

Summary
Enriched tumor epithelium, tumor-associated stroma, and whole tissue were collected by laser microdissection from thin sections across spatially separated levels of ten high-grade serous ovarian carcinomas (HGSOCs) and analyzed by mass spectrometry, reverse phase protein arrays, and RNA sequencing. Unsupervised analyses of protein abundance data revealed independent clustering of an enriched stroma and enriched tumor epithelium, with whole tumor tissue clustering driven by overall tumor “purity.” Comparing these data to previously defined prognostic HGSOC molecular subtypes revealed protein and transcript expression from tumor epithelium correlated with the differentiated subtype, whereas stromal proteins (and transcripts) correlated with the mesenchymal subtype. Protein and transcript abundance in the tumor epithelium and stroma exhibited decreased correlation in samples collected just hundreds of microns apart. These data reveal substantial tumor microenvironment protein heterogeneity that directly bears on prognostic signatures, biomarker discovery, and cancer pathophysiology and underscore the need to enrich cellular subpopulations for expression profiling.
Subject areas: Oncology, Cancer systems biology, Proteomics, Transcriptomics
Graphical abstract
Highlights
-
•
LMD was used to investigate 3-D molecular heterogeneity in ovarian cancer tissue
-
•
Diverse molecular profiles were identified from 3-D spatially separated samples
-
•
Molecular heterogeneity impacts HGSOC prognostic sub-type assignment
-
•
Proteomic heterogeneity analysis web portal deployed at www.lmdomics.org
Oncology; Cancer systems biology; Proteomics; Transcriptomics
Introduction
Ovarian cancer is the fifth leading cause of death in women in the U.S. (Siegel et al., 2020), with nearly 22,000 new cases and 14,000 deaths projected to occur in 2021. Most ovarian cancer cases are diagnosed at an advanced stage and exhibit heterogeneous populations and subpopulations of tumor cells that limit successful therapeutic intervention. The 5-year survival rate of patients diagnosed with metastatic ovarian cancer is less than 30% (Noone Am et al., 2018).
Numerous high-throughput sequencing studies aimed at broadly characterizing the genomic landscape of specific cancer types have been or are being conducted (The Cancer Genome Atlas Research Network, 2011). Although adding substantially to our molecular understanding of cancer, these studies have resulted in limited clinical translation, due in part to an ever increasing body of evidence that points to previously underappreciated levels of heterogeneity in the tumor microenvironment (TME) (Chen et al., 2018).
Substantial molecular and pathologic intra-tumoral differences in high-grade serous ovarian carcinoma (HGSOC) tumors and their relationship with TME dynamics have been described (Jimenez-Sanchez et al., 2017; Schwarz et al., 2015). One study demonstrated that stable and/or regressing tumors lacked common neoepitopes and mutations compared to progressing tumors in the same patient (Jimenez-Sanchez et al., 2017), implicating non-somatic factors within the TME as critical determinants of immune response and overall tumor fate. Multiregional sampling has revealed extensive variation between subpopulations of cells within a single tumor (Shih et al., 2018; Zhang et al., 2018; Bashashati et al., 2013), suggesting individual tumor samples have multiple subtype signatures present with differing levels of activation (Verhaak et al., 2013). Single-cell RNA-seq of HGSOC samples has revealed grade-specific and cell-type-specific transcriptional profiles representative of individual cell types, such as tumor epithelium, stromal cells, and immune cells present within individual tumor specimens (Shih et al., 2018). Further, the presence of subclonal cell populations within primary and/or metastatic tumors has been demonstrated to influence the state of immune infiltration and activation (Zhang et al., 2018).
In this study, we investigated proteomic and transcriptomic 3-dimensional (3D) heterogeneity in the TME of patients with HGSOC tumors using laser microdissection (LMD) of spatially separated tumor-enriched regions, stromal cell populations, as well as whole tumor collections throughout distinct levels from each specimen block. These results reveal stark molecular heterogeneity in the HGSOC TME and underscore the need to account for both compartmental and multi-level heterogeneity in molecular profiling analyses of HGSOCs. The translational relevance of these results for clinical care cannot be understated, as we show variable distribution of disease and/or therapeutic biomarkers in both tumor and stroma, and that the ability to detect these markers is subject to regional variations in local expression levels. We have also created an interactive web portal for interrogation of these proteomic data (https://lmdomics.org/HGSOCheterogeneity/), allowing users to examine the intratumoral heterogeneity of expression for proteins of interest.
Results
Molecular analyses of LMD-enriched cellular populations within HGSOC tumors using locoregional multi-sampling
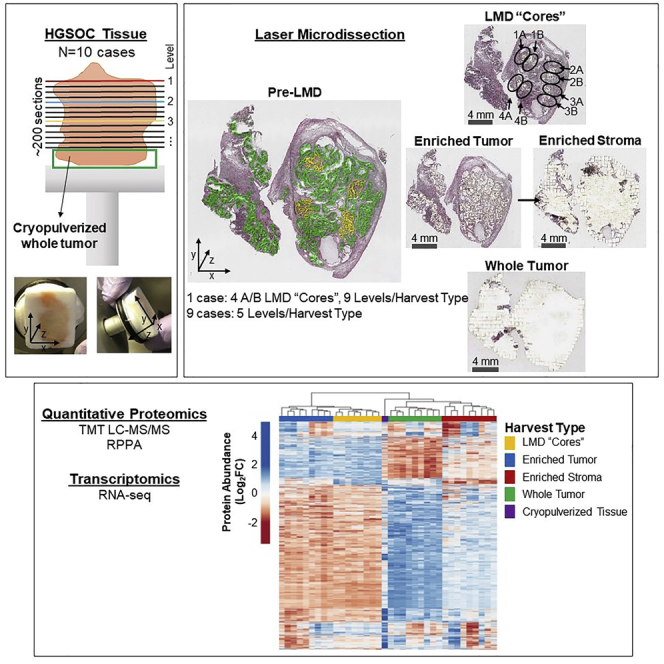
Consecutive thin sections (∼200) were generated from fresh frozen HGSOC tumors (n = 10 patients) to support multi-region sampling by LMD followed by integrated quantitative proteomic and transcriptomic analyses (Figures 1A and S1). All patients had advanced stage (stage III or IV) ovarian or tubo-ovarian disease (Table S1). Specimens were obtained from ovarian and/or tubo-ovarian tissue and other pelvic masses to include the omentum. Some of the patients were chemotherapy naïve (n = 6) at the time of surgical resection, while others had received neoadjuvant chemotherapy (n = 4). Representative hematoxylin and eosin (H&E) stained sections were reviewed by a board-certified pathologist (U.N.M.R.) to confirm histologic characteristics and tumor cellularity within each level (Table S1).
Figure 1.

Study workflow
Illustration of histological tissue preparation, laser microdissection, proteomic analyses, and transcriptomic analysis (A), with representative pre-LMD and post-LMD images from the top and bottom levels of the tissue from Case 343WM (B). (A) Specimen blocks obtained from 10 patients with HGSOC were each cut into ∼200 consecutive thin tissue sections (left ), which were laser microdissected for enrichment of tumor epithelium, stroma, or whole tumor collections (middle panel) for analysis via quantitative proteomics and transcriptomics (right panel). One case (343WM; middle panel, top) was uniquely used for laser microdissection (LMD) enrichment of four tumor epithelium cores with adjacent replicate regions from each of 100 or 50 slides evenly distributed through the depth of the specimen for MS proteomics or transcriptomics, respectively. For 343WM, additional sets of 9 slides from spatially separated levels within the specimen block were each microdissected for all remaining tumor and stroma after collecting the cores by LMD, as well as a nearest neighboring whole tumor collection. The remainder of the specimen was cryopulverized in liquid nitrogen. The specimen blocks from the remaining 9 cases (middle panel, bottom) were divided into 5 levels (quintiles) of equal depth. Within each level, interlaced sections were used for LMD enrichment of tumor epithelium, stroma, and whole tumor collections for each downstream analytical purpose. Proteins and transcripts isolated from each of these distinct collections were analyzed by isobaric tagging and high-resolution liquid chromatography-tandem mass spectrometry, reverse phase protein microarray, and/or next generation sequencing. (B) Representative pre-LMD and post-LMD images are shown for 343WM from tissue sections at the top and bottom levels used for proteomic analysis. The number in the bottom right corner of each micrograph indicates the slide number shown. The scale bar in the bottom left corner of each micrograph indicates a length of 4 mm.
Enriched tumor epithelium, stromal cells, as well as whole tumor collections representing all material on a single tissue section, were collected at spatially distinct intervals (“levels”) from alternating sections throughout the tumor blocks (Figure 1A; Table S2). In case 343WM, additional LMD collections were conducted to generate spatially separated “cores” (with additional technical replicate “cores”) representing defined sub-populations of the tumor epithelium from alternating tissue sections that spanned the entirety of the patient tumor block from four spatially diverse quadrants (i.e., 1–4) and were combined into four independent sample sets, including close biological collection “replicates” (i.e. 1A, 1B, 2A, 2B, etc) to support comparative proteome and transcriptome analyses (Figure 1A). Representative polyethylene naphthalate membrane slides after LMD were imaged, along with adjacent H&E stained tissue sections (Figure 1B), to enable co-registration and automated quantification of tumor and stromal cell populations collected (Table S3). There were marked differences regarding estimation of tumor purity between automated and manual evaluations. Automated digital image analysis estimated 23–56% tumor cellularity at different depths of the block, calculated based on tumor regions of interest collected by LMD/whole tissue area (Table S3). Automated estimates revealed an average cell number of 7,527 ± 172 for tumor cells (n = 15 collections) and 3,899 ± 263 for stroma cells (n = 6 collections) collected by LMD per mm2 of tissue area while manual review estimated 75–95% tumor cellularity (Table S1). Differences between automated and manual review of tumor cellularity can likely be attributed to the presence of small regions of interceding stroma and other cell types in and around the tumor epithelium, which were excluded from the LMD tumor epithelium collections.
Multi-region protein samples were analyzed by mass spectrometry-based, quantitative proteomics using a multiplexed, isobaric tagging methodology (tandem mass tags, TMT-10 or TMTpro-16; Table S4) and reverse phase protein microarray (RPPA). RNA samples were analyzed by targeted RNA-seq analyses. For 343WM, a total of 6,053 proteins (Table S5) and 20,535 transcripts (Table S6) were quantified across all samples. For the remaining 9 cases, an average of 9,223 ± 511 proteins were measured within each patient-specific TMT multiplex, and 6,199 proteins were co-quantified across all 9 patients (Table S7). We further generated RNA-seq analyses for cases 343WB and 343WH and quantified over 19,000 transcripts (Table S8). We made the merged global protein matrix generated from the 9 patients available as a publicly accessible database which is accessible at https://lmdomics.org/HGSOCheterogeneity/. The goal of providing this resource is to support biomarker discovery and investigations into cancer pathophysiology by enabling researchers to investigate protein(s) of interest between LMD-enriched cell populations of tumor and stroma versus whole tumor, as well as relating to local abundance variability spanning the 5 tissue levels from each case, and the conservation of this heterogeneity across these patients.
Tumor and stromal cell populations exhibit diverse molecular profiles and regional variability
Unsupervised hierarchical cluster analysis and principal component analysis (PCA) of variably abundant proteins (Table S9) or transcripts (Table S10) from case 343WM revealed distinct sub-clustering of tumor cores, enriched tumor epithelium and stroma, as well as whole tumor collections (Figures 2A–2D). Two predominant clusters stratifying tumor cores and the tumor epithelium from stroma and whole tumor collections are evident. Notably, the cryopulverized tissue proteome exhibited an intermediate cluster between these two sample groups (Figures 2A and 2B), suggesting this sample type represents a mixture of these cellular populations. The proteins measured in the cryopulverized tissue also reflect contributions from the inclusion of blood, necrosis, and fat within this sample, which had been excluded from the whole tumor collections.
Figure 2.

Unsupervised hierarchical cluster analysis and principal component analysis of LMD-enriched samples by differentially abundant protein and transcript expression
(A, B) 1,928 differentially abundant proteins and (C, D) 3,861 transcripts with median absolute deviation (MAD) > 0.5 from case 343WM and (E, F) 6,199 differentially abundant proteins with MAD>1 in the 9-patient specimen set. (E, F) Protein abundances are represented across n = 123 samples derived from n = 9 patients consisting of LMD-enriched tumor epithelium (n = 45 total; 5 levels/patient), LMD-enriched stroma (n = 33 total; 2–5 levels/patient), and whole tumor (n = 45 total; 5 levels/patient) samples. Highlighted box depicts the median tumor purity values from manual pathology review for each of the whole tumor collections as they appear in the heatmap, ordered from left to right. Dark elongated border line distinguishes the whole tumor collections which clustered with LMD-enriched stroma (left) from those that clustered with LMD-enriched tumor (right). Red and blue “X” marks represent interceding LMD-enriched stroma or tumor collections, respectively.
Similar clustering was seen across tissue cell-type collections of the additional 9 patients with HGSOC. Median Pearson correlations between the LMD-enriched samples from 343WM and the additional 9 cases were 0.68 and 0.24 for the tumor and stroma, respectively. The lower correlation of LMD-enriched stroma samples between these datasets is likely related to the high and variable number of admixed stromal cells, such as cancer-associated fibroblasts (CAFs) and immune cells, within this microenvironment (He et al., 2021; Winterhoff et al., 2017; Verardo et al., 2014; Jiménez-Sánchez et al., 2020). Unsupervised hierarchical cluster analysis of proteins (Table S11; Figures 2E and 2F) and transcripts (Table S12; Figures S2A and S2B) revealed distinct clustering of the LMD-enriched tumor epithelium from the LMD-enriched stroma. Importantly, the extent to which the whole tumor tissue collection more closely associated with tumor epithelium or stroma was directly related to the tumor “purity,” namely, whole tumor tissues with a high percentage of tumor epithelium clustered with LMD-enriched tumor epithelium and those with a lower percentage of tumor epithelium clustered more strongly with LMD-enriched stroma (Table S1).
We next examined the abundance of representative epithelial cell markers, i.e., Keratin Type I Cytoskeletal 19 (KRT19) (Shih et al., 2018), and Cadherin 1 (CDH1) (Rosso et al., 2017), and ovarian cancer-related tumor markers CA125/Mucin-16 (CA-125/MUC16) (Feeley and Wells, 2001), as well as representative stromal markers versican (VCAN) (Ghosh et al., 2010) and fibroblast activation protein alpha (FAP) (Scanlan et al., 1994) across each tissue collection type (Figure 3A). As anticipated, CDH1 and KRT19 protein and transcript abundances were higher in tumor cores and LMD-enriched tumor epithelial collections and comparatively decreased in LMD-enriched stroma and whole tumor collections. VCAN and FAP abundance, in contrast, was elevated in LMD-enriched stroma and whole tumor collections, consistent with these proteins being stromal in origin (Yang et al., 2016; Ghosh et al., 2010). While the overall expression trends were consistent across all levels or cores for a given analyte in the LMD-enriched tumor or stroma collections, substantial regional variability, particularly in protein abundance, between different levels was also apparent. Patient-specific relative standard deviations (RSDs) were calculated to measure the heterogeneity of protein expression between collection levels within each LMD-enriched tumor compartment. The median across all levels/patients of KRT19 and CDH1 protein abundance measurements was 62.3% and 47.4% in the LMD-enriched tumor collections, respectively, and 29.7% and 35.5% for VCAN and FAP in the LMD-enriched stroma collections, respectively. Cell-type enrichment analyses using the transcript and protein data from 343WM were performed (Table S13) (Aran et al., 2017), and except for Cores 4A/B, all of the LMD tumor cores and LMD-enriched tumor epithelium collections correlated most strongly with epithelial cell admixture signatures, while the LMD-enriched stroma and whole tumor collections had the highest correlation with fibroblast and stroma cell signatures, as well as microenvironment scores (Figure 3A). Overall, these highly significant (p < 0.0001) associations between epithelial and stromal marker proteins KRT19, CDH1, VCAN, and FAP with LMD-enriched tumor epithelium and stroma, respectively, were observed using the proteomic data from the additional 9 HGSOC cases assessed (Figure 3B). Further, relative abundance of these proteins was largely more consistent across tumor or stroma collections in comparison with whole tumor collections likely as product intratumoral tumor heterogeneity. Cell type and microenvironment gene signature scores were also significantly different in the additional 9 patients with HGSOC tumors assessed, in which LMD-enriched tumor was again enriched for epithelial cells, while LMD-enriched stroma had the highest scores for fibroblasts, stroma, and microenvironment (p < 0.0001; Figure 3C; Table S14).
Figure 3.

Protein and transcript abundance of epithelial and stromal markers in HGSOC as well as cellular admixture analyses (xCell (Aran et al., 2017))
(A) Heatmaps depicting protein and transcript abundances and cellular admixture enrichment scores from 343WM for replicate tumor cores and by depth within the specimen block for LMD-enriched tumor, stroma, and whole tumor collections. Protein abundance from the cryopulverized tumor is included. Size and color of each shape reflects Spearman correlation. (B) Boxplots depicting relative protein abundances for KRT19, CDH1, FAP, and VCAN and (C) cellular admixture scores in the n = 9 patient specimen set. ES = LMD-enriched stroma; ET = LMD-enriched tumor; WT = whole tumor. p values with (∗) indicate statistically significant differential expression (p < 0.0001) between ES and ET.
We additionally examined the abundance of MUC16 which was significantly enriched in the LMD-enriched tumor relative to stroma (p < 0.01; Figure S3) in the proteomic data overall for the additional 9 patients. Uniquely, however, MUC16 was elevated in LMD-enriched stroma for cases 343VT and 343WQ. CA-125 is monitored as a clinical biomarker in serum for ovarian cancer progression and regression (Vergote et al., 1987; Bast et al., 1983; Canney et al., 1984) and is an epitope of the extracellular domain of the MUC16 protein (Yin et al., 2002; O’Brien et al., 2001, 2002) which can be shed from HGSOC cells via proteolytic cleavage (Fendrick et al., 1993). We hypothesized that the abundance of proteins shed or secreted from tumor cells may exhibit greater variability within the TME due to circulation within interstitial spaces as well as local vasculature. We therefore compared abundance trends of proteins exhibiting a signal peptide sequence targeting a protein for secretion or that are characterized as being localized to the extracellular space by Gene Ontology, relative to proteins lacking these characteristics across all collection levels to assess whether the variance in protein abundance across levels was higher in proteins with signal peptides or which are putatively localized to the extracellular space. Cases 343WM, 343WK, 343WQ, and 343WH were selected for this analysis as proteomic data from all levels/case of each LMD-enriched tumor and stroma were available. We found that proteins with signal peptide sequences had significantly higher variance (p < 0.0001) than proteins lacking these sequences (Table S15) and further that the variance of extracellular proteins was also significantly higher (p < 0.001) than that of those lacking this classification.
Integrated analyses of protein and transcript data revealed 5,742 co-quantified proteins and transcripts from 343WM across all tissue types (Figure 4; Table S16). Proteins and their transcripts for a given LMD collection type showed positive correlation with other samples from the same LMD collection type (e.g. Core 1A protein clustered most strongly with its corresponding transcript and next nearest replicate core collection). Notably, stromal transcripts clustered most strongly with their cognate proteins from the whole tumor collections. In comparison of replicate tumor cores, the strongest correlations, apart from intra-core comparisons, were between the “A” and “B” replicates for each core. The A and B replicates for each core at the proteome-only and transcriptome-only levels had Spearman rho values between 0.65–0.91 and 0.75–0.84, respectively. For replicate cores at the protein and corresponding transcript levels, the Spearman rho values were 0.38–0.51. Correlation of protein and transcript abundance in other patient tumors showed 5,379 co-quantified proteins and transcripts measured from LMD-enriched tumor, stroma, and whole tumor from one level each for cases 343WH and 343WB (Table S17). Correlations were strongest within the same LMD collection type (Figure S4). For 343WH, the LMD-enriched stroma proteins correlated most strongly with the corresponding LMD-enriched stroma transcripts but were also positively correlated with whole tumor transcripts, consistent with 343WM. The tumor cellularity was marginally higher in 343WH (median tumor cellularity 85–95%) relative to 343WM (median tumor cellularity 80–95%) (Table S1), which likely accounts for differences in relatedness between the LMD-enriched stroma protein and either whole tumor RNA or LMD-enriched stroma RNA.
Figure 4.

Protein-RNA Spearman correlation matrix for case 343WM
Spearman correlation analysis of 5,742 genes that were co-measured as proteins and corresponding transcripts in 343WM. Size and color of each circle reflects Spearman correlation.
We further measured 281 antibodies (Table S18) against native and/or post-translationally modified proteins for RPPA analysis (Table S19). PCA clustering of LMD-enriched samples using 63 differentially abundant proteins with median absolute deviation (MAD) >1 revealed similar results as seen using the mass spectrometry (MS)-based proteomic and RNA-seq transcriptomic datasets; the RPPA samples clustered independently by LMD collection type, with the whole tumor collections having mixed relatedness to enriched collections depending on overall tumor purity (Figure S5). Integrated analyses of MS-based and RPPA proteome data (Table S20) revealed 54 proteins as co-quantified between platforms and showed similar findings as above indicating generally positive correlation of protein abundances between LMD-enriched tumor (average Spearman rho: 0.252 ± 0.138; Figure S6A) and stroma (average Spearman rho: 0.400 ± 0.097; Figure S6B) across all cases, with the exceptions of LMD-enriched tumor from 343VT and, to a lesser extent, 343WH showing mixed relatedness to other cases. However, the whole tumor collections (Figure S6C) displayed markedly poorer concordance (average Spearman rho: 0.006 ± 0.156) between the two analytical platforms.
Comparison of proteomic and transcriptomic data from case 343WM revealed common canonical pathways (ingenuity pathway analysis) that were consistently altered within LMD collection types (Table S21). Aryl hydrocarbon receptor signaling and the endocannabinoid cancer inhibition pathway were among the top 5 canonical pathways most activated in LMD-enriched tumor epithelium relative to stroma at based on protein and transcript abundances. Conversely, IL-8 signaling and GP6 signaling were among the pathways predicted to be inhibited, i.e. least activated in enriched tumor epithelium relative to stroma. Additional pathways differentially altered between tumor and stroma included activation of organismal death, morbidity and mortality, bleeding, organization of the cytoplasm and skeleton, as well as cell movement and migration in both proteome and transcriptome data sets (Table S22). Genes encoding known targets of Food and Drug Administration (FDA)-approved drugs (Sun et al., 2017) which were differentially expressed (linear models for microarray data adjusted p value <0.01) between the LMD-enriched tumor and LMD-enriched stroma were identified in the proteomic and transcriptomic data with Log2 fold changes > ±1 (Table S23). Drugs targeting the identified genes include a mixture of those being used currently for clinical management of HGSOC disease, such as bevacizumab (a monoclonal antibody against vascular endothelial growth factor (VEGF) (Krämer and Lipp, 2007)), as well as many drugs that are not.
For the additional 9 HGSOC cases, we examined pathway enrichment using data for 381 proteins which displayed the same pattern of enrichment across all cases and which had Log2 fold changes > ±1 (Table S24). Overall, the canonical pathways (Tables S25 and S26) enriched in LMD tumor epithelium relative to stroma generally overlapped with those from case 343WM suggesting these pathways are highly enriched within tumor or stroma cell populations of HGSOC tumors. The proteomic data from these 9 cases revealed that the protein target of FDA-approved olaparib (PARP1) was enriched in tumor, while targets of estramustine, sorafenib, regorafenib, and pazopanib (MAP1A and PDGFRB) were enriched in stroma (Table S27).
Intratumoral molecular heterogeneity impacts prognostic molecular signatures correlating with altered disease outcome in HGSOC
Several large-scale studies have aimed to identify prognostic molecular signatures for outcomes of patients with HGSOC, most of which have been from genomic and/or transcriptomic data from whole tumor specimen preparations from tumors that are typically qualified for inclusion using tumor cellularity/purity thresholds determined from pathologic inspection (The Cancer Genome Atlas Research Network, 2011; Konecny et al., 2014; Tucker et al., 2014; Liu et al., 2015; Leong et al., 2015; Wang et al., 2017; Verhaak et al., 2013). Per the categorization of tumors into immunoreactive, differentiated, proliferative, and mesenchymal subtypes, patients whose tumors are characterized by mesenchymal signatures have the worst prognosis. Consensus signatures generated by Chen et al (Chen et al., 2018) represent molecular subtype classifications and a random forest probability for composite classifiers, i.e. consensusOV generated from four previously published HGSOC molecular subtype classification systems (Helland et al., 2011; Verhaak et al., 2013; Konecny et al., 2014; Chen et al., 2018; Bentink et al., 2012). We used these consensus signatures to examine correlations between each spatially resolved tissue collection type relative to these HGSOC prognostic subtypes (Figure 5; Table S28; Table S29). This analysis revealed that transcripts from LMD-enriched tumor cores and tumor epithelium from case 343WM were strongly correlated with differentiated and/or proliferative molecular subtypes (Figure 5A). Conversely, transcripts from the LMD-enriched stroma most strongly correlated to the prognostically poor mesenchymal subtype. Molecular subtype profiles for enriched tumor and stroma were largely recapitulated using proteome data for case 343WM (Figure 5A) and in the additional 9 HGSOC cases (Figure 5B). The whole tumor collections showed variable correlations across molecular subtypes, the extent to which seemingly depended on the percent stromal and tumor epithelium admixture contributions to overall tumor purity.
Figure 5.

Protein and transcript abundance of markers correlating with prognostic molecular signatures of altered disease outcome in HGSOC
(A) Transcript abundances for markers correlated with suboptimal surgical debulking identified by Liu et al (2015) (Liu et al., 2015), and protein and transcript abundances correlating with prognostic molecular subtypes identified by Konecny et al (2014) (Konecny et al., 2014) measured in 343WM. (B) Stacked bar graphs depicting the probability of each collection type (LMD-enriched stroma, LMD-enriched tumor, and whole tumor) per level per patient belonging to consensus molecular subtypes identified by Chen et al (2018) (Chen et al., 2018) based on protein abundance. (C) Boxplots depicting the distributions of margin classification thresholds across all levels from the proteomic data from the 9-patient dataset for each collection type. Significance determined by a post hoc Tukey honestly significant difference test indicated that all three groups were significantly different from each other. Both LMD-enriched stroma and LMD-enriched tumor differed from whole tumor (∗∗∗; p < 0.001). Margin means were 0.609, 0.460, and 0.238 for the LMD-enriched stroma, LMD-enriched tumor, and whole tumor, respectively.
The consensusOV classifier also reports a real-valued margin score to discriminate between patients whose tumors possess well-defined versus indeterminate subtypes, in which marginally classifiable tumors are called based on the difference between the top two subtype scores (Chen et al., 2018). We examined the margins of classification using the proteomic data from the 9-patient dataset (Figure 5C). An analysis of variance on the margins indicated significant variation among the tumor tissue collection types (p < 0.001). A post hoc Tukey honestly significant difference test indicated that the LMD-enriched tumor (including the LMD-enriched tumor cores from 343WM) and stroma collections were significantly different from the whole tumor collections (Figure 5C). While the mean of the margin scores from all levels for each of the 9 additional cases demonstrated a significantly improved reliability of margin classification, the proteomic data from LMD-enriched collections from 343WM when examined individually did not show the same improvement in margin classification (data not shown). LMD-enriched stroma and tumor cell populations differed from each other (p < 0.01) and from whole tumor (p < 0.001). Data from the whole tumor collections had the lowest margins of differentiation and thus were frequently indeterminately classified at any threshold. Enrichment by LMD resulted in reliable subtype classification more frequently. Importantly, some LMD-enriched collections within the same patient were reliably marginally classified into different subtypes depending on sampling level, representing intrinsic heterogeneity within the TME.
Methyltransferase nicotinamide N-methyltransferase (NNMT) was recently identified as a master regulator of CAF function, which contributes to ovarian cancer proliferation, migration, and metastasis and results in the presence of a conserved metastasis-associated stromal proteomic signature distinguishing primary lesions from omental metastases (Eckert et al., 2019). The abundance of NNMT was elevated in all LMD-enriched stroma collections relative to LMD-enriched tumor, irrespective of primary (343WK) versus metastatic omental (343VT, 343VX, 343WB, 343WH, 343WJ, 343WQ) or non-omental (343WN, 343WP) origin (Figure S7). Measurement of NNMT in the whole tumor collections was reflective of overall levels of tumor purity, resulting in intermediate abundances dependent on the relative proportions of stroma and tumor in the TME.
We additionally investigated performance of a suboptimal cytoreduction associated network (SCAN) signature (Liu et al., 2015) correlating with stroma activation and tumor invasiveness in transcriptome data from case 343WM. The 11-gene surgical debulking signature from Liu et al most strongly correlated with our LMD-enriched stroma and whole tumor tissue collections (Figure S8), consistent with the correlation of the SCAN signature with stromal involvement.
We next sought to explore molecular heterogeneity within each sample collection type using a phylogenic approach depicting relatedness between samples based on correlation of protein abundance trends (Figure 6; Table S30). In this way, the correlations of samples within the same LMD collection type were calculated and then compared with those from other (as specified) LMD collection types. Comparison of enriched tumor and stroma collections in case 343WM (Figure 6A) suggests that the proteome expression profiles of LMD-enriched stroma are more heterogeneous from a phylogenic perspective (i.e. have lower median Spearman correlations between levels) than either of the LMD-enriched tumor epithelium collections and that the proteomic expression profile of the whole tumor is more closely related to the LMD-enriched stroma. Focusing on LMD-enriched tumor epithelium collections (cores and collection levels across sections [Figure 6B]) illustrates that extent of heterogeneity between all tumor cores is statistically less than that observed between LMD-enriched tumor collections (p < 0.0001). Further, the correlations within and between the LMD-enriched tumor epithelium and stroma samples were calculated for cases 343WM, 343WK, 343WQ, and 343WH. These four cases were again selected as they each have equivalent numbers of LMD-enriched tumor and stroma samples collected per case. In three of the four cases, the degree of heterogeneity between tumor epithelium samples was statistically less (p < 0.01) than the heterogeneity observed in the stroma (Figures 6C–6F).
Figure 6.

Patient-specific dendrograms depicting Spearman correlations between LMD-enriched collections based on differentially expressed proteins
(A) Representative relatedness of all collection types from 343WM, including tumor cores, LMD-enriched tumor epithelium, LMD-enriched stroma, whole tumor, and cryopulverized tissue. (B) Relatedness of tumor cores versus LMD-enriched tumor epithelium from 343WM. (C-F) Relatedness of LMD-enriched tumor versus stroma from 343WM, 343WK, 343WQ, and 343WH, respectively. With the exception of the LMD-enriched tumor cores vs LMD-enriched tumor epithelium comparison in (B) which was calculated using proteins with a median absolute deviation (MAD) > 0.5, all comparisons in (A) and (C-F) were made using proteins with MAD >1. Comparisons marked with (∗) indicate a significant difference (p < 0.01) between groups, with p values of 1.264 × 10−5, 0.0012, 0.0003372, 0.00097, and 0.2205 for the comparisons in (B-F), respectively. The yellow, blue, and red ovals in (B-F) highlight the clusters of LMD-enriched tumor cores, tumor epithelium, and stroma, respectively.
We further investigated regional tissue variations within a single tumor specimen, case 343WQ, to assess how intratumoral heterogeneity can impact molecular subtype classification and measurement of proteomic markers (Figure 7), with similar findings seen across all cases (data not shown). The relative composition of tumor epithelium and stroma is generally unchanged throughout the depth of the specimen block for this patient tumor (Figure 7; left panel). Further, enriched tumor and stroma collections across each of the 5 levels displayed consistent patterns of molecular alterations, correlations with HGSOC consensus molecular subtypes (Chen et al., 2018), and cellular admixture scores (Aran et al., 2017), within tumor and stromal cell populations throughout the tumor (Figure 7; right panel). In contrast, the whole tumor collections displayed widely varied patterns of molecular abundances, subtype classifications, and cellular admixture scores across the 5 levels.
Figure 7.

Representative case (343WQ) depicting variable molecular expression and subtype classification by level
H&E images show the tissue sections mounted on glass slides bounding the top of each level. The scale bar in the bottom left corner of each micrograph indicates a length of 4 mm. The tissue section number is notated in the bottom right corner. The median tumor cellularity with relative standard deviation (%CV), molecular subtype, and protein abundances of representative tumor and stroma markers present in the whole tumor collections are indicated for each level. The median tumor cellularity calculated from review of multiple images per level with the %CV included in parenthesis (from Table S1) is reported as percentages. Correlations with consensus molecular subtypes (Figure 5B and Table S28) are shown. The Log2-transformed protein abundances are shown for tumor/epithelial markers (CDH1 and KRT19) and stroma markers (FAP and VCAN) (from Table S7).
Finally, we selected three patients for whom complete cancer-related medical histories were available (Figure S9) to analyze the heterogeneity of clinical marker expression between LMD levels within each patient’s specimen. Case 343WP was extremely sensitive to chemotherapy and surviving with no evidence of disease as of the last follow-up, more than 5 years after diagnosis (Figure S9A). Cases 343WN (Figure S9B) and 343WQ (Figure S9C) had cancer-related deaths; these patients were moderately sensitive or resistant to chemotherapy, respectively. We generated a composite assessment of 70 genes or gene products analyzed in the clinical setting by Precision Therapeutics, Inc. (now Helomics [Pittsburgh, PA]; a subsidiary of Predictive Oncology [Eagan, MN]), Caris Life Sciences (Irving, TX), Foundation Medicine (Cambridge, MA), Quest Diagnostics (Secaucus, NJ), and/or Ambry Genetics (Alisa Viejo, CA). Of these 70 markers, 31 were measured as proteins in the n = 9 patient specimen set. The variability of protein expression between levels for these markers was examined in cases 343WK, 343WQ, and 343WH, for whom the full complement of 5 levels/cases for all LMD collection types were available. Relative standard deviations of protein expression were calculated and ranked to identify the top 5 most and least variable clinical markers (Table S31). The most variable proteins between levels within a single patient's specimen in our whole tumor samples (comparable with the type of tissue examined in the clinic) were ABL1, NF1, MSH6, MLH1, and PTEN. None of these were among the top 5 most variable in our LMD-enriched tumor samples. TOP1, RAD50, EPCAM, SMARCA4, and APC were the least variable in the whole tumor. TUBB3 and HRAS were among the top 5 most variable proteins in both LMD-enriched tumor and LMD-enriched stroma, while FBN1 was among the least variable in both LMD-enriched compartments.
Discussion
Ovarian cancers are typically diagnosed at an advanced stage after the accumulation of numerous molecular alterations (Torre et al., 2018). In this study, we have demonstrated that LMD enrichment reveals extensive intratumoral molecular heterogeneity, which bears directly on investigations aimed at identifying novel biomarkers or into cancer pathophysiology, and has implications for diagnostic, prognostic, and therapeutic predictive values, as recently described in hepatocellular carcinoma (Buczak et al., 2018).
We found that many molecular alterations were negatively correlated with established diagnostic and prognostic signatures within LMD-enriched cell populations, such as confirmation of several mesenchymal marker genes as emerging from stroma cells in HGSOC tissues recently identified by Zhang et al (Zhang et al., 2019) (Figure 5). Furthermore, we observe a profoundly unique transitioning of molecular subtype assignment based on sampling of the TME using proteomic versus transcriptomic data (Figure 5A). We further note that the correlation of protein and transcript abundance is higher in tumor epithelium versus stroma collections and is likely attributed to the secretory nature of ovarian stroma cells (Furuya, 2012). This was further underscored by our findings that the variance of proteins containing signal peptide sequences or classified as “extracellular” by Gene Ontology across all levels from each LMD-enriched tumor and stroma collection was significantly greater (p < 0.0001) than the variance of proteins lacking these classifications (Table S15).
Comparison across proteomic platforms using proteins co-quantified by MS-based proteomics and RPPA revealed similar findings as seen above with the co-quantified proteins and transcripts in which there was general positive correlation within the same LMD-enriched tumor epithelium or stroma tissues across all cases (Figures S6A and S6B). However, there was poor concordance between proteins co-quantified in whole tumor collections, often even within the same case (Figure S6C). While this is likely partially attributable to the limited number of features (54 proteins) used in this comparison (Ali et al., 2017), it largely recapitulates the findings that whole tumor collections reflect overall degrees of tumor purity and the need for cell-type enrichment for molecular characterization.
consensusOV classification of LMD-enriched tumor, stroma, and whole tumor populations suggests that molecular signatures developed to date have reflected variations in tumor purity that differentially correlate with altered outcome; specifically, lower tumor purity contributes to the assignment of tumors to poorer prognostic outcomes (Figures 5A, 5B, and S8). LMD enrichment of cellular populations statistically improved the reliability of subtype classifications, as measured by comparing consensusOV margin scores (Figure 5C). The predictive value for estimating patient outcomes through molecular subtype assignment (using the molecular prognostic signatures identified by Konecny et al (Konecny et al., 2014)) showed a median survival difference of two years depending on whether the tumor epithelium or stroma and/or mixed tissue is sampled. Specifically, the mesenchymal subtype in the Konecny et al study correlated with our LMD-enriched stroma, whereas the differentiated and proliferative subtypes correlated with our LMD-enriched tumor (Konecny et al., 2014). Notably, the contribution of the stromal microenvironment to the mesenchymal signature has been recently described (Zhang et al., 2019; Izar et al., 2020; Schwede et al., 2020). The observation that poor prognosis molecular subtypes (namely, mesenchymal subtype) are driven by the admixture of stroma cells supports the existence of a pathological ovarian stroma and supports the proposed role of CAFs contributing to disease development and/or progression (Bu et al., 2019; Jia et al., 2016; Narikiyo et al., 2019; Eckert et al., 2019). The presence of a large ovarian stromal compartment is an indicator of poor prognosis (Labiche et al., 2010). Tumors with more interceding stroma have lower resectability, decreasing the likelihood of achieving complete resection (R0) during surgical debulking leading to worse outcomes (Torres et al., 2018; Hamilton et al., 2011). Gene signatures in the ovarian stroma have also been correlated with increased tumor invasiveness and metastatic spread to the blood and lymphatics (Yue et al., 2019). Further, assignment of the cryopulverized tissue in our study to a particular subtype was less clear due to profound signal averaging of the proteome (Figure 5A). The cancer genome atlas and other large-scale tumor characterization efforts to date have applied pre-determined tumor purity cutoff levels to include predominantly high purity tumors in their studies. As a result, molecular drivers of disease progression specific to prognostically poor/low purity tumors are frequently missed (Aran et al., 2015; Chen et al., 2018), as recently realized for colon cancer (Mao et al., 2018) and gliomas (Zhang et al., 2017).
We demonstrate that many markers previously published as correlating with residual disease and/or suboptimal cytoreduction in ovarian cancer are elevated in tumor stroma (Figure S8) (Tucker et al., 2014; Liu et al., 2015). Among these, FABP4 and ADH1B expressions were associated with significantly higher levels of residual disease (Tucker et al., 2014). Expression and shedding of AKAP12 has been recently described in isogenic cell line models of paclitaxel resistance and elevation of AKAP12 transcript also correlated with decreased survival in patients with HGSOC (Bateman et al., 2015). We observe that AKAP12 transcript is elevated in stroma versus tumor epithelium (Tables S5, S6, S7, and S8), suggesting that elevation of AKAP12 in drug-resistant HGSOC cells may be due to chemotherapy-induced epithelial-to-mesenchymal transition and association with poor disease outcome may correlate with patients harboring lower purity tumors, likely mesenchymal subtype tumors. COL11A1 transcripts were also abundant in the stroma and some of the whole tumor collections (Figure S8), consistent with findings that COL11A1 is elevated in CAFs (Jia et al., 2016).
Extensive intratumor heterogeneity was observed in our study within each of the LMD-enriched tumor and stroma compartments, as demonstrated by the tumor cores from case 343WM and between the spatially separated collection levels for each of the 10 HGSOC cases assessed. A comparison of the magnitude of heterogeneity within each LMD-enriched tissue type revealed that overall there was a greater amount of heterogeneity in the stroma tissue relative to tumor epithelium (Figures 6C–6F). Though more related than the stroma collections, we demonstrate heterogeneity within the tumor epithelium resulting from locoregional differences, as replicate tumor cores displayed greater relatedness to each other than to LMD-enriched tumor collections taken from the entire tissue area (Figures 6A and 6B). Consistent with our enriched tumor and stroma cell findings, single-cell RNA sequencing on HGSOCs has previously revealed that individual cells of the same tissue type (tumor epithelium or stroma) differentially clustered with molecular subtypes due to unique gene expression profiles (Winterhoff et al., 2017). Further, heterogeneity within HGSOC molecular subtypes has been partially attributed to activation of distinct transcription factors serving as master regulators of the cellular phenotypes (Zhang et al., 2015; Eckert et al., 2019), as well as re-seeding of the primary site of disease by intraperitoneal metastases (Hu et al., 2020). As mentioned, within the ovarian stromal microenvironment, we observed the highest probability of classification as mesenchymal and/or immunoreactive subtypes (Figure 5).
We prioritized 381 proteins (Table S24) which had the same pattern of expression across all patients in the 9-patient specimen set for pathway and functional analysis. It could be speculated that several candidate proteins from this list represent the most promising targets for future clinical studies as they were consistently significantly enriched in either tumor or stroma across these patients. Compartmentalized expression of genes encoding known FDA-approved anticancer drug targets (Sun et al., 2017) was found in both the LMD-enriched tumor epithelium and stroma collections (Tables S23 and S27). The proteins encoded by VEGFA and FLT1 (also known as VEGFR1) are targets of bevacizumab, a monoclonal antibody often used as an anti-angiogenic therapy for treating patients with HGSOCs (Rossi et al., 2017). In our study, VEGFA transcript expression from case 343WM was significantly enriched in the tumor epithelium, whereas transcript expression of FLT1/VEGFR1 was more abundant in LMD-enriched stroma (Table S23). Dysregulated VEGF/VEGFR expression has been demonstrated to contribute to epithelial ovarian cancer development and/or progression through increased vascularization and improved survival of endothelial cells via anti-apoptotic signaling in the newly formed vessels (Skirnisdottir et al., 2016; Masoumi Moghaddam et al., 2012), though bevacizumab targeting of VEGF/VEGFR has disputed levels of improvement on survival of patients with HGSOCs (Tewari et al., 2019). PARP1 protein was consistently enriched in the tumor epithelium of nine patients in our study (Table S27). Olaparib, rucaparib, and niraparib are poly(ADP-ribose) polymerase (PARP) inhibitors used for treating patients with HGSOCs in either the recurrent or maintenance setting (Jiang et al., 2019). Conversely, MAP1A is a target of estramustine, and PDGFRB is a target of sorafenib, regorafenib, and pazopanib; MAP1A and PDGFRB proteins were both found to be significantly enriched in LMD stroma.
Accurate diagnosis and estimation of prognostic outcome are of utmost interest to patients, so we analyzed the intratumor heterogeneity of protein expression for gene and/or protein markers that are routinely assessed in patients with HGSOCs. Specifically, we examined the variability of protein expression between each of the 5 levels per LMD collection type separately for cases 343WK, 343WQ, and 343WH and reported proteins that were most and least variable within each LMD collection type (Table S31). Our whole tumor samples represent the types of samples analyzed in the clinical setting. ABL1 was among the top 5 most variable proteins in whole tumor samples within each patient's tumor. ABL1 is a target of the drug regorafenib which is currently FDA approved for treatment of colorectal and stomach cancers (Sun et al., 2017) and has been the subject of recent clinical trials for treatment of recurrent epithelial ovarian cancers (Tan et al., 2017). Interim clinical trial results reported regorafenib to be inefficacious in improving progression free survival, overall survival, or overall response rate. Our results suggest that the clinical inefficacy of regorafenib for treating ovarian cancers could, at least in part, be attributed to the high intratumoral heterogeneity of expression of ABL1 or other related proteins. MSH6 and MLH1 were also among the top 5 most variable proteins in this analysis. MSH6 and MLH1 are biomarkers of mismatch repair and microsatellite instability and are used as prognostic indicators of improved survival and clinical response to platinum-based chemotherapy (Zhao et al., 2018). The high levels of intratumoral heterogeneity of MSH6 and MLH1 in our study suggest that the predictive efficacy of platinum-based chemotherapies may not adequately represent true patient response due to the potential for sampling variations of regional protein expression within a tumor biopsy. TUBB3 is the target of the drug ixabepilone, FDA approved for treatment of breast cancer (Sun et al., 2017) and the subject of recent clinical trials for potential use in patients with recurrent or persistent platinum- and taxane-resistant ovarian cancer (De Geest et al., 2010), which showed promising results. Surprisingly, TUBB3 was among the top 5 most variable proteins in both LMD-enriched tumor and LMD-enriched stroma in our analysis.
In conclusion, we demonstrate a critical need to account for cellular subtype and regional TME molecular heterogeneity in cancer molecular profiling efforts that will substantially enable in-depth characterization of spatially distributed subclonal cell populations that have underappreciated roles in driving carcinogenesis. To assist with ongoing research efforts by others, we created an interactive web portal to be used as a community resource, which can be found at https://lmdomics.org/HGSOCheterogeneity/. Users can examine the expression of their protein(s) of interest between LMD-enriched and whole tumor compartments, three-dimensional heterogeneity of protein expression throughout the depth of each specimen block, and the conservation of this variability across our specimen set of 9 patients with HGSOC tumors. Further work to deconstruct the TME will aid clinical diagnosis, improve the efficacy of therapeutic intervention, and increase capabilities to identify druggable molecular markers of disease development and progression.
Limitations of study
In this investigation, we demonstrated significant proteomic and transcriptomic 3D heterogeneity in the TME from tumors of patients with HGSOC using LMD of spatially separated tumor enriched regions, stromal cell populations, as well as whole tumor collections throughout distinct levels from each specimen block. We do acknowledge several limitations of the present study. Additional samples and patients will be critical to elucidate patterns of intra-tumoral heterogeneity that may differ between primary tumors and metastatic lesions or between chemo-naïve specimens as compared to those obtained after neoadjuvant chemotherapy since patients in our study cohort represented a combination of several of these clinical features. Although the present study was limited to HGSOC, the pre-analytical workflows described here, namely using LMD for histology-resolved molecular profiling of the TME, offer a roadmap to generalize these data through multi-region sampling of larger cancer tissue cohorts, other solid tumor cancer types, and to a variety of non-cancerous human tissue pathologies.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Primary antibody information is listed in Table S18 | Various | Various |
| Goat Anti-rabbit IgG | Vector Laboratories, Inc. | Cat# BA1000 |
| Rabbit Anti-mouse IgG | Dako Cytomation | Cat# K1500 |
| Biological samples | ||
| Primary high grade serous ovarian cancer | IFMC/WHIRC Biobank | 343WM |
| High grade serous ovarian cancer omental metastasis | IFMC/WHIRC Biobank | 343WJ |
| High grade serous ovarian cancer omental metastasis | IFMC/WHIRC Biobank | 343VT |
| High grade serous ovarian cancer omental metastasis | IFMC/WHIRC Biobank | 343VX |
| Primary high grade serous ovarian cancer | IFMC/WHIRC Biobank | 343WK |
| High grade serous ovarian cancer adnexal metastasis | IFMC/WHIRC Biobank | 343WN |
| High grade serous ovarian cancer omental metastasis | IFMC/WHIRC Biobank | 343WQ |
| High grade serous ovarian cancer omental metastasis | IFMC/WHIRC Biobank | 343WB |
| High grade serous ovarian cancer pelvic mass metastasis | IFMC/WHIRC Biobank | 343WP |
| High grade serous ovarian cancer omental metastasis | IFMC/WHIRC Biobank | 343WH |
| Chemicals, peptides, and recombinant proteins | ||
| Mayer’s Hematoxylin Solution | Sigma Aldrich | Cat# MHS32 |
| Eosin Y Solution Aqueous | Sigma Aldrich | Cat# HT110216 |
| Phosphatase Inhibitor Cocktail 3 | Sigma Aldrich | Cat# P0044 |
| Phosphatase Inhibitor Cocktail 2 | Sigma Aldrich | Cat# P5726 |
| ProtectRNA RNase Inhibitor 500× Concentrate | Sigma Aldrich | Cat# R7397 |
| cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 11836170001 |
| Mass Spec-Compatible Human Protein Extract, Digest | Promega | Cat# V6951 |
| Pierce Peptide Retention Time Calibration Mixture | Pierce | Cat# 88321 |
| iRT Kit | Biognosys | Cat# Ki-3002-1 |
| Buffer RLT | Qiagen | Cat# 79216 |
| T-PER Tissue Protein Extraction Reagent | Pierce | Cat# 78510 |
| Novex Tris-Glycine SDS Sample Buffer (2X) | Invitrogen | Cat# LC2676 |
| Universal Human Reference RNA | Stratagene | Cat# 740000 |
| AMPure XP | Beckman Coulter | Cat# A63880 |
| SYPRO Ruby Protein Blot Stain | Invitrogen | Cat# S11791 |
| ReBlot Plus Mild Antibody Stripping Solution | Millipore | Cat# 2502 |
| I-Block Protein-Based Blocking Reagent | Applied Biosystems | Cat# T2015 |
| Biotin-Blocking System, Ready-to-use reagents | Dako Cytomation | Cat# X0590 |
| Protein block, serum-free | Dako Cytomation | Cat# K1500 |
| Tyramide-based avidin/biotin amplification system | Dako Cytomation | Cat# K1500 |
| IRDye 680RD Streptavidin | LI-COR Biosciences | Cat# 926-68079 |
| Critical commercial assays | ||
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| Soluble Smart Digest Kit | Thermo Fisher Scientific | Cat# 3251711 |
| TMT10plex Isobaric Label Reagent Set plus TMT11-131C Label Reagent | Thermo Fisher Scientific | Cat# A34808 |
| TMTpro 16plex Label Reagent Set | Thermo Fisher Scientific | Cat# A44520 |
| RNeasy Micro Kit | Qiagen | Cat# 74004 |
| Qubit RNA HS Assay Kit | Thermo Fisher Scientific | Cat# Q32852 |
| Qubit RNA BR Assay Kit | Thermo Fisher Scientific | Cat# Q10210 |
| Agilent RNA 6000 Pico Kit | Agilent | Cat# 5067-1513 |
| SuperScript VILO cDNA Synthesis Kit | Invitrogen | Cat# 11754050 |
| Ion AmpliSeq Kit for Chef DL8 | Thermo Fisher Scientific | Cat# A29024 |
| Ion AmpliSeq Transcriptome Human Gene Expression Panel, Chef-Ready Kit | Thermo Fisher Scientific | Cat# A31446 |
| Ion Library TaqMan Quantitation Kit | Thermo Fisher Scientific | Cat# 4468802 |
| Ion 550 Chip Kit | Thermo Fisher Scientific | Cat# A34538 |
| Ion 550 Kit - Chef | Thermo Fisher Scientific | Cat# A34541 |
| Deposited data | ||
| Raw LC-MS/MS data | This paper | ProteomeXchange Consortium, PRIDE: PXD026570 |
| Raw RNA-seq data | This paper | European Genome-Phenome Archive |
| Software and algorithms | ||
| HALO | Indica Labs | https://indicalab.com/ |
| Torrent Suite version 5.8.0 | Thermo Fisher Scientific | https://www.thermofisher.com/us/en/home/life-science/sequencing/next-generation-sequencing/ion-torrent-next-generation-sequencing-workflow/ion-torrent-next-generation-sequencing-data-analysis-workflow/ion-torrent-suite-software.html |
| Mascot | Matrix Science | https://www.matrixscience.com/ |
| Proteome Discoverer | Thermo Fisher Scientific | https://www.thermofisher.com/us/en/home.html |
| Swiss-Prot | UniProt | http://www.uniprot.org/ |
| R version 2.6.2 | CRAN | https://cran-archive.r-project.org/bin/windows/base/old/2.6.2/ |
| Clustvis version 1.2.0 | BIIT Research Group | https://biit.cs.ut.ee/clustvis/ |
| LIMMA version 3.8 | Bioconductor | https://bioconductor.riken.jp/packages/3.8/bioc/html/limma.html |
| xCell version 1.1.0 | UCSF | http://xcell.ucsf.edu/ |
| ggplot2 version 3.2.1 | CRAN | https://cran.r-project.org/web/packages/ggplot2/index.html |
| consensusOV version 1.8.1 | Bioconductor | http://bioconductor.jp/packages/3.10/bioc/html/consensusOV.html |
| ggtree version 2.0.1 | Bioconductor | https://bioconductor.org/packages/release/bioc/html/ggtree.html |
| ape version 5.3 | CRAN | https://cran.r-project.org/web/packages/ape/index.html |
| MedCalc version 19.0.7 | MedCalc Software Ltd | https://www.medcalc.org/ |
| ComplexHeatmap version 2.2.0 | Bioconductor | https://www.bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| Ingenuity Pathway Analysis | Qiagen | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/ |
| Morpheus version 1.0-1 | Broad Institute | https://software.broadinstitute.org/morpheus/ |
| MicroVigene V5.1.0.0 | Vigenetech | http://www.vigenetech.com/MicroVigene.htm |
| Other | ||
| PEN Membrane Glass Slides | Leica Microsystems | Cat# 11532918 |
| Nitrocellulose-coated slides for RPPA | Grace Bio-Labs | Cat# RD478691-M |
| 96 MicroTubes in bulk (no caps) | Pressure Biosciences Inc | Cat# MT-96 |
| 96 MicroCaps (150uL) in bulk | Pressure Biosciences Inc | Cat# MC150-96 |
| 96 MicroPestles in bulk | Pressure Biosciences Inc | Cat# MP-96 |
| 9mm MS Certified Clear Screw Thread Kits | Fisher Scientific | Cat# 03-060-058 |
| ZORBAX Extend 300 C18, 2.1 x 12.5 mm, 5 μm, guard cartridge (ZGC) | Agilent | Cat# 821125-932 |
| ZORBAX Extend 300 C18, 2.1 x 150 mm, 3.5 μm | Agilent | Cat# 763750-902 |
| EASY-SPRAY C18 2UM 50CM X 75 | Fisher Scientific | Cat# ES903 |
| PM100 C18 3UM 75UMX20MM NV 2PK | Fisher Scientific | Cat# 164535 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Dr. Thomas P. Conrads (conrads@whirc.org).
Materials availability
This study did not generate any unique reagents.
Data and code availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD026570. The RNA-sequencing transcriptomics data have been deposited to the European Genome-Phenome Archive (Lappalainen et al., 2015).
Experimental model and subject details
Flash-frozen high grade serous ovarian tumors were obtained and all study protocols were approved for use under a Western IRB-approved protocol “An Integrated Molecular Analysis of Endometrial and Ovarian Cancer to Identify and Validate Clinically Informative Biomarkers” deemed exempt under US Federal regulation 45 CFR 46.102(f). All experimental protocols involving human data in this study were in accordance with the Declaration of Helsinki and informed consent was obtained from all patients. Relevant clinical information for patients included in the study can be found in Table S1.
Method details
Tissue specimens
Surgically resected fresh-frozen tumor tissue specimens were obtained from ten patients with HGSOC of primary ovarian or tubo-ovarian origin. These comprised a mixture of chemotherapy-naïve specimens and specimens obtained following neoadjuvant chemotherapy (NACT) from patients with advanced stage (stage III or IV) disease. The specific tissues used for the study were from ovary, omentum, and pelvic or adnexa masses. The tissue blocks were sectioned by cryotome into 170 - 210 consecutive 10 μm thin tissue sections (1.7 - 2.1 mm total depth) which were placed on PEN membrane slides (Leica Microsystems). Representative sections were mounted on charged glass slides and stained with hematoxylin and eosin (H&E) after every 10 PEN membrane slide sections (100 μm), except for case 343WM where representative H&E slides were generated after every 20 PEN membrane slide sections (200 μm).
Laser microdissection
The specimen obtained from case 343WM was uniquely prepared for laser microdissection (LMD) and analysis of spatially separated tumor “cores” which represent defined sub-populations of tumor epithelium that were collected as technical replicate areas for comparison with tumor epithelium collected from larger whole slide areas. Four regionally separated areas within the tissue were laser microdissected to enrich tumor epithelium (LMD7, Leica Microsystems) from each of 100 slides for proteomics or 50 slides for transcriptomics. The cores were pooled from a cross-sectional area of approximately 1 mm2 collected by LMD per slide for a depth spanning the entire block (1 mm x 1 mm x 2 mm). An adjacent 1 mm2 was microdissected to serve as a replicate from each core; in total each core plus their respective adjacent replicate regions covered an approximate 2 mm3 aggregate volume of tumor epithelium. The distance between the center of each tumor core and the center of the nearest neighboring core was approximately 4 mm. For a set of 9 sections (200 μm apart) each for proteomics and transcriptomics, all remaining tumor epithelium (between 20 - 44 mm2/section) and stroma (44 - 80 mm2/section) were collected by LMD after the core regions were collected. Whole tumor tissue was collected from a second set of 9 sections (96 - 155 mm2/section), where only regions containing necrosis, blood, and fat were excluded.
For the remaining 9 HGSOC cases, the slides from each block were separated into five equivalent levels (quintiles) assigned by depth within the block. Slides designated for MS proteomics (n=9 patients, 5 levels each), reverse phase protein array (RPPA; n=8 patients, 5 levels each, plus 8 levels from 343WM), and transcriptomics (n=2 patients, 1 level each) within each level were interlaced as much as possible.
All slides were H&E stained prior to LMD. For the slides microdissected for mass spectrometry (MS) proteomics, phosphatase inhibitors (Sigma Aldrich) were added to the 70% ethanol fixative and the first DEPC water; slides for transcriptomics were stained with RNase inhibitors (ProtectRNA; Sigma Aldrich) added into all aqueous solutions. Slide preparation for RPPA did not involve staining with eosin after the hematoxylin but included color development in Scott’s Tap Water (Thermo Fisher Scientific) and two final drying steps in xylenes. Protease inhibitors (Roche) were added to all RPPA staining solutions except for the 100% ethanol and xylene washes.
Within each quintile level per specimen block, LMD was used to enrich cross-sectional areas of 40 mm2 and 15 mm2 (LMD enriched tumor epithelium, LMD enriched stroma, and whole tumor) for analysis via liquid chromatography-tandem mass spectrometry (LC-MS/MS) and reverse phase protein microarray (RPPA), respectively. For two cases (343WH and 343WB), a cross-sectional area of 25 mm2 of each LMD enriched tumor, LMD enriched stroma, and whole tumor was collected from one level for RNA sequencing (RNA-seq). Microdissected tissue was collected into LC-grade water (Fisher Scientific), a tissue extraction/lysis buffer, or Buffer RLT (Qiagen) for analysis via LC-MS/MS, RPPA, and RNA-seq, respectively. The RPPA extraction/lysis buffer consisted of a 1:1 mixture of Tissue Protein Extraction Reagent (T-PER; Pierce), Novex 2x Tris-Glycine SDS Sample Buffer (Invitrogen), plus 2.5% v/v 2-mercaptoethanol which was added to tissue at a ratio of 3 μl buffer per 1 mm2 LMD tissue. For RPPA, each slide spent no more than 30 min dwell time on the LMD microscope. Additional slides were collected into new tubes. Collected tissue was briefly centrifuged and frozen at -80 °C until analysis.
OracleBio image analysis
Pre- and post-LMD images were collected using the Aperio ScanScope XT slide scanner (Leica Microsystems). Image analysis was performed by OracleBio using the Indica Labs HALO platform. Post-LMD images from 343WM were used to develop classification algorithms for identification of the “dissection area” and “all remaining tissue”. A separate algorithm was developed which involved co-registration of post-LMD images with corresponding adjacent H&E-stained sections on glass slides for detection and quantification of the cell nuclei abundance within the LMD regions. The size and number of cells and nuclei collected in each LMD collection was determined using matched sets of reference glass H&E sections and PEN membrane slides following LMD enrichment of tumor (n=15) and stroma (n=6) cell populations.
Peptide preparation for TMT LC-MS/MS
LMD tissue in LC-grade water was dried, re-suspended in 100 mM triethylammonium bicarbonate (TEAB)/10% acetonitrile (ACN), and digested using SMART trypsin (1 μg/30 mm2 tissue; Thermo Fisher) and pressure cycling technology (PCT, Pressure Biosciences, Inc.) (Lee et al., 2020). For 343WM, the remaining tissue in the OCT block was washed with water and cryopulverized in liquid nitrogen in a mortar and pestle. The cryopulverized tissue was re-suspended in 100 mM TEAB/10% ACN and digested by PCT using SMART trypsin (1 μg protease/ 30 mm2 tissue). Peptide concentrations from trypsin digests were determined using the bicinchoninic acid assay (Pierce BCA). Tryptic peptides were labeled with isobaric Tandem Mass Tags (TMT) according to the manufacturer’s instructions using the TMT 10-Plex Kit or TMTpro 16-Plex Kit from Thermo Fisher (Table S4). The TMT 10-Plex reagents and TMTpro 16-plex reagents were used for labeling samples from case 343WM and the remaining nine cases, respectively, to generate patient-specific TMT plexes. Briefly, peptide samples were individually mixed with respective TMT reagents for 1 h at room temperature, then quenched using 5% hydroxylamine. Empty TMT channels were filled using equivalent amounts of peptide mixed from each of the 5 levels of the whole tumor collections in the respective patient-specific plex.
Each TMT-10 or TMTpro-16 multiplex set of samples were loaded onto a C-18 trap column in 10 mM NH4HCO3 (pH 8.0) and fractionated by basic reversed-phase liquid chromatography (bRPLC) into 96 fractions through development of a linear gradient of acetonitrile (0.69% acetonitrile/min). For 343WM, 36 concentrated fractions were generated by pooling in a serpentine manner, each of which were analyzed for LC-MS/MS. For the remaining 9 cases, 24 concentrated fractions were generated following bRPLC which were individually analyzed by LC-MS/MS.
Liquid chromatography-tandem mass spectrometry
The TMT-10 or TMTpro-16 sample multiplex bRPLC fractions were analyzed by LC-MS/MS employing a nanoflow LC system (EASY-nLC 1200, ThermoFisher Scientific, Inc.) coupled online with a Q Exactive HF-X MS (ThermoFisher Scientific, Inc.). In brief, each sample was loaded on a nanoflow HPLC system outfitted with a reversed-phase trap column (Acclaim PepMap100 C18, 2 cm, nanoViper, ThermoFisher Scientific, Inc) and a heated (50 °C) reversed-phase analytical column (Acclaim PepMap RSLC C18, 2 μm, 100 Å, 75 μm × 500 mm, nanoViper, ThermoFisher Scientific, Inc). Peptides were eluted by developing a linear gradient of 2% mobile phase B (95% acetonitrile with 0.1% formic acid) to 32% mobile phase B within 120 min at a constant flow rate of 250 nL/min. High resolution (R=60,000 at m/z 200) broadband (m/z 400 – 1,600) mass spectra (MS) were acquired from which the top 12 most intense molecular ions in each MS scan were selected for high-energy collisional dissociation (HCD, normalized collision energy of 30 for TMT-10 and 34 for TMTpro) acquisition in the orbitrap at high resolution (R=45,000 at m/z 200). Charge state selection was restricted to z = +2, +3 and +4. The RF lens was set to 30% and both MS1 and MS2 spectra were collected in profile mode. Dynamic exclusion (20s) was enabled to minimize redundant selection of peptide molecular ions for HCD.
Quantitative proteomic data processing pipeline for global proteome analysis
Peptide identifications were generated by searching the .raw data files with a publicly-available, non-redundant human proteome database (Swiss-Prot, Homo sapiens, Proteome UP000005640, 20,257 sequences, downloaded 12-01-2017; http://www.uniprot.org/) appended with porcine trypsin (Uniprot: P00761) sequences using Mascot (Matrix Science) and Proteome Discoverer (Thermo Fisher Scientific). Identification, normalization, and Log2 transformation of PSMs was performed as previously described (Lee et al., 2020) for calculation of protein-level abundances.
RNA sequencing
Tissue collected by LMD in Buffer RLT was purified using the RNeasy Micro Kit (Qiagen) per the manufacturer’s instructions. RNA concentrations were determined by fluorescence (Qubit HS and BR kits, Thermo Fisher). RNA integrity numbers (RIN) were calculated using the RNA 6000 Pico Kit 2100 Bioanalyzer (Agilent). All RNA was high-quality with RIN values of 7.2 or greater, except for two samples which had RIN values of 4.9 and 6.5.
RNA samples were reverse transcribed from 10 ng input using the SuperScript VILO cDNA Synthesis Kit. Barcoded cDNA libraries containing 5-6 LMD samples plus a Universal Human Reference RNA (UHR) standard (Stratagene) were prepared on the Ion Chef System using the Ion Ampliseq Chef DL8 materials and the Ion AmpliSeq Transcriptome Human Gene Expression Panel Chef-ready Kit (Thermo Fisher). Libraries were then purified via solid phase reversible immobilization (SPRI) using AMPure XP beads (Beckman Coulter) to remove fragments less than 100 bp, quantified by qPCR (TaqMan Quantitation Kit; Applied Biosystems), and 25 μl of 100 pM diluted library was used for templating, amplification via emulsion PCR, and loaded onto Ion 550 chips on the Ion Chef System.
Sequencing was performed on the Ion Torrent S5 XL (Thermo Fisher) and mapped to the hg19 human reference transcriptome (hg19_Ampliseq_Transcriptome_21K_v1). Successful sequencing runs achieved ≥18M reads/sample (with one exception) and 169-234X AQ20 mean coverage depth. Per the Torrent Suite Software (Torrent Suite v5.8.0), the number of reads aligning to a given gene target represents an expression value referred to as “counts”. The read count per million mapped reads (RPM) for each barcoded sample was calculated by the software as read count x 106 / total number of mapped reads. Normalized RPM-level transcript abundances were calculated relative to the average RPM abundance quantified across all samples for a given transcript and Log2 transformed.
Reverse phase protein microarray
Using an Aushon 2470 arrayer (Aushon BioSystems, Billerica, MA) equipped with 185 μm pins, cell lysates along with internal reference standards were immobilized onto nitrocellulose-coated slides (Grace Bio-labs, Bend, OR) in technical replicates (n=3). To assess the amount of protein in each sample and for normalization purposes, selected arrays were stained with Sypro Ruby Protein Blot Stain (Invitrogen) following manufacturer's instructions. Immunostaining was performed as previously described (Baldelli et al., 2015, Baldelli et al., 2017). In brief, prior to antibody staining, arrays were first treated with Reblot Antibody Stripping solution (Millipore) for 15 minutes at room temperature, washed with PBS and incubated I-block (Applied Biosystems). To reduce unspecific binding between endogenous proteins and the detection system, arrays were then probed with 3% hydrogen peroxide, an avidin/biotin blocking system (Dako Cytomation, Carpinteria, CA), and an additional serum free protein block (Dako Cytomation, Carpinteria, CA) using an automated system (Dako Cytomation, Carpinteria, CA). Each array was then probed with one polyclonal or monoclonal primary antibody targeting a protein of interest. Primary antibodies’ specificity was previously assessed on commercial cell lysates or human tissue lysates by western blotting (Signore and Reeder, 2012). The criteria for validation were the presence of a single band of correct molecular weight in the positive control sample and the absence of a band in the negative control. Antibodies targeting phosphorylated epitopes were additionally validated by ligand induction. A subset of antibodies was further validated against peptide competition. Arrays were probed with total of 281 antibodies targeting native and/or post-translationally modified proteins. Primary antibodies were recognized by a biotinylated anti-rabbit (Vector Laboratories, Inc.) or anti-mouse secondary antibody (Dako Cytomation, Carpinteria, CA). Signal amplification was achieved using a commercially available tyramide-based avidin/biotin amplification system (Dako Cytomation, Carpinteria, CA) coupled with fluorescent detection using the streptavidin-conjugated IRDye680 dye (LI-COR Biosciences, Lincoln, NE), according to the manufacturer's instructions. Selected arrays were incubated with the secondary antibodies alone to capture background and unspecific signal. Images were acquired using a laser scanner (TECAN PowerScanner, Mönnedorf, Switzerland) and analyzed using a commercially available software (MicroVigene V5.1.0.0; Vigenetech, Carlisle, MA). In brief, the software performed spot finding and subtraction of the local background/unspecific signal. Finally, each sample was normalized to the corresponding amount of protein derived from the Sypro Ruby stained slides and triplicates were averaged.
Quantification and statistical analysis
Unsupervised hierarchical clustering and principal component analyses were performed using proteins or transcripts exhibiting a median absolute deviation (MAD) >0.5 or >1, as specified, across all samples and clustered by Pearson correlation as heatmaps using Clustvis (version 1.2.0) in R (version 3.6.2). Differential analyses of global proteome and transcript data matrixes was performed using LIMMA (version 3.8, (Ritchie et al., 2015)) in R (version 3.6.2); features satisfying a LIMMA p-value <0.01 and exhibiting a Log2 fold-change cut-off ±1 were prioritized for downstream analyses. Cell type enrichment analyses were performed using RPM-level RNA-seq data in xCell (http://xcell.ucsf.edu/; version 1.1.0 (Aran et al., 2017)). xCell cell type signature scores of interest were categorized by relative rank from highest to lowest spanning a range of 1 to -1 to enable co-visualization with similarly categorized transcript and protein abundance for candidates of interest. Boxplots of relative protein abundances or cell type signature scores were generated using Ggplot2 (version 3.2.1). Random forest probabilities for classification of consensus molecular subtypes using either global proteome or transcriptome data were calculated using consensusOV (version 1.8.1). Margin scores of molecular subtype classification were calculated by taking the difference between the probability of the highest subtype and the probability of the second highest subtype. ANOVA and post hoc Tukey HSD tests were performed to determine the significance of differences between the margin values for each group. Dendrograms were generated using Ggtree (version 2.0.1) using features with MAD>1 across individual patient samples, with the exception of the comparison between tumor cores and LMD enriched tumor from case 343WM, which used features with MAD>0.5. Tree structure was determined by neighbor-joining tree estimation using Ape (version 5.3) and Spearman correlations between samples were calculated. Molecular subtype classifications were compared by Spearman correlation using MedCalc (version 19.0.7). Functional pathway inference and drug targets were assessed using Ingenuity Pathway Analysis. The heatmap in Figure 7 was generated using ComplexHeatmap (version 2.2.0); the heatmaps in Figures S4 and S6 were generated using Morpheus (https://software.broadinstitute.org/morpheus/; version 1.0-1).
Acknowledgments
This study was supported in part by the U.S. Department of Defense - Uniformed Services University of the Health Sciences (HU0001-16-2-0006 and HU0001-16-2-0014), the Ovarian Cancer Research Program from the Congressionally Directed Medical Research Program (W81XWH-19-1-0183), and the National Cancer Institute (1U01CA233085-01A1). The authors would like to acknowledge Lorcan Sherry and Mark Anderson from OracleBio Limited (Scotland, UK) for histopathology image analysis support. The authors would like to acknowledge Victoria Olowu, Marshé Edwards, Fred Park, Persus Akowuah, Salma Eltahir, Katherine Zhou, Domenic Tommarello, Pang-Ning Teng, Katlin Wilson, Sakiyah TaQee, and Tamara Abulez for histopathology and proteomic sample preparation and informatic analysis support. This manuscript is dedicated to Dr. Uma NM Rao.
Author contributions
N.W.B. and T.P.C. contributed to conception; A.L.H., N.W.B., and T.P.C. contributed to experimental design. G.G., D.M., J.O., G.L.M., and T.P.C. contributed to identification and acquisition of the patient specimens. A.L.H., N.W.B., W.B., S.M.-M., B.L.H., K.A.C., M.Z., V.C., J.L., T.J.L., J.O., D.M., G.G., C.R., B.B., U.N.M.R., E.F.P., G.L.M., and T.P.C. contributed to acquisition, analysis, and/or interpretation of data. A.L.H., N.W.B., Y.C., K.O., A.K.S., K.M.D., C.D.S., U.N.M.R., M.P., E.F.P., G.L.M., and T.P.C. drafted and/or revised the article. Y.C., G.L.M., and T.P.C. acquired funding for the research. All authors read and approved the final manuscript.
Declaration of interests
T.P.C. is a ThermoFisher Scientific, Inc SAB member and receives research funding from AbbVie. E.F.P. receives research funding from Genentech, Pfizer, and AbbVie and is a co-inventor of the RPPA technology described herein and receives royalties on the related license agreements.
Disclaimer
The views expressed herein are those of the authors and do not reflect the official policy of the Department of Army/Navy/Air Force, Department of Defense, or U.S. Government.
Inclusion and diversity
We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. We worked to ensure that the study questionnaires were prepared in an inclusive way. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
Published: July 23, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102757.
Contributor Information
G. Larry Maxwell, Email: george.maxwell@inova.org.
Thomas P. Conrads, Email: conrads@whirc.org.
Supplemental information
Representative H&E stained glass slides from were examined at ∼200 μm intervals for case 343WM or ∼100 μm intervals for the additional 9 cases by a board-certified pathologist for percent by area estimation of tumor cellularity, necrosis, stroma, normal ovarian epithelium, lymphocytes, and polymorphonuclear leukocytes (PMN). Multiple images per level per case were reviewed and the median tumor cellularity and median stroma cellularity are reported with corresponding percent coefficient of variation (%CV) reported in parentheses. Abbreviations: NACT = neoadjuvant chemotherapy; FT = fallopian tube; Ov = ovary; ca = cancer; NOS = not otherwise specified.
Numerical values indicate the number of LMD tissue sections that were used for each collection.
ROI are denoted as millimeter squared (mm2) areas, cell nuclei as μm2 areas or as total cell counts derived from cellular nuclei measured in LMD enriched tumor or stroma ROIs.
. For 343WM, tryptic peptides were labeled with isobaric Tandem Mass Tags (TMT) according to the manufacturer's instructions using the TMT 10-Plex Kit from Thermo Fisher for a total of 4 plexes. 30 μg peptides from the 8 cores, 9 non-discrete whole tumor collections, and cryopulverized tissue sample were individually mixed with respective TMT reagents for 1 hr at room temperature, then quenched using 5% hydroxylamine. 10 μg peptides/sample were similarly TMT labeled from the 9 collections each of remaining LMD-enriched tumor epithelium or stroma. For the remaining 9 cases, digested peptide samples were labeled in patient-specific plexes using the TMTpro 16-Plex Kit (Thermo Fisher). Specifically, 5 μg peptide from up to 5 levels/patient each of LMD enriched tumor epithelium, LMD enriched stroma, and whole tumor collections were incubated with the TMTpro reagents per plex for a total of 80 μg/plex and 1 plex/patient. To replace samples from which <5 μg peptide was recovered, empty TMT channels were filled using equivalent amounts of peptide mixed from each of the 5 levels of the whole tumor collections.
. Log2 transformed fold-change abundances of 6,053 proteins imputed across all samples measured in case 343WM.
Normalized abundances of 20,535 RNA transcripts measured in case 343WM calculated relative to the average RPM abundance quantified across all samples for a given transcript, as reported by the Torrent Suite (v5.8.0) software, before Log2 fold-change transformation.
Log2 transformed fold-change abundances of 6,199 proteins imputed across all samples (n = 123).
Normalized abundances of 19,758 RNA transcripts calculated relative to the average RPM abundance quantified across all samples (n = 6) for a given transcript, as reported by the Torrent Suite (v5.8.0) software, before Log2 fold-change transformation.
http://xcell.ucsf.edu/, [Aran et al., 2017]), related to Figure 3A
http://xcell.ucsf.edu/, [Aran et al., 2017]), related to Figures 3C and 7
P-values indicate the reliability of the presence or absence of a signal peptide or extracellular classification within the indicated LMD-enriched tissue across all levels/case. 9 levels were used for comparison for case 343WM, while 5 levels/case were used each for cases 343WK, 343WQ, and 343WH.
Log2 transformed fold-change abundances of 5,742 imputed proteins that were co-measured at the transcriptome level from case 343WM.
Log2 transformed fold-change abundances of 5,379 imputed proteins that were co-measured at the transcriptome level.
Comparison of the most differential pathways and diseases/functions between LMD enriched tumor and stroma was determined using pairwise supervised analysis of proteomic and transcriptomic data with LIMMA adjusted p-value < 0.01 exhibiting a Log2 fold-change cut-off ± 1.
Comparison of the most differential diseases/functions between LMD enriched tumor and stroma was determined using pairwise supervised analysis of proteomic and transcriptomic data with LIMMA adjusted p-value < 0.01 exhibiting a Log2 fold-change cut-off ± 1.
Comparison of prevalent drug targets present in LMD enriched tumor versus stroma was determined using pairwise supervised analysis of proteomic and transcriptomic data with LIMMA adjusted p-value < 0.01 exhibiting a Log2 fold-change cut-off ± 1. The complete list of all differentially expressed genes that are known drug targets from our proteomic-level and transcriptomic-level data was compared with a list of 150 FDA-approved anticancer drugs as of 2014 analyzed in a study by Sun et al. (2017).
Comparison of the most differential canonical pathways between LMD enriched tumor and stroma was determined using unsupervised analysis of 381 proteins which displayed the same direction of enrichment in all 9 patients with LIMMA adjusted p-value < 0.01 exhibiting a Log2 fold-change cut-off ± 1.
Comparison of the most differential diseases/functions between LMD enriched tumor and stroma was determined using unsupervised analysis of 381 proteins which displayed the same direction of enrichment in all 9 patients with LIMMA adjusted p-value < 0.01 exhibiting a Log2 fold-change cut-off ± 1.
Comparison of prevalent drug targets present in LMD enriched tumor versus stroma was determined using unsupervised analysis of 381 proteins which displayed the same direction of enrichment in all 9 patients with LIMMA adjusted p-value < 0.01 exhibiting a Log2 fold-change cut-off ± 1. The complete list of all differentially expressed genes that are known drug targets from our proteomic-level and transcriptomic-level data was compared with a list of 150 FDA-approved anticancer drugs as of 2014 analyzed in a study by Sun et al., 2017.
A composite list of 70 genes and/or proteins that were examined in the clinical setting for cases 343WP, 343WN, and 343WQ was generated, and variability of protein expression in cases 343WK, 343WQ, and 343WH, for which the full complement of 5 levels/case for each LMD enriched tumor, LMD enriched stroma, and whole tumor were available in our proteomic data. Relative standard deviation (RSD) values of protein expression were calculated between the 5 levels within each collection type within a patient's samples and then ranked. The median rank and median RSD between patients for the top 5 most and least variable proteins within each patients' samples are shown.
References
- Ali M., Khan S.A., Wennerberg K., Aittokallio T. Global proteomics profiling improves drug sensitivity prediction: results from a multi-omics, pan-cancer modeling approach. Bioinformatics. 2017;34:1353–1362. doi: 10.1093/bioinformatics/btx766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D., Hu Z., Butte A.J. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18:220. doi: 10.1186/s13059-017-1349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D., Sirota M., Butte A.J. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 2015;6:8971. doi: 10.1038/ncomms9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldelli E., Bellezza G., Haura E.B., Crinó L., Cress W.D., Deng J., Ludovini V., Sidoni A., Schabath M.B., Puma F. Functional signaling pathway analysis of lung adenocarcinomas identifies novel therapeutic targets for KRAS mutant tumors. Oncotarget. 2015;6:32368–32379. doi: 10.18632/oncotarget.5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldelli E., Calvert V., Hodge A., Vanmeter A., Petricoin E.F., Pierobon M. Reverse phase protein microarrays. In: Espina V., editor. Molecular Profiling: Methods and Protocols. Springer; 2017. pp. 149–169. [DOI] [PubMed] [Google Scholar]
- Bashashati A., Ha G., Tone A., Ding J., Prentice L.M., Roth A., Rosner J., Shumansky K., Kalloger S., Senz J. Distinct evolutionary trajectories of primary high-grade serous ovarian cancers revealed through spatial mutational profiling. J. Pathol. 2013;231:21–34. doi: 10.1002/path.4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bast R.C., Klug T.L., John E.S., Jenison E., Niloff J.M., Lazarus H., Berkowitz R.S., Leavitt T., Griffiths C.T., Parker L. A Radioimmunoassay using a monoclonal antibody to monitor the course of epithelial ovarian cancer. N. Engl. J. Med. 1983;309:883–887. doi: 10.1056/NEJM198310133091503. [DOI] [PubMed] [Google Scholar]
- Bateman N.W., Jaworski E., Ao W., Wang G., Litzi T., Dubil E., Marcus C., Conrads K.A., Teng P.-N., Hood B.L. Elevated AKAP12 in paclitaxel-resistant serous ovarian cancer cells is prognostic and predictive of poor survival in patients. J. Proteome Res. 2015;14:1900–1910. doi: 10.1021/pr5012894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentink S., Haibe-Kains B., Risch T., Fan J.-B., Hirsch M.S., Holton K., Rubio R., April C., Chen J., Wickham-Garcia E. Angiogenic mRNA and microRNA gene expression signature predicts a novel subtype of serous ovarian cancer. PLoS One. 2012;7:e30269. doi: 10.1371/journal.pone.0030269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu L., Baba H., Yoshida N., Miyake K., Yasuda T., Uchihara T., Tan P., Ishimoto T. Biological heterogeneity and versatility of cancer-associated fibroblasts in the tumor microenvironment. Oncogene. 2019;38:4887–4901. doi: 10.1038/s41388-019-0765-y. [DOI] [PubMed] [Google Scholar]
- Buczak K., Ori A., Kirkpatrick J.M., Holzer K., Dauch D., Roessler S., Endris V., Lasitschka F., Parca L., Schmidt A. Spatial tissue proteomics quantifies inter- and intratumor heterogeneity in hepatocellular carcinoma (HCC) Mol. Cell Proteomics. 2018;17:810–825. doi: 10.1074/mcp.RA117.000189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canney P.A., Moore M., Wilkinson P.M., James R.D. Ovarian cancer antigen CA125: a prospective clinical assessment of its role as a tumour marker. Br. J. Cancer. 1984;50:765–769. doi: 10.1038/bjc.1984.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G.M., Kannan L., Geistlinger L., Kofia V., Safikhani Z., Gendoo D.M.A., Parmigiani G., Birrer M., Haibe-Kains B., Waldron L. Consensus on molecular subtypes of high-grade serous ovarian carcinoma. Clin. Cancer Res. 2018;24:5037–5047. doi: 10.1158/1078-0432.CCR-18-0784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Geest K., Blessing J.A., Morris R.T., Yamada S.D., Monk B.J., Zweizig S.L., Matei D., Muller C.Y., Richards W.E. Phase II clinical trial of ixabepilone in patients with recurrent or persistent platinum- and taxane-resistant ovarian or primary peritoneal cancer: a gynecologic oncology group study. J. Clin. Oncol. 2010;28:149–153. doi: 10.1200/JCO.2009.24.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert M.A., Coscia F., Chryplewicz A., Chang J.W., Hernandez K.M., Pan S., Tienda S.M., Nahotko D.A., Li G., Blaženović I. Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature. 2019;569:723–728. doi: 10.1038/s41586-019-1173-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feeley K.M., Wells M. Precursor lesions of ovarian epithelial malignancy. Histopathology. 2001;38:87–95. doi: 10.1046/j.1365-2559.2001.01042.x. [DOI] [PubMed] [Google Scholar]
- Fendrick J.L., Staley K.A., Gee M.K., McDougald S.R., Quirk J.J.G., O'Brien T.J. Characterization of CA 125 synthesized by the human epithelial amnion WISH cell line. Tumor Biol. 1993;14:310–318. doi: 10.1159/000217844. [DOI] [PubMed] [Google Scholar]
- Furuya M. Ovarian cancer stroma: pathophysiology and the roles in cancer development. Cancers. 2012;4:701–724. doi: 10.3390/cancers4030701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S., Albitar L., Lebaron R., Welch W.R., Samimi G., Birrer M.J., Berkowitz R.S., Mok S.C. Up-regulation of stromal versican expression in advanced stage serous ovarian cancer. Gynecol. Oncol. 2010;119:114–120. doi: 10.1016/j.ygyno.2010.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton C.A., Miller A., Miller C., Krivak T.C., Farley J.H., Chernofsky M.R., Stany M.P., Rose G.S., Markman M., Ozols R.F. The impact of disease distribution on survival in patients with stage III epithelial ovarian cancer cytoreduced to microscopic residual: a Gynecologic Oncology Group study. Gynecol. Oncol. 2011;122:521–526. doi: 10.1016/j.ygyno.2011.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He D., Wang D., Lu P., Yang N., Xue Z., Zhu X., Zhang P., Fan G. Single-cell RNA sequencing reveals heterogeneous tumor and immune cell populations in early-stage lung adenocarcinomas harboring EGFR mutations. Oncogene. 2021;40:355–368. doi: 10.1038/s41388-020-01528-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helland Å., Anglesio M.S., George J., Cowin P.A., Johnstone C.N., House C.M., Sheppard K.E., Etemadmoghadam D., Melnyk N., Rustgi A.K. Deregulation of MYCN, LIN28B and LET7 in a molecular subtype of aggressive high-grade serous ovarian cancers. PLoS One. 2011;6:e18064. doi: 10.1371/journal.pone.0018064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y., Taylor-Harding B., Raz Y., Haro M., Recouvreux M.S., Taylan E., Lester J., Millstein J., Walts A.E., Karlan B.Y., Orsulic S. Are epithelial ovarian cancers of the mesenchymal subtype Actually intraperitoneal metastases to the ovary? Front. Cell Dev. Biol. 2020;8:647. doi: 10.3389/fcell.2020.00647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izar B., Tirosh I., Stover E.H., Wakiro I., Cuoco M.S., Alter I., Rodman C., Leeson R., Su M.-J., Shah P. A single-cell landscape of high-grade serous ovarian cancer. Nat. Med. 2020;26:1271–1279. doi: 10.1038/s41591-020-0926-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia D., Liu Z., Deng N., Tan T.Z., Huang R.Y.-J., Taylor-Harding B., Cheon D.-J., Lawrenson K., Wiedemeyer W.R., Walts A.E. A COL11A1-correlated pan-cancer gene signature of activated fibroblasts for the prioritization of therapeutic targets. Cancer Lett. 2016;382:203–214. doi: 10.1016/j.canlet.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X., Li X., Li W., Bai H., Zhang Z. PARP inhibitors in ovarian cancer: sensitivity prediction and resistance mechanisms. J. Cell. Mol. Med. 2019;23:2303–2313. doi: 10.1111/jcmm.14133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Sánchez A., Cybulska P., Mager K.L., Koplev S., Cast O., Couturier D.-L., Memon D., Selenica P., Nikolovski I., Mazaheri Y. Unraveling tumor–immune heterogeneity in advanced ovarian cancer uncovers immunogenic effect of chemotherapy. Nat. Genet. 2020;52:582–593. doi: 10.1038/s41588-020-0630-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Sanchez A., Memon D., Pourpe S., Veeraraghavan H., Li Y., Vargas H.A., Gill M.B., Park K.J., Zivanovic O., Konner J. Heterogeneous tumor-immune microenvironments among differentially growing metastases in an ovarian cancer patient. Cell. 2017;170:927–938 e20. doi: 10.1016/j.cell.2017.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konecny G.E., Wang C., Hamidi H., Winterhoff B., Kalli K.R., Dering J., Ginther C., Chen H.-W., Dowdy S., Cliby W. Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. J. Natl. Cancer Inst. 2014;106:dju249. doi: 10.1093/jnci/dju249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer I., Lipp H.P. Bevacizumab, a humanized anti-angiogenic monoclonal antibody for the treatment of colorectal cancer. J. Clin. Pharm. Ther. 2007;32:1–14. doi: 10.1111/j.1365-2710.2007.00800.x. [DOI] [PubMed] [Google Scholar]
- Labiche A., Heutte N., Herlin P., Chasle J., Gauduchon P., Elie N. Stromal compartment as a survival prognostic factor in advanced ovarian carcinoma. Int. J. Gynecol. Cancer. 2010;20:28–33. doi: 10.1111/IGC.0b013e3181bda1cb. [DOI] [PubMed] [Google Scholar]
- Lappalainen I., Almeida-King J., Kumanduri V., Senf A., Spalding J.D., Ur-Rehman S., Saunders G., Kandasamy J., Caccamo M., Leinonen R. The European Genome-phenome Archive of human data consented for biomedical research. Nat. Genet. 2015;47:692–695. doi: 10.1038/ng.3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Zhao L., Rojas C., Bateman N.W., Yao H., Lara O.D., Celestino J., Morgan M.B., Nguyen T.V., Conrads K.A. Molecular analysis of clinically defined subsets of high-grade serous ovarian cancer. Cell Rep. 2020;31:107502. doi: 10.1016/j.celrep.2020.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong H.S., Galletta L., Etemadmoghadam D., George J., Study T.A.O.C., Köbel M., Ramus S.J., Bowtell D. Efficient molecular subtype classification of high-grade serous ovarian cancer. J. Pathol. 2015;236:272–277. doi: 10.1002/path.4536. [DOI] [PubMed] [Google Scholar]
- Liu Z., Beach J.A., Agadjanian H., Jia D., Aspuria P.-J., Karlan B.Y., Orsulic S. Suboptimal cytoreduction in ovarian carcinoma is associated with molecular pathways characteristic of increased stromal activation. Gynecol. Oncol. 2015;139:394–400. doi: 10.1016/j.ygyno.2015.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y., Feng Q., Zheng P., Yang L., Liu T., Xu Y., Zhu D., Chang W., Ji M., Ren L. Low tumor purity is associated with poor prognosis, heavy mutation burden, and intense immune phenotype in colon cancer. Cancer Manag. Res. 2018;10:3569–3577. doi: 10.2147/CMAR.S171855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masoumi Moghaddam S., Amini A., Morris D.L., Pourgholami M.H. Significance of vascular endothelial growth factor in growth and peritoneal dissemination of ovarian cancer. Cancer Metastasis Rev. 2012;31:143–162. doi: 10.1007/s10555-011-9337-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narikiyo M., Yano M., Kamada K., Katoh T., Ito K., Shuto M., Kayano H., Yasuda M. Molecular association of functioning stroma with carcinoma cells in the ovary: a preliminary study. Oncol. Lett. 2019;17:3562–3568. doi: 10.3892/ol.2019.9992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noone Am H.N., Krapcho M., Miller D., Brest A., Yu M., Ruhl J., Tatalovich Z., Mariotto A., Lewis D.R., Chen H.S. National Cancer Institute; 2018. SEER Cancer Statistics Review, 1975-2015.https://seer.cancer.gov/csr/1975_2015/ based on November 2017 SEER data submission, posted to the SEER web site. [Google Scholar]
- O’Brien T.J., Beard J.B., Underwood L.J., Dennis R.A., Santin A.D., York L. The CA 125 gene: an extracellular superstructure dominated by repeat sequences. Tumor Biol. 2001;22:348–366. doi: 10.1159/000050638. [DOI] [PubMed] [Google Scholar]
- O’Brien T.J., Beard J.B., Underwood L.J., Shigemasa K. The ca 125 gene: a newly discovered extension of the glycosylated N-terminal domain doubles the size of this extracellular superstructure. Tumor Biol. 2002;23:154–169. doi: 10.1159/000064032. [DOI] [PubMed] [Google Scholar]
- Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47 doi: 10.1093/nar/gky1106. D442–d450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi L., Verrico M., Zaccarelli E., Papa A., Colonna M., Strudel M., Vici P., Bianco V., Tomao F. Bevacizumab in ovarian cancer: a critical review of phase III studies. Oncotarget. 2017;8:12389–12405. doi: 10.18632/oncotarget.13310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosso M., Majem B., Devis L., Lapyckyj L., Besso M.J., Llauradó M., Abascal M.F., Matos M.L., Lanau L., Castellví J. E-cadherin: a determinant molecule associated with ovarian cancer progression, dissemination and aggressiveness. PLoS One. 2017;12:e0184439. doi: 10.1371/journal.pone.0184439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlan M.J., Raj B.K., Calvo B., Garin-Chesa P., Sanz-Moncasi M.P., Healey J.H., Old L.J., Rettig W.J. Molecular cloning of fibroblast activation protein alpha, a member of the serine protease family selectively expressed in stromal fibroblasts of epithelial cancers. Proc. Natl. Acad. Sci. U S A. 1994;91:5657–5661. doi: 10.1073/pnas.91.12.5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz R.F., Ng C.K.Y., Cooke S.L., Newman S., Temple J., Piskorz A.M., Gale D., Sayal K., Murtaza M., Baldwin P.J. Spatial and temporal heterogeneity in high-grade serous ovarian cancer: a phylogenetic analysis. PLoS Med. 2015;12:e1001789. doi: 10.1371/journal.pmed.1001789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwede M., Waldron L., Mok S.C., Wei W., Basunia A., Merritt M.A., Mitsiades C.S., Parmigiani G., Harrington D.P., Quackenbush J. The impact of stroma admixture on molecular subtypes and prognostic gene signatures in serous ovarian cancer. Cancer Epidemiol. Biomark. Prev. 2020;29:509–519. doi: 10.1158/1055-9965.EPI-18-1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih A.J., Menzin A., Whyte J., Lovecchio J., Liew A., Khalili H., Bhuiya T., Gregersen P.K., Lee A.T. Identification of grade and origin specific cell populations in serous epithelial ovarian cancer by single cell RNA-seq. PLoS One. 2018;13:e0206785. doi: 10.1371/journal.pone.0206785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2020. CA Cancer J. Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- Signore M., Reeder K.A. Antibody validation by Western blotting. Methods Mol. Biol. 2012;823:139–155. doi: 10.1007/978-1-60327-216-2_10. [DOI] [PubMed] [Google Scholar]
- Skirnisdottir I., Seidal T., Åkerud H. The relationship of the angiogenesis regulators VEGF-A, VEGF-R1 and VEGF-R2 to p53 status and prognostic factors in epithelial ovarian carcinoma in FIGO-stages I-II. Int. J. Oncol. 2016;48:998–1006. doi: 10.3892/ijo.2016.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J., Wei Q., Zhou Y., Wang J., Liu Q., Xu H. A systematic analysis of FDA-approved anticancer drugs. BMC Syst. Biol. 2017;11:87. doi: 10.1186/s12918-017-0464-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan T., Ong W.S., Chan S., Chan J., Chay W.Y., Lim E., Lim S.L., Soh L.T., Tan D.S., Chia J. Interim analysis of A single-arm phase 2 clinical trial of regorafenib in patients with epithelial ovarian cancer. Ann. Oncol. 2017;28:x89–x90. [Google Scholar]
- Tewari K.S., Burger R.A., Enserro D., Norquist B.M., Swisher E.M., Brady M.F., Bookman M.A., Fleming G.F., Huang H., Homesley H.D. Final overall survival of a randomized trial of bevacizumab for primary treatment of ovarian cancer. J. Clin. Oncol. 2019;37:2317–2328. doi: 10.1200/JCO.19.01009. JCO.19.01009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torre L.A., Trabert B., Desantis C.E., Miller K.D., Samimi G., Runowicz C.D., Gaudet M.M., Jemal A., Siegel R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018;68:284–296. doi: 10.3322/caac.21456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres D., Wang C., Kumar A., Bakkum-Gamez J.N., Weaver A.L., McGree M.E., Konecny G.E., Goode E.L., Cliby W.A. Factors that influence survival in high-grade serous ovarian cancer: a complex relationship between molecular subtype, disease dissemination, and operability. Gynecol. Oncol. 2018;150:227–232. doi: 10.1016/j.ygyno.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker S.L., Gharpure K., Herbrich S.M., Unruh A.K., Nick A.M., Crane E.K., Coleman R.L., Guenthoer J., Dalton H.J., Wu S.Y. Molecular biomarkers of residual disease after surgical debulking of high-grade serous ovarian cancer. Clin. Cancer Res. 2014;20:3280–3288. doi: 10.1158/1078-0432.CCR-14-0445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verardo R., Piazza S., Klaric E., Ciani Y., Bussadori G., Marzinotto S., Mariuzzi L., Cesselli D., Beltrami A.P., Mano M. Specific mesothelial signature marks the heterogeneity of mesenchymal stem cells from high-grade serous ovarian cancer. Stem Cells. 2014;32:2998–3011. doi: 10.1002/stem.1791. [DOI] [PubMed] [Google Scholar]
- Vergote I.B., Børmer O.P., Abeler V.M. Evaluation of serum CA 125 levels in the monitoring of ovarian cancer. Am. J. Obstet. Gynecol. 1987;157:88–92. doi: 10.1016/s0002-9378(87)80352-6. [DOI] [PubMed] [Google Scholar]
- Verhaak R.G.W., Tamayo P., Yang J.-Y., Hubbard D., Zhang H., Creighton C.J., Fereday S., Lawrence M., Carter S.L., Mermel C.H., Cancer Genome Atlas Research Network Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J. Clin. Invest. 2013;123:517–525. doi: 10.1172/JCI65833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Armasu S.M., Kalli K.R., Maurer M.J., Heinzen E.P., Keeney G.L., Cliby W.A., Oberg A.L., Kaufmann S.H., Goode E.L. Pooled clustering of high-grade serous ovarian cancer gene expression leads to novel consensus subtypes associated with survival and surgical outcomes. Clin. Cancer Res. 2017;23:4077–4085. doi: 10.1158/1078-0432.CCR-17-0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterhoff B.J., Maile M., Mitra A.K., Sebe A., Bazzaro M., Geller M.A., Abrahante J.E., Klein M., Hellweg R., Mullany S.A. Single cell sequencing reveals heterogeneity within ovarian cancer epithelium and cancer associated stromal cells. Gynecol. Oncol. 2017;144:598–606. doi: 10.1016/j.ygyno.2017.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Lin Y., Shi Y., Li B., Liu W., Yin W., Dang Y., Chu Y., Fan J., He R. FAP promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3–CCL2 signaling. Cancer Res. 2016;76:4124–4135. doi: 10.1158/0008-5472.CAN-15-2973. [DOI] [PubMed] [Google Scholar]
- Yin B.W.T., Dnistrian A., Lloyd K.O. Ovarian cancer antigen CA125 is encoded by the MUC16 mucin gene. Int. J. Cancer. 2002;98:737–740. doi: 10.1002/ijc.10250. [DOI] [PubMed] [Google Scholar]
- Yue H., Wang J., Chen R., Hou X., Li J., Lu X. Gene signature characteristic of elevated stromal infiltration and activation is associated with increased risk of hematogenous and lymphatic metastasis in serous ovarian cancer. BMC Cancer. 2019;19:1266. doi: 10.1186/s12885-019-6470-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A.W., Mcpherson A., Milne K., Kroeger D.R., Hamilton P.T., Miranda A., Funnell T., Little N., De Souza C.P.E., Laan S. Interfaces of malignant and immunologic clonal dynamics in ovarian cancer. Cell. 2018;173:1755–1769.e22. doi: 10.1016/j.cell.2018.03.073. [DOI] [PubMed] [Google Scholar]
- Zhang C., Cheng W., Ren X., Wang Z., Liu X., Li G., Han S., Jiang T., Wu A. Tumor purity as an underlying key factor in glioma. Clin. Cancer Res. 2017;23:6279–6291. doi: 10.1158/1078-0432.CCR-16-2598. [DOI] [PubMed] [Google Scholar]
- Zhang Q., Wang C., Cliby W.A. Cancer-associated stroma significantly contributes to the mesenchymal subtype signature of serous ovarian cancer. Gynecol. Oncol. 2019;152:368–374. doi: 10.1016/j.ygyno.2018.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Jing Y., Zhang M., Zhang Z., Ma P., Peng H., Shi K., Gao W.-Q., Zhuang G. Stroma-associated master regulators of molecular subtypes predict patient prognosis in ovarian cancer. Sci. Rep. 2015;5:16066. doi: 10.1038/srep16066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C., Li S., Zhao M., Zhu H., Zhu X. Prognostic values of DNA mismatch repair genes in ovarian cancer patients treated with platinum-based chemotherapy. Arch. Gynecol. Obstet. 2018;297:153–159. doi: 10.1007/s00404-017-4563-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative H&E stained glass slides from were examined at ∼200 μm intervals for case 343WM or ∼100 μm intervals for the additional 9 cases by a board-certified pathologist for percent by area estimation of tumor cellularity, necrosis, stroma, normal ovarian epithelium, lymphocytes, and polymorphonuclear leukocytes (PMN). Multiple images per level per case were reviewed and the median tumor cellularity and median stroma cellularity are reported with corresponding percent coefficient of variation (%CV) reported in parentheses. Abbreviations: NACT = neoadjuvant chemotherapy; FT = fallopian tube; Ov = ovary; ca = cancer; NOS = not otherwise specified.
Numerical values indicate the number of LMD tissue sections that were used for each collection.
ROI are denoted as millimeter squared (mm2) areas, cell nuclei as μm2 areas or as total cell counts derived from cellular nuclei measured in LMD enriched tumor or stroma ROIs.
. For 343WM, tryptic peptides were labeled with isobaric Tandem Mass Tags (TMT) according to the manufacturer's instructions using the TMT 10-Plex Kit from Thermo Fisher for a total of 4 plexes. 30 μg peptides from the 8 cores, 9 non-discrete whole tumor collections, and cryopulverized tissue sample were individually mixed with respective TMT reagents for 1 hr at room temperature, then quenched using 5% hydroxylamine. 10 μg peptides/sample were similarly TMT labeled from the 9 collections each of remaining LMD-enriched tumor epithelium or stroma. For the remaining 9 cases, digested peptide samples were labeled in patient-specific plexes using the TMTpro 16-Plex Kit (Thermo Fisher). Specifically, 5 μg peptide from up to 5 levels/patient each of LMD enriched tumor epithelium, LMD enriched stroma, and whole tumor collections were incubated with the TMTpro reagents per plex for a total of 80 μg/plex and 1 plex/patient. To replace samples from which <5 μg peptide was recovered, empty TMT channels were filled using equivalent amounts of peptide mixed from each of the 5 levels of the whole tumor collections.
. Log2 transformed fold-change abundances of 6,053 proteins imputed across all samples measured in case 343WM.
Normalized abundances of 20,535 RNA transcripts measured in case 343WM calculated relative to the average RPM abundance quantified across all samples for a given transcript, as reported by the Torrent Suite (v5.8.0) software, before Log2 fold-change transformation.
Log2 transformed fold-change abundances of 6,199 proteins imputed across all samples (n = 123).
Normalized abundances of 19,758 RNA transcripts calculated relative to the average RPM abundance quantified across all samples (n = 6) for a given transcript, as reported by the Torrent Suite (v5.8.0) software, before Log2 fold-change transformation.
http://xcell.ucsf.edu/, [Aran et al., 2017]), related to Figure 3A
http://xcell.ucsf.edu/, [Aran et al., 2017]), related to Figures 3C and 7
P-values indicate the reliability of the presence or absence of a signal peptide or extracellular classification within the indicated LMD-enriched tissue across all levels/case. 9 levels were used for comparison for case 343WM, while 5 levels/case were used each for cases 343WK, 343WQ, and 343WH.
Log2 transformed fold-change abundances of 5,742 imputed proteins that were co-measured at the transcriptome level from case 343WM.
Log2 transformed fold-change abundances of 5,379 imputed proteins that were co-measured at the transcriptome level.
Comparison of the most differential pathways and diseases/functions between LMD enriched tumor and stroma was determined using pairwise supervised analysis of proteomic and transcriptomic data with LIMMA adjusted p-value < 0.01 exhibiting a Log2 fold-change cut-off ± 1.
Comparison of the most differential diseases/functions between LMD enriched tumor and stroma was determined using pairwise supervised analysis of proteomic and transcriptomic data with LIMMA adjusted p-value < 0.01 exhibiting a Log2 fold-change cut-off ± 1.
Comparison of prevalent drug targets present in LMD enriched tumor versus stroma was determined using pairwise supervised analysis of proteomic and transcriptomic data with LIMMA adjusted p-value < 0.01 exhibiting a Log2 fold-change cut-off ± 1. The complete list of all differentially expressed genes that are known drug targets from our proteomic-level and transcriptomic-level data was compared with a list of 150 FDA-approved anticancer drugs as of 2014 analyzed in a study by Sun et al. (2017).
Comparison of the most differential canonical pathways between LMD enriched tumor and stroma was determined using unsupervised analysis of 381 proteins which displayed the same direction of enrichment in all 9 patients with LIMMA adjusted p-value < 0.01 exhibiting a Log2 fold-change cut-off ± 1.
Comparison of the most differential diseases/functions between LMD enriched tumor and stroma was determined using unsupervised analysis of 381 proteins which displayed the same direction of enrichment in all 9 patients with LIMMA adjusted p-value < 0.01 exhibiting a Log2 fold-change cut-off ± 1.
Comparison of prevalent drug targets present in LMD enriched tumor versus stroma was determined using unsupervised analysis of 381 proteins which displayed the same direction of enrichment in all 9 patients with LIMMA adjusted p-value < 0.01 exhibiting a Log2 fold-change cut-off ± 1. The complete list of all differentially expressed genes that are known drug targets from our proteomic-level and transcriptomic-level data was compared with a list of 150 FDA-approved anticancer drugs as of 2014 analyzed in a study by Sun et al., 2017.
A composite list of 70 genes and/or proteins that were examined in the clinical setting for cases 343WP, 343WN, and 343WQ was generated, and variability of protein expression in cases 343WK, 343WQ, and 343WH, for which the full complement of 5 levels/case for each LMD enriched tumor, LMD enriched stroma, and whole tumor were available in our proteomic data. Relative standard deviation (RSD) values of protein expression were calculated between the 5 levels within each collection type within a patient's samples and then ranked. The median rank and median RSD between patients for the top 5 most and least variable proteins within each patients' samples are shown.
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD026570. The RNA-sequencing transcriptomics data have been deposited to the European Genome-Phenome Archive (Lappalainen et al., 2015).