

Abstract
The coupling of protein synthesis and folding is a crucial yet poorly understood aspect of cellular protein folding. Over the past few years, it has become possible to experimentally follow and define protein folding on the ribosome, revealing principles that shape co-translational folding and distinguish it from refolding in solution. Here, we highlight some of these recent findings from biochemical and biophysical studies and their potential significance for cellular protein biogenesis. In particular, we focus on nascent chain interactions with the ribosome, interactions within the nascent protein, modulation of translation elongation rates, and the role of mechanical force that accompanies nascent protein folding. The ability to obtain mechanistic insight in molecular detail has set the stage for exploring the intricate process of nascent protein folding. We believe that the aspects discussed here will be generally important for understanding how protein synthesis and folding are coupled and regulated.
Keywords: co-translational protein folding, folding pathways, mechanical force, misfolding, molecular chaperones, nascent protein, ribosome
Graphical Abstract
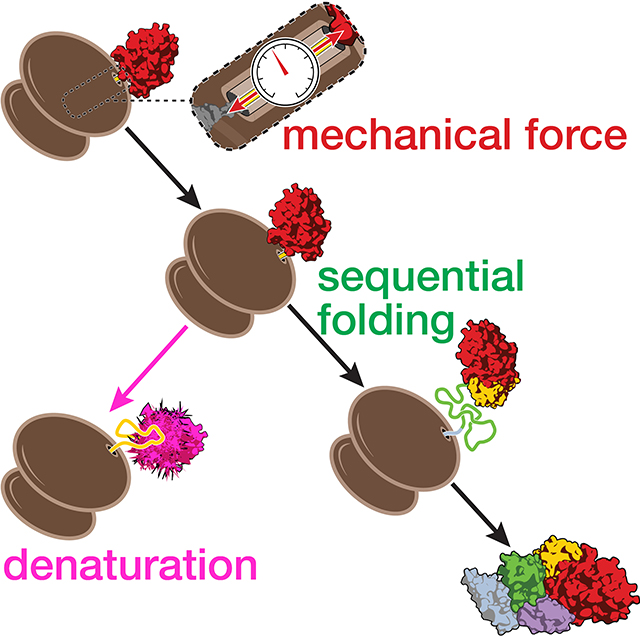
Many proteins begin to fold during translation. This sequential co-translational folding is important to avoid protein aggregation and misfolding. Generation of mechanical force by nascent chain folding and denaturation of co-translationally formed structures are among aspects of co-translational folding that have recently come to light.
INTRODUCTION
How cellular proteins swiftly and robustly fold into their functional native structures remains an outstanding problem in biology. Classic experiments pioneered by Anfinsen have shown that the information contained in the primary sequence is sufficient for proteins to fold into their native structures.[1] The Anfinsen principle posits that folding is driven by minimization of free energy and therefore occurs spontaneously. Given its broad importance for biological function, protein folding has been the subject of intense study for decades, resulting in several breakthroughs that shed light on the mechanisms of protein folding.[2–5] The realization that failure to fold can result in misfolding and aggregation, which have been implicated in many pathologies,[6–8] has further increased interest in understanding the mechanistic underpinnings of protein folding.
As has been pointed out previously,[9] most studies of folding have been carried out with isolated, often relatively small proteins that fold quickly, robustly, and reversibly. Working with a selected subset of proteins that exhibit these properties has greatly facilitated experimental progress. However, this set of folding models is not representative of the majority of proteins found in cellular proteomes.[9] Consequently, the folding of—presumably—most natural proteins remains comparatively poorly understood. These proteins, especially those that are larger and/or possess complex topologies, likely fold along intricate pathways that not only contain important intermediate structures, but are also beset with branch points toward non-native species that compete with productive folding. Additionally, it became clear that, while Anfinsen’s principle holds in general, many cellular proteins require the assistance of molecular chaperones to navigate their complex folding energy landscapes and fold efficiently on biological time scales.[10]
An aspect of protein folding that has received increasing attention over the past decade is its coupling to protein synthesis. Ribosomes synthesize polypeptides by adding one amino acid at a time to the growing nascent chain. The nascent protein emerges vectorially from the ribosome, N-terminus first. Because elongation rates are relatively low (approximately 20 amino acids per second in E. coli[11] and much slower in eukaryotic cells[12,13]), the synthesis of even small proteins requires several seconds. The nascent protein remains firmly anchored in the peptidyl transferase center while it is being extruded through the polypeptide exit tunnel that spans the large subunit of the ribosome. The tunnel is approximately 100 Å long and relatively narrow, restricting the conformations that the C-terminal 30 to 40 amino acids of the nascent chain can adopt. This universally conserved mode of protein synthesis provides ample time for interactions—with in the nascent protein and with surrounding macromolecules—that profoundly affect folding. While still within the polypeptide exit tunnel, the nascent polypeptide can form interactions with protein and RNA residues lining the tunnel walls. Already extruded segments begin to acquire more extensive tertiary structure near the ribosome, just outside the tunnel exit, before the following segments are synthesized. This early, cotranslational folding is thus modulated by interactions with the ribosome itself, as well as with cellular factors recruited to the nascent chain, often through specific binding sites near the tunnel exit.[14]
In this perspective we discuss paradigms of co-translational folding, most of which have resulted from recent experimental work and are still emerging. As such, some of the conclusions are speculative and will need to be scrutinized by experiment and theory. Excellent reviews on the topic have been published recently (see, for instance, these references:[15–20]). Rather than trying to provide a comprehensive review, our aim here is to shine a spotlight on a few aspects that we believe will merit further investigation, with a particular focus on co-translational folding of multi-domain proteins. We apologize in advance for not being able to include all of the seminal work that contributed to firmly establishing nascent protein folding as a key aspect for understanding cellular protein biogenesis.
Temporal sequestration reduces inter-domain misfolding
More than half a century ago, Peter Dunnill conjectured that, in searching for its native structure, a “polypeptide chain undergoing synthesis could [...] have one very great advantage in being extruded sequentially just as it is synthesized”.[21] Dunnill proposed that smaller segments of a nascent protein fold into native-like structures as they emerge from the ribosome, simplifying the conformational search process. It was indeed shown, decades later, that proteins can form native, functional structures before their synthesis is complete,[22] and that co-translational folding of structural units, or domains, provides a mechanism for rapid and efficient folding of large proteins.[23] These seminal studies relied mainly on measuring enzymatic activities and protease protection of nascent proteins still bound to the ribosome as proxies for successful folding.
Biophysically defining co-translational folding pathways has been experimentally challenging for several reasons. Chemical denaturation methods, typically used to study folding in solution, can denature the ribosome, although they have successfully been used within a limited concentration range.[24] Measuring folding of the nascent polypeptide is difficult in the context of the ribosome, a macromolecular machine that is orders of magnitude larger than most proteins. For a number of techniques that have traditionally been used to follow folding (see[25] and references therein), such as tryptophan fluorescence or circular dichroism spectroscopies, the background signal from the ribosome precludes the measurement of nascent polypeptide folding. Moreover, many proteins, including structurally intricate multi-domain proteins, populate transient and potentially heterogeneous on- and off-pathway folding intermediates. Resolving this complexity necessitated the application of novel experimental approaches to characterize their folding. Single-molecule methods in particular have proven useful in dissecting this complexity. Single-molecule force spectroscopy, which permits not only observation of folding, but also manipulation of individual protein molecules to drive conformational transitions,[26] revealed misfolded states in a tandem repeat protein.[27] Single-molecule fluorescence measurements further indicated this misfolding to be sequence-specific, caused by the formation of domain-swapped structures between neighboring domains with high sequence similarity.[28] Subsequently, single-molecule force spectroscopy experiments demonstrated that inter-domain misfolding is prevalent even among dissimilar domains in globular multi-domain proteins.[29–35]
These and other studies strongly suggest that inter-domain misfolding is a general phenomenon that complicates the folding of large multi-domain proteins. The high local concentrations resulting from arranging several domains in one polypeptide likely contributes to the prevalence of forming misfolded states.[36] Clearly, the vectorial mode of cellular protein synthesis enables temporal sequestration of domain folding, so that one domain has the opportunity to fold before the following domain is fully synthesized. This conceptually straightforward mechanism increases the robustness of folding and has been proposed to have enabled the evolution of multi-domain proteins.[36] As will be discussed in the following sections, we are just beginning to understand the complex interplay of numerous effects that potentially modulate nascent protein folding, in addition to the temporal sequestration that is a direct consequence of the mode of cellular protein synthesis.
Interactions with the ribosome surface modulate nascent chain folding
During synthesis, proteins are enzymatically processed, engaged by molecular chaperones, recognized by targeting machinery that guides them to their proper intracellular destination, and monitored by quality control machinery to remove stalled or incomplete products.[19,37] Likewise, assembly of functional oligomers can commence co-translationally.[38] All of these biologically important interactions also have the potential to affect the folding of a newly synthesized protein. Groups of proteins likely interact with these factors in ways that are determined by their sequence properties. For instance, proteins destined for co-translational export from the cytosol contain hydrophobic signal sequences that bind specifically to the signal recognition particle. A more general interaction partner for all cytosolic nascent chains is the ribosome itself. Because the nascent protein remains tethered to the ribosome throughout synthesis, the effective ribosome concentration—from the viewpoint of an amino acid residue in the nascent chain—is in the millimolar range.[39] Even weak interactions may therefore be effective in tuning nascent chain folding.
Steric interactions with the ribosome limit the conformations that a nascent protein can adopt inside or just outside the exit tunnel,[40] which can result in thermodynamic destabilization of folded structures[39,41] (Figure 1). The magnitude of the destabilization has been determined experimentally using pulse proteolysis[24] and NMR spectroscopy[41] (Figure 1). In addition to being sterically destabilized, unfolded nascent chains enter into electrostatic interaction with the surface of the ribosome,[42,43] which carries a high density of negative charges owing to the phosphates in the backbone of ribosomal RNA, the primary constituent of the ribosome. Electrostatic interactions have been found to slow down specific steps along a folding pathway.[42] Attraction of basic nascent chain residues toward the ribosome (and, perhaps, repulsion of acidic ones) can conceivably change the folding energy landscape in various ways that remain to be explored. For instance, simultaneous electrostatic attraction and repulsion of amino acid residues within a nascent polypeptide could energetically favor particular structural states and disfavor others, introducing a conformational bias.
FIGURE 1.

Destabilization of folded domains on the ribosome. (A) Illustration of destabilization by the ribosome. A domain (yellow) is stably folded in isolation (ΔG < 0). In the ribosome-nascent chain complexes shown here, the same domain is fully extruded from the ribosome and connected to the peptidyl transferase center by the subsequent domain (blue) that serves as linker. For relatively short linkers that span just the length of the ribosome exit tunnel (“short linker”, approximately 40 amino acids or less), interactions with the ribosome are destabilizing (ΔG ≥ 0). When translation elongates the linker (“longer linker), the effect of the ribosome decays and stability is restored (ΔG < 0). (B) Experimental determination of nascent chain stability at various chain lengths. Thermodynamic stabilities of the FLN5 domain were determined in isolation (blue) and in ribosome-nascent chain complexes (green). Truncations of the free protein mimic sequestration of the C-terminus in the exit tunnel. The stability of the full domain is approximately −7 kcal/mol. On the ribosome, the domain could, in principle, fold at a linker length of 36 residues from the peptidyl transferase center. However, a further addition of 6–8 amino acids is required for the formation of stable structure. Destabilization by the ribosome effectively delays stable folding until nascent chain elongation provides separation between the domain and the ribosome. Modified from ref.[41]
Even though interactions with the ribosome do not necessarily alter the folding pathway of a given domain,[44,45] they often affect the rates of individual transitions or the stability of folded structures.[31,42,45,46] Intriguingly, the folding of a de novo-designed protein that lacks an evolutionary history appeared completely unaffected by interactions with the ribosome,[47] raising the possibility that rate and stability modulation observed with natural proteins are the result of evolutionary selection. Reduction of folding rates and destabilization of structures formed near the ribosome[24,31,39,41,42,48] might help to delay folding until a complete foldable unit is synthesized, avoiding the formation of stably misfolded states that are difficult to dissolve.
Which part of the emerging nascent multi-domain protein interacts with the ribosome changes as elongation proceeds (Figure 2). Consequently, the effects on folding of a given domain also change during synthesis. When the first domain of a larger protein (Figure 2, red domain) has just emerged, it interacts with the ribosome, reducing its folding rate. As elongation proceeds and the length of the nascent protein increases, the first domain gradually escapes the influence of the ribosome. The following second domain (Figure 2, yellow domain) emerges and begins to interact with the ribosome. This interaction has been found to reduce misfolding between the two domains.[35] Thus, the region of a nascent polypeptide that interacts with the ribosome most strongly shifts in the N- to C-terminal direction during synthesis, and the folding rate of a given domain peaks after it has reached a sufficient distance from the ribosome during elongation (Figure 2).
FIGURE 2.

Ribosome-nascent chain interactions modulate folding rates. (A) Conceptualization illustrating the changing interactions of a nascent multi-domain protein with the ribosome as elongation proceeds. At short nascent chain lengths, strong interactions of the ribosome with the N-terminal domain (red) compete with folding (scenario 1), reducing the folding rate (dashed black line). As elongation proceeds, interactions with the ribosome sequester unfolded segments of the following domain (yellow) that would otherwise form misfolded species with the first domain. Ribosome-nascent chain interactions thus compete with inter-domain misfolding (scenario 2), and the folding rate of the red domain approaches its intrinsic value (dotted red line marked by an asterisk). Further elongation shifts the strong interactions with the ribosome to more C-terminal parts of the yellow domain, reducing its ability to thwart misfolding. Interactions between the unfolded domains promote misfolding and reduce effective folding rate (dotted pink line and scenario 3). (B) Experimental characterization of rate modulation by the ribosome. The folding rate of the G-domain from elongation factor G was determined in single-molecule experiments for various chain lengths. The G-domain folds at a maximal rate when it is separated from the peptidyl transferase center by 93 amino acids of domain II (386RNC). The rate is comparable to that of the G-domain in isolation (Galone). At shorter lengths, the G-domain can fold, but interactions with the ribosome reduce the folding rate (328RNC, 358RNC). Inter-domain misfolding reduced the productive folding rate in longer nascent chains (415RNC, 452RNC). From ref.[35], with permission
It seems likely that the ribosome generally interacts with the proximal segment of a nascent protein, that is, the part that is closest to the ribosomal surface. Sequestering part of a domain slows down its folding. The same domain folds swiftly after elongation has progressed, because the downstream segment of the emerging next domain is sequestered through interactions with the ribosome, which reduces inter-domain misfolding. This scenario provides a relatively simple example of the layering of several effects (temporal sequestration and ribosome interactions) that act simultaneously to change how a nascent protein folds. In addition, how nascent chain binding chaperones engage with their substrates also changes during elongation.[49] Other contributions, discussed in the following sections, may further modulate folding in combination with the effects described in Sections “Temporal sequestration reduces inter-domain misfolding” and “Interactions with the ribosome surface modulate nascent chain folding”.
Nascent chain folding generates mechanical force
Recent studies have found that the folding[47] (and even the presence[50]) of a nascent protein outside the ribosome can result in generation of mechanical forces. We are focusing our discussion here on force associated with nascent chain folding. When a domain of sufficient size folds upon emerging from the ribosome, it becomes sterically excluded from the exit tunnel. At the same time, it is held in immediate proximity to the ribosome because the unfolded C-terminus of the nascent protein within the exit tunnel is anchored in the peptidyl transferase center. This arrangement results in mechanical force on the nascent chain such that the segment within the exit tunnel is under mechanical tension (Figure 3).[47] In the context of continuous nascent chain elongation, forces have been predicted by coarse-grained simulations to be particularly high for fast-folding domains that adopt their structures quickly upon emerging from the ribosome exit tunnel.[51] Steric exclusion is a straightforward mechanism underlying force generation, perhaps similar in concept to the volume exclusion effect that give rise to an entropic force in tethered particle experiments.[52] However, other interactions may be important, and the origin of the “co-translational folding force” is not precisely understood at present.
FIGURE 3.

Nascent chain folding generates mechanical force. (A) Folding of a nascent chain in close proximity to the ribosome sterically excludes it from the exit tunnel. When folding occurs close to the surface of the ribosome, it generates a mechanical force on the unfolded segment of the nascent chain within the tunnel. The force is experienced by and destabilizes the folded domain. It also acts on the C-terminal attachment point. Without stable nascent chain-tunnel interaction, the force propagates to the peptidyl transferase center, where the nascent chain is anchored by a tRNA, potentially affecting ribosome activity. (B) Steric force resulting from nascent protein folding is sufficient to release arrest peptide (AP)-mediated stalling of elongation. Coupling of arrest release to the synthesis of a reporter (NanoLuc luciferase) allows detection of nascent chain folding waypoints in live bacteria. This assay reveals that the main folding event during synthesis of the G-domain of EF-G occurs only after the complete domain (293 amino acids) has been fully extruded from the ribosome, around L = 332 amino acids. Stable intermediates are not detected in shorter chains. RLU: relative light units. From ref.[53](C) The arrest peptide assay also reports on nascent chain folding in vitro, shown here for the R15 domain of spectrin. The signal obtained for the R15 domain without neighboring domains (R15nL) is affected weakly if the upstream R14 domain is present (R14R15nL). However, presence of part of the downstream R16 domain (R15R16nT) shifts the onset and/or magnitude of the signal, indicating that the onset of R15 folding is sensitive to the presence of the N-terminus of R16. fFL: fraction full-length. Note that the length L is defined differently in panels B and C. From ref.[60], with permission
An implication of such a “co-translational folding force” is that it is a universal consequence when the nascent chain forms a stable globular structure close to the ribosome. Indeed, this effect has successfully been exploited to follow nascent chain folding with force-sensing elongation arrest peptides in vivo[47,53,54] and in vitro[44,48,55,56] (Figure 3). These and other examples demonstrate the power of the approach in detecting co-translational folding events, even though the precise mechanisms of overcoming arrest peptide-mediated stalling of elongation remain to be determined.
The “co-translational folding force” results in tension on the region of the nascent polypeptide inside the exit tunnel (Figure 3). It is obvious from Newton’s third law that the folded domain as well as the C-terminal attachment point experience equal and opposite forces. At the folded domain, the force likely contributes to the observed folded state destabilization (see previous section). The effects of force at the C-terminus are less clear. Depending on the sequence, length, and structure of the nascent chain within the exit tunnel, the C-terminal attachment point experiencing force can be formed by interactions of the nascent protein with protein or RNA residues lining the exit tunnel, as is observed in a number of well-characterized arrest peptides.[57] Absent such contacts, the force should be transduced to the 3'-end of the tRNA that anchors the nascent protein in either the A- or the P-site of the peptidyl transferase center. In both cases, it is conceivable that force acting on the nascent polypeptide affects the catalytic activity of the ribosome: either by directly exerting force that distorts the geometry of the peptidyl transferase center, or indirectly by breaking nascent chain-tunnel interactions which modulate ribosome activity.
Regardless of the molecular underpinning, a “co-translational folding force” could provide the basis for a feedback mechanism that adjusts chain elongation rates in response to nascent chain folding events. Such a mechanism of regulating ribosome activity through nascent chain folding is currently not supported by experimental data, except in the case of artificial reporter constructs containing force-sensing arrest peptides (described above). However, for biological processes other than co-translational folding of soluble protein domains, control of translation elongation through the sensing of forces on the nascent chain is a well-established regulatory function of specific arrest peptides.[58] A similar mechanism that regulates elongation in response to folding (and, perhaps, interactions with nascent chain-binding factors) remains speculative to date, but appears physically plausible.
The mechanical tension that some domains can fold against and sustain is considerable.[59] The magnitude of the actual “co-translational folding force” has been estimated experimentally[47] and through modeling.[50,60] However, a quantitative understanding of the effect of force on a folded domain in a nascent protein requires further experimental work. While the mechanical stabilities of a number of proteins have been determined in single-molecule force spectroscopy experiments,[26] the pulling geometry in these experiments differs from the scenario of a nascent protein abutting the surface of the ribosome. Given that unfolding force strongly depend on the direction of the applied force (see, for example, references[61] and[62]) these studies can only provide estimates. Direct measurements are needed to define the magnitude of the “co-translational force” reliably. An interesting conjecture resulting from general force generation by nascent chain folding is that the unfolded polypeptide inside the exit tunnelmay act as a spring that limits the force at the peptidyl transferase center, protecting the active site of the ribosome against mechanical damage. A requirement for such protection could be one reason for the presence of a long and narrow exit tunnel in all extant ribosomes, in addition to its well-established regulatory functions.[58]
Kinetic coupling of nascent chain folding and elongation results in non-equilibrium effects
As described in the previous section, arrest peptides slow down translation elongation through interactions with the ribosome. Even in the absence of these specialized sequences, translation elongation by the ribosome does not occur at a uniform speed across the coding sequence. Instead, it is modulated by several factors, including differential codon usage and mRNA secondary structures.[63,64] Varying local elongation rates in a directed manner can impact nascent chain folding. For instance, clusters of slowly translating codons can further enhance temporal sequestration (see Section "Temporal sequestration reduces inter-domain misfolding") and improve the overall folding efficiency.[65] Indeed,genomicanalysesrevealedthatslowlytranslated clusters of codons are conserved both between and within domains, pointing to important roles for protein biogenesis.[66]
Fast-folding segments of a nascent protein can be expected to conformationally equilibrate during chain elongation and should therefore be insensitive to variations in synthesis rates. However, many folding transitions are known to occur on the timescale of seconds. In this case, an extruded segment of nascent chain may not reach conformational equilibrium before additional segments emerge from the ribosome. It has been shown that the resulting non-equilibrium effects can delay the formation of misfolded species[67] (Figure 4) or result in kinetically stable structures.[68] Strikingly, local changes in elongation rate, elicited by directed synonymous mutations, have been shown to affect the function, stability, and, by inference, the structure of the translation product.[69–71]
FIGURE 4.

Coupling of nascent chain elongation and folding gives rise to non-equilibrium effects. (A) Single-molecule experiments with optical tweezers allow direct observation of non-equilibrium effects on nascent protein folding. Partially synthesized calerythrin nascent chains populate a misfolded state (M, zoom inset). Formation of the misfolded state is significantly delayed (τ) even after the amino acids constituting the misfolded state have been synthesized, owing to non-equilibrium effects of translation and folding. From ref.[67] (B) Characterization of nascent chain dynamics by photon-induced electron transfer (PET) during active elongation shows that the nascent chain interconverts between different compact and elongated structures. As the nascent chain traverses through the exit tunnel, it adopts different conformations as reported by PET efficiencies. When 70 aa of the protein HemK are synthesized (70 aa construct), it adopts a compact structure before rearranging to final native state upon further elongation (112 aa construct). Disrupting the hydrophobic core of the domain (4×A mutants) does not interfere with compaction, but prevents the final rearrangement. From ref.[77], with permission. (C) Variations in the rate of translation elongation alters the final structure of gamma-B crystallin. The protein was heterologously expressed in E. coli, using either “unharmonized” (U) or “harmonized” (H) coding sequences. Even though both coding sequences are synonymous, NMR measurements reveal the presence of two sub-populations of the protein with distinct structures and patterns of disulfide bridges. From ref.[73]
Synonymous codon changes have been implicated in a number of diseases,[72] highlighting their biological importance. Recent experiments have revealed persistent structural changes in the protein product, breaking new ground for studying how changes in codon usage can result in kinetically stable alternative structures[71,73] (Figure 4). Synonymous codon changes can affect not only folding, but also elicit other effects,[18] such as changes in mRNA stability and control protein homeostasis at multiple levels.[74] Dissecting the consequences resulting from kinetic coupling of folding and elongation therefore requires direct measurements of both processes. Measuring local elongation speeds along an open reading frame at biologically relevant rates remains experimentally difficult, with ribosome profiling experiments yielding the most comprehensive, though indirect, view of elongation rates at codon resolution.[75,76] Real-time detection of nascent chain folding in the context of nascent chain elongation is also extremely challenging, but some important progress has been made[67,77] (Figure 4) and may help to develop new strategies for dissecting non-equilibrium effects in co-translational folding.
Directionality determines when nascent chain folding occurs
Implicit in the concept that elongation rates affect the structure of the translation product is the assumption that alternate folding pathways are favored depending on how quickly segments of the nascent chain are extruded from the ribosome. For instance, the N-terminal part of a nascent domain may form local structure to which C-terminal are added once they are extruded during elongation. When the full-length protein is refolded, distal interactions between N- and C-terminal regions may favor a different intermediate that sets up the beginning of an alternative pathway that potentially result in a different final structure. Which pathway the folding protein advances along would then depend on how quickly the C-terminal parts become available during synthesis, relative to the rate at which the N-terminal intermediate forms.
Since most domains longer than about 100 amino acids are thought to populate intermediates,[78] stepwise co-translational folding into successively larger intermediates might be a common phenomenon among proteins above this threshold. However, the directionality of folding is crucial for these considerations to apply. Recent experiments suggested that the G-domain of elongation factor G (EF-G), composed of nearly 300 amino acids, folds through a strictly directional pathway that begins with an obligatory intermediate formed by the extreme C-terminus of the domain.[53] These findings suggest that folding pathways of at least some domains follow a prescribed order, consistent with a “foldon” model.[79] In the case of the G-domain, the pathway starts at the extreme C-terminus of the domain, a restriction that is presumably imposed by the final structure of the domain. Because the C-terminus is synthesized last, the domain does not appear to form stably structured intermediates until it is completely extruded from the ribosome. Similarly, circular permutation experiments with the all-beta MATH domain indicate that co-translational folding is delayed until complete extrusion from the ribosome.[80] In these cases, rate variations during synthesis of the domain would not be expected to alter folding. Thus, directionality is an important aspect of how and when a nascent protein folds during synthesis.
Experiments with the G-domain[53] and several other small proteins or domains[45,48] did not indicate any change in the actual pathway of folding when comparing the ribosome-bound nascent chain to the isolated polypeptide. However, it is worthwhile to keep in mind that absence of proof does not equal proof of absence. Indeed, arrest peptide-experiments indicate that a small domain folds through several early intermediates co-translationally,[56] whereas it exhibits two-state behavior in isolation.
Energetic dependencies among domains determine co-translational folding order
Folded domains in globular proteins are usually arranged in a specific configuration through interactions at the interfaces. These interfaces between domains can significantly contribute to domain stability. For instance, the first two domains in the five-domain protein EF-G share a large interface. The second domain has been suggested to require these interactions for adopting a stable structure, while the first domain appears to be unaffected by the absence of its neighbor.[35] This asymmetric dependency imposes a sequential folding order which, in this case, matches the order of synthesis.
The three C-terminal domains of EF-G serve as an example that such dependencies can also be incompatible with co-translational folding. EF-G adopts an elongated structure in solution,[81] with domain III, the central of the five EF-G domains, forming extensive interfaces with both its C- and N-terminal neighbors. However, while interactions with the N-terminal domains do not contribute to the thermodynamic stability of domain III, those with the C-terminal neighbors are essential for stability.[34] In the absence of the two C-terminal domains, domain III remains essentially unstructured. As a consequence, the domain cannot fold co-translationally, and the C-terminal half of EF-G has to complete folding post-translationally. This particular energetic coupling may enable functionally important flexibility of EF-G, but comes at the expense of efficient autonomous folding and results in the accumulation of unfolded domains during translation. This example of two biological ends, folding and function, coming into conflict during protein biogenesis[34] illustrates that the energetic dependencies among domains have to be taken into account when reconstructing co-translational folding pathways of multi-domain proteins.
Denaturation by unfolded polypeptide is an unexpected challenge during co-translational folding
When domains are autonomously stable, they are expected to fold upon emergence from the ribosome, avoiding extensive accumulation of unfolded polypeptide during synthesis (as discussed in Section "Temporal sequestration reduces inter-domain misfolding"). To reap this benefit of domain-wise co-translational folding, already folded structures must be stable enough to remain folded while the following segments of the protein are synthesized. Modest thermodynamic stability and high cooperativity ensure relatively slow unfolding rates for many domains. However, recent single-molecule experiments revealed that, surprisingly, a folded domain can be destabilized by emerging unfolded polypeptide, effectively undoing co-translationally achieved folding progress[35] (Figure 5). The precise mechanism of this denaturing effect remains to be determined, but might be similar to the well-known destabilization of some proteins by signal sequences[82] or fusion of affinity tags.[83]
FIGURE 5.

Emerging unfolded polypeptide can denature co-translationally folded domains. (A) Unfolded domain II of EF-G denatures the co-translationally folded G-domain on the presence of the ribosome (brown lines) and in the isolated polypeptide (magenta line). The ribosome-binding chaperone trigger factor protects against the denaturation of folded domain (green line). From ref.[35], with permission. (B) As domain II is partially synthesized and extruded from the ribosome, it interacts with the folded N-terminal G-domain and denatures it (386RNC). Because the domain is incomplete, productive folding to the native state is not possible at this chain length. When the complete domain is synthesized and extruded from the ribosome (452RNC), denaturation competes with productive folding. Trigger factor shifts the balance toward the folded state by blocking denaturation. From ref.[35], with permission
Regardless of mechanism, denaturation of a folded domain by adjacent unstructured regions may pose a general challenge for both de novo folding during synthesis and for conformational maintenance of native multi-domain proteins. During synthesis, unfolded polypeptide is inevitably present, and the denaturation effect was observed before the complete, folding-competent domain had been extruded[35] (Figure 5). The bacterial chaperone trigger factor, which is thought to interact with most nascent chains[10] was found to protect against denaturation.[35] The ubiquity of specialized nascent-chain binding chaperones may result from a need to shield emerging polypeptide from wreaking havoc on folded domains during the relatively long time it takes to synthesize a complete, foldable unit. Once a multi-domain protein has successfully reached its fully folded structure, local unfolding within one domain might spread to neighboring ones and cause large-scale misfolding. While the destabilizing effect of an unfolded domain on its structured neighbor during synthesis has so far been described for the one example discussed here, it adds one more potentially crucial aspect of co-translational folding that merits future investigation.
CONCLUSION
Over the past decade, the importance as well as the complexity of co-translational folding has become abundantly clear. The recent work discussed here has provided examples of co-translational folding features that are likely of general significance for the process. However, numerous questions remain to be addressed. For instance, how might structures stabilized in the exit tunnel affect overall folding to the native state? Can nascent chain folding provide a signal for regulating ribosome activity? Which parts of any given protein actually fold co-translationally under physiological conditions in the cell? And, in a cellular context, how do molecular chaperones act, individually or in concert, to help efficient folding?
One challenge along the way to a more complete understanding of co-translational folding, and hence of cellular protein folding more generally, will be to integrate the various aspects that affect protein folding and evaluate their significance. Defining a set of proteins that are good representatives of proteome diversity and studying them with several orthogonal approaches might help to better define individual aspects of co-translational folding and evaluate their general importance. Steps in this direction have been taken by, for example, combining fluorescence spectroscopy and biochemistry techniques with NMR spectroscopy,[73] or combining arrest peptide assays with fluorescence,[56] pulse proteolysis,[48] or single-molecule force spectroscopy.[53]
Experimental investigation of co-translational folding is still a nascent field. Much of the experimental toolkit for investigating the process has become available only recently. While many of these tools have yielded spectacular insights, they collectively still resemble the proverbial blind men appraising the elephant of co-translational folding. Fluorescence spectroscopy, single-molecule manipulation, cryogenic electron microscopy, and nuclear magnetic resonance spectroscopy, among a host of other biochemical and biophysical approaches, are all poised to yield more quantitative insights into nascent chain structure and folding. Combining them with in vivo approaches, such as the arrest peptide assay described above (“Nascent chain folding generates mechanical force”), will further help to pinpoint important steps of co-translational folding pathways in the natural environment of the cytosol, which greatly impacts folding energy landscapes.[84] Understanding folding in the context of synthesis could be particularly important in light of the suggestion that at least some proteins have evolved to be kinetically stable[85] and might, on average, not unfold during their biological lifetime. Proper initial folding during or immediately after synthesis would be a straightforward safeguard against the detrimental effects of misfolded protein accumulation on cellular fitness and viability.
ACKNOWLEDGEMENTS
K.M. acknowledges support through the CMDB Graduate Program (NIH 5T32GM007231). C.M.K. acknowledges support from the NIH (5R01GM121567).
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
DATA AVAILABILITY STATEMENT
Data sharing not applicable – no new data generated.
References
- 1.Anfinsen CB (1973). Principles that govern the folding of protein chains. Science, 181(96), 223–230. [DOI] [PubMed] [Google Scholar]
- 2.Dill KA, & MacCallum JL (2012). The protein-folding problem, 50 years on. Science, 338(6110), 1042–1046. [DOI] [PubMed] [Google Scholar]
- 3.Sosnick TR, & Barrick D (2011). The folding of single domain proteins—Have we reached a consensus? Current Opinion in Structural Biology, 21(1), 12–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dobson CM (2003). Protein folding and misfolding. Nature, 426(6968), 884–890. [DOI] [PubMed] [Google Scholar]
- 5.Hartl FU, & Hayer-Hartl M (2009). Converging concepts of protein folding in vitro and in vivo. Nature Structural & Molecular Biology, 16(6), 574–581. [DOI] [PubMed] [Google Scholar]
- 6.Hartl FU (2017). Protein misfolding diseases. Annual Review of Biochemistry, 86, 21–26. [DOI] [PubMed] [Google Scholar]
- 7.Knowles TP, Vendruscolo M, & Dobson CM (2014). The amyloid state and its association with protein misfolding diseases. Nature Reviews Molecular Cell Biology, 15(6), 384–396. [DOI] [PubMed] [Google Scholar]
- 8.Valastyan JS, & Lindquist S (2014). Mechanisms of protein-folding diseases at a glance. Disease Models & Mechanisms, 7(1), 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braselmann E, Chaney JL, & Clark PL (2013). Folding the proteome. Trends in Biochemical Sciences, 38(7), 337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balchin D, Hayer-Hartl M, & Hartl FU (2016). In vivo aspects of protein folding and quality control. Science, 353(6294), aac4354. [DOI] [PubMed] [Google Scholar]
- 11.Young R, & Bremer H (1976). Polypeptide-chain-elongation rate in Escherichia coli B/r as a function of growth rate. Biochemical Journal, 160(2), 185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bostrom K, Wettesten M, Boren J, Bondjers G, Wiklund O, & Olofsson SO (1986). Pulse-chase studies of the synthesis and intracellular transport of apolipoprotein B-100 in Hep G2 cells. The Journal of Biological Chemistry, 261(29), 13800–13806. [PubMed] [Google Scholar]
- 13.Ingolia NT, Lareau LF, & Weissman JS (2011). Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell, 147(4), 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kramer G, Boehringer D, Ban N, & Bukau B (2009). The ribosome as a platform for co-translational processing, folding and targeting of newly synthesized proteins. Nature Structural & Molecular Biology, 16(6), 589–597. [DOI] [PubMed] [Google Scholar]
- 15.Thommen M, Holtkamp W, & Rodnina MV (2017). Co-translational protein folding: Progress and methods. Current Opinion in Structural Biology, 42: 83–89. [DOI] [PubMed] [Google Scholar]
- 16.Kaiser CM, & Liu K (2018). Folding up and moving on—Nascent protein folding on the ribosome. Journal of Molecular Biology, 430(22), 4580–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cassaignau AME, Cabrita LD, & Christodoulou J (2020). How does the ribosome fold the proteome? Annual Review of Biochemistry, 89: 389–415. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y, Yang Q, & Zhao F (2021). Synonymous but not silent: the codon usage code for gene expression and protein folding. Annual Review of Biochemistry, 90, 10.1146/annurev-biochem-071320-112701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kramer G, Shiber A, & Bukau B (2019). Mechanisms of cotranslational maturation of newly synthesized proteins. Annual Review of Biochemistry, 88: 337–364. [DOI] [PubMed] [Google Scholar]
- 20.Sharma AK, & O’Brien EP (2018). Non-equilibrium coupling of protein structure and function to translation-elongation kinetics. Current Opinion in Structural Biology, 49, 94–103. [DOI] [PubMed] [Google Scholar]
- 21.Dunnill P (1965). How proteins acquire their structure. Science Progress, 53(212), 609–19. [PubMed] [Google Scholar]
- 22.Nicola AV, Chen W, & Helenius A (1999). Co-translational folding of an alphavirus capsid protein in the cytosol of living cells. Nature Cell Biology, 1(6), 341–345. [DOI] [PubMed] [Google Scholar]
- 23.Frydman J, Erdjument-Bromage H, Tempst P, & Hartl FU (1999). Co-translational domain folding as the structural basis for the rapid de novo folding of firefly luciferase. Nature Structural Biology, 6(7), 697–705. [DOI] [PubMed] [Google Scholar]
- 24.Samelson AJ, Jensen MK, Soto RA, Cate JH, & Marqusee S (2016). Quantitative determination of ribosome nascent chain stability. Proceedings of the National Academy of Sciences of the United States of America, 113(47), 13402–13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bartlett AI, & Radford SE (2009). An expanding arsenal of experimental methods yields an explosion of insights into protein folding mechanisms. Nature Structural & Molecular Biology, 16(6), 582–588. [DOI] [PubMed] [Google Scholar]
- 26.Bustamante C, Alexander L, Maciuba K, & Kaiser CM (2020). Single-molecule studies of protein folding with optical tweezers. Annual Review of Biochemistry, 89, 443–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oberhauser AF, Marszalek PE, Carrion-Vazquez M, & Fernandez JM (1999). Single protein misfolding events captured by atomic force microscopy. Nature Structural & Biology, 6(11), 1025–1028. [DOI] [PubMed] [Google Scholar]
- 28.Borgia MB, Borgia A, Best RB, Steward A, Nettels D, Wunderlich B, Schuler B, & Clarke J (2011). Single-molecule fluorescence reveals sequence-specific misfolding in multidomain proteins. Nature, 474(7353), 662–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scholl ZN, Yang W, & Marszalek PE (2014). Chaperones rescue luciferase folding by separating its domains. The Journal of Biological Chemistry, 289(41), 28607–28618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jahn M, Buchner J, Hugel T, & Rief M (2016). Folding and assembly of the large molecular machine Hsp90 studied in single-molecule experiments. Proceedings of the National Academy of Sciences of the United States of America, 113(5), 1232–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu K, Rehfus JE, Mattson E, & Kaiser CM (2017). The ribosome destabilizes native and non-native structures in a nascent multidomain protein. Protein Science, 26(7), 1439–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stigler J, Ziegler F, Gieseke A, Gebhardt JC, & Rief M (2011). The complex folding network of single calmodulin molecules. Science, 334(6055), 512–516. [DOI] [PubMed] [Google Scholar]
- 33.Heidarsson PO, Naqvi MM, Otazo MR, Mossa A, Kragelund BB, & Cecconi C (2014). Direct single-molecule observation of calcium-dependent misfolding in human neuronal calcium sensor-1. Proceedings of the National Academy of Sciences of the United States of America, 111(36), 13069–13074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu K, Chen X, & Kaiser CM (2019). Energetic dependencies dictate folding mechanism in a complex protein. Proceedings of the National Academy of Sciences of the United States of America, 116(51), 25641–25648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu K, Maciuba K, & Kaiser CM (2019). The ribosome cooperates with a chaperone to guide multi-domain protein folding. Molecular Cell, 74(2), 310–319.e7 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han JH, Batey S, Nickson AA, Teichmann SA, & Clarke J (2007). The folding and evolution of multidomain proteins. Nature Reviews Molecular Cell Biology, 8(4), 319–330. [DOI] [PubMed] [Google Scholar]
- 37.Pechmann S, Willmund F, & Frydman J (2013). The ribosome as a hub for protein quality control. Molecular Cell, 49(3), 411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shiber A, Doring K, Friedrich U, Klann K, Merker D, Zedan M, Tippmann F, Kramer G, & Bukau B (2018). Cotranslational assembly of protein complexes in eukaryotes revealed by ribosome profiling. Nature, 561(7722), 268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cabrita LD, Cassaignau AM, Launay HM, Waudby CA, Wlodarski T, Camilloni C, Karyadi ME, Robertson AL, Wang X, Wentink AS, Goodsell LS, Woolhead CA, Vendruscolo M, Dobson CM, & Christodoulou J (2016). A structural ensemble of a ribosome-nascent chain complex during cotranslational protein folding. Nature Structural & Molecular Biology, 23(4), 278–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kudva R, Tian P, Pardo-Avila F, Carroni M, Best RB, Bernstein HD, & von Heijne G (2018). The shape of the bacterial ribosome exit tunnel affects cotranslational protein folding. Elife, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waudby CA, Wlodarski T, Karyadi ME, Cassaignau AME, Chan SHS, Wentink AS, Schmidt-Engler JM, Camilloni C, Vendruscolo M, Cabrita LD, & Christodoulou J (2018). Systematic mapping of free energy landscapes of a growing filamin domain during biosynthesis. Proceedings of the National Academy of Sciences of the United States of America, 115(39), 9744–9749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaiser CM, Goldman DH, Chodera JD, Tinoco I Jr., & Bustamante C (2011). The ribosome modulates nascent protein folding. Science, 334(6063), 1723–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knight AM, Culviner PH, Kurt-Yilmaz N, Zou T, Ozkan SB, & Cavagnero S (2013). Electrostatic effect of the ribosomal surface on nascent polypeptide dynamics. ACS Chemical Biology, 8(6), 1195–1204. [DOI] [PubMed] [Google Scholar]
- 44.Tian P, Steward A, Kudva R, Su T, Shilling PJ, Nickson AA, Hollins JJ, Beckmann R, von Heijne G, Clarke J, & Best RB (2018). Folding pathway of an Ig domain is conserved on and off the ribosome. Proceedings of the National Academy of Sciences of the United States of America, 115(48), E11284–E11293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guinn EJ, Tian P, Shin M, Best RB, & Marqusee S (2018). A small single-domain protein folds through the same pathway on and off the ribosome. Proceedings of the National Academy of Sciences of the United States of America, 115(48), 12206–12211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoffmann A, Becker AH, Zachmann-Brand B, Deuerling E, Bukau B, & Kramer G (2012). Concerted action of the ribosome and the associated chaperone trigger factor confines nascent polypeptide folding. Molecular Cell, 48(1), 63–74. [DOI] [PubMed] [Google Scholar]
- 47.Goldman DH, Kaiser CM, Milin A, Righini M, Tinoco I Jr., Ribosome BC. (2015). Mechanical force releases nascent chain-mediated ribosome arrest in vitro and in vivo. Science, 348(6233), 457–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jensen MK, Samelson AJ, Steward A, Clarke J, & Marqusee S (2020). The folding and unfolding behavior of ribonuclease H on the ribosome. The Journal of Biological Chemistry, 295(33), 11410–11417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stein KC, Kriel A, & Frydman J (2019). Nascent polypeptide domain topology and elongation rate direct the cotranslational hierarchy of Hsp70 and TRiC/CCT. Molecular Cell, 75(6), 1117–1130 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fritch B, Kosolapov A, Hudson P, Nissley DA, Woodcock HL, Deutsch C, & O’Brien EP (2018). Origins of the mechanochemical coupling of peptide bond formation to protein synthesis. Journal of the American Chemical Society, 140(15), 5077–5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leininger SE, Trovato F, Nissley DA, & O’Brien EP (2019). Domain topology, stability, and translation speed determine mechanical force generation on the ribosome. Proceedings of the National Academy of Sciences of the United States of America, 116(12), 5523–5532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Segall DE, Nelson PC, & Phillips R (2006). Volume-exclusion effects in tethered-particle experiments: Bead size matters. Physical Review Letters, 96(8), 088306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen X, Rajasekaran N, Liu K, & Kaiser CM (2020). Synthesis runs counter to directional folding of a nascent protein domain. Nature Communications, 11(1), 5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marino J, von Heijne G, & Beckmann R (2016). Small protein domains fold inside the ribosome exit tunnel. FEBS Letter, 590(5), 655–660. [DOI] [PubMed] [Google Scholar]
- 55.Nilsson OB, Hedman R, Marino J, Wickles S, Bischoff L, Johansson M, Muller-Lucks A, Trovato F, Puglisi JD, O’Brien EP, Beckmann R, & von Heijne G (2015). Cotranslational protein folding inside the ribosome exit tunnel. Cell Reports, 12(10), 1533–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liutkute M, Maiti M, Samatova E, Enderlein J, & Rodnina MV (2020). Gradual compaction of the nascent peptide during cotranslational folding on the ribosome. Elife, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wilson DN, Arenz S, & Beckmann R (2016). Translation regulation via nascent polypeptide-mediated ribosome stalling. Current Opinion in Structural Biology, 37: 123–133. [DOI] [PubMed] [Google Scholar]
- 58.Ito K, Mori H, & Chiba S (2018). Monitoring substrate enables realtime regulation of a protein localization pathway. FEMS Microbiology Letters, 365(11). [DOI] [PubMed] [Google Scholar]
- 59.Eckels EC, Haldar S, Tapia-Rojo R, Rivas-Pardo JA, & Fernandez JM (2019). The mechanical power of titin folding. Cell Reports, 27(6), 1836–1847 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kemp G, Nilsson OB, Tian P, Best RB, & von Heijne G (2020). Cotranslational folding cooperativity of contiguous domains of alpha-spectrin. Proceedings of the National Academy of Sciences of the United States of America, 117(25), 14119–14126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dietz H, Berkemeier F, Bertz M, & Rief M (2006). Anisotropic deformation response of single protein molecules. Proceedings of the National Academy of Sciences of the United States of America, 103(34), 12724–12728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jagannathan B, Elms PJ, Bustamante C, & Marqusee S (2012). Direct observation of a force-induced switch in the anisotropic mechanical unfolding pathway of a protein. Proceedings of the National Academy of Sciences of the United States of America, 109(44), 17820–17825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Samatova E, Daberger J, Liutkute M, & Rodnina MV (2021). Translational control by ribosome pausing in bacteria: How a non-uniform pace of translation affects protein production and folding. Frontiers in Microbiology, 11(3428). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Komar AA (2016). The yin and yang of codon usage. Human Molecular Genetics, 25(R2), R77–R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang G, Hubalewska M, & Ignatova Z (2009). Transient ribosomal attenuation coordinates protein synthesis and co-translational folding. Nature Structural & Molecular Biology, 16(3), 274–280. [DOI] [PubMed] [Google Scholar]
- 66.Chaney JL, Steele A, Carmichael R, Rodriguez A, Specht AT, Ngo K, Li J, Emrich S, & Clark PL (2017). Widespread position-specific conservation of synonymous rare codons within coding sequences. PLOS Computational Biology, 13(5), e1005531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alexander LM, Goldman DH, Wee LM, & Bustamante C (2019). Non-equilibrium dynamics of a nascent polypeptide during translation suppress its misfolding. Nature Communications, 10(1), 2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sander IM, Chaney JL, & Clark PL (2014). Expanding Anfinsen’s principle: contributions of synonymous codon selection to rational protein design. Journal of the American Chemical Society, 136(3), 858–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE, Calcagno AM, Ambudkar SV, & Gottesman MM (2007). A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science, 315(5811), 525–528. [DOI] [PubMed] [Google Scholar]
- 70.Zhou M, Guo J, Cha J, Chae M, Chen S, Barral JM, Sachs MS, & Liu Y (2013). Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature, 495(7439), 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Walsh IM, Bowman MA, Soto Santarriaga IF, Rodriguez A, & Clark PL (2020). Synonymous codon substitutions perturb cotranslational protein folding in vivo and impair cell fitness. Proceedings of the National Academy of Sciences of the United States of America, 117(7), 3528–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sauna ZE, & Kimchi-Sarfaty C (2011). Understanding the contribution of synonymous mutations to human disease. Nature Review Genetics, 12(10), 683–691. [DOI] [PubMed] [Google Scholar]
- 73.Buhr F, Jha S, Thommen M, Mittelstaet J, Kutz F, Schwalbe H, Rodnina MV, & Komar AA (2016). Synonymous codons direct cotranslational folding toward different protein conformations. Molecular Cell, 61(3), 341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stein KC, & Frydman J (2019). The stop-and-go traffic regulating protein biogenesis: How translation kinetics controls proteostasis. Journal of Biological Chemistry, 294(6), 2076–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mohammad F, Green R, & Buskirk AR (2019). A systematically-revised ribosome profiling method for bacteria reveals pauses at single-codon resolution. Elife, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sharma AK, Sormanni P, Ahmed N, Ciryam P, Friedrich UA, Kramer G, & O’Brien EP (2019). A chemical kinetic basis for measuring translation initiation and elongation rates from ribosome profiling data. PLOS Computational Biology, 15(5), e1007070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Holtkamp W, Kokic G, Jager M, Mittelstaet J, Komar AA, & Rodnina MV (2015). Cotranslational protein folding on the ribosome monitored in real time. Science, 350(6264), 1104–1107. [DOI] [PubMed] [Google Scholar]
- 78.Brockwell DJ, & Radford SE (2007). Intermediates: Ubiquitous species on folding energy landscapes? Current Opinion in Structural Biology, 17(1), 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Englander SW, Mayne L, Kan ZY, & Hu W (2016). Protein folding-how and why: By hydrogen exchange, fragment separation, and mass spectrometry. The Annual Review of Biophysics, 45, 135–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Marsden AP, Hollins JJ, O’Neill C, Ryzhov P, Higson S, Mendonca C, Kwan TO, Kwa LG, Steward A, & Clarke J (2018). Investigating the effect of chain connectivity on the folding of a beta-sheet protein on and off the ribosome. Journal of Molecular Biology, 430(24), 5207–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lin J, Gagnon MG, Bulkley D, & Steitz TA (2015). Conformational changes of elongation factor G on the ribosome during tRNA translocation. Cell, 160(1–2), 219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Park S, Liu G, Topping TB, Cover WH, & Randall LL (1988). Modulation of folding pathways of exported proteins by the leader sequence. Science, 239(4843), 1033–1035. [DOI] [PubMed] [Google Scholar]
- 83.Booth WT, Schlachter CR, Pote S, Ussin N, Mank NJ, Klapper V, Offermann LR, Tang C, Hurlburt BK, & Chruszcz M (2018). Impact of an N-terminal polyhistidine tag on protein thermal stability. ACS Omega, 3(1), 760–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gruebele M, Dave K, & Sukenik S (2016). Globular protein folding in vitro and in vivo. The Annual Review of Biophysics, 45: 233–251. [DOI] [PubMed] [Google Scholar]
- 85.Lim SA, Hart KM, Harms MJ, & Marqusee S (2016). Evolutionary trend toward kinetic stability in the folding trajectory of RNases H. Proceedings of the National Academy of Sciences of the United States of America, 113(46), 13045–13050. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable – no new data generated.