

Abstract
Extracellular vesicles (EVs) are emerging in tissue engineering as promising acellular tools, circumventing many of the limitations associated with cell‐based therapies. Epigenetic regulation through histone deacetylase (HDAC) inhibition has been shown to increase differentiation capacity. Therefore, this study aimed to investigate the potential of augmenting osteoblast epigenetic functionality using the HDAC inhibitor Trichostatin A (TSA) to enhance the therapeutic efficacy of osteoblast‐derived EVs for bone regeneration. TSA was found to substantially alter osteoblast epigenetic function through reduced HDAC activity and increased histone acetylation. Treatment with TSA also significantly enhanced osteoblast alkaline phosphatase activity (1.35‐fold), collagen production (2.8‐fold) and calcium deposition (1.55‐fold) during osteogenic culture (P ≤ 0.001). EVs derived from TSA‐treated osteoblasts (TSA‐EVs) exhibited reduced particle size (1‐05‐fold) (P > 0.05), concentration (1.4‐fold) (P > 0.05) and protein content (1.16‐fold) (P ≤ 0.001) when compared to untreated EVs. TSA‐EVs significantly enhanced the proliferation (1.13‐fold) and migration (1.3‐fold) of human bone marrow stem cells (hBMSCs) when compared to untreated EVs (P ≤ 0.05). Moreover, TSA‐EVs upregulated hBMSCs osteoblast‐related gene and protein expression (ALP, Col1a, BSP1 and OCN) when compared to cells cultured with untreated EVs. Importantly, TSA‐EVs elicited a time‐dose dependent increase in hBMSCs extracellular matrix mineralisation. MicroRNA profiling revealed a set of differentially expressed microRNAs from TSA‐EVs, which were osteogenic‐related. Target prediction demonstrated these microRNAs were involved in regulating pathways such as ‘endocytosis’ and ‘Wnt signalling pathway’. Moreover, proteomics analysis identified the enrichment of proteins involved in transcriptional regulation within TSA‐EVs. Taken together, our findings suggest that altering osteoblasts’ epigenome accelerates their mineralisation and promotes the osteoinductive potency of secreted EVs partly due to the delivery of pro‐osteogenic microRNAs and transcriptional regulating proteins. As such, for the first time we demonstrate the potential to harness epigenetic regulation as a novel engineering approach to enhance EVs therapeutic efficacy for bone repair.
Keywords: bone, epigenetics, extracellular vesicles, histone deacetylase, microRNAs, tissue engineering, trichostatin A
Epigenetic regulation promotes the osteogenic potency of osteoblast‐derived EVs for bone repair. The HDAC inhibitor Trichostatin A (TSA) altered osteoblast epigenetic functionality through hyperacetylation, enhancing its mineralisation capacity. EVs derived from TSA treated osteoblasts (TSA‐EVs) were enriched with pro‐osteogenic microRNAs and transcriptional regulating proteins. TSA‐EVs significantly promoted hBMSCs osteogenic differentiation and mineralisation. Epigenetic reprogramming provides a novel engineering approach to enhance EVs therapeutic efficacy as an acellular tool for bone regeneration.
1. INTRODUCTION
There is a tremendous need for bone tissue due to numerous clinical situations (Baroli, 2009; Dimitriou et al., 2011), with currently 10 million people in the UK affected by musculoskeletal disorders costing the National Health Service £4.76 billion annually (Chance‐Larsen et al., 2019). Alarmingly this is anticipated to increase further in the future as a result of the growing ageing population and demand for continued quality of life in the older years. Autografts are the current gold standard treatment for the repair of critical‐sized bone defects, however, they are associated with several concerns such as their limited availability and donor site morbidity (Calori et al., 2011; Djouad et al., 2012). Consequently, there is a critical calling for new approaches to regenerate damaged bone. Hence, extensive research has been conducted within the tissue engineering field to meet the rising demand for clinically‐relevant bone tissue. Although promising cell‐based therapies have been reported (Amini et al., 2012), there has been limited clinical success due to issues associated with intensive cost, scalable manufacture of cells and ethical considerations (Izadpanah et al., 2006). Thus, there is a growing demand to develop acellular approaches for bone repair (Burdick et al., 2013).
Cells are known to secrete a range of bioactive products into the surrounding microenvironment, which have trophic effects on neighbouring cells stimulating numerous biological processes (Driscoll & Patel, 2019; Sun et al., 2019). One of these cell‐secreted factors, extracellular vesicles (EVs), have been acquiring growing interest in recent years as an acellular tool for regenerative medicine (El Andaloussi et al., 2013). EVs are cell‐derived nanoparticles, which contain a diverse biological cargo including proteins, nucleic acids and bioactive molecules, and are heavily involved in intercellular communication, regulating tissue development and homeostasis (Man et al., 2020; Raposo & Stoorvogel, 2013). The beneficial effects once attributed to cells, are now thought to be partially due to the paracrine factors delivered by EVs (Gnecchi et al., 2005; Xin et al., 2014). Hence, there has been intensive investigations into the role these nanoparticles may play as novel acellular tools for bone regeneration (Chen et al., 2019; Qin et al., 2016), overcoming the tremendous regulatory hurdles associated with the clinical translation of cell‐based therapies (Heathman et al., 2015).
In recent years, several studies have reported the considerable utility of EVs to stimulate osteogenesis (Eichholz et al., 2020; Tan et al., 2020). For example, Davies et al. demonstrated the osteoinductive capacity of EVs derived from osteoblasts, eliciting enhanced stem cell mineralisation compared to that of the current gold standard growth factor, bone morphogenic protein 2 (BMP2) (Davies et al., 2017). These osteoblast‐derived EVs may act as extracellular sites of mineral nucleation, due to their enrichment with the calcium‐channelling protein Annexin. Moreover, the role of EVs in facilitating microRNA transfer to recipient cells and promoting their osteogenic differentiation has also been demonstrated (Cui et al., 2016; Tang et al., 2017). Hence, EVs play a critical and multifunctional role in regulating osteogenesis, which is dependent on their biological cargo. Although the potential utility of these nanoparticles have been reported, numerous studies have investigated EV engineering approaches to further promote the therapeutic efficacy of these vesicles beyond their native function (Kim et al., 2020; Man et al., 2020). Of the numerous EV engineering strategies explored in the literature (Man et al., 2020), genetic modification of the EV parental cell has been extensively studied with promising results observed (Kang et al., 2015; Tao et al., 2017). However, there are issues associated with the use of this technology including high associated costs, ineffectual EV loading and increased risk of tumourigenesis (Hanna et al., 2016; Kooijmans et al., 2016). Consequently, there is a tremendous need for alternative methods in augmenting the parental cell phenotype to enhance the therapeutic viability of EVs for bone regeneration.
Altering the cells’ epigenetics via post‐translational modifications has gained increasing attention in regenerative medicine (Dompe et al., 2020; Wijnen & Westendorf, 2019). Epigenetic regulation involves controlling the transcriptional activity of the genome, without altering the underlying nucleotide sequence (Collas et al., 2008; Huynh et al., 2017), therefore providing an alternative safer method of improving EVs therapeutic efficacy through parental cell modification. Histone proteins play a critical role in regulating the structure of the chromatin. Modifying the acetylation state of the histone via augmenting the activity of histone deacetylase (HDAC) and histone acetyltransferase (HAT) have been shown to modify cell transcriptional activity (Lawlor & Yang, 2019). Hyperacetylation of the chromatin induced by the inhibition of HDAC enzymes has been demonstrated to promote osteogenic differentiation, through enhanced osteoblast‐related gene activation (Jonason et al., 2009; Man et al., 2021; Paino et al., 2014). Additionally, hyperacetylation of non‐histone proteins have also been reported to stimulate osteogenesis through transcription factor activation. Jeon et al. reported that BMP2 increased HAT acetylation of the osteogenic transcription factor Runx2 in HEK293 and C2C12 cells, inhibiting Smad specific E3 ubiquitin‐protein ligase 1 (Smurf1)‐mediated degradation of Runx2, ultimately enhancing its stability and transcriptional activity (Jeon et al., 2006). Moreover, several studies have reported the role of HDAC enzymes in binding to Runx2, T‐cell factor, nuclear factor of activated T cells and zinc finger protein 521, which results in modulation of osteogenic differentiation by silencing the expression of key osteogenic‐related genes (Choo et al., 2009; Jensen et al., 2010). HDAC inhibitors (HDACis) are small molecular compounds that have been extensive explored to reprogram the epigenome for cancer therapeutics and regenerative medicine applications (Bolden et al., 2013; Eckschlager et al., 2017; Marks, 2010). Trichostatin A (TSA) is a naturally‐derived hydroxamic acid‐based HDACi that targets class I and II HDAC isoforms (Ma et al., 2015). Several studies have demonstrated TSAs’ efficacy in stimulating both stem/progenitor cells osteogenic differentiation through hyperacetylation induced chromatin remodelling and osteogenic transcription factor activation (Jin et al., 2013; Schroeder & Westendorf, 2005; Schroeder et al., 2004).
Therefore, in this present study, we investigated augmenting the epigenetic functionality of mineralising osteoblasts using the HDACi TSA to promote the therapeutic potency of osteoblast‐derived EVs for bone regeneration. We determined that 5 nM TSA effectively altered osteoblast epigenetic function and promoted its mineralising capacity. EVs isolated from TSA‐treated (TSA‐EVs) and untreated mineralising osteoblasts (MO‐EVs) were administered to human bone marrow‐derived mesenchymal stem cells (hBMSCs) to determine their efficacy in promoting osteogenic differentiation. Additionally, microRNA profiling and proteomics analysis of EVs was performed to elucidate the mechanisms in which TSA‐EVs impart their pro‐osteogenic effects (Figure 1).
FIGURE 1.
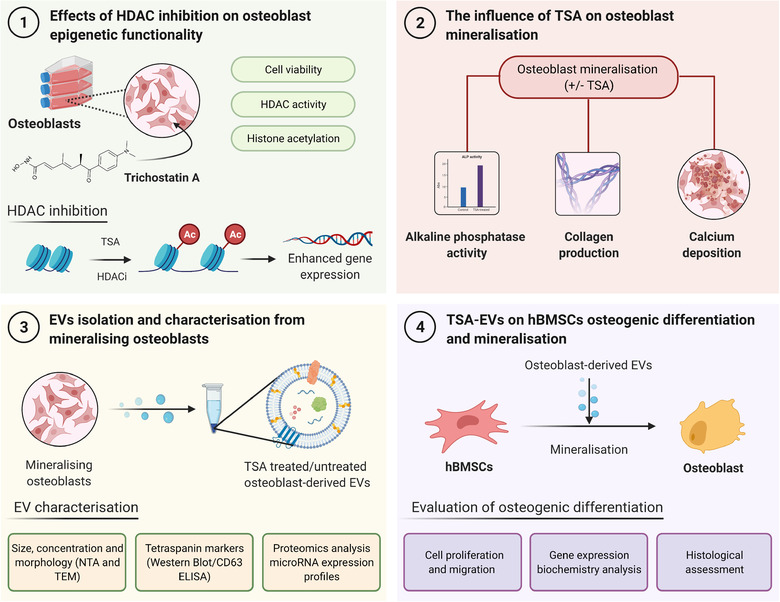
Experimental outline investigating the effects of altering osteoblast epigenetic functionality on the therapeutic potency of their EVs for bone regeneration. 1) The influence of TSA on osteoblast epigenetic functionality was assessed. 2) The effects of TSA on osteoblast mineralisation was evaluated by quantifying ALP activity, collagen production and calcium deposition. 3) EVs were isolated from TSA‐treated and untreated mineralising osteoblast over a 2‐week period, and the nanoparticles were characterised by their size, morphology, protein and microRNA expression. 4) Investigating the effects of TSA‐EV treatment on hBMSCs osteogenic differentiation. Figure created with BioRender.com
2. MATERIALS AND METHODS
2.1. Cell culture and reagents
MC3T3 murine pre‐osteoblasts were purchased from American Type Culture Collection (ATCC, UK) and hBMSCs were acquired from Lonza (Lonza, UK). Basal culture media consisted of minimal essential medium (α‐MEM; Sigma‐Aldrich, UK) supplemented with 10% foetal bovine serum (FBS), 1% penicillin/streptomycin (Sigma‐Aldrich, UK) and L‐glutamine (Sigma‐Aldrich, UK). hBMSCs were used at passage 4. Mineralisation medium comprised of basal culture media supplemented with 10 mM β‐glycerophosphate (Sigma‐Aldrich, UK) and 50 µg/ml L‐ascorbic acid (Sigma‐Aldrich, UK). The synthetic glucocorticoid dexamethasone was excluded due to side effects observed in vivo (Li et al., 2005; Woolf, 2007; Xu et al., 2019) and to standardise EV isolation/dosing protocols as previously reported (Davies et al., 2017). Culture medium utilised for EV isolation and dosing was depleted of FBS‐derived EVs by ultracentrifugation at 120,000 g for 70 min prior to use.
2.2. Cell viability and morphology assessment
Osteoblasts were seeded at 3 × 103 cells/cm2 within a 96‐well plate with basal medium and incubated for 24 h. Media was replaced with fresh basal medium supplemented with/without TSA (Sigma‐Aldrich, UK) (5, 10, 20, 50, 100 nM) and incubated for 1, 3 and 7 days. At each time point, AlamarBlue reagent (Thermo Scientific, UK) was added and incubated for 4 h at 37°C. Fluorescence readings was acquired using a SPARK spectrophotometer (TECAN, CH) at an excitation/emission wavelength of 540/590 nm, respectively. Employing the same protocol, the osteoblast morphology via calcein‐AM staining (Sigma‐Aldrich, UK) after 3 days of culture, observed under an EVOS fluorescent inverted microscope (Thermo Scientific, UK).
2.3. HDAC activity and H3K9 histone acetylation
Cells were cultured in 96‐well plates (3 × 103 cells/cm2) in basal medium for 24 h. Medium was replaced with fresh basal medium supplemented with/without TSA (5, 10, 20, 50, 100 nM). At day 3 and 7, the HDAC activity was quantified using an in situ HDAC activity fluorometric assay kit (BioVision, UK) according to the manufacturer's instructions. Briefly, media was replaced with 100 µl of reaction mix and incubated for 3 h at 37°C. 100 µl of lysine developer was added then further incubated for 30 min at 37°C. Fluorescence was measured in a SPARK spectrophotometer at an excitation/emission wavelength of 368/442 nm. HDAC activity was normalised with DNA content.
Detection of H3K9 acetylation was performed using the EpiQuik™ In Situ Histone H3‐K9 Acetylation Assay Kit (Epigentek, USA) according to the manufacturer's protocol. The absorbance was read in a SPARK spectrophotometer at 450 nm. Histone acetylation was normalised with DNA content.
DNA quantification was determined by Quant‐iT PicoGreen DNA assay (Invitrogen, Life Technologies, UK). Briefly, cells were lysed following three freeze‐thaw cycles in 0.1% Triton™ X‐100 in Phosphate buffered saline (PBS, Lonza, UK). 90 µl of TE (10 mM Tris‐HCl, 1 mM EDTA) buffer was added to 10 µl of cell lysate in a 96‐well plate (Corning, UK). 100 µl of PicoGreen reagent was added to all samples and then incubated at 37°C for 5 min. The fluorescence was then measured in a SPARK spectrophotometer at an excitation/emission wavelength of 480/520 nm.
2.4. Osteoblast mineralisation with TSA
Osteoblasts were cultured in 24‐well plates at a density of 21 × 103 cells/cm2 in basal medium for 24 h. The media was replaced with mineralisation medium supplemented with TSA for 21 days. Medium changes and TSA replenishment was performed every 48 h. Cells cultured in mineralising medium alone were used as control.
2.5. EVs isolation and characterisation
2.5.1. EV isolation
Osteoblasts were cultured at scale in T175 culture flasks (Sarstedt, UK) and medium isolated every two days. Cells were cultured in osteogenic medium supplemented with/without 5 nM TSA for 14 days. EVs were isolated from conditioned medium (400 ml) by differential centrifugation: 2000 g for 20 min, 10,000 g for 30 min and 120,000 g for 70 min to pellet EVs (Davies et al., 2017). The supernatant was removed, and the pellet was washed in sterile PBS and centrifuged at 120,000 g for 70 min and the resultant pellet was re‐suspended in 200 µl PBS. All ultracentrifugation steps were performed utilising the Sorvall WX Ultra Series Ultracentrifuge (Thermo Scientific, UK) and a Fiberlite, F50L‐8×39 fixed angle rotor (Piramoon Technologies Inc., USA). EV characterisation was conducted following guidelines published in the Minimal Information for Studies of Extracellular Vesicles 2018 (Théry et al., 2018).
2.5.2. Particle size and concentration analysis
Total EV protein concentration was determined using the Pierce BCA protein assay kit (Thermo Scientific, UK). Dynamic Light Scattering (Zetasizer Nano ZS, Malvern Instruments, UK) was used to analyse polydispersity index (PDI). Nanoparticle tracking analysis was performed on EV samples to determine particle size and concentration using a ZetaView® instrument (Particle Metrix, Germany). EV samples were diluted 1:100 in PBS and injected into the ZetaView®, where 4 × 40 s videos were obtained of particles in motion. Particle size and concentration was determined with the ZetaView® software. The quantity of CD63 positive particles was confirmed using the ExoELISA‐ULTRA Complete Kit (CD63 Detection) (System Biosciences, USA) following the manufacturers’ protocol.
2.5.3. Transmission electron microscopy (TEM)
EV imaging was conducted via a JEOL JEM1400 transmission electron microscope (TEM) coupled with an AMT XR80 digital acquisition system. Samples were physisorbed to 200 mesh carbon‐coated copper formvar grids (Agar Scientific, UK) and negatively stained with 1% uranyl acetate.
2.5.4. Immunoblotting
Immunoblotting analysis was used to confirm the presence of EV as previously described (Nikravesh et al., 2019). Briefly, following the electrophoretic separation of proteins using precast gels (4%‐15% Mini‐PROTEAN TBX, Biorad, UK), gels were blotted on polyvinylindene disluoride membranes (Fisher Scientific, UK) and blocked with EveryBlot blocking buffer (BioRad, UK). The blots were incubated overnight at 4°C with primary antibodies to Alix (1:1000 dilution, Santa Cruz, USA), Annexin 2 (1:2000 dilution, Abcam, UK), CD9 (1:1000 dilution, Abcam, UK) and calnexin (1:1000 dilution, Abcam, UK). The membranes were incubated with the appropriate secondary antibody, anti‐rabbit for Annexin 2, CD9 and calnexin (1:3000 dilution, Cell Signaling, UK), and anti‐mouse for Alix (1:3000 dilution, Cell Signaling, UK), for 1 h at room temperature. Chemiluminescence detection of bands were images with ChemiDoc XRS+ system (BioRad, UK) by a chemiluminescence reaction using Clarity™ Western ECL substrate (BioRad, UK) and Image Lab software (Life Science Research, BioRad, UK) following supplier's instructions.
2.6. EVs microRNA isolation, analysis and bioinformatics
Total EV RNA was isolated using the Qiagen miRNeasy Mini Kit (Qiagen, UK) according to the manufacturers’ protocol and RNA quantity/purity assessed by measuring 260/280 nm absorbance ratio using a NanoQuant plate and a SPARK spectrophotometer. RNA content was normalised with particle number. Global expression patterns of EV miRNAs were examined by using a microarray chip containing 1963 probes for murine microRNAs (miR‐Base 22). Microarray analysis of EV microRNAs (500 ng of total RNA per EV sample) was performed by LC Sciences (Houston, USA) using the μParaflo™ microRNA microarray biochip technology. Differentially expressed microRNAs were defined by a threshold of P < 0.05 and fold change > 2.0. Significantly altered microRNAs were further analysed to predict their target genes and pathways. Target genes of miRNAs were predicted using two algorithms, DIANA‐Tarbase v 7.0 and DIANA‐microT‐CDS. A microT threshold of 0.8 when microT‐CDS were used for target gene prediction. DIANA‐mirPath v.3 was utilised to perform hierarchical clustering of miRNAs, to assess gene ontology (GO) annotation and Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathways based on interaction level using experimentally validated miRNA interactions derived from DIANA‐Tarbase v7.0 and/or predicted miRNA gene targets provided by DIANA‐microT‐CDS (Vlachos et al., 2012). Benjamini and Hochberg's false discovery rate (FDR) was applied with the significant threshold set at a p‐value of < 0.05.
2.7. Sample preparation for proteomics analysis
Protein extraction for proteomic analysis was performed by adding 400 µl acetone (Thermo Scientific, UK) to 100 µl of EVs previously isolated and resuspended in PBS. Samples were vortexed and incubated at ‐80°C for 1 h. After incubation, the samples were centrifuged at 14,000 g for 10 min. The supernatant was discarded, the pellet dried by inverting and then resuspended in 0.1 M ammonium bicarbonate (Acros Organics, USA), 0.1% RapiGest (Waters Corpo., USA) in LC‐MS grade water (Thermo Scientific, UK) to a final concentration of 1 µg/µl.
Proteins were denatured with 1.5 µl of 1% (w/v) RapiGest in 50 mM ammonium bicarbonate and incubated at 80°C for 45 min. Following incubation, 100 mM DTT (1 µl) was added and incubated for a further 30 min at 60°C to reduce the proteins, before being alkylated with 200 mM iodoacetamide (1 µl) at room temperature for 30 min. Trypsin 1:50 (w/w) (Gold Mass Spectrometry grade, Promega, USA) was added to each sample for proteolytic digestion and left to incubate overnight at 37°C. Trifluoroacetic acid was added to a final concentration of 0.5% (v/v) to hydrolyse the RapiGest and heated for a further 45 min at 37°C, before centrifuging for 25 min at 18,000 g. The supernatant was collected and 5 µl aliquoted for LC‐MS analysis. Aliquoted samples were diluted 1:4 (v/v) with 15 µl of 0.1% formic acid (v/v) to provide a working solution of 200 ng/µl.
2.8. LC‐MS analysis
Extracted peptides obtained from the isolated vesicles were analysed by one dimensional nanoscale reversed‐phase (RP) chromatography using an ACQUITY M‐Class UPLC (Waters Corp., USA) configured for trap and elute. Peptides were loaded (1 µl injection, 200 ng on‐column) onto a Symmetry C18 5 µm, 2 cm × 180 µm pre‐column (Waters Corp., USA) with aqueous 0.1% (v/v) formic acid using a flow rate of 15 µl/min for 2 min. Mobile phases consisted of water with 0.1% (v/v) formic acid (mobile phase A) and acetonitrile with 0.1% (v/v) formic acid (mobile phase B). Peptides were eluted from the pre‐column and separated over a 90 min gradient using a HSS T3 C18 1.7 µm, 15 cm × 75 µm analytical column (Waters Corp., USA). The gradient consisted of 3 ‐ 40% mobile phase B over 60 min at a flow rate of 400 nl/min, whilst maintaining the analytical column temperature at 35°C. Lock mass consisting of [Glu1]‐Fibrinopeptide was delivered to the reference sprayer of the MS source using the M‐Class Auxillary Solvent Manager with a flow rate of 1 µl/min.
MS data were collected on a Synapt XS mass spectrometer (Waters Corp., UK) operated in positive electrospray ionisation (ESI) mode with a nominal resolution of 25,000 FWHM (V optics). The capillary voltage was 3.2 kV, cone voltage was 35 V and source temperature was set at 100°C. Data were acquired over 50 ‐ 2000 Da mass range with a scan time of 0.5 s. All mass spectral data were acquired in continuum mode using UDMSE to obtain fragmentation data simultaneously (Distler et al., 2016; Rodriguez‐Suarez et al., 2013). Function one (low energy) data were collected using a constant trap and transfer energy of 6 eV whilst the second (high energy) function consisted of a transfer collision energy ramp of 19 to 45 eV. For mass accuracy, [Glu1]‐fibrinopeptide (m/z = 785.8426) was acquired as lock mass at a concentration of 100 fmol/µl (in 50:50 CH3CN/H2O, 0.1% formic acid). Lock mass scans were collected every 60 s and averaged over 3 scans to perform mass correction. The time‐of‐flight mass analyser was externally calibrated over the acquisition mass range (50 ‐ 2000 Da) before analysis with a NaCsI mixture (Waters API MS Calibration Solution, 2 µg/µl sodium iodide: 50 ng/µl cesium iodide in 50:50 isopropanol:water, Waters Corp., USA). These data were collected using MassLynx v 4.1 software (Waters Corp., UK) in a randomized order with three technical replicates acquired per sample.
2.9. LC‐MS data analysis
Progenesis QI for Proteomics (Nonlinear Dynamics, UK) was used to process all data. Retention time alignment, peak picking and normalization were conducted to produce peak intensities for retention time (RT) and m/z data pairs. Data were searched against reviewed entries of a Mus musculus UniProt database (17,048 reviewed entries, release 2020_05) to provide protein identifications with a FDR of 1%. A decoy database was generated as previously described (Li et al., 2009) allowing for protein/peptide identification rates to be determined. Peptide and fragment ion tolerances were determined automatically, and searches allowed for one missed cleavage site. Carbamidomethyl of cysteines was applied as a fixed modification, whilst oxidation of methionine and deamidation of asparagine/glutamine were set as variable modifications.
2.10. EV treatment of hBMSCs
2.10.1. EV labelling
EVs were labelled using Cell Mask™ Deep Red Plasma Membrane Stain (1:1000 in PBS) (Thermo Scientific, UK) and incubated for 10 min. Labelled EVs were washed twice with PBS via ultracentrifugation at 120,000 g for 70 min. Cells were seeded at a density of 4 × 103 cells/cm2 in a chamber slide (Corning, UK) for 24 h. Media was replaced with fresh basal media supplemented with labelled EVs. After 2, 6 and 24 h, cells were fixed with 10% (v/v) neutral buffered formalin (NBF, Cellpath, UK), stained with Alexa Fluor 488 phalloidin (1:20) (Cell Signalling Technology, UK) and then mounted with Prolong ™ Gold Antifade Mountant with DAPI (Thermo Scientific, UK) to label the actin cytoskeleton and nuclei respectively. Treated cells were imaged with the aforementioned EVOS fluorescent inverted microscope.
2.10.2. hBMSCs proliferation and migration
The influence of EVs on hBMSCs proliferation was assessed via quantification of DNA content. Briefly, cells were seeded at 1 × 104 cells/cm2 in basal media for 24 h. Then, media was replaced with fresh basal medium supplemented with TSA‐EVs or MO‐EVs (10 µg/ml). Cells cultured in basal medium alone were used as a control. DNA content was evaluated on day 3, 7 and 14 utilising the previously described DNA quantification assay. Migration rate was measured by performing scratch assays. Briefly, cells at a density of 30 × 103 cells/cm2 in a 6‐well plate were seeded and allowed to adhere for 24 h. A scratch was applied with a 200 µl pipette tip and the width was measured as the baseline. Cell were incubated with basal medium with/without EVs (10 µg/ml) for 3 days. Cells cultured in basal medium alone was used as the control. The rate of migration was assessed under a light microscope (EVOS XL Core, Invitrogen, UK).
2.10.3. hBMSC osteogenic culture
hBMSCs were seeded in 24‐well plates (Nunc, UK) at a density of 21 × 103 cells/cm2 in basal medium and incubated for 24 h. The media was replaced with mineralisation medium supplemented with TSA‐EVs or MO‐EVs (10 µg/ml) for 28 days unless stated otherwise. EV‐supplemented mineralisation medium changes were performed every 48 h. Cells cultured in mineralising medium alone was used as the untreated control.
2.11. Quantitative RT‐qPCR analysis
RNase mini kit (Qiagen, UK) was used to extract total RNA from EV treated and untreated hBMSCs according to the manufacturer's protocol. Commercially available primers (Supplementary Table 1) (Primerdesign, UK) were used to quantify levels of alkaline phosphatase (ALP), collagen type I (COL1A), bone sialoprotein (BSP) and osteocalcin (OCN). Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) was used as the internal reference for mRNA. Isolated RNA was amplified in a 20 µl reaction with a 96‐well PCR plate (Starlab, UK). Amplification occurred using the AriaMx Real‐Time PCR System (Agilent Technologies, UK). For each sample, the cycle threshold (Ct) value was acquired and the comparative Ct method (2–∆∆Ct) was utilised to quantify the gene expression levels relative to the housekeeping gene.
2.12. Alkaline phosphatase activity
ALP activity was determined using the 4‐nitrophenyl colourimetric phosphate liquid assay (pNPP, Sigma‐Aldrich, UK). 10 µl of cell lysate was added to 90 µl of pNPP and incubated for 60 min at 37°C. The absorbance at 405 nm was read on a SPARK spectrophotometer. ALP activity was normalised with DNA content.
2.13. In‐Cell Western assay
Intracellular protein expression was assessed with the In‐Cell Western assay (ICW) as previously reported (Man et al., 2021). Briefly, cells were fixed in 10% NBF, permeabilised by 0.1% Triton™ X‐100 in PBS and non‐specific binding was blocked using the Odyssey blocking buffer (Li‐Cor Biosciences, UK). Samples were incubated overnight at 4°C with primary antibodies to ALP (1:300), Col1a (1:200), and OCN (1:400) (Abcam, UK) in Odyssey® buffer. Cells were then incubated with IRDye 800CW secondary antibody (1:800) and CellTag™ 700 stain (1:500; Li‐Cor Biosciences, UK) in Odyssey blocking buffer for 1 h. Prior to scanning, samples were then washed in 0.1% Tween20 in PBS and analysed on an Odyssey SA Imaging System (Li‐Cor Biosciences, UK) at 700 and 800 nm. Quantitative analysis was performed using the Image Studio (Li‐Cor Biosciences: version 5).
2.14. Collagen production
Extracellular matrix collagen deposition was evaluated with picrosirius red staining. Briefly, cells were washed twice in PBS and fixed in 10% NBF for 30 min, prior to staining with 0.1% sirius red in saturated picric acid (Sigma‐Aldrich, UK) for 1 h. The unbound dye was removed by washing in 0.5 M acetic acid followed by distilled water wash and left to air dry prior to imaging using light microscopy (EVOS XL Core, Invitrogen, UK). To quantify collagen staining, 0.5 M sodium hydroxide was used to elute the bound dye and absorbance were read at 590 nm using the SPARK spectrophotometer.
2.15. Mineral deposition
To evaluate mineralisation, calcium deposition was assessed via alizarin red staining. Cells were washed twice in PBS and fixed in 10% NBF for 30 min. Following fixation, cells were washed in distilled water and then incubated with alizarin red solution (Sigma‐Aldrich, UK) for 10 min. The unbound dye was removed by washing is distilled water. Staining was visualised using light microscopy (EVOS XL Core, Invitrogen, UK). For alizarin red quantification, samples were de‐stained with 10% cetylpyridinium chloride (Sigma‐Aldrich, UK) for 1 h and then absorbance were read at 550 nm using the SPARK spectrophotometer.
2.16. Statistical analysis
For all data presented, experiments were performed in triplicate. All statistical analysis was assessed using the IBM SPSS software (IBM Analytics, version 21). The Shapiro‐Wilk test was used to analyse the normality of data. Data that was proven to be normally distributed were analysed using parametric students' T‐test, one‐way ANOVA, or paired T‐test. Non‐normally distributed data were assessed using non‐parametric Mann‐Whitney t‐test or Kruskal‐Wallis ANOVA. P values equal to or lower than 0.05 was considered as significant. *P ≤ 0.05, **P ≤ 0.01 ***P ≤ 0.001.
3. RESULTS
3.1. The effects of TSA on osteoblast epigenetic functionality
To determine the effects of TSA on osteoblast viability, cellular morphology and metabolic activity was assessed. Alterations in osteoblast morphology was observed upon increasing TSA dosages, where cells transitioned from an elongated, fibroblast‐like shape to a larger, flattened morphology (Figure 2a). A time‐dose dependent decrease in osteoblast viability was observed following TSA treatment, with concentrations of ≥ 50 nM for 3 and 7 days significantly reducing osteoblast metabolic activity compared to that in the untreated cells (P ≤ 0.001) (Figure 2b). This reduction in metabolic activity was observed alongside substantial variations in HDAC activity and histone acetylation levels. A time‐dose dependant decrease in HDAC activity was observed upon TSA treatment, where concentrations of ≥ 20 nM (P ≤ 0.05) and ≥ 5 nM (P ≤ 0.001) significantly reduced HDAC activity compared to that in the untreated cells at day 3 and 7 respectively (Figure 2c). Additionally, TSA elicited a time‐dose dependent increase in histone H3K9 acetylation, with concentrations of ≥ 5 nM for both day 3 and 7 significantly enhancing histone acetylation levels compared to that in the untreated cells (P ≤ 0.05 ‐ 0.01) (Figure 2d).
FIGURE 2.
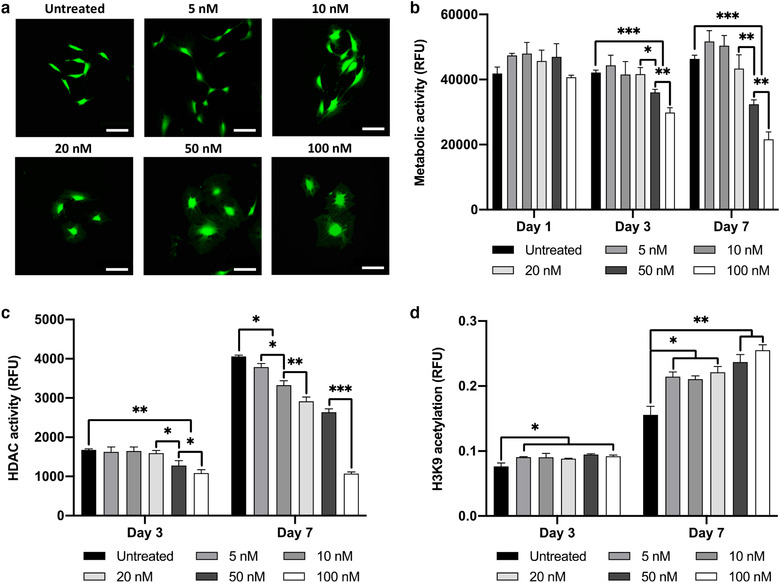
The effect of TSA on osteoblasts viability and epigenetic functionality. TSA caused a time‐dose dependant effect on osteoblast a) morphology (day 3) and b) metabolic activity. Treatment with TSA altered osteoblast c) HDAC activity and d) H3K9 histone acetylation in a time‐dose dependent manner. Data are expressed as mean ± SD (n = 3). *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001. Scale bar = 100 µm
3.2. TSA promotes osteoblast mineralisation
The influence of TSA treatment on osteoblast differentiation was evaluated by quantifying ALP activity, collagen production and calcium deposition (Figure 3a). A time‐dose dependant effect on osteoblast ALP activity was observed following TSA treatment. 5 nM TSA substantially increased ALP activity when compared to cells treated with higher TSA concentrations (≥ 10 nM) (1.1, 1.23‐fold) (P ≤ 0.01 ‐ 0.001) and the untreated cells (1.1, 1.35‐fold) (P ≤ 0.05 ‐ 0.001) at day 7 and 14 (Figure 3b). Variations in ALP activity were followed by significant changes in extracellular matrix collagen production (Figure 3c). TSA at 5 nM significantly enhanced collagen deposition when compared to the other TSA‐treated groups (≥ 10 nM) (≥ 1.1, 1.7‐fold) and the untreated control (1.4, 2.8‐fold) at both day 14 and 21, respectively. TSA treatment induced a time‐dose dependent effect on osteoblast extracellular matrix mineralisation. At day 14 and 21, 5 and 10 nM TSA significantly increased calcium deposition compared to that of the untreated cells and the ≥ 20 nM TSA treated groups (P ≤ 0.001) (Figure 3d, e). 5 nM TSA treatment was utilised for subsequent experiments.
FIGURE 3.
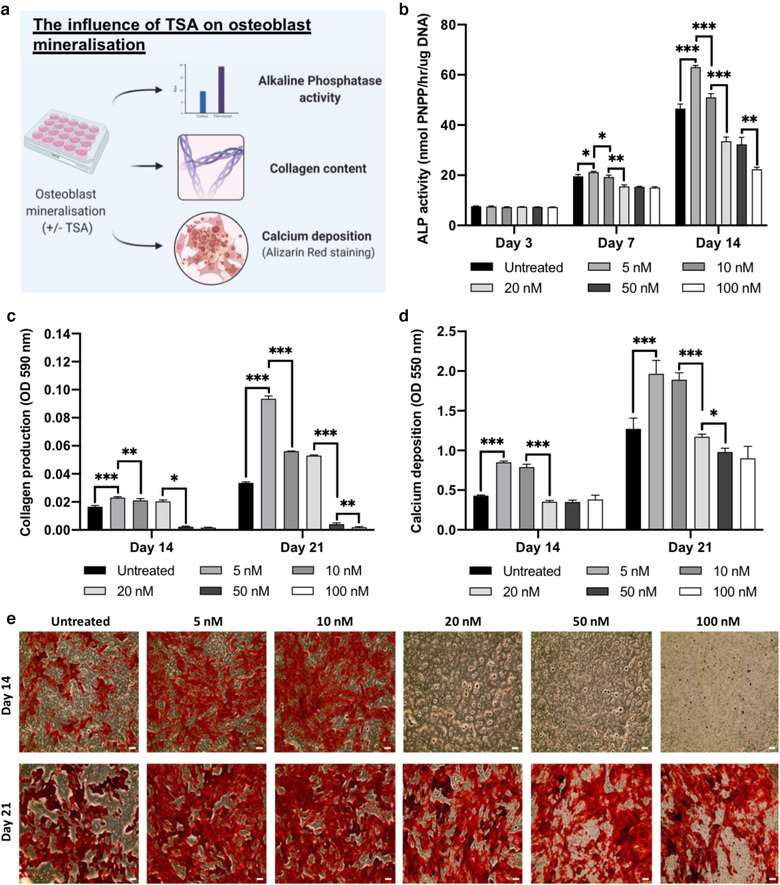
TSA treatment promoted osteoblast mineralisation. a) Schematic representation regarding the assessment of TSA on osteoblast mineralisation. Treatment with TSA elicited a time‐dose dependent effect on osteoblast b) ALP activity, c) extracellular matrix collagen production and d, e) calcium deposition. Data are expressed as mean ± SD (n = 3). *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001. Scale bar = 200 µm
3.3. Characterisation of EVs derived from TSA treated osteoblasts
EVs were isolated from untreated/TSA‐treated osteoblast conditional media over a 2‐week period via differential centrifugation. TEM imaging demonstrated the presence of particles in both groups of a typical size and spherical morphology of EVs, where these nanoparticles displayed heterogeneity in their diameters (Figure 4a and Supplementary Figure 1a). Immunoblotting confirmed the presence of Alix, CD9 and Annexin A2 proteins in both EV groups and the absence of Calnexin expression (Figure 4b). NTA analysis demonstrated that the EVs exhibited an average diameter of 136 and 129 nm for the MO‐EVs and TSA‐EVs respectively (Figure 4c, d) (P > 0.05). There was a 1.4‐fold reduction in the concentration of TSA‐EV particles (4.5 ± 0.82 × 109/ml) compared to MO‐EVs (6.4 ± 0.92 × 109/ml), although not significant (Figure 4d) (P > 0.05). TSA‐EVs exhibited a more monodisperse population (1.34‐fold) when compared to the MO‐EVs (PDI of 0.34 and 0.45, respectively) (Figure 4e) (P ≤ 0.05). Additionally, the TSA‐EVs contained substantially enhanced RNA quantity (3‐fold) when compared to that within the MO‐EVs (Figure 4f) (P ≤ 0.001). A significantly reduced protein content from TSA‐EVs was observed when compared to MO‐EVs (1.16‐fold) (Figure 4g) (P ≤ 0.001). Moreover, there was a slight non‐significant decrease (1.05‐fold) in the quantity of CD63 positive particles in the TSA‐EV group compared to MO‐EVs (Figure 4h) (P > 0.05).
FIGURE 4.
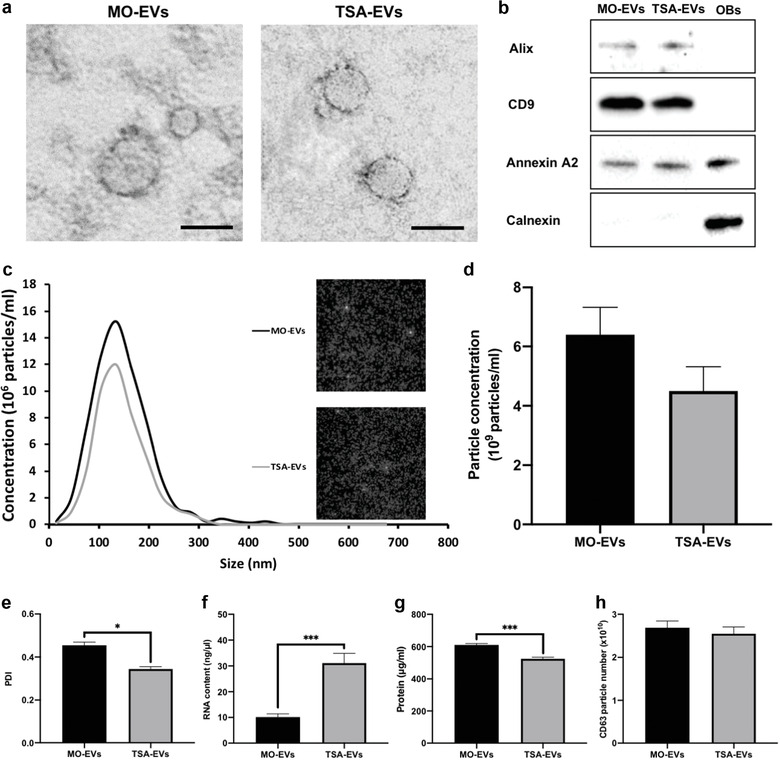
Characterisation of EVs isolated from TSA treated and untreated mineralising osteoblasts. a) TEM image of EVs isolated from TSA‐treated and untreated osteoblasts. Scale bar = 50 nm. b) Western blot analysis confirming the presence of EV markers (Alix, CD9, and Annexin A2) and absence of Calnexin (OBs ‐ osteoblast cell lysate control). c) Particle size distribution of isolated EV samples from NTA. Insert shows snapshot of particles during analysis. d) EV particle size and concentration. e) Polydispersity index of EVs. f) EVs RNA quantification. g) EVs protein content. h) CD63+ particles number. Data are expressed as mean ± SD (n = 3). *P ≤ 0.05 and ***P ≤ 0.001
3.4. TSA‐EVs promoted the proliferation, migration and osteogenic differentiation of hBMSCs
The influence of TSA‐EVs on hBMSCs general behaviour was initially assessed. Cell Mask labelled osteoblast‐derived EVs were successfully internalised by hBMSCs, with labelled EVs situated within the cells’ cytoplasm (Figure 5a). A time‐dependant accumulation of labelled EVs in hBMSCs was observed. Control samples are shown in Supplementary Figure 1b. Treatment with TSA‐EVs significantly increased hBMSCs proliferation in a time‐dependant manner when compared to that of the MO‐EVs treated (P ≤ 0.05) and untreated cells (Figure 5b) (P ≤ 0.01 ‐ 0.001). Additionally, hBMSCs migration was substantially enhanced following treatment with TSA‐EVs when compared to the MO‐EVs treated (1.3‐fold) (P ≤ 0.05) and untreated cells (2.1‐fold) (P ≤ 0.001) (Figure 5c).
FIGURE 5.
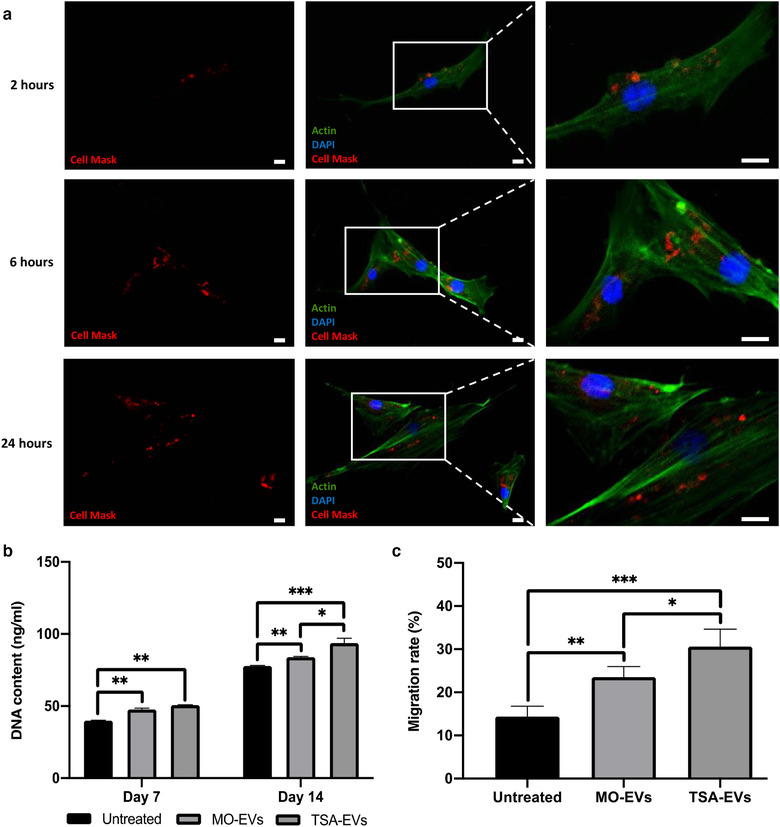
The influence of TSA‐EVs on hBMSCs general behaviour. a) Immunofluorescent images of Cell Mask labelled osteoblast‐derived EVs uptake by hBMSCs were taken at indicated times. Scale bar = 20 µm. The effects of TSA‐EVs on hBMSCs b) proliferation and c) migration. Data are expressed as mean ± SD (n = 3). *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001
The effects of TSA‐EV treatment on hBMSCs osteogenic differentiation was evaluated by quantifying osteoblast‐related gene expression and intracellular protein expression. MO‐EV treatment elicited a slight non‐significant increase in ALP, COL1A, BSP1 and OCN gene expression (P > 0.05) throughout osteogenic culture, whilst displaying a decrease in OCN expression at day 7 and 10 when compared to the untreated group (P > 0.05). Treatment with TSA‐EVs significantly upregulated hBMSCs mRNA expression levels of ALP, COL1A, BSP1 and OCN when compared to MO‐EVs treated (Figure 6a) (P ≤ 0.05 ‐ 0.001). COL1A mRNA expression for MO‐EV and TSA‐EV treated cells was significantly downregulated at day 10 when compared to untreated cells (P ≤ 0.05). Intracellular levels of osteoblast‐related proteins within hBMSCs was analysed by ICW (Figure 6b). Treatment with TSA‐EVs significantly enhanced hBMSCs intracellular protein levels of ALP (1.36, 1.4‐fold), Col1a (1.11, 1.18‐fold) and OCN (1.41, 1.58‐fold) when compared to that of the MO‐EVs treated and the untreated cells at day 14 (P ≤ 0.001) (Figure 6b). The MO‐EV group exhibited a significant increase in hBMSCs Col1a expression at day 14 (P ≤ 0.001), with a slight non‐significant enhancement in expression levels of ALP and OCN compared to the untreated control (P > 0.05).
FIGURE 6.
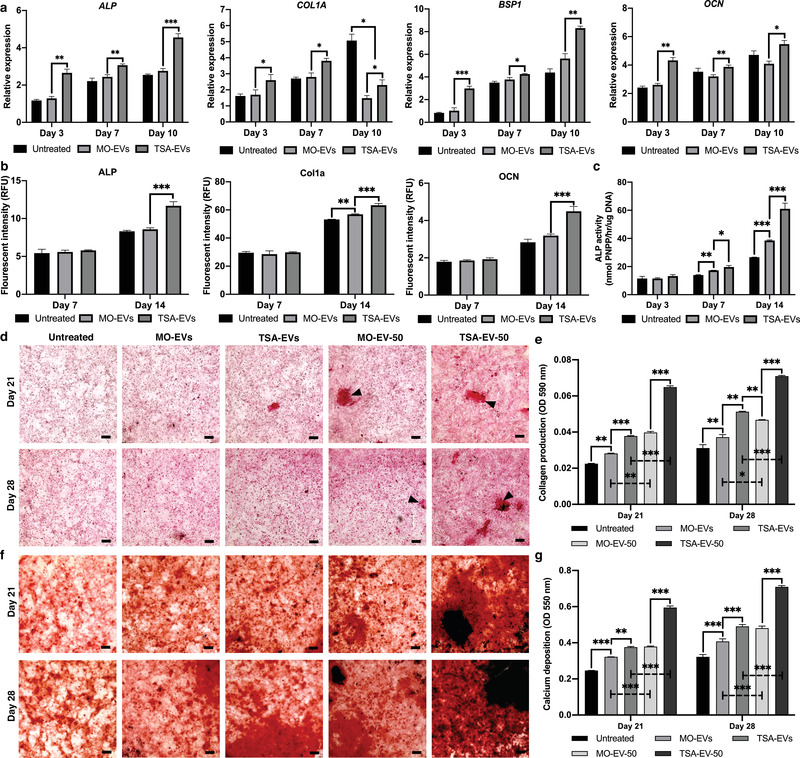
TSA‐EVs promoted hBMSCs osteogenic differentiation and mineralisation. a) Gene expression levels of ALP, COL1A, BSP1 and OCN were measured in TSA‐EV, MO‐EV treated and untreated hBMSCs during osteogenic culture. b) The intracellular protein levels of ALP, Col1a and OCN in EV treated hBMSCs analysed by ICW. c) The effects of TSA‐EVs on hBMSCs ALP activity during osteogenic culture. d) Picrosirius red staining for collagen production of EV‐treated hBMSCs. Black arrows highlight collagen rich mineral nodule‐like structures. e) Quantitative analysis of picrosirius red collagen staining. f) Alizarin red staining for calcium deposition on EV‐treated hBMSCs. Black staining indicates mineral nodule formation. g) Quantitative analysis of alizarin red staining. Scale bars = 200 µm. (MO‐EV, TSA EV; 10 µg/ml) (MO‐EV‐50, TSA‐EV‐50; 50 µg/ml). Data are expressed as mean ± SD (n = 3). *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001
The effects of TSA‐EV treatment on hBMSCs extracellular matrix mineralisation was assessed by quantifying ALP activity, collagen production and calcium deposition. ALP activity was substantially enhanced in hBMSCs treated with TSA‐EVs when compared to that of the MO‐EVs treated (1.2, 1.6‐fold) and untreated cells (1.4, 2.3‐fold) at day 7 and 14 (P ≤ 0.05 ‐ 0.001) (Figure 6c). Alterations in hBMSCs ALP activity were followed by significant changes in extracellular matrix collagen production. Osteoblast‐derived EV treatment elicited a time‐dose dependent increase in hBMSCs collagen deposition during osteogenic culture. The MO‐EVs (1.25, 1.26‐fold) and TSA‐EVs treated (1.68, 1.64‐fold) groups exhibited significantly increased collagen staining compared to the untreated control on day 21 and 28 (P ≤ 0.01 ‐ 0.001) (Figure 6d, e). The TSA‐EV treated cells displayed a 1.34‐ and 1.3‐fold enhancement in collagen deposition when compared to that of the MO‐EVs treated group on day 21 (P ≤ 0.001) and 28 (P ≤ 0.01) (Figure 6e). Moreover, 50 µg/ml EV treatment significantly increased collagen production when compared to 10 µg/ml EVs treated cells on day 21 and 28 (MO‐EV‐50 vs. MO‐EVs (1.41, 1.19‐fold)) (TSA‐EV‐50 vs. TSA‐EVs (1.72, 1.38‐fold)) (P ≤ 0.05 ‐ 0.001). The TSA‐EV‐50 group exhibited a 1.63‐ and 1.51‐fold increase in collagen production when compared to MO‐EV‐50 treated cells (P ≤ 0.001), with substantially increased quantity of collagen rich mineral nodule‐like structures (black arrows). Interestingly, the TSA‐EV treated cells displayed significantly enhanced collagen deposition when compared to the MO‐EV‐50 group on day 28 (1.1‐fold) (P ≤ 0.01). TSA‐EV treatment led to a time‐dose dependent increase in alizarin red staining for calcium deposition when compared to that of the MO‐EV treated and untreated cells, with mineral‐like nodules deposited throughout (Figure 6f). Quantitative analysis revealed that treatment with TSA‐EVs significantly increased extracellular matrix calcium deposition when compared to that of the MO‐EV treated (1.17, 1.2‐fold) (P ≤ 0.01 ‐ 0.001) and untreated cells (1.52, 1.52‐fold) (P ≤ 0.001) after 21 and 28 days of osteogenic culture (Figure 6g). Additionally, 50 µg/ml MO‐EV and TSA‐EV treatment further enhanced hBMSCs calcium deposition when compared to 10 µg/ml EVs treated cells on day 21 and 28 (MO‐EV‐50 vs. MO‐EVs (1.17, 1.18‐fold)) (TSA‐EV‐50 vs. TSA‐EV (1.58, 1.45‐fold)) (P ≤ 0.001). Moreover, the TSA‐EV‐50 group exhibited a significantly increased accumulation of calcium deposits when compared to the MO‐EV‐50 treated cells on day 21 and 28 (1.57, 1.47‐fold) (P ≤ 0.001), with enhanced quantity of mineralised nodule formations (black staining).
3.5. TSA induced hyperacetylation altered the microRNA profile of osteoblast‐derived EVs
To further elucidate the influence of epigenetic modification on osteoblast‐derived EV and its possible role in the enhanced osteoinductive properties, the EVs microRNA expression was profiled. As shown in Figure 7a, hierarchical clustering revealed differential microRNAs expression in TSA‐EVs and MO‐EVs. A total of 27 microRNAs species were upregulated, while 6 were downregulated (fold change cut off, ±2.0; P < 0.05) in the TSA‐EVs compared to that in the MO‐EVs (Figure 7b). Volcano plots showed the variation of microRNAs expression between TSA‐EVs and MO‐EVs (Figure 7c). The differentially expressed microRNAs are identified in Supplementary Table 2.
FIGURE 7.
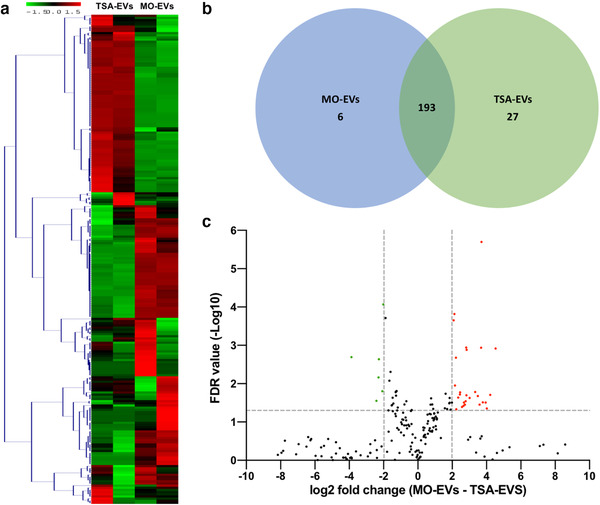
Differential expression of microRNAs derived from TSA‐EVs and MO‐EVs. a) Hierarchical clustering analysis of microRNAs that were differentially expressed between TSA‐EVs and MO‐EVs. b) Venn diagram comparing microRNAs differentially expressed from TSA‐EVs and MO‐EVs. A total of 193 shared microRNAs; 27 microRNAs upregulated in TSA‐EVs and 6 microRNAs upregulated in MO‐EVs. c) Volcano plot displaying Log2 values for the microRNAs fold‐change against Log10 FDR. MicroRNAs with a Log2 fold difference below 2 and a statistical value of > 0.05 were not considered to be statistically significant (vertical and horizontal lines respectively). The red points in the plot represents the significantly upregulated TSA‐EV microRNAs, the green points represent significantly upregulated MO‐EVs microRNAs
3.6. TSA induced differential expression of EV microRNAs related to general regulation mechanisms and osteogenic differentiation
To investigate the functions of the differentially expressed microRNAs, Gene ontology (GO) and Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathways were assessed. MicroRNAs were found to be associated with GO functional annotation of biological processes (e.g., anatomical structural development, cell differentiation), cellular compartments (e.g., organelle, chromosome) and molecular mechanisms (e.g., ion binding, protein binding transcription factor) (Figure 8a‐c). The number of significantly affected KEGG pathways, analysed via experimentally validated and predicted interactions of the differentially expression EV microRNAs are shown in Figure 8d.
FIGURE 8.
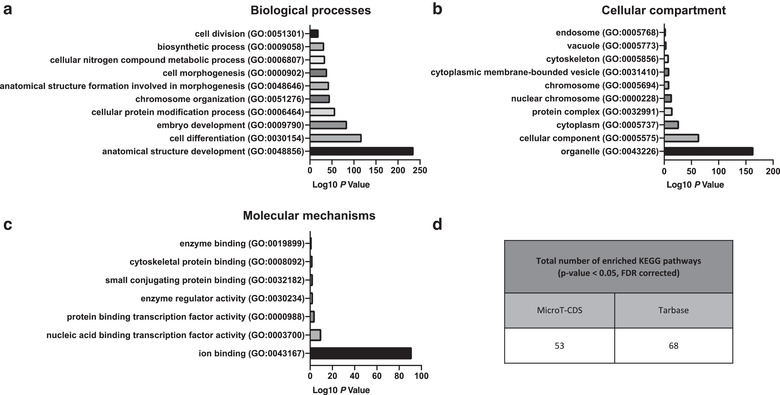
Gene ontology analysis of microRNAs found to be significantly upregulated in TSA‐EVs. Top ten GO prediction scores covering the domains of a) biological processes, b) cellular compartments and c) molecular mechanisms of microRNAs significantly upregulated in TSA‐EVs. d) Total number of experimentally validated and predicted interactions enrich KEGG pathways from MicroT‐CDS and Tarbase databases
All significantly affected KEGG pathways, analysed by predicted and experimental validated interactions of the differentially expressed microRNAs related to osteogenic differentiation and general regulatory mechanisms, are summarised in Table 1. A total of 21 KEGG pathways directly or indirectly related to osteogenic differentiation were enriched within TSA‐EVs. Of note, enriched KEGG pathways of differentially expressed microRNAs including Hippo signalling (mmu04390), MAPK signalling (mmu04010), Wnt signalling (mmu04310), and ECM‐receptor interaction (mmu04512) pathways, are involved in osteogenic differentiation. Additionally, the differentially expressed microRNAs were enriched in pathways related to general regulatory mechanisms including regulation of actin cytoskeleton (mmu04810) and endocytosis (mmu04144), possibly signifying the role of these microRNAs in influencing the internalisation of EVs at the recipient cells. Moreover, enriched pathways of SNARE interactions in vesicular transport (mmu04130), mRNA surveillance pathway (mmu03015) and protein processing in endoplasmic reticulum (mmu04141) may indicate how these microRNAs modulate recipient cell function.
TABLE 1.
Enriched KEGG biological pathways related to osteogenesis and regulatory mechanisms
| KEGG pathways related to osteogenic differentiation | p‐value, FDR corrected | Genes | MiRNAs | Algorithms | |
|---|---|---|---|---|---|
| Hippo signalling pathway | mmu04390 | 1.18E‐05 | 82 | 18 | TarBase |
| 2.68E‐08 | 55 | 25 | MicroT‐CDS | ||
| FoxO signalling pathway | mmu04068 | 3.75E‐07 | 85 | 19 | TarBase |
| 1.04E‐03 | 54 | 28 | MicroT‐CDS | ||
| PI3K‐Akt signalling pathway | mmu04151 | 1.32E‐03 | 111 | 29 | MicroT‐CDS |
| Signalling pathways regulating pluripotency of stem cells | mmu04550 | 1.49E‐03 | 50 | 26 | MicroT‐CDS |
| ECM‐receptor interaction | mmu04512 | 1.49E‐03 | 27 | 27 | MicroT‐CDS |
| MAPK signalling pathway | mmu04010 | 1.71E‐04 | 129 | 18 | TarBase |
| 1.96E‐03 | 86 | 30 | MicroT‐CDS | ||
| cAMP signalling pathway | mmu04024 | 5.22E‐03 | 67 | 30 | MicroT‐CDS |
| cGMP‐PKG signalling pathway | mmu04022 | 3.88E‐02 | 82 | 15 | TarBase |
| 5.51E‐03 | 57 | 29 | MicroT‐CDS | ||
| mTOR signalling pathway | mmu04150 | 1.95E‐03 | 38 | 19 | TarBase |
| 7.73E‐03 | 25 | 22 | MicroT‐CDS | ||
| Estrogen signalling pathway | mmu04915 | 2.09E‐02 | 49 | 15 | TarBase |
| 1.09E‐02 | 29 | 25 | MicroT‐CDS | ||
| AMPK signalling pathway | mmu04152 | 9.07E‐03 | 71 | 19 | TarBase |
| 1.28E‐02 | 43 | 27 | MicroT‐CDS | ||
| Focal adhesion | mmu04510 | 2.44E‐02 | 101 | 17 | TarBase |
| 1.76E‐02 | 66 | 31 | MicroT‐CDS | ||
| Insulin signalling pathway | mmu04910 | 1.60E‐03 | 78 | 18 | TarBase |
| 1.82E‐02 | 48 | 27 | MicroT‐CDS | ||
| TGF‐beta signalling pathway | mmu04350 | 1.60E‐03 | 43 | 18 | TarBase |
| 2.00E‐02 | 32 | 24 | MicroT‐CDS | ||
| Ras signalling pathway | mmu04014 | 4.56E‐02 | 103 | 19 | TarBase |
| 2.03E‐02 | 63 | 30 | MicroT‐CDS | ||
| Wnt signalling pathway | mmu04310 | 4.90E‐02 | 69 | 16 | TarBase |
| 2.74E‐02 | 46 | 30 | MicroT‐CDS | ||
| Hedgehog signalling pathway | mmu04340 | 3.32E‐02 | 20 | 23 | MicroT‐CDS |
| Adherens junction | mmu04520 | 9.90E‐05 | 47 | 17 | TarBase |
| TNF signalling pathway | mmu04668 | 6.48E‐04 | 58 | 16 | TarBase |
| HIF‐1 signalling pathway | mmu04066 | 2.28E‐03 | 62 | 19 | TarBase |
| Rap1 signalling pathway | mmu04015 | 2.16E‐02 | 102 | 18 | TarBase |
| KEGG pathways related to general regulatory mechanism | p‐value, FDR corrected | Genes | MiRNAs | Algorithms | |
|---|---|---|---|---|---|
| Sphingolipid signalling pathway | mmu04071 | 4.39E‐04 | 72 | 18 | TarBase |
| 3.06E‐03 | 46 | 27 | MicroT‐CDS | ||
| Regulation of actin cytoskeleton | mmu04810 | 9.07E‐03 | 108 | 18 | TarBase |
| 3.22E‐03 | 72 | 31 | MicroT‐CDS | ||
| Endocytotsis | mmu04144 | 6.12E‐07 | 128 | 18 | TarBase |
| 7.67E‐03 | 67 | 29 | MicroT‐CDS | ||
| SNARE interactions in vesicular transport | mmu04130 | 4.10E‐02 | 18 | 14 | TarBase |
| mRNA surveillance pathway | mmu03015 | 4.37E‐02 | 31 | 23 | MicroT‐CDS |
| Protein processing in endoplasmic reticulum | mmu04141 | 5.84E‐06 | 101 | 18 | TarBase |
| 2.38E‐02 | 48 | 22 | MicroT‐CDS | ||
3.7. The proteome of TSA‐EVs is enriched in proteins involved in transcriptional regulation
The proteomes of MO‐EVs and TSA‐EVs were compared for three independent sample preparations using label‐free MS‐LC/LC approach. The use of stringent criteria only permitted the inclusion of proteins identified in a least two biological replicates, with > 2 spectral counts in at least one repeat. Protein database searching resulted in the identification of a total of 1325 proteins. Of these, 25 proteins were significantly upregulated in TSA‐EVs, 27 upregulated in MO‐EVs, and 1273 shared proteins (Figure 9a, b). The differentially expressed proteins are identified in Supplementary Table 3. Pearson correlation, comparing all biological samples to one another, show a high average correlation between replicates in the MO‐EV (0.90) and TSA‐EV (0.97) groups (Figure 9c). When comparing MO‐EV and TSA‐EV to one another, an average correlation of 0.85 is observed, revealing a significant degree of similarity in the protein expression between MO‐EVs and TSA‐EVs. To provide an overview of the principal processes, mechanisms and cellular location of proteins significantly upregulated in TSA‐EVs, GO analysis was performed. Significantly enriched TSA‐EV proteins were found to be associated with GO functional annotation of molecular function (e.g., histone acetyltransferase activity, peptide‐lysine‐N‐acetyltransferase activity), cellular components (e.g., histone acetyltransferase complex, pre‐catalytic spliceosome) and biological processes (e.g., histone acetylation, histone modification) (Figure 9d‐f). The functional efficacy of TSA‐EVs on augmenting hBMSCs epigenetic functionality was evaluated by quantifying histone acetylation levels (Supplementary Figure 3). TSA‐EV treated hBMSCs elicited a significant increase in H3K9 acetylation levels in a time‐dependent manner when compared to the MO‐EVs treated (P ≤ 0.01 ‐ 0.001) and the untreated cells (P ≤ 0.001).
FIGURE 9.
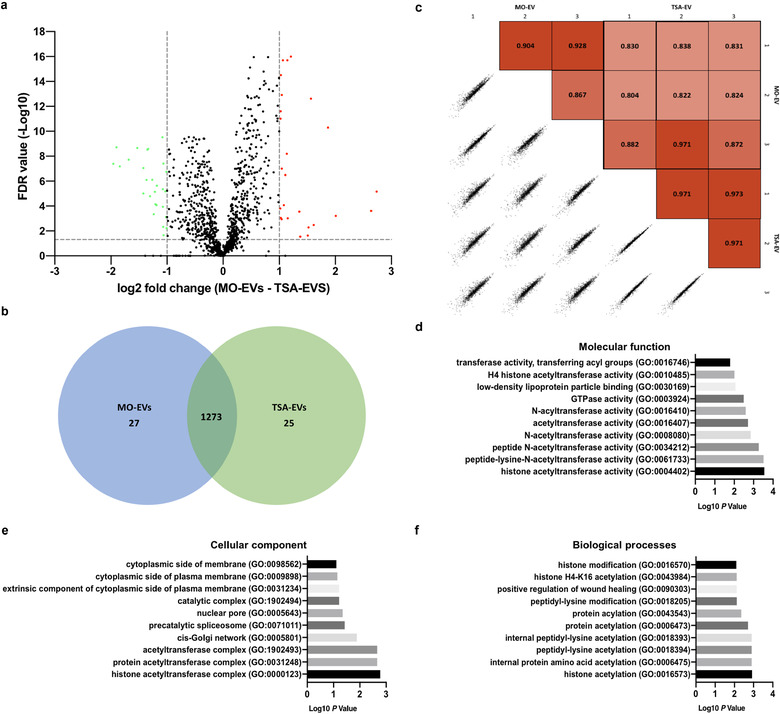
Analysis of differentially expressed proteins derived from TSA‐EVs and MO‐EVs. a) Volcano plot displaying Log2 values for the proteins fold‐change against Log10 FDR. Proteins with a Log2 fold difference below 1 and a statistical value of > 0.05 were not considered to be statistically significant (vertical and horizontal lines respectively). The red points in the plot represents the significantly upregulated TSA‐EV proteins, the green points represent significantly upregulated MO‐EVs proteins. b) Venn diagram comparing proteins differentially expressed from TSA‐EVs and MO‐EVs. A total of 1273 shared proteins; 25 proteins upregulated in TSA‐EVs and 27 proteins upregulated in MO‐EVs. c) Pearson correlations between technical replicates, biological replicates and sample groups were determined. GO analysis of proteins found to be significantly upregulated in TSA‐EVs. Top ten GO prediction scores covering the domains of d) molecular function, e) cellular components and f) biological processes of proteins significantly upregulated in TSA‐EVs
4. DISCUSSION
In the last decade, the field of EVs has rapidly expanded and is constantly unearthing new understanding of the role these nanoparticles play in various biological processes, signifying the potential influence EVs may have on the future direction of healthcare technologies. Several studies have demonstrated the therapeutic efficacy of EVs as novel acellular tools for bone repair (Davies et al., 2017; Wang et al., 2018; Wei et al., 2019). Although the potential utility of these nanosized vesicles has been reported, several approaches have been employed to further improve their therapeutic efficacy (Man et al., 2020). Genetic modification of the EV parent cell is a commonly utilised approach to promote the clinical viability of these nanoparticles (Kang et al., 2015; Tao et al., 2017); however, there are limitations associated with this technique (Hu et al., 2020; Kooijmans et al., 2016). Harnessing post‐translational modifications through altering the cells’ epigenetics has been demonstrated to enhance osteogenic differentiation (Hu et al., 2013; Huynh et al., 2016), hence, providing a potential novel strategy to improve EVs therapeutic efficacy for bone regeneration.
In the present study, we investigated the influence of altering osteoblast's epigenetic functionality to promote the osteoinductive capacity of their secreted EVs. TSA, a naturally‐derived HDACi, has been reported to accelerate osteogenic differentiation through hyperacetylation induced chromatin remodelling and transcription factor activation (Jin et al., 2013; Schroeder & Westendorf, 2005; Schroeder et al., 2004). Chromatin remodelling increases the accessibility to osteoblast‐related genes of interest, while HDAC inhibition and the hyperacetylation of non‐histone proteins promotes the activity/stability of key osteogenic transcriptional factors, such as Runx2 (Huynh et al., 2017). Herein, we demonstrated the successful augmentation of osteoblast epigenetic functionality through TSA‐induced histone hyperacetylation (Figure 10), consistent with several studies in the literature (Hu et al., 2013; Man et al., 2021; Schroeder & Westendorf, 2005). Additionally, we found that TSA induced acetylation resulted in a time‐dose dependent effect on osteoblast mineralisation, where administration of low TSA dosages (≤ 10 nM) increased osteogenesis when compared to higher concentrations (> 20 nM). This has been similarly reported in the literature (Huynh et al., 2016; Schroeder & Westendorf, 2005), likely due to the effects of high HDACi concentrations on the long‐term cellular viability. Hence, from a scalability perspective, administrating TSA at a dose that does not detrimentally affect cell number or functionality is highly advantageous (Rohde et al., 2019). In addition to examining the influence of TSA on osteoblast epigenetic functionality and differentiation, it is important to characterise the effects of this parental cell augmentation on the secreted EVs. It was observed that EVs isolated from TSA‐treated osteoblasts displayed reduced polydispersity (1.34‐fold), particle size (1‐05‐fold), concentration (1.4‐fold) and protein content (1.16‐fold) when compared to MO‐EVs. This profile has been reported to correlate with the degree of osteoblast differentiation (Davies et al., 2019), indicating the potentially enhanced osteoinductive capacity of TSA‐EVs. Moreover, the increased osteoblast extracellular matrix collagen production induced by TSA likely played a critical role in the quantity of EVs isolated from the conditional media, due to collagen‐mediated EV immobilisation (Buzás et al., 2018; Krohn et al., 2016). Interestingly, the TSA‐EVs exhibited a 3‐fold increase in RNA content when compared to that in the MO‐EVs. This suggests that the enhanced osteoblast transcriptional activity induced by TSA mediated hyperacetylation, enriched secreted EVs with elevated levels of RNA species, which may be partly responsible for its improved biological potency at recipient hBMSCs (Figure 10).
FIGURE 10.
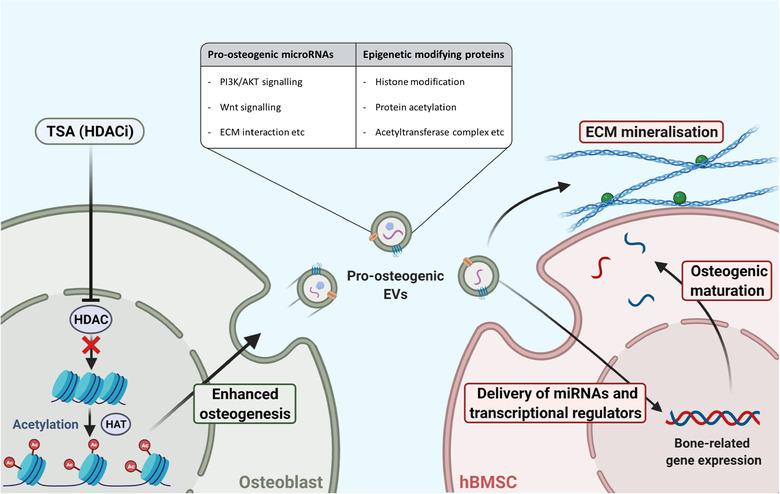
Schematic representation of the mechanism TSA augments osteoblast epigenetic functionality and mineralisation, enhancing the therapeutic potency of their secreted EVs to stimulate hBMSCs osteogenic differentiation. Figure created with BioRender.com
EVs ability to regulate intercellular communication such as stimulating the recruitment of endogenous cells is highly advantageous for bone tissue engineering applications (Su et al., 2018). Our findings demonstrate that osteoblast‐derived EVs significantly promoted hBMSCs proliferation and migration when compared to that of untreated cells, consistent with findings in the literature (Eichholz et al., 2020; Haertinger et al., 2020; Xu et al., 2019). Interestingly, TSA‐EVs elicited a more potent influence on hBMSCs proliferation and migration when compared to that of MO‐EVs. With TSA‐EVs exhibiting a 3‐fold increased RNA content compared to MO‐EVs, it is likely this enrichment plays a role in the enhanced cellular response to TSA‐EVs. It has been reported that microRNAs are the most abundant RNA species within EVs, likely due to their small size (Huang et al., 2013; Kim et al., 2017; O'brien et al., 2020). Hence, microRNA analysis was utilised to further elucidate the role of TSA in promoting osteoblast‐derived EVs paracrine effects. Intriguingly, the SNARE interactions in vesicular transport, endocytosis and regulation of actin cytoskeleton KEGG pathways were enriched by differentially expressed TSA‐EVs microRNAs, indicating the possible microRNA function in regulating EV internalisation at recipient cells. Moreover, TSA‐EVs were significantly enriched with miR‐31‐5p and miR‐143‐3p, microRNAs reported to promote the recruitment of numerous cells types (Li et al., 2015). Wang et al. demonstrated that miR‐143 targets HDAC7 in osteoblasts and endothelial cells, resulting in enhanced osteogenic and angiogenic effects, respectively (Wang et al., 2020). This indicates the role of miR‐143 in TSA‐EVs on promoting target cells differentiation capacity by modifying their epigenetic functionality. Therefore, the enrichment of these microRNAs in TSA‐EVs may be involved in promoting the recruitment and stimulation of endogenous cells through post‐translational modifications for bone regeneration.
Several studies have reported the use of HDACis in modifying the cells’ differentiation capacity (Huynh et al., 2016; Jin et al., 2013), however, there has been limited investigations into their influence in augmenting the cells secretome, particularly the therapeutic efficacy of EVs for bone regeneration. HDACis have been assessed as promising pharmacological agents to halt disease progression or for cancer therapeutics (Minetti et al., 2006; West & Johnstone, 2014), with their influence on EV secretion recently acquiring growing interest. For example, Sandonà et al. reported the utility of TSA in enhancing the therapeutic efficacy of EVs derived from fibro‐adipogenic progenitors (FAPs) to support muscle stem cells‐mediated regeneration of dystrophic muscles. HDACi treatment enriched FAP‐EVs with miR‐206 leading to enhanced regeneration and reduced fibrosis (Sandonà et al., 2020). Wang et al. used of the HDACi B390 to induce apoptosis in pancreatic cancer cells and decreased vascular endothelial growth factor C expression in secreted EVs, inhibiting the nanoparticles role in cancer progression (Wang et al., 2020). Herein, we investigated the influence of epigenetic reprogramming through TSA induced hyperacetylation, on the osteoinductive potency of osteoblast‐derived EVs as a novel engineering approach. We demonstrated that EVs isolated from TSA‐treated osteoblasts substantially promoted the mineralisation capacity of hBMSCs through the early, mid and late‐stages of osteogenic differentiation exhibited by enhanced osteoblast‐related gene/protein expression, ALP activity and extracellular matrix mineralisation compared to MO‐EVs treated and the untreated cells. Additionally, TSA‐EVs elicited an enhanced dose‐dependent increase in hBMSCs extracellular matrix collagen and calcium deposition when compared to MO‐EV treatment, thus providing further evidence into TSA‐EVs increased osteoinductive properties. Interestingly, the MO‐EVs treated hBMSCs presented a disparity in the differentiation status of the cell when compared to the osteogenic maturity of the mineralised matrix. Our findings showed a slight non‐significant increase in the MO‐EV treated hBMSCs osteoblast‐related gene/intracellular protein expression, while also exhibiting a significantly enhanced ALP activity, extracellular matrix collagen production and calcium deposition when compared to the untreated control. This indicates the reduced efficacy of MO‐EVs in stimulating hBMSCs differentiation compared to TSA‐EVs, however, MO‐EVs were able to facilitate the mineralisation of the cell's extracellular matrix, likely due to the enrichment of pro‐mineralising proteins associated with MO‐EVs, including Annexins as reported in the literature and in the proteomics analysis in this present study (Davies et al., 2017, 2019). Moreover, the more mature collagen extracellular matrix from the TSA‐EV treated hBMSCs, likely promoted the sequestering of endogenous EVs secreted from differentiating hBMSCs and TSA‐EV treatment (Buzás et al., 2018; Krohn et al., 2016), thus further facilitating matrix mineralisation. Taken together, the functional analysis of TSA‐EVs osteoinductive efficacy indicates its ability to promote osteogenesis through stimulating hBMSCs osteogenic differentiation and the mineralisation of its extracellular matrix (Figure 10).
To further elucidate the mechanisms in which TSA‐EVs imparts its pro‐osteogenic effects, particularly in stimulating cellular differentiation, microRNA profiling was conducted. Utilising available databases, target prediction of differentially expressed TSA‐EV microRNAs showed substantial enrichment in KEGG pathways related to osteogenic differentiation such as Hippo signalling pathway, MAPK signalling pathway, Wnt signalling pathway, FoxO signalling pathway and ECM‐receptor interaction. MicroRNA profiling highlighted several pro‐osteogenic microRNAs significantly enriched in the TSA‐EVs. Among them, miR‐21 is known to target Spry1, an inhibitor of osteogenic differentiation, and Sox2, a marker of pluripotency, hence resulting in the osteogenic lineage‐specific differentiation (Li et al., 2015; Yang et al., 2013). Furthermore, miR‐21 is involved in the regulation of the phosphatidylinositol 3‐kinase (PI3K)/Akt/glycogen synthase kinase‐3β (GSK‐3β) signalling pathway, leading to an increased accumulation of cytoplasmic β‐catenin, enhanced Runx2 expression and osteogenic differentiation (Meng et al., 2015). Similarly, reports have shown that miR‐26a promotes osteogenic differentiation by targeting GSK‐3β, activating the Wnt signalling pathway (Liu et al., 2018; Su et al., 2015). Zhao et al. demonstrated that miR‐199b‐5p expression was increased during osteogenesis, and when overexpressed it promoted BMSCs osteogenic differentiation through GSK3β/β‐catenin pathway (Zhao et al., 2016). Additionally, miR‐15b, an osteoblast‐specific microRNA (Sainitya et al., 2015), is known to target Smurf1, an inhibitor of osteogenic differentiation (Vimalraj et al., 2014). Smurf1 degrades Runx2 through the proteasomal pathway, therefore, the upregulation of miR‐15b will indirectly increase Runx2 levels (Shimazu et al., 2016). Another miRNA upregulated in TSA‐EVs, miR‐181a, has been reported to promote osteogenesis via inhibiting key transforming growth factor‐β (TGF‐β) signalling molecules such as the TGF‐β type I receptor (TβR‐I/Alk5) and TGF‐β induced (Bhushan et al., 2013; Zheng et al., 2019). Moreover, miR‐125b, a microRNA found to be involved in osteoblast‐osteoclast communication was significantly enriched in TSA‐EVs (Yoshiko & Minamizaki, 2020). Minamizaki et al. reported miR‐125b involvement in regulating bone turnover, where the enrichment of this microRNA in EVs within the bone matrix, inhibits bone resorption via downregulating the transcriptional repressor PRDM1, subsequently upregulating the anti‐osteoclastogenic genes Mafb and Irf8 (Minamizaki et al., 2020). Interestingly, TSA‐EVs were significantly enriched in miR‐22, which has been reported to inhibit the expression of HDAC6 in adipose‐derived stem cells, promoting its osteogenic differentiation (Huang et al., 2012). Similarly, miR‐143 has been shown to stimulate BMSCs and osteoblast mineralisation by inhibiting HDAC7 activity (Wang et al., 2020). This indicates that modifying the osteoblast epigenome through TSA, augments the epigenetic functionality of recipient hBMSCs through EV microRNA delivery, mimicking the mechanism of transgenerational epigenetic inheritance reported in the literature (Sahoo & Losordo, 2014; Sharma, 2014). Together, these findings suggest altering the epigenetic functionality and differentiation of mineralising osteoblast enriches their EVs with pro‐osteogenic microRNAs (Figure 10). Future studies would be required to functionally validate the biological efficacy of these differentially expressed microRNAs. Moreover, it would be of interest to examine the influence on TSA induced epigenetic regulation on the expression and therapeutic efficacy of other RNAs species within the TSA‐EVs.
In order to further investigate the influence of TSA induced epigenetic modification on the composition and biological potency of TSA‐EVs, the vesicles protein composition was analysed via mass spectrometry. GO analysis showed that the differentially expressed TSA‐EV proteins were associated with transcriptional regulation. Among them, Ankyrin repeat domain‐containing protein 11 (AnkRD11) is a chromatin regulator which modulates histone acetylation and gene expression (Gallagher et al., 2015). The introduction of a N‐ethyl‐N‐nitrosourea mutation in the AnkRD11 gene in mice, resulted in craniofacial abnormalities and reduced overall bone mineral density, indicating the role of this protein in normal skeletal development (Barbaric et al., 2008). Another chromatin regulator, KAT8 regulatory NSL complex subunit 3 (Kansl3) was found to be upregulated in TSA‐EVs. Kansl3 is a subunit of the histone acetyltransferase complex, which is involved in histone H4 acetylation and thereby regulating transcriptional activity (Cai et al., 2010). Moreover, histone H4 acetylation has been reported to promote osteogenic maturation, indicating the lineage‐specific effects of Kansl3 on hBMSCs differentiation (Dudakovic et al., 2013). Similarly, the Plant homeodomain finger protein 14 (PHF14) was significantly enriched in TSA‐EVs. Several studies have reported the role of PHD fingers as epigenetic effectors by regulating transcription and chromatin dynamics (Aasland, 1995; Musselman & Kutateladze, 2011) and their role on osteogenesis has been reported in the literature. For example, the PHF20 was shown to positively regulate osteoblast differentiation by increasing H3K4me3 enrichment on the Runx2 promoter, enhancing the transcription factor activity (Yang et al., 2017). In addition to chromatin remodelling proteins, TSA‐EVs were also significantly enriched with proteins involved in regulating RNA processing. For example, the pre‐mRNA‐splicing factor 38B (Prpf38b) was found to be upregulated in TSA‐EVs. Pre‐mRNA splicing factors has been shown to mediate mRNA processing and splicing (Di et al., 2019), indicating the role of Prpf38b in regulating the translation of pre‐mRNA transcripts to mature mRNA in hBMSCs. Similarly, RNA‐binding protein 10 (Rbm10) is known to be involved in mRNA processing and miRNA biogenesis (Zhao et al., 2017). The nuclear carrier protein Ran‐binding protein 17 (Ranbp17) was also enriched in TSA‐EVs, which functions in regulating the transport of proteins and RNAs through the nuclear pore (Lee et al., 2010). Similarly, Washc5 has been shown to mediate endosomal sorting, indicating its role in processing endocytosed EVs (Gilleron et al., 2019). Interestingly, the pro‐osteogenic proteins previously identified in MO‐EVs to be responsible for its pro‐mineralising capacity (Davies et al., 2017, 2019), such as calcium‐channelling annexin proteins, were not significantly altered by TSA treatment. The TSA induced enrichment of EV proteins involved in transcriptional regulation indicates the role of TSA‐EVs in augmenting the epigenetic landscape of the recipient cell (Figure 10), confirmed by the functional assessment of TSA‐EV induced hBMSCs histone hyperacetylation. The delivery of these transcriptional regulating proteins likely facilitate the therapeutic efficacy of TSA‐EV pro‐osteogenic microRNAs and ultimately accelerates the osteogenic maturation of hBMSCs. Moreover, the augmented epigenetic landscape induced by TSA‐EV transcriptional modifiers probably facilitated the enhanced hBMSCs expression of osteoblast‐related genes during the early phase of osteogenesis. It is important to note that due to the diverse biological cargo of EVs, is it likely that the osteoinductive capacity of TSA‐EVs is a combination of changes across all EV components (i.e. metabolites, lipids, proteins, RNA species etc.), although this would require further investigation.
In the present study, we investigated the influence of altering the epigenetic functionality of mineralising osteoblasts on enhancing the therapeutic efficacy of their EVs for bone regeneration. We isolated and characterised EVs from TSA‐treated mineralising osteoblasts and further examined the biological potency of these EVs on hBMSCs. Having demonstrated the enhanced osteoinductive capacity of TSA‐EVs, microRNA profiling and proteomic analysis was conducted to further elucidate the possible mechanism in which these nanoparticles promote osteogenesis. We predicted the targets of the TSA‐EV enriched microRNAs and the signalling pathways to be related to osteogenic differentiation. Moreover, the TSA‐EVs were significantly enriched with transcription regulating proteins. Hence, the TSA‐EV enrichment of these pro‐osteogenic microRNAs and proteins involved in epigenetic regulation may be partially responsible for promoting hBMSCs osteogenic differentiation, however, the functional validation of these augmented components induced by epigenetic reprogramming would be required in future studies.
Having reported the enhanced osteoinductive capacity of TSA‐EVs in vitro, there is growing precedence to elucidate the therapeutic capacity of EVs in more physiologically relevant models in vivo (Man et al., 2020). There are several pertinent issues with the therapeutic delivery of EVs in the field including controlling their release kinetics at the defect site, optimising EV dosing regimen to maximise their therapeutic response in vivo and the development of a biomaterial system that facilitates EV‐induced bone regeneration (Brennan et al., 2020). We believe these factors would have a tremendous impact on the clinical efficacy of EVs, irrespective of the nanoparticle's therapeutic potency. Although the in vivo investigation of TSA‐EVs is beyond the scope of this present study, which is focused on determining whether epigenetic reprogramming is a viable EV engineering approach and elucidating possible mechanisms linked to its enhanced osteoinductive properties, future studies will assess the therapeutic delivery of TSA‐EVs within a biomaterial system to effectively investigate the clinical efficacy of this EV engineering approach for bone augmentation strategies. In addition to conducting in vivo testing, to move this novel approach towards the clinic, demonstration of TSA‐EVs efficacy against a positive treatment control would be a valuable pursuit, for example BMP2.
5. CONCLUSION
In conclusion, these findings demonstrate that altering osteoblasts epigenetic functionality via TSA induced hyperacetylation, enhanced the differentiation capacity of the parental cell and the osteoinductive potency of their EVs. Furthermore, microRNA profiling revealed HDACi treatment enriched TSA‐EVs with microRNAs associated with osteogenic‐related pathways. Proteomics analysis identified the enrichment of epigenetic regulating proteins within TSA‐EVs. These findings demonstrate the considerable utility of epigenetic regulation as a novel engineering approach to enhance the therapeutic efficacy of EVs as an acellular tool for bone repair. To our knowledge, this is the first study to promote EVs regenerative potency for bone augmentation strategies through epigenetic reprogramming.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Kenny Man study conceptualisation, biological laboratory work and manuscript preparation. Mathieu Y. Brunet dynamic light scattering analysis. Soraya Williams sample preparation for proteomic analysis and immunoblotting. Maria Fernandez‐Rhodes immunoblotting. Liam M. Heaney design and planning of proteomic analysis. Lee A. Gethings mass spectrometry analysis. Angelica Federici TEM imaging. Owen G. Davies, David Hoey and Sophie C. Cox contributed with critical revisions and editing. All authors have read and agreed to the published version of the manuscript.
Supporting information
Supporting information.
ACKNOWLEDGEMENTS
The authors acknowledge funding from the EPSRC (EP/S016589/1), the School of Chemical Engineering, University Of Birmingham for M.Y.B's doctoral studentship, and Science Foundation Ireland (SFI) Frontiers for the Future Project Grant (19/FFP/6533), Academy of Medical Sciences, Wellcome Trust, Government Department of Business, Energy and Industrial Strategy, British Heart Foundation, Diabetes UK (SBF004\1090) and the EPSRC/MRC Doctoral Training Centre in Regenerative Medicine.
Man, K. , Brunet, M. Y. , Fernandez‐Rhodes, M. , Williams, S. , Heaney, L. M. , Gethings, L. A. , Federici, A. , Davies, O. G. , Hoey, D. , & Cox, S. C. (2021). Epigenetic reprogramming enhances the therapeutic efficacy of osteoblast‐derived extracellular vesicles to promote human bone marrow stem cell osteogenic differentiation. Journal of Extracellular Vesicles, 10, e12118. 10.1002/jev2.12118
REFERENCES
- Aasland, R. (1995). The phd finger: Implications for chromatin‐mediated transcriptional regulation. Trends in Biochemical Sciences, 20(2), 56–59. [DOI] [PubMed] [Google Scholar]
- Amini, A. R. , Laurencin, C. T. , & Nukavarapu, S. P. (2012). Bone tissue engineering: Recent advances and challenges. Critical Reviews in Biomedical Engineering, 40(5), 363–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbaric, I. , Perry, M. J. , Dear, T. N. , Rodrigues Da Costa, A. , Salopek, D. , Marusic, A. , Hough, T. , Wells, S. , Hunter, A. J. , Cheeseman, M. , & Brown, S. D. M. (2008). An enu‐induced mutation in the ankrd11 gene results in an osteopenia‐like phenotype in the mouse mutant yoda. Physiological Genomics, 32(3), 311–321. [DOI] [PubMed] [Google Scholar]
- Baroli, B. (2009). From natural bone grafts to tissue engineering therapeutics: Brainstorming on pharmaceutical formulative requirements and challenges. Journal of Pharmaceutical Sciences, 98(4), 1317–1375. [DOI] [PubMed] [Google Scholar]
- Bhushan, R. , Grunhagen, J. , Becker, J. , Robinson, P. N. , Ott, C. E. , & Knaus, P. (2013). Mir‐181a promotes osteoblastic differentiation through repression of tgf‐beta signaling molecules. International Journal of Biochemistry & Cell Biology, 45(3), 696–705. [DOI] [PubMed] [Google Scholar]
- Bolden, J. E. , Shi, W. , Jankowski, K. , Kan, C. Y. , Cluse, L. , Martin, B. P. , MacKenzie, K. L. , Smyth, G. K. , & Johnstone, R. W. (2013). Hdac inhibitors induce tumor‐cell‐selective pro‐apoptotic transcriptional responses. Cell Death & Disease, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan, M. Á. , Layrolle, P. , & Mooney, D. J. (2020). Biomaterials functionalized with msc secreted extracellular vesicles and soluble factors for tissue regeneration. Advanced Functional Materials, 30(37), 1909125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdick, J. A. , Mauck, R. L. , Gorman, J. H. , & Gorman, R. C. (2013). Acellular biomaterials: An evolving alternative to cell‐based therapies. Science Translational Medicine 5, 176ps4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzás, E. I. , Tóth, E. Á. , Sódar, B. W. , & Szabó‐Taylor, K. É. (2018). Molecular interactions at the surface of extracellular vesicles. Seminars in Immunopathology, 40(5), 453–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, Y. , Jin, J. , Swanson, S. K. , Cole, M. D. , Choi, S. H. , Florens, L. , Washburn, M. P. , Conaway, J. W. , & Conaway, R. C. (2010). Subunit composition and substrate specificity of a mof‐containing histone acetyltransferase distinct from the male‐specific lethal (msl) complex. Journal of Chemical Biology, 285(7), 4268–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calori, G. M. , Mazza, E. , Colombo, M. , & Ripamonti, C. (2011). The use of bone‐graft substitutes in large bone defects: Any specific needs?” Injury‐International Journal of the Care of the Injured, 42, S56–S63. [DOI] [PubMed] [Google Scholar]
- Chance‐Larsen, K. , Backhouse, M. R. , Collier, R. , Wright, C. , Gosling, S. , Harden, B. , Marsh, S. , Kay, P. , Wyles, H. , Erwin, Jo , & Woolf, A. (2019). Developing a national musculoskeletal core capabilities framework for first point of contact practitioners. Rheumatology Advances in Practice, 3(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Si , Tang, Y. , Liu, Y. , Zhang, P. , Lv, L. , Zhang, X. , Jia, L. , & Zhou, Y. (2019). Exosomes derived from mir‐375‐overexpressing human adipose mesenchymal stem cells promote bone regeneration. Cell Proliferation, 52(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo, M. ‐. K. , Yeo, H. , & Zayzafoon, M. (2009). Nfatc1 mediates hdac‐dependent transcriptional repression of osteocalcin expression during osteoblast differentiation. Bone, 45(3), 579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collas, P. , Noer, A. , & Sorensen, A. L. (2008). Epigenetic basis for the differentiation potential of mesenchymal and embryonic stem cells. Transfusion medicine and hemotherapy : offizielles Organ der Deutschen Gesellschaft fur̈ Transfusionsmedizin und Immunham̈atologie, 35(3), 205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, Y. , Luan, J. , Li, H. , Zhou, X. , & Han, J. (2016). Exosomes derived from mineralizing osteoblasts promote st2 cell osteogenic differentiation by alteration of microrna expression. FEBS Letters, 590(1), 185–192. [DOI] [PubMed] [Google Scholar]
- Davies, O. G. , Cox, S. C. , Azoidis, I. , McGuinness, A. J. A. , Cooke, M. , Heaney, L. M. , & Grover, L. M. (2019). Osteoblast‐derived vesicle protein content is temporally regulated during osteogenesis: Implications for regenerative therapies. Frontiers in Bioengineering and Biotechnology, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, O. G. , Cox, S. C. , Williams, R. L. , Tsaroucha, D. , Dorrepaal, R. M. , Lewis, M. P. , & Grover, L. M. (2017). Annexin‐enriched osteoblast‐derived vesicles act as an extracellular site of mineral nucleation within developing stem cell cultures. Scientific Reports, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di, C. , Syafrizayanti, Zhang, Q. , Chen, Y. , Wang, Y. , Zhang, X. , Liu, Y. , Sun, C. , Zhang, H. , & Hoheisel, J. D. (2019). Function, clinical application, and strategies of pre‐mrna splicing in cancer. Cell Death and Differentiation, 26(7), 1181–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitriou, R. , Jones, E. , Mcgonagle, D. , & Giannoudis, P. V. (2011). Bone regeneration: Current concepts and future directions. BMC Medicine, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Distler, U. , Kuharev, J. , Navarro, P. , & Tenzer, S. (2016). Label‐free quantification in ion mobility‐enhanced data‐independent acquisition proteomics. Nature Protocols, 11(4), 795–812. [DOI] [PubMed] [Google Scholar]
- Djouad, F. , Guérit, D. , Marie, M. , Toupet, K. , Jorgensen, C. , & Noël, D. (2012). Mesenchymal stem cells: New insights into bone regenerative applications. Journal of Biomaterials and Tissue Engineering, 2(1), 14–28. [Google Scholar]
- Dompe, C. , Janowicz, K. , Hutchings, G. , Moncrieff, L. , Jankowski, M. , Nawrocki, M. J. , Józkowiak, M. , Mozdziak, P. , Petitte, J. , Shibli, J. A. , Dyszkiewicz‐Konwińska, M. , Bruska, M. , Piotrowska‐Kempisty, H. , Kempisty, B. , & Nowicki, M. (2020). Epigenetic research in stem cell bioengineering‐anti‐cancer therapy, regenerative and reconstructive medicine in human clinical trials. Cancers, 12, 1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll, J. , & Patel, T. (2019). The mesenchymal stem cell secretome as an acellular regenerative therapy for liver disease. Journal of Gastroenterology, 54(9), 763–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudakovic, A. , Evans, J. M. , Li, Y. , Middha, S. , Mcgee‐Lawrence, M. E. , Van Wijnen, A. J. , & Westendorf, J. J. (2013). Histone deacetylase inhibition promotes osteoblast maturation by altering the histone h4 epigenome and reduces akt phosphorylation. Journal of Chemical Biology, 288(40), 28783–28791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckschlager, T. , Plch, J. , Stiborova, M. , & Hrabeta, J. (2017). Histone deacetylase inhibitors as anticancer drugs. International Journal of Molecular Sciences, 18(7), 1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichholz, K. F. , Woods, I. , Riffault, M. , Johnson, G. P. , Corrigan, M. , Lowry, M. C. , Shen, N. , Labour, M. ‐. N. , Wynne, K. , O'driscoll, L. , & Hoey, D. A. (2020). Human bone marrow stem/stromal cell osteogenesis is regulated via mechanically activated osteocyte‐derived extracellular vesicles. Stem Cells Translational Medicine, 9(11), 1431–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Andaloussi, S. , Mäger, I. , Breakefield, X O. , Wood, M J. A. “Extracellular vesicles: Biology and emerging therapeutic opportunities. Nature Reviews Drug Discovery, 12, 5 (2013). 347–357. [DOI] [PubMed] [Google Scholar]
- Gallagher, D. , Voronova, A. , Zander, M. A. , Cancino, G. I. , Bramall, A. , Krause, M. P. , Abad, C. , Tekin, M. , Neilsen, P. M. , Callen, D. F. , Scherer, S. W. , Keller, G. M. , Kaplan, D. R. , Walz, K. , & Miller, F. D. (2015). Ankrd11 is a chromatin regulator involved in autism that is essential for neural development. Developmental Cell, 32(1), 31–42. [DOI] [PubMed] [Google Scholar]
- Gilleron, J. , Gerdes, J. M. , Zeigerer, A. (2019). Metabolic regulation through the endosomal system. Traffic (Copenhagen, Denmark), 20(8) 552–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnecchi, M. , He, H. , Liang, O. D. , Melo, L. G. , Morello, F. , Mu, H. , Noiseux, N. , Zhang, L. , Pratt, R. E. , Ingwall, J. S. , & Dzau, V. J. (2005). Paracrine action accounts for marked protection of ischemic heart by akt‐modified mesenchymal stem cells. Nature Medicine, 11(4), 367–368. [DOI] [PubMed] [Google Scholar]
- Haertinger, M. , Weiss, T. , Mann, A. , Tabi, A. , Brandel, V. , & Radtke, C. (2020). Adipose stem cell‐derived extracellular vesicles induce proliferation of schwann cells via internalization. Cells, 9(1), 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna, E. , Remuzat, C. , Auquier, P. , & Toumi, M. (2016). Advanced therapy medicinal products: Current and future perspectives. Journal of Market Access & Health Policy, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heathman, T. R.j , Nienow, A. W. , Mccall, M. J. , Coopman, K. , Kara, Bo , & Hewitt, C. J. (2015). The translation of cell‐based therapies: Clinical landscape and manufacturing challenges. Regenerative Medicine, 10(1), 49–64. [DOI] [PubMed] [Google Scholar]
- Hu, Q. , Su, H. , Li, J. , Lyon, C. , Tang, W. , Wan, M. , & Hu, T. Y. (2020). Clinical applications of exosome membrane proteins. Precision Clinical Medicine, 3(1), 54–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, X. , Zhang, X. , Dai, L. , Zhu, J. , Jia, Z. , Wang, W. , Zhou, C. , & Ao, Y. (2013). Histone deacetylase inhibitor trichostatin a promotes the osteogenic differentiation of rat adipose‐derived stem cells by altering the epigenetic modifications on runx2 promoter in a bmp signaling‐dependent manner. Stem Cells and Development, 22(2), 248–255. [DOI] [PubMed] [Google Scholar]
- Huang, S. , Wang, S. , Bian, C. , Yang, Z. , Zhou, H. , Zeng, Y. , Li, H. , Han, Q. , & Zhao, R. C. (2012). Upregulation of mir‐22 promotes osteogenic differentiation and inhibits adipogenic differentiation of human adipose tissue‐derived mesenchymal stem cells by repressing hdac6 protein expression. Stem Cells and Development, 21(13), 2531–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X. , Yuan, T. , Tschannen, M. , Sun, Z. , Jacob, H. , Du, M. , Liang, M. , Dittmar, R. L. , Liu, Y. , Liang, M. , Kohli, M. , Thibodeau, S. N. , Boardman, L. , & Wang, L. (2013). Characterization of human plasma‐derived exosomal RNAs by deep sequencing. Bmc Genomics [Electronic Resource], 14, 319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh, N. C.‐.N. , Everts, V. , & Ampornaramveth, R. S. (2017). Histone deacetylases and their roles in mineralized tissue regeneration. Bone Rep, 7, 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh, N. C.‐.N. , Everts, V. , Pavasant, P. , & Ampornaramveth, R. S. (2016). Inhibition of histone deacetylases enhances the osteogenic differentiation of human periodontal ligament cells. Journal of Cellular Biochemistry, 117(6), 1384–1395. [DOI] [PubMed] [Google Scholar]
- Izadpanah, R. , Trygg, C. , Patel, B. , Kriedt, C. , Dufour, J. , Gimble, J. M. , & Bunnell, B. A. (2006). Biologic properties of mesenchymal stem cells derived from bone marrow and adipose tissue. Journal of Cellular Biochemistry, 99(5), 1285–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen, E. D. , Gopalakrishnan, R. , & Westendorf, J. J. (2010). Regulation of gene expression in osteoblasts. Biofactors, 36(1), 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon, E. ‐. J. , Lee, K. ‐. Y. , Choi, N. ‐. S. , Lee, Mi‐H. , Kim, H. ‐. N. , Jin, Y. ‐. H. , Ryoo, H‐Mo , Choi, Je‐Y. , Yoshida, M. , Nishino, N. , Oh, B. ‐. C. , Lee, K. ‐. S. , Lee, Y. H. , & Bae, S. ‐. C. (2006). Bone morphogenetic protein‐2 stimulates runx2 acetylation. Journal of Biological Chemistry, 281(24), 16502–16511. [DOI] [PubMed] [Google Scholar]
- Jin, H. , Park, J. ‐. Y. , Choi, H. , & Choung, P. ‐. H. (2013). Hdac inhibitor trichostatin a promotes proliferation and odontoblast differentiation of human dental pulp stem cells. Tissue Engineering Part A, 19(5–6), 613–624. [DOI] [PubMed] [Google Scholar]
- Jonason, J. H. , Xiao, G. , Zhang, M. , Xing, L. , & Chen, D. (2009). Post‐translational regulation of runx2 in bone and cartilage. Journal of Dental Research, 88(8), 693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, K. , Ma, R. , Cai, W. , Huang, W. , Paul, C. , Liang, J. , Wang, Y. , Zhao, T. , Kim, Ha W. , Xu, M. , Millard, R. W. , Wen, Z. , & Wang, Y. (2015). Exosomes secreted from cxcr4 overexpressing mesenchymal stem cells promote cardioprotection via akt signaling pathway following myocardial infarction. Stem Cells International, 2015, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H. , Kim, D. , Nam, H. , Moon, S. , Kwon, Y. J. , & Lee, J. B. (2020). Engineered extracellular vesicles and their mimetics for clinical translation. Methods (San Diego, Calif.) 177, 80–94. [DOI] [PubMed] [Google Scholar]
- Kim, K. Mi , Abdelmohsen, K. , Mustapic, M. , Kapogiannis, D. , & Gorospe, M. (2017). RNA in extracellular vesicles. Wiley Interdisciplinary Reviews. RNA, 8(4), e1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooijmans, S. A. A. , Schiffelers, R. M. , Zarovni, N. , & Vago, R. (2016). Modulation of tissue tropism and biological activity of exosomes and other extracellular vesicles: New nanotools for cancer treatment. Pharmacological Research, 111, 487–500. [DOI] [PubMed] [Google Scholar]
- Krohn, J. B. , Hutcheson, J. D. , Martínez‐Martínez, E. , & Aikawa, E. (2016). Extracellular vesicles in cardiovascular calcification: Expanding current paradigms. Journal of Physiology‐London, 594(11), 2895–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor, L. , & Yang, X. B. (2019). Harnessing the hdac‐histone deacetylase enzymes, inhibitors and how these can be utilised in tissue engineering. International Journal of Oral Science, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J‐Ho , Zhou, S. , & Smas, C. M. (2010). Identification of ranbp16 and ranbp17 as novel interaction partners for the bhlh transcription factor e12. Journal of Cellular Biochemistry, 111(1), 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, D. , Li, X. I. , Wang, A. , Meisgen, F. , Pivarcsi, A. , Sonkoly, E. , Ståhle, M. , & Landén, N. X.u (2015). Microrna‐31 promotes skin wound healing by enhancing keratinocyte proliferation and migration. Journal of Investigative Dermatology, 135(6), 1676–1685. [DOI] [PubMed] [Google Scholar]
- Li, G. ‐. Z. , Vissers, J. P. C. , Silva, J. C. , Golick, D. , Gorenstein, M. V. , & Geromanos, S. J. (2009). Database searching and accounting of multiplexed precursor and product ion spectra from the data independent analysis of simple and complex peptide mixtures. Proteomics, 9(6), 1696–1719. [DOI] [PubMed] [Google Scholar]
- Li, H. , Yang, F. , Wang, Z. , Fu, Q. , & Liang, A. (2015). Microrna‐21 promotes osteogenic differentiation by targeting small mothers against decapentaplegic 7. Molecular Medicine Reports, 12(1), 1561–1567. [DOI] [PubMed] [Google Scholar]
- Li, X. , Jin, Li , Cui, Q. , Wang, G. ‐. J. , & Balian, G. (2005). Steroid effects on osteogenesis through mesenchymal cell gene expression. Osteoporosis International, 16(1), 101–108. [DOI] [PubMed] [Google Scholar]
- Liu, Z. , Chang, H. , Hou, Y. H. , Wang, Y. , Zhou, Z. Q. , Wang, M. , Huang, Z. D. , & Yu, B. (2018). Lentivirus‐mediated microrna‐26a overexpression in bone mesenchymal stem cells facilitates bone regeneration in bone defects of calvaria in mice. Molecular Medicine Reports, 18(6), 5317–5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, J. , Guo, X. , Zhang, S. , Liu, H. , Lu, J. , Dong, Z. , Liu, K. , & Ming, L. (2015). Trichostatin a, a histone deacetylase inhibitor, suppresses proliferation and promotes apoptosis of esophageal squamous cell lines. Molecular Medicine Reports, 11(6), 4525–4531. [DOI] [PubMed] [Google Scholar]
- Man, K. , Brunet, M. Y. , Jones, M. ‐. C. , & Cox, S. C. (2020). Engineered extracellular vesicles: Tailored‐made nanomaterials for medical applications. Nanomaterials (Basel), 10, 1838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man, K. , Lawlor, L. , Jiang, L. ‐. H. , & Yang, X. B. (2021). The selective histone deacetylase inhibitor mi192 enhances the osteogenic differentiation efficacy of human dental pulp stromal cells. International Journal of Molecular Sciences, 22(10), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks, P. A. (2010). The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opinion on Investigational Drugs, 19(9), 1049–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng, Yu‐B. , Li, X. , Li, Z. ‐. Y. , Zhao, J. , Yuan, Xu‐Bo , Ren, Yu , Cui, Z. ‐. D. , Liu, Y‐De , & Yang, X. ‐. J. (2015). Microrna‐21 promotes osteogenic differentiation of mesenchymal stem cells by the pi3k/‐catenin pathway. Journal of Orthopaedic Research, 33(7), 957–964. [DOI] [PubMed] [Google Scholar]
- Minamizaki, T. , Nakao, Y. , Irie, Y. , Ahmed, F. , Itoh, S. , Sarmin, N. , Yoshioka, H. , Nobukiyo, A. , Fujimoto, C. , Niida, S. , Sotomaru, Y. , Tanimoto, K. , Kozai, K. , Sugiyama, T. , Bonnelye, E. , Takei, Y. , & Yoshiko, Y. (2020). The matrix vesicle cargo mir‐125b accumulates in the bone matrix, inhibiting bone resorption in mice. Communications Biology, 3(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minetti, G. C. , Colussi, C. , Adami, R. , Serra, C. , Mozzetta, C. , Parente, V. , Fortuni, S. , Straino, S. , Sampaolesi, M. , Di Padova, M. , Illi, B. , Gallinari, P. , Steinkühler, C. , Capogrossi, M. C. , Sartorelli, V. , Bottinelli, R. , Gaetano, C. , & Puri, P. L. (2006). Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nature Medicine, 12(10), 1147–1150. [DOI] [PubMed] [Google Scholar]
- Musselman, C. A. , & Kutateladze, T. G. (2011). Handpicking epigenetic marks with phd fingers. Nucleic Acids Research, 39(21), 9061–9071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'brien, K. , Breyne, K. , Ughetto, S. , Laurent, L. C. , & Breakefield, X. O. (2020). RNA delivery by extracellular vesicles in mammalian cells and its applications. Nature Reviews Molecular Cell Biology, 21(10), 585–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikravesh, N. , Davies, O. G. , Azoidis, I. , Moakes, R. J. A. , Marani, L. , Turner, M. , Kearney, C. J. , Eisenstein, N. M. , Grover, L. M. , & Cox, S. C. (2019). Physical structuring of injectable polymeric systems to controllably deliver nanosized extracellular vesicles. Advanced Healthcare Materials, 8(9), 1801604, 1–11. [DOI] [PubMed] [Google Scholar]
- Paino, F. , La Noce, M. , Tirino, V. , Naddeo, P. , Desiderio, V. , Pirozzi, G. , De Rosa, A. , Laino, L. , Altucci, L. , & Papaccio, G. (2014). Histone deacetylase inhibition with valproic acid downregulates osteocalcin gene expression in human dental pulp stem cells and osteoblasts: Evidence for hdac2 involvement. Stem Cells, 32(1), 279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, Y. H. , Wang, L. , Gao, Z. L. , Chen, G. Y. , & Zhang, C. Q. (2016). Bone marrow stromal/stem cell‐derived extracellular vesicles regulate osteoblast activity and differentiation in vitro and promote bone regeneration in vivo. Scientific Reports, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo, G. , & Stoorvogel, W. (2013). Extracellular vesicles: Exosomes, microvesicles, and friends. Journal of Cell Biology, 200(4), 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Suarez, E. H. C. , Gethings, L. , Giles, K. , Wildgoose, J. , Stapels, M. , Fadgen, K. E. , Geromanos, S. J. , Vissers, J. P.C. , Elortza, F. , & Langridge, J. I. (2013). An ion mobility assisted data independent lc‐ms strategy for the analysis of complex biological samples. Current Analytical Chemistry, 9(2), 199–211. [Google Scholar]
- Rohde, E. , Pachler, K. , & Gimona, M. (2019). Manufacturing and characterization of extracellular vesicles from umbilical cord‐derived mesenchymal stromal cells for clinical testing. Cytotherapy, 21(6), 581–592. [DOI] [PubMed] [Google Scholar]
- Sahoo, S. , & Losordo, D. W. (2014). Exosomes and cardiac repair after myocardial infarction. Circulation Research, 114(2), 333–344. [DOI] [PubMed] [Google Scholar]
- Sainitya, R. , Sriram, M. , Kalyanaraman, V. , Dhivya, S. , Saravanan, S. , Vairamani, M. , Sastry, T. P. , & Selvamurugan, N. (2015). Scaffolds containing chitosan/carboxymethyl cellulose/mesoporous wollastonite for bone tissue engineering. International Journal of Biological Macromolecules, 80, 481–488. [DOI] [PubMed] [Google Scholar]
- Sandonà, M. , Consalvi, S. , Tucciarone, L. , De Bardi, M. , Scimeca, M. , Angelini, D. F. , Buffa, V. , D'amico, A. , Bertini, E. S. , Cazzaniga, S. , Bettica, P. , Bouché, M. , Bongiovanni, A. , Puri, P. L. , & Saccone, V. (2020). Hdac inhibitors tune mirnas in extracellular vesicles of dystrophic muscle‐resident mesenchymal cells. Embo Reports, 21(9), 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder, T. M. , Kahler, R. A. , Li, X. , & Westendorf, J. J. (2004). Histone deacetylase 3 interacts with runx2 to repress the osteocalcin promoter and regulate osteoblast differentiation. Journal of Biological Chemistry, 279(40), 41998–42007. [DOI] [PubMed] [Google Scholar]
- Schroeder, T. M. , & Westendorf, J. J. (2005). Histone deacetylase inhibitors promote osteoblast maturation. Journal of Bone and Mineral Research, 20(12), 2254–2263. [DOI] [PubMed] [Google Scholar]
- Sharma, A. (2014).Novel transcriptome data analysis implicates circulating micrornas in epigenetic inheritance in mammals. Gene, 538(2), 366–372. [DOI] [PubMed] [Google Scholar]
- Shimazu, J. , Wei, J. , & Karsenty, G. (2016). Smurf1 inhibits osteoblast differentiation, bone formation, and glucose homeostasis through serine 148. Cell Reports, 15(1), 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, P. , Tian, Ye , Yang, C. , Ma, X. , Wang, X. , Pei, J. , & Qian, A. (2018). Mesenchymal stem cell migration during bone formation and bone diseases therapy. International Journal of Molecular Sciences, 19(8), 2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, X. , Liao, L. , Shuai, Y. , Jing, H. , Liu, S. , Zhou, H. , Liu, Y. , & Jin, Y. (2015). Mir‐26a functions oppositely in osteogenic differentiation of bmscs and adscs depending on distinct activation and roles of wnt and bmp signaling pathway. Cell Death & Disease, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, D. Z. , Abelson, B. , Babbar, P. , & Damaser, M. S. (2019). Harnessing the mesenchymal stem cell secretome for regenerative urology. Nature Reviews Urology, 16(6), 363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, S. H. S. , Wong, J. R. Y. , Sim, S. J. Y. , Tjio, C. K. E. , Wong, K. L. , Chew, J. R. J. , Hui, J. H. P. , & Toh, W. S. (2020). Mesenchymal stem cell exosomes in bone regenerative strategies‐a systematic review of preclinical studies. Materials Today Bio, 7, 100067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, X. L. , Lin, J. T. , Wang, G. H. , & Lu, J. L. (2017). Microrna‐433‐3p promotes osteoblast differentiation through targeting dkk1 expression. Plos One, 12(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao, S. ‐. C. , Guo, S. ‐. C. , Li, M. , Ke, Q. ‐. F. , Guo, Ya‐P. , & Zhang, C. ‐. Q. (2017). Chitosan wound dressings incorporating exosomes derived from microrna‐126‐overexpressing synovium mesenchymal stem cells provide sustained release of exosomes and heal full‐thickness skin defects in a diabetic rat model. Stem Cells Translational Medicine, 6(3), 736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry, C. , Witwer, K. W. , Aikawa, E. , Alcaraz, M. J. , Anderson, J. D. , Andriantsitohaina, R. , Antoniou, A. , Arab, T. , Archer, F. , Atkin‐Smith, G. K. , Ayre, D. C. , Bach, J. ‐. M. , Bachurski, D. , Baharvand, H. , Balaj, L. , Baldacchino, S. , Bauer, N. N. , Baxter, A. A. , Bebawy, M. … Zuba‐Surma, E. K. (2018). Minimal information for studies of extracellular vesicles 2018 (misev2018): A position statement of the international society for extracellular vesicles and update of the misev2014 guidelines. Journal of Extracellular Vesicles, 7, 1535750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vimalraj, S. , Partridge, N. C. , & Selvamurugan, N. (2014). A positive role of microrna‐15b on regulation of osteoblast differentiation. Journal of Cellular Physiology, 229(9), 1236–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachos, I. S. , Kostoulas, N. , Vergoulis, T. , Georgakilas, G. , Reczko, M. , Maragkakis, M. , Paraskevopoulou, M. D. , Prionidis, K. , Dalamagas, T. , & Hatzigeorgiou, A. G. (2012). Diana mirpath v.2.0: Investigating the combinatorial effect of micrornas in pathways. Nucleic Acids Research, 40(W1), W498–W504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C. ‐ A. , Li, C. ‐ F. , Huang, R. ‐ C. , Li, Y. ‐ H. , Liou, J. ‐ P. , & Tsai, S. ‐ J. (2020). A novel hdac inhibitor suppresses extracellular vesicle vegf‐c‐mediated lymphangiogenesis and pancreatic cancer early dissemination. Research Square. [DOI] [PubMed] [Google Scholar]
- Wang, R. K. , Zhang, H. , Ding, W. B. , Fan, Z. Y. , Ji, B. Y. , Ding, C. , Ji, F. , & Tang, H. (2020). Mir‐143 promotes angiogenesis and osteoblast differentiation by targeting hdac7. Cell Death & Disease, 11(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Omar, O. , Vazirisani, F. , Thomsen, P. , & Ekström, K. (2018). Mesenchymal stem cell‐derived exosomes have altered microrna profiles and induce osteogenic differentiation depending on the stage of differentiation. Plos One, 13(2), e0193059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, Y. , Tang, C. , Zhang, J. , Li, Z. , Zhang, X. , Miron, R. J. , & Zhang, Y. (2019). Extracellular vesicles derived from the mid‐to‐late stage of osteoblast differentiation markedly enhance osteogenesis in vitro and in vivo. Biochemical and Biophysical Research Communications, 514(1), 252–258. [DOI] [PubMed] [Google Scholar]
- West, A. C. , & Johnstone, R. W. (2014). New and emerging hdac inhibitors for cancer treatment. Journal of Clinical Investigation, 124(1), 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijnen, A. J. , & Westendorf, J. J. (2019). Epigenetics as a new frontier in orthopedic regenerative medicine and oncology. Journal of Orthopaedic Research, 37(7), 1465–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf, A. D. (2007). An update on glucocorticoid‐induced osteoporosis. Current Opinion in Rheumatology, 19(4), 370–375. [DOI] [PubMed] [Google Scholar]
- Xin, H. , Li, Yi , & Chopp, M. (2014). Exosomes/mirnas as mediating cell‐based therapy of stroke. Frontiers in Cellular Neuroscience, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, J. , Wang, Y. , Hsu, C. ‐. Y. , Gao, Y. , Meyers, C. A. , Chang, L. , Zhang, L. , Broderick, K. , Ding, C. , Peault, B. , Witwer, K. , & James, A. W. (2019). Human perivascular stem cell‐derived extracellular vesicles mediate bone repair. Elife, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, S. , Guo, R. , Li, P. ‐. Z. , Li, Ke , Yan, Yu , Chen, J. , Wang, G. , Brand‐Saberi, B. , Yang, X. , & Cheng, X. (2019). Dexamethasone interferes with osteoblasts formation during osteogenesis through altering igf‐1‐mediated angiogenesis. Journal of Cellular Physiology, 234(9), 15167–15181. [DOI] [PubMed] [Google Scholar]
- Yang, J. ‐. W. , Jeong, B. ‐. C. , Park, J. , & Koh, J. ‐. T. (2017). Phf20 positively regulates osteoblast differentiation via increasing the expression and activation of runx2 with enrichment of h3k4me3. Science Reports, 7(1), 8060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, N. , Wang, G. , Hu, C. , Shi, Y. , Liao, Li , Shi, S. , Cai, Y. , Cheng, S. , Wang, Xi , Liu, Y. , Tang, L. , Ding, Y. , & Jin, Y. (2013). Tumor necrosis factor suppresses the mesenchymal stem cell osteogenesis promoter mir‐21 in estrogen deficiencyinduced osteoporosis. Journal of Bone and Mineral Research, 28(3), 559–573. [DOI] [PubMed] [Google Scholar]
- Yoshiko, Y. , & Minamizaki, T. (2020). Emerging roles of micrornas as extracellular vesicle cargo secreted from osteoblasts. Journal of Oral Biosciences, 62(3), 228–234. [DOI] [PubMed] [Google Scholar]
- Zhao, J. W. , Sun, Y. , Huang, Y. , Song, F. , Huang, Z. S. , Bao, Y. F. , Zuo, J. , Saffen, D. , Shao, Z. , & Liu, W. et al. (2017). Functional analysis reveals that rbm10 mutations contribute to lung adenocarcinoma pathogenesis by deregulating splicing. Scientific Reports, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, R. , Li, Y. , Lin, Z. , Wan, J. , Xu, C. , Zeng, Y. , & Zhu, Y. (2016). Mir‐199b‐5p modulates bmsc osteogenesis via suppressing gsk‐3 beta/beta‐catenin signaling pathway. Biochemical and Biophysical Research Communications, 477(4), 749–754. [DOI] [PubMed] [Google Scholar]
- Zheng, H. , Liu, J. , Tycksen, E. , Nunley, R. , & Mcalinden, A. (2019). Microrna‐181a/b‐1 over‐expression enhances osteogenesis by modulating pten/pi3k/akt signaling and mitochondrial metabolism. Bone, 123, 92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.