

Abstract
The integrity and function of mitochondria are essential for normal kidney physiology. Mitochondrial DNA (mtDNA) has been widely a concern in recent years because its abnormalities may result in disruption of aerobic respiration, cellular dysfunction, and even cell death. Particularly, aberrant mtDNA copy number (mtDNA-CN) is associated with the development of acute kidney injury and chronic kidney disease, and urinary mtDNA-CN shows the potential to be a promising indicator for clinical diagnosis and evaluation of kidney function. Several lines of evidence suggest that mtDNA may also trigger innate immunity, leading to kidney inflammation and fibrosis. In mechanism, mtDNA can be released into the cytoplasm under cell stress and recognized by multiple DNA-sensing mechanisms, including Toll-like receptor 9 (TLR9), cytosolic cGAS-stimulator of interferon genes (STING) signaling, and inflammasome activation, which then mediate downstream inflammatory cascades. In this review, we summarize the characteristics of these mtDNA-sensing pathways mediating inflammatory responses and their role in the pathogenesis of acute kidney injury, nondiabetic chronic kidney disease, and diabetic kidney disease. In addition, we highlight targeting of mtDNA-mediated inflammatory pathways as a novel therapeutic target for these kidney diseases.
1. Introduction
Mitochondria are double membrane-bound organelles that appear in nearly all eukaryotic cells. In addition to adenosine triphosphate (ATP) production, mitochondria participate in multiple physiological processes, such as heat production, redox homeostasis, calcium signaling, cell growth and death pathway, and antimicrobial immunity [1, 2]. Considering its essential role in providing energy, the integrity and normal function of mitochondria are crucial for the normal function of cells, especially in organs that need a lot of energy, such as the heart and kidney. When the mitochondria are injured, a variety of mitochondrial components will be released into the cytoplasm or extracellular environment and recognized as damage-associated molecular patterns (DAMPs) by pattern recognition receptors (PRRs), thus promoting downstream proinflammatory responses [3, 4]. Although many other mitochondrial components such as N-formyl peptides, ATP, and cardiolipin can act as mitochondrial DAMPs, we focus on mitochondrial DNA (mtDNA) in this review.
mtDNA derives from ancestral bacterial genome and has a double-strand circular structure, 16.5 kb in length. The copy number of mtDNA varies among different cell types, ranging from 100 to 10000 [5]. Mammalian mtDNA encodes 11 messenger RNAs which can be translated to 13 proteins forming four oxidative phosphorylation (OXPHOS) complexes [6]. Although a delicate quality control system has evolved to maintain mitochondrial homeostasis [2], mtDNA is particularly vulnerable to oxidative damage compared with nuclear DNA, due to its subcellular localization close to the electron transport chain where ROS is generated and the lack of protective histones. Mitochondrial genome damage or mutation may lead to aerobic respiration disruption, cellular dysfunction, and even cell death. Accumulating evidence suggests that mtDNA may contribute to the activation of innate immune response which acts as the central hub of the pathogenesis of many diseases [7].
Acute kidney injury (AKI) is characterized by a rapid decline of kidney function, which may progress to chronic kidney disease (CKD) and end-stage renal disease (ESRD). It remains a global challenge for its high morbidity and mortality [8, 9]. The main causes of AKI include renal ischemia, sepsis, and nephrotoxicity. The pathogenesis of AKI and CKD is still unclear, although a key role of prolonged or excessive inflammation has been recognized for a long time. In mechanism, tubular epithelial cell damage has been found pivotal to initiate the inflammatory response via activating resident immune cells such as macrophages and infiltrating leukocytes in the kidney, which release inflammatory mediators to amplify the cascades [10–13]. Moreover, recent studies have demonstrated that mtDNA-associated inflammatory responses were implicated in the pathogenesis of AKI and progression of CKD [14–16].
In this review, we summarize the current understanding of how abnormal mtDNA drives innate immunity and of its role in renal inflammation and in the development of several kidney diseases, including AKI, nondiabetic CKD, and diabetic kidney disease (DKD). In addition, we highlighted the potential of mtDNA as a new indicator as well as a putative therapeutic target for these disorders.
2. mtDNA-Sensing Mechanisms
Although mtDNA is an inherent component of the mitochondria, it can be recognized in the cytosol to trigger innate immunity by different mechanisms because it is relatively isolated, as shown in Figure 1. Moreover, growing evidence also suggests that underlying interactions exist between these DNA-sensing pathways.
Figure 1.
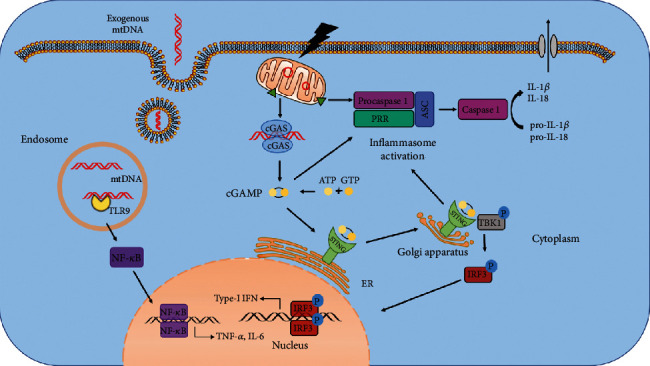
Overview of mtDNA-sensing pathways mediating inflammatory cascades. In conditions of cell injury or stress, aberrant mtDNA is released out of the mitochondria and is recognized by three major sensors to drive innate immune responses. Firstly, TLR9 binds to and is activated by mtDNA in the endosome facilitating downstream NF-κB, leading to upregulated expression of proinflammatory cytokines such as TNF-α and IL-6. Cytosolic mtDNA is also recognized by cGAS which results in the translocation of STING from the ER to the Golgi apparatus, resulting in TBK1-IRF3 activation and increased type-I IFN expression. In addition, mislocalized mtDNA also activates PRRs like NLRP3, which recruits ASC and procaspase 1 to form the inflammasome and contributes to cleavage of IL-1β and IL-18 to their mature forms. ASC, adaptor protein of apoptosis-associated speck-like protein containing a caspase recruitment domain; ATP, adenosine triphosphate; cGAMP, cyclic guanosine monophosphate-adenosine monophosphate; cGAS, cyclic GAS; ER, endoplasmic reticulum; GTP, guanosine triphosphate; IFN, interferon; IRF3, IFN regulatory factor 3; mtDNA, mitochondrial DNA; PRR, pattern recognition receptor; STING, stimulator of interferon genes; TBK1, TANK-binding kinase 1; TLR9, Toll-like receptor 9.
2.1. mtDNA and Toll-Like Receptor 9 (TLR9)
Toll-like receptors (TLRs) belong to highly conserved PRRs that play essential roles in recognizing pathogen-associated molecular patterns (PAMPs) and in triggering innate immune responses and inflammatory cascades [17–19]. Among them, TLR9 has been shown pivotal in sensing bacterial DNA, particularly, unmethylated cytosin-guanosin dinucleotide (CpG) DNA, to provoke innate immunity [20, 21]. Mechanistically, the binding of TLR9 to bacterial CpG DNA is preceded by the endocytosis of foreign bacteria and subsequent translocation of TLR9 from the endoplasmic reticulum to the endocytic vesicles [19, 22, 23]. Evolutionarily derived from bacterial DNA, mtDNA thereby retains unmethylated CpG motifs as well as the ability to activate the TLR9 pathway in a parallel way [24].
The interaction between mtDNA and TLR9 has been shown to be involved in the development of a variety of disorders, such as acute myocardial infarction [25], hepatocellular carcinoma [26, 27], nonalcoholic steatohepatitis [28, 29], and sterile lung injury [30, 31]. Typically, mislocated mtDNA activates TLR9 signaling and downstream myeloid differentiation factor 88 (MyD88), resulting in upregulated expression of nuclear factor- (NF-) κB and other proinflammatory factors such as tumor necrosis factor-α and interleukin 6, to amplify inflammation and exaggerate cell damage [31]. Besides, circulating mtDNA was reported to stimulate TLR9 in polymorphonuclear neutrophils, then facilitate p38-MAPK pathway and contribute to neutrophil secretion [4].
2.2. mtDNA and cGAS-STING Signaling Pathway
Over the past few years, cyclic guanosine monophosphate- (GMP-) adenosine monophosphate (AMP) (cGAMP) synthase (cGAS) has been identified as an important cytosolic DNA sensor which can elicit type-I interferon (IFN) signaling in mammalian cells. Under the condition of cell stress or cell injury, self-DNA from the nucleus or the mitochondria may leak into the cytosol and sensitize cGAS, which further converts ATP and GTP to cyclic GAMP, a second messenger mediating the activation of stimulator of interferon genes (STING). Thereafter, stimulated STING traffics from the endoplasmic reticulum membrane to the Golgi apparatus and interacts with IκB kinase- (IKK-) related kinase TANK-binding kinase 1 (TBK1), which phosphorylates IFN regulatory factor 3 (IRF3) to induce type-I IFN expression [32–35]. The cGAS-STING signaling pathway has been extensively recognized as the predominant pathway of DNA sensing and immune defense in a number of infectious diseases caused by various pathogens. Besides, cGAS acts as the first line of antitumor defense since it can sense the cytosolic DNA of antigen-presenting cells or tumor cells and trigger an antitumor immune response. Cellular senescence, autoimmune diseases, and heart failure are also associated with self-DNA-mediated cGAS-STING activation [36, 37].
Cytosolic mtDNA is one of the main causes for the activation of the cGAS-STING pathway. To date, it has been widely accepted that Bak/Bax-dependent mitochondrial outer membrane permeabilization (MOMP) initiates the release of mtDNA and thus contributes to cGAS-STING-mediated DNA-sensing pathway. In 2018, an Australian group using live-cell lattice light-sheet microscopy found that Bak/Bax pores forming on the outer membrane led to extrusion of the inner mitochondrial membrane into the cytosol, which carries the mitochondrial genome [38]. Later, a group in UK using superresolution imaging showed that during cell death, mitochondrial inner membrane permeabilization (MIMP) occurred following MOMP and allowed mtDNA efflux [39]. On the other hand, a recent study showed that in nonapoptotic cells, small mtDNA fragments were released through pores in the mitochondrial outer membrane (MOM) formed by voltage-dependent anion channel (VDAC) oligomers. Under moderate oxidative stress, the negatively charged backbone of mtDNA interacts directly with positively charged N-terminal domain of VDAC1 to facilitate VDAC1 oligomerization and increase mtDNA release, which drives the IFN signaling response and contributes to the pathogenesis of autoimmune diseases [40, 41]. Additionally, Tigano et al. described that mtDNA double-strand breaks (mtDSBs) triggered type-I IFN response through a novel intrinsic immune surveillance mechanism by which herniation formed by Bak and Bax released mitochondrial RNA into the cytoplasm and activated RIG-I–MAVS signaling pathway [42].
The mitochondrial transcription factor A (TFAM) is an essential protein required for the transcription and replication of mtDNA. Normally, TFAM, along with mtDNA and other proteins, constitutes nucleoid and regulates its architecture, abundance, and segregation. West et al. revealed that TFAM deficiency could promote mitochondrial stress and lead to abnormal mtDNA packaging, which would be released into the cytosol and then trigger cGAS-STING to elicit antiviral signaling [43].
2.3. mtDNA and Inflammasomes
Inflammasomes are multiprotein complexes and are well known for their fundamental roles in caspase activity, innate immunity, and cell death. Canonical inflammasomes consist of PRRs, the adaptor protein of apoptosis-associated speck-like protein containing a caspase recruitment domain (CARD) (ASC) and procaspase 1. Upon activation by PAMPs or DAMPs, PRRs assemble and activate caspase 1, which promotes the maturation of proinflammatory cytokines IL-18 and IL-1β, as well as the cleavage of gasdermin D (GSDMD), leading to pyroptosis, an inflammatory form of regulated necrosis [44, 45]. Nucleotide-binding oligomerization domain- (NOD-) like receptor (NLR) and absent in melanoma 2- (AIM2-) like receptors (ALR) are two out of five PRRs that form inflammasomes. Particularly, NLRP3 inflammasome and AIM2 inflammasome have been frequently linked to mtDNA signaling.
NLRP3 inflammasome constitutes a substantial part of innate immune defense against various infections and participates in the pathophysiology of multiple inflammatory diseases [46, 47]. Leakage of mtDNA [48, 49], as well as other stimuli such as K+ efflux [50] and mitochondrial ROS production [51], is sufficient to trigger the NLRP3 inflammasome cascades. Specifically, cytosolic mtDNA binds to and activates NLRP3 inflammasome in an oxidized form [48]. Further evidence also suggests that mtDNA is released out of mitochondria in a NLRP3 inflammasome-dependent way [52]. Therefore, a positive feedback between mtDNA release and NLRP3 inflammasome activation may reinforce the inflammatory process and enhance tissue damage.
AIM2 senses double-strand DNA (dsDNA), rather than single-strand DNA or RNA, and elicits inflammasome assembly and activation. Endogenous dsDNA derived from irradiation or chemotherapy-induced DNA damage has been implicated in cell death mediated by AIM2 inflammasome [53–56]. In addition to intracellular cytosolic “self-DNA,” exosome secretory and circulatory cell-free mtDNA are also suggested to contribute to AIM2 inflammasome-mediated inflammation [57].
2.4. The Interplay among Different mtDNA-Sensing Pathways
The cGAS-STING pathway and inflammasome activation have been shown to be associated in multiple sets, such as acute lung injury [58] and age-related liver ischemia-reperfusion injury (IRI) [59]. Typically, the stimulated cGAS-STING axis initiates inflammasome assembly and activation via type-I IFN signaling [60, 61] or K+ efflux induced by the translocation of STING to the lysosome and the subsequent lysosomal rupture [62]. In LPS-induced cardiomyopathy, STING-phosphorylated IRF3 traffics to the nucleus and increases the expression of NLRP3, providing the priming signal of inflammasome activation [63]. However, activation of inflammasome has been suggested to suppress the cGAS-STING pathway [64]. Wang et al. found that in response to DNA virus infection, canonical or noncanonical inflammasome activation led to caspase-1 or caspase-4, caspase-5, and caspase-11-dependent cleavage of cGAS and reduced IFN production [65]. Moreover, Banerjee et al. showed that AIM2 inflammasome-activated GSDMD formed pores on the cell membrane and induced K+ efflux, causing a decrease of intracellular K+ which undermined the DNA binding capacity and enzymatic activity of cGAS [66]. Notwithstanding, IL-1β, the product of inflammasome activation and pyroptosis, was found to induce mtDNA release and activate cGAS-STING signaling which protected cells against RNA virus infection [67]. Therefore, the complex positive and negative relationships between the cGAS-STING pathway and inflammasome activation remain elusive and need further investigation.
TLR9-mediated NLRP3 inflammasome activation has been described in several disease models [68–70]. However, the mechanisms of this interrelationship have not been fully elucidated. Besides, the DNA-sensing cGAS-STING and TLR9 signaling pathways were suggested in limited studies to work synergistically in innate immune response [71, 72].
3. mtDNA and Kidney Diseases
3.1. mtDNA and AKI
The significance of the integrity and function of mitochondria for normal kidney function has been universally established. As a key indicator of mitochondrial function, mtDNA copy number (mtDNA-CN) abnormalities have been frequently observed during the development of AKI in both animal models and clinical trials, as shown in Table 1. In the murine model of LPS-induced kidney injury, the mtDNA-CN of whole cell lysates declined [73], while the mtDNA-CN of cytoplasmic extracts increased [15], probably indicating that under cell stress, mtDNA replication was restricted but preexisting mtDNA continued to be released from the mitochondria to the cytosol. Analysis of circulating mtDNA-CN revealed that the concentration of mtDNA in plasma tended to increase although not significantly in bilateral ureteral obstruction (BUO) and ischemia-reperfusion models in mice [16]. Compared with circulating mtDNA-CN, urinary mtDNA- (UmtDNA-) CN has greater potential as an ideal indicator for AKI owing to its accessibility, correlation with renal function, and predictive value for kidney prognosis [74–76]. A case-control study on systemic inflammatory response syndrome (SIRS) showed that increased circulating mtDNA was not related to the renal function, whereas the level of UmtDNA correlated with the severity of AKI. The study also demonstrated that tubular epithelial cells expressed proinflammatory cytokines in response to mtDNA intervention [77].
Table 1.
The alterations of cell-free circulating and urinary mtDNA and their correlations with acute kidney injury and chronic kidney diseases.
| Disease category | Biomarker | Alteration | Disease correlation | Refs |
|---|---|---|---|---|
| AKI | Circulating mtDNA | Not significantly changed | Not significantly correlated with inflammation and renal injury | [16, 75, 77] |
| Urinary mtDNA | Increased | Correlated positively with renal injury, negatively with eGFR and intrarenal mtDNA level | [74–77] | |
| Nondiabetic CKD | Urinary mtDNA | Increased | Correlated positively with renal injury and eGFR decline | [88–90] |
| DKD | Circulating mtDNA | Decreased | Correlated negatively with inflammation and renal injury | [111] |
| Urinary mtDNA | Increased | Correlated positively with inflammation and interstitial fibrosis, negatively with renal function | [108, 111] |
Note: AKI, acute kidney injury; CKD, chronic kidney disease; DKD, diabetic kidney disease; eGFR, estimated glomerular filtration rate.
Several studies have been conducted to assess whether and how aberrant mtDNA contributes to renal inflammation and the onset of AKI. Early in 2008, Yasuda et al. showed that TLR9 deficiency or TLR9 suppression by a selective inhibitor H154 protected mice from septic AKI as evidenced by increased survival, improved kidney function, and decreased inflammatory cytokine release and splenic apoptosis [78]. In 2016, the same group found that mice intravenously injected with exogenous mitochondrial debris presented with kidney injury, mitochondrial damage, and cytokine production, which were reversed in Tlr9 KO mice or by pretreatment with DNase [14]. Their results suggested that mtDNA facilitated TLR9 activation and contributed to septic AKI. However, global Tlr9 deletion in mice had no protective effect on ischemic kidney injury [79, 80], while renal proximal tubule specific deficiency or inhibition of TLR9 significantly ameliorated renal damage and dysfunction after renal ischemia [81, 82]. The different outcomes imply diverse functions of TLR9 depending on the specific cell types which merits further investigation. In cisplatin-induced AKI, the expression of cGAS and STING is enhanced, accompanied by increased phosphorylation of downstream TBK1 and p65, and translocation of STING to the Golgi apparatus. Depletion of STING utilizing knockout mice and pharmacological inhibition of STING by C-176 both alleviated inflammatory responses and improved renal dysfunction. However, the classic downstream molecules including IRF3 and type-I IFNs remained unchanged, which needed to be further clarified [15]. In addition, mitochondrial damage and NLRP3 inflammasome activation were described in AKI models caused by contrast media. Inhibition of PINK1-parkin pathway-mediated mitophagy enhanced the generation of mt-ROS and NLRP3 inflammasome activation in human renal proximal tubular cell line (HK2 cell), which could be attenuated by the administration of MitoTEMPO, a mitochondria-targeted antioxidant. However, only oxidized nuclear DNA but not mtDNA was analyzed in this experimental setting [83].
3.2. mtDNA and Nondiabetic CKD
Increasing evidence suggests that mtDNA-CN is closely correlated with the progression of CKD (Table 1). Decreased mtDNA content was observed in the renal cortex of partially nephrectomized rats, a commonly used CKD model [84]. The Atherosclerosis Risk in Communities (ARIC) study showed that higher mtDNA-CN level in the buffy coat was associated with decreased incidence of CKD independent of traditional risk factors such as diabetes and hypertension [85]. In agreement, a recent cohort study involving 4812 CKD patients demonstrated that decreased mtDNA-CN in whole blood correlated with increased all-cause mortality and infection-related deaths [86]. The level of mtDNA-CN in blood cells is negatively correlated with the occurrence and prognosis of CKD, whereas cell-free circulating mtDNA-CN tends to be positively correlated to kidney injury [16]. Of note, cell-free circulating mtDNA was also detected in abundance in healthy individuals [87]. Basically, the role of cell-free circulating mtDNA is not well understood, since the quality other than the quantity of mtDNA is rarely evaluated, and damages to mtDNA such as oxidation, fragmentation, and break may be more direct triggers to act as DAMPs. Moreover, elevated UmtDNA was detected in patients with hypertension compared with that in healthy individuals, and this elevation was associated to markers of kidney damage [88]. In a longitudinal study, analysis of 131 CKD patients showed that lower UmtDNA at baseline was linked to favorable renal outcomes at 6 months [89]. Similarly, an observational study involving 32 nondiabetic CKD patients showed that the level of UmtDNA correlated with the rate of renal function decline and predicted the risk of serum creatinine doubling or need of dialysis [90]. However, in a larger cohort of 102 nondiabetic CKD patients, there was no significant correlation between the UmtDNA level and the rate of eGFR decline, though the UmtDNA level was associated with baseline eGFR, proteinuria, and pathological damage [91]. Based on these results, whether UmtDNA can serve as a reliable indicator of CKD progression remains to be determined.
Abnormalities in mtDNA may also enable renal inflammation and fibrosis and promote CKD progression. TFAM-associated mitochondrial dysfunction is involved in the development of various kidney diseases including cisplatin-induced AKI [92], CKD [93], and kidney cystic disease [94]. Chung et al. demonstrated that mice with conditional knockout of Tfam in kidney tubule cells presented with mtDNA depletion and bioenergetic impairment at 6 weeks and renal fibrosis, immune cells infiltration, and azotemia at 12 weeks. Mechanistically, TFAM deficiency causes mtDNA mispackaging and leaking into the cytosol resulting in the activation of cGAS-STING pathway and the upregulation of downstream NF-κB which underlies the renal fibrosis and inflammation in CKD progression [93].
Mitochondrial dysfunction and the subsequent NLRP3 inflammasome activation have been linked to renal tubular injury and tubulointerstitial fibrosis in albumin-overload mouse models and aldosterone-treated human tubular epithelial cells [95, 96]. In the CKD models of nephrectomy and unilateral ureteral obstruction (UUO), Nlrp3 knockout ameliorated mitochondrial morphological abnormalities and mtDNA-CN reduction, thus attenuating renal fibrosis [97, 98]. Similarly, administration of cyclosporin A (CsA), a mitochondrial permeability transition pore (mPTP) inhibitor, also attenuated mitochondrial damage and NLRP3 inflammasome activation [95]. Early studies demonstrated that mtDNA fragments could release into the cytosol through mPTP, and CsA prevented pore opening and subsequent mtDNA release [99, 100]. Taken together, these findings suggest that cytosolic mtDNA contributes to CKD progression via activation of NLRP3 inflammasome.
3.3. mtDNA and Diabetic Kidney Disease (DKD)
DKD is the leading cause of CKD and ESRD worldwide. DKD patients are predisposed to cardiovascular diseases, infections, and mortality [101]. Nevertheless, the pathogenesis of DKD is still elusive. Complications of diabetes in organs other than the kidney, such as diabetic retinopathy [102], diabetic peripheral neuropathy [103], and skin conditions [104] have been associated with mitochondrial dysfunction and mtDNA changes. The involvement of oxidative stress and mtDNA damage is also gradually recognized as a key factor underlying the development of DKD. About 20 years ago, hyperglycemia-induced oxidative mtDNA damage was found implicated in diabetic nephropathy (DN), as evidenced by increased 8-OHdG expression and subsequent mtDNA deletion [105, 106]. Using gas chromatography-mass spectrometry, Sharma et al. found that most of the differentially expressed urine metabolites in DKD patients in comparison with those in healthy individuals were linked to mitochondrial functions. Reduced mtDNA in urine exosomes, which reflected intrarenal mtDNA levels, further confirmed mitochondrial damage in DKD [107]. Moreover, the mtDNA levels in urine supernatants negatively correlated with intrarenal mtDNA levels and might serve as a potential indicator of the severity of interstitial fibrosis in patients pathologically diagnosed with DN [108]. However, the alteration of peripheral blood mtDNA in DKD patients is controversial. An early study found increased peripheral blood mtDNA-CN in type-2 diabetic patients with nephropathy, compared with that in those patients without nephropathy and in healthy controls [109], whereas another result recently showed a low peripheral blood mtDNA-CN in DN patients [110]. Therefore, large-scale, long-term studies are still needed to determine the significance of mtDNA changes in peripheral blood in DKD patients.
High glucose downregulated intracellular mtDNA in murine endothelial cells and podocytes and facilitated the release of mtDNA into circulation which filtered through the kidney and further triggered chronic renal inflammation [111]. However, human mesangial cells treated by high glucose showed elevated cellular mtDNA content, accumulation of ROS, and increased mitochondrial fragmentation [112]. After that, excessive ROS caused mtDNA damage and activated TLR9 pathway as evidenced by increased expression of NF-κB and MYD88 [113]. It was also suggested that altered mtDNA content and mtDNA damage occurred earlier than bioenergetic dysfunction. These results indicate that mtDNA change in mesangial cells may contribute to the development of DKD.
4. Therapeutic Targets and Future Perspectives
A series of intracellular mechanisms, including mt-ROS scavenging, mitochondrial biogenesis, mitophagy, and mtDNA repair, are among the mitochondrial quality control system, which work synergistically to maintain mitochondrial homeostasis [2]. Since the common priming step of mtDNA-sensing pathways mediating inflammatory responses is mtDNA damage or release, it can be theorized that protective strategies specific to mtDNA or mitochondria may be preferred choices for the treatment of kidney injury, as shown in Table 2. Firstly, multiple mitochondria-targeted antioxidants, such as mitoquinol mesylate (MitoQ), SS-31, or plastoquinol decylrhodamine 19 (SkQR1), have been shown to effectively attenuate ROS accumulation and kidney injury and favor recovery of kidney function [114–117]. In IR-induced AKI, administration of MitoTEMPO, a specific scavenger of mitochondrial superoxide, alleviated mitochondrial damage and inflammation, partially by rescuing TFAM transcription decrease and mtDNA depletion caused by excess mt-ROS [118]. Additionally, treatment with diazoxide, a mitochondrial KATP channel opener, was also found to reduce ROS accumulation and mtDNA oxidization and thereby ameliorate renal ischemic injury [119].
Table 2.
Characteristics and implications of mtDNA-sensing pathways targeted by pharmacological modulators in acute and chronic kidney diseases.
| Agent | Mechanism | Model | Administration | Effect | Refs |
|---|---|---|---|---|---|
| MitoTEMPO | Mitochondria-targeted antioxidant | Iohexol (HK-2 cell) | Preincubation for 4 h before culture with iohexol | Prevented RTEC apoptosis | [83] |
| IRI (mouse) | One direct injection into each kidney after reperfusion followed by five daily i.p. injections | Ameliorated AKI | [118] | ||
| MitoQ | Mitochondria-targeted antioxidant | LPS (rat) | i.v. injection after LPS injection | Ameliorated AKI | [115] |
| IRI (mouse) | i.v. injection 15 min before ischemia | Ameliorated AKI | [116] | ||
| SS-31 | Mitochondria-targeted antioxidant | IRI (rat) | Subcutaneously injection 30 min before ischemia and at the onset of reperfusion | Ameliorated AKI | [117] |
| SkQR1 | Mitochondria-targeted antioxidant | LPS (newborn rat) | i.p. administration 3 h before LPS treatment | Ameliorated AKI | [114] |
| Diazoxide | KATP channel opener, reducing ROS accumulation | IRI (rat) | Muscle injection before ischemia | Ameliorated AKI | [119] |
| Formoterol | β2-Adrenergic agonist, inducing MB | IRI (mouse) | i.p. injection starting at 24 h after reperfusion, daily for five days | Promoted AKI recovery | [122] |
| LY344864 | 5-HT1F receptor agonist, inducing MB | IRI (mouse) | i.p. injection starting at 24 h after reperfusion, daily for five days | Promoted AKI recovery | [123] |
| DNase | Enzyme for mtDNA degradation | MTD (mouse) | Incubation with MTD | Abolished renal mitochondrial injury | [14] |
| Cyclosporin A | mPTP inhibitor | Albumin (mPTC) | Preincubation for 30 min before albumin treatment | Attenuated renal tubular injury in CKD | [95] |
| H154 | TLR9 inhibitor | CLP (mouse) | i.p. administration immediately after CLP surgery | Ameliorated AKI | [78] |
| C-176 | STING inhibitor | Cisplatin (mouse) | i.p. injection 1 h before cisplatin injection | Ameliorated AKI | [15] |
Note: 5-HT1F, 5-hydroxytryptamine 1F; AKI, acute kidney injury; CKD, chronic kidney disease; CLP, cecal ligation and puncture; i.p., intraperitoneal; i.v., intravenous; IRI, ischemia/reperfusion injury; MB, mitochondrial biogenesis; mPTC, mouse proximal tubular cells; mPTP, mitochondrial permeability transition pore; MTD, mitochondrial debris; mtDNA, mitochondrial DNA; RTEC, renal tubular epithelial cells; STING, stimulator of interferon; TLR9, Toll-like receptor 9.
Mitochondria are constantly renewed through eliminating the old or damaged ones by mitophagy and producing new, functional mitochondria by mitochondrial biogenesis. Peroxisome proliferator-activated receptor gamma coactivator (PGC-1α) is the master regulator of mitochondrial biogenesis and is highly expressed in the kidney, making it a potential therapeutic target for different kidney diseases [120, 121]. Treatment with formoterol, a specific β2-adrenergic agonist, stimulated mitochondrial biogenesis and facilitated the recovery of renal function via upregulation of PGC1-α following IR-induced AKI in mice [122]. In addition, agonism of 5-hydroxytryptamine 1F (5-HT1F) receptors also increased PGC1-α transcript levels and restored the expression of mitochondrial proteins and mtDNA-CN altered by AKI [123, 124].
However, there is still a lack of treatments directly targeting mtDNA [125, 126]. In spite of limited self-repair capacity of mtDNA compared with that of nucleus DNA, several mtDNA repair mechanisms have already been determined, including base excision repair, DNA break repair, mismatch repair, and homologous recombination (HR) [127, 128]. In the kidney, some key molecules involved in mtDNA maintenance have also been identified, such as polymerase β and TWNK [129, 130]. In addition, 8-hydroxyguanine DNA glycosylase (OGG1), an mtDNA repair protein, was elevated in the kidneys of mice with septic AKI [131]. Besides, Y-box-binding protein 1 (YBX1), which mediated mismatch mtDNA repair, was upregulated in the kidneys of CKD and DKD patients and UUO mouse models [132]. Moreover, a recent study on rheumatoid arthritis revealed that inhibition of MRE11A, a DNA repair nuclease located in mitochondria, resulted in mtDNA oxidization and translocation, triggering inflammasome assembly and tissue inflammation, which were reversed by MRE11A overexpression [133]. However, whether interventions on the mtDNA repair proteins would make a difference in the progression of AKI and CKD remains to be investigated. In addition, suppressing the release of mtDNA appears to be another option to restrain the downstream inflammation, taking into account the already unveiled mechanisms, including Bak/Bax-dependent MOMP, VDAC oligomers-formed pores, and mPTP.
The detection technology of mtDNA-CN is another noteworthy issue. Until now, the most widely used method to measure mtDNA-CN is quantitative polymerase chain reaction (qPCR), by calculating the ratio of copy number of a mitochondrial gene to that of a nuclear gene [134]. The limitation of qPCR for mtDNA-CN measurement is that it can only quantify a relative copy number. Recently, digital PCR (dPCR) has emerged as a new method to quantify absolute mtDNA-CN [90, 91]. Other methods like plasmid-normalized PCR-based assay and DNA microarrays have also been tried to estimate mtDNA-CN [85, 86]. Therefore, more technological advances are needed in the future for high accuracy and convenience of mtDNA-CN measurement.
In conclusion, mitochondrial dysfunction and mtDNA abnormality are associated with AKI and CKD, and mtDNA-CN may be a potential biomarker for the assessment of kidney injury and for the prediction of renal prognosis. Moreover, mtDNA leaking into the cytoplasm may trigger innate immunity via several DNA-sensing mechanisms including TLR9, cGAS-STING, and NLRP3/AIM2 inflammasome signaling as well as the interactions among these pathways, leading to kidney inflammation and fibrosis which are implicated in the pathogenesis of AKI, nondiabetic CKD, and DKD. Further, strategies targeting mtDNA-sensing pathway-mediated inflammation, including using mitochondria-targeted antioxidants, STING, or TLR9 inhibitors, enhancing mitochondrial biogenesis or mtDNA degradation, and reducing mtDNA release, may be promising therapies for these kidney diseases.
Acknowledgments
This study was supported by the funds from the National Natural Science Foundation of China (81770674) and the Primary Research and Development Plan of Zhejiang Province (2020C03034) to Fei Han.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Authors' Contributions
Lini Jin and Binfeng Yu reviewed the references and drafted the manuscript. Ines Armando helped in the revision of the manuscript. Fei Han generated the research idea and finally revised the manuscript. Lini Jin and Binfeng Yu contributed equally.
References
- 1.Galvan D. L., Green N. H., Danesh F. R. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney International. 2017;92(5):1051–1057. doi: 10.1016/j.kint.2017.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tang C., Cai J., Yin X. M., Weinberg J. M., Venkatachalam M. A., Dong Z. Mitochondrial quality control in kidney injury and repair. Nature Reviews Nephrology. 2021;17(5):299–318. doi: 10.1038/s41581-020-00369-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.West A. P., Shadel G. S., Ghosh S. Mitochondria in innate immune responses. Nature Reviews Immunology. 2011;11(6):389–402. doi: 10.1038/nri2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Q., Raoof M., Chen Y., et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wai T., Ao A., Zhang X., Cyr D., Dufort D., Shoubridge E. A. The role of mitochondrial DNA copy number in mammalian Fertility1. Biology of Reproduction. 2010;83(1):52–62. doi: 10.1095/biolreprod.109.080887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wallace D. C. Mitochondrial genetic medicine. Nature Genetics. 2018;50(12):1642–1649. doi: 10.1038/s41588-018-0264-z. [DOI] [PubMed] [Google Scholar]
- 7.Collins L. V., Hajizadeh S., Holme E., Jonsson I. M., Tarkowski A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. Journal of Leukocyte Biology. 2004;75(6):995–1000. doi: 10.1189/jlb.0703328. [DOI] [PubMed] [Google Scholar]
- 8.Lameire N. H., Bagga A., Cruz D., et al. Acute kidney injury: an increasing global concern. Lancet. 2013;382(9887):170–179. doi: 10.1016/S0140-6736(13)60647-9. [DOI] [PubMed] [Google Scholar]
- 9.on behalf of the Acute Disease Quality Initiative Workgroup 16, Chawla L. S., Bellomo R., et al. Acute kidney disease and renal recovery: consensus report of the Acute Disease Quality Initiative (ADQI) 16 Workgroup. Nature Reviews. Nephrology. 2017;13(4):241–257. doi: 10.1038/nrneph.2017.2. [DOI] [PubMed] [Google Scholar]
- 10.Thurman J. M. Triggers of inflammation after renal ischemia/reperfusion. Clinical Immunology. 2007;123(1):7–13. doi: 10.1016/j.clim.2006.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venkatachalam M. A., Griffin K. A., Lan R., Geng H., Saikumar P., Bidani A. K. Acute kidney injury: a springboard for progression in chronic kidney disease. American Journal of Physiology. Renal Physiology. 2010;298(5):F1078–F1094. doi: 10.1152/ajprenal.00017.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Basile D. P., Anderson M. D., Sutton T. A. Pathophysiology of acute kidney injury. Comprehensive Physiology. 2012;2(2):1303–1353. doi: 10.1002/cphy.c110041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guzzi F., Cirillo L., Roperto R. M., Romagnani P., Lazzeri E. Molecular mechanisms of the acute kidney injury to chronic kidney disease transition: an updated view. International Journal of Molecular Sciences. 2019;20(19):p. 4941. doi: 10.3390/ijms20194941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsuji N., Tsuji T., Ohashi N., Kato A., Fujigaki Y., Yasuda H. Role of mitochondrial DNA in septic AKI via Toll-like receptor 9. Journal of the American Society of Nephrology. 2016;27(7):2009–2020. doi: 10.1681/ASN.2015040376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maekawa H., Inoue T., Ouchi H., et al. Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Reports. 2019;29(5):1261–1273.e6. doi: 10.1016/j.celrep.2019.09.050. e6. [DOI] [PubMed] [Google Scholar]
- 16.Homolová J., Janovičová Ľ., Konečná B., et al. Plasma concentrations of extracellular DNA in acute kidney injury. Diagnostics. 2020;10(3):p. 152. doi: 10.3390/diagnostics10030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takeda K., Kaisho T., Akira S. Toll-Like Receptors. Annual Review of Immunology. 2003;21(1):335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 18.Anders H. J., Banas B., Schlondorff D. Signaling danger: Toll-like receptors and their potential roles in kidney disease. Journal of the American Society of Nephrology. 2004;15(4):854–867. doi: 10.1097/01.ASN.0000121781.89599.16. [DOI] [PubMed] [Google Scholar]
- 19.Majer O., Liu B., Barton G. M. Nucleic acid-sensing TLRs: trafficking and regulation. Current Opinion in Immunology. 2017;44:26–33. doi: 10.1016/j.coi.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hemmi H., Takeuchi O., Kawai T., et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408(6813):740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 21.Takeshita F., Leifer C. A., Gursel I., et al. Cutting edge: role of Toll-like receptor 9 in CpG DNA-induced activation of human cells. Journal of Immunology. 2001;167(7):3555–3558. doi: 10.4049/jimmunol.167.7.3555. [DOI] [PubMed] [Google Scholar]
- 22.Leifer C. A., Kennedy M. N., Mazzoni A., Lee C. W., Kruhlak M. J., Segal D. M. TLR9 is localized in the endoplasmic reticulum prior to stimulation. Journal of Immunology. 2004;173(2):1179–1183. doi: 10.4049/jimmunol.173.2.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Latz E., Schoenemeyer A., Visintin A., et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nature Immunology. 2004;5(2):190–198. doi: 10.1038/ni1028. [DOI] [PubMed] [Google Scholar]
- 24.Cardon L. R., Burge C., Clayton D. A., Karlin S. Pervasive CpG suppression in animal mitochondrial genomes. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(9):3799–3803. doi: 10.1073/pnas.91.9.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bliksøen M., Mariero L. H., Torp M. K., et al. Extracellular mtDNA activates NF-κB via toll-like receptor 9 and induces cell death in cardiomyocytes. Basic Research in Cardiology. 2016;111(4):p. 42. doi: 10.1007/s00395-016-0553-6. [DOI] [PubMed] [Google Scholar]
- 26.Liu Y., Yan W., Tohme S., et al. Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular carcinoma through Toll-like receptor 9. Journal of Hepatology. 2015;63(1):114–121. doi: 10.1016/j.jhep.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bao D., Zhao J., Zhou X., et al. Mitochondrial fission-induced mtDNA stress promotes tumor-associated macrophage infiltration and HCC progression. Oncogene. 2019;38(25):5007–5020. doi: 10.1038/s41388-019-0772-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garcia-Martinez I., Santoro N., Chen Y., et al. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. The Journal of Clinical Investigation. 2016;126(3):859–864. doi: 10.1172/JCI83885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao Y., Wang Y., Liu H., Liu Z., Zhao J. Mitochondrial DNA from hepatocytes induces upregulation of interleukin-33 expression of macrophages in nonalcoholic steatohepatitis. Digestive and Liver Disease. 2020;52(6):637–643. doi: 10.1016/j.dld.2020.03.021. [DOI] [PubMed] [Google Scholar]
- 30.Zhang J. Z., Liu Z., Liu J., Ren J. X., Sun T. S. Mitochondrial DNA induces inflammation and increases TLR9/NF-κB expression in lung tissue. International Journal of Molecular Medicine. 2014;33(4):817–824. doi: 10.3892/ijmm.2014.1650. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Jing R., Hu Z. K., Lin F., et al. Mitophagy-mediated mtDNA release aggravates stretching-induced inflammation and lung epithelial cell injury via the TLR9/MyD88/NF-κB pathway. Frontiers in Cell and Development Biology. 2020;8:p. 819. doi: 10.3389/fcell.2020.00819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun L., Wu J., du F., Chen X., Chen Z. J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339(6121):786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu J., Sun L., Chen X., et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339(6121):826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ishikawa H., Ma Z., Barber G. N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461(7265):788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cai X., Chiu Y. H., Chen Z. J. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Molecular Cell. 2014;54(2):289–296. doi: 10.1016/j.molcel.2014.03.040. [DOI] [PubMed] [Google Scholar]
- 36.Motwani M., Pesiridis S., Fitzgerald K. A. DNA sensing by the cGAS-STING pathway in health and disease. Nature Reviews. Genetics. 2019;20(11):657–674. doi: 10.1038/s41576-019-0151-1. [DOI] [PubMed] [Google Scholar]
- 37.Ablasser A., Chen Z. J. cGAS in action: expanding roles in immunity and inflammation. Science. 2019;363(6431) doi: 10.1126/science.aat8657. [DOI] [PubMed] [Google Scholar]
- 38.McArthur K., Whitehead L. W., Heddleston J. M., et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science. 2018;359(6378) doi: 10.1126/science.aao6047. [DOI] [PubMed] [Google Scholar]
- 39.Riley J. S., Quarato G., Cloix C., et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. The EMBO Journal. 2018;37(17) doi: 10.15252/embj.201899238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim J., Gupta R., Blanco L. P., et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science. 2019;366(6472):1531–1536. doi: 10.1126/science.aav4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crow M. K. Mitochondrial DNA promotes autoimmunity. Science. 2019;366(6472):1445–1446. doi: 10.1126/science.aaz9308. Retraction in: Science. 2019 Dec 20;366(6472):1531-1536. [DOI] [PubMed] [Google Scholar]
- 42.Tigano M., Vargas D. C., Tremblay-Belzile S., Fu Y., Sfeir A. Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance. Nature. 2021;591(7850):477–481. doi: 10.1038/s41586-021-03269-w. [DOI] [PubMed] [Google Scholar]
- 43.West A. P., Khoury-Hanold W., Staron M., et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520(7548):553–557. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lamkanfi M., Dixit V. M. Mechanisms and functions of inflammasomes. Cell. 2014;157(5):1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 45.Broz P., Dixit V. M. Inflammasomes: mechanism of assembly, regulation and signalling. Nature Reviews. Immunology. 2016;16(7):407–420. doi: 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- 46.Martinon F., Burns K., Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–426. doi: 10.1016/S1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 47.Srinivasula S. M., Poyet J. L., Razmara M., Datta P., Zhang Z. J., Alnemri E. S. The PYRIN-CARD Protein ASC Is an Activating Adaptor for Caspase-1. The Journal of Biological Chemistry. 2002;277(24):21119–21122. doi: 10.1074/jbc.C200179200. [DOI] [PubMed] [Google Scholar]
- 48.Shimada K., Crother T. R., Karlin J., et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36(3):401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhong Z., Liang S., Sanchez-Lopez E., et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560(7717):198–203. doi: 10.1038/s41586-018-0372-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Muñoz-Planillo R., Kuffa P., Martínez-Colón G., Smith B. L., Rajendiran T. M., Núñez G. K+ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity. 2013;38(6):1142–1153. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou R., Yazdi A. S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 52.Nakahira K., Haspel J. A., Rathinam V. A. K., et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nature Immunology. 2011;12(3):222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hornung V., Ablasser A., Charrel-Dennis M., et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458(7237):514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fernandes-Alnemri T., Yu J. W., Datta P., Wu J., Alnemri E. S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458(7237):509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hu B., Jin C., Li H. B., et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science. 2016;354(6313):765–768. doi: 10.1126/science.aaf7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lian Q., Xu J., Yan S., et al. Chemotherapy-induced intestinal inflammatory responses are mediated by exosome secretion of double-strand DNA via AIM2 inflammasome activation. Cell Research. 2017;27(6):784–800. doi: 10.1038/cr.2017.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bae J. H., Jo S. I. I., Kim S. J., et al. Circulating cell-free mtDNA contributes to AIM2 inflammasome-mediated chronic inflammation in patients with type 2 diabetes. Cell. 2019;8(4):p. 328. doi: 10.3390/cells8040328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ning L., Wei W., Wenyang J., Rui X., Qing G. Cytosolic DNA-STING-NLRP3 axis is involved in murine acute lung injury induced by lipopolysaccharide. Clinical and Translational Medicine. 2020;10(7, article e228) doi: 10.1002/ctm2.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhong W., Rao Z., Rao J., et al. Aging aggravated liver ischemia and reperfusion injury by promoting STING- mediated NLRP3 activation in macrophages. Aging Cell. 2020;19(8, article e13186) doi: 10.1111/acel.13186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kerur N., Fukuda S., Banerjee D., et al. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nature Medicine. 2018;24(1):50–61. doi: 10.1038/nm.4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu F., Niu Q., Fan X., et al. Priming and activation of inflammasome by canarypox virus vector ALVAC via the cGAS/IFI16-STING-type I IFN pathway and AIM2 sensor. Journal of Immunology. 2017;199(9):3293–3305. doi: 10.4049/jimmunol.1700698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gaidt M. M., Ebert T. S., Chauhan D., et al. The DNA inflammasome in human myeloid cells is initiated by a STING-cell death program upstream of NLRP3. Cell. 2017;171(5):1110–1124.e18. doi: 10.1016/j.cell.2017.09.039. e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li N., Zhou H., Wu H., et al. STING-IRF3 contributes to lipopolysaccharide-induced cardiac dysfunction, inflammation, apoptosis and pyroptosis by activating NLRP3. Redox Biology. 2019;24:p. 101215. doi: 10.1016/j.redox.2019.101215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Corrales L., Woo S. R., Williams J. B., McWhirter S. M., Dubensky T. W., Jr., Gajewski T. F. Antagonism of the STING pathway via activation of the AIM2 inflammasome by intracellular DNA. Journal of Immunology. 2016;196(7):3191–3198. doi: 10.4049/jimmunol.1502538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Y., Ning X., Gao P., et al. Inflammasome activation triggers caspase-1-mediated cleavage of cGAS to regulate responses to DNA virus infection. Immunity. 2017;46(3):393–404. doi: 10.1016/j.immuni.2017.02.011. [DOI] [PubMed] [Google Scholar]
- 66.Banerjee I., Behl B., Mendonca M., et al. Gasdermin D restrains type I interferon response to cytosolic DNA by disrupting ionic homeostasis. Immunity. 2018;49(3):413–426.e5. doi: 10.1016/j.immuni.2018.07.006. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aarreberg L. D., Esser-Nobis K., Driscoll C., Shuvarikov A., Roby J. A., Gale M., Jr. Interleukin-1beta induces mtDNA release to activate innate immune signaling via cGAS-STING. Mol Cell. 2019;74(4):801–815. doi: 10.1016/j.molcel.2019.02.038. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu G., Zhu Q., Zeng J., et al. Extracellular mitochondrial DNA promote NLRP3 inflammasome activation and induce acute lung injury through TLR9 and NF-κB. Journal of Thoracic Disease. 2019;11(11):4816–4828. doi: 10.21037/jtd.2019.10.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhao C. C., Xie Q. M., Xu J., Yan X. B., Fan X. Y., Wu H. M. TLR9 mediates the activation of NLRP3 inflammasome and oxidative stress in murine allergic airway inflammation. Molecular Immunology. 2020;125:24–31. doi: 10.1016/j.molimm.2020.06.016. [DOI] [PubMed] [Google Scholar]
- 70.Kim S. K., Park K. Y., Choe J. Y. Toll-like receptor 9 is involved in NLRP3 inflammasome activation and IL-1beta production through monosodium urate-induced mitochondrial DNA. Inflammation. 2020;43(6):2301–2311. doi: 10.1007/s10753-020-01299-6. [DOI] [PubMed] [Google Scholar]
- 71.Temizoz B., Kuroda E., Ohata K., et al. TLR9 and STING agonists synergistically induce innate and adaptive type-II IFN. European Journal of Immunology. 2015;45(4):1159–1169. doi: 10.1002/eji.201445132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu L., Mao Y., Xu B., et al. Induction of neutrophil extracellular traps during tissue injury: involvement of STING and Toll-like receptor 9 pathways. Cell Proliferation. 2019;52(3, article e12579) doi: 10.1111/cpr.12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ding Y., Zheng Y., Huang J., et al. UCP2 ameliorates mitochondrial dysfunction, inflammation, and oxidative stress in lipopolysaccharide-induced acute kidney injury. International Immunopharmacology. 2019;71:336–349. doi: 10.1016/j.intimp.2019.03.043. [DOI] [PubMed] [Google Scholar]
- 74.Whitaker R. M., Stallons L. J., Kneff J. E., et al. Urinary mitochondrial DNA is a biomarker of mitochondrial disruption and renal dysfunction in acute kidney injury. Kidney International. 2015;88(6):1336–1344. doi: 10.1038/ki.2015.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hu Q., Ren J., Ren H., et al. Urinary mitochondrial DNA identifies renal dysfunction and mitochondrial damage in sepsis-induced acute kidney injury. Oxidative Medicine and Cellular Longevity. 2018;2018:14. doi: 10.1155/2018/8074936.8074936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hu Q., Ren J., Wu J., et al. Urinary mitochondrial DNA levels identify acute kidney injury in surgical critical illness patients. Shock. 2017;48(1):11–17. doi: 10.1097/SHK.0000000000000830. [DOI] [PubMed] [Google Scholar]
- 77.Jansen M. P. B., Pulskens W. P., Butter L. M., et al. Mitochondrial DNA is released in urine of SIRS patients with acute kidney injury and correlates with severity of renal dysfunction. Shock. 2018;49(3):301–310. doi: 10.1097/SHK.0000000000000967. [DOI] [PubMed] [Google Scholar]
- 78.Yasuda H., Leelahavanichkul A., Tsunoda S., et al. Chloroquine and inhibition of Toll-like receptor 9 protect from sepsis-induced acute kidney injury. American Journal of Physiology. Renal Physiology. 2008;294(5):F1050–F1058. doi: 10.1152/ajprenal.00461.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bakker P. J., Scantlebery A. M., Butter L. M., et al. TLR9 mediates remote liver injury following severe renal ischemia reperfusion. PLoS One. 2015;10(9) doi: 10.1371/journal.pone.0137511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li X., Yun Z., Tan Z., et al. The role of Toll-like receptor (TLR) 2 and 9 in renal ischemia and reperfusion injury. Urology. 2013;81(6):1379.e15–1379.e20. doi: 10.1016/j.urology.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 81.Han S. J., Li H., Kim M., Shlomchik M. J., Lee H. T. Kidney proximal tubular TLR9 exacerbates ischemic acute kidney injury. Journal of Immunology. 2018;201(3):1073–1085. doi: 10.4049/jimmunol.1800211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Han S. J., Williams R. M., D’Agati V., Jaimes E. A., Heller D. A., Lee H. T. Selective nanoparticle-mediated targeting of renal tubular Toll-like receptor 9 attenuates ischemic acute kidney injury. Kidney International. 2020;98(1):76–87. doi: 10.1016/j.kint.2020.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lin Q., Li S., Jiang N., et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biology. 2019;26:p. 101254. doi: 10.1016/j.redox.2019.101254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fedorova L. V., Tamirisa A., Kennedy D. J., et al. Mitochondrial impairment in the five-sixth nephrectomy model of chronic renal failure: proteomic approach. BMC Nephrology. 2013;14(1) doi: 10.1186/1471-2369-14-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tin A., Grams M. E., Ashar F. N., et al. Association between mitochondrial DNA copy number in peripheral blood and incident CKD in the Atherosclerosis Risk in Communities study. Journal of the American Society of Nephrology. 2016;27(8):2467–2473. doi: 10.1681/ASN.2015060661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fazzini F., Lamina C., Fendt L., et al. Mitochondrial DNA copy number is associated with mortality and infections in a large cohort of patients with chronic kidney disease. Kidney International. 2019;96(2):480–488. doi: 10.1016/j.kint.2019.04.021. [DOI] [PubMed] [Google Scholar]
- 87.Meddeb R., Dache Z. A. A., Thezenas S., et al. Quantifying circulating cell-free DNA in humans. Scientific Reports. 2019;9(1):p. 5220. doi: 10.1038/s41598-019-41593-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Eirin A., Saad A., Tang H., et al. Urinary mitochondrial DNA copy number identifies chronic renal injury in hypertensive patients. Hypertension. 2016;68(2):401–410. doi: 10.1161/HYPERTENSIONAHA.116.07849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chang C. C., Chiu P. F., Wu C. L., et al. Urinary cell-free mitochondrial and nuclear deoxyribonucleic acid correlates with the prognosis of chronic kidney diseases. BMC Nephrology. 2019;20(1):p. 391. doi: 10.1186/s12882-019-1549-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wei P. Z., Kwan B. C. H., Chow K. M., et al. Urinary mitochondrial DNA level in non-diabetic chronic kidney diseases. Clinica Chimica Acta. 2018;484:36–39. doi: 10.1016/j.cca.2018.05.036. [DOI] [PubMed] [Google Scholar]
- 91.Wei Z., Kwan B. C. H., Chow K. M., et al. Urinary mitochondrial DNA level as a biomarker of tissue injury in non-diabetic chronic kidney diseases. BMC Nephrology. 2018;19(1):p. 367. doi: 10.1186/s12882-018-1178-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Guo Y., Ni J., Chen S., et al. MicroRNA-709 mediates acute tubular injury through effects on mitochondrial function. Journal of the American Society of Nephrology. 2018;29(2):449–461. doi: 10.1681/ASN.2017040381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chung K. W., Dhillon P., Huang S., et al. Mitochondrial damage and activation of the STING pathway Lead to renal inflammation and fibrosis. Cell Metabolism. 2019;30(4):784–799.e5. doi: 10.1016/j.cmet.2019.08.003. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ishii K., Kobayashi H., Taguchi K., et al. Kidney epithelial targeted mitochondrial transcription factor A deficiency results in progressive mitochondrial depletion associated with severe cystic disease. Kidney International. 2021;99(3):657–670. doi: 10.1016/j.kint.2020.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhuang Y., Yasinta M., Hu C., et al. Mitochondrial dysfunction confers albumin-induced NLRP3 inflammasome activation and renal tubular injury. American Journal of Physiology. Renal Physiology. 2015;308(8):F857–F866. doi: 10.1152/ajprenal.00203.2014. [DOI] [PubMed] [Google Scholar]
- 96.Bi X., Wang J., Liu Y., Wang Y., Ding W. MnTBAP treatment ameliorates aldosterone-induced renal injury by regulating mitochondrial dysfunction and NLRP3 inflammasome signalling. American Journal of Translational Research. 2018;10(11):3504–3513. [PMC free article] [PubMed] [Google Scholar]
- 97.Gong W., Mao S., Yu J., et al. NLRP3 deletion protects against renal fibrosis and attenuates mitochondrial abnormality in mouse with 5/6 nephrectomy. American Journal of Physiology. Renal Physiology. 2016;310(10):F1081–F1088. doi: 10.1152/ajprenal.00534.2015. [DOI] [PubMed] [Google Scholar]
- 98.Guo H., Bi X., Zhou P., Zhu S., Ding W. NLRP3 deficiency attenuates renal fibrosis and ameliorates mitochondrial dysfunction in a mouse unilateral ureteral obstruction model of chronic kidney disease. Mediators of Inflammation. 2017;2017:10. doi: 10.1155/2017/8316560.8316560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ekstrand M. I., Falkenberg M., Rantanen A., et al. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Human Molecular Genetics. 2004;13(9):935–944. doi: 10.1093/hmg/ddh109. [DOI] [PubMed] [Google Scholar]
- 100.Patrushev M., Kasymov V., Patrusheva V., Ushakova T., Gogvadze V., Gaziev A. I. Release of mitochondrial DNA fragments from brain mitochondria of irradiated mice. Mitochondrion. 2006;6(1):43–47. doi: 10.1016/j.mito.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 101.Alicic R. Z., Rooney M. T., Tuttle K. R. Diabetic kidney disease: challenges, progress, and possibilities. Clinical Journal of the American Society of Nephrology. 2017;12(12):2032–2045. doi: 10.2215/CJN.11491116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Malik A. N., Parsade C. K., Ajaz S., et al. Altered circulating mitochondrial DNA and increased inflammation in patients with diabetic retinopathy. Diabetes Research and Clinical Practice. 2015;110(3):257–265. doi: 10.1016/j.diabres.2015.10.006. [DOI] [PubMed] [Google Scholar]
- 103.Chandrasekaran K., Anjaneyulu M., Choi J., et al. Role of mitochondria in diabetic peripheral neuropathy: influencing the NAD+-dependent SIRT1-PGC-1α-TFAM pathway. International Review of Neurobiology. 2019;145:177–209. doi: 10.1016/bs.irn.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rizwan H., Pal S., Sabnam S., Pal A. High glucose augments ROS generation regulates mitochondrial dysfunction and apoptosis via stress signalling cascades in keratinocytes. Life Sciences. 2020;241:p. 117148. doi: 10.1016/j.lfs.2019.117148. [DOI] [PubMed] [Google Scholar]
- 105.Suzuki S., Hinokio Y., Komatu K., et al. Oxidative damage to mitochondrial DNA and its relationship to diabetic complications. Diabetes Research and Clinical Practice. 1999;45(2-3):161–168. doi: 10.1016/S0168-8227(99)00046-7. [DOI] [PubMed] [Google Scholar]
- 106.Kakimoto M., Inoguchi T., Sonta T., et al. Accumulation of 8-hydroxy-2'-deoxyguanosine and mitochondrial DNA deletion in kidney of diabetic rats. Diabetes. 2002;51(5):1588–1595. doi: 10.2337/diabetes.51.5.1588. [DOI] [PubMed] [Google Scholar]
- 107.Sharma K., Karl B., Mathew A. V., et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. Journal of the American Society of Nephrology. 2013;24(11):1901–1912. doi: 10.1681/ASN.2013020126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wei P. Z., Kwan B. C. H., Chow K. M., et al. Urinary mitochondrial DNA level is an indicator of intra-renal mitochondrial depletion and renal scarring in diabetic nephropathy. Nephrology, Dialysis, Transplantation. 2018;33(5):784–788. doi: 10.1093/ndt/gfx339. [DOI] [PubMed] [Google Scholar]
- 109.Malik A. N., Shahni R., Iqbal M. M. Increased peripheral blood mitochondrial DNA in type 2 diabetic patients with nephropathy. Diabetes Research and Clinical Practice. 2009;86(2):e22–e24. doi: 10.1016/j.diabres.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 110.al-Kafaji G., Aljadaan A., Kamal A., Bakhiet M. Peripheral blood mitochondrial DNA copy number as a novel potential biomarker for diabetic nephropathy in type 2 diabetes patients. Experimental and Therapeutic Medicine. 2018;16(2):1483–1492. doi: 10.3892/etm.2018.6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cao H., Wu J., Luo J., Chen X., Yang J., Fang L. Urinary mitochondrial DNA: a potential early biomarker of diabetic nephropathy. Diabetes/Metabolism Research and Reviews. 2019;35(4, article e3131) doi: 10.1002/dmrr.3131. [DOI] [PubMed] [Google Scholar]
- 112.Al-Kafaji G., Golbahar J. High glucose-induced oxidative stress increases the copy number of mitochondrial DNA in human mesangial cells. BioMed Research International. 2013;2013:8. doi: 10.1155/2013/754946.754946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Czajka A., Ajaz S., Gnudi L., et al. Altered mitochondrial function, mitochondrial DNA and reduced metabolic flexibility in patients with diabetic nephropathy. eBioMedicine. 2015;2(6):499–512. doi: 10.1016/j.ebiom.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Plotnikov E. Y., Pevzner I., Zorova L., et al. Mitochondrial damage and mitochondria-targeted antioxidant protection in LPS-induced acute kidney injury. Antioxidants. 2019;8(6):p. 176. doi: 10.3390/antiox8060176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lowes D. A., Thottakam B. M. V., Webster N. R., Murphy M. P., Galley H. F. The mitochondria-targeted antioxidant MitoQ protects against organ damage in a lipopolysaccharide-peptidoglycan model of sepsis. Free Radical Biology & Medicine. 2008;45(11):1559–1565. doi: 10.1016/j.freeradbiomed.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 116.Dare A. J., Bolton E. A., Pettigrew G. J., Bradley J. A., Saeb-Parsy K., Murphy M. P. Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox Biology. 2015;5:163–168. doi: 10.1016/j.redox.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Birk A. V., Liu S., Soong Y., et al. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. Journal of the American Society of Nephrology. 2013;24(8):1250–1261. doi: 10.1681/ASN.2012121216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhao M., Wang Y., Li L., et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics. 2021;11(4):1845–1863. doi: 10.7150/thno.50905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sun Z., Zhang X., Ito K., et al. Amelioration of oxidative mitochondrial DNA damage and deletion after renal ischemic injury by the KATP channel opener diazoxide. American Journal of Physiology. Renal Physiology. 2008;294(3):F491–F498. doi: 10.1152/ajprenal.00263.2007. [DOI] [PubMed] [Google Scholar]
- 120.Whitaker R. M., Corum D., Beeson C. C., Schnellmann R. G. Mitochondrial biogenesis as a pharmacological target: a new approach to acute and chronic diseases. Annual Review of Pharmacology and Toxicology. 2016;56(1):229–249. doi: 10.1146/annurev-pharmtox-010715-103155. [DOI] [PubMed] [Google Scholar]
- 121.Tran M., Tam D., Bardia A., et al. PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. The Journal of Clinical Investigation. 2011;121(10):4003–4014. doi: 10.1172/JCI58662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jesinkey S. R., Funk J. A., Stallons L. J., et al. Formoterol restores mitochondrial and renal function after ischemia-reperfusion injury. Journal of the American Society of Nephrology. 2014;25(6):1157–1162. doi: 10.1681/ASN.2013090952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Garrett S. M., Whitaker R. M., Beeson C. C., Schnellmann R. G. Agonism of the 5-hydroxytryptamine 1F receptor promotes mitochondrial biogenesis and recovery from acute kidney injury. The Journal of Pharmacology and Experimental Therapeutics. 2014;350(2):257–264. doi: 10.1124/jpet.114.214700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gibbs W. S., Collier J. B., Morris M., Beeson C. C., Megyesi J., Schnellmann R. G. 5-HT1Freceptor regulates mitochondrial homeostasis and its loss potentiates acute kidney injury and impairs renal recovery. American Journal of Physiology. Renal Physiology. 2018;315(4):F1119–F1128. doi: 10.1152/ajprenal.00077.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Che R., Yuan Y., Huang S., Zhang A. Mitochondrial dysfunction in the pathophysiology of renal diseases. American Journal of Physiology. Renal Physiology. 2014;306(4):F367–F378. doi: 10.1152/ajprenal.00571.2013. [DOI] [PubMed] [Google Scholar]
- 126.Szeto H. H. Pharmacologic approaches to improve mitochondrial function in AKI and CKD. Journal of the American Society of Nephrology. 2017;28(10):2856–2865. doi: 10.1681/ASN.2017030247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kazak L., Reyes A., Holt I. J. Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nature Reviews. Molecular Cell Biology. 2012;13(10):659–671. doi: 10.1038/nrm3439. [DOI] [PubMed] [Google Scholar]
- 128.Dahal S., Dubey S., Raghavan S. C. Homologous recombination-mediated repair of DNA double-strand breaks operates in mammalian mitochondria. Cellular and Molecular Life Sciences. 2018;75(9):1641–1655. doi: 10.1007/s00018-017-2702-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sykora P., Kanno S., Akbari M., et al. DNA polymerase beta participates in mitochondrial DNA repair. Molecular and Cellular Biology. 2017;37(16) doi: 10.1128/MCB.00237-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Herbers E., Kekäläinen N. J., Hangas A., Pohjoismäki J. L., Goffart S. Tissue specific differences in mitochondrial DNA maintenance and expression. Mitochondrion. 2019;44:85–92. doi: 10.1016/j.mito.2018.01.004. [DOI] [PubMed] [Google Scholar]
- 131.Bartz R. R., Fu P., Suliman H. B., et al. Staphylococcus aureus sepsis induces early renal mitochondrial DNA repair and mitochondrial biogenesis in mice. PLoS One. 2014;9(7, article e100912) doi: 10.1371/journal.pone.0100912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bhreathnach U., Griffin B., Brennan E., Ewart L., Higgins D., Murphy M. Profibrotic IHG-1 complexes with renal disease associated HSPA5 and TRAP1 in mitochondria. Biochimica et Biophysica Acta - Molecular Basis of Disease. 2017;1863(4):896–906. doi: 10.1016/j.bbadis.2017.01.015. [DOI] [PubMed] [Google Scholar]
- 133.Li Y., Shen Y., Jin K., et al. The DNA repair nuclease MRE11A functions as a mitochondrial protector and prevents T cell pyroptosis and tissue inflammation. Cell Metabolism. 2019;30(3):477–492.e6. doi: 10.1016/j.cmet.2019.06.016. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Castellani C. A., Longchamps R. J., Sun J., Guallar E., Arking D. E. Thinking outside the nucleus: mitochondrial DNA copy number in health and disease. Mitochondrion. 2020;53:214–223. doi: 10.1016/j.mito.2020.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]