

Abstract
Oxidative stress is a major component of most major retinal diseases. Many extrinsic anti-oxidative strategies have been insufficient at counteracting one of the predominant intrinsic sources of reactive oxygen species (ROS), mitochondria. The proton gradient across the inner mitochondrial membrane is a key driving force for mitochondrial ROS production, and this gradient can be modulated by members of the mitochondrial uncoupling protein (UCP) family. Of the UCPs, UCP2 shows a widespread distribution and has been shown to uncouple oxidative phosphorylation, with concomitant decreases in ROS production. Genetic studies using transgenic and knockout mice have documented the ability of increased UCP2 activity to provide neuroprotection in models of a number of diseases, including retinal diseases, indicating that it is a strong candidate for a therapeutic target. Molecular studies have identified the structural mechanism of action of UCP2 and have detailed the ways in which its expression and activity can be controlled at the transcriptional, translational and posttranslational levels. These studies suggest a number of ways in control of UCP2 expression and activity can be used therapeutically for both acute and chronic conditions. The development of such therapeutic approaches will greatly increase the tools available to combat a broad range of serious retinal diseases.
Keywords: Retina, Reactive oxygen species (ROS), Uncoupling, UCP, Mitochondria, Neuroprotection
1. Introduction
The socioeconomic cost of retinal degenerative diseases is large, and growing. Blindness, low vision, age-related macular degeneration, glaucoma, and diabetic retinopathy affect over 15.4 million people in the USA, a number that is expected to be 24.3 million by 2050 (Varma et al., 2016; Wittenborn & Rein, 2014). As of 2013, the economic burden of eye disease was estimated to be $139 billion (Wittenborn et al., 2013). The impact of these diseases will only increase, with no solution in sight. While many of them have unique genetic or environmental origins, they also share significant pathophysiological abnormalities, including an imbalance in the production of reactive oxygen species (ROS) and their detoxification, which is referred to as oxidative stress (Altomare et al., 1997; Kimura et al., 2017; Lefevere et al., 2017; Totan et al., 2009; Wang et al., 2008). In the eye and in other systems, oxidative stress can damage a wide variety of molecules, and may be a shared principal cause of degeneration.
This review discusses oxidative stress in a broad context, and then focuses on a group of proteins that we propose hold great promise for therapeutic intervention to ameliorate the effects of oxidative stress in multiple eye diseases. Mitochondrial uncoupling proteins (UCPs) are endogenous proteins that exhibit anti-oxidative activity in a variety of conditions (Diano et al., 2003; Hass & Barnstable, 2019a; Mailloux & Harper, 2011; Mehta & Li, 2009; Nègre-Salvayre et al., 1997; Qin et al., 2019). These proteins partially dissipate the electrochemical gradient in mitochondria, leading to a partial ‘uncoupling’ of electron transport from proton translocation, a critical metabolic process. By doing so, UCPs can also decrease the driving force by which mitochondria generate ROS.
In this review we will discuss sources of energy in the retina, how these relate to the generation of ROS, and a more specific mechanism by which UCPs are thought to decrease ROS. This function is invariably tied to structure, and we will further describe what is known of the gene and protein structure of a particular member of the UCP, UCP2. Of all UCPs, UCP2 is expressed in the greatest diversity of tissues, and appears to be under the tightest regulation, making it a prime candidate as a therapeutic target, the effect of which could be applied to a number of tissues. We will further discuss factors that regulate UCP2 abundance and function, as well as the roles of this protein in normal physiology and disease.
2. Retinal energetics and mitochondria
2.1. Anatomical and Metabolic Compartments of the Eye
The eye is divided into two main compartments along its anterior-posterior axis, each having distinct anatomical and metabolic properties that contribute to differing energetic requirements and burdens of oxidative stress. The anterior chamber is bounded by the cornea and lens. At the anterior aspect of the eye, the avascular cornea is directly exposed to atmospheric molecular oxygen (O2) diffusing through the tear film. Despite high levels of O2, oxygen consumption is surprisingly low in multiple parts of the anterior chamber, including both the cornea and lens (Freeman, 1972; Trayhurn & Van Heyningen, 1971). Instead of utilizing mitochondrial metabolism, most glucose is converted to lactate in these tissues during the process of anaerobic glycolysis (Freeman, 1972). This process likely limits generation of mitochondria-derived ROS in the cornea. However, a high concentration of atmospheric O2 itself can be a source of ROS through non-mitochondrial pathways, and unsurprisingly human corneal endothelial cells express high levels of the anti-oxidant protein peroxiredoxin, encoded by the PRDX6 gene, on their cell surface (Lovatt et al., 2018).
In the posterior chamber of the eye, the neural retina, retinal pigment epithelium (RPE), the choroicapillaris, and sclera are more distant from the cornea and have more restricted access to O2. Alongside O2, glucose, lactate, and other circulating nutrients collectively power the generation of adenosine triphosphate (ATP). While anaerobic glycolysis appears to be the principal form of metabolism in the anterior chamber, cells in the posterior chamber exhibit a more varied mixture of glucose utilization in aerobic glycolysis and anaerobic glycolysis.
The retina is the most complex functional unit of the posterior pole, and the distinct layers of the retina are served by blood from both the central retinal artery and choroidal vasculature. Astrocytes, müller glia, microglia, bipolar cells, amacrine cells, and ganglion cells of the inner retina are supplied with metabolic fuel through the central retinal artery, whereas photoreceptors, horizontal cells, and the RPE are supplied by the choriocapillaris. Müller glia and retinal astrocytes in combination with capillary endothelial cells comprise a ‘blood-retina barrier’ that regulates the diffusion of serum macro-molecules to the retina. RPE cells form a similar barrier between the retina and fenestrated capillaries of the choroidal blood supply. There are substantial physiological differences between the retinal and choroidal vasculature. Retinal arterial circulation is characterized by low blood flow and high oxygen extraction, whereas blood flow in choroidal vessels is higher, but oxygen extraction is low (Campochiaro, 2000). In retinal vessels, efficient autoregulation is mainly influenced by local factors, whereas choroidal circulation is controlled by sympathetic innervation (Boussery et al., 2002; Delaey & Van De Voorde, 2000; Steinle et al., 2000). The difference in blood flow regulation is also reflected in the differences between the expression of angiogenic regulators and cytokines in the endothelial cells of these barriers (Brylla et al., 2003).
2.2. The Retina Has a Particularly High Energy Demand
Blood flow disproportionately serves neural tissues, including the retina. A sampling of blood flow to a several neural and non-neural tissues in the Antarctic seal found that the energy supply to the visual system is uniquely high (Niven & Laughlin, 2008), as it is in the brains of many species (Niven & Laughlin, 2008). The high energy demand in the retina arises in part from the electrophysiological properties of photoreceptors. In darkness the photoreceptor plasma membrane is continuously depolarized due to tonic activity of cyclic nucleotide-gated cation channels. These channels allow entry of Na+ and Ca2+ ions along their concentration gradient (Fesenko et al., 1985; Hagins et al., 1970). These ions are pumped out of cells by Na+/K+- and Ca2+-ATPases (Krizaj & Copenhagen, 1998; Okawa et al., 2008; Stahl & Baskin, 1984). Based on the contribution these ATPases make to electrophysiological responses of photoreceptors and the stoichiometry of ion currents to ATP hydrolysis, the metabolic costs of maintaining a depolarized plasma membrane potential in darkness are estimated to outweigh any of the other events in photoreceptor signal transduction, particularly in darkness (Okawa et al., 2008). The energy requirements of cone photoreceptors may be even higher than that of rods, given that rod phototransduction can be saturated at higher intensities of light but cone phototransduction does not saturate (Burkhardt, 1994; Fain, 1976). These atypical cone energy requirements may explain their extremely dense cluster of mitochondria that outstrips even that of rods (Hoang et al., 2002).
RPE phagocytizes and recycles photoreceptor outer segments and is critical for photoreceptor biology. Being located close to choroidal blood supply, the RPE has more immediate access to nutrients than photoreceptors. While a it has become clear that RPE is able to utilize a broad spectrum of metabolic substrates (Bisbach et al., 2020b; Chao et al., 2017; Kanow et al., 2017), it is not yet clear whether the energetic demands of RPE are comparable to those of photoreceptors, or whether RPE metabolic flexibility exists primarily to spare glucose for utilization by photoreceptors (Kanow et al., 2017).
Retinal ganglion cells (RGCs) in the inner retina are another critical consumer of ATP. These projection neurons integrate outer retinal signals and transmit them along the optic nerve, which in humans can be ~45 mm long (Bernstein et al., 2016). It is likely that the length of the axons of these cells proportionally increases their mass and according to Kleiber’s allometric scaling law, their demand for ATP (West et al., 2002; Yu et al., 2013). A significant portion of these axons are unmylinated, which may increase the energetic cost of signal transduction roughly five-fold per axon (Neishabouri & Faisal). To supply the energy that can meet this high demand RGCs are outfitted with dense clusters of mitochondria, much like photoreceptors in the outer retina (Rueda et al., 2016; Yu et al., 2013).
2.3. Mitochondrial Energy Generation
Cells generate ATP primarily by two metabolic pathways, anaerobically by converting glucose to lactate in glycolysis, or aerobically through the mitochondrial electron transport chain (ETC). Of the two, the ETC is more efficient and can generate 30-32 mol ATP per mol glucose, whereas glycolysis can only generate 2-3 mol ATP per mol glucose (Alberts et al., 2008).
The ETC generates ATP using a combination of four distinct multiprotein complexes (Complex I-IV) and two electron carriers (coenzyme Q and cytochrome C) to extract electrons from NADH or succinate and use those electrons to reduce molecular oxygen (O2) to water (Figure 1) (Alberts et al., 2002). The affinity of mitochondrial components for electrons increases in each sequential reaction of the electron transport chain, and the terminal reaction at cytochrome C oxidase (½ O2 + 2H+ + 2e− → H2O) is associated with an energetically favorable redox potential (E°≈820 mV). The favorability of ETC redox reactions drives translocation of protons (H+) from the mitochondrial matrix to the intermembrane space at complexes I, III, and IV. H+ translocation ‘charges’ mitochondria with a H+-derived electrochemical gradient, known as the proton-motive force (PMF) (Mitchell, 1961). The PMF favors the flow of H+ from the intermembrane space back to the matrix, and this flow powers rotary movement of the F1F0-ATP synthase, forming ATP from adenosine diphosphate (ADP) and inorganic phosphate (Abrahams et al., 1994). The PMF can be dissected into two distinct but interacting gradients – an electrical gradient known as the mitochondrial membrane potential (ΨM), and a concentration gradient, which for H+ is the pH.
Figure 1. Main Components of the Electron Transport Chain and their Roles in Forming ROS.

(A) In this simplified model of the electron transport chain, electrons are either derived from oxidation of NADH by complex I (NADH:CoQ Oxidoreductase) or succinate by complex II (succinate dehydrogenase). These electrons are transported through complex III (Coenzyme Q: Cytochrome C reductase) and complex IV (Cytochrome C Oxidase) where they power translocation of protons from the mitochondrial matrix to the intermembrane space, before finally being used to reduce O2 to H2O at complex IV. (B) Should more NADH be supplied than complex I can quickly oxidize, electrons from NADH will interact with O2 to form a superoxide molecule (a form of ROS) (C) Uncoupling, mediated by a protonophore or an uncoupling protein, can decrease production of superoxide by NADH. (D) Should more succinate be supplied than complex III or IV can transport electrons from, electrons from succinate can be transported ‘in reverse’ to complex I through the shared complex I and complex II substrate coenzyme Q10. Reverse electron transport through complex I further aggravates superoxide production. (E) Uncoupling mediated by a protonophore or an uncoupling protein may allow complex I, III, or IV to transport electrons more quickly in the conventional ‘forward direction’, preventing reverse electron transport and thus aggravated superoxide production.
Measures of enzyme activity show that inner retinal layers are enriched for mitochondrial cytochrome C oxidase (COX) activity (Hevner & Wong-Riley, 1990; Kageyama & Wong-Riley, 1984). This enzyme is a key component of the ETC, and COX enrichment suggests an enhanced capacity for generating ATP that supports high metabolic requirements associated with the continuous transmission of visual signals by inner retinal neurons such as RGCs. In a mouse model of glaucoma, we have found significant impairment of retinal COX activity and a decrease in mitochondrial DNA – both are likely the result of mitochondrial damage related to elevated intra-ocular pressure (Figure 2). Additionally, retinas from mice lacking the complex I component gene Ndufs4 exhibit less light-stimulated electrical activity in the retina, and possess fewer retinal ganglion cells (Yu et al., 2015). Similarly, dysfunction in the mitochondrial electron transfer chain proteins ND1, ND4 and ND6 give rise to Leber’s hereditary optic neuropathy, a disease that only affects retinal ganglion cells (Yen et al., 2006). This further emphasizes the high dependence of these cells on mitochondrial function. These data also support the hypothesis that in neurodegenerative disorders like glaucoma, oxidative damage impairs mitochondrial energy generation, and in turn an impaired ability of the retina to generate ATP alone is sufficient for further degeneration (Kong et al., 2009). It will be interesting to see whether mitochondrial defects can be compensated by modulating the activity of UCPs, a potentially more effective and more general approach than individualized gene therapy.
Figure 2. Elevated intraocular pressure impairs mitochondrial function and decreases mtDNA/nDNA in a mouse model of glaucoma.

Impaired mitochondrial function in microbead-injected Ucp2flox/flox retinal tissue. (A) Representative histochemical labeling of cytochrome oxidase activity in retinal tissue sections 3 days following bead injections, and (B) measurement of 3,3’-diaminobenzidine intensity (n=5). (C) mtDNA/nDNA was also measured in this tissue, and this measure of mitochondrial mass was unchanged 3 days after bead injection (n=3), but was significantly decreased 30 days following bead injection (n=8). *p<0.05. Figure and legend were reproduced and modified with permission from (Hass & Barnstable, 2019b).
2.4. ROS, and Their Sources in Mitochondria
A high density of mitochondria enables retinal neurons to generate significant amounts of ATP, but these mitochondria are among the most prevalent cellular sources reactive oxygen species (ROS) (Kudin et al., 2008; Wong et al., 2019). ROS are reactive oxygen-containing molecules generated from a progenitor superoxide (), which itself is formed through the direct interaction of O2 and an electron in or outside of the ETC (Murphy, 1989; St-Pierre et al., 2002). ROS themselves are often free radicals (for example HO•, ) or their derivatives (for example H2O2). Each of these molecules has unique properties that governs which molecules they are more likely to react with and which cellular barriers they are more likely to cross, reviewed more extensively in (Collin, 2019). ROS can react with and damage nuclear or mitochondria DNA (Abu-Amero et al., 2006; Izzotti et al., 2003), carbohydrates, lipids (Esterbauer et al., 1991), or proteins (Tezel et al., 2005; Yang et al., 2016; Zhao et al., 2014) to form semi-permanent adducts that impair cell and tissue function (Hashizume et al., 2008; Sela et al., 1993), or may alternatively participate in cell signaling (D'Autréaux & Toledano, 2007). As much as 0.15% of ETC flux can form superoxide radicals, though production of this and other ROS are dependent on the type of ETC substrate used by mitochondria (ex. NADH vs. succinate) (St-Pierre et al., 2002), polarity of the PMF (Korshunov et al., 1997; Murphy, 2009), and the amount of mitochondrial material (Brand, 2016; Murphy, 1989, 2009) (Figure 1B,C).
Substrates can determine the extent of ROS generation because different electron donors interact with distinct mitochondrial complexes, with each complex possessing a different capability to generate ROS. There are at least five different small molecule mitochondrial electron donors that feed electrons to the ETC (NADH, succinate, glycerol-3-phosphate, fattyacyl-CoA and dihydroorotate) through a combination of conventional ETC complexes and other proteins which reduce coenzyme Q10 (Brand, 2016). Among the mitochondrial components which handle these electrons, complex I (NADH-Ubiquinone Oxidoreductase) appears to ‘leak’ electrons that form ROS to the greatest extent, though it is clear that complex III (the Ubiquinone-Cytochrome C Oxidoreductase) is another important source and that ROS are not exclusively produced from a single site in mitochondria (Hirst et al., 2008; Kudin et al., 2008; St-Pierre et al., 2002; Wong et al., 2019). The production rate of ROS from a given site or substrate is modulated by the balance between the delivery and utilization of that substrate to mitochondria. For example, a high [NADH] reduces the flavin site of complex I, and electrons from this site interact with O2 to form molecules (Figure 1B) (Hirst et al., 2008; Pryde & Hirst, 2011). The polarity of the PMF is another important factor in the generation of ROS. At complex I, the PMF is an electrochemical gradient against which H+ are pumped. Without that thermodynamic barrier, NADH may be oxidized more quickly so that [NADH] does not build up or interact with O2 through the complex I flavin site.
ROS may also be formed through a combination of supraphysiological levels of the complex II substrate succinate and a high PMF. Together, these conditions can ‘reverse’ the direction of mitochondrial electron transport, and causing electrons to flow ‘backwards’ towards complex I (Murphy, 2009). This happens because the flow of electrons from succinate to coenzyme Q10 can push reduced coenzyme Q10 to not only participate in the electron transport at complex III, but also to drive complex I in reverse (Figure 1C) (Murphy, 2009). Reverse electron transport at complex I can result in up to 2% of electron flux forming ROS - provided that these circumstances are set using the complex III inhibitor antimycin A (Chance et al., 1979). Such inhibitors, as well as hypoxia or anoxia, can assist in causing ETC reversal by blocking the exit of electrons from the ETC and ‘backing up’ their flow (Chouchani et al., 2014). Just as a hyperpolarized PMF may contribute to reversing electron transport, dissipation of the PMF can decrease the generation of ROS by reverse electron transport (Pryde & Hirst, 2011; Robb et al., 2018). Chemical uncouplers and uncoupling proteins are able to dissipate PMF, ‘uncoupling’ mitochondrial respiration from PMF-powered ATP synthesis, and this hypothesized mechanism of action has propelled studies in their possible use to decrease the generation of mitochondrial ROS.
One limitation on studies of how mitochondria can generate ROS is the question of whether a high PMF or reverse electron transport-stimulated H2O2 are generated in vivo. Recent advances in high-resolution imaging approaches have revealed substantial variance in ΨM between cristae of the same mitochondrion, which could suggest that in a subset of cristae, ΨM may be sufficiently polarized to drive ROS formation (Wolf et al., 2019). Increasing evidence also suggests that mitochondrial reverse electron transport at complex I drives macrophage polarization (Mills et al., 2016), the formation of ROS during ischemia-reperfusion injury (Chouchani et al., 2014), and may even increase fruit fly lifespan (Scialò et al., 2016). These all suggest that increases in PMF and thus ROS are possible under physiological and/or pathological circumstances.
Whether this unique form of ‘reversed’ mitochondrial metabolism occurs physiologically or pathologically in the retina is less clear, though a recent study of metabolic flux in the mouse retina has shown that a metabolic pathway unique to hypoxic tissues (Chouchani et al., 2014; Hochachka & Dressendorfer, 1976; Hochachka et al., 1975) likely operates in the retina – instead of complex II activity forming fumarate from succinate, it ‘reverses‘ and forms succinate from fumarate (Bisbach et al., 2020a). Unlike the reverse mode of complex I activity, it is unclear what impact this unique process has on the generation of ROS within the retina, though low levels of O2 in the photoreceptor layers in general may mitigate ROS formation from complex II reversal (Linsenmeier & Zhang, 2017; Yu & Cringle, 2006).
The final causal factor that regulates mitochondrial ROS production is simply the mitochondrial content of a cell or tissue type. Each mitochondrion has a non-zero chance to produce ROS from any given mode of ETC function, so the abundance of mitochondria in a cell will amplify whichever of these modes of ROS generation is most dominant (Boveris & Chance, 1973). Mitochondria are particularly dense in photoreceptors and retinal ganglion cells, and this fact alone may suggest a high burden of ROS generation in the retina. However, due to high levels of oxygen consumption by a combination of photoreceptor and RPE mitochondria, O2 tension is low close to photoreceptor nuclei and inner segments (Lau & Linsenmeier, 2012; Linsenmeier & Zhang, 2017; Yu & Cringle, 2001, 2006), suggesting that effects of high mitochondrial density on ROS production may be moderated by lower concentrations of oxygen available to form those ROS (Maddalena et al., 2017). Alternately the influence of mitochondrial density on ROS may be more prevalent in the inner retina, where oxygen tension appears higher closer to where retinal ganglion cell mitochondria are localized, at least in vascularized mammalian retinas (Yu & Cringle, 2001).
Most of the discussion above is applicable to all tissues. The retina, however, has additional factors influencing ROS production. Retinal cells are affected in a continuous manner by light-induced oxidative stress. This is particularly true for photoreceptors transforming light in the phototransduction process and the presence of many visual pigment derivatives generating ROS. There is also growing evidence that light can influence ROS generation in retinal ganglion cells and this may be a contributing factor in glaucoma (Zhao & Shen, 2020).
2.5. What are the consequences of mitochondrial ROS, and how are they circumvented?
ROS are important contributors to disease in many tissues and organs. Within the CNS, ROS thought to be central components of Alzheimer’s Disease (Baldeiras et al., 2010; Massaad, 2011; Polidori et al., 2007), Parkinson’s Disease (Alam et al., 1997; Floor & Wetzel, 1998; Yoritaka et al., 1996), Huntington’s disease (Chen et al., 2007; Rotblat et al., 2014; Sorolla et al., 2008), glaucoma (Carter-Dawson et al., 2004; Izzotti et al., 2003; Tezel et al., 2005), age-related macular degeneration (Frank et al., 1999; Totan et al., 2009; Venza et al., 2012), diabetic retinopathy (Altomare et al., 1997; Paget et al., 1998), and Leber’s hereditary optic neuropathy (Wang et al., 2008; Wang et al., 2005; Yen et al., 2004), amongst others. In all of these conditions, excessive generation of ROS that is not counterbalanced by antioxidant systems appears to be a mechanism leading to cellular dysfunction and death. This is exemplified by genetic deletion of antioxidant proteins, which can disrupt the balance between ROS generation and detoxification in the retina. More specifically, deletion of either cytosolic Sod1 or mitochondrial Sod2 genes coding for the anti-oxidative superoxide dismutase enzyme results in degeneration of both the inner and outer retina, demonstrating that constant cytosolic and mitochondrial detoxification of ROS is necessary for maintenance of retina health (Hashizume et al., 2008; Sandbach et al., 2001).
Exogenous antioxidants are thought to prevent the accumulation of ROS-mediated damage. Cell culture and animal studies have shown that different small molecule antioxidants or transcription factors which code for anti-oxidative proteins can limit cell death and degeneration (Noh et al., 2013; Xiong et al., 2015; Yang et al., 2016). The factor RDCVF, a trophic factor of the thioredoxin family, can protect rod photoreceptors against a number of oxidative stresses including light damage (Elachouri et al., 2015). RDCVF also enhances glucose consumption in cone photoreceptors as well as preventing oxidative stress (Léveillard & Sahel, 2010). This provides an interesting illustration that cell survival at a high metabolic rate requires antioxidant strategies. Despite successful studies in animals, studies of anti-oxidants have not yet yielded a therapeutic that consistently prevents neurodegeneration in humans (Garcia-Medina et al., 2015). One reason for this may be insufficient penetration to target tissues, particularly to the subcellular compartment that act as a source of ROS (i.e. mitochondria). Research is underway to mitigate this issue by developing compounds that better penetration to mitochondria, such as triphenylphosphonium derivatives of preexisting antioxidants (Anders et al., 2006; Murphy & Smith, 2007; Zielonka et al., 2017). Other reasons for insufficient antioxidant efficacy in the clinic may include modest ROS buffering due to poor antioxidative capacity, duel antioxidant/pro-oxidant activities that are dependent on the cellular microenvironment, or an inability for antioxidants to localize to critical cellular compartments (Carr & Frei, 1999). As drugs with antioxidant activity continue to be discovered, we expect that it is only a matter of time until these latter issues are also addressed.
However, while neutralizing ROS is an important component of the body’s defenses, the central thesis of this review is that it is more effective to block their production, particularly using endogenous factors that have already been optimized by evolution. Because mitochondria with a high PMF produce more ROS, depolarization of the mitochondrial membrane can attenuate the production of ROS, a concept that is well established in isolated mitochondria and submitochondrial particles (Korshunov et al., 1997; Pryde & Hirst, 2011; Robb et al., 2018). By providing an alternative route for hydrogen ions to pass through the inner mitochondrial membrane, depolarization of ΨM also ‘uncouples’ ATP synthesis from electron transport. Uncoupling, and consequent reductions in the generation of ROS may be accomplished in several ways.
The first of these is through the use of exogenous compounds such as 2,4-dinitrophenol (DNP), carbonyl cyanide m-chlorophenyl hydrazone (CCCP), carbonyl cyanide-p-trifluoromethoxyphenyl hydrazone (FCCP). These are chemical uncouplers, and their activity induces uncontrolled depolarization of the mitochondrial membrane. These compounds have been used (sometimes illegally) to decrease the efficiency of ATP synthesis to promote weight loss, though at higher doses they are lethal (Grundlingh et al., 2011). Such unregulated compounds are too toxic to be a likely means of decreasing the production of ROS.
A more physiological form of uncoupling results from activity of proteins or complexes such as the F1F0-ATP synthase, adenine nucleotide translocase, aspartate-glutamate carrier, and phosphate carrier (Kunji & Robinson, 2010). These transport reactions cause a ‘leak’ of protons across the inner mitochondrial membrane that powers solute transport. Because these processes are intertwined, manipulation of transporter levels or activity that may decrease ROS could also affect the distribution of metabolites inside mitochondria and thus have unforeseen consequences on the generation of ROS.
A third mechanism of uncoupling in mitochondria, and one that offers strong therapeutic potential, is catalyzed by mitochondrial uncoupling proteins, or UCPs (Parker et al., 2009; Sluse, 2012; Vozza et al., 2014). UCPs mediate an inducible H+ leak that is more tightly regulated than that of DNP, FCCP, or CCCP (Parker et al., 2009). By regulating the PMF, UCPs are thought to regulate production of ROS. The next sections are dedicated to broadly introducing UCPs by describing important aspects of UCP genetics, physiology, regulation, as well as how these govern the generation of ROS in the retina.
3. Mitochondrial Uncoupling Proteins
3.1. Phylogeny of Uncoupling Protein
Uncoupling proteins (UCPs) belong to the SLC25 superfamily of mitochondrial solute carrier genes. In humans, this superfamily has 53 members, all of which are encoded in nuclear DNA. Within this superfamily, the 5 membered UCP family exists in the mouse and human genomes (UCP1-UCP5), with each UCP numbered in the order of their discovery.
Different forms of UCPs can be found at many branches of the phylogenetic tree, with UCP paralogs in vertebrates, invertebrates, and even plants (Figure 3) (Gaudry & Jastroch, 2019; Nogueira et al., 2005). This phylogeny largely agrees with other studies where initial sequence comparisons between the human UCP family and putative UCPs from Drosophila and C. elegans suggested that UCP4 may be the ancestral form of uncoupling protein from which the others are derived (Hanák & Jezek, 2001). Later studies have revised this model and found an early divergence of the ancestral UCP into three branches that preceded the divergence of protostomes and deuterostomes (Figure 4) (Sokolova & Sokolov, 2005). Two of these branches evolved into UCP4s and UCP5s found in both vertebrates and invertebrates (Hughes & Criscuolo, 2008). The third gave rise to an invertebrate UCP (UCP6) and the precursor of vertebrate UCP1, UCP2 and UCP3 (Hughes & Criscuolo, 2008; Sokolova & Sokolov, 2005). Comparison of nucleotide sequences and gene locations suggest that UCP2 and UCP3 are closely related to each other. They are adjacent to each other on human chromosome 11 (mouse chromosome 7), and are generally thought to be the result of an evolutionarily recent gene duplication event (Pecqueur et al., 1999). As the members of this third branch were the first to be identified and characterized, they are also foundational to our understanding of UCP structure and function.
Figure 3. Unrooted phylogenetic tree for mammalian UCPs, plant uncoupling proteins (PUMPs) and other mitochondrial carrier protein sequences.
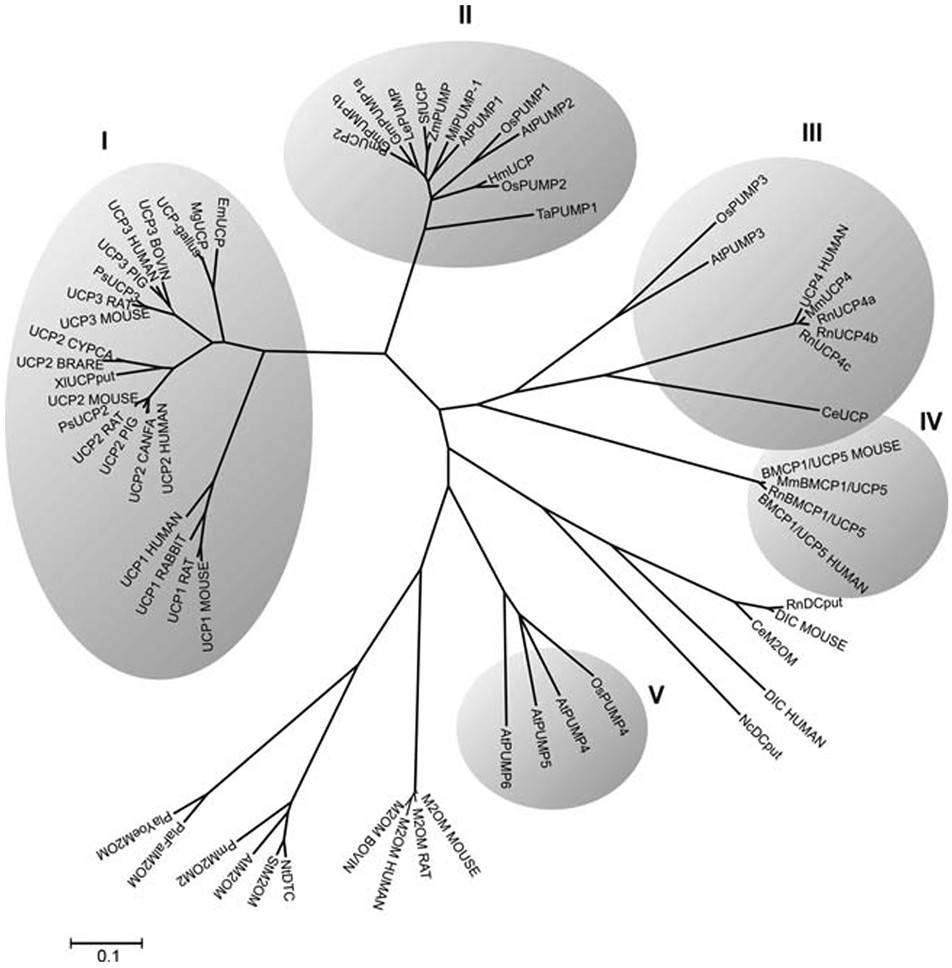
The lengths of the lines connecting genes corresponds to the relative evolutionary distance (sequence divergence) between them. The clusters (shaded) represent groups of genes that are much more closely related to each other than to any others on the tree. UCP1, UCP2 and UCP3 appear in cluster I, UCP4 in cluster III and UCP5 in cluster IV. Figure reproduced from (Nogueira et al., 2005) with permission. The tree indicates an early separation of UCP4 and 5, a second separation on non-vertebrate uncoupling proteins and then the later separation into the UCP1,2, and 3 forms of uncoupling protein. The following sequences (accession numbers in parentheses) were used for the phylogenetic analysis: UCP1_MOUSE (P12242), UCP1_RAT (P04633), UCP1_RABBIT (P14271), UCP1_HUMAN (P25874), UCP2_HUMAN (P55851), UCP2_CANFA (Q9N2J1), UCP2_RAT (P56500), PsUCP2 (AAG33984), UCP2_MOUSE (P70406), UCP2_PIG (097562), X1UCPput (AAH44682), UCP2_BRARE (Q9W720), UCP2_CYPCA (Q9W725), UCP3_MOUSE (P56501), UCP3_RAT (P56499), PsUCP3 (AAG33985), UCP3_CANFA (Q9N2I9), UCP3_PIG (097649), UCP3_HUMAN (P55916), UCP3_BOVIN (077792), UCP-gallus (AAL35325.2), MgUCP (AAL28138), EmUCP (AAK16829), PmUCP2 (AAL92117), GmPUMPlb (AAL68563), GmPUMPla (AAL68562), MiPUMP-1 (AAK70939), LePUMP (AAL82482), AtPVMP1 (CAA11757), SfUCP (BAA92172), HmUCP (BAC06495), ZmPUMP (AAL87666), OsPUMP1 (BAB40657), OsPUMP2 (BAB40658), OsPUMP3 (CAE01569), OsPUMP4 (BAD09745), TaPUMP1 (BAB16385), AtPUMP2 (NP_568894), AtPUMP3 (F7A19_22), AtPUMP4 (F22K18_230), AtPUMP5 (F14M13_10), AtPUMP6 (T5E8_270), DIC_MOUSE (Q9QZD8), RnDCput (NP_596909), DIC_HUMAN (Q9UBX3), CeM2OM (NP_509133), NcDCput (XP_327953), M2OM_MOUSE (Q9CR62), M2OM_RAT (P97700), M2OM_HUMAN (Q02978), M2OM_BOVIN (P22292), NtDTC (CAC84545), StM2OM (CAA68164), AtM2OM (NP_197477), PmM2OM2 (S65042), P1aFa1M2OM (CAD51134), P1aYoeM2OM (EAA21506), BMCP1/UCP5 MOUSE (Q9Z2B2), MmBMCP1/UCP5 (NP_035528), RnBMCP1/UCP5 (NP_445953), BMCP1/UCP5_HUMAN (095258), UCP4_HUMAN (095847), MmUCP4 (BAC66453), RnUCP4a (CAC20898), RnUCP4b (CAC20899), RnUCP4c (CAC20900), and CeUCP (NP_505414). Protein abbreviations are from the SWISPROT database.
Figure 4. Simplified phylogenetic tree of human UCPs.
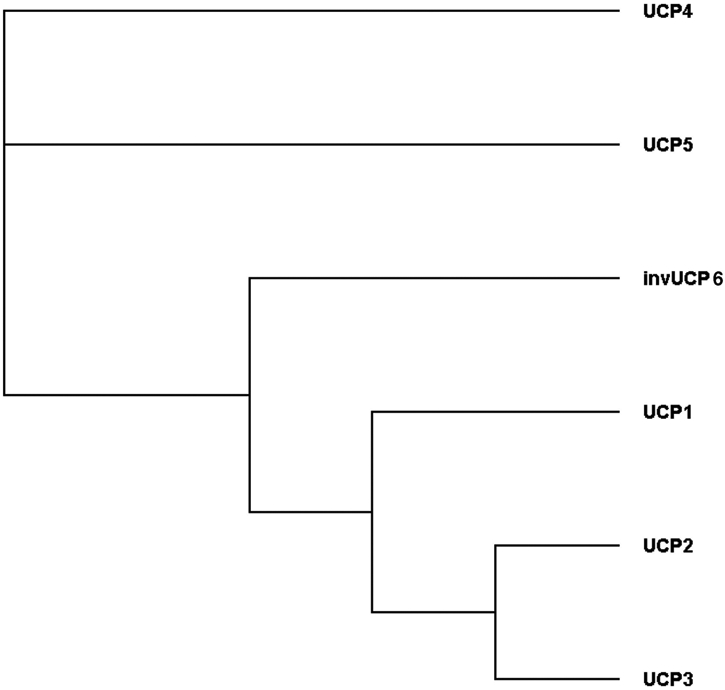
In agreement with the larger tree of Figure 2, we compared protein sequence homologies using the Clustal Omega program (Madeira et al., 2019) give rise to a tree indicating the early divergence of UCP4 and 5 and a later divergence into invertebrate UCPs and a precursor of UCP1, 2 and 3.
As we have mentioned, the primary function of UCPs is to uncouple ATP synthesis from electron transport in mitochondria – this function was derived from the first uncoupling protein to be characterized in detail, UCP1. UCP1 is strongly expressed in brown adipose tissue, where it generates heat by dissipating the PMF, to such a high extent as to induce thermogenesis (Nicholls et al., 1978). UCP2 and UCP3 similarly dissipate the PMF, but as we will discuss, this may not be the only function of these molecules (Bouillaud et al., 2016; Esteves & Brand, 2005). Compared to these other uncoupling proteins however, the functions of UCP4 and UCP5 are less well-understood. While they are able to transport H+ when reconstituted in liposomes (Hoang et al., 2012), mixed evidence suggests that they may (Perreten Lambert et al., 2014) or may not (Chu et al., 2009; Ho et al., 2006) do the same in cells. Once a larger cohort of studies support a unified function for UCP4 and UCP5, it will likely become clear whether the ancestral function of every member of this family is to uncouple, or whether evolutionary changes allow some to act more specifically as H+-coupled metabolite transporters.
3.2. Uncoupling Protein Structure and Mechanism of Uncoupling
Protein function is constrained by structure, and these features often provide hints about each other. Alignment of the protein sequences of the five human uncoupling proteins clearly indicates their homology and identifies key residues that are invariant (Figure 5). The core UCP structure has three repeating units, each with a loop of amino acids passing through the membrane twice, resulting in six transmembrane helices (H1 to H6 – blue bars in Figure 5), In the odd-numbered helices a conserved proline residue introduces a kink in the helix (highlighted in red), and a conserved glycine allows movement of the polypeptide chain (highlighted in yellow). Conserved prolines are also found in helices 4 and 6, further altering the helical structures. There is no crystal structure of any uncoupling protein. However, by fitting the amino acid sequences of uncoupling proteins to the known crystal structure of other SLC25 members it has been possible to obtain high-resolution models of UCP1 and UCP2. These studies used crystal structures of the bovine and Saccharomyces cerevisiae adenine nucleotide translocator proteins (ANT1; Figure 6A) (Pebay-Peyroula et al., 2003). These macromolecules have only a <20% sequence identity but a very similar domain structure and high conservation of certain key amino acid residues (Berardi et al., 2011). The structures have been refined by more recent NMR analyses that also identified the binding sites of several regulatory ligands (Berardi et al., 2011). The six transmembrane helices line a cavity through the membrane (Figure 6B), the motifs after proline-induced kinks (PX[D/E]XX[R/K]X[K/R] generate a salt-bridge network that can close the cavity on the matrix side (Cytosolic gate area or C-state)(Palmieri & Pierri, 2010). A second salt-bridge network is formed by the motif [F/Y][D/E]XX[R/K] at the C-termini of the even numbered helices and this can close the cavity on the intermembrane space, or cytoplasmic, side of the molecule (Matrix gate area or M-state) (Berardi et al., 2011; Palmieri & Pierri, 2010). For most of the carriers of the SLC25 family it is thought that a substrate enters this cavity from the intermembrane space with the protein in the C-state and binding of this substrate induces a conformation changes such that the salt bridges switch to the M-state and the substrate can then exit to the matrix side. These movements are illustrated in Figure 7A, with the actual transitions of one loop repeat shown in Figure 7B and 7C. Relative to ANT1, UCP2 has a reduction in the characteristic 3-fold symmetry of SLC25 members, and has a wider opening to the mitochondrial matrix – though the biophysical implications of structural divergence from other SLC25 family members are unclear, aside from implying a distinct substrate binding preference, with the likely substrate being H+-bound fatty acids (Berardi et al., 2011).
Figure 5. Sequence alignment of the five human UCP proteins.

Sequences used for alignment were the RefSeqs UCP1 (NP_068605.1), UCP2 (NP_003346.2), UCP3 (NP_003347.1), UCP4 (NP_004268.3) and UCP5 (XP_016885426.1). Alignment was carried out using the Clustal Omega program (Madeira et al., 2019). Conserved residues are marked with “*”, and conserved residues with “.” or “:”. The transmembrane helices are underlined in blue, key prolines in these helices outlined in red, and highly conserved glycines in yellow.
Figure 6.

(A) Crystal structure of the Adenine Nucleotide Transporter from which most of the structural models of UCPs are derived. The 3D crystal structure of the carboxyatractyloside-ADP/ATP carrier complex (devoid of the inhibitor) is shown with the following regions in color: cytosolic gate (blue); substrate-binding site (orange); and matrix gate (purple). In P-G level 1 and P-G level 2, prolines are shown in red and glycines in yellow. From (Palmieri & Pierri, 2010) with permission. (B) When the crystal structures are rotated 90 degrees and viewed end on, UCP2 and other members of the SLC25 transporter family have a clear central pore.
Figure 7. Schematic of conformational changes of the six MC transmembrane a-helices occurring during the catalytic transport cycle.

In (A) the structural changes occurring during one transport cycle are shown. At the top left the protein is in the C-state and substrate (proton or protonated fatty acid for UCP2) can enter the pore from the intermembrane space. Following a conformation change, the M-state has an open gate at the gate at the matrix side and release the proton or protonated fatty acid. This cycle is reversible and if the fatty acid molecule is transported back without its proton, the net results is the movement of one proton down its concentration gradient. (B) shows the crystal structure of the first repeat with two transmembrane helices. (C) represents the conformational changes undergone by this repeat in going from the C-state to the M-state. The yellow (glycine) and red (proline) residues are those identified in Figures 4 and 5. Figure reproduced from (Nogueira et al., 2005) with permission.
Uncoupling involves a net movement of hydrogen ions from the intermembrane space into the mitochondrial matrix. The consensus mechanism of transport is that H+ are bound to and carried across the mitochondrial membrane by fatty acids, which are ‘flipped’ by the conformational change between the C- and M-states. UCP2 (Berardi & Chou, 2014). Both mutation studies and NMR experiments have suggested that fatty acid from the intermembrane space binds to a hydrophobic groove between transmembrane helices H1 and H6, and the charged head group interacts with a group of positively charged amino acids. Binding induces a conformational change in the uncoupling protein and the fatty acid is then exposed to the matrix environment where it is deprotonated. Unless the fatty acid flips back, this process will lead to the accumulation of charged fatty acids in the mitochondrial matrix, and the extent to which this may influence mitochondrial physiology is unclear, though it seems likely that a greater concentration of fatty acids at the matrix side of the inner mitochondrial membrane would support an increase in β-oxidation (Pecqueur et al., 2008).
There is still considerable debate about whether fatty acid and H+ transport are the full range of functions for UCP2. Plenty of evidence suggests a capability to transport fatty-acid bound protons, and this transport partially decreases the PMF/ΨM. However, when reconstituted in liposomes, UCP2 will also catalyze transport of phosphate, malate, and aspartate (Vozza et al., 2014). Just as we do not yet understand the complete roles of UCP4 and UCP5 due to the artificial nature of their activities after reconstitution in liposomes, we also cannot yet fully support the concept that UCP2 is a solute carrier of more than H+ and fatty acids. Our skepticism is supported by the evidence that in the brain, UCP2-deficiency does not substantially increase flux of 13C-labeled aspartate (Contreras et al., 2017), which is more likely occur if it acted as an aspartate transporter in vivo. This study does not rule out the possibility of a variety of UCP2-mediated transport mechanisms, but we anticipate that the full function of UCP2 will be addressed in future studies using UCP2-knockout cells or animals.
3.3. Tissue Distribution of Uncoupling Proteins
Each of the five UCPs has a distinct pattern of expression among tissues. UCP1 is predominantly found in brown fat, although it has also been found in T lymphocytes and may be present at lower levels in other tissues (Rupprecht et al., 2012). Of the UCP family, UCP2 appears to have the most ubiquitous tissue distribution of UCP transcripts, though protein expression under normal circumstances is highest in thymus and T-cells (Pecqueur et al., 2001; Rupprecht et al., 2012). UCP3 expression is largely restricted to skeletal, smooth, and cardiac muscle. UCP4 and UCP5 protein has been detected in neurons and glia – with much broader expression of their corresponding transcripts. Transcripts for UCP2, UCP3, UCP4 and UCP5 can also be detected in the mouse retina, though the corresponding amounts of protein are unknown (Barnstable et al., 2003). It is also important to be cautious in interpretation of quantitation of uncoupling protein content in tissues; aside from UCP1, commercially available antibodies are notoriously non-specific and difficult to validate. This may be due to some combination of low expression or poor antibody specificity, but these difficulties with conventional methods of detection can be potentially overcome through the use of home-made (Rupprecht et al., 2012) antibodies or by profiling polyribosome-bound RNA to accurately profile actively translating RNA molecules, which can reveal drastically different patterns of expression relative to total transcript levels (Nie et al., 2015).
These RNA and protein expression patterns determine the extent to which UCPs affect mitochondrial physiology. The highest levels of expression are seen for UCP1 in brown adipose tissue, where it may account for 4-10% of total mitochondrial protein (Heaton et al., 1978; Rousset et al., 2007). Estimates of UCP1 activity suggest that can depolarize the ΨM by as much as 55 mV, which is a drastic amount considering the approximately −120 to −140 mV resting ΨM (Gerencser et al., 2012; Gerencser et al., 2016). In many tissues, UCP2 is approximately two orders of magnitude less abundant than UCP1 is in brown adipose tissue, and its maximal ability to uncouple is approximately 10-15 mV (Ježek et al., 2018). The effect of this uncoupling on generation of ROS is dependent on ΨM. At the resting ΨM or lower potentials, uncoupling has little effect on ROS. However, ROS generation increases exponentially as a function of ΨM past conventional physiological levels (Korshunov et al., 1997), and at a supraphysiological ΨM, changes in the range of 10 mV are sufficient to decrease production of ROS by 30-40% (Korshunov et al., 1997; Lambert & Brand, 2004). Therefore, UCP-mediated mitochondrial uncoupling may decrease ROS under circumstances when ΨM is polarized past the physiological norm. Unfortunately, this theory is difficult to test, as ΨM is not easily quantified in vivo, so whether it is hyperpolarized during certain stages of disease and requires mitochondrial uncoupling is unknown. Thankfully, there have been attempts to develop in vivo probes of ΨM (Logan et al., 2016), though they are not yet in common use.
From here onward we will focus more specifically on one member of the UCP family, UCP2. This UCP has not only the broadest tissue distribution, but is subject to extensive regulation, and regulates of oxidative stress and cell survival, making it a promising target for therapeutic intervention to alleviate numerous diseases across many tissues, including the eye. In particular, how UCP2 expression is regulated will be essential knowledge if it is to be targeted to particular tissues and utilized for a therapeutic purpose.
3.4. Structure and transcriptional regulation of the UCP2 gene
The Ucp2 gene spans over 8.4 kb and contains 8 exons and 7 introns. The exon/intron boundaries are highly conserved with those of other UCPs, although variable intron sizes suggest differences in the capacity for these genes to contain transcriptional activators and repressors. The Ucp2 gene contains two small 5’ untranslated exons separated from the remaining sequence by introns of 1.4 kb and 3.0 kb respectively. Exons 3/4, 5/6 and 7/8 encode the three structural repeats of each molecule respectively. After the coding sequence there is a 339 bp 3’ untranslated region. Ucp2 is expressed in most tissues, where transcription can be regulated through numerous mechanisms (Figure 8A). The Ucp2 promoter contains a serum response element (SRE) and this may mediate the observed enhancement of Ucp2 transcription by growth factors such as LIF and PEDF, both of which appear to activate a signal pathway that involves STAT3 (He et al., 2014; Lapp et al., 2014). The promoter also contains an SP1 site and two E-boxes, and though the factors binding to these have not yet been identified, the E-box motifs appear to be necessary for the binding of other transcriptional regulators, such as PPAR-family transcription factors (Medvedev et al., 2001). These factors broadly regulate expression of genes critical for metabolism and mitochondrial activity in different tissues and species (33-36). This family consists of three members; PPARγ, PPARα and δ/β, all of which heterodimerize with the retinoid X receptor (RXR) to transactivate target genes through binding to regulatory elements in the promoter regions. Members of the PPAR family are expressed throughout different regions of the eye and retina (Braissant et al., 1996; Castelli et al., 2018; Zhang et al., 2015), and their activity is particularly triggered by omega-3-fatty acids as well as during oxidative stress. This explains why these proteins are often upregulated during different forms of disease (Castelli et al., 2018). One component of their disease response may be to enhance expression of UCP2.
Figure 8. Factors Regulating UCP2 Transcription, Translation, and Activity.

(A) The UCP2 gene contains several upstream regulatory elements that serve as binding sites for transcription factors that either enhance or repress gene expression. (B) Abundance and translation of the UCP2 transcript is further regulated by the presence of an upstream open reading frame (uORF), microRNAs, and RNA-binding proteins such as hnRNP-K. (C) Following translation, UCP2 activity is modulated by the abundance of fatty acids, superoxide levels, and lipid peroxides. Activity is inhibited by the nucleotide GDP and the redox-active dipeptide glutathione (GSH).
Treatment of rodent hepatocytes with the PPAR activators WY14,643 and fenofibrate led to a 5–10 fold increase in UCP2 mRNA (37,38). In hypertensive rats, both PPARα and PPARγ were upregulated in the nodose ganglion and treatment with fenofibrate led to upregulation of UCP2 and a decrease in blood pressure. Deletion mapping of the mouse UCP2 promoter indicated that PPAR binding sites could be responsible for both basal and stimulated UCP2 expression (35). In humans, PPAR-stimulated transcription depends on the pair of E-boxes in the proximal promoter although PPAR itself does not bind to these motifs. In mouse, an equivalent pair of E-boxes is found closer to the transcription start site. In the mouse, an additional PPAR binding site has been found in an intron of UCP3, a gene that is immediately adjacent to UCP2 gene (39). This site loops out to interact with the UCP2 promoter and thus provides an additional mechanism for direct stimulation of UCP2 transcription. A cofactor of PPAR, PGC-1α, also regulates UCP2 transcription through several pathways. PGC-1α contains an N-terminal activation domain that interacts with both a large group of transcription factors and with chromatin remodeling complexes. In addition to acting as a cofactor for PPAR family transcription factors, it may also potentiate binding of other transcription factors such as thyroid hormone and stimulate the production of other factors that interact with the UCP2 promoter such as sterol regulatory element binding protein (34).
In addition to these positive regulatory elements, there are several mechanisms by which UCP2 transcription may be repressed. The transcription factor Foxa1 binds to the UCP2 promoter and, when the repressor sirtuin1 is bound at an adjacent site, can decrease UCP2 transcription (Vatamaniuk et al., 2006). The growth factor TGF-β can also decrease UCP2 transcription by inducing binding of SMAD4 to repressive SMAD elements in the proximal promoter (Sayeed et al., 2010).
This multiplicity of transcriptional control mechanisms integrate UCP2 transcription with many other pathways controlling growth and metabolism and emphasize the key role that this protein plays in cell physiology. Changes in UCP2 transcription, induced through disease, altered metabolism, or direct immune stimulation generally occurs on a time scale of days (Pecqueur et al., 2001; Rupprecht et al., 2012), however post-transcriptional regulation of UCP2 appears to act on a much faster time scale, which likely reflects a large pool of untranslated UCP2 message that may exist to respond more immediately to mitochondria-derived oxidative damage.
3.5. Post-transcriptional regulation of UCP2 expression
Although transcriptional regulation of UCP2 is important, particularly in chronic disease, post-transcriptional regulation may be both more important and more immediate in controlling the levels of UCP2 in mitochondria (Figure 8B) (Pecqueur et al., 2001). One of the first mechanisms found to regulate UCP2 at the post-transcriptional level was identified in a yeast three-hybrid screen (Ostrowski et al., 2004). A widely expressed RNA binding protein, hnRNP-K, binds to sites in the 3’ UTR of UCP2 RNA, and binding increases translation (Hurtaud et al., 2006; Ostrowski et al., 2004). This process can be enhanced by several hormones including insulin, angiopoietin, and adiponectin (Ostrowski et al., 2004; Tahir et al., 2014; Zhou et al., 2012) that can cross the blood-brain or blood-retina barrier (Greco et al., 1970; Lin et al., 2012) and thus may participate in the regulation of retinal UCP2 levels. These findings also emphasize a link between signals that regulate systemic glucose and fatty acid metabolism and more direct regulation of mitochondrial function through UCP2.
A second regulatory mechanism has been proposed following the identification of a 36 amino acid open reading frame in the 5’ untranslated region of Ucp2 RNA (Hurtaud et al., 2006). Mutations of this upstream open reading frame sequence lead to increased UCP2 protein expression, indicating an inhibitory role for this sequence (Hurtaud et al., 2006). Activity of this uORF and thus Ucp2 translation are regulated by the metabolite glutamine, an amino acid and neurotransmitter precursor (Hurtaud et al., 2007; Rupprecht et al., 2019). It has been proposed that glutamine-stimulated dynamic changes in UCP2 translation enhance the metabolic ‘flexibility’ of fast-growing neuroblastoma cells (Rupprecht et al., 2019). Neural tissue is constantly importing glutamine in the glutamine/glutamate cycle and the relationship between glutamine, UCP2, and metabolic flexibility in the context of postmitotic or quiescent tissue in the retina or CNS has not to our knowledge been explored (Rupprecht et al., 2019).
A third element that regulates UCP2 translation are small noncoding RNAs that regulate mRNA levels and translation. These micro RNAs (miRNAs) are ubiquitous in tissues and biofluids, but as they bind loosely to target transcript, miRNA specificity is low (as is our ability to predict interactions between RNAs from sequence alone). Numerous publications link changes in UCP2 expression to specific miRNAs (Donadelli et al., 2014). These publications cover studies in a variety of tissues and few distinguish direct effects on the UCP2 RNA from indirect effects on another regulatory molecule. Most studies have shown that disease or stress lead to a decrease in one or more miRNAs and an increase in UCP2 protein that, in turn, decreases oxidative stress (Rubattu et al., 2017). In a few cases a miRNA has been shown to bind to the 3’ UTR of Ucp2 RNA and inhibit its translation (Sun et al., 2011). Some of the same miRNAs are also expressed in the eye, including mir214, 133a, and 503 (Karali et al., 2010), though to date there are no studies of the impact these regulators have on retinal physiology. Notably, there is growing evidence that miRNAs act over long distances, and tissue-intrinsic expression of a miRNA is not a prerequisite for it to have activity in that tissue. While manipulating systemic miRNAs for tissue specific effects may present an attractive approach to controlling UCP2 levels, their broad specificity indicate a high likelihood for additionally controlling expression of off-target transcripts.
3.6. UCP2 turnover
The complex transcriptional and translational regulation of UCP2 expression has rapid functional consequences because it (and UCP3) have unusually short half-lives. In a range of tissues, the half-life of UCP2 has been measured as 1 hour, considerably shorter than that of other inner mitochondrial membrane proteins which have reported half-lives of between 4 and 17 days (Giardina et al., 2008; Rousset et al., 2007). Import into the mitochondria involves a series of specific chaperones and a recognition sequence within the uncoupling protein itself (Hansen & Herrmann, 2019; Schleiff & McBride, 2000). Since the levels of UCP2 in isolated mitochondria are stable, it is thought that degradation of UCP2 is due to a cytoplasmic factors rather than a mitochondrial protease (Azzu et al., 2008). The abundance of factors governing Ucp2 transcription, translation, and protein stability suggest that UCP2 levels are sensitive to a wide range of local and systemic influences. This argues for a central role in regulating a variety of mitochondrial functions beyond simple control of ROS production.
4. Regulation and function of UCP2 in Normal Tissue
4.1. Uncoupling and ATP production
In addition to the effects that uncoupling has on ROS in a number of disease models, the influence of uncoupling is not exclusive to a single cellular phenotype. By uncoupling mitochondria, the efficiency of ATP production is decreased. When this uncoupling is mediated by high concentrations of a chemical protonophore, uncoupling will permeabilized the mitochondrial membrane to H+ and abolish the drive for ATP synthesis. At lower concentrations of such drugs, the H+ ‘leak’ across the mitochondrial membrane is lower, and there still exists a lower-magnitude PMF that can drive ATP production at a lower rate (Brennan, Southworth, et al., 2006). However, the chemiosmotic gradient also negatively regulates activity of electron transport chain complexes I and IV. If uncoupling relieves this negative regulation, complexes I and IV consume NADH and consume O2 at a greater rate. This pumps more H+ to the mitochondrial intermembrane space, and thus opposes uncoupling dependent mitochondrial depolarization. Therefore depending on the degree of uncoupling, the cellular ATP demand, and the maximal rate of substrate oxidation, a mitochondrion exposed to a protonophore or an uncoupling protein may or may not become depolarized.
This partially explains why genetic manipulation of UCP2 yields disparate effects on mitochondria of different cells. In some, UCP2 overexpression decreases [ATP] (Chan et al., 2001), while in others it or may not affect [ATP]/[ADP] (Perreten Lambert et al., 2014). These differences could be accounted for by measuring mitochondrial substrate oxidation. Alternately, UCP2 knockdown or knockout may increase (Vozza et al., 2014) or decrease (Ho et al., 2010) [ATP]/[ADP]. In some cases, these differences may be due to changes in the number or quality of mitochondria. UCP2 overexpression alters the number and size of mitochondria, and in some cases this effect may buffer total cellular [ATP] by increasing the number of mitochondria (Diano et al., 2003). Simply put, UCP2 may affect ATP production, though this effect likely occurs to different magnitudes in distinct cell-types depending on other critical metabolic parameters.
4.2. Regulation of UCP2 activity
Although a correlation exists between UCP2 expression and activity (overexpression of UCP2 in mice leads to increased uncoupling activity), the activity of UCP2 is controlled by several factors acting as allosteric modulators (Figure 8C). These UCP2 regulators include posttranslational modification of cysteine residues by glutathione, mitochondrial matrix superoxide, fatty acids, peroxidized fatty acids such as 4-hydroxynonenal (4-HNE), and the nucleotide GDP. The allosteric interactions of UCP2 with GDP or glutathione are inhibitory, whereas superoxides, fatty acids, and 4-HNE enhance uncoupling activity – though it is not yet clear whether superoxides and fatty acids act independently of peroxidized lipids or whether these molecules increase the supply of peroxidized lipids that in turn activate UCP2 and possibly other UCPs. There is also debate about whether fatty acids act as allosteric modulators or more directly as substrates. Since fatty acids carry the protons across the inner mitochondrial membrane, increasing their concentration may help drive this process (Berardi et al., 2011). It has also been suggested that facilitated fatty acid transport may enhance fatty acid utilization by mitochondrial metabolism, shifting the balance of glucose and fatty acid oxidation (Pecqueur et al., 2008).
How does UCP2 function in normal conditions?
Since our central thesis is that UCP2 activity can reduce ROS production, it follows that increasing UCP2 activity should provide protection against oxidative damage. In one of our first experiments we carried out a simple in vitro test. A neuronal cell line was transfected with a UCP2 gene and the resulting cells subjected to either hydrogen peroxide or nitric oxide. In each case the transfected cells showed greatly reduced cell death when compared with the transfection controls (Figure 9). In a second set of experiments we compared the phenotype and responses of UCP2 overexpressing and knockout mice. The transgenic overexpressing mice were produced by inserting an 80kb human BAC clone containing both UCP2 and UCP3 genes and promoters (Horvath et al., 2003). Transgenic animals showed overexpression of UCP2 by RNA, protein and functional (coupling) measurements. No expression of UCP3 was detected in retina or other CNS regions. The UCP2 knockout line was generated by Dr. Bradford Lowell by insertional mutagenesis (Zhang et al., 2001). Counts of cells in the ganglion cell layer detected 48% more cells in the overexpressors and 20% fewer cells in the knockout animals compared to wild-type (Reddy et al., 2002). Electroretinogram measurements of both transgenic and knockout mice show no difference from wildtype in either b-wave or oscillatory potentials when compared with wild type, but both showed a decrease in the a-wave (18.5% and 19.0% respectively). Further studies are needed to show whether this effect on the a-wave was due to altered properties of photoreceptors or of the overlying RPE cells. We also tested whether UCP2 to protect against excitotoxic stress in the intact eye. We injected NMDA kainic acid or vehicle into the eyes of wild type or UCP2 transgenic mice and then counted the number of surviving cells in the ganglion cell layer (Barnstable et al., 2016). The UCP2 transgenic mice showed much greater cell survival against both excitotoxic agents, further supporting the protective role of this uncoupling protein (Table 1).
Figure. 9. UCP2 is protective against H2O2 and NO in PC12 cells.

A, RT-PCR amplification of UCP2 from a plasmid positive control (hUCP2), PC12 cells transfected with GFPalone (Con), and PC12 cells transfected with GFP plus hUCP2 (GFP_hUCP2), confirming expression only in the transfected cells. B–D, Images of cells transfected with GFP alone before (B) and after (C) treatment with H2O2 or transfected with both GFP and hUCP2 (D). All fluorescent spots represent cells, with the larger spots represent multicellular aggregates. Many more cells survive after hUCP2 transfection. E, Summary of three independent experiments, in each of which six separate wells were counted. Cell death is presented as the mean ± SE. After treatment with either H2O2 or NO, there is significantly less (P <0.05) cell death in cultures transfected with hUCP2. (Adapted from ref 11)
Table 1.
Survival of neurons in the ganglion cell layer of mouse eyes injected with NMDA or kainic acid
| NMDA (160 nmol) | KA (10 nmol) | |
|---|---|---|
| Wild type | 61.66 ± 2.1 | 21.19 ± 1.4 |
| Transgenic | 103.66 ± 3.6*** | 39.96 ± 1.8*** |
Numbers represent cells counted per 1000 μm length of retina. In all experiments, n=6
p<0.0001. Table adapted from ref 123.
The various mechanisms of UCP2 activation show that the protein is sensitive to oxidative damage, and responds both by increasing its levels and activity. The burden of oxidative damage is theoretically low in most tissues possessing intact anti-oxidative enzymes (SODs, thioredoxins, peroxiredoxins, etc.), and with NAD-linked metabolic substrates, brain mitochondria can even deplete exogenous ROS rather than allow them to accumulate (Starkov et al., 2014). This implies that the utility of UCP2 is for the most part a countermeasure against damaging insults or disease. The ideal test for this hypothesis would be to determine whether mice lacking Ucp2 show an overt disease phenotype. In fact, mitochondria in mice lacking the Ucp2 gene do not behave identically to those in Ucp2-sufficient mice. Our own work to catalogue a portion of the pleiotropic Ucp2 functions found that deletion of this gene in retinal glia increases the formation of ROS and ROS-derived lipid peroxides (Hass & Barnstable, 2019b), which suggests that to some extent, the basal activity of UCP2 decreases the accumulation of ROS in the retina and other tissues. That mice lacking Ucp2 do not display an obvious disease phenotype does not necessarily speak to its ability as an antioxidative protein, and rather may indicate a high capacity of other antioxidant systems to compensate for Ucp2 deficiency.
ROS are not only damaging factors, but also important signals of mitochondrial stress. Part of the ROS-dependent response to mitochondrial stress is an increase in fission and subsequent disposal of stressed or damaged mitochondria, also known as mitophagy (Frank et al., 2012). By increasing ROS, Ucp2 deletion can increase mitochondrial fragmentation (fission) in lung endothelial cells following intermittent hypoxia (Haslip et al., 2015), ischemia/reperfusion injury in kidney tissue (Andrews et al., 2005; Qin et al., 2019), or in undamaged primary cortical astrocytes (Hass & Barnstable, 2019b). Fragmentation of the mitochondrial network in the absence of Ucp2 is likely a precursor to mitophagy, which has been observed in both lung endothelial cells and primary cortical astrocytes (Haslip et al., 2015; Hass & Barnstable, 2019b). We’ve also found that UCP2-deletion induces a strong increase in LC3 puncta throughout the mouse retina following elevated intraocular pressure (Figure 10). Labeled LC3 takes a punctate form when it becomes aggregated membranes bound for autophagy (Kabeya et al., 2000), and that this autophagic marker is enhanced by deletion of a mitochondrial protein suggests that UCP2 is a regulator of mitochondrial autophagy.
Figure 10. Layer Specific Ucp2-deletion dependent retinal autophagy.

We performed an immunohistochemistry experiment to label the autophagy marker LC3B (green) and DNA (Hoechst-33258, blue) in fixed frozen retinal tissue sections from Ucp2flox/flox (n=4), Ucp2flox/flox; Gfap-creERT2 (n=3), and Ucp2flox/flox; Thy1- creERT2 (n=3) mice. In mice expressing Gfap-creERT2, Ucp2 was deleted in GFAP-expressing astrocytes and müller glia, and in Thy1-creERT2-expressing mice, Ucp2 was deleted in projection neurons, which in the retina are retinal ganglion cells. Example images are on the left, and quantifications of LC3B puncta density are on the right. Compared to the same regions of Ucp2flox/flox retinas, LC3B puncta significantly increase in the outer retinas of Ucp2flox/flox; Gfap-creERT2, and in the inner retinas of Ucp2flox/flox; Thy1- creERT2 mice. *p<0.05.
Increased mitophagy can enhance mitochondrial quality and potentially protect against cell death, particularly in glaucoma and possibly in other forms of retinal degeneration (Dai et al., 2018; Hass & Barnstable, 2019b). Other groups have found alternate relationships between UCP2 and cellular or mitochondrial integrity. In one such study, UCP2 deletion was found to decrease mitochondrial number in tyrosine hydroxylase containing neurons of the substantia nigra (Andrews et al., 2005), yet Ucp2 sufficiency induces mitochondrial fission following exposure to glucose in the ventromedial nucleus of the hypothalamus (Toda et al., 2016). The disparate effects of UCP2 presence and absence on mitochondrial dynamics show that the interactions between UCP2, ROS, and fission are not consistent across different cell types.
While Ucp2-dependant changes in mitochondrial quality control encompass one class of means through which this protein can influence physiology, the roles of UCP2 in ATP and ROS generation also play critical roles in systemic metabolism. For example, due to effects of UCP2 on ATP production, Ucp2 knock-out mice have elevated pancreatic beta cell glucose sensitivity in response to diet (Joseph et al., 2002). Due to the effects of UCP2 in mitigating ROS production, Ucp2 knock-out mice produce more ROS within immune cells, and are thus invulnerable to parasitic infection by Toxoplasma gondii (Arsenijevic et al., 2000). Alongside these positive effects of knockout come a variety of vulnerabilities, including enhanced damage in different models of neurodegenerative disease (Andrews et al., 2005; Haines et al., 2010).
Dissipation of the PMF by UCP2 action releases energy in the form of heat. Normally the magnitude of this is unlikely to perturb normal metabolism. It has, however, been noted that UCP2 is discretely expressed in axons of neurons involved in homeostatic regulation, such that this could allow local uncoupling activity and heat production (Horvath et al., 1999). It has been suggested that the heat produced by the axonal UCP2 might be sufficient to modulate synaptic transmission in these homeostatic centers.
5. Uncoupling proteins and disease.
The ROS and redox-dependent feedback loops that regulate UCP activity imply that although protein levels in the mitochondrial membrane are low relative to UCP1, UCP2 is still able to regulate ROS homeostasis. As we have discussed, the formation of mitochondrial ROS can be highly dependent on the PMF, so UCP-dependent dissipation of the PMF likely serves to decrease the generation of ROS (Korshunov et al., 1997; Miwa et al., 2003). From the examples below, it is clear that by decreasing levels of ROS, UCP-dependent dissipation of the PMF regulates cellular health and homeostasis in the heart, pancreas, kidney, and central nervous system.
In cardiac disease ischemic heart cells are under oxidative stress and show elevated ROS levels. UCP2 and UCP3 can protect against ischemic injury and reduce ROS production (Cadenas, 2018). In animal models UCP2 and UCP3 were able to counteract the lipotoxic effects of obesity in the heart and reduce cell death and subsequent heart failure (Ruiz-Ramírez et al., 2016). In a study of healthy young Japanese men Ucp2 and Ucp3polymorphisms were associated with heart rate variability, suggesting a link between autonomic cardiovascular regulation and uncoupling (Matsunaga et al., 2009). A similar protective effect of UCP2 has been found in cerebral ischemia (Contreras et al., 2017). For example, in mice overexpressing UCP2, damage was reduced following either experimental stroke or traumatic brain injury (Ruiz-Ramírez et al., 2016).
The link between UCP2 and diabetes or obesity is well characterized. In both animal and human studies links between UCP2 and body mass index have been described (Horvath et al., 2003; Schonfeld-Warden & Warden, 2001). It is not clear whether these effects are directly on tissues or due to an observed regulatory role of UCP2 on insulin secretion by pancreatic beta cells, though whether this is due to a decrease in ROS production or possible lowered ATP levels is not clear (Chan et al., 2004; Fisler & Warden, 2006; Zhang et al., 2001). Some of the beneficial effects of UCP2 in peripheral tissues damaged in diabetes are probably direct since mitochondrial uncoupling has been identified as a key defensive strategy in kidney. Diabetes led to increased UCP2 levels and increased uncoupling in kidney proximal tubule cells (Friederich et al., 2008; Friederich-Persson et al., 2012). Interestingly, after reduction of Ucp2 by siRNA, uncoupling of kidney mitochondria detected and was achieved through increased activity of the adenine nucleotide transporter (Friederich-Persson et al., 2012). In the absence of uncoupling, the efficiency of ATP synthesis will likely increase, and additional ANT-mediated ATP export may be sufficient to stimulate an ANT-mediated H+ current. The interplay between UCP2 and the ANTs indicates some of the complexity of the regulation of mitochondrial membrane potential and ROS production.
Within the CNS there is also abundant evidence for the protective role of UCP2 is a number of disease models. In mice, increased UCP2-mediated uncoupling led to significantly better cell survival of substantia nigra neurons from MPTP-induced Parkinsonism (Figure 11) (Andrews et al., 2005). Overexpression of Ucp2 in mice reduced cell loss in hippocampus following pilocarpine-induced seizures (Figure 11) (Diano et al., 2003; Dutra et al., 2018).
Figure 11. UCP2 Protects CNS neurons from degeneration.
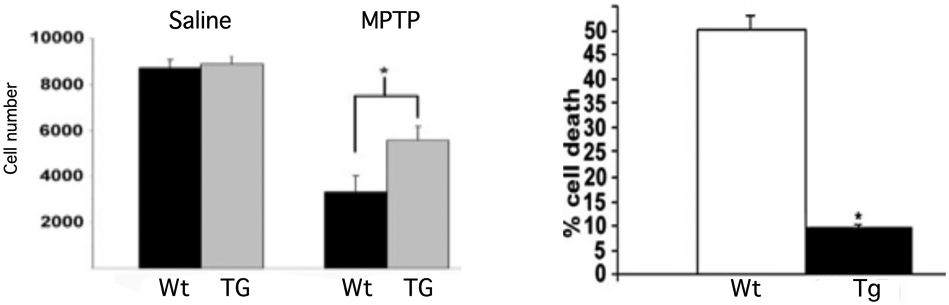
A. Stereological quantification of dopamine cell number revealed that UCP2 TG mice had significantly (p<0.05) more surviving neurons than their WT littermates after MPTP treatment. *Significant with respect to wild-type controls. B. Epileptic seizure-induced cell death is diminished in hUCP2- expressing mice. Cell death in the CA1 area of the hippocampus was significantly reduced in surviving transgenic animals compared with surviving wild-type littermates 24 h after seizures. Figure adapted form refs 160 and 11.
Oxidative stress is also a major pathological component of many of the other major degenerative retinal diseases. In the following sections we briefly survey the data known on the roles of oxidative stress and uncoupling proteins (primarily UCP2) in each disease.
5.1. Diabetic retinopathy.
Diabetic retinopathy (DR) is one of the most common complications of diabetes and is a leading cause of blindness among working age people as well as the elderly (Sabanayagam et al., 2019; Tombran-Tink et al., 2012). The more severe form of the disease, proliferative DR, is caused by abnormal growth of new retinal blood vessels and diabetic macular edema. Earlier, non-proliferative stages of the disease can be recognized and there is still debate as to whether DR is a vascular disease or the vascular complications are a result of earlier neuronal and inflammatory aspects of the disease (Antonetti et al., 2006; Barber et al., 2011).
There is no comprehensive treatment to prevent the progression of the disease, and it is clear that there is an urgent need for new therapeutic approaches. Oxidative stress induced by hyperglycemia is a major cause of microvascular complications in diabetic retinopathy, and may be intimately involved in the neurodegenerative and inflammatory processes that may precede these vascular effects (Barber et al., 1998; Gardner et al., 2002; Kowluru & Abbas, 2003; Madsen-Bouterse et al., 2010). Preventing increased ROS production by mitochondria blocks other pathways of hyperglycemic damage in this disease (Brownlee, 2001). Mitochondrial ROS production is increased in bovine retinal endothelial cells and pericytes cultured with elevated (23 or 30 mM) glucose (relative to 5 mM controls) (Cui et al., 2006). Ucp2 transcript increased with 23 mM glucose, but further increases led to a return of levels to control values, suggesting that UCP2 can respond to hyperglycemia within a range of glucose concentrations (Cui et al., 2006).
To determine more directly whether UCP2 might be involved in diabetic retinopathy, several groups have carried out genetic screens of patients to look for an association of Ucp2 variants with the disease. These studies have identified a small but significant linkage between several UCP2 polymorphisms and the DR phenotype (Crispim et al., 2010; Shen et al., 2014; Zhou et al., 2018). Because this linkage is small, Ucp2 is not a causative risk factor diabetic retinopathy. When segregated into proliferative and non-proliferative forms of the disease, the genetic association was stronger with proliferative diabetic retinopathy. Since at least one of the associated polymorphisms is in the UCP2 promoter, it is thought that these variations may change UCP2 expression in response to elevated glucose (Crispim et al., 2019).
It is likely that DR affects UCP2 activity in the retina as well. When rats are exposed to the pancreatic beta cell-selective toxin streptozotocin, there is a loss in GDP-sensitive respiration in retinal mitochondria (Osorio-Paz et al., 2015). GDP is an inhibitor of UCP2, which suggests that an initial consequence of diabetes in the retina is to diminish UCP2 activity, though this activity reappears 45 days following streptozotocin administration (Osorio-Paz et al., 2015). Diminished UCP2 expression or activity could therefore be an early response in diabetic retinopathy.
The db/db mouse has frequently been used as a model of diabetes and its complications. It has been shown that the level of retinal cell death and damage in the db/db mouse is mimicked by UCP2 deletion (He et al., 2016). A transcriptomic analysis of the db/db mouse found a significant increase in UCP2 expression as compared with heterozygous controls (Bogdanov et al., 2014). Interestingly, treatment of db/db mice with fenofibrate, an activator of PPAR and thus of UCP2 transcription, restored many aspects of a normal phenotype to the db/db (Bogdanov et al., 2015).
Ucp2 knockdown in human vascular endothelial (HUVEC) cells increased high glucose induced apoptosis and, conversely, virally-mediated overexpression of UCP2 inhibited high glucose induced caspase 3 activation and apoptosis (He et al., 2016; Koziel et al., 2015). A variety of other responses in endothelial cells, pericytes and Muller glial cells, and a number of studies of antioxidants in cell culture all indicate the broad effects of elevated glucose on retinal pathophysiology and suggest that reducing oxidative stress will be beneficial (Liu et al., 2017; Rani et al., 2016). Yet to date, studies of antioxidant therapy in diabetic retinopathy have only shown limited effects (Kowluru & Kennedy, 2001). As indicated earlier, part of the problem with these studies may be the access of the compounds to the mitochondria, the major site of ROS production. Given the clear role of UCP2 in regulating ROS production, a more targeted approach developing compounds that selectively activate this uncoupler may provide benefit for diabetic retinopathy patients.
5.2. Glaucoma
Glaucoma is a group of chronic progressive optic neuropathies that are associated with ganglion cell loss (Tombra-Tink et al., 2008; Sun et al., 2017). While there are juvenile forms of glaucoma, the most common forms of the disease affect adults over the age of 40. There are four generally recognized categories of adult glaucoma: primary open-angle and angle-closure, and the secondary open and angle-closure glaucoma. The most common type found in the United States is primary, open-angle glaucoma (POAG). Patients with POAG often show no symptoms until the optic nerve damage and loss of ganglion cells is severe. Although many patients with POAG have increased intraocular pressure, some do not and the causes of such normal tension glaucoma are still under investigation (Tezel, 2020).
Reduced anti-oxidant defenses (Mousa et al., 2015), increases in DNA oxidation (Izzotti et al., 2003), protein carbonyl formation (Tezel et al., 2005), lipid peroxidation (Moreno et al., 2004), and reduced mitochondrial function (Figure 2, (Abu-Amero et al., 2006; Hass & Barnstable, 2019b)) have been detected in tissue from glaucoma patients or in animal models of glaucoma. These phenotypes indicate that oxidative damage is a central phenotype in glaucoma. A wide variety of anti-oxidant compounds can provide some protection of ganglion cells in primary open angle glaucoma (Shen et al., 2015; Yang et al., 2016), and uncoupling proteins may be an alternative therapeutic.
In addition to direct oxidative stress damage in retinal ganglion cells, there is evidence for oxidative injury to a set of specialized glia at the optic nerve head (Feilchenfeld et al., 2008; Govindarajan et al., 2009; Kuehn et al., 2005). One response of these cells results in local T-cell activation that stimulates an adaptive immune response increasing neurodegeneration (Tezel & Group, 2009; Tezel et al., 2007). Ucp2 expression can alter the phenotype of inflammatory immune cells and mediate a switch to an anti-inflammatory state (De Simone et al., 2015). Enhanced Ucp2 expression in microglia and T-cells may, therefore, relieve inflammatory burdens associated with glaucoma. We initially hypothesized that we could induce similar perturbations in retinal and optic nerve head glia by overexpressing Ucp2 in GFAP+ cells to reduce a glial inflammatory phenotype (Hass & Barnstable, 2016).
To approach this and other hypotheses, we used multiple transgenic mouse lines that took advantage of Thy1 expression in RGCs (Young et al., 2008) and GFAP expression in astrocytes and müller glia (Ganat et al., 2006; Hass & Barnstable, 2019b). By combining promoters for these genes with a creERT2 construct, we could recombine LoxP sites either in introns of the endogenous mouse Ucp2 gene to delete it (Hass & Barnstable, 2019b), or surrounding a stop site which precedes an exogenous mouse Ucp2 sequence inserted in the Rosa26 locus (Hass & Barnstable, 2019a; Kim et al., 2015).
We determined the impact of RGC or glial Ucp2 overexpression or deletion on cell death in an experimental model of glaucoma (Hass & Barnstable, 2019a, 2019b), wherein we injected polystyrene microbeads injected through the cornea to impede the flow of aqueous humor through trabecular meshwork in the anterior chamber (Cone et al., 2012; Sappington et al., 2010). The impeded flow of aqueous humor increases intra-ocular pressure (IOP), an elevation in which is a risk factor in human glaucoma. Raising IOP in mice resulted in retinal ganglion cell death, though the degree to which IOP was raised does not necessarily predict retinal ganglion cell death as well as it does dysfunction (Calkins, 2012; Della Santina et al., 2013; Hass & Barnstable, 2019a, 2019b).
Once we established a model of glaucoma in which we altered Ucp2 expression, we determined whether manipulation of RGC or glial Ucp2 altered cell death in glaucoma (Hass & Barnstable, 2019a, 2019b). Overexpression of Ucp2 in ganglion cells was able to significantly decrease the cell death induced by elevated intra-ocular pressure (Hass & Barnstable, 2019a). On the other hand, overexpression of Ucp2 in glial cells had no major effect on cell death (Figure 12) (Hass & Barnstable, 2019a). These results suggest that Ucp2 activity in retinal glia is either insufficient to overcome the oxidative burden in glaucoma or possibly that the glial oxidative burden in glaucoma is not causal of RGC death. This is not meant to suggest that glia are not critical regulators of RGC and retina health or oxidative status, only that the oxidative state of a glial cell may not directly impact the oxidative state of a RGC. Additionally, these results account for the role of Ucp2 in müller cells and astrocytes of the retina, but not microglia of the retina. It has been suggested that cells from an immune lineage express higher levels of UCP2 (Rupprecht et al., 2012), and because microglia originate from this lineage, altering Ucp2 expression in these cells may yield a more drastic impact on RGC survival.
Figure 12. Increased Ucp2 Expression in RGCs but not Astrocytes and Müller Glia Decreases Retinal Ganglion Cell Loss.

(A) Difference tonometric intra-ocular pressure (IOP) readings between bead- and PBS-injected eyes before and following intraocular surgery. Injection of polystyrene microbeads through the conea elevated IOP over a 30 day period in Ucp2KI (n=11), Ucp2KI; Gfap-creERT2 (n=6), Ucp2KI; Thy1- creERT2 (n=9) eyes. (B) We performed an immunohistochemistry experiment to visualize the retinal ganglion cell marker RBPMS in fixed, flat-mounted retinal tissue from the mice in (A), and determined RGC loss due to microbead- and PBS-injected eyes of the indicated genotypes, 30 days following microbead injection. (C) Representative images of retinal whole-mounts labeled for RBPMS, quantified in (B). In mice expressing Gfap-creERT2, Ucp2 was overexpressed in GFAP-expressing astrocytes and müller glia, and in Thy1- creERT2-expressing mice, Ucp2 was overexpressed in projection neurons, which in the retina are retinal ganglion cells. Figure reproduced from (Hass & Barnstable, 2019a) with permission. *p<0.05.
Deletion of Ucp2 was protective against oxidative damage and RGC death in the same model of glaucoma, regardless of whether the genetic deletion occurred in Thy1-positive retinal ganglion cells or in GFAP-positive astroglia and müller glia (Figure 13) (Hass & Barnstable, 2019b). This result was initially puzzling, because while Ucp2 deletion increased ROS generation in isolated glia, there was a lower ROS burden in the retina. We eventually found research showing that ROS can act as a signal capable of promoting mitophagy (Frank et al., 2012), and found an enhancement of mitophagy both in müller glia and RGCs lacking Ucp2 (Hass & Barnstable, 2019b). Interventions that enhance mitophagy also appear to preserve retinal ganglion cells following IOP elevation in rat eyes, suggesting that increased mitochondrial quality control is neuroprotective (Dai et al., 2018). We reasoned that because astroglia are capable of internalizing damaged mitochondria from retinal ganglion cells (Davis et al., 2014), Ucp2 knockout, which enhances glial mitophagy, may also enhance trans-cellular degradation of damaged RGC mitochondria or other components to protect against mitochondrial damage in glaucoma.
Figure 13. Ucp2 Deletion Protects Against Glaucoma.

(A) intra-ocular pressure (IOP) difference between microbead and PBS-injected Ucp2flox/flox, Ucp2flox/flox; Gfap-creERT2, and Ucp2flox/flox; Thy1- creERT2 eyes over 30 days following a single microbead injection. (B) Example RBPMS-labeled RGCs in PBS- and Bead-injected eyes from Ucp2flox/flox (n=29), Ucp2flox/flox; Gfap-creERT2 (n=10), and Ucp2flox/flox; Thy1- creERT2 (n=13). Scale bar (red) = 100 μm. Differences in RGC density between PBS- and Bead-injected eyes are quantified in (C). A difference of “0” indicated no retinal ganglion cell death. **p<0.01.
The other major locus at which oxidative stress appears to act is the trabecular meshwork where altered cell adhesion and extracellular matrix secretion may alter tissue permeability (Zhou et al., 1999). In all these cases there is a strong argument for the involvement of mitochondrial ROS and thus a case for directing therapies to reducing this at its source.
5.3. Age-Related Macular Degeneration
Age-Related Macular Degeneration (ARMD) is a major cause of blindness in the aging population and is causing growing societal and economic problems in many countries. The causes of macular degeneration are not understood but the sequence of pathological development has been well documented.
Changes in the RPE and the formation of drusen are early signs that are accompanied by death of cone photoreceptors (Algvere et al., 2016). While this dry form of ARMD can continue to geographic atrophy in many patients, a significant number of patients progress to the wet form of ARMD in which choroidal neovascularization invades the retina and causes rapid loss of photoreceptors and vision. Genetic screens identified loci strongly associated with the dry and wet forms of ARMD respectively (Dewan et al., 2006; Klein et al., 2005). Subsequent studies have confirmed these findings and identified additional risk and protective factors associated with ARMD (Gehrs et al., 2010; Gorin & daSilva, 2020). Given the lack of understanding of ARMD causes, it is not surprising that there are no good therapies for the disease. While anti-VEGF therapies can prevent the effects of neovascular growth, they do not treat the underlying loss of cells, and vision continues to decline (Tao et al., 2010).
The role of oxidative stress and its importance to cone photoreceptors and RPE cells continues to be debated (Totan et al., 2009). Both cell types are constitutively assaulted by a range of harmful agents, including sunlight, smoking and some dietary components. These extrinsic factors can be ameliorated by an array of cytoplasmic anti-oxidant enzymes and compounds in the eye. Several features of photoreceptor-RPE interactions, however, provide additional oxidative stress and risk of damage. As discussed earlier, photoreceptors are extremely active metabolically and this may result in higher levels of ROS production by mitochondria (Rueda et al., 2016). RPE cells in particular must also handle a high burden of ROS, possibly due in part to their close proximity to O2-rich choroidal vessels(Jarrett & Boulton, 2012). There is also extensive evidence of ROS-mediated damage to mitochondrial DNA in RPE cells, together with alterations in mitochondrial structure and function (Fisher & Ferrington, 2018; Kaarniranta et al., 2020).
Reduction of anti-oxidant capacity alone is sufficient to recapitulate the effects of aging and ARMD on mice, including altered cell morphology, a fragmented mitochondrial structure, and impaired bioenergetic function which led to altered gene expression and mitochondrial fragmentation in cone photoreceptors (Brown et al., 2019). Similar changes in RPE mitochondria are seen in primary cultures of RPE cells from aged donors (He & Tombran-Tink, 2010). One of the changes that occurs with age in RPE cells is a decrease in the expression of Ucp2, a correlation that suggests a decreased ability to control ROS production (He et al., 2019). More direct evidence for a role of UCP2 comes from experiments in which expression of this uncoupling protein was enhanced by the neuroprotective factor PEDF and allowed the RPE cells to better resist oxidative stress (He et al., 2014; Wang et al., 2019). Similarly, the Ucp2 regulator PGC-1α can reduce oxidative stress and improve mitochondrial biogenesis (Kaarniranta et al., 2018).Animals hemizygous for PGC-1α show symptoms that mirror ARMD when fed a high fat diet (Zhang et al., 2018).
It is clear that oxidative stress plays an important role in ARMD, that UCP2 is less able to modulate ROS formation in aging, and that many very different factors that all reduce oxidative damage in models of ARMD share a common feature of enhancing expression and activity of UCP2. We suggest that a concerted effort to experimentally modulate UCP2 in ARMD models will lead to novel therapeutic strategies to combat this disease.
6. UCPs as therapeutic targets.
If we accept the premise that oxidative stress is a major factor in the pathophysiology of retinal disease, it follows that therapeutic approaches that reduce this stress will have positive effects on disease magnitude and progression. There is an overabundance of evidence linking oxidative stress and disease, so therapeutic interventions for neurodegeneration in the CNS or retina should target production or detoxification of ROS. As discussed earlier, the low bioavailability of the active compound at the source from which ROS are generated is one issue preventing anti-oxidant therapy being fully effective, but efforts are underway to solve this issue by designing anti-oxidant compounds targeted to ROS-forming sites in mitochondria (Smith & Murphy, 2010; Watson et al., 2019).
At the same time, evolution has optimized endogenous antioxidant molecules to function within the context of organelles such as mitochondria, and there is also sufficient motive to explore the full potential of modulating endogenous proteins as a source of protection. In this review we make the case that mitochondrial uncoupling can modulate the levels of oxidative stress and this can prevent cell death in a variety of disease models (Andrews et al., 2005; Hass & Barnstable, 2019a; Mehta & Li, 2009). When considering ‘uncoupling’ as a therapeutic approach, we can either develop chemical uncouplers where the effect is determined by the dose used, genetic methods of enhancing the expression of uncoupling proteins, or develop compounds that can modulate the activity of uncoupling proteins.
There are already a number of compounds that can uncouple mitochondria by dissipating the proton gradient across the inner mitochondrial membrane. FCCP is one such agent, and low doses as low as 100 nM FCCP significantly improves post-ischemic functional recovery in isolated rat hearts (Brennan, Berry, et al., 2006). Similarly, 1 mg/L dinitrophenol added to the drinking water of mice led to increased mitochondrial biogenesis, animal lifespan and a number of biochemical changes associated with health (Cerqueira et al., 2011). More recently, a cyanotriazole derivative, OPC-163493, has been shown to provide mild uncoupling and to be relatively safe (Kanemoto et al., 2019). However, higher concentrations of these uncoupling agents can collapse of the proton gradient and halt ATP synthesis. Unfortunately, patients inevitably attempt to enhance the effect of a drug by taking a larger dose, and for exogenous uncouplers such a decision would be fatal (Grundlingh et al., 2011). This class of compounds therefore represents a useful proof of principle for the benefits of mild uncoupling on oxidative stress, but with limited translatability to the clinic. Alternately, the effects of these compounds serve as a warning for studies in which overexpression of uncoupling proteins is desired – a substantial increase in mitochondrial uncoupling could be dangerous if not controlled, and while there are substantial control mechanisms in place to regulate UCP activity, the dose of this protein in mitochondria is nonetheless an critical consideration for studies aimed at translation.
The other approach is to alter the activity of endogenous uncoupling mechanisms and the most potent of these are the uncoupling proteins. We have described numerous disease models in which increased activity of UCP2 provides protection against cell death, and in most of these examples UCP2 expression was set through genetic manipulation. The same concept can be carried forward to a human population by using viral vectors to deliver Ucp2. AAV serotypes are able to preferentially infect specific types of ocular cells, and the use of cell type-specific promoters can restrict expression to particular cell types within the eye. Ideally, viral gene expression will be inducible and tunable to account for disease-specific, disease stage-specific, or patient-specific differences in ROS metabolism. For example, in one theoretical scenario where a glaucoma patient with high IOP was treated with an AAV containing a Ucp2 gene, it might be necessary to induce high levels of expression in the early phases of treatment. If the IOP was reduced, perhaps by the use of appropriate drugs, then the stress on optic nerve head and ganglion cells would be less and the levels of UCP2 could be reduced. There are a number of inducible AAV systems being tested (Le Guiner et al., 2014; Stieger et al., 2006; Strobel et al., 2020). Tetracycline, doxycycline and cumate have all been shown to be effective in model systems but to date none have been fully tested in clinical trials. As we learn more about the role of microRNAs in regulating Ucp2 expression, gene therapies supplying these molecules may also be possible.
While sophisticated gene therapy approaches my become reality in the future, there is currently a need for drugs that can modulate the levels or activity of uncoupling proteins. The optimal therapeutic agents might vary according to whether the insult is chronic or acute. Most retinal diseases are chronic and slowly progressing. For these, a low but stable increase in UCP2 activity achieved by any mechanism may be adequate to counteract an ongoing oxidative insult. Alternatively, acute injury such as trauma or ischemia needs a rapid response and may only be served by compounds that change UCP2 activity.
As described above, PPAR activators lead to enhance Ucp2 transcription. These drugs have historically demonstrated a range of other beneficial effects, particularly for diabetes and diabetic complications. In some countries, the PPARα agonist fenofibrate is an approved drug for diabetic retinopathy. PPARα activation reduces oxidative stress and apoptosis (Pearsall et al., 2019), and fenofibrate has been shown to reduce retinal inflammation and reduce nerve fiber layer loss (Abcouwer, 2013; Hu et al., 2013). Another approach is to increase the expression or activity of the transcriptional co-activator PGC-1α. Pharmacological upregulation of PGC-1α enhances respiration and decreases H2O2 levels in retinal pigment epithelial cells (Satish et al., 2018). Neither PPARs, PGC-1α, or any other transcriptional regulator is specific for Ucp2, and whether the protective effects of these compounds requires UCP2 activity will only be determined through the use of these drugs in Ucp2 -deficient animal models.
The same lack of specificity for UCP2 is also true for post-transcriptional regulators of Ucp2. As we have discussed, miRNAs bind promiscuously to transcripts and thus like PPARs and PGC-1α, miRNAs used for a therapeutic effect are likely to have pleiotropic effect. The same is true for other regulators of translation, such as hnRNP-K. However, each of these molecules were discovered more recently, and don’t benefit from same the history of characterization as PPARs and PGC-1α. To date there has yet to be a detailed exploration into whether manipulating the mechanisms that regulate Ucp2 translation (or degradation) is protective against oxidative damage, and we feel that this field is ripe for further study.
Perhaps the best hope for therapeutic agents to regulate UCP2 can be found in the natural regulators of its activity. For example, 4-hydroxy-2-nonenal (4-HNE) is produced from lipids by the actions of ROS and can activate UCP2. A number of natural products that can reduce ROS production have structural features in common with 4-HNE and may have as part of their action an activation of UCP2. One example of this is curcumin, a natural product derived from the rhizome of Curcuma longa L. This polyphenol has been used in traditional Chinese medicine for thousands of years. Recent studies have shown that it is beneficial in a number of retinal diseases (reviewed in (Radomska-Leśniewska et al., 2019)).
A major problem with curcumin and related compounds is their poor solubility and hence poor bioavailability. Efforts to encapsulate curcumin in liposomes have met with some success in peripheral tissues, though the blood retina barrier may prevent systemic delivery of lipophilic molecules related to 4-HNE. However, several methods of drug delivery avoid the complication of the blood-retina barrier, such as direct injections into the vitreous, or drug delivery through eyedrops. The latter technique has been used to deliver peptides that prevent the retinal effects in an animal model of diabetes (Liu et al., 2012; Sheibani et al., 2019). Whether the permeability of a given molecule to the blood-retina barrier is identical for mice and humans remains unknown.
In a complex disease like diabetic retinopathy it probably does not matter if any effective drug has multiple effects including effects on UCP2. In other cases, such as ischemic injury, it would probably be advantageous to have a drug that only activated the UCP2 pathway. While there are tantalizing clues about such drugs, urgently needed systematic studies of actions on and specificity for UCP2 have yet to be carried out, and are anticipated.
7. Future Directions and Conclusions
While the initial triggers for a variety of retinal degenerative diseases show wide variation, there is increasing evidence that oxidative stress and the perturbations in tissue function caused by oxidative damage are common sequelae and critical therapeutic targets. Though there are multiple sources of oxidative stress, mitochondria are one of the most prominent sources, particularly when the proton motive force is high. Uncoupling can reduce this force and thus reduce the formation of ROS. Mitochondrial uncoupling proteins, or UCPs, provide modest uncoupling, which consistently decreases ROS production. Of the UCPs, UCP2 appears to play an important role many tissues, including the retina. The multiple mechanisms that control UCP2 suggest that it is an important player in cell metabolism and highlight its potential utility as a drug target.
It remains to be seen whether alteration of UCP2 protein levels by some form of gene regulation or alteration of UCP2 activity by small molecule compounds will be the most therapeutically advantageous. As in many other cases, the unique structure and properties of the eye make it a valuable model to test such therapeutics and, because of the local delivery methods, also make therapeutics less likely to have unwanted side effects in other tissues. With the efforts now being directed towards understanding the basic properties and therapeutic effects of UCP2 and the other uncoupling proteins, we can look forward to have a set of novel therapeutics that will greatly increase the tools available to combat retinal degenerative diseases.
Acknowledgements
This work was supported by National Institutes of Health grants NS100508 and EY 029992
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors declare no competing or financial interests.
References
- Abcouwer SF (2013). Direct effects of PPARα agonists on retinal inflammation and angiogenesis may explain how fenofibrate lowers risk of severe proliferative diabetic retinopathy. Diabetes, 62(1), 36–38. 10.2337/db12-1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahams JP, Leslie AG, Lutter R, & Walker JE (1994). Structure at 2.8 A resolution of F1-ATPase from bovine heart mitochondria. Nature, 370(6491), 621–628. 10.1038/370621a0 [DOI] [PubMed] [Google Scholar]
- Abu-Amero KK, Morales J, & Bosley TM (2006). Mitochondrial abnormalities in patients with primary open-angle glaucoma. Invest Ophthalmol Vis Sci, 47(6), 2533–2541. 10.1167/iovs.05-1639 [DOI] [PubMed] [Google Scholar]
- Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, Marsden CD, Jenner P, & HalliweN B (1997). Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem, 69(3), 1196–1203. 10.1046/j.1471-4159.1997.69031196.x [DOI] [PubMed] [Google Scholar]
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, & Walter P (2002). Molecular Biology of the Cell (4th ed.). Garland Science. [Google Scholar]
- Alberts B, Wilson JH, & Hunt T (2008). Molecular biology of the cell (5th ed.). Garland Science. [Google Scholar]
- Algvere PV, Kvanta A, & Seregard S (2016). Drusen maculopathy: a risk factor for visual deterioration. Acta Ophthalmol, 94(5), 427–433. 10.1111/aos.13011 [DOI] [PubMed] [Google Scholar]
- Altomare E, Grattagliano I, Vendemaile G, Micelli-Ferrari T, Signorile A, & Cardia L (1997). Oxidative protein damage in human diabetic eye: evidence of a retinal participation. Eur J Clin Invest, 27(2), 141–147. 10.1046/j.1365-2362.1997.780629.x [DOI] [PubMed] [Google Scholar]
- Anders MW, Robotham JL, & Sheu SS (2006). Mitochondria: new drug targets for oxidative stress-induced diseases. Expert Opin Drug Metab Toxicol, 2(1), 71–79. 10.1517/17425255.2.1.71 [DOI] [PubMed] [Google Scholar]
- Andrews ZB, Horvath B, Barnstable CJ, Elsworth J, Elseworth J, Yang L, Beal MF, Roth RH, Matthews RT, & Horvath TL (2005). Uncoupling protein-2 is critical for nigral dopamine cell survival in a mouse model of Parkinson's disease. J Neurosci, 25(1), 184–191. 10.1523/JNEUROSCI.4269-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonetti DA, Barber AJ, Bronson SK, Freeman WM, Gardner TW, Jefferson LS, Kester M, Kimball SR, Krady JK, LaNoue KF, Norbury CC, Quinn PG, Sandirasegarane L, Simpson IA, & Group JDRC (2006). Diabetic retinopathy: seeing beyond glucose-induced microvascular disease. Diabetes, 55(9), 2401–2411. 10.2337/db05-1635 [DOI] [PubMed] [Google Scholar]
- Arsenijevic D, Onuma H, Pecqueur C, Raimbault S, Manning BS, Miroux B, Couplan E, Alves-Guerra MC, Goubern M, Surwit R, Bouillaud F, Richard D, Collins S, & Ricquier D (2000). Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet, 26(4), 435–439. 10.1038/82565 [DOI] [PubMed] [Google Scholar]
- Azzu V, Affourtit C, Breen EP, Parker N, & Brand MD (2008). Dynamic regulation of uncoupling protein 2 content in INS-1E insulinoma cells. Biochim Biophys Acta, 1777(10), 1378–1383. 10.1016/j.bbabio.2008.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldeiras I, Santana I, Proença MT, Garrucho MH, Pascoal R, Rodrigues A, Duro D, & Oliveira CR (2010). Oxidative damage and progression to Alzheimer's disease in patients with mild cognitive impairment. J Alzheimers Dis, 21(4), 1165–1177. 10.3233/jad-2010-091723 [DOI] [PubMed] [Google Scholar]
- Barber AJ, Gardner TW, & Abcouwer SF (2011). The significance of vascular and neural apoptosis to the pathology of diabetic retinopathy. Invest Ophthalmol Vis Sci, 52(2), 1156–1163. 10.1167/iovs.10-6293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, & Gardner TW (1998). Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest, 102(4), 783–791. 10.1172/JCI2425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnstable CJ, Li M, Reddy R, & Horvath TL (2003). Mitochondrial uncoupling proteins: regulators of retinal cell death. Adv Exp Med Biol, 533, 269–275. [DOI] [PubMed] [Google Scholar]
- Barnstable CJ, Reddy R, Li H, & Horvath TL (2016). Mitochondrial Uncoupling Protein 2 (UCP2) Regulates Retinal Ganglion Cell Number and Survival. J Mol Neurosci, 58(4), 461–469. 10.1007/s12031-016-0728-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardi MJ, & Chou JJ (2014). Fatty acid flippase activity of UCP2 is essential for its proton transport in mitochondria. Cell Metab, 20(3), 541–552. 10.1016/j.cmet.2014.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardi MJ, Shih WM, Harrison SC, & Chou JJ (2011). Mitochondrial uncoupling protein 2 structure determined by NMR molecular fragment searching. Nature, 476(7358), 109–113. 10.1038/nature10257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein SL, Meister M, Zhuo J, & Gullapalli RP (2016). Postnatal growth of the human optic nerve. Eye (Lond), 30(10), 1378–1380. 10.1038/eye.2016.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisbach CM, Hass DT, Robbings BM, Rountree AM, Sadilek M, Sweet IR, & Hurley JB (2020a). Succinate Can Shuttle Reducing Power from the Hypoxic Retina to the O. Cell Rep, 31(5), 107606. 10.1016/j.celrep.2020.107606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisbach CM, Hass DT, Robbings BM, Rountree AM, Sadilek M, Sweet IR, & Hurley JB (2020b). Succinate Can Shuttle Reducing Power from the Hypoxic Retina to the O2 Rich Pigment Epithelium. Cell Rep, 31(5), 107606. 10.1016/jcelrep.2020.107606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov P, Corraliza L, Villena JA, Carvalho AR, Garcia-Arumí J, Ramos D, Ruberte J, Simó R, & Hernández C (2014). The db/db mouse: a useful model for the study of diabetic retinal neurodegeneration. PLoS One, 9(5), e97302. 10.1371/journal.pone.0097302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov P, Hernández C, Corraliza L, Carvalho AR, & Simó R (2015). Effect of fenofibrate on retinal neurodegeneration in an experimental model of type 2 diabetes. Acta Diabetol, 52(1), 113–122. 10.1007/s00592-014-0610-2 [DOI] [PubMed] [Google Scholar]
- Bouillaud F, Alves-Guerra MC, & Ricquier D (2016). UCPs, at the interface between bioenergetics and metabolism. Biochim Biophys Acta, 1863(10), 2443–2456. 10.1016/j.bbamcr.2016.04.013 [DOI] [PubMed] [Google Scholar]
- Boussery K, Delaey C, & Van de Voorde J (2002). Rat retinal tissue releases a vasorelaxing factor. Invest Ophthalmol Vis Sci, 43(10), 3279–3286. [PubMed] [Google Scholar]
- Boveris A, & Chance B (1973). The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J, 134(3), 707–716. 10.1042/bj1340707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braissant O, Foufelle F, Scotto C, Dauça M, & Wahli W (1996). Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology, 137(1), 354–366. 10.1210/endo.137.1.8536636 [DOI] [PubMed] [Google Scholar]
- Brand MD (2016). Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med, 100, 14–31. 10.1016/j.freeradbiomed.2016.04.001 [DOI] [PubMed] [Google Scholar]
- Brennan JP, Berry RG, Baghai M, Duchen MR, & Shattock MJ (2006). FCCP is cardioprotective at concentrations that cause mitochondrial oxidation without detectable depolarisation. Cardiovasc Res, 72(2), 322–330. 10.1016/jxardiores.2006.08.006 [DOI] [PubMed] [Google Scholar]
- Brennan JP, Southworth R, Medina RA, Davidson SM, Duchen MR, & Shattock MJ (2006). Mitochondrial uncoupling, with low concentration FCCP, induces ROS-dependent cardioprotection independent of KATP channel activation. Cardiovasc Res, 72(2), 313–321. 10.1016/j.cardiores.2006.07.019 [DOI] [PubMed] [Google Scholar]
- Brown EE, DeWeerd AJ, Ildefonso CJ, Lewin AS, & Ash JD (2019). Mitochondrial oxidative stress in the retinal pigment epithelium (RPE) led to metabolic dysfunction in both the RPE and retinal photoreceptors. Redox Biol, 24, 101201. 10.1016/j.redox.2019.101201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlee M (2001). Biochemistry and molecular cell biology of diabetic complications. Nature, 414(6865), 813–820. 10.1038/414813a [DOI] [PubMed] [Google Scholar]
- Brylla E, Tscheudschilsuren G, Santos AN, Nieber K, Spanel-Borowski K, & Aust G (2003). Differences between retinal and choroidal microvascular endothelial cells (MVECs) under normal and hypoxic conditions. Exp Eye Res, 77(5), 527–535. 10.1016/s0014-4835(03)00219-7 [DOI] [PubMed] [Google Scholar]
- Burkhardt DA (1994). Light adaptation and photopigment bleaching in cone photoreceptors in situ in the retina of the turtle. J Neurosci, 14(3 Pt 1), 1091–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadenas S (2018). Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim Biophys Acta Bioenerg, 1859(9), 940–950. 10.1016/j.bbabio.2018.05.019 [DOI] [PubMed] [Google Scholar]
- Calkins DJ (2012). Critical pathogenic events underlying progression of neurodegeneration in glaucoma. Prog Retin Eye Res, 31(6), 702–719. 10.1016/j.preteyeres.2012.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campochiaro PA (2000). Retinal and choroidal neovascularization. J Cell Physiol, 184(3), 301–310. [DOI] [PubMed] [Google Scholar]
- Carr A, & Frei B (1999). Does vitamin C act as a pro-oxidant under physiological conditions? FASEB J, 13(9), 1007–1024. 10.1096/fasebj.13.9.1007 [DOI] [PubMed] [Google Scholar]
- Carter-Dawson L, Shen FF, Harwerth RS, Crawford ML, Smith EL, & Whitetree A (2004). Glutathione content is altered in Mü cells of monkey eyes with experimental glaucoma. Neurosci Lett, 364(1), 7–10. 10.1016/j.neulet.2004.03.082 [DOI] [PubMed] [Google Scholar]
- Castelli V, d’Angelo M, Antonosante A, Catanesi M, Benedetti E, Desideri G, & Cimini A (2018). Physiology and Pathophysiology of PPARs in the Eye. Nuclear Receptor Research(5), 17. [Google Scholar]
- Cerqueira FM, Laurindo FR, & Kowaltowski AJ (2011). Mild mitochondrial uncoupling and calorie restriction increase fasting eNOS, akt and mitochondrial biogenesis. PLoS One, 6(3), e18433. 10.1371/journal.pone.0018433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CB, De Leo D, Joseph JW, McQuaid TS, Ha XF, Xu F, Tsushima RG, Pennefather PS, Salapatek AM, & Wheeler MB (2001). Increased uncoupling protein-2 levels in beta-cells are associated with impaired glucose-stimulated insulin secretion: mechanism of action. Diabetes, 50(6), 1302–1310. 10.2337/diabetes.50.6.1302 [DOI] [PubMed] [Google Scholar]
- Chan CB, Saleh MC, Koshkin V, & Wheeler MB (2004). Uncoupling protein 2 and islet function. Diabetes, 53 Suppl 1, S136–142. 10.2337/diabetes.53.2007.s136 [DOI] [PubMed] [Google Scholar]
- Chance B, Sies H, & Boveris A (1979). Hydroperoxide metabolism in mammalian organs. Physiol Rev, 59(3), 527–605. 10.1152/physrev.1979.59.3.527 [DOI] [PubMed] [Google Scholar]
- Chao JR, Knight K, Engel AL, Jankowski C, Wang Y, Manson MA, Gu H, Djukovic D, Raftery D, Hurley JB, & Du J (2017). Human retinal pigment epithelial cells prefer proline as a nutrient and transport metabolic intermediates to the retinal side. J Biol Chem, 292(31), 12895–12905. 10.1074/jbc.M117.788422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CM, Wu YR, Cheng ML, Liu JL, Lee YM, Lee PW, Soong BW, & Chiu DT (2007). Increased oxidative damage and mitochondrial abnormalities in the peripheral blood of Huntington's disease patients. Biochem Biophys Res Commun, 359(2), 335–340. 10.1016/j.bbrc.2007.05.093 [DOI] [PubMed] [Google Scholar]
- Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, Eyassu F, Shirley R, Hu CH, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa ASH, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, & Murphy MP (2014). Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature, 515(7527), 431–435. 10.1038/nature13909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu AC, Ho PW, Kwok KH, Ho JW, Chan KH, Liu HF, Kung MH, Ramsden DB, & Ho SL (2009). Mitochondrial UCP4 attenuates MPP+ - and dopamine-induced oxidative stress, mitochondrial depolarization, and ATP deficiency in neurons and is interlinked with UCP2 expression. Free Radic Biol Med, 46(6), 810–820. 10.1016/j.freeradbiomed.2008.12.015 [DOI] [PubMed] [Google Scholar]
- Collin F (2019). Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int J Mol Sci, 20(10). 10.3390/ijms20102407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cone FE, Steinhart MR, Oglesby EN, Kalesnykas G, Pease ME, & Quigley HA (2012). The effects of anesthesia, mouse strain and age on intraocular pressure and an improved murine model of experimental glaucoma. Exp Eye Res, 99, 27–35. 10.1016/j.exer.2012.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras L, Rial E, Cerdan S, & Satrustegui J (2017). Uncoupling Protein 2 (UCP2) Function in the Brain as Revealed by the Cerebral Metabolism of (1-. Neurochem Res, 42(1), 108–114. 10.1007/s11064-016-1999-5 [DOI] [PubMed] [Google Scholar]
- Crispim D, Fagundes NJ, dos Santos KG, Rheinheimer J, Bouças AP, de Souza BM, Macedo GS, Leiria LB, Gross JL, & Canani LH (2010). Polymorphisms of the UCP2 gene are associated with proliferative diabetic retinopathy in patients with diabetes mellitus. Clin Endocrinol (Oxf), 72(5), 612–619. 10.1111/j.1365-2265.2009.03684.x [DOI] [PubMed] [Google Scholar]
- Crispim D, Rodrigues M, da Silva LPA, Bouças AP, Canani LH, Carlessi R, & de Souza BM (2019). The A allele of the UCP2 -866G/A polymorphism changes UCP2 promoter activity in HUVECs treated with high glucose. Mol Biol Rep, 46(5), 4735–4741. 10.1007/s11033-019-04918-0 [DOI] [PubMed] [Google Scholar]
- Cui Y, Xu X, Bi H, Zhu Q, Wu J, Xia X, Qiushi Ren, & Ho PC (2006). Expression modification of uncoupling proteins and MnSOD in retinal endothelial cells and pericytes induced by high glucose: the role of reactive oxygen species in diabetic retinopathy. Exp Eye Res, 83(4), 807–816. 10.1016/j.exer.2006.03.024 [DOI] [PubMed] [Google Scholar]
- D'Autréaux B, & Toledano MB (2007). ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol, 8(10), 813–824. 10.1038/nrm2256 [DOI] [PubMed] [Google Scholar]
- Dai Y, Hu X, & Sun X (2018). Overexpression of parkin protects retinal ganglion cells in experimental glaucoma. Cell Death Dis, 9(2), 88. 10.1038/s41419-017-0146-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CH, Kim KY, Bushong EA, Mills EA, Boassa D, Shih T, Kinebuchi M, Phan S, Zhou Y, Bihlmeyer NA, Nguyen JV, Jin Y, Ellisman MH, & Marsh-Armstrong N (2014). Transcellular degradation of axonal mitochondria. Proc Natl Acad Sci U S A, 111(26), 9633–9638. 10.1073/pnas.1404651111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simone R, Ajmone-Cat MA, Pandolfi M, Bernardo A, De Nuccio C, Minghetti L, & Visentin S (2015). The mitochondrial uncoupling protein-2 is a master regulator of both M1 and M2 microglial responses. J Neurochem. 135(1), 147–156. 10.1111/jnc.13244 [DOI] [PubMed] [Google Scholar]
- Delaey C, & Van De Voorde J (2000). Regulatory mechanisms in the retinal and choroidal circulation. Ophthalmic Res, 32(6), 249–256. 10.1159/000055622 [DOI] [PubMed] [Google Scholar]
- Della Santina L, Inman DM, Lupien CB, Horner PJ, & Wong RO (2013). Differential progression of structural and functional alterations in distinct retinal ganglion cell types in a mouse model of glaucoma. J Neurosci, 33(44), 17444–17457. 10.1523/JNEUROSCI.5461-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewan A, Liu M, Hartman S, Zhang SS, Liu DT, Zhao C, Tam PO, Chan WM, Lam DS, Snyder M, Barnstable C, Pang CP, & Hoh J (2006). HTRA1 promoter polymorphism in wet age-related macular degeneration. Science, 314(5801), 989–992. 10.1126/science.1133807 [DOI] [PubMed] [Google Scholar]
- Diano S, Matthews RT, Patrylo P, Yang L, Beal MF, Barnstable CJ, & Horvath TL (2003). Uncoupling protein 2 prevents neuronal death including that occurring during seizures: a mechanism for preconditioning. Endocrinology, 144(11), 5014–5021. 10.1210/en.2003-0667 [DOI] [PubMed] [Google Scholar]
- Donadelli M, Dando I, Fiorini C, & Palmieri M (2014). UCP2, a mitochondrial protein regulated at multiple levels. Cell Mol Life Sci, 71(7), 1171–1190. 10.1007/s00018-013-1407-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutra MRH, Feliciano RDS, Jacinto KR, Gouveia TLF, Brigidio E, Serra AJ, Morris M, Naffah-Mazzacoratti MDG, & Silva JA (2018). Protective Role of UCP2 in Oxidative Stress and Apoptosis during the Silent Phase of an Experimental Model of Epilepsy Induced by Pilocarpine. Oxid Med Cell Longev, 2018, 6736721. 10.1155/2018/6736721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elachouri G, Lee-Rivera I, Clérin E, Argentini M, Fridlich R, Blond F, Ferracane V, Yang Y, Raffelsberger W, Wan J, Bennett J, Sahel JA, Zack DJ, & Léveillard T (2015). Thioredoxin rod-derived cone viability factor protects against photooxidative retinal damage. Free Radic Biol Med, 81, 22–29. 10.1016/j.freeradbiomed.2015.01.003 [DOI] [PubMed] [Google Scholar]
- Esterbauer H, Schaur RJ, & Zollner H (1991). Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med, 11(1), 81–128. 10.1016/0891-5849(91)90192-6 [DOI] [PubMed] [Google Scholar]
- Esteves TC, & Brand MD (2005). The reactions catalysed by the mitochondrial uncoupling proteins UCP2 and UCP3. Biochim Biophys Acta, 1709(1), 35–44. 10.1016/j.bbabio.2005.06.002 [DOI] [PubMed] [Google Scholar]
- Fain GL (1976). Sensitivity of toad rods: Dependence on wave-length and background illumination. J Physiol, 261(1), 71–101. 10.1113/jphysiol.1976.sp011549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feilchenfeld Z, Yücel YH, & Gupta N (2008). Oxidative injury to blood vessels and glia of the prelaminar optic nerve head in human glaucoma. Exp Eye Res, 87(5), 409–414. 10.1016/j.exer.2008.07.011 [DOI] [PubMed] [Google Scholar]
- Fesenko EE, Kolesnikov SS, & Lyubarsky AL (1985). Induction by cyclic GMP of cationic conductance in plasma membrane of retinal rod outer segment. Nature, 313(6000), 310–313. 10.1038/313310a0 [DOI] [PubMed] [Google Scholar]
- Fisher CR, & Ferrington DA (2018). Perspective on AMD Pathobiology: A Bioenergetic Crisis in the RPE. Invest Ophthalmol Vis Sci, 59(4), AMD41–AMD47. 10.1167/iovs.18-24289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisler JS, & Warden CH (2006). Uncoupling proteins, dietary fat and the metabolic syndrome. Nutr Metab (Lond), 3, 38. 10.1186/1743-7075-3-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floor E, & Wetzel MG (1998). Increased protein oxidation in human substantia nigra pars compacta in comparison with basal ganglia and prefrontal cortex measured with an improved dinitrophenylhydrazine assay. J Neurochem, 70(1), 268–275. 10.1046/j.1471-4159.1998.70010268.x [DOI] [PubMed] [Google Scholar]
- Frank M, Duvezin-Caubet S, Koob S, Occhipinti A, Jagasia R, Petcherski A, Ruonala MO, Priault M, Salin B, & Reichert AS (2012). Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim Biophys Acta, 1823(12), 2297–2310. 10.1016/j.bbamcr.2012.08.007 [DOI] [PubMed] [Google Scholar]
- Frank RN, Amin RH, & Puklin JE (1999). Antioxidant enzymes in the macular retinal pigment epithelium of eyes with neovascular age-related macular degeneration. Am J Ophthalmol, 127(6), 694–709. 10.1016/s0002-9394(99)00032-x [DOI] [PubMed] [Google Scholar]
- Freeman RD (1972). Oxygen consumption by the component layers of the cornea. J Physiol, 225(1), 15–32. 10.1113/jphysiol.1972.sp009927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friederich M, Fasching A, Hansell P, Nordquist L, & Palm F (2008). Diabetes-induced up-regulation of uncoupling protein-2 results in increased mitochondrial uncoupling in kidney proximal tubular cells. Biochim Biophys Acta, 1777(7–8), 935–940. 10.1016/j.bbabio.2008.03.030 [DOI] [PubMed] [Google Scholar]
- Friederich-Persson M, Aslam S, Nordquist L, Welch WJ, Wilcox CS, & Palm F (2012). Acute knockdown of uncoupling protein-2 increases uncoupling via the adenine nucleotide transporter and decreases oxidative stress in diabetic kidneys. PLoS One, 7(7), e39635. 10.1371/journal.pone.0039635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganat YM, Silbereis J, Cave C, Ngu H, Anderson GM, Ohkubo Y, Ment LR, & Vaccarino FM (2006). Early postnatal astroglial cells produce multilineage precursors and neural stem cells in vivo. J Neurosci, 26(33), 8609–8621. 10.1523/JNEUROSCI.2532-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Medina JJ, Garcia-Medina M, Garrido-Fernandez P, Galvan-Espinosa J, Garcia-Maturana C, Zanon-Moreno V, & Pinazo-Duran MD (2015). A two-year follow-up of oral antioxidant supplementation in primary open-angle glaucoma: an open-label, randomized, controlled trial. Acta Ophthalmol, 93(6), 546–554. 10.1111/aos.12629 [DOI] [PubMed] [Google Scholar]
- Gardner TW, Antonetti DA, Barber AJ, LaNoue KF, & Levison SW (2002). Diabetic retinopathy: more than meets the eye. Surv Ophthalmol, 47 Suppl 2, S253–262. 10.1016/s0039-6257(02)00387-9 [DOI] [PubMed] [Google Scholar]
- Gaudry MJ, & Jastroch M (2019). Molecular evolution of uncoupling proteins and implications for brain function. Neurosci Lett, 696, 140–145. 10.1016/j.neulet.2018.12.027 [DOI] [PubMed] [Google Scholar]
- Gehrs KM, Jackson JR, Brown EN, Allikmets R, & Hageman GS (2010). Complement, age-related macular degeneration and a vision of the future. Arch Ophthalmol, 128(3), 349–358. 10.1001/archophthalmol.2010.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerencser AA, Chinopoulos C, Birket MJ, Jastroch M, Vitelli C, Nicholls DG, & Brand MD (2012). Quantitative measurement of mitochondrial membrane potential in cultured cells: calcium-induced de- and hyperpolarization of neuronal mitochondria. J Physiol, 590(12), 2845–2871. 10.1113/jphysiol.2012.228387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerencser AA, Mookerjee SA, Jastroch M, & Brand MD (2016). Measurement of the Absolute Magnitude and Time Courses of Mitochondrial Membrane Potential in Primary and Clonal Pancreatic Beta-Cells. PLoS One, 11(7), e0159199. 10.1371/journal.pone.0159199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardina TM, Steer JH, Lo SZ, & Joyce DA (2008). Uncoupling protein-2 accumulates rapidly in the inner mitochondrial membrane during mitochondrial reactive oxygen stress in macrophages. Biochim Biophys Acta, 1777(2), 118–129. 10.1016/j.bbabio.2007.11.006 [DOI] [PubMed] [Google Scholar]
- Gorin MB, & daSilva MJ (2020). Predictive genetics for AMD: Hype and hopes for genetics-based strategies for treatment and prevention. Exp Eye Res, 191, 107894. 10.1016/j.exer.2019.107894 [DOI] [PubMed] [Google Scholar]
- Govindarajan B, Junk A, Algeciras M, Salomon RG, & Bhattacharya SK (2009). Increased isolevuglandin-modified proteins in glaucomatous astrocytes. Mol Vis, 15, 1079–1091. [PMC free article] [PubMed] [Google Scholar]
- Greco AV, Ghirlanda G, Fedeli G, & Gambassi G (1970). Insulin in the cerebro spinal fluid of man. Eur Neurol, 3(5), 303–307. 10.1159/000113983 [DOI] [PubMed] [Google Scholar]
- Grundlingh J, Dargan PI, El-Zanfaly M, & Wood DM (2011). 2,4-dinitrophenol (DNP): a weight loss agent with significant acute toxicity and risk of death. J Med Toxicol, 7(3), 205–212. 10.1007/s13181-011-0162-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagins WA, Penn RD, & Yoshikami S (1970). Dark current and photocurrent in retinal rods. Biophys J, 10(5), 380–412. 10.1016/S0006-3495(70)86308-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines BA, Mehta SL, Pratt SM, Warden CH, & Li PA (2010). Deletion of mitochondrial uncoupling protein-2 increases ischemic brain damage after transient focal ischemia by altering gene expression patterns and enhancing inflammatory cytokines. J Cereb Blood Flow Metab, 30(11), 1825–1833. 10.1038/jcbfm.2010.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KG, & Herrmann JM (2019). Transport of Proteins into Mitochondria. Protein J, 38(3), 330–342. 10.1007/s10930-019-09819-6 [DOI] [PubMed] [Google Scholar]
- Hanák P, & Jezek P (2001). Mitochondrial uncoupling proteins and phylogenesis--UCP4 as the ancestral uncoupling protein. FEBS Lett, 495(3), 137–141. [DOI] [PubMed] [Google Scholar]
- Hashizume K, Hirasawa M, Imamura Y, Noda S, Shimizu T, Shinoda K, Kurihara T, Noda K, Ozawa Y, Ishida S, Miyake Y, Shirasawa T, & Tsubota K (2008). Retinal dysfunction and progressive retinal cell death in SOD1-deficient mice. Am J Pathol, 172(5), 1325–1331. 10.2353/ajpath.2008.070730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslip M, Dostanic I, Huang Y, Zhang Y, Russell KS, Jurczak MJ, Mannam P, Giordano F, Erzurum SC, & Lee PJ (2015). Endothelial uncoupling protein 2 regulates mitophagy and pulmonary hypertension during intermittent hypoxia. Arterioscler Thromb Vasc Biol, 35(5), 1166–1178. 10.1161/ATVBAHA.114.304865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hass DT, & Barnstable CJ (2016). Uncoupling protein 2 in the glial response to stress: implications for neuroprotection. Neural Regen Res, 11(8), 1197–1200. 10.4103/1673-5374.189159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hass DT, & Barnstable CJ (2019a). Cell Autonomous Neuroprotection by the Mitochondrial Uncoupling Protein 2 in a Mouse Model of Glaucoma. Front Neurosci, 13, 201. 10.3389/fnins.2019.00201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hass DT, & Barnstable CJ (2019b). Mitochondrial Uncoupling Protein 2 Knock-out Promotes Mitophagy to Decrease Retinal Ganglion Cell Death in a Mouse Model of Glaucoma. J Neurosci, 39(18), 3582–3596. 10.1523/JNEUROSCI.2702-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Leung KW, Ren Y, Pei J, Ge J, & Tombran-Tink J (2014). PEDF improves mitochondrial function in RPE cells during oxidative stress. Invest Ophthalmol Vis Sci, 55(10), 6742–6755. 10.1167/iovs.14-14696 [DOI] [PubMed] [Google Scholar]
- He Y, Luan Z, Fu X, & Xu X (2016). Overexpression of uncoupling protein 2 inhibits the high glucose-induced apoptosis of human umbilical vein endothelial cells. Int J Mol Med, 37(3), 631–638. 10.3892/ijmm.2016.2478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, & Tombran-Tink J (2010). Mitochondrial decay and impairment of antioxidant defenses in aging RPE cells. Adv Exp Med Biol, 664, 165–183. 10.1007/978-1-4419-1399-9_20 [DOI] [PubMed] [Google Scholar]
- He Y, Wang X, Liu X, Ji Z, & Ren Y (2019). Decreased uncoupling protein 2 expression in aging retinal pigment epithelial cells. Int J Ophthalmol, 12(3), 375–380. 10.18240/ijo.2019.03.04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton GM, Wagenvoord RJ, Kemp A, & Nicholls DG (1978). Brown-adipose-tissue mitochondria: photoaffinity labelling of the regulatory site of energy dissipation. Eur J Biochem, 82(2), 515–521. 10.1111/j.1432-1033.1978.tb12045.x [DOI] [PubMed] [Google Scholar]
- Hevner RF, & Wong-Riley MT (1990). Regulation of cytochrome oxidase protein levels by functional activity in the macaque monkey visual system. J Neurosci, 10(4), 1331–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J, King MS, & Pryde KR (2008). The production of reactive oxygen species by complex I. Biochem Soc Trans, 36(Pt 5), 976–980. 10.1042/BST0360976 [DOI] [PubMed] [Google Scholar]
- Ho PW, Chu AC, Kwok KH, Kung MH, Ramsden DB, & Ho SL (2006). Knockdown of uncoupling protein-5 in neuronal SH-SY5Y cells: Effects on MPP+-induced mitochondrial membrane depolarization, ATP deficiency, and oxidative cytotoxicity. J Neurosci Res, 84(6), 1358–1366. 10.1002/jnr.21034 [DOI] [PubMed] [Google Scholar]
- Ho PW, Liu HF, Ho JW, Zhang WY, Chu AC, Kwok KH, Ge X, Chan KH, Ramsden DB, & Ho SL (2010). Mitochondrial uncoupling protein-2 (UCP2) mediates leptin protection against MPP+ toxicity in neuronal cells. Neurotox Res, 17(4), 332–343. 10.1007/s12640-009-9109-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang QV, Linsenmeier RA, Chung CK, & Curcio CA (2002). Photoreceptor inner segments in monkey and human retina: mitochondrial density, optics, and regional variation. Vis Neurosci, 19(4), 395–407. 10.1017/s0952523802194028 [DOI] [PubMed] [Google Scholar]
- Hoang T, Smith MD, & Jelokhani-Niaraki M (2012). Toward understanding the mechanism of ion transport activity of neuronal uncoupling proteins UCP2, UCP4, and UCP5. Biochemistry, 51(19), 4004–4014. 10.1021/bi3003378 [DOI] [PubMed] [Google Scholar]
- Hochachka PW, & Dressendorfer RH (1976). Succinate accumulation in man during exercise. Eur J Appl Physiol Occup Physiol, 35(4), 235–242. 10.1007/BF00423282 [DOI] [PubMed] [Google Scholar]
- Hochachka PW, Owen TG, Allen JF, & Whittow GC (1975). Multiple end products of anaerobiosis in diving vertebrates. Comp Biochem Physiol B, 50(1), 17–22. 10.1016/0305-0491(75)90292-8 [DOI] [PubMed] [Google Scholar]
- Horvath TL, Warden CH, Hajos M, Lombardi A, Goglia F, Diano S. (1999). Brain uncoupling protein 2: uncoupled neuronal mitochondria predict thermal synapses in homeostatic centers. J Neurosci., 19(23):10417–10427. DOI: 10.1523/JNEUROSCI.19-23-10417.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath TL, Diano S, Miyamoto S, Barry S, Gatti S, Alberati D, Livak F, Lombardi A, Moreno M, Goglia F, Mor G, Hamilton J, Kachinskas D, Horwitz B, & Warden CH (2003). Uncoupling proteins-2 and 3 influence obesity and inflammation in transgenic mice. Int J Obes Relat Metab Disord, 27(4), 433–442. 10.1038/sj.ijo.0802257 [DOI] [PubMed] [Google Scholar]
- Hu Y, Chen Y, Ding L, He X, Takahashi Y, Gao Y, Shen W, Cheng R, Chen Q, Qi X, Boulton ME, & Ma JX (2013). Pathogenic role of diabetes-induced PPAR-α down-regulation in microvascular dysfunction. Proc Natl Acad Sci U S A, 110(38), 15401–15406. 10.1073/pnas.1307211110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes J, & Criscuolo F (2008). Evolutionary history of the UCP gene family: gene duplication and selection. BMC Evol Biol, 8, 306. 10.1186/1471-2148-8-306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtaud C, Gelly C, Bouillaud F, & Lévi-Meyrueis C (2006). Translation control of UCP2 synthesis by the upstream open reading frame. Cell Mol Life Sci, 63(15), 1780–1789. 10.1007/s00018-006-6129-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtaud C, Gelly C, Chen Z, Lévi-Meyrueis C, & Bouillaud F (2007). Glutamine stimulates translation of uncoupling protein 2mRNA. Cell Mol Life Sci, 64(14), 1853–1860. 10.1007/s00018-007-7039-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzotti A, Saccà SC, Cartiglia C, & De Flora S (2003). Oxidative deoxyribonucleic acid damage in the eyes of glaucoma patients. Am J Med, 114(8), 638–646. [DOI] [PubMed] [Google Scholar]
- Jarrett SG, & Boulton ME (2012). Consequences of oxidative stress in age-related macular degeneration. Mol Aspects Med, 33(4), 399–417. 10.1016/j.mam.2012.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ježek P, Holendová B, Garlid KD, & Jabůrek M (2018). Mitochondrial Uncoupling Proteins: Subtle Regulators of Cellular Redox Signaling. Antioxid Redox Signal, 29(7), 667–714. 10.1089/ars.2017.7225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph JW, Koshkin V, Zhang CY, Wang J, Lowell BB, Chan CB, & Wheeler MB (2002). Uncoupling protein 2 knockout mice have enhanced insulin secretory capacity after a high-fat diet. Diabetes, 51(11), 3211–3219. 10.2337/diabetes.51.11.3211 [DOI] [PubMed] [Google Scholar]
- Kaarniranta K, Kajdanek J, Morawiec J, Pawlowska E, & Blasiak J (2018). PGC-1α Protects RPE Cells of the Aging Retina against Oxidative Stress-Induced Degeneration through the Regulation of Senescence and Mitochondrial Quality Control. The Significance for AMD Pathogenesis. Int J Mol Sci, 19(8). 10.3390/ijms19082317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaarniranta K, Uusitalo H, Blasiak J, Felszeghy S, Kannan R, Kauppinen A, Salminen A, Sinha D, & Ferrington D (2020). Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog Retin Eye Res, 79, 100858. 10.1016/j.preteyeres.2020.100858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, & Yoshimori T (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J, 19(21), 5720–5728. 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama GH, & Wong-Riley MT (1984). The histochemical localization of cytochrome oxidase in the retina and lateral geniculate nucleus of the ferret, cat, and monkey, with particular reference to retinal mosaics and ON/OFF-center visual channels. J Neurosci, 4(10), 2445–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanemoto N, Okamoto T, Tanabe K, Shimada T, Minoshima H, Hidoh Y, Aoyama M, Ban T, Kobayashi Y, Ando H, Inoue Y, Itotani M, & Sato S (2019). Antidiabetic and cardiovascular beneficial effects of a liver-localized mitochondrial uncoupler. Nat Commun, 10(1), 2172. 10.1038/s41467-019-09911-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanow MA, Giarmarco MM, Jankowski CS, Tsantilas K, Engel AL, Du J, Linton JD, Farnsworth CC, Sloat SR, Rountree A, Sweet IR, Lindsay KJ, Parker ED, Brockerhoff SE, Sadilek M, Chao JR, & Hurley JB (2017). Biochemical adaptations of the retina and retinal pigment epithelium support a metabolic ecosystem in the vertebrate eye. Elife, 6. e28899, 10.7554/eLife.28899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karali M, Peluso I, Gennarino VA, Bilio M, Verde R, Lago G, Dollé P, & Banfi S (2010). miRNeye: a microRNA expression atlas of the mouse eye. BMC Genomics, 11, 715. 10.1186/1471-2164-11-715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JG, Sun BH, Dietrich MO, Koch M, Yao GQ, Diano S, Insogna K, & Horvath TL (2015). AgRP Neurons Regulate Bone Mass. Cell Rep, 13(1), 8–14. 10.1016/j.celrep.2015.08.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A, Namekata K, Guo X, Noro T, Harada C, & Harada T (2017). Targeting Oxidative Stress for Treatment of Glaucoma and Optic Neuritis. Oxid Med Cell Longev, 2017, 2817252. 10.1155/2017/2817252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, & Hoh J (2005). Complement factor H polymorphism in age-related macular degeneration. Science, 308(5720), 385–389. 10.1126/science.1109557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong GY, Van Bergen NJ, Trounce IA, & Crowston JG (2009). Mitochondrial dysfunction and glaucoma. J Glaucoma, 18(2), 93–100. 10.1097/IJG.0b013e318181284f [DOI] [PubMed] [Google Scholar]
- Korshunov SS, Skulachev VP, & Starkov AA (1997). High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett, 416(1), 15–18. [DOI] [PubMed] [Google Scholar]
- Kowluru RA, & Abbas SN (2003). Diabetes-induced mitochondrial dysfunction in the retina. Invest Ophthalmol Vis Sci, 44(12), 5327–5334. 10.1167/iovs.03-0353 [DOI] [PubMed] [Google Scholar]
- Kowluru RA, & Kennedy A (2001). Therapeutic potential of anti-oxidants and diabetic retinopathy. Expert Opin Investig Drugs, 10(9), 1665–1676. 10.1517/13543784.10.9.1665 [DOI] [PubMed] [Google Scholar]
- Koziel A, Sobieraj I, & Jarmuszkiewicz W (2015). Increased activity of mitochondrial uncoupling protein 2 improves stress resistance in cultured endothelial cells exposed in vitro to high glucose levels. Am J Physiol Heart Circ Physiol, 309(1), H147–156. 10.1152/ajpheart.00759.2014 [DOI] [PubMed] [Google Scholar]
- Krizaj D, & Copenhagen DR (1998). Compartmentalization of calcium extrusion mechanisms in the outer and inner segments of photoreceptors. Neuron, 21(1), 249–256. 10.1016/s0896-6273(00)80531-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudin AP, Malinska D, & Kunz WS (2008). Sites of generation of reactive oxygen species in homogenates of brain tissue determined with the use of respiratory substrates and inhibitors. Biochim Biophys Acta, 1777(7–8), 689–695. 10.1016/j.bbabio.2008.05.010 [DOI] [PubMed] [Google Scholar]
- Kuehn MH, Fingert JH, & Kwon YH (2005). Retinal ganglion cell death in glaucoma: mechanisms and neuroprotective strategies. Ophthalmol Clin North Am, 18(3), 383–395, vi. 10.1016/j.ohc.2005.04.002 [DOI] [PubMed] [Google Scholar]
- Kunji ER, & Robinson AJ (2010). Coupling of proton and substrate translocation in the transport cycle of mitochondrial carriers. Curr Opin Struct Biol, 20(4), 440–447. 10.1016/j.sbi.2010.06.004 [DOI] [PubMed] [Google Scholar]
- Lambert AJ, & Brand MD (2004). Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem J, 382(Pt 2), 511–517. 10.1042/BJ20040485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapp DW, Zhang SS, & Barnstable CJ (2014). Stat3 mediates LIF-induced protection of astrocytes against toxic ROS by upregulating the UPC2 mRNA pool. Glia, 62(2), 159–170. 10.1002/glia.22594 [DOI] [PubMed] [Google Scholar]
- Lau JC, & Linsenmeier RA (2012). Oxygen consumption and distribution in the Long-Evans rat retina. Exp Eye Res, 102, 50–58. 10.1016/j.exer.2012.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guiner C, Stieger K, Toromanoff A, Guilbaud M, Mendes-Madeira A, Devaux M, Guigand L, Cherel Y, Moullier P, Rolling F, & Adjali O (2014). Transgene regulation using the tetracycline-inducible TetR-KRAB system after AAV-mediated gene transfer in rodents and nonhuman primates. PLoS One, 9(9), e102538. 10.1371/journal.pone.0102538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefevere E, Toft-Kehler AK, Vohra R, Kolko M, Moons L, & Van Hove I (2017). Mitochondrial dysfunction underlying outer retinal diseases. Mitochondrion, 36, 66–76. 10.1016/j.mito.2017.03.006 [DOI] [PubMed] [Google Scholar]
- Lin T, He F, & Lei B (2012). [Expressions of adiponectin and its receptors in the retina of normal and type 1 diabetic mice]. Nan Fang Yi Ke Da Xue Xue Bao, 32(11), 1543–1547. [PubMed] [Google Scholar]
- Linsenmeier RA, & Zhang HF (2017). Retinal oxygen: from animals to humans. Prog Retin Eye Res, 58, 115–151. 10.1016/j.preteyeres.2017.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu WY, Liou SS, Hong TY, & Liu IM (2017). Protective Effects of Hesperidin (Citrus Flavonone) on High Glucose Induced Oxidative Stress and Apoptosis in a Cellular Model for Diabetic Retinopathy. Nutrients, 9(12). 10.3390/nu9121312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Leo LF, McGregor C, Grivitishvili A, Barnstable CJ, & Tombran-Tink J (2012). Pigment epithelium-derived factor (PEDF) peptide eye drops reduce inflammation, cell death and vascular leakage in diabetic retinopathy in Ins2(Akita) mice. Mol Med, 18, 1387–1401. 10.2119/molmed.2012.00008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan A, Pell VR, Shaffer KJ, Evans C, Stanley NJ, Robb EL, Prime TA, Chouchani ET, Cochemé HM, Fearnley IM, Vidoni S, James AM, Porteous CM, Partridge L, Krieg T, Smith RA, & Murphy MP (2016). Assessing the Mitochondrial Membrane Potential in Cells and In Vivo using Targeted Click Chemistry and Mass Spectrometry. Cell Metab, 23(2), 379–385. 10.1016/j.cmet.2015.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovatt M, Adnan K, Peh GSL, & Mehta JS (2018). Regulation of Oxidative Stress in Corneal Endothelial Cells by Prdx6. Antioxidants (Basel), 7(12), 180, 10.3390/antiox7120180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Léveillard T, & Sahel JA (2010). Rod-derived cone viability factor for treating blinding diseases: from clinic to redox signaling. Sci Transl Med, 2(26), 26ps16. 10.1126/scitranslmed.3000866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddalena LA, Selim SM, Fonseca J, Messner H, McGowan S, & Stuart JA (2017). Hydrogen peroxide production is affected by oxygen levels in mammalian cell culture. Biochem Biophys Res Commun, 493(1), 246–251. 10.1016/j.bbrc.2017.09.037 [DOI] [PubMed] [Google Scholar]
- Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, Basutkar P, Tivey ARN, Potter SC, Finn RD, & Lopez R (2019). The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res, 47(W1), W636–W641. 10.1093/nar/gkz268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen-Bouterse SA, Mohammad G, Kanwar M, & Kowluru RA (2010). Role of mitochondrial DNA damage in the development of diabetic retinopathy, and the metabolic memory phenomenon associated with its progression. Antioxid Redox Signal, 13(6), 797–805. 10.1089/ars.2009.2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailloux RJ, & Harper ME (2011). Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic Biol Med, 51(6), 1106–1115. 10.1016/j.freeradbiomed.2011.06.022 [DOI] [PubMed] [Google Scholar]
- Massaad CA (2011). Neuronal and vascular oxidative stress in Alzheimer's disease. Curr Neuropharmacol, 9(4), 662–673. 10.2174/157015911798376244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga T, Gu N, Yamazaki H, Tsuda M, Adachi T, Yasuda K, Moritani T, Tsuda K, Nonaka M, & Nishiyama T (2009). Association of UCP2 and UCP3 polymorphisms with heart rate variability in Japanese men. J Hypertens, 27(2), 305–313. 10.1097/HJH.0b013e32831ac967 [DOI] [PubMed] [Google Scholar]
- Medvedev AV, Snedden SK, Raimbault S, Ricquier D, & Collins S (2001). Transcriptional regulation of the mouse uncoupling protein-2 gene. Double E-box motif is required for peroxisome proliferator-activated receptor-gamma-dependent activation. J Biol Chem, 276(14), 10817–10823. 10.1074/jbc.M010587200 [DOI] [PubMed] [Google Scholar]
- Mehta SL, & Li PA (2009). Neuroprotective role of mitochondrial uncoupling protein 2 in cerebral stroke. J Cereb Blood Flow Metab, 29(6), 1069–1078. 10.1038/jcbfm.2009.4 [DOI] [PubMed] [Google Scholar]
- Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Däbritz JHM, Gottlieb E, Latorre I, Corr SC, McManus G, Ryan D, Jacobs HT, Szibor M, Xavier RJ, Braun T, Frezza C, Murphy MP, & O'Neill LA (2016). Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell, 167(2), 457–470.e413. 10.1016/j.cell.2016.08.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P (1961). Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature, 191, 144–148. 10.1038/191144a0 [DOI] [PubMed] [Google Scholar]
- Miwa S, St-Pierre J, Partridge L, & Brand MD (2003). Superoxide and hydrogen peroxide production by Drosophila mitochondria. Free Radic Biol Med, 35(8), 938–948. [DOI] [PubMed] [Google Scholar]
- Moreno MC, Campanelli J, Sande P, Sánez DA, Keller Sarmiento MI, & Rosenstein RE (2004). Retinal oxidative stress induced by high intraocular pressure. Free Radic Biol Med, 37(6), 803–812. [DOI] [PubMed] [Google Scholar]
- Mousa A, Kondkar AA, Al-Obeidan SA, Azad TA, Sultan T, Osman E, & Abu-Amero KK (2015). Association of total antioxidants level with glaucoma type and severity. Saudi Med J, 36(6), 671–677. 10.15537/smj.2015.6.10697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP (1989). Slip and leak in mitochondrial oxidative phosphorylation. Biochim Biophys Acta, 977(2), 123–141. 10.1016/s0005-2728(89)80063-5 [DOI] [PubMed] [Google Scholar]
- Murphy MP (2009). How mitochondria produce reactive oxygen species. Biochem J, 417(1), 1–13. 10.1042/BJ20081386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP, & Smith RA (2007). Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol, 47, 629–656. 10.1146/annurev.pharmtox.47.120505.105110 [DOI] [PubMed] [Google Scholar]
- Neishabouri AM, Faisal AA (2011). The metabolic efficiency of myelinated vs unmyelinated axons. BMC Neurosci 12, P100. 10.1186/1471-2202-12-S1-P100 [DOI] [Google Scholar]
- Nicholls DG, Bernson VS, & Heaton GM (1978). The identification of the component in the inner membrane of brown adipose tissue mitochondria responsible for regulating energy dissipation. Experientia Suppl, 32, 89–93. 10.1007/978-3-0348-5559-4_9 [DOI] [PubMed] [Google Scholar]
- Nie D, Chen Z, Ebrahimi-Fakhari D, Di Nardo A, Julich K, Robson VK, Cheng YC, Woolf CJ, Heiman M, & Sahin M (2015). The Stress-Induced Atf3-Gelsolin Cascade Underlies Dendritic Spine Deficits in Neuronal Models of Tuberous Sclerosis Complex. J Neurosci, 35(30), 10762–10772. 10.1523/JNEUROSCI.4796-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niven JE, & Laughlin SB (2008). Energy limitation as a selective pressure on the evolution of sensory systems. J Exp Biol, 211(Pt 11), 1792–1804. 10.1242/jeb.017574 [DOI] [PubMed] [Google Scholar]
- Nogueira FT, Borecký J, Vercesi AE, & Arruda P (2005). Genomic structure and regulation of mitochondrial uncoupling protein genes in mammals and plants. Biosci Rep, 25(3-4), 209–226. 10.1007/s10540-005-2886-5 [DOI] [PubMed] [Google Scholar]
- Noh YH, Kim KY, Shim MS, Choi SH, Choi S, Ellisman MH, Weinreb RN, Perkins GA, & Ju WK (2013). Inhibition of oxidative stress by coenzyme Q10 increases mitochondrial mass and improves bioenergetic function in optic nerve head astrocytes. Cell Death Dis, 4, e820. 10.1038/cddis.2013.341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nègre-Salvayre A, Hirtz C, Carrera G, Cazenave R, Troly M, Salvayre R, Pénicaud L, & Casteilla L (1997). A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J, 11(10), 809–815. [PubMed] [Google Scholar]
- Okawa H, Sampath AP, Laughlin SB, & Fain GL (2008). ATP consumption by mammalian rod photoreceptors in darkness and in light. Curr Biol, 18(24), 1917–1921. 10.1016/j.cub.2008.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio-Paz I, Uribe-Carvajal S, & Salceda R (2015). In the Early Stages of Diabetes, Rat Retinal Mitochondria Undergo Mild Uncoupling due to UCP2 Activity. PLoS One, 10(5), e0122727. 10.1371/journal.pone.0122727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski J, Klimek-Tomczak K, Wyrwicz LS, Mikula M, Schullery DS, & Bomsztyk K (2004). Heterogeneous nuclear ribonucleoprotein K enhances insulin-induced expression of mitochondrial UCP2 protein. J Biol Chem, 279(52), 54599–54609. 10.1074/jbc.M406753200 [DOI] [PubMed] [Google Scholar]
- Paget C, Lecomte M, Ruggiero D, Wiernsperger N, & Lagarde M (1998). Modification of enzymatic antioxidants in retinal microvascular cells by glucose or advanced glycation end products. Free Radic Biol Med, 25(1), 121–129. 10.1016/s0891-5849(98)00071-9 [DOI] [PubMed] [Google Scholar]
- Palmieri F, & Pierri CL (2010). Structure and function of mitochondrial carriers - role of the transmembrane helix P and G residues in the gating and transport mechanism. FEBS Lett, 584(9), 1931–1939. 10.1016/j.febslet.2009.10.063 [DOI] [PubMed] [Google Scholar]
- Parker N, Crichton PG, Vidal-Puig AJ, & Brand MD (2009). Uncoupling protein-1 (UCP1) contributes to the basal proton conductance of brown adipose tissue mitochondria. J Bioenerg Biomembr, 41(4), 335–342. 10.1007/s10863-009-9232-8 [DOI] [PubMed] [Google Scholar]
- Pearsall EA, Cheng R, Matsuzaki S, Zhou K, Ding L, Ahn B, Kinter M, Humphries KM, Quiambao AB, Farjo RA, & Ma JX (2019). Neuroprotective effects of PPARα in retinopathy of type 1 diabetes. PLoS One, 14(2), e0208399. 10.1371/journal.pone.0208399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trézéguet V, Lauquin GJ, & Brandolin G (2003). Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature, 426(6962), 39–44. 10.1038/nature02056 [DOI] [PubMed] [Google Scholar]
- Pecqueur C, Alves-Guerra MC, Gelly C, Levi-Meyrueis C, Couplan E, Collins S, Ricquier D, Bouillaud F, & Miroux B (2001). Uncoupling protein 2, in vivo distribution, induction upon oxidative stress, and evidence for translational regulation. J Biol Chem, 276(12), 8705–8712. 10.1074/jbc.M006938200 [DOI] [PubMed] [Google Scholar]
- Pecqueur C, Bui T, Gelly C, Hauchard J, Barbot C, Bouillaud F, Ricquier D, Miroux B, & Thompson CB (2008). Uncoupling protein-2 controls proliferation by promoting fatty acid oxidation and limiting glycolysis-derived pyruvate utilization. FASEB J, 22(1), 9–18. 10.1096/fj.07-8945com [DOI] [PubMed] [Google Scholar]
- Pecqueur C, Cassard-Doulcier AM, Raimbault S, Miroux B, Fleury C, Gelly C, Bouillaud F, & Ricquier D (1999). Functional organization of the human uncoupling protein-2 gene, and juxtaposition to the uncoupling protein-3 gene. Biochem Biophys Res Commun, 255(1), 40–46. 10.1006/bbrc.1998.0146 [DOI] [PubMed] [Google Scholar]
- Perreten Lambert H, Zenger M, Azarias G, Chatton JY, Magistretti PJ, & Lengacher S (2014). Control of mitochondrial pH by uncoupling protein 4 in astrocytes promotes neuronal survival. J Biol Chem, 289(45), 31014–31028. 10.1074/jbc.M114.570879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polidori MC, Griffiths HR, Mariani E, & Mecocci P (2007). Hallmarks of protein oxidative damage in neurodegenerative diseases: focus on Alzheimer's disease. Amino Acids, 32(4), 553–559. 10.1007/s00726-006-0431-x [DOI] [PubMed] [Google Scholar]
- Pryde KR, & Hirst J (2011). Superoxide is produced by the reduced flavin in mitochondrial complex I: a single, unified mechanism that applies during both forward and reverse electron transfer. J Biol Chem, 286(20), 18056–18065. 10.1074/jbc.M110.186841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin N, Cai T, Ke Q, Yuan Q, Luo J, Mao X, Jiang L, Cao H, Wen P, Zen K, Zhou Y, & Yang J (2019). UCP2-dependent improvement of mitochondrial dynamics protects against acute kidney injury. J Pathol, 247(3), 392–405. 10.1002/path.5198 [DOI] [PubMed] [Google Scholar]
- Radomska-Leśniewska DM, Osiecka-Iwan A, Hyc A, Góźdź A, Dąbrowska AM, & Skopiński P (2019). Therapeutic potential of curcumin in eye diseases. Cent Eur J Immunol, 44(2), 181–189. 10.5114/ceji.2019.87070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rani V, Deep G, Singh RK, Palle K, & Yadav UC (2016). Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci, 148, 183–193. 10.1016/j.lfs.2016.02.002 [DOI] [PubMed] [Google Scholar]
- Reddy R, Horvath T, & Barnstable C (2002). Uncoupling Protein UCP2 Regulates Programmed Cell Death in the Ganglion Cell Layer of Mouse Retina. Investigative Ophthalmology & Visual Science, 43(13), 1994–1994.12037010 [Google Scholar]
- Robb EL, Hall AR, Prime TA, Eaton S, Szibor M, Viscomi C, James AM, & Murphy MP (2018). Control of mitochondrial superoxide production by reverse electron transport at complex I. J Biol Chem, 293(25), 9869–9879. 10.1074/jbc.RA118.003647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotblat B, Southwell AL, Ehrnhoefer DE, Skotte NH, Metzler M, Franciosi S, Leprivier G, Somasekharan SP, Barokas A, Deng Y, Tang T, Mathers J, Cetinbas N, Daugaard M, Kwok B, Li L, Carnie CJ, Fink D, Nitsch R, Galpin JD, Ahern CA, Melino G, Penninger JM, Hayden MR, & Sorensen PH (2014). HACE1 reduces oxidative stress and mutant Huntingtin toxicity by promoting the NRF2 response. Proc Natl Acad Sci U S A, 111(8), 3032–3037. 10.1073/pnas.1314421111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset S, Mozo J, Dujardin G, Emre Y, Masscheleyn S, Ricquier D, & Cassard-Doulcier AM (2007). UCP2 is a mitochondrial transporter with an unusual very short half-life. FEBS Lett, 581(3), 479–482. 10.1016/j.febslet.2007.01.010 [DOI] [PubMed] [Google Scholar]
- Rubattu S, Stanzione R, Bianchi F, Cotugno M, Forte M, Della Ragione F, Fioriniello S, D'Esposito M, Marchitti S, Madonna M, Baima S, Morelli G, Sciarretta S, Sironi L, Gelosa P, & Volpe M (2017). Reduced brain UCP2 expression mediated by microRNA-503 contributes to increased stroke susceptibility in the high-salt fed stroke-prone spontaneously hypertensive rat. Cell Death Dis, 8(6), e2891. 10.1038/cddis.2017.278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda EM, Johnson JE, Giddabasappa A, Swaroop A, Brooks MJ, Sigel I, Chaney SY, & Fox DA (2016). The cellular and compartmental profile of mouse retinal glycolysis, tricarboxylic acid cycle, oxidative phosphorylation, and ~P transferring kinases. Mol Vis, 22, 847–885. [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Ramírez A, López-Acosta O, Barrios-Maya MA, & El-Hafidi M (2016). Cell Death and Heart Failure in Obesity: Role of Uncoupling Proteins. Oxid Med Cell Longev, 2016, 9340654. 10.1155/2016/9340654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupprecht A, Bräuer AU, Smorodchenko A, Goyn J, Hilse KE, Shabalina IG, Infante-Duarte C, & Pohl EE (2012). Quantification of uncoupling protein 2 reveals its main expression in immune cells and selective up-regulation during T-cell proliferation. PLoS One, 7(8), e41406. 10.1371/journal.pone.0041406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupprecht A, Moldzio R, Mödl B, & Pohl EE (2019). Glutamine regulates mitochondrial uncoupling protein 2 to promote glutaminolysis in neuroblastoma cells. Biochim Biophys Acta Bioenerg, 1860(5), 391–401. 10.1016/j.bbabio.2019.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabanayagam C, Banu R, Chee ML, Lee R, Wang YX, Tan G, Jonas JB, Lamoureux EL, Cheng CY, Klein BEK, Mitchell P, Klein R, Cheung CMG, & Wong TY (2019). Incidence and progression of diabetic retinopathy: a systematic review. Lancet Diabetes Endocrinol, 7(2), 140–149. 10.1016/S2213-8587(18)30128-1 [DOI] [PubMed] [Google Scholar]
- Sandbach JM, Coscun PE, Grossniklaus HE, Kokoszka JE, Newman NJ, & Wallace DC (2001). Ocular pathology in mitochondrial superoxide dismutase (Sod2)-deficient mice. Invest Ophthalmol Vis Sci, 42(10), 2173–2178. [PubMed] [Google Scholar]
- Sappington RM, Carlson BJ, Crish SD, & Calkins DJ (2010). The microbead occlusion model: a paradigm for induced ocular hypertension in rats and mice. Invest Ophthalmol Vis Sci, 51(1), 207–216. 10.1167/iovs.09-3947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satish S, Philipose H, Rosales MAB, & Saint-Geniez M (2018). Pharmaceutical Induction of PGC-1. Oxid Med Cell Longev, 2018, 9248640. 10.1155/2018/9248640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayeed A, Meng Z, Luciani G, Chen LC, Bennington JL, & Dairkee SH (2010). Negative regulation of UCP2 by TGFβ signaling characterizes low and intermediate-grade primary breast cancer. Cell Death Dis, 1, e53. 10.1038/cddis.2010.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleiff E, & McBride H (2000). The central matrix loop drives import of uncoupling protein 1 into mitochondria. J Cell Sci, 113 (Pt 12), 2267–2272. [DOI] [PubMed] [Google Scholar]
- Schonfeld-Warden NA, & Warden CH (2001). Physiological effects of variants in human uncoupling proteins: UCP2 influences body-mass index. Biochem Soc Trans, 29(Pt 6), 777–784. 10.1042/0300-5127:0290777 [DOI] [PubMed] [Google Scholar]
- Scialò F, Sriram A, Fernández-Ayala D, Gubina N, Lõhmus M, Nelson G, Logan A, Cooper HM, Navas P, Enríquez JA, Murphy MP, & Sanz A (2016). Mitochondrial ROS Produced via Reverse Electron Transport Extend Animal Lifespan. Cell Metab, 23(4), 725–734. 10.1016/j.cmet.2016.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sela S, Shasha SM, Mashiach E, Haj M, Kristal B, & Shkolnik T (1993). Effect of oxygen tension on activity of antioxidant enzymes and on renal function of the postischemic reperfused rat kidney. Nephron, 63(2), 199–206. 10.1159/000187183 [DOI] [PubMed] [Google Scholar]
- Sheibani N, Wang S, Darjatmoko SR, Fisk DL, Shahi PK, Pattnaik BR, Sorenson CM, Bhowmick R, Volpert OV, Albert DM, Melgar-Asensio I, & Henkin J (2019). Novel anti-angiogenic PEDF-derived small peptides mitigate choroidal neovascularization. Exp Eye Res, 188, 107798. 10.1016/j.exer.2019.107798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C, Chen L, Jiang L, & Lai TY (2015). Neuroprotective effect of epigallocatechin-3-gallate in a mouse model of chronic glaucoma. Neurosci Lett. 600, 132–136. 10.1016/j.neulet.2015.06.002 [DOI] [PubMed] [Google Scholar]
- Shen Y, Wen Z, Wang N, Zheng Z, Liu K, Xia X, Gu Q, Shi Y, & Xu X (2014). Investigation of variants in UCP2 in Chinese type 2 diabetes and diabetic retinopathy. PLoS One, 9(11), e112670. 10.1371/journal.pone.0112670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluse FE (2012). Uncoupling proteins: molecular, functional, regulatory, physiological and pathological aspects. Adv Exp Med Biol, 942, 137–156. 10.1007/978-94-007-2869-1_6 [DOI] [PubMed] [Google Scholar]
- Smith RA, & Murphy MP (2010). Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann N Y Acad Sci, 1201, 96–103. 10.1111/j.1749-6632.2010.05627.x [DOI] [PubMed] [Google Scholar]
- Sokolova IM, & Sokolov EP (2005). Evolution of mitochondrial uncoupling proteins: novel invertebrate UCP homologues suggest early evolutionary divergence of the UCP family. FEBS Lett, 579(2), 313–317. 10.1016/jiebslet.2004.11.103 [DOI] [PubMed] [Google Scholar]
- Sorolla MA, Reverter-Branchat G, Tamarit J, Ferrer I, Ros J, & Cabiscol E (2008). Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic Biol Med, 45(5), 667–678. 10.1016/j.freeradbiomed.2008.05.014 [DOI] [PubMed] [Google Scholar]
- St-Pierre J, Buckingham JA, Roebuck SJ, & Brand MD (2002). Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem, 277(47), 44784–44790. 10.1074/jbc.M207217200 [DOI] [PubMed] [Google Scholar]
- Stahl WL, & Baskin DG (1984). Immunocytochemical localization of Na+,K+ adenosine triphosphatase in the rat retina. J Histochem Cytochem, 32(2), 248–250. 10.1177/32.2.6319483 [DOI] [PubMed] [Google Scholar]
- Starkov AA, Andreyev AY, Zhang SF, Starkova NN, Korneeva M, Syromyatnikov M, & Popov VN (2014). Scavenging of H2O2 by mouse brain mitochondria. J Bioenerg Biomembr, 46(6), 471–477. 10.1007/s10863-014-9581-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinle JJ, Krizsan-Agbas D, & Smith PG (2000). Regional regulation of choroidal blood flow by autonomic innervation in the rat. Am J Physiol Regul Integr Comp Physiol, 279(1), R202–209. 10.1152/ajpregu.2000.279.1.R202 [DOI] [PubMed] [Google Scholar]
- Stieger K, Le Meur G, Lasne F, Weber M, Deschamps JY, Nivard D, Mendes-Madeira A, Provost N, Martin L, Moullier P, & Rolling F (2006). Long-term doxycycline-regulated transgene expression in the retina of nonhuman primates following subretinal injection of recombinant AAV vectors. Mol Ther, 13(5), 967–975. 10.1016/j.ymthe.2005.12.001 [DOI] [PubMed] [Google Scholar]
- Strobel B, Düchs MJ, Blazevic D, Rechtsteiner P, Braun C, Baum-Kroker KS, Schmid B, Ciossek T, Gottschling D, Hartig JS, & Kreuz S (2020). A Small-Molecule-Responsive Riboswitch Enables Conditional Induction of Viral Vector-Mediated Gene Expression in Mice. ACS Synth Biol, 9(6), 1292–1305. 10.1021/acssynbio.9b00410 [DOI] [PubMed] [Google Scholar]
- Sun LL, Jiang BG, Li WT, Zou JJ, Shi YQ, & Liu ZM (2011). MicroRNA-15a positively regulates insulin synthesis by inhibiting uncoupling protein-2 expression. Diabetes Res Clin Pract, 91(1), 94–100. 10.1016/j.diabres.2010.11.006 [DOI] [PubMed] [Google Scholar]
- Sun X, Dai Y, Chen Y, Yu DY, Cringle SJ, Chen J, Kong X, Wang X, & Jiang C (2017). Primary angle closure glaucoma: What we know and what we don't know. Prog Retin Eye Res, 57, 26–45. 10.1016/j.preteyeres.2016.12.003 [DOI] [PubMed] [Google Scholar]
- Tahir TA, Singh H, & Brindle NP (2014). The RNA binding protein hnRNP-K mediates post-transcriptional regulation of uncoupling protein-2 by angiopoietin-1. Cell Signal, 26(7), 1379–1384. 10.1016/j.cellsig.2014.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y, Libondi T, & Jonas JB (2010). Long-term follow-up after multiple intravitreal bevacizumab injections for exudative age-related macular degeneration. J Ocul Pharmacol Ther, 26(1), 79–83. 10.1089/jop.2009.0095 [DOI] [PubMed] [Google Scholar]
- Tezel G (2020). A broad perspective on the molecular regulation of retinal ganglion cell degeneration in glaucoma. Prog Brain Res, 256(1), 49–77. 10.1016/bs.pbr.2020.05.027 [DOI] [PubMed] [Google Scholar]
- Tezel G, & Group, F. A. P. O. R. I. C. W. (2009). The role of glia, mitochondria, and the immune system in glaucoma. Invest Ophthalmol Vis Sci, 50(3), 1001–1012. 10.1167/iovs.08-2717 [DOI] [PubMed] [Google Scholar]
- Tezel G, Yang X, & Cai J (2005). Proteomic identification of oxidatively modified retinal proteins in a chronic pressure-induced rat model of glaucoma. Invest Ophthalmol Vis Sci, 46(9), 3177–3187. 10.1167/iovs.05-0208 [DOI] [PubMed] [Google Scholar]
- Tezel G, Yang X, Luo C, Peng Y, Sun SL, & Sun D (2007). Mechanisms of immune system activation in glaucoma: oxidative stress-stimulated antigen presentation by the retina and optic nerve head glia. Invest Ophthalmol Vis Sci, 48(2), 705–714. 10.1167/iovs.06-0810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda C, Kim JD, Impellizzeri D, Cuzzocrea S, Liu ZW, & Diano S (2016). UCP2 Regulates Mitochondrial Fission and Ventromedial Nucleus Control of Glucose Responsiveness. Cell, 164(5), 872–883. 10.1016/j.cell.2016.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombran-Tink J, Barnstable CJ, & Shields MB (Eds.) (2008). Mechanisms of the Glaucomas: Disease Processes and Therapeutic Modalities pp762 Springer/Humana Press. [Google Scholar]
- Tombran-Tink J, Barnstable CJ, & Gardner TW (Eds.) (2012). Visual Dysfunction in Diabetes: The Science of Patient Impairment and Health Care. pp379, Springer/Humana Press. [Google Scholar]
- Totan Y, Yağci R, Bardak Y, Ozyurt H, Kendir F, Yilmaz G, Sahin S, & Sahin Tiğ U (2009). Oxidative macromolecular damage in age-related macular degeneration. Curr Eye Res, 34(12), 1089–1093. 10.3109/02713680903353772 [DOI] [PubMed] [Google Scholar]
- Trayhurn P, & Van Heyningen R (1971). Aerobic metabolism in the bovine lens. Exp Eye Res, 12(3), 315–327. 10.1016/0014-4835(71)90156-4 [DOI] [PubMed] [Google Scholar]
- Varma R, Vajaranant TS, Burkemper B, Wu S, Torres M, Hsu C, Choudhury F, & McKean-Cowdin R (2016). Visual Impairment and Blindness in Adults in the United States: Demographic and Geographic Variations From 2015 to 2050. JAMA Ophthalmol, 134(7), 802–809. 10.1001/jamaophthalmol.2016.1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatamaniuk MZ, Gupta RK, Lantz KA, Doliba NM, Matschinsky FM, & Kaestner KH (2006). Foxa1-deficient mice exhibit impaired insulin secretion due to uncoupled oxidative phosphorylation. Diabetes, 55(10), 2730–2736. 10.2337/db05-0470 [DOI] [PubMed] [Google Scholar]
- Venza I, Visalli M, Cucinotta M, Teti D, & Venza M (2012). Association between oxidative stress and macromolecular damage in elderly patients with age-related macular degeneration. Aging Clin Exp Res, 24(1), 21–27. 10.3275/7659 [DOI] [PubMed] [Google Scholar]
- Vozza A, Parisi G, De Leonardis F, Lasorsa FM, Castegna A, Amorese D, Marmo R, Calcagnile VM, Palmieri L, Ricquier D, Paradies E, Scarcia P, Palmieri F, Bouillaud F, & Fiermonte G (2014). UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc Natl Acad Sci U S A, 111(3), 960–965. 10.1073/pnas.1317400111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JY, Gu YS, Wang J, & Tong Y (2008). Oxidative stress in Chinese patients with Leber's hereditary optic neuropathy. J Int Med Res, 36(3), 544–550. 10.1177/147323000803600320 [DOI] [PubMed] [Google Scholar]
- Wang JY, Zhang HY, Li YM, Yu HN, Wang J, Shen SR, & Tong Y (2005). Study on total cell antioxidant capacity in Leber's hereditary optic neuropathy. Shi Yan Sheng Wu Xue Bao, 38(6), 555–558. [PubMed] [Google Scholar]
- Watson MA, Wong HS, & Brand MD (2019). Use of S1QELs and S3QELs to link mitochondrial sites of superoxide and hydrogen peroxide generation to physiological and pathological outcomes. Biochem Soc Trans, 47(5), 1461–1469. 10.1042/BST20190305 [DOI] [PubMed] [Google Scholar]
- West GB, Woodruff WH, & Brown JH (2002). Allometric scaling of metabolic rate from molecules and mitochondria to cells and mammals. Proc Natl Acad Sci U S A, 99 Suppl 1, 2473–2478. 10.1073/pnas.012579799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittenborn J, & Rein D (2014). The future of vision: Forecasting the prevalence and costs of vision problems. Prevent Blindness. [Google Scholar]
- Wittenborn JS, Zhang X, Feagan CW, Crouse WL, Shrestha S, Kemper AR, Hoerger TJ, Saaddine JB, & Group VC-ES (2013). The economic burden of vision loss and eye disorders among the United States population younger than 40 years. Ophthalmology, 120(9), 1728–1735. 10.1016/j.ophtha.2013.01.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf DM, Segawa M, Kondadi AK, Anand R, Bailey ST, Reichert AS, van der Bliek AM, Shackelford DB, Liesa M, & Shirihai OS (2019). Individual cristae within the same mitochondrion display different membrane potentials and are functionally independent. EMBO J, 38(22), e101056. 10.15252/embj.2018101056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong HS, Benoit B, & Brand MD (2019). Mitochondrial and cytosolic sources of hydrogen peroxide in resting C2C12 myoblasts. Free Radic Biol Med, 130, 140–150. 10.1016/j.freeradbiomed.2018.10.448 [DOI] [PubMed] [Google Scholar]
- Xiong W, MacColl Garfinkel AE, Li Y, Benowitz LI, & Cepko CL (2015). NRF2 promotes neuronal survival in neurodegeneration and acute nerve damage. J Clin Invest, 125(4), 1433–1445. 10.1172/JCI79735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Hondur G, & Tezel G (2016). Antioxidant Treatment Limits Neuroinflammation in Experimental Glaucoma. Invest Ophthalmol Vis Sci, 57(4), 2344–2354. 10.1167/iovs.16-19153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen MY, Kao SH, Wang AG, & Wei YH (2004). Increased 8-hydroxy-2'-deoxyguanosine in leukocyte DNA in Leber's hereditary optic neuropathy. Invest Ophthalmol Vis Sci, 45(6), 1688–1691. 10.1167/iovs.03-0568 [DOI] [PubMed] [Google Scholar]
- Yen MY, Wang AG, & Wei YH (2006). Leber's hereditary optic neuropathy: a multifactorial disease. Prog Retin Eye Res, 25(4), 381–396. 10.1016/j.preteyeres.2006.05.002 [DOI] [PubMed] [Google Scholar]
- Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, & Mizuno Y (1996). Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci U S A, 93(7), 2696–2701. 10.1073/pnas.93.7.2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young P, Qiu L, Wang D, Zhao S, Gross J, & Feng G (2008). Single-neuron labeling with inducible Cre-mediated knockout in transgenic mice. Nat Neurosci, 11(6), 721–728. 10.1038/nn.2118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu AK, Song L, Murray KD, van der List D, Sun C, Shen Y, Xia Z, & Cortopassi GA (2015). Mitochondrial complex I deficiency leads to inflammation and retinal ganglion cell death in the Ndufs4 mouse. Hum Mol Genet, 24(10), 2848–2860. 10.1093/hmg/ddv045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu DY, & Cringle SJ (2001). Oxygen distribution and consumption within the retina in vascularised and avascular retinas and in animal models of retinal disease. Prog Retin Eye Res, 20(2), 175–208. [DOI] [PubMed] [Google Scholar]
- Yu DY, & Cringle SJ (2006). Oxygen distribution in the mouse retina. Invest Ophthalmol Vis Sci, 47(3), 1109–1112. 10.1167/iovs.05-1118 [DOI] [PubMed] [Google Scholar]
- Yu DY, Cringle SJ, Balaratnasingam C, Morgan WH, Yu PK, & Su EN (2013). Retinal ganglion cells: Energetics, compartmentation, axonal transport, cytoskeletons and vulnerability. Prog Retin Eye Res, 36, 217–246. 10.1016/j.preteyeres.2013.07.001 [DOI] [PubMed] [Google Scholar]
- Zhang CY, Baffy G, Perret P, Krauss S, Peroni O, Grujic D, Hagen T, Vidal-Puig AJ, Boss O, Kim YB, Zheng XX, Wheeler MB, Shulman GI, Chan CB, & Lowell BB (2001). Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell, 105(6), 745–755. [DOI] [PubMed] [Google Scholar]
- Zhang M, Chu Y, Mowery J, Konkel B, Galli S, Theos AC, & Golestaneh N (2018). Pgc-1a repression and high-fat diet induce age-related macular degeneration-like phenotypes in mice. Dis Model Mech, 11(9), dmm032698. 10.1242/dmm.032698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Gu H, & Hu N (2015). Role of Peroxisome Proliferator-Activated Receptor γ in Ocular Diseases. J Ophthalmol, 2015, 275435. 10.1155/2015/275435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Miriyala S, Miao L, Mitov M, Schnell D, Dhar SK, Cai J, Klein JB, Sultana R, Butterfield DA, Vore M, Batinic-Haberle I, Bondada S, & St Clair DK (2014). Redox proteomic identification of HNE-bound mitochondrial proteins in cardiac tissues reveals a systemic effect on energy metabolism after doxorubicin treatment. Free Radic Biol Med, 72, 55–65. 10.1016/j.freeradbiomed.2014.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, & Shen Y (2020). Light-Induced Retinal Ganglion Cell Damage and the Relevant Mechanisms. Cell Mol Neurobiol, 40(8), 1243–1252. 10.1007/s10571-020-00819-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Li Y, & Yue BY (1999). Oxidative stress affects cytoskeletal structure and cell-matrix interactions in cells from an ocular tissue: the trabecular meshwork. J Cell Physiol, 180(2), 182–189. [DOI] [PubMed] [Google Scholar]
- Zhou M, Xu A, Tam PK, Lam KS, Huang B, Liang Y, Lee IK, Wu D, & Wang Y (2012). Upregulation of UCP2 by adiponectin: the involvement of mitochondrial superoxide and hnRNP K. PLoS One, 7(2), e32349. 10.1371/journal.pone.0032349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou TC, Yang L, Liu YY, Qin Y, Li YP, Zhang L, Yang K, & Yang Y (2018). Polymorphisms in the Uncoupling Protein 2 Gene Are Associated with Diabetic Retinopathy in Han Chinese Patients with Type 2 Diabetes. Genet Test Mol Biomarkers, 22(11), 637–643. 10.1089/gtmb.2018.0115 [DOI] [PubMed] [Google Scholar]
- Zielonka J, Joseph J, Sikora A, Hardy M, Ouari O, Vasquez-Vivar J, Cheng G, Lopez M, & Kalyanaraman B (2017). Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem Rev, 117(15), 10043–10120. 10.1021/acs.chemrev.7b00042 [DOI] [PMC free article] [PubMed] [Google Scholar]