

Abstract
Infantile neuronal ceroid lipofuscinosis (INCL) is a devastating neurodegenerative lysosomal storage disease (LSD) caused by inactivating mutations in the CLN1 gene. CLN1 encodes palmitoyl-protein thioesterase-1 (PPT1), a lysosomal enzyme that catalyzes the deacylation of S-palmitoylated proteins to facilitate their degradation and clearance by lysosomal hydrolases. Despite the discovery more than two decades ago that CLN1 mutations causing PPT1-deficiency underlies INCL, the precise molecular mechanism(s) of pathogenesis has remained elusive. Here we report that autophagy is dysregulated in Cln1−/− mice, which mimic INCL and in postmortem brain tissues as well as cultured fibroblasts from INCL patients. Moreover, Rab7, a small GTPase, critical for autophagosome-lysosome fusion, requires S-palmitoylation for trafficking to the late endosomal/lysosomal membrane where it interacts with RILP (Rab-interacting lysosomal protein), essential for autophagosome-lysosome fusion. Notably, Ppt1-deficiency in Cln1−/− mice, dysregulated Rab7-RILP interaction and preventing autophagosome-lysosome fusion, which impaired degradative functions of the autolysosome leading to INCL pathogenesis. Importantly, treatment of Cln1−/− mice with a brain-penetrant, PPT1-mimetic, small molecule, N-tert (Butyl)hydroxylamine (NtBuHA), ameliorated this defect. Our findings reveal a previously unrecognized role of CLN1/PPT1 in autophagy and suggest that small molecules functionally mimicking Ppt1 may have therapeutic implications.
Keywords: Neurodegeneration, Lysosomal storage disease, Infantile neuronal ceroid lipofuscinosis, S-palmitoylation, Palmitoyl-protein thioesterases-1
1. INTRODUCTION
Autophagy is an essential cellular degradative process in which dysfunctional cytoplasmic contents are digested by lysosomal hydrolases1. There are several autophagic pathways, which include macroautophagy (hereafter called autophagy)2, microautophagy3 and chaperone-mediated autophagy4. During autophagy several vesicle fusion events including those of autophagosome and lysosome lead to the eventual degradation and clearance of the cargo within a hybrid structure called autophagolysosome. Several autophagy genes (called Atg) encoding components of the autophagic process are required for the initiation of autophagy. One of these events is marked by the de novo formation of double-membrane structures called phagophores. Two ubiquitin-like conjugation systems involving the Atg5–Atg12–Atg16 complex and phosphatidylethanolamine-conjugated microtubule‐associated protein 1 light chain 3 (LC3-II) are required for the initiation of autophagic process5, 6. Emerging evidence indicates that dysregulation of autophagy is one of the central pathogenic mechanisms underlying many human diseases including the LSDs7, 8.
In the majority of >60 LSDs, neurodegeneration is a devastating manifestation9. Infantile neuronal ceroid lipofuscinosis (INCL)10 is a uniformly fatal neurodegenerative LSD caused by inactivating mutations in the CLN1 gene11. CLN1 encodes palmitoyl-protein thioesterase-1 (PPT1)12, a lysosomal enzyme13 that catalyzes depalmitoylation of S-palmitoylated proteins (constituents of ceroid), required for their degradation by lysosomal hydrolases14. Thus, it has been suggested that PPT1-deficiency impairs degradation of these lipid-modified proteins causing lysosomal ceroid accumulation leading to INCL. Despite this knowledge, for more than two decades the precise molecular mechanism of INCL pathogenesis has remained elusive.
S-palmitoylation (also called S-acylation)15, 16 is a reversible posttranslational lipid-modification in which a 16-carbon saturated fatty acid (generally palmitic acid) is attached to specific cysteine residues in polypeptides via thioester linkage. While numerous proteins in the brain have been reported to undergo S-palmitoylation16, 17, these proteins must also be depalmitoylated for recycling or degradation by lysosomal hydrolases14. S-palmitoylation has been reported to promote protein-protein interaction, membrane affinity, protein trafficking, stability and function16, 18. S-palmitoylation in mammals is catalyzed by a family of 23 palmitoyl acyltransferases (PATs), which are zinc finger proteins containing a DHHC (Asp-His-His-Cys) motif and these enzymes are commonly called ZDHHCs or ZDHHC-PATs19, 20. In contrast, depalmitoylation of S-acylated proteins is catalyzed by thioesterases21, some of which are localized in the lysosome and the others in the cytoplasm. Recently, a group of protein depalmitoylases called ABHD17 have been characterized that catalyzes the turnover of N-Ras22. Emerging evidence indicates that S-palmitoylation plays critical roles in endosomal sorting and trafficking of subcellular proteins to their destinations, which are especially important in the nervous system16, 17.
In eukaryotic cells, vesicular transport facilitates intracellular proteins to reach their destinations23. In this process, a large superfamily of Ras-like GTPases (called Rabs) play pivotal roles in vesicle formation, cargo selection, sorting, transport and vesicular fusion, all of which are critical for endocytic and autophagic degradation. One of the proteins belonging to the Rab-superfamily of GTPases, is Rab724. It directly or indirectly performs multiple important functions in vesicular trafficking and membrane fusion events that occur between early endosome and late endosome/lysosome25, 26. Rab7 (also known as RAB7A) upon interaction with RILP (Rab-interacting lysosomal protein) on late endosomal/lysosomal membrane facilitates the autophagosome-lysosome fusion generating a hybrid organelle called autolysosome, which facilitates the degradation of cargo from intracellular sources by lysosomal acid hydrolases8, 27. Thus, the impaired autophagosome-lysosome fusion28 is one of the suggested mechanisms for the accumulation of undegraded cargo in the lysosome leading to the pathogenesis of LSDs8, 29. In the present study, we sought to determine whether autophagy is dysregulated in INCL, and if so, what might be the underlying mechanism(s).
2. MATERIALS AND METHODS
2.1. Animals
Wild type (WT) mice (C57BL/6J; Stock No. 000664) was purchased from the Jackson Laboratory (Bar Harbor, Maine). Cln1−/− mice30 were a generous gift from Dr Sandra L. Hofmann, University of Texas Southwestern Medical Center, Dallas, TX. All animals were housed in a pathogen-free facility with 12 h light/12 h dark cycles with access to water and food ad libitum. All mice were genotyped according to the protocol of Jackson Laboratory. All animal procedures were carried out in accordance with institutional guidelines after the approval of an animal study protocol (ASP#16-012) by the Animal Care and Use Committee (ACUC) of the NICHD, NIH. The animals were age- and sex-matched for each experiment. To study the age dependent changes 1-, 3- and 6-month old animals were used and for most of the other experiments we used 6-month old animals.
2.2. Chemicals
N-tert (Butyl) hydroxylamine (NtBuHA) was purchased from Sigma-Aldrich (St Louis, MO): NtBuHA (Cat# 479675).
2.3. Postmortem brain tissues from control and INCL patients
Brain tissues from postmortem patients with INCL were obtained from the Human Brain and Spinal Fluid Resource Center at the VA West Los Angeles Healthcare Center (Los Angeles, CA). We also obtained postmortem brain tissue samples from age- and sex-matched control subjects from the Brain and Tissue Bank for Developmental Disorders at the University of Maryland (Baltimore, Maryland, USA). These tissues were obtained under a clinical protocol (#01-CH-0086) approved by the NICHD Institutional Review Board (IRB)31. For details go to: http://www.clinicaltrials.gov; NCT00028262.
2.4. Fibroblast culture and treatment with NtBuHA
Normal fibroblasts (GM00498) were obtained from Coriell Institute of Medical Research, USA. PPT1-deficient INCL fibroblasts were derived from skin biopsy of an INCL patient admitted to a “bench-to-bedside” clinical protocol (#01-CH-0086) approved by the Institutional Review Board (IRB) of NICHD, NIH. Fibroblasts were cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM glutamine, 100 U/ml penicillin and streptomycin at 37° C in humidified atmosphere with 5% CO2. The human embryonic kidney (HEK) 293T and COS-1 cells were also cultured and maintained in DMEM containing 10% FBS at 37°C in a humidified incubator. The fibroblasts from normal subjects and INCL patients were a generous gift from late Dr. K.E. Wisniewski.
Normal and INCL fibroblasts were plated at equal density such that they reach 40%−50% confluence in 24 hours. The cells were then treated with without 0.5 mM NtBuHA daily for 3 days until the cultures were 80% to 90% confluent as previously reported32.
2.5. Serum-starvation experiments
Since serum starvation increases autophagy, we sought to determine the effects of serum-starvation on the level of LC3-II positive puncta in normal and INCL cells. For starvation study, semi-confluent normal and INCL fibroblasts were harvested with 0.025% trypsin and 0.52mM EDTA and plated at a density of 4 × 105 cells/well in 6-well plates. Next day, the cells were incubated with Opti-MEM for 3 hrs.
2.6. Realtime RT-PCR
Realtime RT-PCR was performed using total RNA to determine mRNA levels in the brain cortical tissues from WT and Cln1−/− mice as well as normal and INCL fibroblasts as previously reported33. Primers in this study are presented in Table S1.
2.7. Transfection
Fibroblasts, COS-1 cells, NIH3T3 cells or HEK293T cells were transfected with the plasmids using lipofectamine 2000 (Invitrogen) or lipofectamine 3000 as per the manufacturer’s protocol. Briefly, confluent cells were incubated with plasmid lipofectamine mixture for 6 h in media without serum and then cultured with DMEM containing 10% FBS. 2 days after transfection cells were processed further as per experimental protocol.
2.8. Fractionation of cortical tissues and fibroblasts
Brain cortical tissues were homogenized in 0.25 M sucrose buffer containing Tris, EDTA, 1x protease and phosphatase inhibitor (Pierce, USA) using Dounce homogenizer. Homogenates were then centrifuged at 800 × g for 10 min at 4° C to pellet down nuclei and debris. Post-nuclear supernatants were collected and used for further analysis. Fibroblast lysates were prepared in buffer containing 20 mM Tris, 150 mM NaCl, 2% Triton X-100, 10% Glycerol (Sigma), 1X protease inhibitor and 1X phosphatase inhibitor (Pierce). Membrane and cytosolic fractions of cortical tissues and fibroblasts were prepared using S-PEK (Calbiochem) according to manufacturer’s specifications.
2.9. Isolation and purification of lysosomes
Lysosome isolation kit (LYSISO1, Sigma Aldrich) was used to purify lysosomes from mouse brain cortex33. Briefly, freshly isolated cortical tissues were homogenized in 4 volumes of 1x extraction buffer containing Halt Protease Inhibitor (PI) (ThermoFisher; Cat#78430) and centrifuged for 1000 × g for 10 minutes. The resultant supernatant was then centrifuged at 20,000 × g for 20 minutes at 4°C. he pellet containing the Crude Lysosomal Fraction was resuspended in 1x extraction buffer and layered over the multistep Optiprep gradient consisting of 27%, 22.5%, 19%, 16%, 12% and 8% Optiprep. The samples were then centrifuged for 4 h at 150,000 × g in a swing out rotor. The sample was resolved into 5 layers, F1 (top layer) to F5 (bottom layer), formed at the junction of each gradient. Fraction 2 and fraction 3 were found to be enriched in lysosomal content determined by Western blot analyses using the lysosomal markers, LAMP1 or LAMP2 and by measuring the β-N-acetylglucosaminidase activity. Both fraction 2 and 3 were combined for all experiments.
2.10. Western blot analysis
Western blot analyses were performed according to a previously described method33. Briefly, 30 μg of protein was resolved in 4–12% Bis Tris gel (Invitrogen) by SDS-PAGE and the protein bands were electro-transferred to PVDF membrane. The membranes were blocked with 5% fat-free dried milk for 1 h at room temperature and were then probed overnight with primary antibody. After thorough washing, the blots were incubated with horseradish peroxidase- conjugated secondary antibody (Santa Cruz Biotechnology) for 1 h at room temperature and developed using enhanced chemiluminescence detection reagents (Pierce). Primary antibodies used were: LC3B, Rab7 (Cell Signaling Technology), ATG 12, Beclin-1, Pancadherin (Cell signaling Technology), GAPDH (Santa Cruz Biotechnology), Rab7-interacting lysosomal protein (RILP) (Santacruz Biotechnology; Abcam), β-Actin (US Biologicals), ubiquitin (Covance), p62 (BD Bioscience) and Myc (Sigma). The LAMP2 (H4B4) antibody, developed by Drs. J. Thomas August and James E.K. Hildreth of Department of Pharmacology and Molecular Sciences, Johns Hopkins University School of Medicine, was obtained from the Developmental Studies Hybridoma Bank generated under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242.
2.11. Transmission electron microscopy
Transmission electron microscopy of postmortem brain tissues from an INCL patient and those of an age- and Sex-matched normal subject was performed according to previously published method34. Briefly, after fixation with 2.5 % glutaraldehyde in sodium phosphate buffer the brain tissues were washed once with Millonig’s phosphate buffer and kept in the same buffer at 4°C until final processing. Ultra-thin tissue sections were then stained with lead citrate and uranyl acetate and examined with a LEO 912 electron microscope (JFE Enterprises, Brookeville, MD).
2.12. Immunohistochemistry of brain tissues
The mouse brain cortical tissues were fixed in 3.7% paraformaldehyde, embedded in paraffin and processed for histological analyses. Briefly, tissues were incubated in xylene followed by successive passage through concentrations (100 to 0%) of ethanol in PBS. Tissue sections were blocked with 5% BSA in PBS. The sections were then incubated with TrueBlack® Lipofuscin Autofluorescence Quencher solution (Biotium # 23007) for 2 mins and rinsed with PBS to block the background autofluorescence. Sections were then incubated overnight with either anti-LC3B (Abcam), anti-ubiquitin (Zymed) or p62 (BD Bioscience) followed by incubation with Alexa Fluor 594-conjugated anti-rabbit and anti-mouse secondary antibodies (Invitrogen). Sections were mounted with DAPI containing mounting medium and images were captured using Zeiss LSM 510 Inverted Meta confocal microscope (Carl Zeiss) or in fluorescence Nikon Ti-E inverted microscope. The identical region of the cerebral cortex was chosen for imaging for comparison of data in different samples32.
2.13. Immunocytochemistry
Immunofluorescence analysis was performed according to a previously reported method33. Briefly cells (neurons or fibroblasts) were grown in chamber slide (Lab-Tek) and fixed with methanol or 4% paraformaldehyde (PFA). The PFA fixed cells were permeabilized with 0.1% Triton X in PBS for 10 min in room temperature. Non-specific interactions of antibodies were blocked using 2% BSA and 5% serum in PBS for 1 h at room temperature. The cells were then incubated overnight with primary antibody at 4° C, followed by incubation with secondary antibodies at room temperature for 1 h. Primary antibodies used are anti-LC3B (Cell signaling Technology), anti-LAMP2 (Hybridoma Technology), anti-Rab7 (Cell signaling Technology; Abcam), anti-RILP (Abcam), anti-ubiquitin (Zymed) and p62 (BD Bioscience), Pancadherin (Cell signaling). Secondary antibodies used are Alexa fluor-488 conjugated anti-rabbit, Alexafluor-conjugated 594 anti-mouse (Invitrogen). Nuclei were stained with DAPI (Sigma-Aldrich). Fluorescence was visualized using Zeiss LSM 510 Inverted Meta confocal microscope (Carl Zeiss), and the image obtained was processed with the LSM image software (Carl Zeiss) to determine the overlapping coefficient for colocalization analysis.
2.14. Potential S-palmitoylated cysteine residues in Rab7 predicted by CSS-Palm analysis
Peptide sequences of Human (Accession: AAD02565.1), Mouse (Accession: CAA61797.1) and Dog (Accession: NM_001003316.1) Rab7-proteins were compared. This was followed by S-palmitoylation site-prediction by a computer program called CSS-Palm35.
2.15. S-Palmitoylation Assays
We used three different assays for confirming S-palmitoylation of Rab7: (a). Incorporation of 14[C]palmitate instead of 3[H]palmitate as previously reported15. (b) S-palmitoylated proteins were identified by acyl-biotinyl exchange method (ABE) as previously reported36 with minor modifications. Briefly, COS-1 cells were transfected with Myc-DDK-tagged RAB7 (Origen) using Lipofectamine 2000 (Invitrogen), lysed with RIPA buffer (Pierce) and incubated overnight with 10 mM N-ethylmalemide (NEM, Pierce) and 1x protease inhibitor cocktail (PI, Pierce) at 4°C with gentle mixing. The next day, NEM was removed by three sequential precipitation by the chloroform-methanol (CM) method as previously described. Following third precipitation, the protein was divided into two parts. One part was treated with 1 M hydroxylamine (Sigma) pH 7.4 (freshly prepared), 1 mM HPDP-biotin (Pierce), 0.2% Triton X-100 (Sigma), and 1 × PI and the other part was treated with similar mixture without hydroxylamine for 1 h at room temperature. The protein was then precipitated by CM method and treated with 200 μM HPDP-biotin, 0.2% Triton X-100, and 1xPI at room temperature 25°C for 1 h. HPDP-biotin was then removed by three sequential CM precipitations. Following the third precipitation, protein was dissolved in 50 μl 2% SDS, 50 mM Tris/Cl, 5 mM EDTA pH 7.4, diluted into 950 μl 50 mM Tris/5 mM EDTA buffer containing 0.2% Triton X-100 and 1xPI and then incubated at room temperature for 30 min with gentle shaking. The sample was then centrifuged at 16,000 × g for 1 min to remove particulates and incubated overnight with 25μl streptavidin-agarose (Pierce) at 4°C. After incubation the resin was washed four times with phosphate buffered saline containing 0.3% NP-40. Bound proteins were eluted by boiling the resins with a SDS-PAGE loading buffer containing β-mercaptoethanol for 5 min. Eluted sample was then subjected to Western blot analysis with Rab7 antibody. (c) Acyl-Rac assay: To further confirm the palmitoylation of Rab7, we performed the Acyl-Rac Assay37 with minor modifications. Briefly, 1 mg of protein (cell lysate/tissue lysate) was diluted to 1 ml in blocking buffer (100 mM HEPES, 1.0 mM EDTA, 2.5% SDS, 50mM N-ethylmaleimide, pH 7.5) and incubated at 50°C for 60 min with frequent vortexing (after each 15 mins). Three volume of ice-cold acetone was then added and kept at −20°C for 20 min for protein precipitation. Then it was centrifuged at 5,000 g for 10 min and the pellet was extensively washed with 70% acetone at least 4 times. Thereafter the pellet was resuspended in 250 μl of binding buffer (100 mM HEPES, 1.0 mM EDTA, 1% SDS, pH 7.5). Approximately 50 μl of resuspended protein was saved as the total input. The remaining protein was divided in two aliquots (100ul each). One aliquot was treated with 100 μl of 2 M NH2OH (freshly prepared, pH 7.5) while the other aliquot was treated with 100ul PBS (for control). Thiopropyl Sepharose (GE-Amersham) was activated by letting it swell in ultrapure water for 15 −20 mins and washed (2–3 times) with ultrapure water. Approximately 40 μl of prewashed and activated thiopropyl Sepharose was added to each of these aliquots and the binding reactions were carried out on a rotator at room temperature for around 4 hours. The unbound fraction was collected carefully to anther tube to consider as a non-palmitoylated protein fraction. Resins were washed at least five times with binding buffer. For immunoblot analysis, resin was boiled (95° C for 5 min) with 60 μl of 4x loading buffer (NuPage 4x LD). Then both bound and unbound fraction were separated via SDS-PAGE (Bolt 4–12% Bis Tris Plus) on a Mini-Gel apparatus (Invitrogen).
2.16. Proximity Ligation Assay
Duolink Proximity Ligation Assay38 was used to study interaction between Rab7 and RILP. Briefly the primary neurons from WT and Cln1−/− mice were grown in chamber slides and fixed with methanol for 15 to 20 min at −20°C. The cells were blocked with goat serum and incubated with Rab7 antibody (Abcam; Cat#ab50533; dilution 1:1000) and RILP antibody (Abcam; Cat#ab140188; dilution 1: 3000) overnight at 4°C. After three washes with PBS the cells were incubated with anti-rabbit-MINUS (Sigma–Aldrich; Cat#DUO92005 Anti-RILP) and anti-mouse–PLUS (Sigma–Aldrich; Cat#DUO92001-for Rab7 antibody) PLA probes and subjected to ligation and amplification reaction using Duolink® In Situ Detection Reagents Green (Sigma–Aldrich; DUO9201) according to manufacturer’s protocol. For controls, cells were similarly processed but without primary antibody or with one primary antibody only. When the primary antibodies were in close proximity (<40 nm apart) fluorescent signals were detected as green dots under the FITC channel. The cells were mounted with DAPI-Fluoromount G (Thermo Fisher, Cat#010020) and visualized using an LSM710 confocal microscope. High resolution z stack images were captured at 40X, the z stacks were merged by maximum intensity projection using Zen Desk software to include all the PLA signals and quantitated using Duolink image tool.
2.17. Rab7 GTPase Assay
Rab7 GTPase activity was determined in the normal and INCL fibroblasts. Cells were transiently transfected with myc-flag-tagged Rab7. 48 hours after transfection Rab7 was immunoprecipitated using c-Myc-Tag IP/Co-IP Kit antibody (Thermo Fisher, Cat # 23620). GTPase activity was then measured in the immunoprecipitated Rab7 using GTPase activity assay kit (Novus, Cat # 602-0120). Briefly, the pH of the eluates from IP was adjusted to 7.4 and then incubated with GTPase-substrate (supplied in the assay kit; Novus, Cat # 602-0120) for 1h and allowed to develop the color, which was quantified spectrophotometrically at 600 nm. Changes in absorbance was then normalized by the density of the Rab7 protein band in the Western blot of the eluates.
2.18. Proteasome assay
Proteasome activity was determined using lysates of cortical tissues from WT and Cln1−/− mice and lysates of cultured cells from normal and INCL patients using a proteasome assay kit (Chemicon) according to the instructions provided by the manufacturer.
2.19. Statistics
All quantitative results were statistically analyzed using Graph Pad Prism (Version 4) and are expressed as the mean of at least three independent experiments ± S.D. For in vivo experiments, the numbers reported (n) are biological replicates. For in vitro studies ‘n’ represents total number of repeats from at least 3 independent experiments. Unpaired 2-tailed Student’s t-test was performed for experiments with only 2 groups. For experiment with multiple groups one-way or two-way ANOVA was performed followed by appropriate post-hoc test (Tukey’s or Newman-Keuls) as specified in the figure legends. p< 0.05 was considered statistically significant.
3. RESULTS
3.1. Autophagy is dysregulated in Cln1−/− mouse brain
In eukaryotic cells, macroautophagy is the major autophagic process that predominantly degrades long-lived proteins, protein aggregates, and aged organelles in the cytoplasm and its regulation is mediated by several cellular proteins1, 5, 27. Thus, we determined markers of macroautophagy (hereafter will be referred to as autophagy) in Cln1−/− mouse brain to assess how autophagy is regulated in the absence of Ppt1. At the initial stages of autophagosome formation, two ubiquitin-like conjugations occur. In one, ATG5 and ATG12 form a covalently bound complex, which is essential for the elongation of pre-autophagosomal structure1, 5. In the other, conjugation with microtubule-associated protein light chain 3-I (LC3-I) is converted to LC3-II by the addition of phosphatidylethanolamine. Therefore, while LC3-II is a reliable autophagosome marker, the elevated level of ATG12-ATG5 conjugate may also indicate increased autophagosome formation39, 40. Accordingly, we performed Western blot analyses of homogenates from cortical tissues of Cln1−/− mice and those of their WT littermates, which showed markedly higher levels of LC3-II in Cln1−/− mice (Figure 1A and 1B) even though the levels of ATG12-ATG5 conjugates were virtually identical (Figure 1A and 1C). The level of beclin-1, a component of PI3K that also plays crucial role in autophagosome biogenesis1, 5 also remained unaltered in the cortical tissues of both WT and Cln1−/− mice (Figure 1A and 1D). These results indicated that although autophagy per se was not upregulated in Cln1−/− mice, the number of autophagosomes have increased in the cortical tissues. We also detected time-dependent increase in LC3-II protein levels in cortical tissues from Cln1−/− mice and those from heir WT littermates (Supplementary Figure S1A). The results of immunofluorescence studies also showed that PPT1-deficiency caused increased levels of LC3-positive puncta in the cortical tissues of Cln1−/− mice (Figure 1E) suggesting the accumulation of autophagosomes. These results were further confirmed by electron microscopic analysis of cortical tissues from WT mice and those from their Cln1−/− littermates, which showed accumulation of autophagic structures in the brain of Cln1−/− mice (Figure 1F). Taken together, these results demonstrated that there is progressive accumulation of autophagosomes in the brain of Cln1−/− mice.
Figure 1. Dysregulation of autophagy in Cln1−/− mice and in INCL patient’s brain and fibroblasts.

Western blots (A) and densitometric analyses of LC3-II (B), ATG12-ATG5 conjugate (C) and beclin-1 (D) in cortical tissue homogenates prepared from WT and Cln1−/− mice. Data are presented as mean ± SD (n=4), *p<0.05, NS, non-significant. (E) Immunohistochemical detection of autophagosomal structures using LC3 antibody in the cortical tissue sections of WT and Cln1−/− mice. (F) Electron microscopic detection of double membrane autophagosomal structure in the cortical tissue sections from WT and Cln1−/− mice. Arrows indicate an autophagosome in Cln1−/− mouse cortex. (G) Western blots of total homogenates prepared from postmortem brain of a normal [N] subject and that of an INCL patient for ATG12-ATG5 conjugate, beclin-1 and LC3-II. (H) Western blots and densitometric analyses of (I) LC3-II levels in normal-(NF) and INCL-fibroblasts; (J) beclin-1 and (K) ATG12-ATG5 conjugate. Data are presented as mean ± SD (n=4), *p<0.001. NS, non-significant; (L) Transmission electron micrographs of normal and INCL fibroblasts. Note the accumulation of dense dark structures (GRODs) in INCL fibroblasts but not detectable in normal fibroblasts. N, nucleus.
3.2. Defective autophagy in brain tissues from a postmortem INCL patient
To determine whether the results from the brain of Cln1−/− mice are replicable in human INCL, we analyzed LC3-II levels in postmortem brain tissues from an INCL patient and those from an age- and sex-matched normal control by Western blot analysis. The results showed an increased level of LC3-II in postmortem brain tissues of the INCL patient, but not in those of the normal control (Figure 1G). Since PPT1 is ubiquitously expressed and cultured fibroblasts from INCL patients develop the same pathology found in the INCL brain, we performed Western blot analyses of cultured fibroblasts from INCL patients and those from their normal counterparts. The results showed that the levels of LC3-II in INCL fibroblasts were higher (Figure 1H and 1I) than those in normal fibroblasts although no significant differences in ATG12-ATG5 (Figure 1H and 1J) and Beclin 1 (Figure 1H–1K)) were apparent. We also performed electron microscopic analyses of normal and INCL fibroblasts, which showed the presence of granular osmiophilic deposits (GRODs) in INCL fibroblasts only (Figure 1L). Cumulatively, these results suggested that unlike the normal cells, increased numbers of autophagosomes accumulate in PPT1-deficient cells from INCL patients.
3.3. Blocked autophagy in Cln1−/− mice and in cultured INCL fibroblasts
In contrast to autophagy, intracellular short-lived proteins are primarily degraded by the ubiquitin-proteasome pathway. Polyubiquitinated proteins can also be entrapped within autophagosomes through their interaction with p62/sequestosome1(SQSTM1)41. It has also been reported that impaired autophagy can cause accumulation of ubiquitinated proteins in LSDs42. Accordingly, we determined the level of polyubiquitinated proteins in cortical tissues from Cln1−/− mice and those of their WT littermates by Western blot analysis. The results showed markedly higher levels of polyubiquitinated proteins in the brain tissues of Cln1−/− mice as compared with those of their WT littermates (Figure 2A, upper panel and 2B). Interestingly, we also found an increased level of autophagic adapter protein p62/SQSTM1 in Cln1−/− mouse brain (Figure 2A, lower panel and 2C)). Consistent with these results, immunofluorescence study also revealed markedly higher accumulation of ubiquitinated proteins and p62/SQSTM1 in cortical tissues of Cln1−/− mice (Figure 2D; Supplementary Figures S1B and S1C). We also observed that p62/SQSTM1 mostly accumulated in the cells with increased level of autophagosomes in the cortex of Cln1−/− mice (Supplementary Figure S1B). Accumulation of p62/SQSTM1 in Cln1−/− mouse brain also colocalized with ubiquitinated proteins (Supplementary Figure S1C) suggesting that accumulation of polyubiquitinated proteins might be due to the block of autophagic process in Cln1−/− mice.
Figure 2. Autophagy cargos accumulate in Cln1−/− mouse brain cortex.
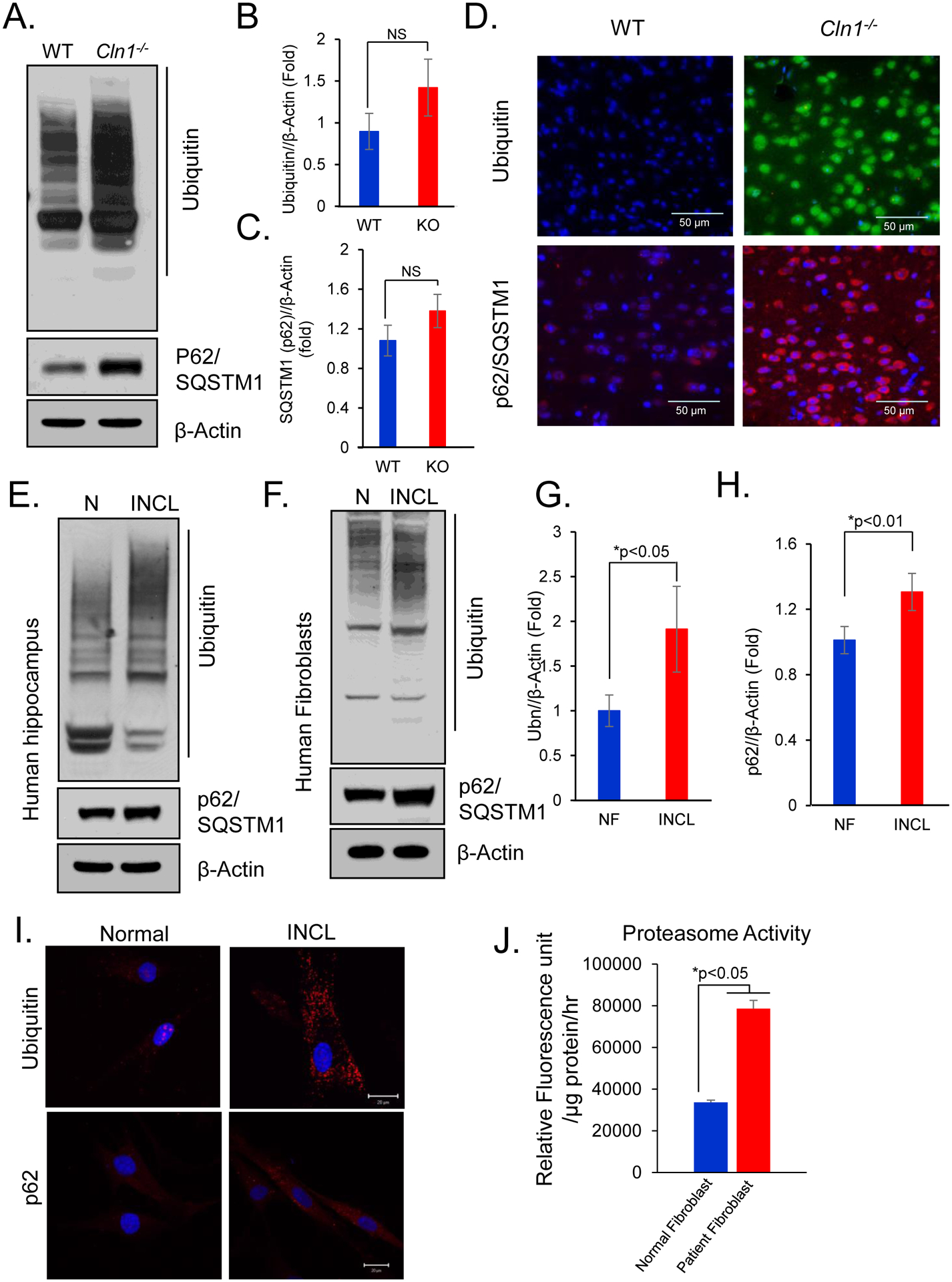
(A)Western blots of polyubiquitinated proteins and p62/SQSTM1 and corresponding densitometric analyses of (B) polyubiquitinated protein and (C) p62/SQSTM1 in cortical tissue homogenates of brain tissues from WT and Cln1−/− mice. Data are presented as mean ± SD, n=4, *p<0.05. (D) Immunofluorescence of polyubiquitinated proteins and p62/SQSTM1 in cortical tissue sections of WT and Cln1−/− mice. (E, F) Western blots analysis and (G, H) corresponding densitometric quantitation of homogenates prepared from postmortem brain of normal individual and INCL patients for polyubiquitinated proteins and p62. Data are presented as mean ± SD (n=4), *p<0.05, **p<0.01. (I) Immunofluorescence of polyubiquitinated proteins and p62 in normal and INCL fibroblasts. (J) Proteasome activity in normal and INCL patients’ fibroblasts. Data are presented as mean ± SD, n=4, *p<0.05.
Like the findings in Cln1−/− mice, higher levels of polyubiquitinated proteins and p62/SQSTM1 were also detected by Western blot analyses of postmortem brain tissues from an INCL patient as compared with those of an age- and sex-matched normal control subject (Figure 2E).The Western blot analysis of homogenates of INCL fibroblasts showed higher levels of polyubiquitinated proteins as well as p62/SQSTM1 (Figures 2F to 2H). Consistent with these results, markedly higher accumulation of ubiquitinated proteins and p62/SQSTM1 in INCL fibroblasts were also detected by immunocytochemistry (Figure 2I). Notably, the proteasome activity was also substantially increased in cultured INCL cells as compared with that in the normal controls (Figure 2J). Taken together, these results demonstrated that ubiquitinated proteins accumulate within the cells lacking PPT1, most likely due to the impairment of autophagic degradation.
3.4. Dysregulated autophagosome-lysosome fusion in cultured INCL fibroblasts
To further characterize autophagy, we cultured normal and INCL fibroblasts in presence or absence of serum. Since serum starvation causes induction of autophagy, we determined the level of LC3-II in those cells by Western blot analysis in order to detect any differences that might exist in autophagy induction between normal and INCL cells. The results showed elevated level of LC3-II in both normal and INCL fibroblasts cultured in serum deprived condition (Figure 3A). However, we observed appreciably higher LC3-II level in INCL cells as compared to normal cells in both culture conditions. This might be due to the defect in autophagosomal clearance by lysosomes as observed in the autophagy flux assay in which we treated both normal and INCL cells with vacuolar (lysosomal) H+-ATPase-inhibitor, bafilomycin. We detected higher level of LC3-II in INCL cells as compared to normal cells, although no appreciable difference in LC3-II level was observed in bafilomycin A1-treated normal and INCL fibroblasts (Figure 3B). These results suggest that autophagosome formation is not affected in INCL cells and the increased accumulation of autophagosomes in these cells is mainly due to the reduced turnover. Interestingly, our data also showed markedly elevated level of LC3-II in INCL fibroblasts following bafilomycin treatment, which indicates that autophagosomal degradation is not completely impaired, rather it is partially blocked in those cells. One of the reasons for this may be due to the disruption in autophagosome-lysosome fusion, which has been reported previously in other LSDs28. Thus, to determine if autophagosome-lysosome fusion is dysregulated in INCL cells, we assessed colocalization of autophagosomal marker LC3 and lysosomal marker LAMP1 using immunofluorescence. Accordingly, we cultured normal and INCL fibroblasts in serum-free medium to induce autophagy and then immunostained them with the antibodies against lysosomal marker, LAMP1. and autophagosomal marker LC3. We observed more pronounced colocalization of lysosomal LAMP1-signals with those of the LC3-positive autophagosomal structures in normal fibroblasts compared with those in INCL fibroblasts (Figure 3C–D) suggesting impairment in autophagosome-lysosome fusion in INCL cells. To further confirm these results, we induced autophagy by serum-starvation in normal and INCL cells transfected with RFP-tagged LC3 (RFP-LC3) and then immunostained them with lysosomal marker, LAMP2 to assess autophagosome-lysosome colocalization in these cells. The results showed significantly lower colocalization of LAMP2-signals with those of the RFP-LC3 INCL fibroblasts as compared to normal cells (Figure 3E–F). Taken together, these results clearly suggested that the increased numbers of autophagosomes in INCL cells are most likely due to impaired autophagosome-lysosome fusion.
Figure 3. Autophagosome-lysosome fusion in normal and INCL fibroblasts.
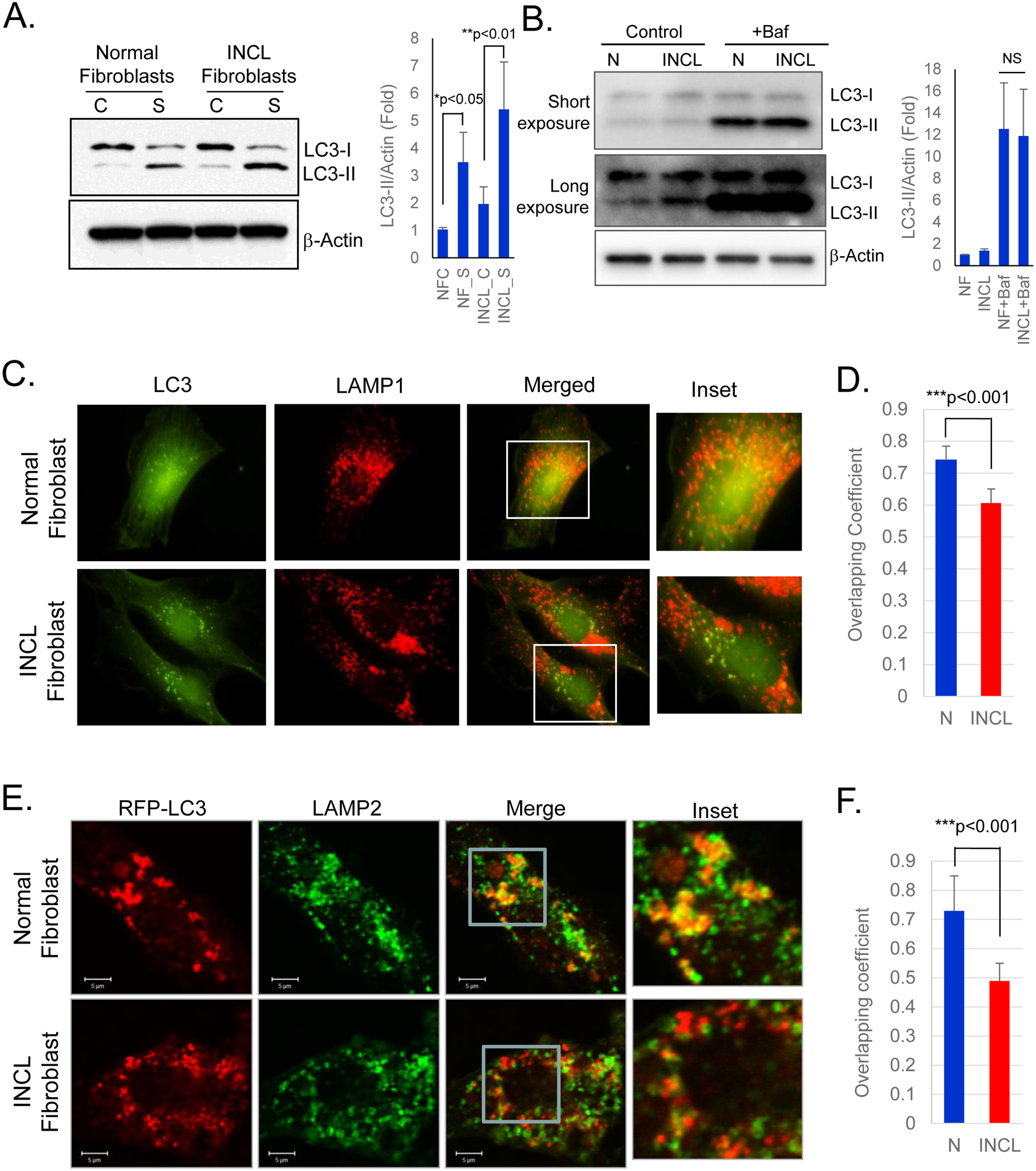
(A) Western blot analysis of normal and INCL fibroblasts cultured in either serum-supplemented [C] or serum-deprived [S] media; Data are presented as mean ± SD, n=3; *p<0.01 and **p<0.01 (Two-way ANOVA); (B) Western blot of cell lysates prepared from normal and INCL fibroblasts treated with or without v-ATPase-inhibitor, bafilomycin A1; Data are presented as mean ± SD, n=3 (Two-way ANOVA); (C) Colocalization of autophagic marker LC3 (green) and lysosomal marker LAMP1 (red) detected by fluorescence microscopy in normal and INCL cells; (D) Corresponding colocalization analysis determined by Mander’s coefficient. Cells were cultured in media without serum and then fixed and immunostained with LC3 and LAMP1 antibody. Note markedly reduced level of colocalization was detected in INCL cells (magnified inset); (E) Immunofluorescence confocal images of RFP-LC3 (red) transfected normal and INCL fibroblasts stained with lysosomal marker LAMP2 (green) detected by confocal microscopic analysis in normal and INCL cells. Cells were cultured in media without serum and then fixed and immunostained with LAMP2 antibody. Note markedly reduced level of colocalization was detected in INCL cells (magnified inset); (F) Colocalization analysis of LC3 and Lamp2 immunfluorescence quantified by Mander’s coefficient. Data are presented as mean ± SD, *p<0.001.
3.5. Lysosomal-localization of Rab7 is defective in Cln1−/− mouse brain and in INCL fibroblasts
A small GTPase, Rab7, plays important role in late endosome and lysosome trafficking and transport25. It has also been reported to mediate the autophagosome-lysosome fusion generating a hybrid organelle, autolysosome25, 26 where the undigested cargo in the autophagosome meets the lysosomal hydrolases. Moreover, autophagosomes have been shown to accumulate in cells in which Rab7 is knocked down27. Based on our findings that autophagosome-lysosome fusion is defective in INCL cells, we reasoned that Rab7 localization on late endosome/lysosome may be impaired in PPT1-deficient cells. To test this hypothesis, we first determined the level of Rab7 in total cortical lysates from Cln1−/− mice and those in the INCL fibroblasts by Western blot analysis. The results showed there was virtually no difference in Rab7-protein levels in total lysates between cortical tissues from WT and Cln1−/− mice or between those from normal and INCL fibroblasts (Figure 4A). We also determined the Rab7-mRNA levels in brain tissues from WT and Cln1−/− mice and those in cultured fibroblasts from normal and INCL patients and the results showed no significant differences (Supplementary Figures S2A and S2B).
Figure 4. Mislocalization of Rab7 in Cln1−/− mouse brain and INCL cells.
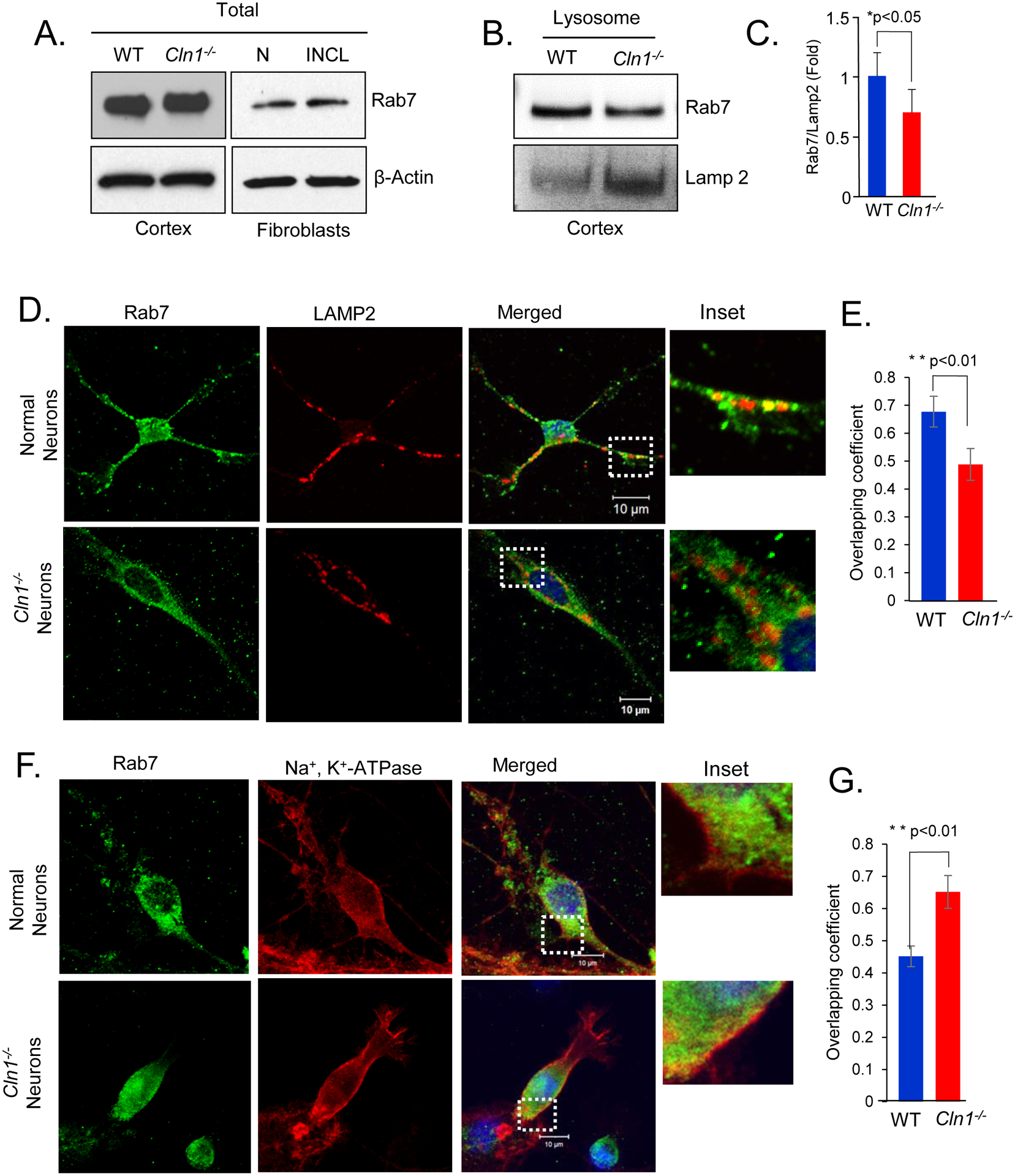
(A) Western blot analysis of WT and Cln1−/− mouse cortical tissue homogenates and the lysates of normal and INCL fibroblasts for Rab7. (B) Western blots of lysosomal fractions prepared from WT and Cln1−/− mouse cortex for Rab7 and lysosomal marker LAMP2 and corresponding (C) densitometric quantification. Data are presented as mean ± SD (n=4), *p<0.05. (D) Confocal immunofluorescent images of WT and Cln1−/− neurons stained with antibodies for Rab7 with lysosomal marker, LAMP2 and (E) corresponding colocalization analysis determined by Mander’s coefficient. Data are presented are the mean ± SD, n=22 (WT neurons), n=14 (Cln1−/− neurons) **p<0.01; (F) Confocal images of cultured WT and Cln1−/− neurons showing colocalization of Rab7 with the plasma membrane marker, Na+, K+- ATPase and (G) corresponding colocalization analysis determined by Mander’s coefficient. Data are presented are the mean ± SD, n=13 (WT neurons), n=17 (Cln1−/− neurons) **p<0.01.
Since Rab7 localizes predominantly to the late endosomal and lysosomal membranes25, we determined Rab7-protein levels in purified lysosomes from cortical tissues of WT and Cln1−/− mice by Western blot analysis. The results showed that the levels of Rab7 in LAMP2-positive (lysosomal) membrane fractions from the brains of Cln1−/− mice were appreciably lower compared with those of their WT littermates (Figure 4B and 4C). The level of lysosomal marker, Lamp 2 was found to be increased in Cln1−/− mice brain lysates but remained unaltered in INCL fibroblasts compared with that in normal control (Supplementary Figures S2C). To further confirm these results, we performed immunostaining of primary cultured neurons from WT and Cln1−/− mouse brain using Rab7- and LAMP2-antibodies to determine whether the signals from two proteins colocalized. We found that compared WT neurons, a markedly lower level of Rab7-LAMP2 colocalization occurred in Cln1−/− neurons (Figure 4D and 4E). Similarly, confocal imaging of normal and INCL fibroblasts also showed that compared with normal fibroblasts, a markedly lower level of Rab7-LAMP2 colocalization occurred in INCL fibroblasts (Supplementary Figures S3A and S3B). These results suggested reduced colocalization of Rab7 on the lysosomes in PPT1-deficient cells.
Next, we determined the levels of Rab7-protein in the membrane- and cytosolic-fractions of brain tissues from WT and Cln1−/− mice, respectively. The results showed that the levels of Rab7-protein were appreciably higher in the membrane fractions from brain tissues of Cln1−/− mice compared with those of their WT littermates (Supplementary Figure S3C Fig, left panels). In contrast, the lower level of Rab7 was detected in the cytosolic fractions of the brain from Cln1−/− mice compared with those from their WT littermates (Supplementary Figure S3C Fig, right panels). Similarly, increased membrane association of Rab7 was also detected in INCL fibroblasts compared with those in normal fibroblasts (Supplementary Figure S3D). Furthermore, immunofluorescence data also showed that compared with WT neurons substantially higher level of membrane-associated Rab7 was present in PPT1-deficient cultured Cln1−/− neurons (Figure 4F). These results were confirmed by analyzing Mander’s colocalization coefficients (Figure 4G). Similarly, normal fibroblasts showed substantially lower level of membrane-associated Rab7 compared to PPT1-deficient cultured INCL fibroblasts (Supplementary Figure S3E and S3F). Taken together, these data clearly showed higher level of membrane localization of Rab7 in PPT1-deficient cells.
3.6. Rab7-RILP interaction is suppressed in Cln1−/− mouse brain and in cultured INCL cells
Rab7 is recruited to the membrane in its GDP-bound form and the exchange of GDP with GTP is facilitated by guanine nucleotide exchange factor (GEF), which mediates its activation. Active Rab7 then interacts with its effector molecule, RILP, for transport to the lysosomal membrane where its GTPase activity is manifested43, 44. Thus, the amount of RILP interacting with Rab7 is an indicator of the level of active-Rab7. Because of the altered membrane distribution of Rab7, we reasoned that its function may be impaired in PPT1-deficient INCL cells. Accordingly, we first determined the level of RILP in cortical tissues from Cln1−/− mice and in cultured fibroblasts from INCL patients. The results showed no significant difference in RILP-protein levels in the cortical tissues of Cln1−/− mice and in those of their WT littermates (Figure 5A, left panel). Similarly, total RILP levels in INCL fibroblasts were also virtually identical to those in normal fibroblasts (Figure 5A. right panel). Moreover, no significant differences in the mRNA levels of RILP in the brain tissues of Cln1−/− mice, their WT littermates as well as those in INCL and normal fibroblasts were found (Supplementary Figures S4A and S4B). Notably, the RILP levels in lysosomal fractions of brain cortical tissues from Cln1−/− mice were significantly lower compared with those from their WT littermates (Figure 5B) suggesting that interaction between Rab7 and RILP may be adversely affected in PPT1-deficient cells. To confirm these results, we immunostained normal and INCL cells using antibodies to Rab7 and RILP and analyzed the cells by confocal microscopy to determine the level of colocalization of these two proteins. The results showed that the level of Rab7-RILP colocalization in INCL cells was markedly lower compared with that in normal fibroblasts (Supplementary Figure S5A). To further confirm these results, we performed proximity ligation assay (PLA)38. The results showed that there were significantly reduced Rab7-RILP PLA-signals in INCL fibroblasts compared with those in normal controls (Figure 5C) indicating that Rab7-RILP interaction in INCL fibroblasts was significantly reduced compared with that in normal fibroblasts (Figure 5D). Notably, colocalization of Rab7-RILP with lysosomal marker, LAMP2 by PLA showed appreciably reduced levels of colocalization signals in INCL fibroblasts compared with those in normal fibroblasts (Supplementary Figure S5B). Cumulatively, these results strongly suggested that the interaction between Rab7 and RILP was suppressed in PPT1-deficient cells.
Figure 5. Impaired Rab7-RILP interaction and GTPase activity in INCL.
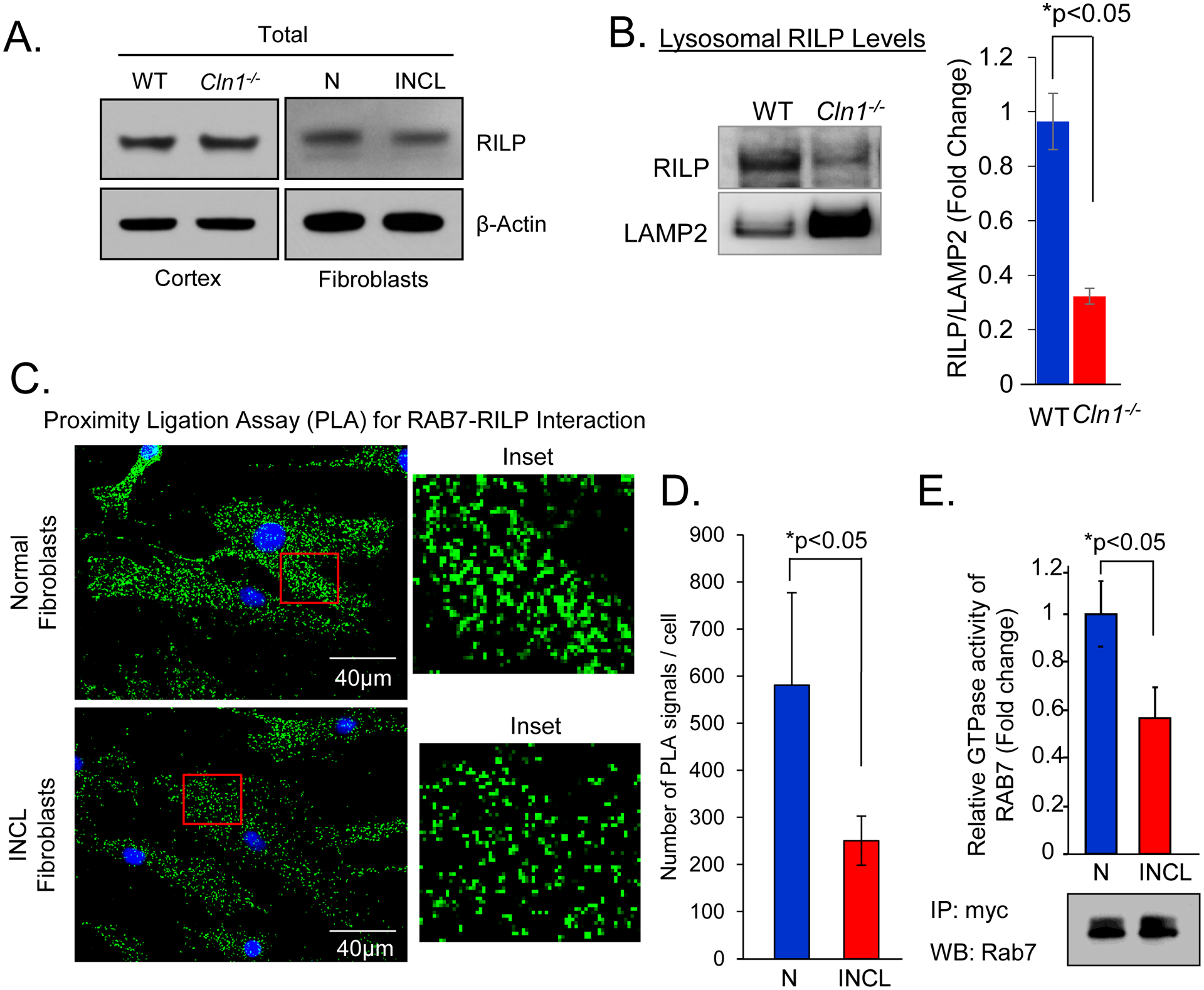
(A)Western blot analysis of RILP in cortical tissue homogenates of WT and Cln1−/− mice. (B) Western blot analysis of RILP in purified lysosomal fractions from the cortical tissues from WT and Cln1−/− mice. (C) Proximity ligation assay to detect the interaction of Rab7 and RILP in normal and INCL fibroblasts and (D) corresponding quantification. Data are presented as mean ± SD (n=25 (Normal fibroblast), n=37 (INCL fibroblast) *p<0.05. (E) Relative GTPase activity in the immunoprecipitates of lysates of normal and INCL fibroblasts transfected with myc-tagged Rab7 with myc-antibody and Western blot to show the input using Rab7-antibody. N, Normal; INCL, Fibroblasts from an INCL patient. Data are presented as mean ± SD (n=4), *p<0.05.
3.7. Suppression of Rab7 GTPase activity in PPT1-defcient INCL cells
It has been reported that RILP interacts with GTP-bound active Rab7 for transport to the lysosomal surface43,44. We reasoned that reduced Rab7-RILP interaction may inhibit autophagosome-lysosome fusion in PPT1-deficient INCL fibroblasts compared with that in normal control fibroblasts. We also compared Rab7 GTPase activity in cultured normal and INCL fibroblasts. To achieve this, we transfected both normal and INCL cells with a Myc-tagged Rab7-cDNA construct, immunoprecipitated Rab7 using Myc-antibody and then determined the GTPase activity in the immunoprecipitate. The results showed a significantly reduced GTPase activity in INCL cells compared with that in their normal counterparts (Figure 5E). Taken together these results demonstrated that suppression of Rab7 GTPase activity in PPT1-deficient cells impairs autophagosome-lysosome fusion.
3.8. S-palmitoylation of Rab7 and its membrane-localization in INCL fibroblasts
Numerous proteins, especially in the brain, undergo S-palmitoylation for membrane-anchorage, required for their function [15–17]. While S-palmitoylation serves a very important role in regulating protein stability, protein-protein interaction, membrane affinity for proteins and protein trafficking [15–17], the S-palmitoylated proteins also require depalmitoylation for recycling and/or degradation by lysosomal hydrolases14. Thus, dynamic S-palmitoylation45 is essential for the steady-state membrane localization and function of these proteins. Recently, it has been reported that canine Rab7 is S-palmitoylated on Cys-83 and Cys-84 and this post-translational modification is required for its endosome-to-TGN trafficking46. Previously, we reported that Ppt1-deficiency caused mislocalization of a critical subunit (V0a1) of v-ATPase to the plasma membrane instead of its normal localization on lysosomal membrane33. We hypothesized that S-palmitoylation of Rab7 may cause its mislocalization in Ppt1-deficient cells. To test this hypothesis, we first analyzed the mouse and human Rab7 peptide sequences by CSS-palm35, a computer program that predicts the cysteine residues in a protein, which are the potential S-palmitoylation sites. Our results showed that Cys-83, Cys-205 and Cys-207 in Rab7 proteins from Homo sapiens (Supplementary Figure S6A), Mus musculus (Supplementary Figure S6B) and Canis lupus (Supplementary Figure S6C) are the potential S-palmitoylation sites and are conserved in several mammalian species.
To confirm if Rab7 undergoes S-palmitoylation, we transfected myc-tagged mouse Rab7-cDNA construct in COS-1 cells and determined S-Palmitoylation using 3 different methods: (i) incorporation of [14C]-palmitate in presence and absence of hydroxylamine [15], which specifically cleaves the thioester linkage that attaches palmitate to specific cysteine residues in polypeptides; (ii) Acyl-biotin exchange (ABE) method36 and (iii) Acyl-RAC (resin-assisted capture) assay37. The results of all three of these assays unequivocally demonstrated that Rab7 is an S-palmitoylated protein (Figure 6A, 6B and 6C)) as previously reported46. However, when we performed mutagenesis studies using myc-tagged-Rab7 cDNA-construct in which all the cysteine residues in Rab7, conserved in 3 different species indicated above (i.e. Cys-83, Cys-84, Cys-143, Cys-205 and Cys-207), were mutated one at a time to either serine or alanine, we found that only Cys-205Ser or Ala) and Cys-207Ser or Ala) mutations completely abrogated S-palmitoylation of Rab7 (Figure 6D and 6E)). These results from mouse Rab7 are in contrast to the previous report demonstrating that in canine Rab7, Cys-83 and Cys-84 are both S-palmitoylated and mutation of the two C-terminal cysteines (Rab7 C205,207S) resulted in a reduction in the level of Rab7 S- palmitoylation46. The reason(s) for this discrepancy remains unclear and needs further investigation. Interestingly, we found that the level of S-palmitoylated Rab7 in PPT1-deficient INCL fibroblasts was substantially higher compared with that in the normal control cells (Figure 6F). Taken together, these results demonstrated that Rab7 undergoes S-palmitoylation and PPT1-deficiency suppresses its localization on late endosomal/lysosomal membrane.
Figure 6. S-palmitoylation of Rab7.
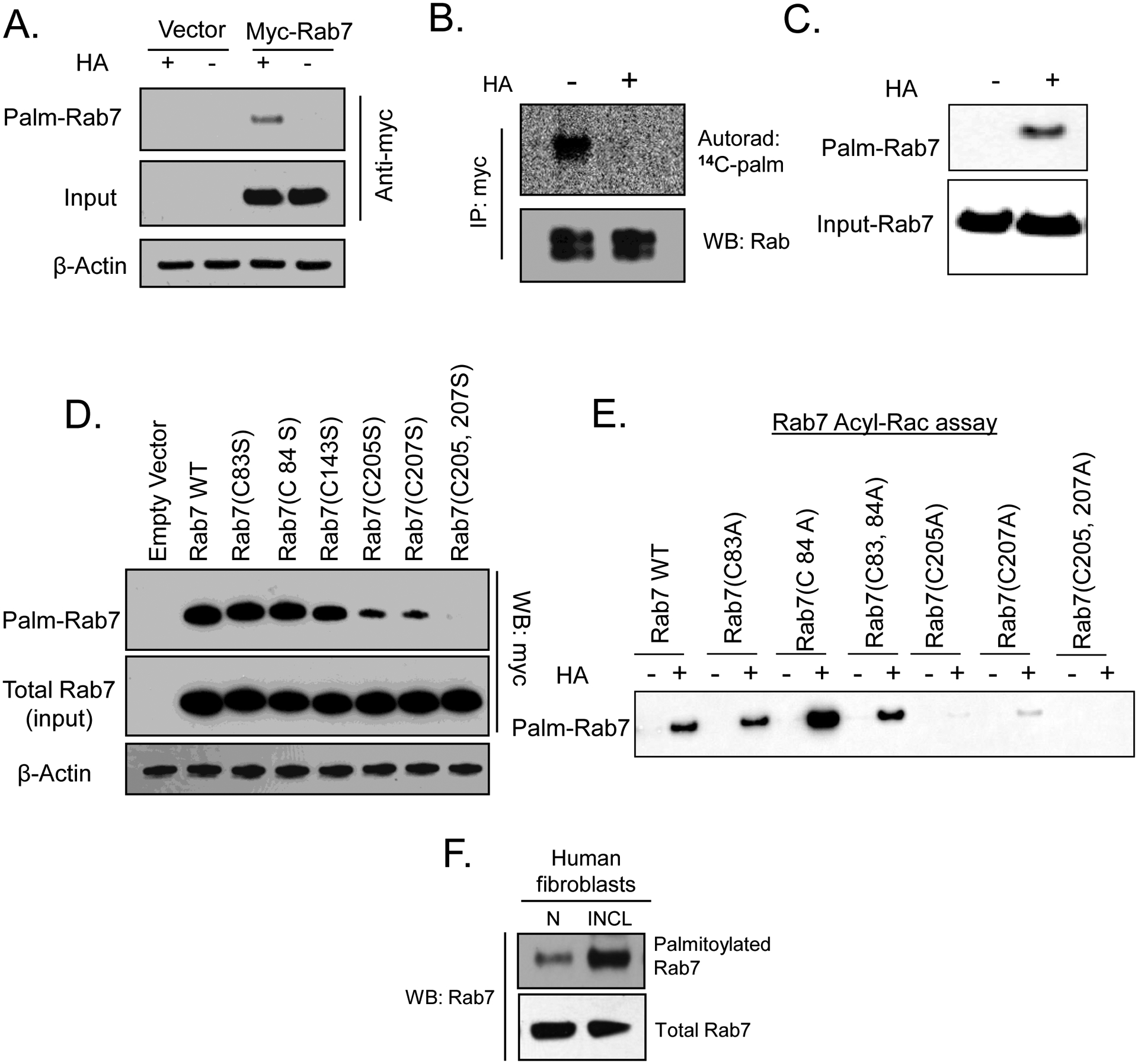
(A) S-palmitoylation of Rab7 was detected by acyl-biotinyl exchange (ABE) method in COS-1 cells transfected with myc-tagged Rab7. (B) Incorporation of 14C palmitic acid in Rab7 in the absence and presence of hydroxylamine (HA). COS-1 cells were transfected with myc-tagged Rab7, labeled with [14C] palmitic acid, immunoprecipitated using myc-antibody, resolved by SDS-PAGE and radioactive palmitate incorporation into Rab7 removed by HA was detected by autoradiography. (C) HEK-293T cells were transfected with myc-Rab7 and Acyl-RAC assay was performed to confirm S-palmitoylation; (D) Detection of S-palmitoylation signal in WT- and mutant-Rab7 transfected in COS-1 cells by ABE method. (E) Detection of S-palmitoylation in WT- and mutant-Rab7 by Acyl RAC Assay; (F) Levels of S-palmitoylated Rab7 in normal and INCL fibroblasts; N, Normal fibroblast, INCL, Fibroblasts from INCL patient.
3.9. NtBuHA-treatment enhances Rab7-RILP interaction and ameliorates defective autophagy
Previously, we identified a brain-penetrant, PPT1-mimetic, small molecule, N-tert (Butyl) hydroxylamine (NtBuHA), which showed neuroprotective and lifespan expanding properties in Cln1−/− mice32. Since Rab7 requires S-palmitoylation for intracellular trafficking and since the results of our present study showed that Rab7-RILP interaction is suppressed in PPT1-deficient fibroblasts from INCL patient, we sought to determine whether NtBuHA ameliorated this defect. Accordingly, we treated cultured INCL fibroblasts with NtBuHA and determined Rab7-RILP interaction by PLA reaction. The results showed that compared with normal fibroblasts (Figure 7A, left panel and Figure 7B, blue bar) those from INCL patient contained significantly lower signal for Rab7-RILP interaction (Figure 7A, middle panel and Figure 7B, red bar). Importantly, the INCL fibroblasts treated with NtBuHA showed significantly increased signal for Rab7-RILP interaction (Figure 7A, right panel and Figure 7B, light blue bar). Taken together, these results demonstrated that NtBuHA-treatment can significantly improve Rab7-RILP interaction and hence Rab7 function that is essential for autophagosome-lysosome fusion.
Figure 7: Amelioration of defective autophagy by NtBuHA.
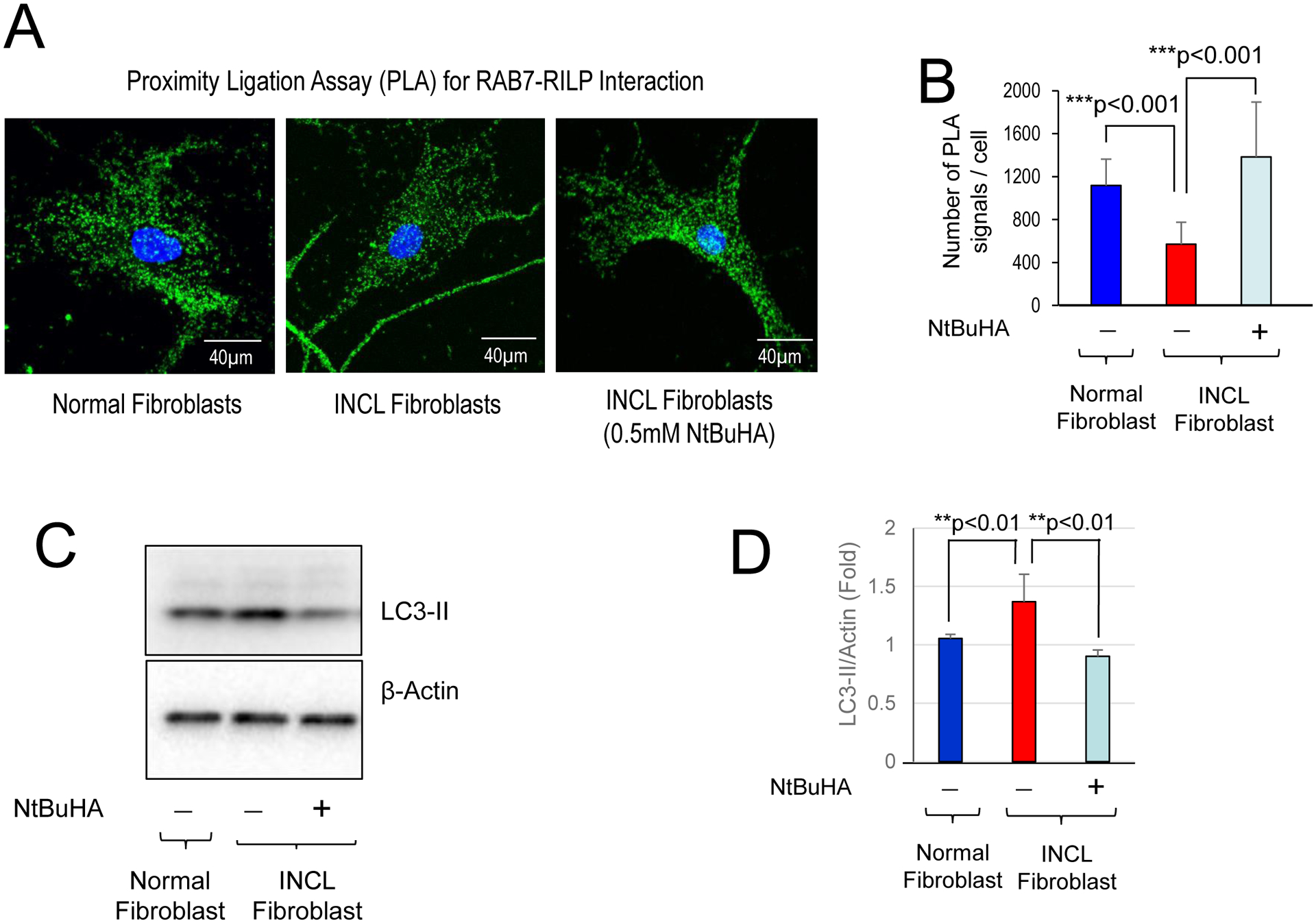
Cultured fibroblasts from an INCL patient were either untreated or treated with 0.5mM NtBuHA for 4 days before fixation with methanol. Proximity ligation assay (PLA) demonstrating the interaction of RAB7 with RILP. (A) Quantitation of PLA in Normal fibroblast and INCL fibroblasts treated with or without 0.5 mM NtBuHA. Data are presented as mean ± SD (n=20), *p<0.001. (B) Western blots of normal and INCL fibroblasts treated with or without NtBuHA and (C) corresponding densitometric analysis for quantification. The results are presented as the mean ± SD (n=4) and statistical significance was determined by One-Way ANOVA test followed by Tukey’s post hoc test. **p<0.01.
Next, we sought to determine whether NtBuHA treatment can reduce autophagosome accumulation in INCL fibroblasts. Accordingly, we treated INCL fibroblasts for 3 days with NtBuHA and then determined the level of autophagosomal marker LC3-II by Western blot analysis. We observed significantly higher level of LC3-II in INCL cells as compared to normal fibroblasts. Importantly, treatment of the INCL cells with a PPT1-mimetic small molecule like NtBuHA decreased the level of LC3-II in INCL cells (Figure 7C and 7D). These data demonstrate that treatment with NtBuHA is effective in ameliorating the defective Rab7-RILP interaction and autophagy in INCL fibroblasts.
4.0. DISCUSSION
In this study, we presented evidence from postmortem brain tissues and Cln1−/− mouse brain that autophagy is dysregulated. We also demonstrated that Rab7 undergoes S-palmitoylation and this posttranslational lipid-modification is essential for its trafficking to the late endosomal/lysosomal surface to interact with RILP. Moreover, we showed that Ppt1-deficiency in Ppt1-deficient Cln1−/− mice the localization of Rab7 on late endosomal/lysosomal compartment is dysregulated. Furthermore, this abnormality suppressed Rab7-RILP interaction inhibiting Rab7 GTPase activity required for autophagosome-lysosome fusion. Since autophagosome-lysosome fusion is essential for the formation of a hybrid organelle, autolysosome, in which the lysosomal hydrolases encounter the undigested cargo so that it can be degraded. Thus, the disruption of this process leads to the accumulation of undegraded cargo (ceroid) in the lysosome leading to INCL pathogenesis. Importantly, we demonstrate that a PPT1-mimetic, brain-penetrant small molecule, NtBuHA, which has been reported to be neuroprotective32 ameliorated the defective Rab7-RILP interaction and autophagy suggesting that it may have therapeutic implications for INCL.
Neuronal Ceroid Lipofuscinoses (NCLs), commonly known as Batten disease47–50, constitute a group of the most common neurodegenerative lysosomal storage diseases (LSDs), which mostly affect children. Mutations in at least 13 different genes (called CLNs) underlie various forms of NCLs49, 51. The infantile NCL (or INCL) is one of the most devastating neurodegenerative diseases [10], caused by inactivating mutations in the CLN1 gene [11], which encodes palmitoyl-protein thioesterase-1 (PPT1) [12]. PPT1 is a lysosomal enzyme [12, 13] that catalyzes the removal of palmitate residues from S-palmitoylated proteins (constituents of ceroid), required for their degradation and clearance by lysosomal hydrolases [14]. The Cln1−/− mice, generated by targeted disruption of the Cln1 gene30, recapitulates virtually all clinical and pathological features of INCL52 including lysosomal dysfunction.
Lysosomes are the major subcellular organelles, which degrade both endocytosed material from extracellular sources as well as dysfunctional intracellular macromolecules and organelles accumulated in autophagosomes1, 3, 5, 53. Lysosomal storage disorders have been called the disorders of dysregulated autophagy [7, 8], because autophagy is a degradative process, which is dysregulated in most of the LSDs [8]. Although defective autophagy has been reported in a juvenile NCL(CLN3-disease) disease model54, to our knowledge, it has not been reported in INCL. In the present study, we report that autophagy is dysregulated in Cln1−/− mice, a reliable animal model of INCL. Moreover, we also found this defect in postmortem brain tissues from an INCL patient as well as in cultured INCL fibroblasts. We demonstrated that in the brain of Cln1−/− mice, the lysosomal localization of Rab7 is markedly reduced. Furthermore, both mouse and human Rab7 undergoes S-palmitoylation and this modification is essential for its localization on late endosomal/lysosomal membrane where Rab7-RILP interaction activates Rab7 GTPase activity, required for autophagosome-lysosome fusion.
In contrast to a previous report that Cys-83 and Cys-84 in canine Rab7 are the S-palmitoylated residues46, our results show that Cys-205 and Cys-207 in human Rab7 undergo S-palmitoylation. We used three different methods to confirm that Rab7 is an S-palmitoylated protein. These are: (a) incorporation of 14C-palmitate; (b) Acyl-biotin exchange (ABE) method and (c) Resin-assisted capture (Acyl-Rac) assay. To confirm our results that Rab7 is S-palmitoylated on Cys-205 and Cys-207, we performed experiments in HEK293T and COS-1 cells, which were transfected with constructs in which each of the five cysteine residues in Rab7 (i.e. Cys-83, Cys-84, Cys-143, Cys-205 and Cys-207) were mutated one Cys at a time to Ser or Ala (Supplementary Figures S7A and S7B). We tested the WT HEK293T cells as well as each of the transfected cell populations for Rab7 S-palmitoylation. The results showed that the cells transfected either with a construct containing Cys205Ser mutant or Cys207Ser mutant of Rab7 yielded significantly reduced S-palmitoylation of Rab7. However, cells transfected with both mutant constructs showed complete loss of Rab7 S-palmitoylation. To further confirm these results, we also used constructs in which Cys-205 and Cys-207 were mutated to Ala instead of Ser. Here again, HEK293T cells transfected with the construct containing double mutant (i.e. Cys205Ala and Cys207Ala) showed that S-palmitoylation of Rab7 was completely abrogated. We interpret these results to suggest that in each cell population, a certain percentage of the cells may contain Rab7 S-palmitoylation on Cys-205 and in the remainder of the cells, Cys-207 may be S-palmitoylated, even though one of these residues (Cys-207) has been reported to undergo prenylation55.
How might Cys-205 or Cys-207 in Rab7 be S-palmitoylated when one of these Cys residues have been reported to undergo prenylation, which is an irreversible lipid modification? We speculate that Cys-205 is the authentic S-palmitoylation site in Rab7. However, our results show that in cells expressing Cys205Ala or Cys205Ser mutant of Rab7, Cys-207 is S-palmitoylated. This result is expected, if we assume that in cells expressing Cys205Ala or Cys205Ser mutant, a portion of the newly synthesized Rab7 may be S-palmitoylated on Cys-207, before prenylation can occur on this residue. Similarly, in cells transfected with Cys207Ala or Cys2017Ser construct, Cys-205 becomes the sole S-palmitoylation site. Alternatively, all Cys-207 residues in Rab7 at any given time may not be prenylated and those that are not S-palmitoylated. This notion is further supported by the finding that cells in which both Cys-205 and Cys-207 are mutated to Ser or Ala completely abrogated S-palmitoylation of Rab7. Recently, it has been reported that canine Rab7 is S-palmitoylated on Cys-83 and Cys-8446. However, the CSS-Palm analysis of human, mouse and dog Rab7 predicted that Cys-83, Cys-205 and Cys-207 are the potential palmitoylation sites (Supplementary Figure S6A, S6B and S6C). In our experiments, we were unable to confirm that Cys-83 or Cys-84 in murine Rab7 undergo S-palmitoylation. Moreover, our results showed that only the double mutation in Rab7 (i.e. Cys205Ser or Ala and Cys207Ser or Ala) in the same construct completely abrogated S-palmitoylation.
We have demonstrated that in the brain tissues of Cln1−/− mice as well as in cultured cells from INCL patients Rab7-RILP interaction is suppressed and this may be the reason for impaired autophagosome-lysosome fusion in the PPT1-deficient cells. Although it has been reported that impaired autophagosome-lysosome fusion is one of the fundamental defects in LSDs, the molecular mechanism(s) of this abnormality has remained poorly understood. The results of our present study demonstrate that lack of Ppt1, in Cln1−/− mice and in cultured cells from INCL patients, altered extra-lysosomal membrane localization of Rab7 reduced its interaction with RILP, which most likely impairs its GTPase activity that is critically important for autophagosome-lysosome fusion.
While our results show that suppression of Rab7-RILP interaction, at least in part, may dysregulate autophagy in our experimental model, there might be other factors that contribute to this defect. It has been shown that RILP interacts with the V1G1 subunit of v-ATPase and thereby regulates the v-ATPase activity56, 57. Since our previous work demonstrated that PPT1-deficiency causes a misrouting of the v-ATPase subunit VOa1 causing lysosomal acidification defect33, it is possible that reduced Rab7-RILP interaction may affect the v-ATPase function reducing autophagosome lysosome fusion. However, it requires further experiments to prove this. Even though our findings provide an insight into one of the potential mechanisms that underlies defective autophagy in a mouse model of INCL, we propose that varying factors suppressing Rab7-RILP interaction may contribute to pathogenesis of other LSDs unrelated to INCL. Our results revealed a previously unrecognized role of PPT1 in autophagy and provided strong evidence that disruption of Rab7-RILP interaction causes dysregulated autophagy in PPT1-deficient cells and suggest that brain-penetrant, Ppt1-mimetic, small molecules like NtBuHA32 ameliorating these defects may have therapeutic implications.
Supplementary Material
Figure S1. Impaired autophagy flux causes accumulation of p62/SQSTM1 and polyubiquitinated proteins in Cln1−/− mouse brain and in INCL cells. (A) Western blot and corresponding densitometric analysis of WT and Cln1−/− cortical tissue lysate for autophagosomal marker LC3-II. Images (20x) of WT and Cln1−/− mouse cortical sections stained with (B) p62/SQSTM1 and LC3 and (C) p62/SQSTM1 and ubiquitin antibodies.
Figure S2. RAB7-mRNA in WT and Cln1−/− mouse brain and normal and INCL fibroblasts Levels of RAB7-mRNA were determined by real-time RT-PCR analysis using total RNA in: (A) cortical tissues from WT and Cln1−/− mice and (B) normal and INCL fibroblasts. (C) Lamp2 levels in total cortical homogenate from WT and Cln1−/− mice (left panel) as well as from WT and INCL fibroblast (right panel).
Figure S3. Lysosomal and membrane localization of RAB7 in WT and Cln1−/− mouse brain and normal and INCL fibroblasts. (A) Confocal immunofluorescence images of normal and INCL fibroblasts stained with antibodies for Rab7 with lysosomal marker, LAMP2 and (B) corresponding colocalization analysis determined by Manders coefficient. Data are presented as mean ± SD, n=22 (Normal fibroblast), n=25 (INCL fibroblast) *p<0.05. (C) Detection of RAB7 level in membrane and cytosolic fractions prepared from WT and Cln1−/− mouse cortex by Western blot analysis. (D) Level of RAB7 in the membrane and cytosolic fractions was also determined in normal and INCL cells by Western blot analysis. (F) Confocal images of cultured fibroblasts from normal and INCL patient showing colocalization of Rab7 with the membrane marker, pancadherin and (G) corresponding colocalization analysis determined by Mander’s coefficient. Data are presented as mean ± SD, n=25 (Normal fibroblast), n=26 (INCL fibroblast) *p<0.05.
Figure S4. RILP-mRNA in WT and Cln1−/− mouse brain and normal and INCL fibroblasts Expression of RILP-mRNA was determined by qRT-PCR analysis in (A) cortical tissues from WT and Cln1−/− mice and (B) normal and INCL fibroblasts.
Figure S5. Detection of RILP-Rab7 colocalization with LAMP2 in normal and INCL cells. (A) Detection of Rab7-RILP colocalization in normal and INCL patient cells using confocal imaging. (B) Detection of colocalization of Rab7-RILP PLA signals in lysosomal compartment in normal and INCL patient cells using confocal imaging.
Figure S6. Cysteine residues in Rab7 predicted to be S-palmitoylated by CSS-Palm analyses CSS-palm analyses were performed using Rab7 peptide sequences from (A). human; (B). Mouse and (C) dog. Note that in all three species Cys-83, Cys-205 and Cys-207 were predicted to be S-palmitoylated.
Figure S7. S-palmitoylation site-mutants in human Rab7 to confirm palmitoylation site(s) Human Rab7 peptide sequences were first analyzed by CSS-Palm to predict potential S-palmitoylation sites. In each construct one of the 5-cystine residues (i.e. Cys-83, Cys-84, Cys-143, Cys-205 and Cys-207) were mutated to serine (marked in red) and in one construct both Cys-205 and Cys-207 were mutated to serine. These constructs were used to transfect HEK-293 cells and to determine Rab7 S-palmitoylation by Acyl-Rac assay.
Synopsis:
Palmitoyl-protein thioesterase 1-deficiency leads to the dysregulation of autophagy due to defective Rab7-RILP interaction impairing autophagosome-lysosome fusion, which contributes to INCL pathogenesis.
Acknowledgements:
We also thank Drs. J.Y. Chou for critical review of the manuscript and helpful suggestions. We also thank Dr. V. Schram (Microscopy and Imaging Core, Eunice Kennedy-Shriver National Institute of Child Health and Human Development [NICHD]) for his help with confocal microscopy. This research was supported in full by the Intramural Research Program of the Eunice Kennedy Shriver NICHD, NIH. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- LSD
Lysosomal storage disease
- NCLs
neuronal ceroid lipofuscinoses
- INCL
Infantile neuronal ceroid lipofuscinosis
- CLN1
Ceroid lipofuscinosis neuronal 1
- PPT1
Palmitoyl-protein thioesterases-1
- ATGs
Autophagy-related genes
- RILP
Rab-interacting lysosomal protein
- NtBuHA
N-tert(Butyl) hydroxylamine
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest with the contents of this article.
REFERENCES
- [1].Klionsky D Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007; 8:931–937. [DOI] [PubMed] [Google Scholar]
- [2].Yu L, Chen Y, Tooze SA. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018; 14: 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Li WW, Li J, Bao JK. Microautophagy: lesser-known self-eating. Cell Mol Life Sci 2012; 69:1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 2012; 22: 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mizushima N, Noda T, Yoshimori T, et al. A protein conjugation system essential for autophagy. Nature 1998; 395: 395–398. [DOI] [PubMed] [Google Scholar]
- [6].Klionsky DJ, Schulman BA. Dynamic regulation of macroautophagy by distinctive ubiquitin- like proteins. Nat Struct Mol Biol 2014; 21: 336–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Seranova E, Connolly KJ, Zatyka M, et al. Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem 2017; 61: 733–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Platt FM. Emptying the stores: lysosomal diseases and therapeutic strategies, Nat Rev Drug Discov 2018; 17: 133–150. [DOI] [PubMed] [Google Scholar]
- [9].Proia RL, Wu YP. Blood to brain to the rescue. J Clin Invest 2004; 13(8):1108–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Santavuori P Neuronal ceroid-lipofuscinoses in childhood, Brain Dev 1988; 10: 80–83. [DOI] [PubMed] [Google Scholar]
- [11].Vesa J, Hellsten E, Verkruyse LA, Camp LA, Rapola J, Santavuori P. et al. Mutations in the palmitoyl protein thioesterases gene causing infantile neuronal ceroid lipofuscinosis, Nature 1995; 376: 584–87. [DOI] [PubMed] [Google Scholar]
- [12].Camp LA, Hofmann SL. Purification and properties of a palmitoyl-protein thioesterase that cleaves palmitate from H-Ras. J Biol Chem 1993; 268: 22566–22574. [PubMed] [Google Scholar]
- [13].Hellsten E, Vesa J, Olkkonen VM, Jalanko A, Peltonen L. Human palmitoyl protein thioesterase: evidence for lysosomal targeting of the enzyme and disturbed cellular routing in infantile neuronal ceroid lipofuscinosis. EMBO J 1996; 15:5240–5245. [PMC free article] [PubMed] [Google Scholar]
- [14].Lu JY, Verkruyse LA, Hofmann SL. Lipid thioesters derived from acylated proteins accumulate in infantile neuronal ceroid lipofuscinosis: correction of the defect in lymphoblasts by recombinant palmitoyl-protein thioesterases. Proc Natl Acad Sci USA 1996; 93:10046–10050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Schmidt MFG. Fatty acylation of proteins. Biochim Biophys Acta 1989; 988: 411–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fukata Y, Fukata M. Protein palmitoylation in neuronal development and synaptic plasticity, Nat. Rev. Neurosci 11 (2010) 161–175. [DOI] [PubMed] [Google Scholar]
- [17].El-Husseini AE, Craven SE, Chetkovich DM, et al. Dual palmitoylation of PSD95 mediates its vesicular sorting, postsynaptic targeting, and ion channel clustering. J Cell Biol 2000; 148:159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhang MM, Hang HC. Protein S-palmitoylation in cellular differentiation. Biochem Soc Trans 2017; 45:275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tsutsumi R, Fukata Y, Fukata M. Discovery of protein-palmitoylating enzymes, Pflugers Arch 2008; 456: 1199–1206. [DOI] [PubMed] [Google Scholar]
- [20].Greaves J, Chamberlain LH. DHHC palmitoyl transferases: substrate interactions and (patho) physiology. Trends Biochem Sci 2011; 36: 245–253. [DOI] [PubMed] [Google Scholar]
- [21].Zeidman R, Jackson CS, Magee AI. Protein acyl thioesterases (Review), Mol Membr Biol 2009; 26: 32–41. [DOI] [PubMed] [Google Scholar]
- [22].Lin DT, Conibear E. ABHD17 proteins are novel protein depalmitoylases that regulate N-Ras palmitate turnover and subcellular localization. Elife 2015; 4: e11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell 2004; 166: 153–166. [DOI] [PubMed] [Google Scholar]
- [24].Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol 2001; 2: 107–117. [DOI] [PubMed] [Google Scholar]
- [25].Guerra F, Bucci C. Multiple roles of the small GTPase Rab7, Cell 2016; 5 pii: E34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jäger S, Bucci C, Tanida I, et al. Role of Rab7 in maturation of late autophagic vacuoles. J Cell Sci 2004; 117:4837–4848. [DOI] [PubMed] [Google Scholar]
- [27].Ravikumar B, Sarkar S, Davies JE, et al. Regulation of mammalian autophagy in physiology and pathophysiology, Physiol. Rev 2010; 90: 1383–1435. [DOI] [PubMed] [Google Scholar]
- [28].Luzio JP, Pryor PR, Bright NA, Lysosomes: fusion and function, Nat Rev Mol Cell Biol 2007; 8: 622–32. [DOI] [PubMed] [Google Scholar]
- [29].Shen HM, Mizushima N. At the end of the autophagic road: an emerging understanding of lysosomal functions in autophagy. Trends Biochem Sci 2014; 39:61–71. [DOI] [PubMed] [Google Scholar]
- [30].Gupta P, Soyombo AA, Atashband A, et al. Disruption of PPT1 or PPT2 causes neuronal Ceroid lipofuscinosis in knockout mice. Proc Natl Acad Sci USA 2001; 98:13566–13571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Levin SW, Baker EH, Zein WM, Zhang Z, Quezado ZM, Miao N, et al. Oral cysteamine bitartrate and N-acetylcysteine for patients with infantile neuronal ceroid lipofuscinosis: a pilot study. Lancet Neurol 2014; 13:777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sarkar C, Chandra G, Peng S, Zhang Z, Liu A, Mukherjee AB. Neuroprotection and lifespan extension in Ppt1(−/−) mice by NtBuHA: therapeutic implications for INCL. Nat Neurosci 2013; 16:1608–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bagh MB, Peng S, Chandra G, et al. Misrouting of v-ATPase subunit V0a1 dysregulates lysosomal acidification in a neurodegenerative lysosomal storage disease model. Nat Commun 2017; 8:14612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang Z, Butler JD, Levin SW, et al. Lysosomal ceroid depletion by drugs: therapeutic implications for a hereditary neurodegenerative disease of childhood, Nat Med 2001;7: 478–484. [DOI] [PubMed] [Google Scholar]
- [35].Ren J, Wen L, Gao X, et al. CSS-Palm 2.0: an updated software for palmitoylation sites prediction, Prot Eng Design Select 2008; 21: 639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wan J, Roth AF, Bailey AO, Davis NG. Palmitoylated proteins: purification and identification, Nat Protoc 2007; 2: 1573–1784. [DOI] [PubMed] [Google Scholar]
- [37].Forrester MT, Hess DT, Thompson JW, et al. Site-specific analysis of protein S-acylation by resin-assisted capture. J Lipid Res 2011; 52:393–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Söderberg O, Gullberg M, Jarvius M, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation, Nat Meth 2006; 3: 995–1000. [DOI] [PubMed] [Google Scholar]
- [39].Levine B, Klionsky DJ. Development by self-digestion: Molecular mechanisms and biological functions of autophagy Cell 2004; 6:463–477. [DOI] [PubMed] [Google Scholar]
- [40].Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3:452–60. [DOI] [PubMed] [Google Scholar]
- [41].Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. http://www.pnas.org/content/105/52/20567. abstract-aff-1#aff-1 Proc Natl Acad Sci USA 2008; 105: 20567–20574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Settembre C, Fraldi A, Jahreiss L, et al. A block of autophagy in lysosomal storage disorders. Hum Mol Genet 2008;17: 119–129. [DOI] [PubMed] [Google Scholar]
- [43].Jordens I, Fernandez-Borja M, Marsman M, et al. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors, Curr Biol 2001; 11:1680–1685. [DOI] [PubMed] [Google Scholar]
- [44].Cantalupo G, Alifano P, Roberti V, et al. Rab-interacting lysosomal protein (RILP): the Rab effector required for transport to lysosomes. EMBO J 2001; 20: 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Salaun C, Greaves J, Chamberlain LH. The intracellular dynamic of protein palmitoylation. J Cell Biol 2010; 191:1229–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Modica G, Skorobogata O, Sauvageau E, et al. Rab7 palmitoylation is required for efficient endosome-to-TGN trafficking, J Cell Sci 2017; 130: 2579–2590. [DOI] [PubMed] [Google Scholar]
- [47].Anderson GW, Goebel HH, Simonati A. Human pathology of NCL, Biochim Biophys Acta 2013; 1832:1807–1826. [DOI] [PubMed] [Google Scholar]
- [48].Mole SE, Cotman SL. Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochim Biophys Acta 2015; 1852: 2237–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Cooper JD, Tarczyluk MA, Nelvagal HR. Towards a new understanding of NCL pathogenesis, Biochim Biophys Acta 2015; 1852: 2256–2261. [DOI] [PubMed] [Google Scholar]
- [50].Mukherjee AB, Appu AP, Sadhukhan T, et al. Emerging new roles of the lysosome and neuronal ceroid lipofuscinoses, Mol. Neurodegener 2018; 14(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kousi M, Lehesjoki AE, Mole SE. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses, Hum Mol Genet 2012; 33: 42–63. [DOI] [PubMed] [Google Scholar]
- [52].Bible E, Gupta P, Hofmann SL, et al. Regional and cellular neuropathology in the palmitoyl protein thioesterase-1 null mutant mouse model of infantile neuronal ceroid lipofuscinosis, Neurobiol Dis 2004; 16: 346–359. [DOI] [PubMed] [Google Scholar]
- [53].Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol 2009; 10: 623–635. [DOI] [PubMed] [Google Scholar]
- [54].Cao Y, Espinola JA, Fossale E, et al. Autophagy is disrupted in a knock-in mouse model of juvenile neuronal ceroid lipofuscinosis. J Biol Chem 2006; 281: 20483–93. [DOI] [PubMed] [Google Scholar]
- [55].Joberty G, Tavitian A, Zahraoui A. Isoprenylation of Rab proteins possessing a C-terminal CaaX motif, FEBS Lett 1993; 330: 323–338. [DOI] [PubMed] [Google Scholar]
- [56].De Luca M, Cogli L, Progida C, et al. RILP regulates vacuolar ATPase through interaction with the V1G1 subunit. J. Cell Sci 2015; 127:2597–2708. [DOI] [PubMed] [Google Scholar]
- [57].De Luca M, Bucci C. A new V-ATPase regulatory mechanism mediated by the Rab interacting lysosomal protein (RILP). Commun Integr Biol 2014; 7: e971572. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Impaired autophagy flux causes accumulation of p62/SQSTM1 and polyubiquitinated proteins in Cln1−/− mouse brain and in INCL cells. (A) Western blot and corresponding densitometric analysis of WT and Cln1−/− cortical tissue lysate for autophagosomal marker LC3-II. Images (20x) of WT and Cln1−/− mouse cortical sections stained with (B) p62/SQSTM1 and LC3 and (C) p62/SQSTM1 and ubiquitin antibodies.
Figure S2. RAB7-mRNA in WT and Cln1−/− mouse brain and normal and INCL fibroblasts Levels of RAB7-mRNA were determined by real-time RT-PCR analysis using total RNA in: (A) cortical tissues from WT and Cln1−/− mice and (B) normal and INCL fibroblasts. (C) Lamp2 levels in total cortical homogenate from WT and Cln1−/− mice (left panel) as well as from WT and INCL fibroblast (right panel).
Figure S3. Lysosomal and membrane localization of RAB7 in WT and Cln1−/− mouse brain and normal and INCL fibroblasts. (A) Confocal immunofluorescence images of normal and INCL fibroblasts stained with antibodies for Rab7 with lysosomal marker, LAMP2 and (B) corresponding colocalization analysis determined by Manders coefficient. Data are presented as mean ± SD, n=22 (Normal fibroblast), n=25 (INCL fibroblast) *p<0.05. (C) Detection of RAB7 level in membrane and cytosolic fractions prepared from WT and Cln1−/− mouse cortex by Western blot analysis. (D) Level of RAB7 in the membrane and cytosolic fractions was also determined in normal and INCL cells by Western blot analysis. (F) Confocal images of cultured fibroblasts from normal and INCL patient showing colocalization of Rab7 with the membrane marker, pancadherin and (G) corresponding colocalization analysis determined by Mander’s coefficient. Data are presented as mean ± SD, n=25 (Normal fibroblast), n=26 (INCL fibroblast) *p<0.05.
Figure S4. RILP-mRNA in WT and Cln1−/− mouse brain and normal and INCL fibroblasts Expression of RILP-mRNA was determined by qRT-PCR analysis in (A) cortical tissues from WT and Cln1−/− mice and (B) normal and INCL fibroblasts.
Figure S5. Detection of RILP-Rab7 colocalization with LAMP2 in normal and INCL cells. (A) Detection of Rab7-RILP colocalization in normal and INCL patient cells using confocal imaging. (B) Detection of colocalization of Rab7-RILP PLA signals in lysosomal compartment in normal and INCL patient cells using confocal imaging.
Figure S6. Cysteine residues in Rab7 predicted to be S-palmitoylated by CSS-Palm analyses CSS-palm analyses were performed using Rab7 peptide sequences from (A). human; (B). Mouse and (C) dog. Note that in all three species Cys-83, Cys-205 and Cys-207 were predicted to be S-palmitoylated.
Figure S7. S-palmitoylation site-mutants in human Rab7 to confirm palmitoylation site(s) Human Rab7 peptide sequences were first analyzed by CSS-Palm to predict potential S-palmitoylation sites. In each construct one of the 5-cystine residues (i.e. Cys-83, Cys-84, Cys-143, Cys-205 and Cys-207) were mutated to serine (marked in red) and in one construct both Cys-205 and Cys-207 were mutated to serine. These constructs were used to transfect HEK-293 cells and to determine Rab7 S-palmitoylation by Acyl-Rac assay.