. IL-13 variants were associated with challenge-proven FA (OR=1.75, 95%CI=1.20-2.53) to a greater degree than FS (OR=1.48, 95%CI=0.98-2.23) in the Australian HealthNuts cohort 36, along with association for plasma total IgE levels. While this is the only CGAS on IL-13, the discoveries were replicated in an independent replication cohort within this single report 59.
Gawron ska-Szklarz and colleagues 60 studied the alleles in NAT2, which encodes the N-acetyltransferase 2 enzyme that participates in the detoxication of many drugs and arylamine xenobiotics. Polymorphisms in NAT2 result in slow and fast acetylation phenotypes, and studies have reported a dominance of slow acetylation activity in allergic 61, 62 and atopic patients 63. This study found that the risk of developing FA was almost 3 times higher in slow acetylators than controls (OR=2.8, 95%CI=1.6-4.9). While this is a single CGAS on NAT2, it is backed up by the orthogonal evidence from prior studies connecting slow acetylation with allergic phenotypes, and together supports the role for acetylation genotypes as factors of individual susceptibility to FA 60.
NLRP3 encodes pyrin protein that controls the activity of inflammatory caspase-1 by forming complexes called inflammasomes 64 and has been associated with autoinflammatory diseases and Crohn’s disease 65. A study in a Japanese population assessed the association of NLRP3 polymorphisms with susceptibility to FA and FIA 66, a life-threatening condition that can lead to various cutaneous, respiratory, gastrointestinal and cardiovascular symptoms including hypotension, vascular collapse, and cardiac dysrhythmia. While no significant association were observed for FA, significant associations were noted with FIA (OR=1.53-1.81, 95%CI=1.09-1.27 to 2.16-2.56), and the risk alleles were found to increase the enhancer activity of NLRP3 expression, and increase NLRP3 mRNA stability 66. In a case-control study of 30 Algerian cases with IgE-mediated CMA and 28 non-allergic controls 67 a SNP in C-REL was found to be associated with CMA (OR=4.11, 95%CI=1.82-9.29). C-Rel encodes transcription factor c-REL, a subunit of NF-KB transcription factor and has been implicated in inflammatory diseases such as ulcerative colitis 68 and celiac disease 69.
There have been CGAS examining biological candidates with negative results for FcGRIIa 70, Toll like receptors (TLRs, TLR-2 and TLR-4) 71, FOXP3 67, and two closely related genes, IDO1 and IDO2 72. It should be noted that all these studies may have been severely underpowered (sample sizes <200), making the exclusion of these candidate genes for FA somewhat difficult. Finally, while these FA CGAS typically focus on one or two biological candidate genes, some studies have targeted GWAS-identified loci as the set of candidate genes. Using this parallel approach, associations with CMA have been documented for TLR10/1/6 and IL2 29, FA with C11orf30/LRRC32, TMEM232/SLC25A46, TNFRSF6B/ZGPAT, OVOL1, KIF3A/IL13, GLB1, CCDC80, ZNF365, OR10A3/NLRP10, IL2/IL21, CLEC16A/DEXI, ZNF652, TSLP/WDR36, and STAT6 28, and FA with C11orf30/LRRC32 73.
An examination of the broad functional categories of these CGAS-identified genes offers support to the Dual Allergen hypothesis on the pathophysiology of FA with a prominent role for barrier and immunity related genes. A disrupted skin barrier may facilitate cutaneous exposure to food allergens, and immunological processes mediated by immune related molecules during sensitization may lead to the progression to FA subsequent to sensitization. In fact, the genes identified through the CGAS for FA can be broadly classified into four functional categories described in Table 1: (i) Genes playing roles in skin barrier integrity; (ii) Genes associated with innate immunity; (iii) Genes associated with adaptive immunity; and (iv) Immune-regulatory and immune-modulatory genes. These candidate genes often have more than one study with direct genetic evidence, or some orthogonal evidence from non-genetic data (Table E2). The CGAS approach is limited in that it does not have the capacity to identify novel genes that may add to our understanding of the pathophysiology of FA as each gene is selected based on prior knowledge. On the other hand, the advantage to this approach is that it is narrow in hypothesis and not limited by the stringent thresholds set in place with significance testing in the genome-wide approaches described below.
GENOME-WIDE ASSOCIATION STUDIES OF FOOD ALLERGY
The unbiased GWAS approach takes advantage of the genetic architecture of human populations wherein SNPs are often found within blocks of linkage disequilibrium (LD, i.e. where all SNPs within a block are highly correlated to each other). GWAS measures a reduced set of SNPs that can be used as a proxy for all remaining variants within the block. Importantly, the foundation for the GWAS approach is the “common disease, common variant” hypothesis, wherein common diseases are hypothesized to be attributable to many common genetic variants 74, 75. It is important to point out that the GWAS approach does not in fact implicate genes (unlike the CGAS above), but rather identifies genetic loci, and relies on fine-mapping (Box 1) follow-up approaches to hone in on causal genetic variants and specific genes within the loci. With no studies devoted to fine-mapping FA genetic loci, and the assignment of loci to genes by GWAS studies is driven by physical proximity of genes to the region of association for the summaries below.
There have been eight GWAS of FA (Table 1, Table E3) and overall, these highlight four points: (i) As expected, GWAS loci are generally common in frequency. (ii) They have modest effect sizes on average (ORs<1.5). (iii) The single most replicated region maps to the HLA region and HLA Class II genes (HLA-DR/DQ); and (iv) with the exception of HLA and FLG, there is no overlap in genes identified from the CGAS and GWAS approaches. As with CGAS, there is a great degree of variability in the definition of FA in the GWASs, and therefore difficulty in our ability to truly disentangle FA from FS, based on the ascertainment of cases and unaffected controls across these studies as outlined in Table E1. In fact, with some studies using unphenotyped population-based controls, we may also be limited in discerning FA genes from general atopy genes. There is also variability in definitions of GWAS thresholds, and for the purposes of this review we report all loci meeting at least a suggestive threshold defined by the authors (Table E3), but only summarize in detail those that pass the stronger, standard GWAS threshold (p<5x10−8) below. Unlike CGAS, CIs are not often reported for GWAS, and therefore not presented in this section.
The first GWAS locus for FA implicated the HLA region in the US-based Chicago Family Cohort Food Allergy Study 76 where they performed GWAS for any FA among nine foods (Table E1) and the three most common types of FA individually (peanut, egg white and cow’s milk). No genome-wide significant associations were found for FA overall, EA or CMA. Only one GWAS signal was identified for PA mapping to HLA class II genes. Two identified SNPs rs9275596 (OR=1.7, mapping to an intergenic region between HLA-DQB1 and HLA-DQA2 genes) and rs7192 (OR=1.7, a p.Leu242Val change in HLA-DRA) were highly correlated and represent a single risk factor for PA. In an extended approach looking at maternal effects, which serves as a proxy for the genetic interaction with the intrauterine environment 77, significant maternal effects were noted with FA at rs4235235 (RR=0.36, p=4.82x10−8) located in a noncoding RNA gene LOC101927947. However, no maternal effects were detected for PA, EA or CMA.
The second GWAS of FA was done on challenge-proven pediatric PA cases and controls from the HealthNuts cohort 78. Despite a small sample size (73 PA cases and 148 non-allergic controls), this multi-ethnic GWAS including European, Mixed (European/Asian) and Asian ancestries was able to identify a locus (rs10018666, OR=5.86) mapping to the SLC2A9 gene reaching genome-wide significance but the authors were unable to replicate this finding.
The European-based Genetics of Food Allergy (GOFA) Study 79 identified two loci associated with FA. The first of these mapped to the epidermal differentiation complex (EDC, rs12123821, OR=2.55) near the FLG gene and, second to the cytokine gene cluster (rs11949166, OR=0.60) on chromosome 5. Both loci had robust replication in the Chicago Family Cohort Food Allergy Study. Given that both the EDC and cytokine gene cluster region are known AD loci, they were specifically examined for their independent effects on FA, and both were found to retain independent effects of FA risk beyond AD. Combining the GOFA and Chicago Family Cohort Food Allergy Study revealed two additional FA susceptibility loci. The peak SNP on chromosome 11 (rs2212434, OR=1.29) is an intergenic variant between C11orf30 and LRRC32 genes that reached genome-wide significance for FA, but not any specific FA. The peak SNP on chromosome18 (rs12964116, OR=1.90) is located in intron1 of SERPINB and reached genome-wide significance for FA and was also associated with PA. A second SNP mapping to SERPINB (rs1243064, OR=1.65) was found for EA. This study was the second GWAS to map the HLA locus with a 3’UTR variant in HLA-DQB1 gene (rs9273440, OR=0.66) showing association with PA, but not with other specific foods or FA in general.
A more recent study by Asai et al 80 combined PA cases from the Canadian Peanut Allergy Registry (CanPAR) study with controls from the Busselton Health Study in Australia in a discovery analysis. An additional six cohorts were included in two approaches: (i) as replication datasets to follow up on the discovery results in CanPAR; and (ii) full meta-analysis framework including the (1) Chicago Food Allergy study, (2) Genetic Epidemiology Research on Aging (GERA) study, (3) HealthNuts study, (4) German UFA study, (5) Dutch IDEAL study and (6) Dutch GENEVA study. This meta-analysis reflects the single largest GWAS approach with ~36,000 samples. From the discovery/replication approach for PA, a locus (rs115218289, OR=0.18) mapping to the ITGA6 gene was identified to be associated with PA. This was also the third GWAS study to identify that SNPs mapping to the HLA region determine risk for PA (OR=0.51-2.11). The HLA SNPs identified in the discovery analysis were found to have a trend with reaction severity of PA, specificity to PA, and independence from asthma comorbidity 81.
In the only GWAS focused entirely in a non-European ancestry population, Khor and colleagues studied self-reported FA to seven foods (kiwi, peach, Chinese yam, eggs, mackerel, crab, and shrimp) in 11,011 adult Japanese women from the Luna Luna cohort 82. Once again, the HLA region the most strongly associated; in fact, it was the only GWAS signal for peach (rs28359885, OR=1.80) and shrimp (rs74995702, OR=1.9). While both foods had peak associations mapping to HLA, they are distinct, independent signals from each other, supporting the role of allergen specificity of the HLA region for FA. While this study is the largest single-study GWAS analysis, it should be noted that this is based entirely on a health questionnaire, and has limitations in its robustness of FA definition, and an inability to appropriately classify FS from FA.
The most recent GWAS of PA 31 tested for genetic determinants of PA in the Learning Early About Peanut Allergy (LEAP) study, and was the only GWAS to ‘control’ for environmental exposure by limiting the GWAS to only the children randomized to the peanut avoidance arm. They identified a genome-wide significant association signal on chromosome 18q21.32 mapping to the MALT1 gene with strong regulatory signatures for MALT1 gene expression. MALT1 gene functions as a critical part of the CARMA1-BCL10-MALT1 (CBM) complex, causing NFkB activation in B and T cells in response to antigen binding to their receptors and plays crucial role for both innate and adaptive immune responses 83. Next to the strong reported effect sizes for FLG as summarized in the CGAS section, this association with MALT1 was double in its effect size (OR=10.99, with 58.6% of carriers developing PA in contrast to 12.7% of non-carriers). Importantly, the authors provide evidence supporting its role as a genetic risk factor for progression to PA following sensitization. MALT1 has not been implicated in prior genetic studies, and the study was unable to identify FLG or HLA as determinants of PA despite strong prior evidence for these genes from CGAS and GWAS. One of the key features of the LEAP study is its ascertainment criteria; the inclusion of peanut sensitized participants at baseline facilitated the ability to test specifically for the risk of PA (beyond sensitization). This is in contrast to prior GWAS that may very well represent risk of sensitization and not specifically allergy.
Overall, the GWAS approach for FA has demonstrated convincing evidence for the CGAS documented HLA locus albeit with modest effect sizes (OR=0.47-2.11). In contrast, FLG, one of the strongest candidate genes does not have much support from the larger GWAS despite its large effect sizes from CGAS (OR=1.63-5.3, p=0.04-5.12x10−7), with the exception of the EDC locus in the GOFA GWAS 79. This is potentially because the LOF mutations are not available on the array used, the association did not meet traditional GWAS thresholds and were not reported within the GWAS framework or the nature of the unphenotyped population-based controls in GOFA. Despite the successes, the cumulative heritability in FA explained by these GWAS loci is limited; the five genome-wide significant loci identified by Marenholtz et al. 79 account for less than 50% of heritability estimated from whole-genome data suggesting other loci beyond these discoveries. A limitation with the GWAS approach is the SNPs identified from the GWAS may not be the true causal variants themselves but generally a proxy for some unmeasured disease-causing variant that needs additional follow-up for discovery, and thus far there has been very limited follow up of these loci. These GWAS have been plagued with small sample sizes, and heterogeneity in case definition similar to that described for the CGAS. In fact, the only attempted meta-analysis revealed sensitivity to FA definition criteria where the inclusion of studies with questionnaire-only diagnosis weakened results 80. Finally, and perhaps most important in the case of FA, these GWAS have all ignored GxE interactions with the exception of the most recent study in LEAP, and for a disease model where heterogeneity in exposure leads to heterogeneity in disease outcome, this could be an added limitation in power and the reason for limited results from the GWAS of FA, and residual unexplained heritability.
In addition to the four functional categories largely captured within the CGAS approach, the FA associated genes/loci identified through the unbiased GWAS approach additionally point out one the functional category of vascular and endothelial cell factors (Table 1). Genes related to vascular and endothelial cell factors could be involved in the pathophysiology of FA through 2 putative mechanisms: (i) An endothelial barrier defect promoting sensitization, and (ii) Endothelial cells acting as antigen presenting cells. In fact, across the landscape of FA genetic loci identified from both the GWAS and CGAS approaches there is a marked enrichment for both adaptive and innate immunity pathways (Fig S1).
HUMAN LEUKOCYTE ANTIGEN (HLA) GENES AND FA
The Major Histocompatibility Complex (MHC), lies on chromosome 6p21.3, and is rich in immune-response genes, including the HLA genes which encode families of cell-surface proteins that function as key determinants of antigen recognition by the adaptive immune system. Despite the HLA region being associated with more diseases than any other region of the genome 84, particularly with immune, infectious, and allergic diseases, it has been challenging to dissect its role in disease etiology. This is due in part to extremely high levels of genomic polymorphism (referred to as alleles) at several of these loci (e.g. the class I HLA-A, - B, and -C genes, and the class II -DRB1, -DQB1, -DQA1, -DPB1, and -DPA1 genes) and strong LD across the region. There are >26,000 alleles at the HLA genes identified to date 85, most of which are within the peptide-binding groove of the HLA molecule or in the region that interacts with the T-cell receptor 86. The result is a diverse array of HLA molecules that selectively bind peptides derived from degraded proteins in a process known as antigen presentation. T lymphocytes that contain clonally-restricted receptors for these peptide-HLA ligands undergo a differentiation and maturation cascade upon binding that ultimately determines whether the resulting immune response is inflammatory, regulatory, or – as is often the case – both. Depending on the particular peptide-HLA combination and on tissue microenvironmental cues, such as concurrent cytokine recognition, these T cell responses can direct B lymphocytes (a process known as ‘T cell help’) to produce antigen-specific immunoglobulins of various isotypes, such as the IgE molecules that have critical effector functions in allergy. This sequence of events offers several steps in which genetic polymorphisms within HLA genes may influence allergic outcomes, including binding of specific peptides to HLA molecules, initial recognition or lack of recognition of particular allergens, the type of initial T cell response 87, 88, the balance between regulatory and helper T cells 89, 90, and interactions between T and B cells that influence immunoglobulin isotype production 91, 92.
For FA, the HLA locus is by far the strongest evidence from the GWAS, but as noted above, these GWAS associations are at the locus- and not gene-level. Given the importance of HLA in antigen presentation, and the extensive work looking specifically at HLA alleles, we provide a detailed overview of the associations at the allelic level with FA in this section (Table E4). As only some studies report CIs, we only summarize ORs in this section. HLA nomenclature is a standardized process 85, but has changed over the course of the available publications for phenotypes in the atopic march. To simplify interpretation, we only report results where the HLA allele has up to two digits resolution (sets of digits separated by a colon ‘:’). The digits before the first colon (e.g. HLA-DQA1*01) references the serological antigen type, and the next set of digits (e.g. HLA-DQA1*01:02) is the subtype. Alleles whose numbers differ in the two sets of digits differ in one or more nucleotide substitutions that change the amino acid sequence of the encoded protein. The HLA region is also widely implicated across the atopic march (Figure 1), therefore, we provide a contrast of these associations at the allelic level for FA and across other atopic march phenotypes (asthma 93, AD 94 and AR 9) in Figure 2.
Fig 2: HLA risk alleles associated with asthma 93, AD 94, AR 9 and four different food allergens.
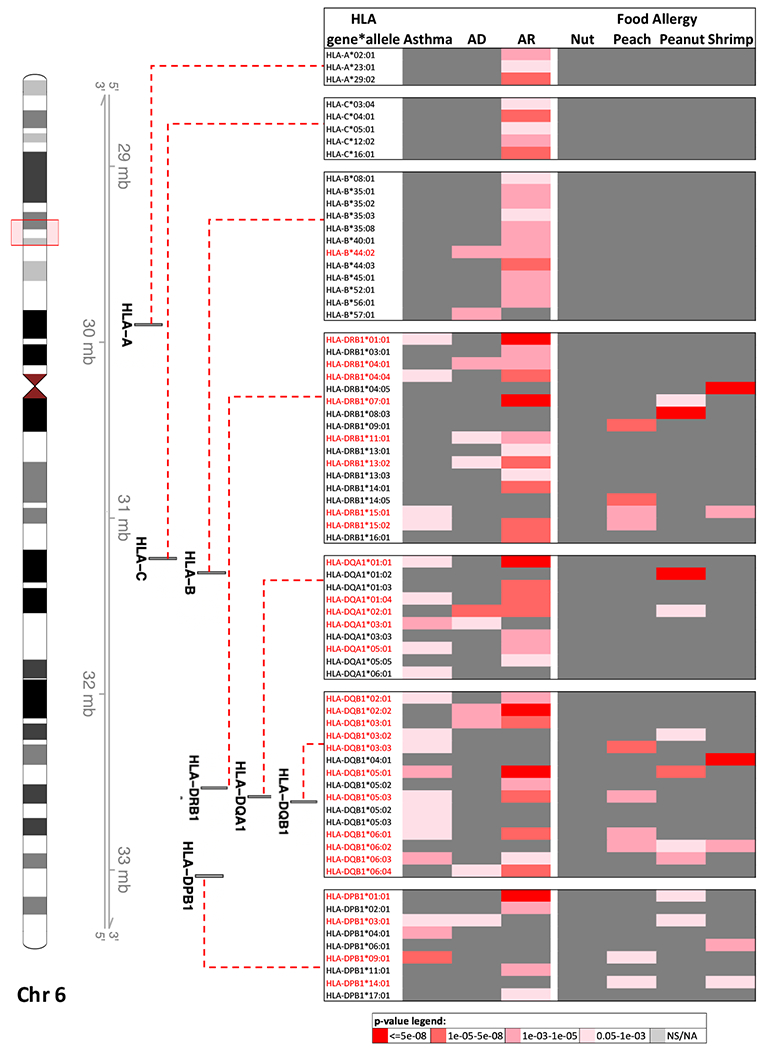
Red box on the ideogram of chromosome 6 highlights the HLA region in the human genome. The association between four-digit alleles of three HLA class I genes (HLA-A, B and C), and four HLA class II genes (HLA-DQA1, DQB1, DRB1 and DPB1) and the atopic phenotypes are presented. Alleles of HLA class II genes are associated with all phenotypes whereas alleles of class I genes are largely associated with AR.
In the first report on HLA alleles, Boehncke et al 95 compared FA cases with pollinosis to healthy controls. They tested for association with 8 food allergens and reported that patients with the HLA-DRB1*08 allele had a high risk of PA (RR=6.04). In a family-based cohort, Howell et al 96 reported an increased risk for PA for DRB1*08 (OR=4.37) and DQB1*04 (OR=6.79). In 2006, Shreffler et al 97 studied sibling pairs for 7 HLA-DQ and 18 HLA-DR alleles, and reported nominal associations between PA and DQ7 (*03:01, *03:04, *03:05, *03:09, *03:10, *03:12 and *03:13, ORs=NR). In a study of peanut-allergic subjects and their peanut-tolerant full siblings 98, Dreskin et al looked at how differences in levels of peanut-specific (ps) IgE and psIgG might be related to differences in the ability to present peanut allergens in a thorough evaluation of 59 HLA class II alleles of HLA-DRB1, HLA-DQB1 and HLA-DPB1 genes. No associations were noted between psIgG and psIgE levels and HLA class II alleles. However, it is highly likely that the quantitative level of psIgE and psIgG4 is a function of GxE interaction between exposure to peanut antigen and the specific HLA alleles that are involved in the presentation of peanut peptides. Therefore, this negative finding has major limitations because interactions with exposure were not considered.
The largest CGAS study on HLA was performed by Madore and colleagues 99. They found associations between PA and five HLA-DQB1 alleles. DQB1*02 (OR=0.12), DQB1*03:02 (OR=0.52), DQB1*05 (OR=0.21), DQB1*05:01 (OR=0.25) alleles were protective, and DQB1*06:03 (OR=2.59) was a risk allele for PA. Given that HLA-DQB1 gene is also a susceptibility gene for asthma they adjusted their analysis for asthma and two alleles DQB1*02 (OR=0.09) and DQB1*06:03 (OR=2.82) continued to show their association with PA. HLA associations are not limited to PA; nominal associations have also been observed for HLA-B*07 and HLA-DRB1*11 for nut allergic patients compared to atopic controls, and HLA-DRB1*13 and HLA-DQB1*06 comparing patients to blood donor controls 100. HLA class II haplotypes carrying HLA II alleles DQB1*06:02 and DQB1*05:01 were also found to modulate antibody responses to CM proteins 30 revealing both risk and protective alleles at DQB1 for CMA related traits.
With the availability of bioinformatic approaches to leverage SNP GWAS data to impute HLA alleles 101–103, several of the GWAS studies have used this approach to look beyond the SNPs (described in the GWAS review above) to examine specific HLA alleles with FA risk. In the first GWAS of FA, 50 HLA class II alleles were analyzed for PA in a European ancestry population 76. They identified two alleles (HLA-DQA1*01:02, OR=NR and HLA-DQB1*06, OR=NR) as risk alleles for PA. In the second GWAS of PA, Martino et al performed an in-depth exploration of HLA alleles for PA. Although no result reached genome-wide significance for PA, they were able to replicate both these finding (HLA-DQA1*01:02, OR=1.81 and HLA-DQB1*06, OR=2.26) reported by Hong et al. 76 with similar directions of effect. HLA alleles have also been examined for peach and shrimp allergies 82. HLA-DRB1 (*09:01, *14:05, *15:01, and *15:02), HLA-DQB1 (*03:03, *06:02, *05:03 and *06:01) and HLA-DPB1 (*14:01 and *09:01) were all found to increase risk for peach allergy (OR=1.4-2.5, Table E4). For shrimp allergy, HLA-DRB1 (*04:05 and *15:01), HLA-DQB1 (*04:01 and *06:02) and HLA-DPB1 (*06:01 and *14:01) were found to increase risk (OR=1.55-5.24, Table E4). It is important to note the alleles for the two foods are different from each other, and also different from HLA-DQA1*01:02 identified for PA, reflecting a specificity for allergen.
It is challenging to dissect independence between the associations from the GWAS SNPs and the HLA alleles/haplotypes, limiting our ability to test if there are quantitative (SNPs identified are often strong regulatory SNPs for HLA gene expression across multiple HLA genes) or qualitative (alleles representing binding of specific allergenic peptides forming ligands for specific T cell recognition) effects or both. Notably, extensive LD within the HLA region results in the co-segregation of combinations of HLA allelic variants at multiple loci. Indeed, several of the frequent allergy-associated HLA alleles mentioned above, such as DQB1*06:02 and DQA1*01:02, co-exist on a single haplotype together with particular DRB1 and DRB5 alleles, raising the additional question whether a single gene or a combination of these genes are driving the association with allergy. Nonetheless, the summary in Figure 2 highlights the following key features of HLA associations across phenotypes in the atopic march: (i) HLA-DR/DQ associations have consistent effects on FA when it comes to the atopic march. Often, the alleles are associated with one but not multiple phenotypes within the atopic march, and unlike FLG where the same LOF alleles convey risk across the atopic march phenotypes, here the specific HLA associations with FAs are often distinct from those with asthma 93 and AD 94. (ii) even within FA, HLA allelic associations often differ between the specific allergen, for example the HLA-DQA1*01:02 is noted specifically for peanut and no other FA or atopic march phenotype. While for many of the HLA alleles, associations across phenotypes of the atopic march are unique, alleles at the HLA-DQB1 gene tend to be shared between atopic asthma and FA. While some part of this could be reflective of tight LD with non-coding regulatory variants described above, we acknowledge that some part of this may also reflect cross reactivity across allergens manifesting as associations noted across allergic phenotypes.
GENE-ENVIRONMENT INTERACTIONS IN FOOD ALLERGY
The role of environmental factors as key determinants of FA risk has been well established 104. Many of these environment factors are thought to impact the development and regulation of the immune system either directly or indirectly. However, there is very limited understanding of how these factors affect the development of FA in the context of genetics. Despite the importance of the environment in FA risk, and the need to consider GxE interaction in a systematic fashion, very few such studies have been conducted to date.
Brough et al 105 tested the hypothesis that early life environmental peanut exposure may be a risk for PA development, and tested whether this relationship was affected by FLG LOF mutations in European children from the Manchester Asthma and Allergy Study (MAAS). In carriers of FLG LOF mutations, there was a dose-response relationship between early-life environmental exposure to peanut protein in household dust and subsequent peanut sensitization (PS) and PA. Each 2.7-fold increase in house dust peanut exposure during infancy was associated with a more than 6-fold increase in the odds of PS and a 3.3-fold increase in the odds of PA. Koplin et al 2015 106 tested whether polymorphisms in the DBP/GC gene that lower the vitamin D binding proteins (DBP) of the offspring could compensate for adverse effects of low serum vitamin D on FA risk at 1 and 2 years of age from the HealthNuts Study. The results from the study showed that low 25[OH]D3 serum levels (OR=6, 95%CI=0.9-38.9) and high vitamin D levels (OR=4, 95%CI=1.3-12.9) of the children were associated with FA only in participants with GG genotype at the rs7041 SNP at 1 year of age. The relationship between maternal use of vitamin D supplements during pregnancy and FA risk was also modified by the variant, with a significantly reduced risk of FA in infants with the GT/TT genotype (OR=0.1, 95%CI=0.03-0.41, P=0.001) but not those with GG genotype (OR=0.34, 95%CI=0.04-2.79, P=0.32). In contrast, associations between infant formula use with added vitamin D and FA was not modified by genotype. They also found that vitamin D insufficiency at 2 years was associated with persistent EA (OR=12.96) among infants with GG genotype.
The only GWAS allowing for an unbiased genome wide evaluation of GxE effects on PA is the LEAP study 31. The setting of the clinical trial with high risk participants randomized to peanut exposure or avoidance allowed for a stratified GWAS approach, and a strong GxE interaction is noted for the MALT1 risk allele for PA when analysis was extended to quantitative psIgE levels across the full set of consumers and avoiders as described above 31.
GENETICS OF EOSINOPHILIC ESOPHAGITIS (EOE)
EoE is a chronic inflammatory condition characterized by esophageal eosinophilia and dysphagia 107. The food related inflammation during EoE pathogenesis is classified as a mixed immune reaction 6, i.e. both IgE and cell-mediated immune responses. Candidate gene based studies of EoE have shown that CCL26 108 and FLG 109 are associated with disease susceptibility, and several loci including TSLP|WDR36 110, CAPN14 111,112, C11orf30|LRRC32 112, STAT6 112, ANKRD27 112, CLEC16A 113 have been identified through GWAS. Interestingly, genetic analysis study of EoE based on Immunochip platform found lack of association of EoE with the genetic variants in the HLA region genes 113. While there are dissimilarities between immune reactions occurring during FA and EoE pathogenesis, there are overlapping genes and genetic loci (C11ORF30|LRRC32 28, 73, 79, 80, CLEC16A|DEXI 28, FLG 23–26,28, STAT6 28, 40, 42, TSLP|WDR36 28) between the two that may suggest partly overlapping mechanism. For example, given similarities in the mucosal structure of the skin and the esophagus 114, disrupted barrier integrity due to FLG LOF mutations in the skin may cause cutaneous exposure to food allergen leading to FA, and in the esophagus may cause allergen-induced eosinophilic infiltration resulting in EoE.
CONCLUDING REMARKS
This review systematically collates the genetic association studies of FA where we have compiled the list of genetic loci, genes and specific variants that show associations with FA and FS. It is useful to summarize these observations in the context of the RR to sibs (λs), which for monogenic diseases or largely genetic disorders, tends to be high (e.g. λs ~ 500 for Cystic Fibrosis); but for FA is 1.3-fold to 12 with a complex interplay between environment and genes – a far more daunting challenge for discovery. While the reviewed studies are plagued by low power, heterogeneity in definition of FA, variability in the comparison control group, and difficulty in our ability to truly disentangle FA from FS and general atopy genetics, they have no doubt led to a set of loci, genes and variants that explain risk for FA in part. With respect to biological mechanism in FA, the genetic loci point strongly to the critical role of barrier and immune function both at individual gene levels (Table 1) and at pathway-based levels (Fig S1). In fact, the largest reported genetic effects on FA are from MALT1 (OR=10.99), FLG LOF (average OR~2.9) and HLA alleles (average OR~2.03) pointing to immune and barrier related mechanism. Taken together with the striking overlap between FA genetics and AD, asthma and AR genetics these loci offer collective support of prior hypotheses such as the atopic march and Dual Allergen Hypothesis. Despite these successes, the greatest challenge in the field of FA genetics is to elucidate the effect of associated variant on expression, regulation and function of associated gene(s) and to better understand the specific mechanism of action on FA risk and pathogenesis. The integration of genetic studies with other omics studies such as transcriptomics, proteomics and metabolomics hold great promise in understanding mechanism as discussed in our companion review 115, and this needs to be considered within study designs that better define outcome, enable the dissection of FA from FS, distinguish FA genetics from general atopy genes, and most importantly consider environmental interactions.
Supplementary Material
Acknowledgements:
We thank Drs Gerald Nepom, Karen Cerosaletti and Carole Ober for their review and feedback on the manuscript.
Funding:
Drs. Kanchan and Mathias are supported in part by the Immune Tolerance Network, an international clinical research consortium headquartered at the Benaroya Research Institute, and by the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH) under Award Number UM1AI109565. Drs. Bunyavanich and Irizar are supported in part by NIH grants: NIH R01 AI 147028, U19 AI 136053, R01 AI118833. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
LIST OF ABBREVIATIONS
- FA
Food allergy
- FS
Food sensitization
- FIA
Food induced anaphylaxis
- PA
Peanut allergy
- EA
Egg allergy
- CMA
Cow’s milk allergy
- SPT
Skin prick test
- IgE
Immunoglobulin E
- IgG4
Immunoglobulin G4
- OFC
Oral food challenge
- DBPCFC
Double blinded placebo controlled oral food challenge
- AD
Atopic dermatitis
- AR
Allergic rhinitis
- CGAS
Candidate gene association studies
- GWAS
Genome-wide association studies
- GxE
Gene by environment interactions
- SNP
Single nucleotide polymorphism
- OR
Odds ratio
- RR
Relative risk
- LD
Linkage Disequilibrium
Footnotes
Conflicts of Interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Tham EH, Leung DYM. How Different Parts of the World Provide New Insights Into Food Allergy. Allergy Asthma Immunol Res. 2018;10(4):290–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sicherer SH, Sampson HA. Food allergy. J Allergy Clin Immunol. 2010;125(2 Suppl 2):S116–25. [DOI] [PubMed] [Google Scholar]
- 3.du Toit G, Tsakok T, Lack S, Lack G. Prevention of food allergy. J Allergy Clin Immunol. 2016;137(4):998–1010. [DOI] [PubMed] [Google Scholar]
- 4.Spergel JM. From atopic dermatitis to asthma: the atopic march. Ann Allergy Asthma Immunol. 2010;105(2):99–106; quiz 7-9, 17. [DOI] [PubMed] [Google Scholar]
- 5.Wahn U What drives the allergic march? Allergy. 2000;55(7):591–9. [DOI] [PubMed] [Google Scholar]
- 6.Sicherer SH, Sampson HA. Food allergy: A review and update on epidemiology, pathogenesis, diagnosis, prevention, and management. J Allergy Clin Immunol. 2018;141(1):41–58. [DOI] [PubMed] [Google Scholar]
- 7.Paternoster L, Standl M, Waage J, Baurecht H, Hotze M, Strachan DP, et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat Genet. 2015;47(12):1449–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pividori M, Schoettler N, Nicolae DL, Ober C, Im HK. Shared and distinct genetic risk factors for childhood-onset and adult-onset asthma: genome-wide and transcriptome-wide studies. Lancet Respir Med. 2019;7(6):509–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waage J, Standl M, Curtin JA, Jessen LE, Thorsen J, Tian C, et al. Genome-wide association and HLA fine-mapping studies identify risk loci and genetic pathways underlying allergic rhinitis. Nat Genet. 2018;50(8):1072–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tham EH, Leung DY. Mechanisms by Which Atopic Dermatitis Predisposes to Food Allergy and the Atopic March. Allergy Asthma Immunol Res. 2019;11(1):4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davidson WF, Leung DYM, Beck LA, Berin CM, Boguniewicz M, Busse WW, et al. Report from the National Institute of Allergy and Infectious Diseases workshop on “Atopic dermatitis and the atopic march: Mechanisms and interventions”. J Allergy Clin Immunol. 2019;143(3):894–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hourihane JO, Dean TP, Warner JO. Peanut allergy in relation to heredity, maternal diet, and other atopic diseases: results of a questionnaire survey, skin prick testing, and food challenges. BMJ. 1996;313(7056):518–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sicherer SH, Furlong TJ, Maes HH, Desnick RJ, Sampson HA, Gelb BD. Genetics of peanut allergy: a twin study. J Allergy Clin Immunol. 2000;106(1 Pt 1):53–6. [DOI] [PubMed] [Google Scholar]
- 14.Liu X, Zhang S, Tsai HJ, Hong X, Wang B, Fang Y, et al. Genetic and environmental contributions to allergen sensitization in a Chinese twin study. Clin Exp Allergy. 2009;39(7):991–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liem JJ, Huq S, Kozyrskyj AL, Becker AB. Should Younger Siblings of Peanut-Allergic Children Be Assessed by an Allergist before Being Fed Peanut? Allergy Asthma Clin Immunol. 2008;4(4):144–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsai HJ, Kumar R, Pongracic J, Liu X, Story R, Yu Y, et al. Familial aggregation of food allergy and sensitization to food allergens: a family-based study. Clin Exp Allergy. 2009;39(1):101–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koplin JJ, Allen KJ, Gurrin LC, Peters RL, Lowe AJ, Tang ML, et al. The impact of family history of allergy on risk of food allergy: a population-based study of infants. Int J Environ Res Public Health. 2013;10(11):5364–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupta RS, Walkner MM, Greenhawt M, Lau CH, Caruso D, Wang X, et al. Food Allergy Sensitization and Presentation in Siblings of Food Allergic Children. J Allergy Clin Immunol Pract. 2016;4(5):956–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Irvine AD, McLean WH, Leung DY. Filaggrin mutations associated with skin and allergic diseases. N Engl J Med. 2011;365(14):1315–27. [DOI] [PubMed] [Google Scholar]
- 20.Rodriguez E, Baurecht H, Herberich E, Wagenpfeil S, Brown SJ, Cordell HJ, et al. Meta-analysis of filaggrin polymorphisms in eczema and asthma: robust risk factors in atopic disease. J Allergy Clin Immunol. 2009;123(6):1361–70 e7. [DOI] [PubMed] [Google Scholar]
- 21.van den Oord RA, Sheikh A. Filaggrin gene defects and risk of developing allergic sensitisation and allergic disorders: systematic review and meta-analysis. BMJ. 2009;339:b2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lack G Update on risk factors for food allergy. J Allergy Clin Immunol. 2012;129(5):1187–97. [DOI] [PubMed] [Google Scholar]
- 23.Brown SJ, Asai Y, Cordell HJ, Campbell LE, Zhao Y, Liao H, et al. Loss-of-function variants in the filaggrin gene are a significant risk factor for peanut allergy. J Allergy Clin Immunol. 2011;127(3):661–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tan HT, Ellis JA, Koplin JJ, Matheson MC, Gurrin LC, Lowe AJ, et al. Filaggrin loss-of-function mutations do not predict food allergy over and above the risk of food sensitization among infants. J Allergy Clin Immunol. 2012;130(5):1211–3 e3. [DOI] [PubMed] [Google Scholar]
- 25.Asai Y, Greenwood C, Hull PR, Alizadehfar R, Ben-Shoshan M, Brown SJ, et al. Filaggrin gene mutation associations with peanut allergy persist despite variations in peanut allergy diagnostic criteria or asthma status. J Allergy Clin Immunol. 2013;132(1):239–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Venkataraman D, Soto-Ramirez N, Kurukulaaratchy RJ, Holloway JW, Karmaus W, Ewart SL, et al. Filaggrin loss-of-function mutations are associated with food allergy in childhood and adolescence. J Allergy Clin Immunol. 2014;134(4):876–82 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ziyab AH, Karmaus W, Yousefi M, Ewart S, Schauberger E, Holloway JW, et al. Interplay of filaggrin loss-of-function variants, allergic sensitization, and eczema in a longitudinal study covering infancy to 18 years of age. PLoS One. 2012;7(3):e32721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirota T, Nakayama T, Sato S, Yanagida N, Matsui T, Sugiura S, et al. Association study of childhood food allergy with genome-wide association studies-discovered loci of atopic dermatitis and eosinophilic esophagitis. J Allergy Clin Immunol. 2017;140(6):1713–6. [DOI] [PubMed] [Google Scholar]
- 29.Henneman P, Petrus NCM, Venema A, van Sinderen F, van der Lip K, Hennekam RC, et al. Genetic susceptibility for cow’s milk allergy in Dutch children: the start of the allergic march? Clin Transl Allergy. 2015;6:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Savilahti EM, Ilonen J, Kiviniemi M, Saarinen KM, Vaarala O, Savilahti E. Human leukocyte antigen (DR1)-DQB1*0501 and (DR15)-DQB1*0602 haplotypes are associated with humoral responses to early food allergens in children. Int Arch Allergy Immunol. 2010;152(2):169–77. [DOI] [PubMed] [Google Scholar]
- 31.Winters A, Bahnson HT, Ruczinski I, Boorgula MP, Malley C, Keramati AR, et al. The MALT1 locus and peanut avoidance in the risk for peanut allergy. J Allergy Clin Immunol. 2019;143(6):2326–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38(4):441–6. [DOI] [PubMed] [Google Scholar]
- 33.Marenholz I, Nickel R, Ruschendorf F, Schulz F, Esparza-Gordillo J, Kerscher T, et al. Filaggrin loss-of-function mutations predispose to phenotypes involved in the atopic march. J Allergy Clin Immunol. 2006;118(4):866–71. [DOI] [PubMed] [Google Scholar]
- 34.Magert HJ, Standker L, Kreutzmann P, Zucht HD, Reinecke M, Sommerhoff CP, et al. LEKTI, a novel 15-domain type of human serine proteinase inhibitor. J Biol Chem. 1999;274(31):21499–502. [DOI] [PubMed] [Google Scholar]
- 35.Kusunoki T, Okafuji I, Yoshioka T, Saito M, Nishikomori R, Heike T, et al. SPINK5 polymorphism is associated with disease severity and food allergy in children with atopic dermatitis. J Allergy Clin Immunol. 2005;115(3):636–8. [DOI] [PubMed] [Google Scholar]
- 36.Ashley SE, Tan HT, Vuillermin P, Dharmage SC, Tang MLK, Koplin J, et al. The skin barrier function gene SPINK5 is associated with challenge-proven IgE-mediated food allergy in infants. Allergy. 2017;72(9):1356–64. [DOI] [PubMed] [Google Scholar]
- 37.Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996;4(3):313–9. [DOI] [PubMed] [Google Scholar]
- 38.Takeda K, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura S, et al. Essential role of Stat6 in IL-4 signalling. Nature. 1996;380(6575):627–30. [DOI] [PubMed] [Google Scholar]
- 39.Takeda K, Kamanaka M, Tanaka T, Kishimoto T, Akira S. Impaired IL-13-mediated functions of macrophages in STAT6-deficient mice. J Immunol. 1996;157(8):3220–2. [PubMed] [Google Scholar]
- 40.Amoli MM, Hand S, Hajeer AH, Jones KP, Rolf S, Sting C, et al. Polymorphism in the STAT6 gene encodes risk for nut allergy. Genes Immun. 2002;3(4):220–4. [DOI] [PubMed] [Google Scholar]
- 41.Tamura K, Suzuki M, Arakawa H, Tokuyama K, Morikawa A. Linkage and association studies of STAT6 gene polymorphisms and allergic diseases. Int Arch Allergy Immunol. 2003;131(1):33–8. [DOI] [PubMed] [Google Scholar]
- 42.van Ginkel CD, Pettersson ME, Dubois AEJ, Koppelman GH. Association of STAT6 gene variants with food allergy diagnosed by double-blind placebo-controlled food challenges. Allergy. 2018;73(6):1337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koppelman GH, Reijmerink NE, Colin Stine O, Howard TD, Whittaker PA, Meyers DA, et al. Association of a promoter polymorphism of the CD14 gene and atopy. Am J Respir Crit Care Med. 2001;163(4):965–9. [DOI] [PubMed] [Google Scholar]
- 44.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2(8):675–80. [DOI] [PubMed] [Google Scholar]
- 45.Woo JG, Assa’ad A, Heizer AB, Bernstein JA, Hershey GK. The-159 C-->T polymorphism of CD14 is associated with nonatopic asthma and food allergy. J Allergy Clin Immunol. 2003;112(2):438–44. [DOI] [PubMed] [Google Scholar]
- 46.Campos E, Shimojo N, Inoue Y, Arima T, Suzuki S, Tomiita M, et al. No association of polymorphisms in the 5’ region of the CD14 gene and food allergy in a Japanese population. Allergol Int. 2007;56(1):23–7. [DOI] [PubMed] [Google Scholar]
- 47.Dreskin SC, Ayars A, Jin Y, Atkins D, Leo HL, Song B. Association of genetic variants of CD14 with peanut allergy and elevated IgE levels in peanut allergic individuals. Ann Allergy Asthma Immunol. 2011;106(2):170–2. [DOI] [PubMed] [Google Scholar]
- 48.Rigoli L, Briuglia S, Caimmi S, Ferrau V, Gallizzi R, Leonardi S, et al. Gene-environment interaction in childhood asthma. Int J Immunopathol Pharmacol. 2011;24(4 Suppl):41–7. [DOI] [PubMed] [Google Scholar]
- 49.Borish L, Aarons A, Rumbyrt J, Cvietusa P, Negri J, Wenzel S. Interleukin-10 regulation in normal subjects and patients with asthma. J Allergy Clin Immunol. 1996;97(6):1288–96. [DOI] [PubMed] [Google Scholar]
- 50.Aubert JD, Dalal BI, Bai TR, Roberts CR, Hayashi S, Hogg JC. Transforming growth factor beta 1 gene expression in human airways. Thorax. 1994;49(3):225–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Campos Alberto EJ, Shimojo N, Suzuki Y, Mashimo Y, Arima T, Matsuura T, et al. IL-10 gene polymorphism, but not TGF-beta1 gene polymorphisms, is associated with food allergy in a Japanese population. Pediatr Allergy Immunol. 2008;19(8):716–21. [DOI] [PubMed] [Google Scholar]
- 52.Jacob CM, Pastorino AC, Okay TS, Castro AP, Gushken AK, Watanabe LA, et al. Interleukin 10 (IL10) and transforming growth factor beta1 (TGFbeta1) gene polymorphisms in persistent IgE-mediated cow’s milk allergy. Clinics (Sao Paulo). 2013;68(7):1004–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen TK, Lee JH, Yu HH, Yang YH, Wang LC, Lin YT, et al. Association between human IL-10 gene polymorphisms and serum IL-10 level in patients with food allergy. J Formos Med Assoc. 2012;111(12):686–92. [DOI] [PubMed] [Google Scholar]
- 54.Gaudieri S, Lucas M, Lucas A, McKinnon E, Albloushi H, Rauch A, et al. Genetic variations in IL28B and allergic disease in children. PLoS One. 2012;7(1):e30607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McKenzie GJ, Emson CL, Bell SE, Anderson S, Fallon P, Zurawski G, et al. Impaired development of Th2 cells in IL-13-deficient mice. Immunity. 1998;9(3):423–32. [DOI] [PubMed] [Google Scholar]
- 56.Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363(13):1211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsunemi Y, Saeki H, Nakamura K, Sekiya T, Hirai K, Kakinuma T, et al. Interleukin-13 gene polymorphism G4257A is associated with atopic dermatitis in Japanese patients. J Dermatol Sci. 2002;30(2):100–7. [DOI] [PubMed] [Google Scholar]
- 58.Ying XJ, Zhao SW, Wang GL, Xie J, Xu HM, Dong P. Association of interleukin-13 SNP rs20541 with allergic rhinitis risk: a meta-analysis. Gene. 2013;521(2):222–6. [DOI] [PubMed] [Google Scholar]
- 59.Ashley SE, Tan HT, Peters R, Allen KJ, Vuillermin P, Dharmage SC, et al. Genetic variation at the Th2 immune gene IL13 is associated with IgE-mediated paediatric food allergy. Clin Exp Allergy. 2017;47(8):1032–7. [DOI] [PubMed] [Google Scholar]
- 60.Gawronska-Szklarz B, Pawlik A, Czaja-Bulsa G, Gornik W, Luszawska-Kutrzeba T, Wrzesniewska J. Genotype of N-acetyltransferase 2 (NAT2) polymorphism in children with immunoglobulin E-mediated food allergy. Clin Pharmacol Ther. 2001;69(5):372–8. [DOI] [PubMed] [Google Scholar]
- 61.Orzechowska-Juzwenko K, Milejski P, Patkowski J, Nittner-Marszalska M, Malolepszy J. Acetylator phenotype in patients with allergic diseases and its clinical significance. Int J Clin Pharmacol Ther Toxicol. 1990;28(10):420–5. [PubMed] [Google Scholar]
- 62.Zielinska E, Niewiarowski W, Bodalski J, Stanczyk A, Bolanowski W, Rebowski G. Arylamine N-acetyltransferase (NAT2) gene mutations in children with allergic diseases. Clin Pharmacol Ther. 1997;62(6):635–42. [DOI] [PubMed] [Google Scholar]
- 63.Gawronska-Szklarz B, Luszawska-Kutrzeba T, Czaja-Bulsa G, Kurzawski G. Relationship between acetylation polymorphism and risk of atopic diseases. Clin Pharmacol Ther. 1999;65(5):562–9. [DOI] [PubMed] [Google Scholar]
- 64.Ting JP, Willingham SB, Bergstralh DT. NLRs at the intersection of cell death and immunity. Nat Rev Immunol. 2008;8(5):372–9. [DOI] [PubMed] [Google Scholar]
- 65.Villani AC, Lemire M, Fortin G, Louis E, Silverberg MS, Collette C, et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat Genet. 2009;41(1):71–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hitomi Y, Ebisawa M, Tomikawa M, Imai T, Komata T, Hirota T, et al. Associations of functional NLRP3 polymorphisms with susceptibility to food-induced anaphylaxis and aspirin-induced asthma. J Allergy Clin Immunol. 2009;124(4):779–85 e6. [DOI] [PubMed] [Google Scholar]
- 67.Rahmoun N, El Mecherfi KE, Bouchetara A, Lardjem Hetraf S, Dahmani Amira C, Adda Neggaz L, et al. Association of REL Polymorphism with Cow’s Milk Proteins Allergy in Pediatric Algerian Population. Fetal Pediatr Pathol. 2018;37(1):74–83. [DOI] [PubMed] [Google Scholar]
- 68.Cho JH, Brant SR. Recent insights into the genetics of inflammatory bowel disease. Gastroenterology. 2011;140(6):1704–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Varade J, Palomino-Morales R, Ortego-Centeno N, Diaz-Rubio M, Fernandez-Gutierrez B, Gonzalez-Gay MA, et al. Analysis of the REL polymorphism rs13031237 in autoimmune diseases. Ann Rheum Dis. 2011;70(4):711–2. [DOI] [PubMed] [Google Scholar]
- 70.Pawlik A, Carlsson L, Meisel P, Czaja-Bulsa G, Mokrzycka M, Gawronska-Szklarz B. The FcgammaRIIa polymorphism in children with atopic diseases. Int Arch Allergy Immunol. 2004;133(3):233–8. [DOI] [PubMed] [Google Scholar]
- 71.Galli E, Ciucci A, Cersosimo S, Pagnini C, Avitabile S, Mancino G, et al. Eczema and food allergy in an Italian pediatric cohort: no association with TLR-2 and TLR-4 polymorphisms. Int J Immunopathol Pharmacol. 2010;23(2):671–5. [DOI] [PubMed] [Google Scholar]
- 72.Buyuktiryaki B, Sahiner UM, Girgin G, Birben E, Soyer OU, Cavkaytar O, et al. Low indoleamine 2,3-dioxygenase activity in persistent food allergy in children. Allergy. 2016;71(2):258–66. [DOI] [PubMed] [Google Scholar]
- 73.Marenholz I, Grosche S, Ruschendorf F, Kalb B, Blumchen K, Schlags R, et al. Evaluation of food allergy candidate loci in the Genetics of Food Allergy study. J Allergy Clin Immunol. 2018;142(4):1368–70 e2. [DOI] [PubMed] [Google Scholar]
- 74.Collins FS, Guyer MS, Charkravarti A. Variations on a theme: cataloging human DNA sequence variation. Science. 1997;278(5343):1580–1. [DOI] [PubMed] [Google Scholar]
- 75.Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet. 2001;17(9):502–10. [DOI] [PubMed] [Google Scholar]
- 76.Hong X, Hao K, Ladd-Acosta C, Hansen KD, Tsai HJ, Liu X, et al. Genome-wide association study identifies peanut allergy-specific loci and evidence of epigenetic mediation in US children. Nat Commun. 2015;6:6304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu X, Hong X, Tsai HJ, Mestan KK, Shi M, Kefi A, et al. Genome-wide association study of maternal genetic effects and parent-of-origin effects on food allergy. Medicine (Baltimore). 2018;97(9):e0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Martino DJ, Ashley S, Koplin J, Ellis J, Saffery R, Dharmage SC, et al. Genomewide association study of peanut allergy reproduces association with amino acid polymorphisms in HLA-DRB1. Clin Exp Allergy. 2017;47(2):217–23. [DOI] [PubMed] [Google Scholar]
- 79.Marenholz I, Grosche S, Kalb B, Ruschendorf F, Blumchen K, Schlags R, et al. Genome-wide association study identifies the SERPINB gene cluster as a susceptibility locus for food allergy. Nat Commun. 2017;8(1):1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Asai Y, Eslami A, van Ginkel CD, Akhabir L, Wan M, Ellis G, et al. Genome-wide association study and meta-analysis in multiple populations identifies new loci for peanut allergy and establishes C11orf30/EMSY as a genetic risk factor for food allergy. J Allergy Clin Immunol. 2018;141(3):991–1001. [DOI] [PubMed] [Google Scholar]
- 81.Asai Y, Eslami A, van Ginkel CD, Akhabir L, Wan M, Yin D, et al. A Canadian genome-wide association study and meta-analysis confirm HLA as a risk factor for peanut allergy independent of asthma. J Allergy Clin Immunol. 2018;141(4):1513–6. [DOI] [PubMed] [Google Scholar]
- 82.Khor SS, Morino R, Nakazono K, Kamitsuji S, Akita M, Kawajiri M, et al. Genome-wide association study of self-reported food reactions in Japanese identifies shrimp and peach specific loci in the HLA-DR/DQ gene region. Sci Rep. 2018;8(1):1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Thome M CARMA1, BCL-10 and MALT1 in lymphocyte development and activation. Nat Rev Immunol. 2004;4(5):348–59. [DOI] [PubMed] [Google Scholar]
- 84.MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017;45(D1):D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Robinson J, Halliwell JA, Hayhurst JD, Flicek P, Parham P, Marsh SG. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res. 2015;43(Database issue):D423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dendrou CA, Petersen J, Rossjohn J, Fugger L. HLA variation and disease. Nat Rev Immunol. 2018;18(5):325–39. [DOI] [PubMed] [Google Scholar]
- 87.Murray JS, Pfeiffer C, Madri J, Bottomly K. Major histocompatibility complex (MHC) control of CD4 T cell subset activation. II. A single peptide induces either humoral or cell-mediated responses in mice of distinct MHC genotype. Eur J Immunol. 1992;22(2):559–65. [DOI] [PubMed] [Google Scholar]
- 88.Gutierrez-Arcelus M, Baglaenko Y, Arora J, Hannes S, Luo Y, Amariuta T, et al. Allele-specific expression changes dynamically during T cell activation in HLA and other autoimmune loci. Nat Genet. 2020;52(3):247–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nishimura Y, Kamikawaji N, Fujisawa K, Yoshizumi H, Yasunami M, Kimura A, et al. Genetic control of immune response and disease susceptibility by the HLA-DQ gene. Res Immunol. 1991;142(5-6):459–66. [DOI] [PubMed] [Google Scholar]
- 90.Constant SL, Bottomly K. Induction of Th1 and Th2 CD4+ T cell responses: the alternative approaches. Annu Rev Immunol. 1997;15:297–322. [DOI] [PubMed] [Google Scholar]
- 91.Oseroff C, Sidney J, Tripple V, Grey H, Wood R, Broide DH, et al. Analysis of T cell responses to the major allergens from German cockroach: epitope specificity and relationship to IgE production. J Immunol. 2012;189(2):679–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wataya M, Sano T, Kamikawaji N, Tana T, Yamamoto K, Sasazuki T. Comparative analysis of HLA restriction and cytokine production in hepatitis B surface antigen-specific T cells from low- and high-antibody responders in vaccinated humans. J Hum Genet. 2001;46(4):197–206. [DOI] [PubMed] [Google Scholar]
- 93.Kontakioti E, Domvri K, Papakosta D, Daniilidis M. HLA and asthma phenotypes/endotypes: a review. Hum Immunol. 2014;75(8):930–9. [DOI] [PubMed] [Google Scholar]
- 94.Weidinger S, Willis-Owen SA, Kamatani Y, Baurecht H, Morar N, Liang L, et al. A genome-wide association study of atopic dermatitis identifies loci with overlapping effects on asthma and psoriasis. Hum Mol Genet. 2013;22(23):4841–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Boehncke WH, Loeliger C, Kuehnl P, Kalbacher H, Bohm BO, Gall H. Identification of HLA-DR and -DQ alleles conferring susceptibility to pollen allergy and pollen associated food allergy. Clin Exp Allergy. 1998;28(4):434–41. [DOI] [PubMed] [Google Scholar]
- 96.Howell WM, Turner SJ, Hourihane JO, Dean TP, Warner JO. HLA class II DRB1, DQB1 and DPB1 genotypic associations with peanut allergy: evidence from a family-based and case-control study. Clin Exp Allergy. 1998;28(2):156–62. [DOI] [PubMed] [Google Scholar]
- 97.Shreffler WG, Charlop-Powers Z, Sicherer SH. Lack of association of HLA class II alleles with peanut allergy. Ann Allergy Asthma Immunol. 2006;96(6):865–9. [DOI] [PubMed] [Google Scholar]
- 98.Dreskin SC, Tripputi MT, Aubrey MT, Mustafa SS, Atkins D, Leo HL, et al. Peanut-allergic subjects and their peanut-tolerant siblings have large differences in peanut-specific IgG that are independent of HLA class II. Clin Immunol. 2010;137(3):366–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Madore AM, Vaillancourt VT, Asai Y, Alizadehfar R, Ben-Shoshan M, Michel DL, et al. HLA-DQB1*02 and DQB1*06:03P are associated with peanut allergy. Eur J Hum Genet. 2013;21(10):1181–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hand S, Darke C, Thompson J, Stingl C, Rolf S, Jones KP, et al. Human leucocyte antigen polymorphisms in nut-allergic patients in South Wales. Clin Exp Allergy. 2004;34(5):720–4. [DOI] [PubMed] [Google Scholar]
- 101.Dilthey AT, Moutsianas L, Leslie S, McVean G. HLA*IMP--an integrated framework for imputing classical HLA alleles from SNP genotypes. Bioinformatics. 2011;27(7):968–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jia X, Han B, Onengut-Gumuscu S, Chen WM, Concannon PJ, Rich SS, et al. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS One. 2013;8(6):e64683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zheng X, Shen J, Cox C, Wakefield JC, Ehm MG, Nelson MR, et al. HIBAG--HLA genotype imputation with attribute bagging. Pharmacogenomics J. 2014;14(2):192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hong X, Wang X. Epigenetics and development of food allergy (FA) in early childhood. Curr Allergy Asthma Rep. 2014;14(9):460. [DOI] [PubMed] [Google Scholar]
- 105.Brough HA, Simpson A, Makinson K, Hankinson J, Brown S, Douiri A, et al. Peanut allergy: effect of environmental peanut exposure in children with filaggrin loss-of-function mutations. J Allergy Clin Immunol. 2014;134(4):867–75 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Koplin JJ, Suaini NH, Vuillermin P, Ellis JA, Panjari M, Ponsonby AL, et al. Polymorphisms affecting vitamin D-binding protein modify the relationship between serum vitamin D (25[OH]D3) and food allergy. J Allergy Clin Immunol. 2016;137(2):500–6 e4. [DOI] [PubMed] [Google Scholar]
- 107.Davis BP, Rothenberg ME. Mechanisms of Disease of Eosinophilic Esophagitis. Annu Rev Pathol. 2016;11:365–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Blanchard C, Wang N, Stringer KF, Mishra A, Fulkerson PC, Abonia JP, et al. Eotaxin-3 and a uniquely conserved gene-expression profile in eosinophilic esophagitis. J Clin Invest. 2006;116(2):536–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Blanchard C, Stucke EM, Burwinkel K, Caldwell JM, Collins MH, Ahrens A, et al. Coordinate interaction between IL-13 and epithelial differentiation cluster genes in eosinophilic esophagitis. J Immunol. 2010;184(7):4033–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rothenberg ME, Spergel JM, Sherrill JD, Annaiah K, Martin LJ, Cianferoni A, et al. Common variants at 5q22 associate with pediatric eosinophilic esophagitis. Nat Genet. 2010;42(4):289–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kottyan LC, Davis BP, Sherrill JD, Liu K, Rochman M, Kaufman K, et al. Genome-wide association analysis of eosinophilic esophagitis provides insight into the tissue specificity of this allergic disease. Nat Genet. 2014;46(8):895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sleiman PM, Wang ML, Cianferoni A, Aceves S, Gonsalves N, Nadeau K, et al. GWAS identifies four novel eosinophilic esophagitis loci. Nat Commun. 2014;5:5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kottyan LC, Maddox A, Braxton JR, Stucke EM, Mukkada V, Putnam PE, et al. Genetic variants at the 16p13 locus confer risk for eosinophilic esophagitis. Genes Immun. 2019;20(4):281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.De Benedetto A, Qualia CM, Baroody FM, Beck LA. Filaggrin expression in oral, nasal, and esophageal mucosa. J Invest Dermatol. 2008;128(6):1594–7. [DOI] [PubMed] [Google Scholar]
- 115.Irizar H, Kanchan K, Mathias RA, Bunyavanich S. Advancing Food Allergy through Omics Sciences. J Allergy Clin Immunol Pract. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.