

ABSTRACT
The iron-sulfur cluster coordinating transcription factor IscR is important for the virulence of Yersinia pseudotuberculosis and a number of other bacterial pathogens. However, the IscR regulon has not yet been defined in any organism. To determine the Yersinia IscR regulon and identify IscR-dependent functions important for virulence, we employed chromatin immunoprecipitation sequencing (ChIP-Seq) and RNA sequencing (RNA-Seq) of Y. pseudotuberculosis expressing or lacking iscR following iron starvation conditions, such as those encountered during infection. We found that IscR binds to the promoters of genes involved in iron homeostasis, reactive oxygen species metabolism, and cell envelope remodeling and regulates expression of these genes in response to iron depletion. Consistent with our previous work, we also found that IscR binds in vivo to the promoter of the Ysc type III secretion system (T3SS) master regulator LcrF, leading to regulation of T3SS genes. Interestingly, comparative genomic analysis suggested over 93% of IscR binding sites were conserved between Y. pseudotuberculosis and the related plague agent Yersinia pestis. Surprisingly, we found that the IscR positively regulated sufABCDSE Fe-S cluster biogenesis pathway was required for T3SS activity. These data suggest that IscR regulates the T3SS in Yersinia through maturation of an Fe-S cluster protein critical for type III secretion, in addition to its known role in activating T3SS genes through LcrF. Altogether, our study shows that iron starvation triggers IscR to coregulate multiple, distinct pathways relevant to promoting bacterial survival during infection.
KEYWORDS: Yersinia, Yersinia pseudotuberculosis, ChIP-Seq, IscR, RNA-Seq, Yersinia pestis
INTRODUCTION
Iron is an important cofactor for many proteins involved in respiration, oxidative stress resistance, gene regulation, and other processes (1). Most bacteria require ∼10−6 M iron to support optimal growth. Yet, in the mammalian host, the level of free iron is only 10−18 M due to the concerted action of mammalian iron storage and carrier proteins (2, 3). During infection, the amount of available iron decreases even further as a result of nutritional immunity, a process through which inflammatory mediators lead to further sequestration of iron (4). Yersinia pseudotuberculosis and the related enteropathogen Yersinia enterocolitica cause self-limiting mesenteric lymphadenitis and gastroenteritis in immunocompetent people as well as serious disseminated infection in iron-overloaded individuals (5, 6). The plague agent Yersinia pestis is closely related to Y. pseudotuberculosis, emerging as a distinct species ∼1,500 to 6,400 years ago (7). The elevated susceptibility of iron-overloaded individuals to disseminated Yersinia infection underscores the low iron bioavailability experienced by Yersinia in mammalian tissues other than the intestinal lumen, despite expression of multiple iron uptake systems (8–10). In vitro, enteropathogenic Yersinia can utilize both inorganic and heme iron sources through a number of iron uptake systems typical of many pathogens (11–14). Although the importance of iron availability in Yersinia infection is well accepted, the transcription regulatory networks that operate under iron starvation conditions have not been firmly established.
In many pathogenic bacteria, expression of iron uptake systems and virulence factors is controlled by the conserved global iron regulator Fur (15). Although multiple iron uptake systems are controlled by Fur in Yersinia, including yersiniabactin, the key siderophore in pathogenic Yersinia, Fur has not been shown to directly regulate known virulence factors other than those involved in metal acquisition (16, 17). Rather, we discovered that expression of a key virulence factor, the Yersinia secretion (Ysc) type III secretion system (T3SS), encoded on a 70-kb virulence plasmid pYV/pCD1 (18), was controlled by a different iron-regulated transcription factor, IscR (9, 19). Subsequently, IscR has been shown to coordinate virulence factor expression in multiple pathogens (20–24).
In Escherichia coli where IscR was discovered, DNA binding is regulated by ligation of an Fe-S cluster (25), providing a linkage between iron availability and activity of this transcription factor. E. coli IscR controls the expression of more than 40 genes, including anaerobic metabolism and respiration, and the Isc and Suf Fe-S cofactor biogenesis systems (26). IscR exists in either a holo form, when it is bound to a [2Fe-2S] cluster, or in an apo form, which is clusterless (25). While holo-IscR binds to so-called type I motif sequences with a significantly higher affinity than apo-IscR, both holo-IscR and apo-IscR can bind to type II motif sequences (25, 27, 28). IscR can both activate and repress transcription depending on the position of its binding site relative to a promoter. Bioavailability of iron, oxygen tension, and reactive oxygen species have all been inferred to affect the relative ratio of holo- to apo-IscR (25, 29–35). In turn, holo-IscR negatively regulates iscR transcription through two type I motif sequences in the isc operon promoter, leading to an increase in overall IscR levels under aerobic and/or iron-starved conditions (9, 25), and derepression of the Isc and Suf biogenesis pathways to maintain Fe-S cluster homeostasis (25, 26).
In Yersinia, IscR binds to a type II motif in the promoter of the gene encoding LcrF, the master regulator of the Ysc T3SS (9, 19). IscR, and subsequently LcrF, levels increase with oxygen tension through derepression of the isc operon, driving T3SS expression (9). While the T3SS is important for Yersinia virulence, Y. pseudotuberculosis lacking T3SS expression can still colonize the intestinal lumen as well as the mesenteric lymph nodes (9, 36–38). In contrast, Y. pseudotuberculosis lacking iscR is defective in colonization of all mouse tissues tested (9, 19). This suggests that IscR regulates additional virulence factors in Yersinia. In order to assess further how IscR contributes to Yersinia virulence, we used whole transcriptome RNA sequencing (RNA-Seq) and chromatin immunoprecipitation with massively parallel DNA sequencing (ChIP-Seq) to identify genes directly regulated by IscR following iron starvation, which Yersinia experiences during disseminated infection. Interestingly, comparative genomics revealed very high predicted conservation of the IscR regulon in Y. pestis. One highly conserved IscR binding site was found in the promoter of the suf Fe-S cluster biogenesis operon. Surprisingly, our data show that IscR direct regulation of the Y. pseudotuberculosis suf operon is critical for T3SS activity under iron-depleted conditions.
RESULTS
Identification of IscR and iron-regulated genes in Y. pseudotuberculosis.
Yersinia experience iron starvation during extraintestinal infection (39, 40). Since IscR levels and activity are regulated by iron, we chose to assess the bacterial transcriptional response following iron starvation. Wild-type (WT) and ΔiscR Y. pseudotuberculosis bacteria were starved for iron using previously published methods (see Materials and Methods) (12). Cultures either remained iron limited (no iron added back to Chelex-treated media) or were supplemented with organic iron (5 μM hemin) or inorganic iron (3.6 μM FeSO4), which can both be utilized by Yersinia via distinct uptake systems (11–14), and incubated for 3 h at 37°C. RNA-Seq analysis showed that levels of IscR mRNA and protein were highest following prolonged iron starvation compared to after iron supplementation (Fig. 1A to C). As holo-IscR negatively regulates isc operon transcription, these data are consistent with a decrease in holo-IscR activity following iron starvation and suggest that these growth conditions modulate IscR-regulated gene expression and provide a mimic of conditions found in the host. Indeed, we previously found that even adding back only 0.036 μM FeSO4 prevented derepression of iscR expression (9).
FIG 1.

IscR mRNA and protein levels decrease upon iron supplementation. (A) Expression of iscR in Y. pseudotuberculosis under various iron conditions, as measured by RNA-Seq. Reads are represented by trimmed mean of M-values (TMM) of WT in Chelex-treated M9 minimal medium with no iron source added back (Chelex), supplemented with 5 μM hemin (+Heme), or supplemented with 3.6 μM FeSO4 (+FeSO4). ****, P < 0.0001; n.s., not significant (EdgeR with a corrected FDR post hoc test). (B) Expression of iscR mRNA relative to 16S rRNA, as measured by quantitative reverse transcription-PCR (qRT-PCR). *, P < 0.05 (one-way ANOVA with Dunnett’s post hoc test). (C) IscR protein levels in whole-cell extracts from WT Y. pseudotuberculosis, as detected using an anti-IscR antibody (αIscR). RpoA served as a loading control. Shown is the average of three biological replicates ± standard deviation. **, P < 0.01; *, P < 0.05 (one-way ANOVA with Dunnett’s post hoc test).
Global RNA-Seq analysis revealed a number of genes that respond to iron availability. We used clustering analysis to sort these differentially expressed genes into seven groups based on the relative changes in expression following prolonged iron starvation compared to supplementation with inorganic iron or heme. The two largest clusters were genes that were either downregulated (cluster VI) or upregulated (cluster II) in response to iron limitation. (Fig. 2; see Data Set S1 in the supplemental material). Cluster VI and II genes also showed similar expression between the two iron sources. Cluster II genes contained iscR in addition to genes involved in iron uptake (ex-hemPR, hmuSTUV, iutA/iucABCD, and feoB), iron sulfur cluster biogenesis (sufABCDSE), lipopolysaccharide (LPS) biosynthesis and modification (arnABCDE, lpxABDTL, rfaQ, and pagP), lipoprotein and outer membrane protein targeting (lolCDE, bamA, surA, and skp), cell wall remodeling and defense, the dusB-fis virulence-associated operon, and the T3SS. Upregulation of iron uptake genes and the suf operon in response to iron starvation was expected given previous studies (12, 14, 33), but regulation of LPS modification, periplasmic protein targeting, and cell wall remodeling by iron has not been previously reported in Yersinia. Cluster VI included genes involved in energy metabolism (sucABCD, fdoGHI, and dmsAD), antioxidants (sodB, katAG, and ompW), and the AI-2 autoinducer pathway (lsrR regulator and lsr operon). Altogether, these results suggest that Y. pseudotuberculosis increases iron metabolism and cell envelope remodeling during iron starvation, a condition known to be experienced by Yersinia during extraintestinal infection (39, 40).
FIG 2.
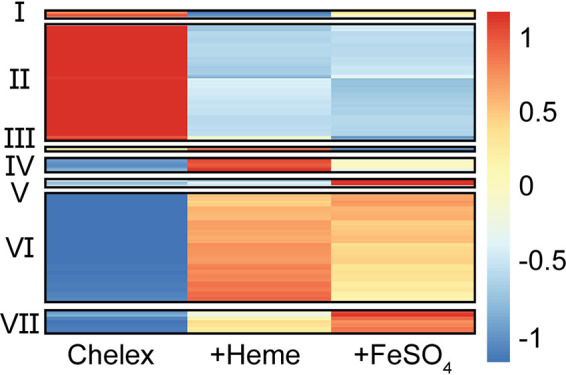
Iron supplementation modulates the expression of 1,373 genes in Yersinia. RNA-Seq was carried out in WT Y. pseudotuberculosis after iron starvation (Chelex) or followed by supplementation with heme or FeSO4, as in Fig. 1. Cluster analysis was performed on the average RNA-Seq reads normalized by trimmed mean of M-values (TMM) for all 1,373 differentially expressed genes. Expression values were transformed and scaled by gene before cluster analysis. The color bar shows relative gene expression.
Transcriptomic data of the Y. pseudotuberculosis wild type and the ΔiscR mutant under both iron-starved and non-iron-starved conditions. Download Data Set S1, XLSX file, 3.6 MB (3.6MB, xlsx) .
Copyright © 2021 Balderas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Genes that are differentially regulated in response to changes in iron availability may be controlled by IscR, Fur, and possibly other transcription factors or may be indirectly regulated by these factors. In order to determine which genes are affected by IscR, we carried out the same RNA-Seq analysis as above with the ΔiscR mutant. Under prolonged iron starvation (Chelex condition), when IscR levels are highest in the WT strain, a total of 324 genes were differentially expressed in the wild-type strain versus the ΔiscR strain (Fig. 3; Data Set S1). Of the 127 genes whose expression was greater in the wild type than in the ΔiscR mutant, 48 (∼38%) were carried on pYV and were among the genes with the largest fold changes between the wild-type and ΔiscR strains. As our previous data showed that apo-IscR directly regulates the T3SS master regulator LcrF, the majority of these pYV-encoded genes are likely to be indirectly regulated by IscR. In iron-limited conditions, such as those encountered by Yersinia during extraintestinal infection, the dominant form of IscR is predicted to be apo-IscR. Genes induced by apo-IscR would be expected to be more highly expressed during iron starvation compared to after iron supplementation as well as have decreased expression upon deletion of iscR. Genes whose expression patterns were consistent with apo-IscR induction of their promoters included the T3SS genes yscCDEFGHK, the sufABCDSE operon, metal transport genes iucABCDiutA, alcC, cirA, fhuC_1, and oprC, and cell envelope biosynthesis and remodeling genes pagP, amiD, and ydhO (Fig. 4A and Data Set S1). As expected, the 197 genes expressed at lower levels in the wild type compared to the ΔiscR strain under prolonged iron starvation included those in the isc operon. Genes repressed by apo-IscR would be expected to have higher mRNA levels following iron supplementation compared to iron starvation in wild-type bacteria as well as have increased expression upon deletion of iscR. Genes whose expression patterns are consistent with apo-IscR repression include sodB, katA, ompW, and the [4Fe-4S] cluster protein napF (Fig. 4B).
FIG 3.
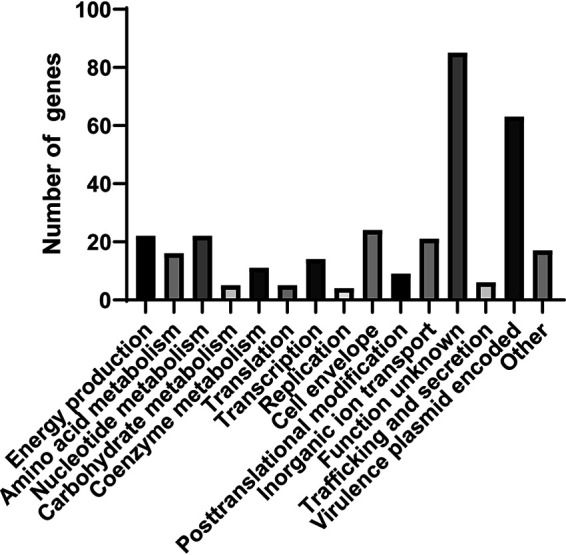
Deletion of iscR leads to expression changes in genes involved in virulence, ion transport, cell envelope, and other processes following prolonged iron starvation. Clusters of Orthologous Groups of proteins (COG) analysis of genes differentially expressed between the WT and ΔiscR strains following iron starvation.
FIG 4.

Several Yersinia genes follow expression patterns consistent with activation or repression by apo-IscR. Expression of various genes as measured by RNA-Seq. Reads are represented by trimmed mean of M-values (TMM) of WT (black) and ΔiscR strains (gray). ****, P < 0.0001; n.s. not significant (EdgeR with a corrected FDR post hoc test). (A) Genes predicted to be activated by apo-IscR. (B) Genes predicted to be repressed by apo-IscR.
ChIP-Seq analysis of in vivo IscR binding in Y. pseudotuberculosis.
In order to identify the genes directly regulated by IscR, we carried out in vivo genome-wide detection of IscR binding sites via ChIP-Seq, using an anti-FLAG antibody and a Y. pseudotuberculosis strain expressing FLAG-tagged IscR (see Fig. S1 in the supplemental material; see Materials and Methods). Wild-type Y. pseudotuberculosis was used as a negative control since the FLAG antibody should not pull down IscR-DNA complexes in the absence of the affinity tag. A total of 295 unique regions of the genome were enriched during the FLAG pulldown (Fig. 5A and B). Of these ChIP-Seq peaks, 176 fell within 500 nucleotides upstream of an open reading frame (ORF) start codon and no more than 100 nucleotides downstream of a start codon, potentially within a regulatory region controlling transcription. Out of these 176 peaks, 173 are found on the chromosome, and three on the pYV virulence plasmid. A total of 37 peaks fell in between divergent genes, and such ChIP-Seq peaks were assigned to both genes (encoded on the sense and antisense strands) for our preliminary analysis. Therefore, the 176 identified peaks fell within the predicted regulatory regions of 213 transcription units (TUs) encoding 401 individual genes (Data Set S2). Of these 213 TUs, 46 contain genes found in cluster II (upregulated by iron starvation) and 40 contain genes found in cluster VI and VII (downregulated by iron starvation). The remaining 127 transcription units associated with a ChIP-Seq peak were not found in our cluster analysis, as they were not differentially expressed in response to iron. These data show that 40% of the IscR binding sites identified by our ChIP-Seq analysis were upstream of genes responsive to iron under aerobic conditions.
FIG 5.

ChIP-Seq reveals IscR to be a global regulator in Yersinia. (A) ChIP-Seq analysis reveals enrichment of IscR binding sites throughout the genome following anti-FLAG pulldown. The first track (blue) represents read coverage of IscR-bound sequences throughout the Y. pseudotuberculosis IscR-3-xFLAG chromosome from three biological replicates, shown as count per million reads (CPM). The second track (pink) represents sequencing reads from WT Y. pseudotuberculosis (negative control). (B) ChIP-Seq plots illustrating read coverage of the pYV virulence plasmid (not scaled to the size of the genome). The y axis for the pYV plasmid is significantly higher than for the chromosome because pYV copy number is expected to be high under these conditions (96). (C) ChIP-Seq sequence read peaks mapped to the yscW-lcrF promoter. The x axis indicates the genomic position of the ChIP-Seq peaks. Dashed lines correspond to the center of the known 30-bp IscR type II binding site. The arrow indicates the transcriptional start site (39). (D) Motif that is overrepresented within 100 nucleotides of the zenith of IscR ChIP-Seq sequence read peaks. This motif was identified in 175 peaks out of 176 peaks. The motif representing the type II motif characterized in E. coli K-12 MG1655 is shown as reference. (E) The motif illustrated in Fig. 5D has a high probability of being located at or near the peak summit (Centri-Mo).
3xFLAG-tagged IscR rescues an iscR deletion. (A) Whole-cell extracts from WT Y. pseudotuberculosis or a strain harboring a chromosomally encoded 3xFLAG-tagged IscR were visualized using anti-FLAG or anti-RpoA antibodies. (B) To measure the relative efficiency of the Ysc T3SS, Yersinia strains were grown under T3SS-inducing conditions, and secreted proteins precipitated by trichloroacetic acid were visualized using Coomassie blue. Relative amounts of the T3SS effector protein YopE were quantified by densitometry compared to a spiked-in BSA protein control. The averages of three biological replicates ± standard deviations are shown. (C) Yersinia strains were grown under T3SS-inducing conditions, and relative iscS mRNA levels were evaluated by qPCR and normalized to 16s rRNA. The average of three biological replicates ± standard deviation is shown. ***, P < 0.001; **, P < 0.01; n.s., not significant (one-way ANOVA with Dunnett’s post hoc test). Download FIG S1, PDF file, 0.2 MB (219.5KB, pdf) .
Copyright © 2021 Balderas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
ChIP-Seq data assessing genome wide enrichment of IscR binding sites in Y. pseudotuberculosis. Download Data Set S2, XLSX file, 0.1 MB (147.6KB, xlsx) .
Copyright © 2021 Balderas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The most well-documented IscR binding site in Yersinia is the type II site found 367 nucleotides upstream of the yscW start codon (19). A ChIP-Seq peak was detected in this region, with the pinnacle of the peak centering 364 nucleotides upstream of yscW-lcrF (Fig. 5C). We used MEME suite tools to probe for overrepresented sequences near the center of ChIP-Seq peaks (41). MEME analysis revealed identifiable IscR type II binding sequences in 175 out of the 176 ChIP-Seq peaks positioned within a putative regulatory region, with the predicted binding sequences highly correlated with the center of the ChIP-Seq peak (CentriMo P value, 8.4E−1028; Fig. 5D). The overrepresented sequence from these IscR-enriched sites strongly resembles the consensus IscR type II site from E. coli (Fig. 5E) (28). No significant ChIP-Seq peaks were detected at predicted IscR type I sites in Yersinia (Fig. S2A and B). This may be because holo-IscR does not cross-link as well or bind with high enough affinity as apo-IscR, and the resulting peaks are difficult to detect over the signals of apo-IscR binding. Taken together, these data indicate that our 3xFLAG-IscR ChIP was able to enrich for IscR type II binding sites, but not type I binding sites, under the conditions used. Importantly, genes driven by type II motif-containing promoters are predicted to be regulated by IscR under iron-limited and aerobic conditions, such as those encountered by Yersinia during disseminated infection and therefore potential virulence factors.
Global IscR ChIP-seq analysis fails to identify predicted IscR type I sites. (A) Known E. coli IscR type I binding sites were used to generate an IscR type I binding motif using MEME suite tools. The Y. pseudotuberculosis IP2666 genome was scanned for IscR type I sites using FIMO specifically upstream of the nfuA, iscRSUA, erpA, and DN756_20960_cysE promoters. The predicted sequences were aligned to the E. coli consensus IscR type I binding motif. (B) IscR ChIP-seq plots illustrating read coverage of IscR binding peaks assigned to the promoter of nfuA, iscRSUA, erpA, and DN756_20960_cysE from all three replicates combined. Dashed lines correspond to the zenith of the predicted 29-bp IscR type I motifs. Download FIG S2, PDF file, 0.1 MB (128.4KB, pdf) .
Copyright © 2021 Balderas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
In order to validate our in vivo IscR binding results, we carried out in vitro binding studies on the identified IscR binding sites upstream of the katA and sodB antioxidants as well as the known virulence genes fis and ail (Fig. 6A). IscR has been shown to regulate antioxidant genes in other bacterial pathogens (23, 42, 43). The gene fis, which is part of the dusB-fis operon, encodes a nucleoid-associated protein involved in resistance to oxidative stress (44). The protein Ail is important for tight attachment to host cells, type III secretion, and resistance against serum killing (45, 46). Although, unlike the katA and sodB genes, expression of fis and ail was not responsive to iron, we included them in our analysis because of their known roles in extraintestinal infection (44, 46). Since the promoters of these IscR targets are predicted to encode an IscR type II site, we used purified IscR-C92A (apo-locked IscR) to assess binding in vitro. In vitro electrophoretic mobility shift assays (EMSAs) showed that apo-IscR binds to these promoters, although the binding to the ail promoter appeared weaker (Fig. 6B). Indeed, the binding site predicted upstream of ail is missing key residues known to be important for IscR binding. A very large band shift was observed when assessing IscR binding to the katA promoter fragment (unpublished observations), indicating the possibility of more than one IscR binding site in this region. Indeed, when the katA promoter fragment was split into two, each with a bioinformatically identifiable IscR type II motif, binding to both fragments was observed. Collectively, these data demonstrate that IscR binds both in vitro and in vivo to the promoters of genes known or predicted to be involved in virulence.
FIG 6.

IscR binds to the promoters of dusB-fis, ail, sodB, and katA in vivo and in vitro. (A) IscR-enriched sequence reads located within the promoter regions of dusB-fis, ail, sodB, and katA. The y axis indicates read count, while the x axis indicates the genomic position of the ChIP-Seq peaks. Dashed lines correspond to the identified 30-bp IscR type II motif sequence. The arrows indicate previously identified transcriptional start sites (39). (B) Purified E. coli IscR-C92A was used for electrophoretic mobility shift assays (EMSAs) using DNA from the promoter regions of dusB-fis, ail, sodB, and katA. The predicted IscR type II binding site is noted below each EMSA, and critical nucleotides are highlighted in color. Note the katA promoter template was split into two distinct fragments.
IscR binding sites are conserved in human-pathogenic Yersinia.
We previously showed that IscR regulated the T3SS in both Y. pestis and Y. pseudotuberculosis, suggesting conservation of IscR regulation between these two species (9). To assess the degree of conservation of the identified IscR binding sites in the human-pathogenic Yersinia species, we carried out a comparative genomics analysis. We first searched for orthologs of the first gene of each of the 213 TUs containing an upstream IscR in vivo binding site in Y. pseudotuberculosis IP2666 (Data Set S3). Out of these 213 TUs, we could identify 203 orthologs of the first gene in each TU in Y. pseudotuberculosis IP32953, 203 in Y. pestis CO92, and 152 in Y. enterocolitica 8081 (Fig. 7A), which is more distantly related to Y. pseudotuberculosis compared to Y. pestis. In order to assess conservation of IscR binding sites among these four strains, MEME suite was utilized to assess the similarity of identified IscR binding motifs to the IscR type II consensus motif generated from this study (Fig. 5E). Of the TUs for which an ortholog could be identified in at least one of the other Yersinia strains, the majority contained an upstream identifiable IscR type II binding motif. If orthologous type II motif sequences in two different Yersinia strains are functionally conserved in terms of IscR regulatory control, then the distance between the motif and the downstream gene should be similar between the two species. Indeed, the predicted IscR binding site for orthologous genes were of similar distance from the downstream start codon, with more similarity among the Y. pseudotuberculosis and Y. pestis strains than between Y. pseudotuberculosis and Y. enterocolitica (Fig. S3A). In addition, the IscR binding sites overall are highly conserved among strains IP2666, IP32953, and CO92, although many diverge in strain 8081 (Fig. S3B). In order to visualize the conservation of IscR type II motifs in Yersinia, a heatmap was generated illustrating the retainment of critical nucleotides that have been shown to be important for IscR binding (Fig. 7B) (28). Importantly, there was very high conservation of IscR type II motifs between Y. pseudotuberculosis and Y. pestis. Out of the 203 transcription units whose putative regulatory regions were bound by IscR in Y. pseudotuberculosis IP2666 and conserved in Y. pestis CO92, 173 (∼85%) had IscR type II motif sequences in their promoters that were 100% conserved between strains IP2666 and CO92, while 17 (∼8%) contained only differences in nucleotides not critical for IscR binding. Interestingly, the IscR binding sites that were among the most conserved between all three Yersinia species included those within the promoters of sodB, katA, the suf operon, pagP, ompW, yscW-lcrF, dusB-fis, and ail.
FIG 7.

IscR binding site sequences are highly conserved in human-pathogenic Yersinia. (A) The first gene of the 213 transcription units identified in this study as being targets of IscR in Y. pseudotuberculosis IP2666 were used to identify orthologs in Y. pseudotuberculosis IP32953, Yersinia pestis CO92, and Yersinia enterocolitica 8081. (B) Heatmap showing similarity of IscR binding sites in Y. pseudotuberculosis IP32953, Y. pestis CO92, and Y. enterocolitica 8081 compared to Y. pseudotuberculosis IP2666, in the promoters of the 213 IscR transcription units. Blue boxes indicate a bioinformatically identified IscR type II binding motif predicted to enable stronger binding in the IP32953, CO92, or 8081 strain compared to the IP2666 strain. Yellow boxes indicate a motif predicted to bind IscR more weakly in strain IP32953, CO92, or 8081 compared to IP2666. White boxes indicate the predicted IscR binding site is similar in IP32953, CO92, or 8081 compared to IP2666. Gray boxes indicate no ortholog was found in the strain. Black boxes indicate an ortholog was identified but no IscR binding site was predicted by MEME suite tools. Values represent absolute log difference between MEME-FIMO-pvalue of known IscR binding site to orthologous IscR binding site.
Conservation of IscR binding sites in Y. pseudotuberculosis (IP2666, IP32953), Y. pestis (CO92), and Y. enterocolitica (8081). (A) Distance of the identified IscR binding site in Y. pseudotuberculosis (IP2666) from the start codon of each transcription unit member of the IscR regulon is plotted versus the distance of the predicted IscR binding site from the start codon of the identified ortholog in Y. pseudotuberculosis (IP32953), Y. pestis (CO92), or Y. enterocolitica (8081). (B) The log10 FIMO P value of the identified IscR binding site in Y. pseudotuberculosis (IP2666) is plotted versus the log10 FIMO P value of the predicted IscR binding site upstream of identified orthologs in Y. pseudotuberculosis (IP32953), Y. pestis (CO92), or Y. enterocolitica (8081). Download FIG S3, PDF file, 0.3 MB (297.9KB, pdf) .
Copyright © 2021 Balderas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Comparative genomics of IscR binding sites in Y. pseudotuberculosis IP2666, Y. pseudotuberculosis IP32953, Y. pestis CO92, and Y. enterocolitica 8081. Download Data Set S3, XLSX file, 0.08 MB (82.9KB, xlsx) .
Copyright © 2021 Balderas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Integration of transcriptome profiling with identified IscR binding sites.
To determine which genes are directly regulated by IscR in Y. pseudotuberculosis, we collated the ChIP-Seq results with our published RNA-Seq data carried out under the same aerobic, non-iron-starved conditions used for the IscR chromatin precipitation (19) (Data Set S2). Under this condition, only 18 out of the 213 putative IscR regulatory sites found to be associated with an IscR binding site drove IscR-dependent changes in expression of predicted transcription units. Three of these 18 TUs, DN756_21815-DN756_21820, yscW-lcrF, and yscIJKL, are carried on the pYV virulence plasmid and were downregulated in the iscR mutant. We have extensively validated direct regulation of the yscW-lcrF operon by IscR (9, 19) (Fig. 5C). The yscIJKL T3SS genes are carried within the virC operon, yscABCDEFGHIJKL (47), which is controlled by the T3SS master regulator LcrF (48). Since lcrF transcription is directly regulated by IscR (19), further experiments are needed to determine whether alternative transcriptional start sites that are regulated by LcrF or IscR exist for the virC operon. DN756_21815 and DN756_21820 are a hypothetical gene and pseudogene, respectively, and are carried on the pYV virulence plasmid. Interestingly, deletion of DN756_21815 and DN756_21820 had a small but significant effect on secretion of the T3SS effector protein YopE into culture supernatant (Fig. S4). The remaining 15 TUs are encoded on the chromosome and include sodB, ail, and fis. A number of ChIP-Seq studies have shown that transcription factors exhibit binding to promoters whose genes are differentially regulated by the transcription factor under some but not all conditions tested, including the condition used for ChIP-Seq (49, 50). Therefore, it was not surprising to see limited overlap between the ChIP-Seq data and RNA-Seq from only one culture condition.
Deletion of DN756_21815 and DN756_21820, identified gene targets of IscR, results in a small but significant decrease in type III secretion. (A) Read coverage of IscR-binding peaks proximal to DN756_21815_DN756_21820. Counts per million reads (CPM) are plotted versus the genomic position. Dashed lines correspond to the predicted 30-bp IscR type II motif. Arrows indicate transcriptional start sites (48). (B) Expression of DN756_21815, and DN756_21820 genes under various iron conditions as measured by RNA-seq. Reads are represented by trimmed mean of M-values (TMM) of WT (black) and ΔiscR strains (gray) grown in M9 minimal medium containing FeSO4 (non-iron-starved [NIS]), iron starved in Chelex-treated M9 minimal medium with no iron source added back (Chelex) or supplemented with 5 mM hemin (+Heme) or FeSO4 (+FeSO4). **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (EdgeR with a corrected FDR post hoc test). (C) Yersinia strains were grown under rich media, T3SS-inducing conditions. The secretome of these cultures was visualized with Coomassie blue. The effector protein, YopE, was quantified by densitometry relative to the WT control to measure the relative efficiency of the Ysc T3SS. The average of five biological replicates ± standard deviation is shown. ****, P < 0.0001; *, P < 0.05 (one-way ANOVA with Dunnett’s post hoc test). Download FIG S4, PDF file, 0.3 MB (286.6KB, pdf) .
Copyright © 2021 Balderas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We assessed whether our transcriptome analysis of iron-starved Yersinia might reveal additional genes associated with in vivo IscR binding whose expression is regulated by IscR. Indeed, 86 TUs with an upstream IscR binding site were found to contain genes differentially expressed in the presence and absence of IscR under at least one of the RNA-Seq conditions (Data Set S2). We carried out a cluster of orthologous genes (COG) analysis on genes of the IscR direct regulon whose promoters contain an IscR enrichment site (Fig. S5A), functional regulon genes associated with a ChIP-Seq peak that showed differential expression by IscR in at least one condition tested (Fig. S5B), and the indirect regulon whose expression depends on IscR but are not associated with a ChIP-Seq peak (Fig. S5C). The direct regulon was enriched for coenzyme and lipid metabolism as well as inorganic iron uptake compared to the indirect regulon, while the indirect regulon was enriched for biosynthesis (amino acid, nucleotide, and carbohydrate metabolism), energy production, and the T3SS (virulence plasmid). Included in the functional regulon were genes whose expression patterns are consistent with apo-IscR induction or repression in our RNA-Seq analysis and are therefore predicted to be directly controlled by IscR during disseminated infection: the suf operon; the metal transporters iucABCDiutA, alcC, cirA, fhuC_1, oprC, and yiuA; the cell envelope enzymes pagP, amiD, and ydhO; and the oxidative stress associated sodB, katA, and ompW. However, many genes within the functional regulon did not display an expression pattern consistent with apo-IscR activation or repression yet showed differential expression in response to IscR under at least one RNA-Seq condition. This is consistent with other reports that identified ChIP-Seq peaks for genes differentially regulated by their respective transcription factors only under certain environmental conditions (49, 50).
COG analysis of the IscR regulon, functional regulon, and indirect regulon. Clusters of Orthologous Groups of proteins (COG) analysis of genes with an IscR ChIP-Seq peak (A), genes with an IscR ChIP-Seq peak and are differentially expressed between WT and the iscR mutant under at least one tested condition (B), and genes differentially expressed between WT and the iscR mutant but have no IscR ChIP-Seq peak (C). Download FIG S5, PDF file, 0.3 MB (294.1KB, pdf) .
Copyright © 2021 Balderas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
IscR directly regulates the suf operon, which contributes to type III secretion under iron-limiting conditions.
The validated IscR binding site in the suf promoter exhibited 100% conservation among all human-pathogenic Yersinia (Fig. 7B), yet the role of the suf operon has never been studied in Yersinia. We observed a ChIP-Seq peak in the suf promoter with an identifiable IscR type II binding motif (Fig. 8A), as well as in vitro binding of IscR to the suf promoter using an EMSA (Fig. 8B). Furthermore, the suf operon was differentially expressed by IscR following iron starvation (Fig. S6), when IscR levels are high and Fur repression is predicted to decrease (51, 52). Indeed, sufABCDSE were found to be expressed 3- to 11-fold more in wild-type Y. pseudotuberculosis than in the ΔiscR mutant during iron starvation (Fig. 8C and Fig. S6). Together, these data strongly suggest that apo-IscR binds to a type II binding motif sequence in the suf promoter, activating suf operon expression.
FIG 8.

IscR directly regulates the Suf Fe-S cluster biogenesis pathway, which is important for type III secretion activity specifically under aerobic conditions. (A) IscR-enriched sequence reads located within the promoter regions of sufABCDSE. The y axis indicates read count, while the x axis indicates the genomic position of the ChIP-Seq peaks. Dashed lines correspond to the identified 30-bp IscR type II motif sequence. Arrows indicate previously identified transcriptional start site (39). (B) Purified E. coli IscR-C92A was used for electrophoretic mobility shift assays (EMSAs) using DNA from the promoter regions of sufABCDSE. The predicted IscR type II binding site is noted below the EMSA, and critical nucleotides are highlighted in color. (C) Expression of sufA relative to 16s rRNA as measured by qRT-PCR of WT (black) and ΔiscR strains (gray). **, P < 0.01 (one-way ANOVA with Dunnett’s post hoc test). Average of three independent experiments ± standard deviation is shown. (D and E) WT and ΔsufABCDSE cultures were iron starved in Chelex-treated M9 minimal medium and supplemented with either 3.6 μM FeSO4 (high [hi]) or 0.036 μM FeSO4 (low [lo]) before type III secretion was induced under aerobic (D) or anaerobic (E) conditions. Proteins secreted into culture supernatant (Sup) and precipitated with trichloroacetic acid were probed with antibodies for YopE and YopD T3SS cargo proteins. Cell lysates were probed with antibodies for RpoA, YopE, and YopD. One representative experiment out of three biological replicates is shown.
Expression of the suf operon is modulated by iron and requires IscR. Expression of sufA, sufB, sufC, sufD, sufS, and sufE genes under various iron conditions as measured by RNA-seq. Reads are represented by trimmed mean of M-values (TMM) of WT (black) and ΔiscR strains (gray) grown in M9 minimal medium containing 3.6 μM FeSO4 (non-iron-starved [NIS]), iron starved in Chelex-treated M9 minimal medium with no iron source added back (Chelex) or supplemented with 5 mM hemin (+Heme) or 3.6 μM FeSO4 (+FeSO4).**, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (EdgeR with a corrected FDR post hoc test). Download FIG S6, PDF file, 0.1 MB (146.5KB, pdf) .
Copyright © 2021 Balderas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We constructed a ΔsufABCDSE Y. pseudotuberculosis mutant, which exhibited normal growth and motility (Fig. S7A and B). Surprisingly, this mutant had a significant type III secretion defect under aerobic conditions, when the Suf system is the dominant Fe-S cluster biogenesis pathway (Fig. 8D and E). These data suggest that an Fe-S cluster-containing protein is important for T3SS expression or activity. As the suf mutant did not exhibit a defect in T3SS gene expression (Fig. S7C), these data indicate that the Suf pathway contributes to type III secretion in a posttranslational manner. The Isc pathway is the dominant Fe-S cluster biogenesis pathway under anaerobic conditions. However, we were unable to test whether the Isc pathway was required for type III secretion under anaerobic conditions because we were unsuccessful in deleting iscSUA, suggesting these are essential genes. Together, these data suggest that IscR regulates the Yersinia T3SS in two distinct ways: through direct control of the LcrF T3SS master regulator and through direct control of the Suf pathway that matures an Fe-S cluster protein critical for T3SS activity.
The Suf pathway does not affect motility or growth. (A) Iron-starved Y. pseudotuberculosis were either kept in iron-starved Chelex-treated M9 minimal medium with no iron source added back (Chelex) or supplemented with 3.6 μM FeSO4 (+FeSO4). Cultures were grown at 26°C for 10 h, and optical density at 600 nm was measured every hour. Non-iron-starved (NIS) samples were never exposed to Chelex-treated M9. (B) The indicated strains were spotted onto motility agar (1% tryptone, 0.25% agar) and were grown at 26°C. The diameters of the colonies were measured 24 h and 48 h later and used to calculate percent motility. Graphs represent three biological replicates. ****, P < 0.0001 (one-way ANOVA with Dunnett’s post hoc test). (C) Expression of lcrF, iscR, and yopE, relative to 16S rRNA, following iron-starved conditions in M9 minimal medium at 37°C, as measured by qPCR. Black bars represent cultures which were iron starved and supplemented with 3.6 μM FeSO4 before induction of the type III secretion system, while gray bars represent cultures that received 0.036 μM FeSO4 before induction of the type III secretion system. Average of three independent experiments ± standard deviation is shown. ***, P < 0.001; **, P < 0.01; *, P < 0.05 (one-way ANOVA with Dunnett’s post hoc test). Download FIG S7, PDF file, 0.2 MB (213.5KB, pdf) .
Copyright © 2021 Balderas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DISCUSSION
IscR and iron are both critical factors in the pathogenesis of Yersinia and a number of other bacteria (9, 14, 20, 21, 53). In this study, we present the first characterization of the IscR direct regulon. We used ChIP-Seq and RNA-Seq analysis to identify Yersinia genes directly regulated by IscR in response to changes in iron availability. Our data suggest that IscR allows Yersinia to couple sensing of iron starvation, a condition experienced by the bacterium during extraintestinal infection, to control of genes involved in iron homeostasis, cell envelope modification, oxidative stress response, as well as T3SS expression and activity (Fig. 9). Over 93% of in vivo IscR binding sites identified in Y. pseudotuberculosis were conserved in Y. pestis, suggesting that Y. pestis retained IscR as an important regulator after its evolution from foodborne pathogen to plague agent.
FIG 9.

Model of how iron modulates IscR levels and leads to differential expression of IscR-dependent virulence factors. (A) The Suf system loads Fe-S clusters onto IscR under aerobic conditions, while the Isc system is more active under anaerobic conditions. Iron bioavailability and oxygen tension affect the holo-/apo-IscR ratio. (B) Low iron and oxidative stress, which are encountered by Yersinia during disseminated infection, lead to increased Fe-S cluster demand. This leads to a decrease in holo-IscR, subsequent derepression of overall IscR expression, and an increase in apo-IscR levels. Apo-IscR directly regulates genes involved in the T3SS, oxidative stress resistance, cell envelope remodeling, iron homeostasis, and other processes, enabling virulence.
In this study, we show that complete iron starvation induces IscR mRNA and protein levels in Yersinia under aerobic conditions. Previously, iron limitation was shown to induce IscR levels in both E. coli and Vibrio vulnificus (33, 54). Iron starvation not only limits the amount of free iron that can incorporate into Fe-S cofactors but can also affect the function of the Fe-S cluster biogenesis machinery (55). Ultimately, iron starvation leads to a reduction of Fe-S cluster availability causing a shift to more apo-IscR compared to holo-IscR (Fig. 9). Since holo-IscR represses transcription of the isc operon, this shift leads to derepression of the isc operon under iron-starved conditions (31). We previously found that supplementing Yersinia with as little as 0.036 μM FeSO4 prevented this derepression of iscR, suggesting why in our earlier study IscR levels were not iron responsive under aerobic conditions (9). However, we show here that following prolonged iron starvation, IscR and IscR target genes are indeed responsive to either inorganic or heme iron. These data suggest that during infection, when the availability of free iron is vanishingly low and is further reduced by inflammatory pathways (39, 40), IscR levels should fluctuate in response to the ability of Yersinia to scavenge iron.
Two important categories of genes found to be regulated by iron and IscR were involved in cell envelope modification and the oxidative stress response. Yersinia modifies its LPS during growth at 37°C such that its LPS is less stimulatory to the endotoxin receptor Toll-like receptor 4 (TLR4) (56–59). Interestingly, we found in this study that a number of Yersinia LPS biosynthesis and modification enzymes genes are upregulated upon iron starvation. Of these genes, only pagP, which encodes an outer membrane lipid A palmitoyltransferase, was found to be directly regulated by IscR. The pagP promoter contained an IscR type II motif that bound to IscR in vivo, and pagP expression was consistent with induction by apo-IscR. Salmonella PagP, which is regulated by magnesium through the PhoP/Q regulatory system (60), enables production of LPS lipid A that is less stimulatory to TLR4 (61–63). In contrast, Y. pestis has an inactive pagP allele, but when the Y. pseudotuberculosis functional pagP gene is inserted into Y. pestis, the LPS that is produced is more immunostimulatory (64). Therefore, how IscR affects LPS stimulation of TLR4 and other LPS sensors remains to be determined. To our knowledge, iron availability has not been previously shown to affect Yersinia LPS gene expression nor has IscR been shown to regulate LPS modifying genes in other bacterial species. In contrast, IscR has been implicated in controlling genes involved in the response to free radical stress in Vibrio vulnificus, Pseudomonas aeruginosa, and Burkholderia mallei (21, 23, 24, 43, 54). The Yersinia T3SS, which is positively regulated by IscR, inhibits phagocytic cell oxidative burst. Therefore, IscR induction of the T3SS should lead to a decrease in the amount of reactive oxygen species (ROS) encountered by Yersinia during infection. In this study, we also found that IscR directly repressed the genes encoding catalase, katA, superoxide dismutase, sodB, and the oxidative stress-associated OmpW (65). It is possible that IscR upregulation of the T3SS may be coupled to lower expression of KatA and SodB, since the bacteria would be less likely to encounter ROS stress when the T3SS is active.
We showed that IscR binds to the sufABCDSE promoter both in vitro and in vivo and that suf expression is consistent with apo-IscR induction of the suf promoter. This is in line with a previous study showing that IscR activates the E. coli suf operon upon exposure to the iron chelator dipyridyl (33). However, surprisingly, our data also showed that deletion of the suf operon leads to reduced Ysc T3SS activity. Loss of the suf operon did not affect T3SS gene expression or protein levels but instead affected secretion of T3SS cargo. This phenotype was observed under standard aerobic non-iron-starved conditions (data not shown) and was even more pronounced under iron-limited conditions where the suf operon becomes more important for synthesizing Fe-S clusters. Loss of the suf operon may affect the electron transport chain and lead to a proton motive force defect, which is required for T3SS activity (66). However, the suf mutant had no motility defect, which also requires the proton motive force. Under anaerobic iron-replete conditions, when the Isc pathway mediates Fe-S cluster biogenesis for the cell, the suf mutant displays normal type III secretion. Collectively, these results suggest that a Yersinia Fe-S cluster protein, matured by the Suf pathway under aerobic conditions, promotes T3SS activity. Interestingly, the Salmonella Isc pathway, but not the Suf pathway, was shown to be important for IscR-mediated repression of the SPI-1 T3SS and for virulence following oral infection. In contrast to the Yersinia T3SS, Salmonella expresses its SPI-1 T3SS in the intestine where it is required for entry into intestinal epithelial cells (20).
IscR positively regulates the T3SS master regulator LcrF, and type III secretion occurs only under anaerobic conditions in the absence, not the presence, of iron (9, 19). Therefore, it was surprising that only the virC structural operon (yscCDEFGHKL) but not other LcrF-regulated genes were significantly upregulated under our aerobic iron starvation conditions. Indeed, while mRNA levels of the yscW-lcrF operon trended up in the absence of iron, this difference was not statistically significant. In fact, some T3SS effector protein and chaperone genes (yopO, yopK, yopH, yopJ, and sycDH), the yscM T3SS regulatory gene, and the needle tip protein lcrV were expressed approximately two- to fourfold more after iron supplementation. However, importantly, most of these genes were downregulated ∼10- to 50-fold by deletion of iscR, demonstrating that the presence of an iscR gene had a much stronger effect on T3SS gene expression than changes in iron availability under aerobic conditions. Y. pseudotuberculosis transits from the small intestine to lymph tissue and vital organs during infection. In the intestines, Yersinia experiences an anaerobic iron-replete environment where it does not require its T3SS (67–69). In contrast, once Yersinia crosses the intestinal barrier and the T3SS becomes important for virulence, oxygen tension increases and host iron sequestration causes a drastically smaller amount of bioavailable iron. Collectively, our data show that IscR represses the Yersinia T3SS in the intestines and induces the T3SS once Yersinia disseminate.
Only about 40% of genes associated with an IscR binding site were found to be differentially regulated by IscR in at least one condition tested, and even fewer had expression patterns consistent with apo-IscR activation or repression. However, a recent study examining the regulons associated with five different two-component regulatory systems in E. coli noted the existence of genes whose promoters directly bound a transcription factor (TF) but showed differential expression by that TF only under certain conditions (50). The authors speculated that this is due to other regulators controlling such promoters and referred to these genes as exhibiting “hypothetical functional binding” by the TF, as other studies have also suggested (70). We postulate that ail and dusB-fis may fall into this category, as they are differentially expressed by IscR only under non-iron-starved conditions but show IscR binding to their promoters in vitro and in vivo. The ail gene encodes an adhesin that contributes to delivery of T3SS effector proteins into target host cells and mediates complement resistance (45, 46, 71, 72). The factor for inversion (Fis) nucleoid-associated protein influences the topological state of DNA and affects gene expression (73). Fis regulates virulence in a number of pathogens, including Y. pseudotuberculosis (44). In some organisms, Fis expression is regulated by growth phase, while in others it is regulated by specific environmental signals (73). For example, Salmonella Fis regulates the supercoiling of DNA encoding the SPI-1 and SPI-2 T3SSs in an opposite manner in response to oxygen (74, 75). In our study, we show that the Yersinia fis gene is expressed at higher levels under iron starvation conditions than after iron supplementation. In Y. pseudotuberculosis, Fis has been shown to be crucial for resistance to oxidative stress and colonization of the murine spleen and liver (44). Additional environmental signals, such as oxygen tension, should be examined for their ability to regulate Yersinia ail and fis to explore whether IscR regulation of these genes may be important under anaerobic conditions such as those found in the intestinal lumen.
The remaining 60% of genes associated with an IscR binding site were not found to be differentially expressed by IscR under any condition tested. Choudhary et al. categorized such genes as having “potentially nonfunctional binding” of the TF (50). The in vivo IscR binding associated with these genes either represents spurious, nonfunctional binding, or another regulator is preventing IscR from exerting an effect on gene expression under the conditions tested. Indeed, there is evidence that IscR regulates promoters also targeted by other TFs. For example, out of the 213 TUs targeted by IscR, 65 had a predicted Fur box in their corresponding regulatory region (unpublished observations). Additional RNA-Seq analysis of WT and ΔiscR Yersinia will help determine whether the “potentially nonfunctional binding” genes identified in this study are differentially expressed under certain conditions, for example anaerobic iron-replete conditions that mimic those encountered by Yersinia in the intestinal lumen where IscR is also necessary for colonization.
Fur in uropathogenic E. coli (UPEC) regulates genes found in pathogenicity islands and siderophores not present in commensal E. coli K-12 strain MG1655 (76). This difference in Fur regulon members from the two species could be due to the lack of the pathogenicity islands in commensal E. coli. Interestingly, UPEC Fur directly regulated genes that were also present in commensal E. coli, yet only UPEC Fur directly regulated this subset of genes. These data suggest that as species evolve and find new niches, regulons and transcription regulatory networks change to adapt to the environment. Current models suggest that Y. pestis evolved as recently as ∼1,500 years ago from Y. pseudotuberculosis through a series of DNA element acquisitions along with gene loss (77), making Yersinia an interesting model to study the IscR regulon. These genomic changes enabled a previously facultative foodborne pathogen to colonize and be transmitted through a flea vector. However, once inside the host, both Y. pseudotuberculosis and Y. pestis colonize lymph nodes and spread to deeper tissues (5, 36). In addition, while the two pathogens exhibit very different life cycles, there is evidence that, within lymph nodes, they both experience iron starvation (39, 40). For example, Y. pseudotuberculosis in the Peyer’s patches upregulate metal acquisition genes such as hmuR, alcC, znuABC, mntH, as well as others (39). These same genes are upregulated in our transcriptomic data upon iron starvation. The results of our comparative genomic analysis suggest that the IscR regulon is highly conserved in Y. pseudotuberculosis and Y. pestis. We speculate that IscR control of genes involved in iron homeostasis, virulence, stress response, and other pathways has provided a fitness benefit to Y. pestis during its evolution as the plague agent and was therefore retained. Furthermore, as a number of the IscR binding sites identified in Y. pseudotuberculosis are conserved in the Y. enterocolitica genome, we also speculate that IscR-regulated genes important for pathogenesis were present in the shared ancestor of Y. enterocolitica and Y. pseudotuberculosis/Y. pestis.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
All strains used in this study are listed in Table 1. Y. pseudotuberculosis strains were grown in M9 minimal medium supplemented with Casamino Acids, referred to here as M9+3.6 μM FeSO4, at 26°C with shaking at 250 rpm, unless otherwise indicated (78). Non-iron-starved conditions were achieved by subculturing an M9+3.6 μM FeSO4 overnight culture to an optical density at 600 nm (OD600) of 0.2 in fresh M9+3.6 μM FeSO4, and shaking for 3 h at 37°C.
TABLE 1.
Strains used in this study
| Strain | Relevant genotype | Reference |
|---|---|---|
| IP2666 (WT) | Naturally lacks full-length YopT | 97 |
| IP2666 (ΔiscR) | iscR in-frame deletion of codons 2 to 156 | 19 |
| IP2666 (IscR 3xFLAG) | In-frame C terminus 3xFLAG tag of chromosomal IscR | This work |
| IP2666 (ΔsufABCDSE) | sufABCDSE in-frame deletion retains first 10 codons of SufA and last 20 codons of SufE | This work |
| IP2666 (ΔDN756_21815- DN756_21820) |
DN756_21815-DN756_21820 in-frame deletion retains first 10 codons of DN756_21815 and last 10 codons of DN756_21820 |
This work |
| IP2666 (Apo-locked IscR) | IscR-C92A/C98A/C104A | 19 |
| IP2666 (ΔflhD) | WT strain with inactive flhDC from Yersinia pestis | 19 |
Iron starvation was achieved by growing Y. pseudotuberculosis aerobically in M9 medium lacking iron treated with Chelex 100 resin to remove all traces of iron in acid-washed glassware, as previously described (9, 12). Specifically, iron-replete overnight cultures (M9+3.6 μM FeSO4) grown at 26°C aerobically were diluted to an OD600 of 0.1 into Chelex-treated M9 medium and grown for 8 h at 26°C aerobically with agitation. Cultures were then subcultured a second time to an OD600 of 0.1 in fresh Chelex-treated M9 and grown for 12 h at 26°C with agitation. Cultures were then subcultured to an OD600 of 0.1 into 20-ml Chelex-treated M9 medium with either no iron, 5 μM hemin, or 3.6 μM FeSO4 and grown for 3 h at 37°C with agitation.
Construction of Yersinia mutant strains.
A 3xFLAG affinity tag was placed at the C terminus of IscR encoded at the native iscR chromosomal locus to facilitate detection of IscR with FLAG monoclonal antibody (see Fig. S1A in the supplemental material). The 3xFLAG affinity tag was chromosomally added to the C terminus of iscR through splicing by overlap extension (79). The primer pair FiscR_cds/RiscR_cds (Table 2) was used to amplify ∼500 bp upstream of iscR plus the iscR coding region excluding the stop codon. The primer pair F3xFLAG/R3xFLAG was used to amplify the 3xFLAG tag. The primer pair F3’iscR/R3’iscR was used to amplify the ∼500-bp downstream region of iscR including the stop codon. These amplified PCR fragments were cloned into a BamHI- and SacI-digested pSR47s suicide plasmid (λpir-dependent replicon, kanamycin resistant [Kanr], sacB gene conferring sucrose sensitivity) using the NEBuilder HiFi DNA Assembly kit (New England Biolabs, Inc.). Recombinant plasmids were transformed into E. coli S17-1 λpir competent cells and later introduced into Y. pseudotuberculosis IP2666 via conjugation. The resulting Kanr, irgansan-resistant (Yersinia selective antibiotic) integrants were grown in the absence of antibiotics and plated on sucrose-containing media to select for clones that had lost sacB (and by inference, the linked plasmid DNA). Kans, sucrose-resistant, Congo red-positive colonies were screened by PCR and sequenced. Yersinia carrying only the 3xFLAG-IscR allele secreted levels of YopE similar to those secreted by the wild-type strain, suggesting that the 3xFLAG tag does not affect the ability of IscR to regulate T3SS gene expression (Fig. S1B). In addition, derepression of iscS mRNA was not observed in the 3xFLAG-IscR strain, suggesting the 3xFLAG-IscR retains the ability to repress iscS (25) (Fig. S1C). These data demonstrate that the 3xFLAG affinity tag does alter IscR function and thus is suitable for ChIP-Seq experiments.
TABLE 2.
Y. pseudotuberculosis primers used in this study
| Primer | Primer sequencea | Reference |
|---|---|---|
| FiscR_cds | gatatcgaattcctgcagcccggggGCTCCTTAAATTTAGCCATGGC | This work |
| RiscR_cds | ggtctttgtagtcTGCGCGCAGATTGACGTTAATC | This work |
| F3xFLAG | cgtcaatctgcgcgcaGACTACAAAGACCATGAC | This work |
| R3xFLAG | ccgcaaattctgcttaCTCGAGTCCACCTTTATC | This work |
| F3’iscR | aggtggactcgagTAAGCAGAATTTGCGGAATTTTAC | This work |
| R3’iscR | ggtggcggccgctctagaactagtgCCTTCAACATAGTTGAAGC | This work |
| F5’ΔsufABCDSE | cgaattcctgcagcccggggTCAATCCTCGTTTTGCGC | This work |
| R5’ΔsufABCDSE | cctccagaccAGAAAATGTCCCAACTGATTC | This work |
| F5’ΔDN756_21815- DN756_21820 |
cgaattcctgcagcccggggGTTGAGTTCTGAAAGAACAATTGTG | This work |
| R5’ΔDN756_21815_21820 | ggcgaccttcTTTGCGGTTGGCTTCGATG | This work |
| F3’ΔsufABCDSE | gacattttctGGTCTGGAGGCGATGATC | This work |
| R3’ΔsufABCDSE | agggaacaaaagctggagctATAGTCAGTACTGACCCCG | This work |
| F3’ΔDN756_21815-21820 | caaccgcaaaGAAGGTCGCCTCGATAAAG | This work |
| R3’ΔDN756_21815_21820 | agggaacaaaagctggagctTTAACTGATCTGCCAGAATTAC | This work |
| qPCR_lcrF_F | GGAGTGATTTTCCGTCAGTA | 8 |
| qPCR_lcrF_R | CTCCATAAATTTTTGCAACC | 8 |
| qPCR_iscR_F | CAGGGCGGAAATCGCTGCCT | 9 |
| qPCR_iscR_R | ATTAGCCGTTGCGGCGCCTAT | 9 |
| qPCR_sufA_F | CGCAAATTACGCGGCTTATGC | This work |
| qPCR_sufA_R | GGCAGGCTCTTTAGCCATATC | This work |
| qPCR_iscS_F | CGACGCCAGTAGATCCGCGT | 19 |
| qPCR_iscS_R | ACGAGGGTCTGCACCCACCA | 19 |
| qPCR_yopE_F | CCATAAACCGGTGGTGAC | 98 |
| qPCR_yopE_R | CTTGGCATTGAGTGATACTG | 98 |
| qPCR_16s_F | AGCCAGCGGACCACATAAAG | 25 |
| qPCR_16s_R | AGTTGCAGACTCCAATCCGG | 25 |
| EMSA_suf_F | tttttCTCGAGGAGTGTTTTTTCTTTTAGACC | This work |
| EMSA_suf_R | tttttGGATCCTAAACGTTTTCAGGTTGAA | This work |
| EMSA_dusBfis_F | tttttCTCGAGGTATCGATAATATTTCAGTATTAAC | This work |
| EMSA_dusBfis_R | tttttGGATCCGTAGAAATATATCTGTCACACATC | This work |
| EMSA_ail_F | tttttCTCGAGCTGTCACCGTCCTGG | This work |
| EMSA_ail_R | tttttGGATCCGACTAAAGTGGCCAGCC | This work |
| EMSA_sodB_F | tttttCTCGAGTAACTGACGGTCCG | This work |
| EMSA_sodB_R | tttttGGATCCGGTGTGGGGTGAA | This work |
| EMSA_katA1_F | ccgCTCGAGTATTGCTCTCTCATTGTTCGTTATG | This work |
| EMSA_katA1_R | cgcGGATCCATTTGAAATTATGATCATTTGAAATACTGTGC | This work |
| EMSA_katA2_F | ccgCTCGAGGATCGTCAGTTCCCACAG | This work |
| EMSA_katA2_R | cgcGGATCCCCCCACTAGTTGTAGTCG | This work |
| NdeI-iscR_F | GGAATTCCATATGAGACTGACATCTAAAGGGCGC | This work |
| BamHI-His6-IscR_R | GCGGGATCCTTAGTGGTGGTGGTGGTGGTGAGCGCGTAACTTAACGTCGATCGC | This work |
Uppercase nucleotides specify primer that anneals to target for molecular cloning; lowercase nucleotides depict complementary sequence for NEB Gibson Assembly or extra nucleotides to facilitate efficient restriction digestion. The XhoI and BamHI restriction sites are underlined.
Generation of the ΔsufABCDSE and ΔDN756_21815-21820 mutants were generated via splicing by overlap extension (79). Primer pairs F5/R5ΔsufABCDSE and F5/R5ΔDN756_21815-21820 (Table 2) were used to amplify ∼1,000 bp 5′ of sufA and DN756_21815, respectively. Primer pairs F3/R3ΔsufABCDSE and F3/R3ΔDN756_21815-21820 were used to amplify ∼1,000 bp 3′ of sufE and DN756_21820, respectively. Amplified PCR fragments were cloned into a BamHI- and SacI-digested pSR47s suicide plasmid (λpir-dependent replicon, Kanr, sacB gene conferring sucrose sensitivity) using the NEBuilder HiFi DNA Assembly kit (New England Biolabs, Inc.). Mutant strains were generated as described above for the 3xFLAG-IscR strain.
Chromatin immunoprecipitation followed by high-throughput sequencing.
ChIP-Seq experiments were performed in the IscR-3xFLAG strain and the parental wild-type IP2666 strain after growth at 37°C for 3 h in M9+3.6 μM FeSO4, using the IP2666plB1 genome (GenBank accession no. CP032566.1 and CP032567.1) for analysis. ChIP assays were performed as previously described (80) using a monoclonal mouse anti-FLAG antibody (Sigma-Aldrich) that enriched for IscR-3XFLAG. Immunoprecipitated DNA was sheared to 200 to 500 bp via probe-based sonication. DNA quantification was carried out using both Agilent High Sensitivity DNA kit and Invitrogen Qubit dsDNA HS assay kit once DNA was immunoprecipitated and purified. For ChIP-Seq experiments, 10 ng of immunoprecipitated and purified DNA fragments from IscR-3XFLAG (three biological replicates) and WT non-FLAG-tagged IscR (one biological replicate), along with 10 ng of input control, were used to generate libraries using Illumina TruSeq ChIP Library Preparation kit. This kit was used following the manufacturer’s instructions, except that the purification of the ligation products was performed using an Invitrogen E-Gel Power Snap system with Invitrogen E-Gel Size Select II 2% agarose gels. Final library validation was performed using the Qubit and Bioanalyzer. This resulted in eight libraries for sequencing: triplicate immunoprecipitated samples and a single negative-control sample, each with a paired input sample. Libraries were sequenced by the University of Wisconsin at Madison Biotechnology Center DNA Sequencing Facility on an Illumina HiSeq to produce 51-bp single-end reads.
Initial quality control checks were performed on FASTQ files with FastQC v0.11.5 (81). Trimmomatic v0.36 (82) was used to remove low-quality reads from the FATSQ files and to also remove Illumina adapters. The reference files for the Y. pseudotuberculosis IP26666pIB1 chromosome and plasmid were downloaded from the NCBI (GenBank accession no. CP032566.1 and CP032567.1) and concatenated into a single file. The trimmed FASTQ files were then aligned to the Y. pseudotuberculosis genome using Bowtie2 v2.3.3.1 (83) with the default settings to generate SAM files. To remove all unaligned reads and all reads which aligned ambiguously, the SAM files were filtered with Samtools v1.6 (84) with the flags “view -q 10.”
Sequences associated with IscR enrichment following precipitation, or “peaks,” were identified using three separate programs: QuEST v2.4 (85), MOSAiCS v2.18.0 (86), and MACS2 (87). QuEST was run according to the manual with the following options: perform false-discovery rate (FDR) analysis, search for punctate peaks (option 1), and use relaxed peak calling criteria (option 3). QuEST was run separately for each of the three immunoprecipitated samples and the negative-control sample using the paired input sample as a background file. From the generated output, the “max_pos” of each identified enriched region was used at the peak summit. Wiggle files were outputted as a default function of QuEST. MOSAiCS was run as a two-sample analysis as described in the vignette (2 May 2019 version) chapters 3 and 5.1 with the same sample pairs as used with QuEST. During the use of MOSAiCS, a fragment and bin size of 175 nucleotides was used. This number was derived by multiplying the average peak shift, as calculated by QuEST from the immunoprecipitated samples, by two. Peak summits and wiggle files were outputted by MOSAiCS as the last steps of the analysis. MACS2 was run according to the manual with a criterion cutoff of FDR value of <E−5, P value of <−log1070.
Sequences with IscR enrichment following immunoprecipitation were considered bona fide ChIP-Seq peaks if, for each peak-calling program, a peak was called in at least two of three samples. Peaks were considered to be the same if their summits fell within 175 bp of each other. At every location where a negative-control peak overlapped an immunoprecipitated sample peak, we manually examined the negative-control data to determine whether the peak should be removed from our analysis. Specifically, we visually looked for negative peaks which were centered on the immunoprecipitated sample peaks and also confirmed that the negative peaks were composed of both forward and reverse mapping reads. MochiView (88) and Tablet (89) were used to visualize the Wig files and binary SAM (BAM) files, respectively. Peaks confirmed to be arising from background noise were removed from our analysis. The average position of the immunoprecipitated sample peak summits was used as the new peak summit moving forward. This process was repeated for each peak-calling program independently, thus yielding three lists of confirmed peak summits. Finally, confirmed peak summits were clustered as described above. All peaks that were identified by at least two of three peak-calling programs were marked as bona fide peaks, and other peaks were discarded. Each peak in the final list was examined by eye with MochiView, and particular care was taken to confirm the validity of peaks called by only two of three algorithms. For visualization and graphing purposes, BAM files were displayed on tracks with Integrative Genomics Viewer using BAMCoverage from DeepTools2 (90). Of the 295 peaks, 80% were called by all three peak-calling programs.
RNA isolation and RNA-Seq.
For RNA isolated from samples subjected to iron starvation, Y. pseudotuberculosis WT and ΔiscR strains were grown in 5 ml of M9+3.6 μM FeSO4 overnight. Cultures were then diluted to an OD600 of 0.1 in 20 ml of M9 medium lacking iron treated with Chelex (Bio-Rad) to remove trace amounts of iron and allowed to grow for 8 h at 26°C (12). The cultures were then diluted again to an OD600 of 0.1 into 20-ml Chelex-treated M9 medium and allowed to grow for 12 h at 26˚C. Cultures were then diluted to an OD600 of 0.1 into 20-ml Chelex-treated M9 medium with either no iron, 5 μM hemin, or 3.6 μM FeSO4. Stock hemin solutions were Chelex treated overnight. After 3 h of growth at 37°C, 5 ml of culture from each condition was pelleted by centrifugation for 5 min at 4,000 rpm. The supernatant was removed, and pellets were resuspended in 500 μl media and treated with 1 ml Bacterial RNA Protect reagent (Qiagen) according to the manufacturer’s protocol. Total RNA was isolated using the RNeasy minikit (Qiagen) per the manufacturer’s protocol. Contaminating DNA was removed using the TURBO DNA-free kit (Life Technologies/Thermo Fischer). rRNA was removed using the RiboMinus Transcriptome isolation kit bacteria (Invitrogen). The cDNA library was prepared using the NEB Ultra Directional RNA Library Prep kit for Illumina. The quality of RNA and cDNA libraries was assessed using an Agilent 2000 Bioanalyzer. Libraries were sequenced using the HiSeq2500 Illumina sequencing platform for 50-bp single reads (University of California [UC] Davis Genome Center).
Trimmomatic (82) version 0.36 was used to trim off low-quality bases and adapter sequences. Trimmed reads were then aligned to a concatenated chromosome (GenBank accession no. CP032566.1) and virulence plasmid (CP032567.1) through Bowtie2 (83). Low-quality aligned reads were removed by filtering out mapped reads with a mapQC score of <10. The BAM file with the aligned reads with a mapQC score of >10 was then split to separate chromosomal mapped reads from virulence plasmid mapped reads. Mapped reads were then normalized to trimmed mean of M-values (TMM), and differential expression analysis was performed using EdgeR (91). Genes were called differentially expressed if the log2 fold change (FC) was ≥1 or ≤−1 with an FDR value of <0.01.
Motif identification and in silico search.
IscR binding motif analyses were carried out using the MEME tool from the MEME software suite with default settings (41). The E. coli IscR type I and type II binding motifs were compiled from a training set consisting of known IscR binding sites in E. coli (25, 28). The FIMO tool from the MEME software suite was then used to search for an IscR type I binding motif upstream of Yersinia iscRSUA, cysE, erpA, and nfuA.
In order to identify a Yersinia IscR consensus binding site from sequences enriched during IscR-FLAG immunoprecipitation, 50 nucleotides within the center of all 176 enriched DNA regions were extracted, and the MEME suite was used to generate a Yersinia IscR type II motif. The CentiMo tool from the MEME suite was utilized to assess whether the predicted IscR type II consensus motif was often found near the center of each IscR ChIP-Seq peak.
Western blot analysis.
Bacterial pellets were resuspended in final sample buffer plus 0.2 M dithiothreitol (FSBS+DTT) and boiled for 15 min. At the time of loading, samples were normalized to the same number of cells by OD600. Protein samples were run on a 12.5% sodium dodecyl sulfate-polyacrylamide gel (SDS-PAG) and transferred to a blotting membrane (Immobilon-P) with a wet mini trans-blot cell (Bio-Rad). Blots were blocked for an hour in Tris-buffered saline with Tween 20 and 5% skim milk and probed with rabbit anti-IscR (28), rabbit anti-YopD (gift from Alison Davis and Joan Mecsas), goat anti-YopE (Santa Cruz Biotechnology), rabbit anti-RpoA (gift from Melanie Marketon), and horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotech). Gels were imaged by Image Lab software (Bio-Rad).
Type III secretion assay under non-iron-starved conditions.
Visualization of T3SS cargo secreted in broth culture was performed as previously described (92). For standard Y. pseudotuberculosis T3SS induction (Fig. S1B and Fig. S4C), bacteria were grown overnight in LB medium, subcultured in LB plus 20 mM sodium oxalate (to chelate calcium and induce type III secretion) and 20 mM MgCl2 to an OD600 of 0.2, grown at 26°C for 1.5 h followed by 37°C for another 1.5 h. Cultures were normalized by OD600 and pelleted at 13,200 rpm for 10 min at room temperature. Supernatants were removed, and proteins were precipitated by addition of trichloroacetic acid (TCA) to a final concentration of 10%. Samples were incubated on ice for 20 min and pelleted at 13,200 rpm for 15 min at 4°C. Resulting pellets were washed twice with ice-cold 100% acetone and subsequently resuspended in FSB+DTT. Samples were boiled for 5 min prior to running on a 12.5% SDS-PAG.
Type III secretion assay under iron-starved aerobic or anaerobic conditions.
We previously found that small amounts of iron are needed to obtain sufficient Yersinia growth under anaerobic conditions to detect T3SS activity (9); therefore, we added 0.036 μM to the “low iron” samples for these experiments rather than continuing to iron starve them. Cultures were grown aerobically in Chelex-treated M9 minimal medium plus 0.9% glucose in acid-washed glassware, as previously described (9). Specifically, iron-replete overnight cultures (M9+3.6 μM FeSO4) grown at 26°C aerobically were subcultured to an OD600 of 0.1 into Chelex-treated M9 medium plus 0.9% glucose and grown for 8 h at 26°C aerobically with agitation. Cultures were then subcultured a second time to OD600 0.1 in fresh Chelex-treated M9 media plus 0.9% glucose and grown for 12 h at 26°C with agitation. Subsequently, cultures were then subcultured a third time to an OD600 of 0.2 in M9 medium plus 0.9% glucose supplemented with 3.6 μM FeSO4 (iron replete) or with 0.036 μM FeSO4 (iron limitation), grown for 2 h at 26°C with agitation, and then shifted to 37°C for 4 h with agitation to induce type III secretion. For anaerobic cultures, the cultures were instead diluted a second time to an OD600 of 0.1 in M9 medium plus 0.9% glucose supplemented with 3.6 μM FeSO4 (iron replete) or with 0.036 μM FeSO4 (iron limitation), and transferred to a vinyl anaerobic chamber where they were grown at 26°C for 12 h. Cultures were then shifted to 37°C for another 4 h to induce type III secretion. Samples were then processed as described above for standard secretion assay conditions.
Quantitative PCR (qPCR) analysis.
A total of 5 ml of culture from each condition were pelleted by centrifugation for 5 min at 4,000 rpm (4 krpm). The supernatant was removed, and pellets were resuspended in 500 μl of media and treated with 1 ml Bacterial RNA Protect reagent (Qiagen) according to the manufacturer’s protocol. Total RNA was isolated using the RNeasy minikit (Qiagen) per the manufacturer’s protocol. After harvesting total RNA, genomic DNA was removed via the TURBO-DNA-free kit (Life Technologies/Thermo Fisher). cDNA was generated for each sample by using the Moloney murine leukemia virus (M-MLV) reverse transcriptase (Invitrogen) according to the manufacturer’s instructions, as we previously described (19). Power SYBR green PCR master mix (Thermo Fisher Scientific) and primers (Table 2) with optimized concentrations were used to measure target gene levels. The expression levels of each target gene were normalized to that of 16S rRNA present in each sample and calculated by the ΔΔCT method. Three independent biological replicates were collected for each tested condition. For each target transcript, significant differential expression between different bacterial strains was defined by P value of <0.05 of two-way analysis of variance (ANOVA) (one-way ANOVA with Tukey posttest).
Cluster of Orthologous Groups (COG) analysis.
The GenBank file of the Y. pseudotuberculosis chromosome (GenBank accession no. CP032566.1) and virulence plasmid (CP032567.1) were downloaded from NCBI. These files were opened using the Artemis Genome Browser (93), where the proteome was exported to a FASTA file. EggNOG 4.5.1 (94) was used to determine the following gene information based on ortholog databases: preferred gene name, COG category, and gene function.
Cluster analysis.
Cluster analysis was used to cluster gene expression data from the RNA-Seq experiments. The elbow method was first used to determine the appropriate number of clusters. Gene expression data were inputted as normalized reads (TMM) values. These gene expression values were then scaled by gene. R-package pheatmap was used to create clusters dependent on Euclidean distances and a complete method.
Protein purification and electrophoretic mobility shift assays (EMSAs).
The iscR coding sequence was PCR amplified from the E. coli K-12 MG1655 chromosome using primers that incorporated a NdeI restriction site at the 5′ end of the gene, and a BamHI site and His6 tag (order listed in 5′–3′ direction) at the 3′ end. The NdeI- and BamHI-digested fragment was cloned into pET11a to generate pPK14263, which was subsequently transformed into strain PK7878 (28). IscR-C92A-His6 was purified as previously described for untagged IscR (26, 28). DNA fragments containing the predicted IscR binding region for ail (−386 to +14 bp relative to the +1 transcription start site [TSS]), dusB-fis (−447 to −247 bp relative to the +1 TSS), sodB (−116 to +84 bp relative to the +1 TSS), katA1 (−240 to −93 bp relative to the +1 TSS), katA2 (−92 to +1 bp relative to the +1 TSS), and sufA (−144 to +56 bp relative to the +1 TSS) were amplified from Y. pseudotuberculosis genomic DNA using primers (Table 2). Amplified products were digested with XhoI and BamHI and subsequently ligated into the pPK7179 plasmid. DNA templates for EMSAs were isolated from plasmid DNA after restriction digestion with XhoI and BamHI. These fragments and linearized plasmid (which served as competitor DNA in the EMSAs) were purified using the QIAquick PCR purification kit (Qiagen). IscR-C92A was incubated with DNA fragments (∼5 to 10 nM) for 30 min at 37°C in 40 mM Tris (pH 7.9), 30 mM KCl, 100 μg/ml bovine serum albumin (BSA), and 1 mM DTT. Glycerol was added to 10%, and samples were loaded onto a nondenaturing 6% polyacrylamide gel in 0.5× Tris-borate-EDTA (TBE) buffer and run at 200 V for 3.5 h. The gel was stained with SYBR green EMSA nucleic acid gel stain (Molecular Probes) and visualized using a Typhoon FLA 900 imager (GE).
Comparative genomics.
Orthologs of the 213 IscR targets were determined in Y. pseudotuberculosis IP32953 (NC_006155.1), Y. pestis CO92 (NC_003143.1), and Y. enterocolitica 8081 (NC_008800.1) through a combination of BLAST and Mauve, a multiple genome alignment tool (95). Paralogs were avoided by using Mauve. For all orthologs, 1,000 nucleotides upstream and downstream of the start codon were extracted. An IscR binding site was predicted using MEME suite tools and was compared to the known IscR binding site in Y. pseudotuberculosis IP2666. The orthologous IscR binding site was identified based on sequence similarity and distance of the IscR binding site from the start codon. Sequence similarity was calculated by computing the absolute distance between logMEME-FIMO-pvalue in Y. pseudotuberculosis IP2666 (known IscR binding site) compared to logMEME-FIMO-pvalue of orthologous binding site in the indicated strain of Yersinia (binding site in question).
Motility assay.
A total of 1-μl Y. pseudotuberculosis overnight culture was spotted onto motility medium containing 1% tryptone/0.25% agar. The plates were incubated at 26°C for 24 h or 48 h before the diameter of the motile colony was measured.
Microbial genome browser.
Both ChIP-Seq and RNA-Seq tracks were deposited to the UCSC Microbial Genome browser. For RNA-Seq data, final BAM file alignments were converted to bigwig files using bamCoverage-deepTools (90). Chromosomal alignments were normalized by counts per million mapped reads (CPM), while alignments to the virulence plasmid were not normalized due to the known increase in plasmid copy number at 37°C relative to the chromosome (96). For ChIP-Seq data, both BAM files that were mapped to the chromosome or virulence plasmid were normalized by CPM. The RNA-Seq and ChIP-Seq data are available for viewing, and the track hub data can be found at http://zam.soe.ucsc.edu/hubs/StoneYersinia/hub.txt.
ACKNOWLEDGMENTS
This study was supported by National Institutes of Health (www.NIH.gov) grant R01AI119082 (to V.A. and P.J.K.). D.B. and P.A. received support from the National Human Genome Research Institute of the National Institutes of Health under award number 4R25HG006836. D.H.R. received support from the Ford Foundation (www.fordfoundation.org). We thank Patricia Chan and Todd Lowe for assistance with the Microbial Genome Browser. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Citation Balderas D, Mettert E, Lam HN, Banerjee R, Gverzdys T, Alvarez P, Saarunya G, Tanner N, Zoubedi A, Wei Y, Kiley PJ, Auerbuch V. 2021. Genome scale analysis reveals IscR directly and indirectly regulates virulence factor genes in pathogenic Yersinia. mBio 12:e00633-21. https://doi.org/10.1128/mBio.00633-21.
Contributor Information
Victoria Auerbuch, Email: vastone@ucsc.edu.
Gisela Storz, National Institute of Child Health and Human Development (NICHD).
REFERENCES
- 1.Heroven AK, Dersch P. 2014. Coregulation of host-adapted metabolism and virulence by pathogenic yersiniae. Front Cell Infect Microbiol 4:146. doi: 10.3389/fcimb.2014.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Litwin CM, Calderwood SB. 1993. Role of iron in regulation of virulence genes. Clin Microbiol Rev 6:137−149. doi: 10.1128/cmr.6.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carpenter C, Payne SM. 2014. Regulation of iron transport systems in Enterobacteriaceae in response to oxygen and iron availability. J Inorg Biochem 133:110−117. doi: 10.1016/j.jinorgbio.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rohmer L, Hocquet D, Miller SI. 2011. Are pathogenic bacteria just looking for food? Metabolism and microbial pathogenesis. Trends Microbiol 19:341−348. doi: 10.1016/j.tim.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Putzker M, Sauer H, Sobe D. 2001. Plague and other human infections caused by Yersinia species. Clin Lab 47:453−466. [PubMed] [Google Scholar]
- 6.Dube P. 2009. Interaction of Yersinia with the gut: mechanisms of pathogenesis and immune evasion. Curr Top Microbiol Immunol 337:61−91. doi: 10.1007/978-3-642-01846-6_3. [DOI] [PubMed] [Google Scholar]
- 7.Sun YC, Jarrett CO, Bosio CF, Hinnebusch BJ. 2014. Retracing the evolutionary path that led to flea-borne transmission of Yersinia pestis. Cell Host Microbe 15:578−586. doi: 10.1016/j.chom.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller HK, Schwiesow L, Au-Yeung W, Auerbuch V. 2016. Hereditary hemochromatosis predisposes mice to Yersinia pseudotuberculosis infection even in the absence of the type III secretion system. Front Cell Infect Microbiol 6:69. doi: 10.3389/fcimb.2016.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hooker-Romero D, Mettert E, Schwiesow L, Balderas D, Alvarez PA, Kicin A, Gonzalez AL, Plano GV, Kiley PJ, Auerbuch V. 2019. Iron availability and oxygen tension regulate the Yersinia Ysc type III secretion system to enable disseminated infection. PLoS Pathog 15:e1008001. doi: 10.1371/journal.ppat.1008001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quenee LE, Hermanas TM, Ciletti N, Louvel H, Miller NC, Elli D, Blaylock B, Mitchell A, Schroeder J, Krausz T, Kanabrocki J, Schneewind O. 2012. Hereditary hemochromatosis restores the virulence of plague vaccine strains. J Infect Dis 206:1050−1058. doi: 10.1093/infdis/jis433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thompson JM, Jones HA, Perry RD. 1999. Molecular characterization of the hemin uptake locus (hmu) from Yersinia pestis and analysis of hmu mutants for hemin and hemoprotein utilization. Infect Immun 67:3879−3892. doi: 10.1128/IAI.67.8.3879-3892.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwiesow L, Mettert E, Wei Y, Miller HK, Herrera NG, Balderas D, Kiley PJ, Auerbuch V. 2018. Control of hmu heme uptake genes in Yersinia pseudotuberculosis in response to iron sources. Front Cell Infect Microbiol 8:47. doi: 10.3389/fcimb.2018.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stojiljkovic I, Hantke K. 1992. Hemin uptake system of Yersinia enterocolitica: similarities with other TonB-dependent systems in gram-negative bacteria. EMBO J 11:4359−4367. doi: 10.1002/j.1460-2075.1992.tb05535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perry RD, Bobrov AG, Fetherston JD. 2015. The role of transition metal transporters for iron, zinc, manganese, and copper in the pathogenesis of Yersinia pestis. Metallomics 7:965–978. doi: 10.1039/C4MT00332B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Troxell B, Hassan HM. 2013. Transcriptional regulation by Ferric Uptake Regulator (Fur) in pathogenic bacteria. Front Cell Infect Microbiol 3:59. doi: 10.3389/fcimb.2013.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perry RD, Fetherston JD. 2011. Yersiniabactin iron uptake: mechanisms and role in Yersinia pestis pathogenesis. Microbes Infect 13:808−817. doi: 10.1016/j.micinf.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao H, Zhou D, Li Y, Guo Z, Han Y, Song Y, Zhai J, Du Z, Wang X, Lu J, Yang R. 2008. The iron-responsive fur regulon in Yersinia pestis. J Bacteriol 190:3063−3075. doi: 10.1128/JB.01910-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gemski P, Lazere JR, Casey T, Wohlhieter JA. 1980. Presence of a virulence-associated plasmid in Yersinia pseudotuberculosis. Infect Immun 28:1044−1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller HK, Kwuan L, Schwiesow L, Bernick DL, Mettert E, Ramirez HA, Ragle JM, Chan PP, Kiley PJ, Lowe TM, Auerbuch V. 2014. IscR is essential for Yersinia pseudotuberculosis type III secretion and virulence. PLoS Pathog 10:e1004194. doi: 10.1371/journal.ppat.1004194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vergnes A, Viala JPM, Ouadah-Tsabet R, Pocachard B, Loiseau L, Méresse S, Barras F, Aussel L. 2017. The iron–sulfur cluster sensor IscR is a negative regulator of Spi1 type III secretion system in Salmonella enterica. Cell Microbiol doi: 10.1111/cmi.12680. [DOI] [PubMed] [Google Scholar]
- 21.Choi G, Jang KK, Lim JG, Lee ZW, Im H, Choi SH. 2020. The transcriptional regulator IscR integrates host-derived nitrosative stress and iron starvation in activation of the vvhBA operon in Vibrio vulnificus. J Biol Chem 295:5350−5361. doi: 10.1074/jbc.RA120.012724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lim JG, Choi SH. 2014. IscR is a global regulator essential for pathogenesis of Vibrio vulnificus and induced by host cells. Infect Immun 82:569−578. doi: 10.1128/IAI.01141-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones-Carson J, Laughlin J, Hamad MA, Stewart AL, Voskuil MI, Vázquez-Torres A. 2008. Inactivation of [Fe-S] metalloproteins mediates nitric oxide-dependent killing of Burkholderia mallei. PLoS One 3:e1976. doi: 10.1371/journal.pone.0001976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi YS, Shin DH, Chung IY, Kim SH, Heo YJ, Cho YH. 2007. Identification of Pseudomonas aeruginosa genes crucial for hydrogen peroxide resistance. J Microbiol Biotechnol 17:1344−1352. [PubMed] [Google Scholar]
- 25.Schwartz CJ, Giel JL, Patschkowski T, Luther C, Ruzicka FJ, Beinert H, Kiley PJ. 2001. IscR, an Fe-S cluster-containing transcription factor, represses expression of Escherichia coli genes encoding Fe-S cluster assembly proteins. Proc Natl Acad Sci U S A 98:14895−14900. 10.1073/pnas.251550898. doi: 10.1073/pnas.251550898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giel JL, Rodionov D, Liu M, Blattner FR, Kiley PJ. 2006. IscR-dependent gene expression links iron-sulphur cluster assembly to the control of O2-regulated genes in Escherichia coli. Mol Microbiol 60:1058–1075. doi: 10.1111/j.1365-2958.2006.05160.x. [DOI] [PubMed] [Google Scholar]
- 27.Rajagopalan S, Teter SJ, Zwart PH, Brennan RG, Phillips KJ, Kiley PJ. 2013. Studies of IscR reveal a unique mechanism for metal-dependent regulation of DNA binding specificity. Nat Struct Mol Biol 20:740–747. doi: 10.1038/nsmb.2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nesbit AD, Giel JL, Rose JC, Kiley PJ. 2009. Sequence-specific binding to a subset of IscR-regulated promoters does not require IscR Fe-S cluster ligation. J Mol Biol 387:28−41. doi: 10.1016/j.jmb.2009.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Py B, Moreau PL, Barras F. 2011. Fe-S clusters, fragile sentinels of the cell. Curr Opin Microbiol 14:218−223. doi: 10.1016/j.mib.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 30.Miller HK, Auerbuch V. 2015. Bacterial iron-sulfur cluster sensors in mammalian pathogens. Metallomics 7:943–956. doi: 10.1039/C5MT00012B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giel JL, Nesbit AD, Mettert EL, Fleischhacker AS, Wanta BT, Kiley PJ. 2013. Regulation of iron-sulphur cluster homeostasis through transcriptional control of the Isc pathway by [2Fe-2S]-IscR in Escherichia coli. Mol Microbiol 87:478−492. doi: 10.1111/mmi.12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng M, Wang X, Templeton LJ, Smulski DR, LaRossa RA, Storz G. 2001. DNA microarray-mediated transcriptional profiling of the Escherichia coli response to hydrogen peroxide. J Bacteriol 183:4562−4570. doi: 10.1128/JB.183.15.4562-4570.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Outten FW, Djaman O, Storz G. 2004. A suf operon requirement for Fe-S cluster assembly during iron starvation in Escherichia coli. Mol Microbiol 52:861−872. doi: 10.1111/j.1365-2958.2004.04025.x. [DOI] [PubMed] [Google Scholar]
- 34.Imlay JA. 2006. Iron-sulphur clusters and the problem with oxygen. Mol Microbiol 59:1073–1082. doi: 10.1111/j.1365-2958.2006.05028.x. [DOI] [PubMed] [Google Scholar]
- 35.Yeo WS, Lee JH, Lee KC, Roe JH. 2006. IscR acts as an activator in response to oxidative stress for the suf operon encoding Fe-S assembly proteins. Mol Microbiol 61:206−218. doi: 10.1111/j.1365-2958.2006.05220.x. [DOI] [PubMed] [Google Scholar]
- 36.Balada-Llasat JM, Mecsas J. 2006. Yersinia has a tropism for B and T cell zones of lymph nodes that is independent of the type III secretion system. PLoS Pathog 2:e86. doi: 10.1371/journal.ppat.0020086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crimmins GT, Mohammadi S, Green ER, Bergman MA, Isberg RR, Mecsas J. 2012. Identification of MrtAB, an ABC transporter specifically required for Yersinia pseudotuberculosis to colonize the mesenteric lymph nodes. PLoS Pathog 8:e1002828. doi: 10.1371/journal.ppat.1002828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simonet M, Mazigh D, Berche P. 1984. Growth of Yersinia pseudotuberculosis in mouse spleen despite loss of a virulence plasmid of mol. wt 47 x 106. J Med Microbiol 18:371−375. doi: 10.1099/00222615-18-3-371. [DOI] [PubMed] [Google Scholar]
- 39.Nuss AM, Beckstette M, Pimenova M, Schmühl C, Opitz W, Pisano F, Heroven AK, Dersch P. 2017. Tissue dual RNA-seq allows fast discovery of infection-specific functions and riboregulators shaping host-pathogen transcriptomes. Proc Natl Acad Sci U S A 114:E791−E800. doi: 10.1073/pnas.1613405114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sebbane F, Lemaître N, Sturdevant DE, Rebeil R, Virtaneva K, Porcella SF, Hinnebusch BJ. 2006. Adaptive response of Yersinia pestis to extracellular effectors of innate immunity during bubonic plague. Proc Natl Acad Sci U S A 103:11766−11771. doi: 10.1073/pnas.0601182103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME Suite: tools for motif discovery and searching. Nucleic Acids Res 37:W202−W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Somprasong N, Jittawuttipoka T, Duang-Nkern J, Romsang A, Chaiyen P, Schweizer HP, Vattanaviboon P, Mongkolsuk S. 2012. Pseudomonas aeruginosa thiol peroxidase protects against hydrogen peroxide toxicity and displays atypical patterns of gene regulation. J Bacteriol 194:3904−3912. doi: 10.1128/JB.00347-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim SH, Lee BY, Lau GW, Cho YH. 2009. IscR modulates catalase A (KatA) activity, peroxide resistance, and full virulence of Pseudomonas aeruginosa PA14. J Microbiol Biotechnol 19:1520−1526. doi: 10.4014/jmb.0906.06028. [DOI] [PubMed] [Google Scholar]
- 44.Green ER, Clark S, Crimmins GT, Mack M, Kumamoto CA, Mecsas J. 2016. Fis is essential for Yersinia pseudotuberculosis virulence and protects against reactive oxygen species produced by phagocytic cells during infection. PLoS Pathog 12:e1005898. doi: 10.1371/journal.ppat.1005898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamashita S, Lukacik P, Barnard TJ, Noinaj N, Felek S, Tsang TM, Krukonis ES, Hinnebusch BJ, Buchanan SK. 2011. Structural insights into ail-mediated adhesion in Yersinia pestis. Structure 19:1672–1682. doi: 10.1016/j.str.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pierson DE, Falkow S. 1993. The ail gene of Yersinia enterocolitica has a role in the ability of the organism to survive serum killing. Infect Immun 61:1846−1852. doi: 10.1128/IAI.61.5.1846-1852.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Michiels T, Vanooteghem JC, Lambert de Rouvroit C, China B, Gustin A, Boudry P, Cornelis GR. 1991. Analysis of virC, an operon involved in the secretion of Yop proteins by Yersinia enterocolitica. J Bacteriol 173:4994−5009. doi: 10.1128/jb.173.16.4994-5009.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schwiesow L, Lam H, Dersch P, Auerbuch V. 2015. Yersinia type III secretion system master regulator LcrF. J Bacteriol 198:604−614. doi: 10.1128/JB.00686-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kleinman CL, Sycz G, Bonomi HR, Rodriguez RM, Zorreguieta A, Sieira R. 2017. ChIP-seq analysis of the LuxR-type regulator VjbR reveals novel insights into the Brucella virulence gene expression network. Nucleic Acids Res 45:5757−5769. doi: 10.1093/nar/gkx165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choudhary KS, Kleinmanns JA, Decker K, Sastry AV, Gao Y, Szubin R, Seif Y, Palsson BO. 2020. Elucidation of regulatory modes for five two-component systems in Escherichia coli reveals novel relationships. mSystems 5:e00980-20. [CrossRef] doi: 10.1128/mSystems.00980-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee JH, Yeo WS, Roe JH. 2003. Regulation of the sufABCDSE operon by Fur. J Microbiol 41:109−114. [Google Scholar]
- 52.Lee KC, Yeo WS, Roe JH. 2008. Oxidant-responsive induction of the suf operon, encoding a Fe-S assembly system, through Fur and IscR in Escherichia coli. J Bacteriol 190:8244−8247. doi: 10.1128/JB.01161-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Palmer LD, Skaar EP. 2016. Transition metals and virulence in bacteria. Annu Rev Genet 50:67−91. doi: 10.1146/annurev-genet-120215-035146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lim JG, Bang YJ, Choi SH. 2014. Characterization of the Vibrio vulnificus 1-Cys peroxiredoxin prx3 and regulation of its expression by the Fe-S cluster regulator IscR in response to oxidative stress and iron starvation. J Biol Chem 289:36263−36274. doi: 10.1074/jbc.M114.611020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Santos JA, Pereira PJB, Macedo-Ribeiro S. 2015. What a difference a cluster makes: the multifaceted roles of IscR in gene regulation and DNA recognition. Biochim Biophys Acta 1854:1101−1112. doi: 10.1016/j.bbapap.2015.01.010. [DOI] [PubMed] [Google Scholar]
- 56.Rebeil R, Ernst RK, Jarrett CO, Adams KN, Miller SI, Hinnebusch BJ. 2006. Characterization of late acyltransferase genes of Yersinia pestis and their role in temperature-dependent lipid A variation. J Bacteriol 188:1381−1388. doi: 10.1128/JB.188.4.1381-1388.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krasikova IN, Khotimchenko SV, Solov’eva TF, Ovodov YS. 1995. Mutual influence of plasmid profile and growth temperature on the lipid composition of Yersinia pseudotuberculosis bacteria. Biochim Biophys Acta 1257:118−124. doi: 10.1016/0005-2760(95)00061-g. [DOI] [PubMed] [Google Scholar]
- 58.Kawahara K, Tsukano H, Watanabe H, Lindner B, Matsuura M. 2002. Modification of the structure and activity of lipid A in Yersinia pestis lipopolysaccharide by growth temperature. Infect Immun 70:4092−4098. doi: 10.1128/iai.70.8.4092-4098.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Montminy SW, Khan N, McGrath S, Walkowicz MJ, Sharp F, Conlon JE, Fukase K, Kusumoto S, Sweet C, Miyake K, Akira S, Cotter RJ, Goguen JD, Lien E. 2006. Virulence factors of Yersinia pestis are overcome by a strong lipopolysaccharide response. Nat Immunol 7:1066–1073. doi: 10.1038/ni1386. [DOI] [PubMed] [Google Scholar]
- 60.Dalebroux ZD, Miller SI. 2014. Salmonellae PhoPQ regulation of the outer membrane to resist innate immunity. Curr Opin Microbiol 17:106−113. doi: 10.1016/j.mib.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bishop RE, Gibbons HS, Guina T, Trent MS, Miller SI, Raetz CRH. 2000. Transfer of palmitate from phospholipids to lipid A in outer membranes of Gram-negative bacteria. EMBO J 19:5071−5080. doi: 10.1093/emboj/19.19.5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kawasaki K, Ernst RK, Miller SI. 2004. Deacylation and palmitoylation of lipid A by Salmonellae outer membrane enzymes modulate host signaling through Toll-like receptor 4. J Endotoxin Res 10:439−444. doi: 10.1179/096805104225006264. [DOI] [PubMed] [Google Scholar]
- 63.Bishop RE. 2005. The lipid A palmitoyltransferase PagP: molecular mechanisms and role in bacterial pathogenesis. Mol Microbiol 57:900−912. doi: 10.1111/j.1365-2958.2005.04711.x. [DOI] [PubMed] [Google Scholar]
- 64.Chandler CE, Harberts EM, Pelletier MR, Thaipisuttikul I, Jones JW, Hajjar AM, Sahl JW, Goodlett DR, Pride AC, Rasko DA, Trent MS, Bishop RE, Ernst RK. 2020. Early evolutionary loss of the lipid A modifying enzyme PagP resulting in innate immune evasion in Yersinia pestis. Proc Natl Acad Sci U S A 117:22984−22991. doi: 10.1073/pnas.1917504117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang P, Ye Z, Ye C, Zou H, Gao Z, Pan J. 2020. OmpW is positively regulated by iron via Fur, and negatively regulated by SoxS contribution to oxidative stress resistance in Escherichia coli. Microb Pathog 138:103808. doi: 10.1016/j.micpath.2019.103808. [DOI] [PubMed] [Google Scholar]
- 66.Wilharm G, Lehmann V, Krauss K, Lehnert B, Richter S, Ruckdeschel K, Heesemann J, Trülzsch K. 2004. Yersinia enterocolitica type III secretion depends on the proton motive force but not on the flagellar motor components MotA and MotB. Infect Immun 72:4004−4009. doi: 10.1128/IAI.72.7.4004-4009.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zheng L, Kelly CJ, Colgan SP. 2015. Physiologic hypoxia and oxygen homeostasis in the healthy intestine. A review in the theme: cellular responses to hypoxia. Am J Physiol Cell Physiol 309:C350−C360. doi: 10.1152/ajpcell.00191.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jacobi CA, Gregor S, Rakin A, Heesemann J. 2001. Expression analysis of the yersiniabactin receptor gene fyuA and the heme receptor hemR of Yersinia enterocolitica in vitro and in vivo using the reporter genes for green fluorescent protein and luciferase. Infect Immun 69:7772−7782. doi: 10.1128/IAI.69.12.7772-7782.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Avican K, Fahlgren A, Huss M, Heroven AK, Beckstette M, Dersch P, Fällman M. 2015. Reprogramming of Yersinia from virulent to persistent mode revealed by complex in vivo RNA-seq analysis. PLoS Pathog 11:e1004600. doi: 10.1371/journal.ppat.1004600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sastry AV, Gao Y, Szubin R, Hefner Y, Xu S, Kim D, Choudhary KS, Yang L, King ZA, Palsson BO. 2019. The Escherichia coli transcriptome mostly consists of independently regulated modules. Nat Commun 10:5536. doi: 10.1038/s41467-019-13483-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miller VL, Falkow S. 1988. Evidence for two genetic loci in Yersinia enterocolitica that can promote invasion of epithelial cells. Infect Immun 56:1242−1248. doi: 10.1128/IAI.56.5.1242-1248.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kirjavainen V, Jarva H, Biedzka-Sarek M, Blom AM, Skurnik M, Meri S. 2008. Yersinia enterocoliticaserum resistance proteins YadA and Ail bind the complement regulator C4b-binding protein. PLoS Pathog 4:e1000140. doi: 10.1371/journal.ppat.1000140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Duprey A, Reverchon S, Nasser W. 2014. Bacterial virulence and Fis: adapting regulatory networks to the host environment. Trends Microbiol 22:92−99. doi: 10.1016/j.tim.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 74.Ó Cróinín T, Carroll RK, Kelly A, Dorman CJ. 2006. Roles for DNA supercoiling and the Fis protein in modulating expression of virulence genes during intracellular growth of Salmonella enterica serovar Typhimurium. Mol Microbiol 62:869−882. doi: 10.1111/j.1365-2958.2006.05416.x. [DOI] [PubMed] [Google Scholar]
- 75.Ó Cróinín T, Dorman CJ. 2007. Expression of the Fis protein is sustained in late-exponential- and stationary-phase cultures of Salmonella enterica serovar Typhimurium grown in the absence of aeration. Mol Microbiol 66:237–251. doi: 10.1111/j.1365-2958.2007.05916.x. [DOI] [PubMed] [Google Scholar]
- 76.Banerjee R, Weisenhorn E, Schwartz KJ, Myers KS, Glasner JD, Perna NT, Coon JJ, Welch RA, Kiley PJ. 2020. Tailoring a global iron regulon to a uropathogen. mBio 11:e00351-20. doi: 10.1128/mBio.00351-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McNally A, Thomson NR, Reuter S, Wren BW. 2016. ‘Add, stir and reduce’: Yersinia spp. as model bacteria for pathogen evolution. Nat Rev Microbiol 14:177–190. doi: 10.1038/nrmicro.2015.29. [DOI] [PubMed] [Google Scholar]
- 78.Cheng LW, Anderson DM, Schneewind O. 1997. Two independent type III secretion mechanisms for YopE in Yersinia enterocolitica. Mol Microbiol 24:757−765. doi: 10.1046/j.1365-2958.1997.3831750.x. [DOI] [PubMed] [Google Scholar]
- 79.Warrens AN, Jones MD, Lechler RI. 1997. Splicing by over-lap extension by PCR using asymmetric amplification: an improved technique for the generation of hybrid proteins of immunological interest. Gene 186:29–35. doi: 10.1016/S0378-1119(96)00674-9. [DOI] [PubMed] [Google Scholar]
- 80.Myers KS, Yan H, Ong IM, Chung D, Liang K, Tran F, Keleş S, Landick R, Kiley PJ. 2013. Genome-scale analysis of Escherichia coli FNR reveals complex features of transcription factor binding. PLoS Genet 9:e1003565. doi: 10.1371/journal.pgen.1003565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Andrews S. 2015. FASTQC: a quality control tool for high throughput sequence data. Babraham Institute, Cambridge, United Kingdom.
- 82.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Valouev A, Johnson DS, Sundquist A, Medina C, Anton E, Batzoglou S, Myers RM, Sidow A. 2008. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat Methods 5:829–834. doi: 10.1038/nmeth.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kuan PF, Chung D, Pan G, Thomson JA, Stewart R, Keleş S. 2011. A statistical framework for the analysis of ChIP-Seq data. J Am Stat Assoc 106:891−903. doi: 10.1198/jasa.2011.ap09706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, Shirley XS. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Homann OR, Johnson AD. 2010. MochiView: versatile software for genome browsing and DNA motif analysis. BMC Biol 8:49. doi: 10.1186/1741-7007-8-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Milne I, Stephen G, Bayer M, Cock PJA, Pritchard L, Cardle L, Shawand PD, Marshall D. 2013. Using tablet for visual exploration of second-generation sequencing data. Brief Bioinform 14:193−202. doi: 10.1093/bib/bbs012. [DOI] [PubMed] [Google Scholar]
- 90.Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T. 2016. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44:W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kwuan L, Adams W, Auerbuch V. 2013. Impact of host membrane pore formation by the Yersinia pseudotuberculosis type III secretion system on the macrophage innate immune response. Infect Immun 81:905−914. doi: 10.1128/IAI.01014-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Carver T, Harris SR, Berriman M, Parkhill J, McQuillan JA. 2012. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28:464–469. doi: 10.1093/bioinformatics/btr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Huerta-Cepas J, Szklarczyk D, Forslund K, Cook H, Heller D, Walter MC, Rattei T, Mende DR, Sunagawa S, Kuhn M, Jensen LJ, Von Mering C, Bork P. 2016. EGGNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res 44:D286–D293. doi: 10.1093/nar/gkv1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Darling ACE, Mau B, Blattner FR, Perna NT. 2004. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14:1394−1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang H, Avican K, Fahlgren A, Erttmann SF, Nuss AM, Dersch P, Fallman M, Edgren T, Wolf-Watz H. 2016. Increased plasmid copy number is essential for Yersinia T3SS function and virulence. Science 353:492–495. doi: 10.1126/science.aaf7501. [DOI] [PubMed] [Google Scholar]
- 97.Bliska JB, Guan K, Dixon JE, Falkow S. 1991. Tyrosine phosphate hydrolysis of host proteins by an essential Yersinia virulence determinant. Proc Natl Acad Sci U S A 88:1187−1191. doi: 10.1073/pnas.88.4.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Morgan JM, Duncan MC, Johnson KS, Diepold A, Lam H, Dupzyk AJ, Martin LR, Wong WR, Armitage JP, Linington RG, Auerbuch V. 2017. Piericidin A1 blocks Yersinia Ysc type III secretion system needle assembly. mSphere 2:e00030-17. [CrossRef] doi: 10.1128/mSphere.00030-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transcriptomic data of the Y. pseudotuberculosis wild type and the ΔiscR mutant under both iron-starved and non-iron-starved conditions. Download Data Set S1, XLSX file, 3.6 MB (3.6MB, xlsx) .
Copyright © 2021 Balderas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
3xFLAG-tagged IscR rescues an iscR deletion. (A) Whole-cell extracts from WT Y. pseudotuberculosis or a strain harboring a chromosomally encoded 3xFLAG-tagged IscR were visualized using anti-FLAG or anti-RpoA antibodies. (B) To measure the relative efficiency of the Ysc T3SS, Yersinia strains were grown under T3SS-inducing conditions, and secreted proteins precipitated by trichloroacetic acid were visualized using Coomassie blue. Relative amounts of the T3SS effector protein YopE were quantified by densitometry compared to a spiked-in BSA protein control. The averages of three biological replicates ± standard deviations are shown. (C) Yersinia strains were grown under T3SS-inducing conditions, and relative iscS mRNA levels were evaluated by qPCR and normalized to 16s rRNA. The average of three biological replicates ± standard deviation is shown. ***, P < 0.001; **, P < 0.01; n.s., not significant (one-way ANOVA with Dunnett’s post hoc test). Download FIG S1, PDF file, 0.2 MB (219.5KB, pdf) .
Copyright © 2021 Balderas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
ChIP-Seq data assessing genome wide enrichment of IscR binding sites in Y. pseudotuberculosis. Download Data Set S2, XLSX file, 0.1 MB (147.6KB, xlsx) .
Copyright © 2021 Balderas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Global IscR ChIP-seq analysis fails to identify predicted IscR type I sites. (A) Known E. coli IscR type I binding sites were used to generate an IscR type I binding motif using MEME suite tools. The Y. pseudotuberculosis IP2666 genome was scanned for IscR type I sites using FIMO specifically upstream of the nfuA, iscRSUA, erpA, and DN756_20960_cysE promoters. The predicted sequences were aligned to the E. coli consensus IscR type I binding motif. (B) IscR ChIP-seq plots illustrating read coverage of IscR binding peaks assigned to the promoter of nfuA, iscRSUA, erpA, and DN756_20960_cysE from all three replicates combined. Dashed lines correspond to the zenith of the predicted 29-bp IscR type I motifs. Download FIG S2, PDF file, 0.1 MB (128.4KB, pdf) .
Copyright © 2021 Balderas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Conservation of IscR binding sites in Y. pseudotuberculosis (IP2666, IP32953), Y. pestis (CO92), and Y. enterocolitica (8081). (A) Distance of the identified IscR binding site in Y. pseudotuberculosis (IP2666) from the start codon of each transcription unit member of the IscR regulon is plotted versus the distance of the predicted IscR binding site from the start codon of the identified ortholog in Y. pseudotuberculosis (IP32953), Y. pestis (CO92), or Y. enterocolitica (8081). (B) The log10 FIMO P value of the identified IscR binding site in Y. pseudotuberculosis (IP2666) is plotted versus the log10 FIMO P value of the predicted IscR binding site upstream of identified orthologs in Y. pseudotuberculosis (IP32953), Y. pestis (CO92), or Y. enterocolitica (8081). Download FIG S3, PDF file, 0.3 MB (297.9KB, pdf) .
Copyright © 2021 Balderas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Comparative genomics of IscR binding sites in Y. pseudotuberculosis IP2666, Y. pseudotuberculosis IP32953, Y. pestis CO92, and Y. enterocolitica 8081. Download Data Set S3, XLSX file, 0.08 MB (82.9KB, xlsx) .
Copyright © 2021 Balderas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Deletion of DN756_21815 and DN756_21820, identified gene targets of IscR, results in a small but significant decrease in type III secretion. (A) Read coverage of IscR-binding peaks proximal to DN756_21815_DN756_21820. Counts per million reads (CPM) are plotted versus the genomic position. Dashed lines correspond to the predicted 30-bp IscR type II motif. Arrows indicate transcriptional start sites (48). (B) Expression of DN756_21815, and DN756_21820 genes under various iron conditions as measured by RNA-seq. Reads are represented by trimmed mean of M-values (TMM) of WT (black) and ΔiscR strains (gray) grown in M9 minimal medium containing FeSO4 (non-iron-starved [NIS]), iron starved in Chelex-treated M9 minimal medium with no iron source added back (Chelex) or supplemented with 5 mM hemin (+Heme) or FeSO4 (+FeSO4). **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (EdgeR with a corrected FDR post hoc test). (C) Yersinia strains were grown under rich media, T3SS-inducing conditions. The secretome of these cultures was visualized with Coomassie blue. The effector protein, YopE, was quantified by densitometry relative to the WT control to measure the relative efficiency of the Ysc T3SS. The average of five biological replicates ± standard deviation is shown. ****, P < 0.0001; *, P < 0.05 (one-way ANOVA with Dunnett’s post hoc test). Download FIG S4, PDF file, 0.3 MB (286.6KB, pdf) .
Copyright © 2021 Balderas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
COG analysis of the IscR regulon, functional regulon, and indirect regulon. Clusters of Orthologous Groups of proteins (COG) analysis of genes with an IscR ChIP-Seq peak (A), genes with an IscR ChIP-Seq peak and are differentially expressed between WT and the iscR mutant under at least one tested condition (B), and genes differentially expressed between WT and the iscR mutant but have no IscR ChIP-Seq peak (C). Download FIG S5, PDF file, 0.3 MB (294.1KB, pdf) .
Copyright © 2021 Balderas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Expression of the suf operon is modulated by iron and requires IscR. Expression of sufA, sufB, sufC, sufD, sufS, and sufE genes under various iron conditions as measured by RNA-seq. Reads are represented by trimmed mean of M-values (TMM) of WT (black) and ΔiscR strains (gray) grown in M9 minimal medium containing 3.6 μM FeSO4 (non-iron-starved [NIS]), iron starved in Chelex-treated M9 minimal medium with no iron source added back (Chelex) or supplemented with 5 mM hemin (+Heme) or 3.6 μM FeSO4 (+FeSO4).**, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (EdgeR with a corrected FDR post hoc test). Download FIG S6, PDF file, 0.1 MB (146.5KB, pdf) .
Copyright © 2021 Balderas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The Suf pathway does not affect motility or growth. (A) Iron-starved Y. pseudotuberculosis were either kept in iron-starved Chelex-treated M9 minimal medium with no iron source added back (Chelex) or supplemented with 3.6 μM FeSO4 (+FeSO4). Cultures were grown at 26°C for 10 h, and optical density at 600 nm was measured every hour. Non-iron-starved (NIS) samples were never exposed to Chelex-treated M9. (B) The indicated strains were spotted onto motility agar (1% tryptone, 0.25% agar) and were grown at 26°C. The diameters of the colonies were measured 24 h and 48 h later and used to calculate percent motility. Graphs represent three biological replicates. ****, P < 0.0001 (one-way ANOVA with Dunnett’s post hoc test). (C) Expression of lcrF, iscR, and yopE, relative to 16S rRNA, following iron-starved conditions in M9 minimal medium at 37°C, as measured by qPCR. Black bars represent cultures which were iron starved and supplemented with 3.6 μM FeSO4 before induction of the type III secretion system, while gray bars represent cultures that received 0.036 μM FeSO4 before induction of the type III secretion system. Average of three independent experiments ± standard deviation is shown. ***, P < 0.001; **, P < 0.01; *, P < 0.05 (one-way ANOVA with Dunnett’s post hoc test). Download FIG S7, PDF file, 0.2 MB (213.5KB, pdf) .
Copyright © 2021 Balderas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.