

Abstract
Apatinib is an oral tyrosine kinase inhibitor that targets VEGFR2 signaling and shows potent antitumor effects in various cancers. In this study, we explored the efficacy of apatinib against oral squamous cell carcinoma (OSCC). The relationships between VEGFR2 protein expression and clinical variables were investigated in OSCC patients. OSCC tissues had higher VEGFR2 levels than paracancerous tissues. Compared to patients with low VEGFR2 expression, patients with high VEGFR2 expression had poorer overall survival (OS) and disease-free survival (DFS). Apatinib significantly induced G0/G1 phase arrest and apoptosis, inhibited cell growth and colony formation ability, and blocked autophagic flux by downregulating p-AKT and p-mTOR signaling via the VEGFR2/AKT/mTOR pathway in vitro. Moreover, the inhibition of ERK phosphorylation increased apatinib-induced apoptosis in vitro and in vivo. Apatinib synergized with SCH772984 to achieve a more significant suppression of tumor growth than individual treatment, suggesting the combination of apatinib and SCH772984 as a potent OSCC therapy.
Keywords: Apatinib, VEGFR2, ERK, OSCC, autophagy
Introduction
Of the tumors that affect the oral and maxillofacial region, oral squamous cell carcinoma (OSCC) represents one of the most commonly occurring malignancies. With continual progress in diagnosis and treatment strategies, the 5-year OS of OSCC patients has been improved to 50%-60%, while that of patients with advanced disease decreases to 30%-40% [1-3]. Local and distant tumor recurrence is the main cause of death in locally advanced OSCC. Our research focuses on improving the survival of these patients. Owing to the toxic side effects and low efficacy of chemotherapeutic drugs, the development of targeted therapy has opened up a new approach for the treatment of oral cancer, particularly for patients with advanced, recurrent and metastatic oral cancer.
Therapeutic strategies targeting vascular endothelial growth factor receptors (VEGFRs) have been extensively studied because of the important roles of VEGFRs in carcinogenesis [4,5]. Apatinib is a tyrosine kinase inhibitor targeting VEGFR-2, which can induce apoptosis and autophagy, and inhibit tumorigenesis and development in anaplastic thyroid cancer [6], hepatocellular carcinoma [7] and osteosarcoma [8]. As the first small-molecule antiangiogenic agent, apatinib has been approved by the State Food and Drug Administration as a third-line treatment for gastric cancer [9]. In addition, clinical trials in other tumors have confirmed the efficacy and safety of apatinib [10-12]. However, whether apatinib has similar antitumor effects in OSCC patients has not been retrieved.
In this study, we evaluated the antitumor effect of apatinib and its potential mechanism in OSCC in vitro and in vivo. We further explored potential treatment combinations based on the mechanism of apatinib.
Materials and methods
Clinical samples
The study included 57 patients from January 2011 to June 2014 in Shanghai Jiaotong University Affiliated Ninth People’s Hospital diagnosed with locally advanced OSCC. All patients underwent standard treatment (surgery + postoperative radiotherapy). Cancer tissue and adjacent normal tissue specimens were collected for western blot analysis from 13 OSCC patients. The Ethics Committee of the Ninth People’s Hospital approved this study. All patients agreed to the use of their tumor tissue for the study. The primary outcome variable of this study was overall survival (OS), defined as the rate of death from any cause. Disease-free survival (DFS) was defined as the time until tumor recurrence or death for any reason.
Cells and chemicals
Four OSCC cell lines (HB96, CAL27, HN6, and HN4) were applied in the present study. The CAL27 cells were purchased from the American Type Culture Collection (ATCC) (Manassas, USA). HB96 cell line was derived from our previous OSCC in vitro cell carcinogenesis model, the HN6 and HN4 cells were a gift from the National Institutes of Health (USA). Defined keratinocyte serum-free medium (Invitrogen, USA) were used to culture HIOEC cells. Apatinib was supplied by Hengrui Medicine Co., Ltd. (Jiangsu, China), and chloroquine (CQ), Bafilomycin A1 (Baf A1) and SCH772984 (SCH) were obtained from Selleck Chemicals (Houston, USA).
Cell viability assay and colony formation assay
A density of 1000 OSCC cells (HB96, CAL27, HN6, and HN4) per well was cultured in medium for 12 h before being exposed to apatinib and SCH. The supernatant of each well was removed, and fresh medium containing 10% CCK-8 solution (Dojindo, Japan) was added to each well. The absorbance (450 nm) was measured to determine the cell viability after 2 h of incubation at 37°C. For the colony formation assay, 500 cells (HB96, CAL27, and HN6) per well were seeded in 6-well plates. Neutral paraformaldehyde and stained with crystal violet solution was used to fix the cells after 2 weeks. Colonies containing 50 to 100 cells were counted. This experiment was performed three times.
Wound healing assay
OSCC cells (HB96, CAL27, and HN6) were seeded in plates. When the cell density of each well reached 90%-95% confluence, the cells were starved in DMEM (no fetal bovine serum) for 12 h. A scratch was then made using a pipette tip, and fresh medium or apatinib (20 µM) was added to each well. The wound was photographed at 0, 6 and 12 h. Cell migration was determined by calculating the percentage of the wound closure area using ImageJ software.
Cell cycle analysis and apoptosis assay
After treatment, the OSCC cells (HB96, CAL27 and HN6) were washed and fixed according to the standard protocols. PI/RNase staining buffer (BD Pharmingen, San Diego, CA, USA) was used for cell cycle labeling. The cell samples were assayed by flow cytometer (FACSCalibur™, BD Biosciences, USA), then FlowJo software (BD Biosciences) was used to analyze the results.
For the apoptosis assay, a FITC-annexin V apoptosis detection kit (BD Biosciences, USA) was applied to double-stain the apoptotic and necrotic cells. Apoptosis of OSCC was evaluated by flow cytometry and FlowJo software.
Western blot assay and antibodies
Total protein extraction according to a standard procedure. Briefly speaking, the concentration of the extracted proteins (20 µg/lane) was determined using a Bradford assay (BCATM, USA) and separated on 6% to 15% SDS-PAGE. The primary antibody at a 1:1000 dilution and the fluorescently labeled anti-rabbit or anti-mouse IgG secondary antibodies (7076 and 7704, Cell Signaling Technology, USA) at a 1:10000 dilution. β-Actin (1226, Cell Signaling Technology) was used as an internal control. The primary antibodies: rabbit anti-cleaved fragment of human PARP (5625, Cell Signaling Technology), anti-VEGF receptor 2 (2478, Cell Signaling Technology), anti-cleaved fragment of caspase-3 (9664, Cell Signaling Technology), anti-caspase-3 (9662, Cell Signaling Technology), anti-p21 (2947, Cell Signaling Technology), anti-cyclind 1 (2947, Cell Signaling Technology), anti-p62 (ab109012, Abcam, Cambridge, UK), and anti-LC3B (43566, Cell Signaling Technology), mTOR pathway antibody sampler kit (9964, Cell Signaling Technology), phospho-AKT pathway antibody sampler kit (9916, Cell Signaling Technology) and p-Erk1/2 pathway sampler kit were purchased from Cell Signaling Technology (9911, Cell Signaling Technology).
Autophagic flux assay
OSCC cells (CAL27, HB96, HN6 and HN4) were inoculated in 6-well plates, then transfected with the GFP-mRFP-LC3 construct for 24 h. Apatinib and CQ were then added. After culturing for a further 12 h, PBS was used to wash the cultured cells three times before observing with a confocal microscope (Zeiss, Baden-Wurttemberg, Germany).
Transmission electron microscopy
After treatment with apatinib, 2% glutaraldehyde was used to immobilize the CAL27, HN6, HN30 and HB96 cells. The cells were then fixed with 1% OsO4 for 2 h, dehydrated with ethanol, and injected with epoxypropane. The samples were prepared as previously described [13]. JEM 1230 transmission electron microscope (JEOL, USA) was used to analyze those samples at 60 kV. The image analysis mainly used a 5000× or 20000× angle of view.
Tumor xenografts
The detailed method used was consistent with that in our previous report [14]. Briefly, A total of 2 × 106 CAL27 cells were suspended in mixed liquor (DMEM plus Matrigel), and a total volume of 0.1 mL per injection site. The mice with 100-150 mm3 tumor xenografts were randomly divided into the following four groups (n=6 each): control group, apatinib group, SCH group, and apatinib + SCH group. In the control group, the mice were treated with saline each day; in the apatinib group, the mice were orally administered apatinib solution (150 mg/kg) each day; in the SCH group, the mice were treated with i.p. injections of SCH (5 mg/kg); and in the apatinib + SCH group, the mice were treated with apatinib solution (150 mg/kg) and SCH (5 mg/kg) each day. The development and progression of solid tumors were monitored every 2 days. The mice were humanely euthanized when the tumor length over 1.5 cm. The tumor volume (V) = L (tumor length) × W (tumor width)2/2. Xenograft samples were collected and underwent next test.
Immunohistochemistry
Microscopic examination of Ki67 and VEGFR2 immunohistochemical staining was performed by two pathologists. The detailed method used was consistent with that in our previous report [14]. Results can be evaluated by a semiquantitative method that assigns an H-score to OSCC samples [15]. In brief, the immunohistochemical staining score was defined by multiplying the percentage of positive cells by the staining intensity, then the assessment of protein expression was defined as negative (≤ median score) or positive (> median score).
Statistical analysis
All data were analyzed with IBM SPSS Statistics 23 (IBM Corporation, Armonk, NY, USA) and GraphPad Prism 6 (GraphPad Software, San Diego, CA, USA). Continuous variables are represented as the mean ± SD. The means of the samples were compared using either an unpaired, two-tailed Student’s t-test or one-way ANOVA. A threshold value P<0.05 indicated statistical significance.
Results
VEGFR2 is highly expressed in oral squamous cell carcinoma and is associated with poor survival
The western blot results of tissues from 13 OSCC patients showed higher levels of VEGFR2 in OSCC tissues than in paracancerous tissues (P<0.005) (Figure 1A, 1B). The VEGFR2 protein level was higher in the four OSCC cell lines than in HIOEC cells (Figure 1C). Among the four OSCC cell lines tested, the CAL27, HB96 and HN6 cell lines showed higher VEGFR-2 levels than the HN4 cell line.
Figure 1.
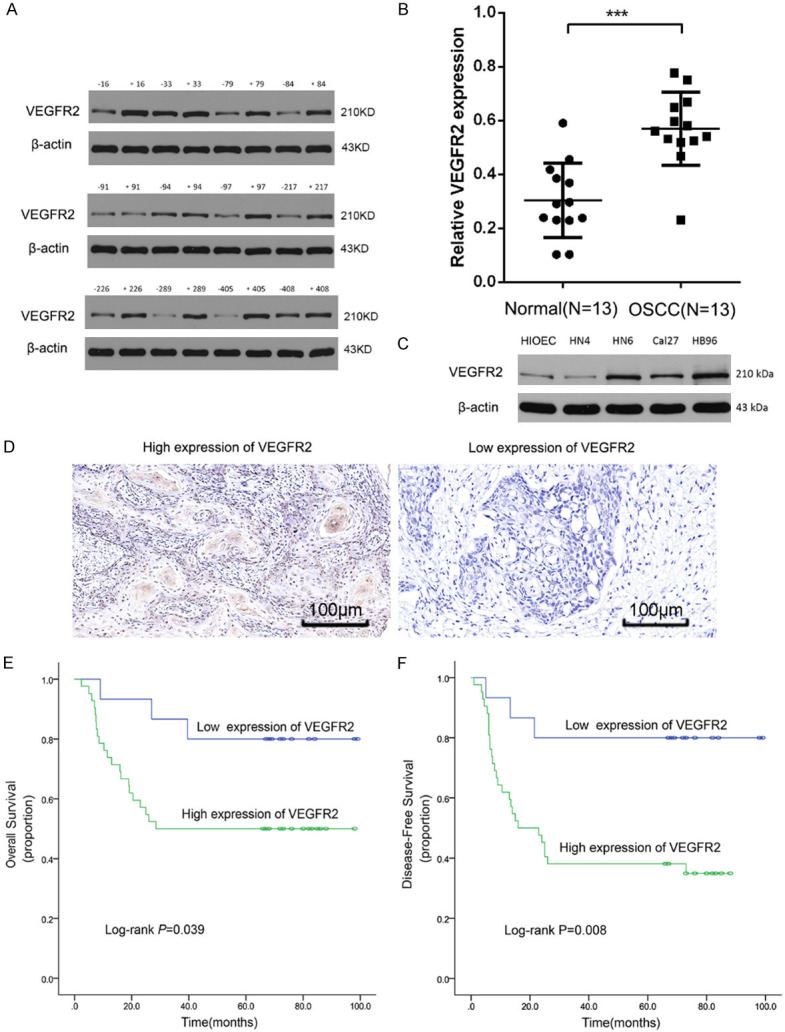
VEGFR2 expression was elevated in OSCC tissue and was associated with a poor prognosis. A, B: Western blot showing higher VEGFR2 expression in OSCC tissue compared to adjacent healthy tissue. ***P<0.001. C: Western blot assay of VEGFR2 expression in OSCC cell lines. β-Actin was used as a control. D: Immunohistochemistry staining of VEGFR2 in OSCC samples (200×). E, F: Kaplan-Meier curves showing the relation between VEGFR2 expression and overall survival and disease-free survival in 53 OSCC patients.
To evaluate the correlation between the level of VEGFR2 expression and prognosis in OSCC patients, VEGFR2 expression was estimated by immunohistochemical staining in the 57 OSCC patient cohort (Figure 1D). No obvious differences were detected in VEGFR2 expression based on clinical variables (Supplementary Table 1). We investigated the relationship between VEGFR2 levels and OS/DFS in OSCC patients, and Kaplan-Meier analysis indicated that high VEGFR2 expression indicated poor OS (P<0.039) and poor DFS (P<0.008) (Figure 1E, 1F).
Apatinib inhibited the proliferation, clone formation, invasion and migration of OSCC cells
To assess the effects of apatinib on OSCC cells in vitro, cell viability assays, colony formation assays, wound healing assays and transwell matrix assays were performed. Our results showed that the growth of CAL27 and HB96 cells was suppressed by apatinib in a time- and dose-dependent manner (Figure 2A). However, in HN6 and HN4 cells, the suppression of cell viability was unchanged at 48 h and 72 h. The IC50 (48 h) values of apatinib in CAL27, HB96, HN6 and HN4 cells are shown in Figure 2A; these values were used for subsequent assays. Compared with the negative control, apatinib treatment suppressed colony formation in a dose-dependent manner; 20 µM apatinib significantly inhibited over 80% number of CAL27 and HB96 cell colonies, and 10 µM apatinib effectively decreased over 80% number of HN6 cell colonies (Figure 2B). Transwell matrix assays and wound healing assays were used to assess the effect of 20 µM apatinib on invasion and migration of Cal27, HB96 and HN6 cells. It was observed that 20 µM was sufficient to inhibit the invasion and migration of the OSCC cell lines. The wound area (Figure 2C) and the number of invading cells (Figure 2D) were significantly different between the apatinib and negative control groups.
Figure 2.
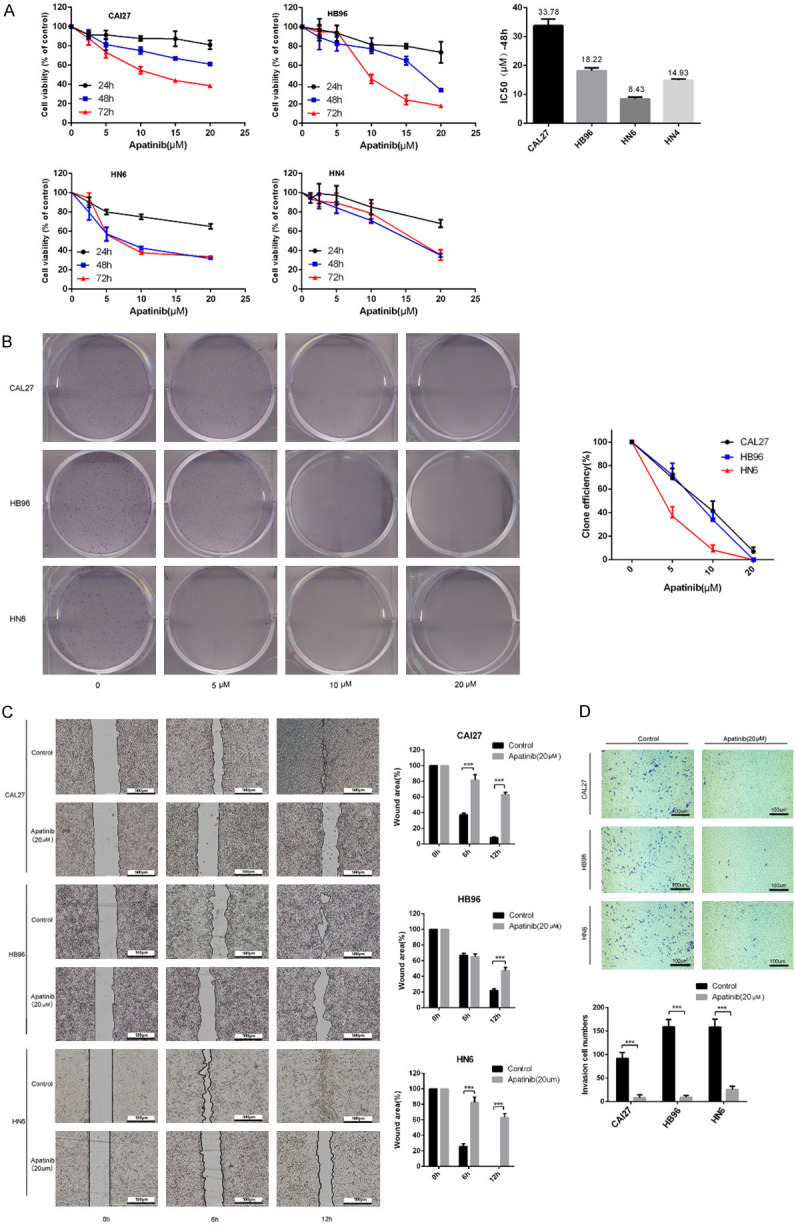
Apatinib inhibited the proliferation, migration and invasion of OSCC cells. A: CAL27, HB96, HN6 and HN4 cells were cultured in media containing various concentrations of apatinib for 24, 48, and 72 h. Cell viability was detected by CCK8 assay to identify the apatinib IC50 value at 48 h in CAL27, HB96, HN6 and HN4 cells. B: Colony formation assay of CAL27, HB96 and HN6 cells. Each datapoint is representative of three independent experiments. The data are representative of three independent experiments. C: Wound healing assay in CAL27 and HB96 cells treated with or without 20 μM apatinib for 6 h and 12 h. The migration index (the ratio of migration distance to total distance) was used to measure the movement ability. D: Apatinib suppressed the invasion of OSCC cells (CAL27, HB96 and HN6); the invasion index was measured by transwell assay. ***P<0.001 vs control group.
Apatinib promoted cell apoptosis and induced cell cycle arrest in CAL27, HB96 and HN6 cells
The expression levels of cleaved PARP, total caspase 3 and cleaved caspase 3 (key apoptotic proteins) in CAL27, HB96 and HN6 cells after treatment with gradient concentrations of apatinib were analyzed by western blotting. Compared to the negative control group, the levels of total caspase 3 was decreased, but the cleaved caspase3 and PARP were markedly increased after treatment with apatinib in a dose-dependent manner in CAL27, HB96 and HN6 cells (Figure 3A). Moreover, compared with the negative control group, the 20 µM apatinib-treated group had an increased proportion of annexin-V-positive OSCC cells (Figure 3B, 3C), suggesting that apatinib inhibited the growth of OSCC cells by promoting apoptosis. Flow cytometry analysis of the cell cycle revealed that the G0/G1 phase was mainly arrested after apatinib treatment, while the proportion of G2/M and S phase cells was decreased (Figure 3D, 3E). Consistent with the cell cycle alterations, we observed significant upregulation of P21 and downregulation of cyclin D1 (Figure 3A). These results showed that apatinib promoted OSCC cell apoptosis and G0/G1 phase arrest.
Figure 3.

Apatinib-induced apoptosis and cell-cycle arrest. A: The cells were treated with apatinib (5, 10, 15, 20 μM) for 24 h. Protein expression of cleaved-PARP, caspase 3, cleaved caspase 3, P21 and cyclinD1 in CAL27, HB96 and HN6 cells was measured by western blot. β-Actin was used as a control. B, C: Apatinib-induced apoptosis in OSCC cells. Apoptosis were detected by annexin V-FITC and propidium iodide (PI) staining. D, E: Apatinib causes G0/G1 cell-cycle arrest in OSCC cells. Apatinib (20 μM) or dimethyl sulfoxide (DMSO) was added to the culture medium of CAL27, HB96 and HN6 cells. Cell-cycle distribution assessment was performed after 70% ethanol fixing and PI staining. The data are representative of three independent experiments. **P<0.01, ***P<0.001.
Apatinib inhibited autophagy in OSCC cells
During the development of autophagy, LC3 (map-lc3) in the cytoplasm enzymatically dissociates a small segment of polypeptide to form lc3-I, which binds to phosphatidylethanolamine and becomes the autophagosome membrane form, lc3-II. P62 is a marker of autophagic flux, which closely related to the initiation and inhibition of autophagic flux. As shown in Figure 4A, 4B, LC3II expression gradually increased in a time- and dose-dependent manner, and P62 expression also showed an increasing trend in CAL27, HB96, HN6 and HN4 cells. The increase in LC3II and P62 expression indicated that autophagy initiation was normal while its downstream steps were inhibited, and phagocytes and lysosomes were unable to fuse. Moreover, we observed that apatinib-induced autophagy flux was maintained when autophagosome degradation was inhibited with bafilomycin A1 (Baf A1), no significant changes have been seen in the increasing level of LC3II (Figure 4C). To further detect autophagic flux (Figure 4E), the cells underwent adenoviral infection with a vector carrying mCherry-GFP-LC3B. Under fluorescence microscopy, mCherry-GFP-LC3B was observed in the cytoplasm in the form of diffuse yellow fluorescence (the combined effect of mCherry and GFP) when no autophagy was occurring, while clusters on the autophagosome membrane were visible as yellow puncta (LC3B puncta) in the presence of autophagy. After the fusion of autophagosomes and lysosomes, GFP fluorescence was partially quenched and was visible as red puncta. The fluorescence microscopy images revealed that the number of yellow puncta was more obviously increased in CAL27 and HB96 cells in the apatinib group (20 µM, 24 h) than in the negative control group (Figure 4F). Furthermore, the western blot and immunofluorescence results indicated that the function of apatinib was consistent with the traditional autophagy inhibitor CQ (Figure 4D, 4F). Compared to the starvation group, the number of yellow puncta was increased, but the number of red puncta was decreased in the merged image in the apatinib group. In addition, electron microscopy images showed that CAL27 and HB96 cells treated with apatinib had higher numbers of autophagosomes compared to the negative control (Figure 4G). These findings revealed that apatinib inhibited autophagy by suppressing autophagosome-lysosome fusion.
Figure 4.

Apatinib inhibits autophagy in OSCC cells. A: OSCC cells were incubated in medium containing 5, 10, 20 and 40 μM apatinib for 24 h, and the expression of P62 and LC3-II was measured by western blot. B: OSCC cells were incubated in medium containing 20 μM apatinib for 12, 24 and 48 h, and the expression of P62 and LC3-II was measured by western blot. C: OSCC cells were incubated in medium containing 20 μM apatinib for 24 h with or without Baf A1 (50 nM), and the expression of LC3-II was measured by western blot. D: OSCC cells were incubated in medium containing 20 μM apatinib for 24 h with or without CQ (50 nM), and the expression of P62 and LC3-II was measured by western blot. β-Actin was used as the control. E, F: Representative images of early autophagosomes (yellow dots generated from overlapping GFP and mCherry puncta), shown as yellow points, and late autolysosomes (red dots generated from RFP puncta), shown as red points. CAL27-GFP-mcherry-LC3 and HB96-GFP-mcherry-LC3 cells (transfected with the GFP-mcherry-LC3 lentivirus) treated with 20 μM apatinib for 24 h, starvation, or 50 nM CQ for 12 h. ***P<0.001. G: Autophagic vacuoles (red arrows) were seen in CAL27 and HB96 cells incubated in apatinib (20 μM) for 48 h. Few autophagic vacuoles were seen in the control group. These experiments were repeated three times.
Apatinib suppresses VEGFR2 signaling through the VEGFR2/AKT/m-TOR signaling pathway
Apatinib reportedly inhibits cancer growth by interfering in the VEGFR2/AKT/mTOR and RAF/MEK/ERK signaling pathways [6,16]. In the current study, western blotting revealed that VEGFR2, p-mTOR and p-AKT expression in CAL27, HB96 and HN6 cells was decreased in a dose-dependent manner after treatment with apatinib for 24 h (Figure 5A). The amount of total AKT and mTOR protein in HB96 and HN6 cells did not change after treatment with low concentrations of apatinib but decreased after treatment with high concentrations. Interestingly, exposure to apatinib increased the level of p-ERK in CAL27, HB96 and HN6 cells in a dose-dependent manner but had little influence on the total level of ERK protein.
Figure 5.

Apatinib induced an antitumor effect through the AKT/mTOR pathway and was enhanced by SCH. A: CAL27, HB96 and HN6 cells treated with or without apatinib (0, 5, 10, 20 and 40 μM) for 24 h. The expression of total VEGFR2, total AKT, phosphorylated AKT, total mTOR, phosphorylated mTOR, total ERK, phosphorylated ERK and GAPDH was detected by western blot. B: Cells were treated with apatinib (20 μM) with or without SCH (10 μM) for 24 h. Western blots of p-AKT, and p-ERK extracts from CAL27, HB96 and HN6 cells. C: CAL27, HB96 and HN6 cells were treated with apatinib (20 μM), SCH (10 μM), or apatinib + SCH for 24 h. The ratio of apoptotic cells was measured by annexin V-FITC and propidium iodide (PI) staining. These experiments were repeated three times.
Tumor suppression induced by apatinib is increased by an ERK inhibitor in vivo and in vitro
The ERK signaling pathway is reportedly associated with the occurrence, development and metastasis of tumors. Based on our results showing that apatinib inhibits activation of the Akt/mTOR signaling pathway and enhances ERK phosphorylation in OSCC cells, we combined apatinib with ERK inhibitors to enhance the antitumor effect in OSCC. Low concentrations of apatinib (10 µM) significantly reduced the IC50 of SCH in CAL27, HB96 and HN6 cells (Supplementary Table 2). Western blotting revealed that apatinib combined with SCH significantly inhibited the phosphorylation of AKT and ERK (Figure 5B). Compared with the effect of apatinib or ERK inhibitors alone, the effect of combination therapy on apoptosis was dramatically increased (Figure 5C).
To evaluate whether apatinib and ERK inhibitors have similar anticancer effects on OSCC tumors in vivo, we established xenograft models by subcutaneously injecting CAL27 cells. Apatinib treatment with or without SCH led to obvious tumor suppression, while no significant tumor suppression was observed in the SCH-treated group compared with control (Figure 6A). Furthermore, apatinib combined with SCH treatment exerted a greater inhibitory effect on the tumor volume than either drug alone (Figure 6A, 6B). No significant difference in body weight between groups (Figure 6C). The xenograft immunohistochemical results showed that treatment with apatinib plus SCH resulted in the most significant decrease in Ki67 expression of all tested groups (Figure 6D, 6E). Taken together, these findings strongly indicate that the ERK inhibitor SCH enhances the antitumor effect of apatinib in vivo.
Figure 6.
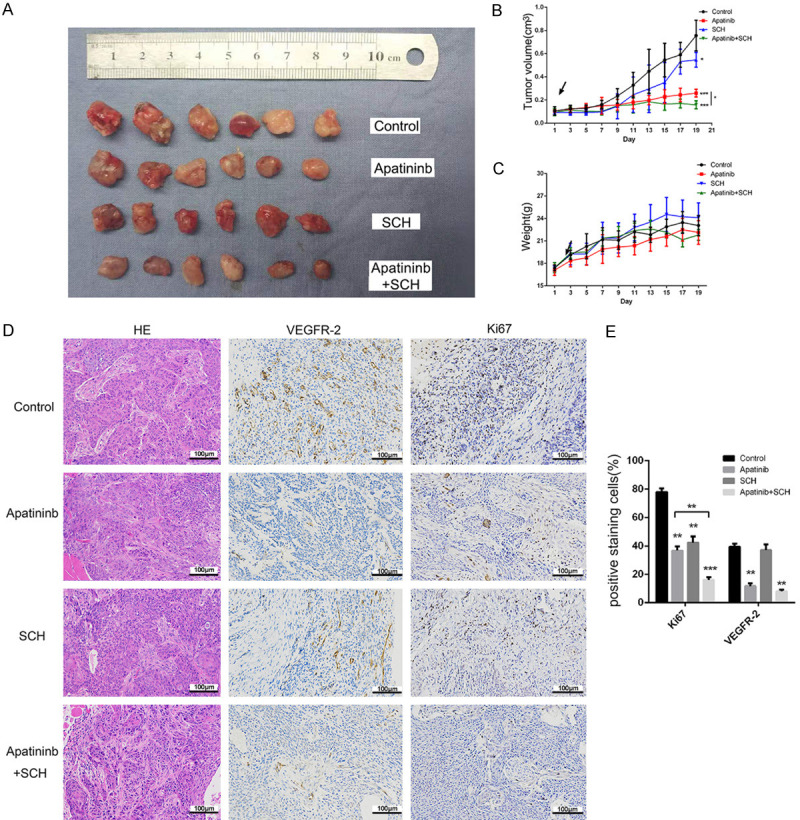
The ERK inhibitor SCH enhanced the in vivo antitumor effect of apatinib. A: Apatinib plus SCH showed a more significant antitumor effect than treatment with apatinib or SCH alone. B, C: Comparison of the tumor growth curves and mouse weights between the groups treated with and without apatinib and SCH (tumor volumes and weights were determined every 2 days after treatment initiation). D, E: Representative HE and IHC staining for Ki67 and VEGFR2, performed in the four tumor sample groups. *P<0.05, **P<0.01, ***P<0.001.
Discussion
Apatinib is a highly selective tyrosine kinase inhibitor of VEGFR2 whose antitumor effects on a variety of tumors have been confirmed in multiple studies [6,18-20]. In the present study, apatinib was found to activate apoptosis and inhibit autophagy by suppressing the AKT/mTOR signaling pathway. Apatinib-induced apoptosis was enhanced by cotreatment with an ERK inhibitor, both in vitro and in vivo.
It has been reported that apatinib can promote autophagy and apoptosis in osteosarcoma [8], colorectal cancer [21] and anaplastic thyroid cancer [6]. As the two main mechanisms leading to programmed cell death [22], recent studies have shown that apoptosis and autophagy are interrelated in signaling pathways [23]. Here, we found that apatinib inhibited OSCC cell proliferation in vitro while inducing apoptosis in a time- and dose-dependent manner. The role of autophagy in cancer is complex. Macroautophagy (hereafter referred to as autophagy) is a cell survival pathway that can help prevent bioenergy failure caused by metabolic stress, maintain the quality and quantity of proteins and organelles, and improve many aspects of tumor growth, including the occurrence, development and maintenance of a malignant state. As cancer cells grow, spread, and form solid tumors, they face harsh conditions such as hypoxia and nutritional deficiencies, which may hinder their growth and lead to cell death. Under these conditions, autophagy maintains or even enhances tumorigenesis [24]. The inhibition of autophagy has been shown to enhance apoptosis in anaplastic thyroid cancer [6] and osteosarcoma cells [8]. However, in this study, we found that the expression of LC3II increased gradually in a time- and dose-dependent manner, while P62 expression also showed an increasing trend in four OSCC cell lines, indicating that autophagy initiation was normal while its downstream steps were inhibited, suggesting that phagocytes and lysosomes could not fuse. Compared with the starvation and CQ groups, the apatinib group showed a similar level of autophagic flux inhibition in OSCC cells. Our study demonstrated that apatinib inhibited VEGF-mediated cell proliferation, migration and invasion in the OSCC cell lines, possibly by inducing apoptosis and reducing autophagy through blocking the VEGFR2-dependent AKT/mTOR pathway.
Signaling through the Akt/mTOR pathway can be initiated by several mechanisms. This signaling pathway is one of the major survival gateways in tumor cells, and accumulating evidence has shown that an overactive AKT/mTOR pathway promotes tumor cell survival [25]. As a downstream target of VEGFR, the PI3K-AKT-mTOR signaling pathway is closely related to the induction of apoptosis and autophagy and is the focus of VEGFR-targeted therapy in other cancers [6,7]. The current study found that apatinib decreased p-AKT and p-mTOR levels in a dose-dependent manner in four OSCC cell lines. SC79, a specific agonist of AKT, restored apatinib-induced autophagy in OSCC cells.
The ERK signaling pathway is involved in endothelial cell proliferation and VEGF-mediated cancer cell survival [4]. However, in our study, we found that apatinib markedly enhanced ERK phosphorylation but did not affect the level of total ERK protein. A possible reason for this is that a single inhibitor of the AKT signaling pathway activates the ERK pathway, similar to the AKT inhibitor honokiol, which inhibits activation of Akt while enhancing the phosphorylation of ERK1/ERK2 [26]. Moreover, MEK/ERK upregulation is related to radiotherapy, cisplatin and cedoximab resistance, suggesting the combined use with MEK inhibitors for HSCNN patients [27,28]. Thus, combining inhibition of the Akt/mTOR pathway and ERK signaling pathway may be a feasible method [25]. Drug resistance is a universal and important phenomenon in cancer treatment. Many chemotherapies and targeting drugs lead to drug resistance, which can decrease their therapeutic efficacy. Activation of the ERK signaling pathway may be one of the causes of apatinib resistance, and combination with ERK inhibitors may enhance the antitumor effect of apatinib. We applied the ERK inhibitor SCH and detected its combined effect with apatinib on OSCC cells and found that apatinib-induced apoptosis was enhanced by SCH both in vitro and in vivo. Thus, ERK inhibitors as potential combination of apatinib might show an attractive therapeutic strategy for OSCC.
Conclusions
This is the first study to show that apatinib exhibits antitumor effects on OSCC cells in vitro and in vivo, and provides a reliable mechanism by which apatinib induces apoptosis and suppresses autophagy via the AKT/mTOR pathway. Moreover, blocking the ERK signaling pathway enhanced the antitumor effect of apatinib in OSCC cell lines both in vitro and in vivo. These findings indicate that targeting ERK combined with apatinib may be an attractive therapeutic strategy for OSCC.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81972525, 81602370 and 81672660), the Shuguang Program of the Shanghai Municipal Education Commission (17SG18), the Shanghai Municipal Commission of Health and Family Planning (2018BR41), and the Program of Shanghai Academic/Technology Research Leader (19XD1422300).
All laboratory procedures were approved by the laboratory animal care and use committee of the hospital.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Chinn SB, Myers JN. Oral cavity carcinoma: current management, controversies, and future directions. J. Clin. Oncol. 2015;33:3269–76. doi: 10.1200/JCO.2015.61.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singhvi HR, Malik A, Chaturvedi P. The role of chronic mucosal trauma in oral cancer: a review of literature. Indian J Med Paediatr Oncol. 2017;38:44–50. doi: 10.4103/0971-5851.203510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Q, Yu C, Peng S, Xu H, Wright E, Zhang X, Huo X, Cheng E, Pham TH, Asanuma K, Hatanpaa KJ, Rezai D, Wang DH, Sarode V, Melton S, Genta RM, Spechler SJ, Souza RF. Autocrine VEGF signaling promotes proliferation of neoplastic Barrett’s epithelial cells through a PLC-dependent pathway. Gastroenterology. 2014;146:461–72. doi: 10.1053/j.gastro.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chatterjee S, Heukamp LC, Siobal M, Schöttle J, Wieczorek C, Peifer M, Frasca D, Koker M, König K, Meder L, Rauh D, Buettner R, Wolf J, Brekken RA, Neumaier B, Christofori G, Thomas RK, Ullrich RT. Tumor VEGF: VEGFR2 autocrine feed-forward loop triggers angiogenesis in lung cancer. J Clin Invest. 2013;123:1732–40. doi: 10.1172/JCI65385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feng H, Cheng X, Kuang J, Chen L, Yuen S, Shi M, Liang J, Shen B, Jin Z, Yan J, Qiu W. Apatinib-induced protective autophagy and apoptosis through the AKT-mTOR pathway in anaplastic thyroid cancer. Cell Death Dis. 2018;9:1030. doi: 10.1038/s41419-018-1054-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang H, Cao Y, Chen Y, Li G, Yu H. Apatinib promotes apoptosis of the SMMC-7721 hepatocellular carcinoma cell line via the PI3K/Akt pathway. Oncol Lett. 2018;15:5739–43. doi: 10.3892/ol.2018.8031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu K, Ren T, Huang Y, Sun K, Bao X, Wang S, Zheng B, Guo W. Apatinib promotes autophagy and apoptosis through VEGFR2/STAT3/BCL-2 signaling in osteosarcoma. Cell Death Dis. 2017;8:e3015. doi: 10.1038/cddis.2017.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang S. Problematic analysis and inadequate toxicity data in phase III apatinib trial in gastric cancer. J. Clin. Oncol. 2016;34:3821. doi: 10.1200/JCO.2016.67.3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li F, Zhu T, Cao B, Wang J, Liang L. Apatinib enhances antitumour activity of EGFR-TKIs in non-small cell lung cancer with EGFR-TKI resistance. Eur J Cancer. 2017;84:184–92. doi: 10.1016/j.ejca.2017.07.037. [DOI] [PubMed] [Google Scholar]
- 11.Song Z, Lin Y, Zhang X, Feng C, Lu Y, Gao Y, Dong C. Cyclic RGD peptide-modified liposomal drug delivery system for targeted oral apatinib administration: enhanced cellular uptake and improved therapeutic effects. Int J Nanomedicine. 2017;12:1941–58. doi: 10.2147/IJN.S125573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan M, Zhang J, Wang Z, Wang B, Zhang Q, Zheng C, Li T, Ni C, Wu Z, Shao Z, Hu X. Phosphorylated VEGFR2 and hypertension: potential biomarkers to indicate VEGF-dependency of advanced breast cancer in anti-angiogenic therapy. Breast Cancer Res Treat. 2014;143:141–51. doi: 10.1007/s10549-013-2793-6. [DOI] [PubMed] [Google Scholar]
- 13.Yang W, Jiang C, Xia W, Ju H, Jin S, Liu S, Zhang L, Ren G, Ma H, Ruan M, Hu J. Blocking autophagy flux promotes interferon-alpha-mediated apoptosis in head and neck squamous cell carcinoma. Cancer Lett. 2019;451:34–47. doi: 10.1016/j.canlet.2019.02.052. [DOI] [PubMed] [Google Scholar]
- 14.Yang CZ, Ma J, Zhu DW, Liu Y, Montgomery B, Wang LZ, Li J, Zhang ZY, Zhang CP, Zhong LP. GDF15 is a potential predictive biomarker for TPF induction chemotherapy and promotes tumorigenesis and progression in oral squamous cell carcinoma. Ann Oncol. 2014;25:1215–22. doi: 10.1093/annonc/mdu120. [DOI] [PubMed] [Google Scholar]
- 15.Hirsch FR, Varella-Garcia M, Bunn PJ, Di Maria MV, Veve R, Bremmes RM, Baron AE, Zeng C, Franklin WA. Epidermal growth factor receptor in non-small-cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis. J. Clin. Oncol. 2003;21:3798–807. doi: 10.1200/JCO.2003.11.069. [DOI] [PubMed] [Google Scholar]
- 16.Huang M, Huang B, Li G, Zeng S. Apatinib affect VEGF-mediated cell proliferation, migration, invasion via blocking VEGFR2/RAF/MEK/ERK and PI3K/AKT pathways in cholangiocarcinoma cell. Bmc Gastroenterol. 2018;18:169. doi: 10.1186/s12876-018-0870-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun Y, Liu W, Liu T, Feng X, Yang N, Zhou H. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J Recept Sig Transd. 2015;35:600–604. doi: 10.3109/10799893.2015.1030412. [DOI] [PubMed] [Google Scholar]
- 18.Fathi Maroufi N, Rashidi MR, Vahedian V, Akbarzadeh M, Fattahi A, Nouri M. Therapeutic potentials of apatinib in cancer treatment: possible mechanisms and clinical relevance. Life Sci. 2020;241:117106. doi: 10.1016/j.lfs.2019.117106. [DOI] [PubMed] [Google Scholar]
- 19.Zhao S, Ren S, Jiang T, Zhu B, Li X, Zhao C, Jia Y, Shi J, Zhang L, Liu X, Qiao M, Chen X, Su C, Yu H, Zhou C, Zhang J, Camidge DR, Hirsch FR. Low-dose apatinib optimizes tumor microenvironment and potentiates antitumor effect of PD-1/PD-L1 blockade in lung cancer. Cancer Immunol Res. 2019;7:630–643. doi: 10.1158/2326-6066.CIR-17-0640. [DOI] [PubMed] [Google Scholar]
- 20.Song Y, Ma T, Zhang X, Cheng X, Olajuyin A, Sun Z, Zhang X. Apatinib preferentially inhibits PC9 gefitinib-resistant cancer cells by inducing cell cycle arrest and inhibiting VEGFR signaling pathway. Cancer Cell Int. 2019;19:117. doi: 10.1186/s12935-019-0836-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng X, Feng H, Wu H, Jin Z, Shen X, Kuang J, Huo Z, Chen X, Gao H, Ye F, Ji X, Jing X, Zhang Y, Zhang T, Qiu W, Zhao R. Targeting autophagy enhances apatinib-induced apoptosis via endoplasmic reticulum stress for human colorectal cancer. Cancer Lett. 2018;431:105–114. doi: 10.1016/j.canlet.2018.05.046. [DOI] [PubMed] [Google Scholar]
- 22.Sperandio S, de Belle I, Bredesen DE. An alternative, nonapoptotic form of programmed cell death. Proc Natl Acad Sci U S A. 2000;97:14376–81. doi: 10.1073/pnas.97.26.14376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Djavaheri-Mergny M, Maiuri MC, Kroemer G. Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin 1. Oncogene. 2010;29:1717–9. doi: 10.1038/onc.2009.519. [DOI] [PubMed] [Google Scholar]
- 24.Li X, He S, Ma B. Autophagy and autophagy-related proteins in cancer. Mol Cancer. 2020;19:12. doi: 10.1186/s12943-020-1138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA. Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist Update. 2008;11:32–50. doi: 10.1016/j.drup.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhai H, Nakade K, Oda M, Mitsumoto Y, Akagi M, Sakurai J, Fukuyama Y. Honokiol-induced neurite outgrowth promotion depends on activation of extracellular signal-regulated kinases (ERK1/2) Eur J Pharmacol. 2005;516:112–7. doi: 10.1016/j.ejphar.2005.04.035. [DOI] [PubMed] [Google Scholar]
- 27.Kong LR, Chua KN, Sim WJ, Ng HC, Bi C, Ho J, Nga ME, Pang YH, Ong WR, Soo RA, Huynh H, Chng WJ, Thiery JP, Goh BC. MEK inhibition overcomes cisplatin resistance conferred by SOS/MAPK pathway activation in squamous cell carcinoma. Mol Cancer Ther. 2015;14:1750–60. doi: 10.1158/1535-7163.MCT-15-0062. [DOI] [PubMed] [Google Scholar]
- 28.Rong C, Muller MF, Xiang F, Jensen A, Weichert W, Major G, Plinkert PK, Hess J, Affolter A. Adaptive ERK signalling activation in response to therapy and in silico prognostic evaluation of EGFR-MAPK in HNSCC. Br J Cancer. 2020;123:288–297. doi: 10.1038/s41416-020-0892-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.