


Abstract
Experiments that are planned using accurate prediction algorithms will mitigate failures in recombinant protein production. We have developed TISIGNER (https://tisigner.com) with the aim of addressing technical challenges to recombinant protein production. We offer three web services, TIsigner (Translation Initiation coding region designer), SoDoPE (Soluble Domain for Protein Expression) and Razor, which are specialised in synonymous optimisation of recombinant protein expression, solubility and signal peptide analysis, respectively. Importantly, TIsigner, SoDoPE and Razor are linked, which allows users to switch between the tools when optimising genes of interest.
Graphical Abstract
Graphical Abstract.
Optimising recombinant protein production using TISIGNER.com
INTRODUCTION
Recombinant protein production is a key process for life science research and the development of biotherapeutics. However, low protein expression and aggregation are the two major bottlenecks of recombinant protein production (1–7). Since mRNA abundance alone is insufficient to explain protein abundance (8–12), several features of mRNA sequence have been proposed to affect protein expression. These features are mostly related to codon usage, such as the codon adaptation index and tRNA adaptation index (13–17), or measures of mRNA secondary structure, such as G+C content, minimum free energy (MFE) of RNA secondary structure, and mRNA:ncRNA interaction avoidance (18–23). Many of these features are not independent, making it challenging to distinguish the impacts of individual features (24). This, in turn, hinders the development of accurate prediction/optimisation tools. Recent systematic studies suggest that MFE is the most important feature in protein expression (24,25). However, more recent work shows that the mRNA accessibility of translation initiation sites outperforms MFE in predicting relative protein levels from mRNA sequences (26,27). Accessibility is computed by considering all possible structures for a region, weighted by free energy, not just the single structure with the MFE (28).
In addition to high protein expression level, high solubility is preferable for the purification and long-term storage of recombinant proteins. However, almost half of the successfully expressed proteins are insoluble (http://targetdb.rcsb.org/metrics), which makes the recombinant protein production process challenging. A number of methods have been suggested to improve protein solubility, for example, truncation, mutagenesis, and the use of solubility-enhancing tags (2,29–31). Nevertheless, accurate solubility prediction could save resources and aid in designing soluble proteins before the experiments. With these in mind, we have recently formulated the solubility-weighted index (SWI), which outperforms recent solubility prediction tools based on machine-learning algorithms (32).
Besides, many recombinant proteins of interest are secretory. The intracellular accumulation of heterologous secretory proteins may be toxic to the host cells. Therefore, the translocation efficiency of these proteins plays an important role in the yield quantity and quality. Secretory proteins usually have a short peptide at the N-terminus called signal peptide (SP), which is responsible for the translocation of secretory proteins via the Sec, signal recognition particle (SRP) or twin arginine transport (Tat) pathways (33–36). Detection of SPs or fusion of a suitable SP at the N-terminus is useful for optimising protein production (37–40). In addition, different pathways have different advantages, for example, the SRP dependent pathway can be used for rapidly folding proteins (41). However, the Sec dependent pathway, which is common across all forms of life, has been widely used for recombinant protein expression because of higher protein production capacity and quality (41,42). In addition, the presence of SPs should almost always be checked when planning the expression experiments for uncharacterised proteins.
Existing web tools predict or optimise either protein expression or solubility alone (43–51). Several web tools exist for predicting SPs (52–56). Only a very few tools can detect toxic proteins, for example, SpiderP, ClanTox and ToxinPred (57–59). These tools are either limited to predicting the venoms of certain organisms, such as spiders, or they are not designed to predict the signal peptides of toxins, rather to predict the toxicity of mature peptides. Moreover, these tools are offered through different independent services. We reasoned these functionalities should be integrated in order to assist not only in choosing appropriate expression systems, but also in optimising the expression and solubility levels of recombinant proteins. Here we present TISIGNER.com that integrates the optimisation tools TIsigner (translation initiation coding region designer), SoDoPE (soluble domain for protein expression) for protein expression and solubility, respectively, and Razor for detecting SPs (26,32,60). Our web application provides easy, fast and interactive ways to assist users in planning and designing their experiments.
WEB SERVICES
TIsigner
TIsigner offers tunable protein expression by optimising the mRNA accessibility of translation initiation sites (26). The regions used to calculate accessibility (opening energy) are specific to the expression hosts, which is calculated using RNAplfold (28,61,62). For Escherichia coli, Saccharomyces cerevisiae, and Mus musculus expression hosts, the optimal regions relative to the start codon for optimisation are −24:24, −7:89, −8:11, respectively. For other expression hosts, we provide an option ‘Other’, which optimises the accessibility of the region −24:89. Since E. coli is the most popular expression host, the default settings aim to optimise protein expression in E. coli with the T7 lac promoter system (see below). In this case, only the protein coding sequence is required for input where the 5′UTR (5′ untranslated region) sequence used as default is the most popular, truncated version of the T7 promoter (63) (Figure 1). Otherwise, the 5′UTR sequence is also required. For 5′UTRs shorter than 71 nucleotides, upstream sequences can be used to extend the UTRs.
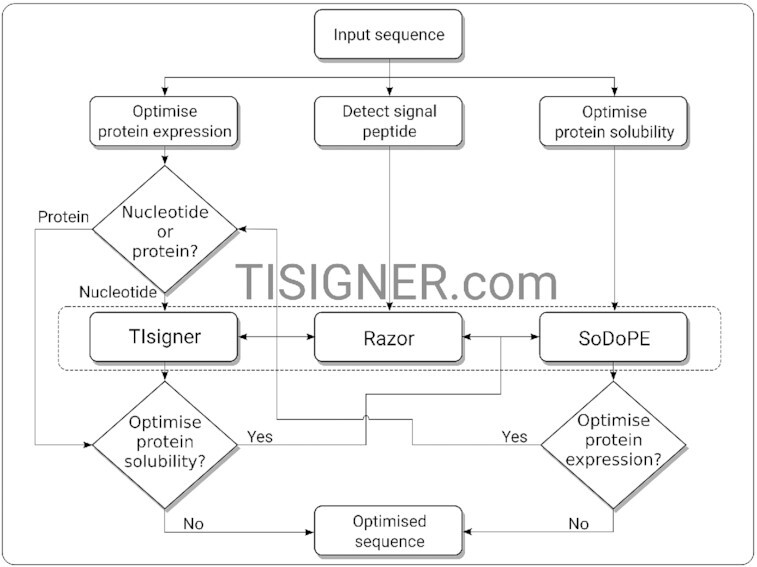
Figure 1.

Flow chart for optimising recombinant protein production using the TISIGNER web application. TIsigner, SoDoPE and Razor are linked so that protein expression and solubility can be seamlessly optimised. TIsigner accepts a nucleotide sequence as input, whereas SoDoPE and Razor accept either a nucleotide or protein sequence. SoDoPE, soluble domain for protein expression; TIsigner, translation initiation coding region designer.
The settings for TIsigner are grouped by complexity (i.e. general, extra, and advanced). The general settings include the options to modify the expression host, promoter and target expression score. The target expression score ranges from 0 to 100 (i.e. from the minimum to maximum predicted level), which is derived from a logistic regression of the opening energy distribution of 11 430 expression experiments in Escherichia coli from the ‘Protein Structure Initiative: Biology’ (PSI:Biology) (64,65). Hence, this scoring system is only applicable to the E. coli T7 lac promoter system. Since, there is a non-linear relationship between opening energy and expression score, an interactive plot is also displayed along with the slider to set the target expression score. For other expression hosts and promoters, the target expression level can be either maximised or minimised (i.e. binary). The extra settings have the options to optimise sequence within the translation initiation region or the full-length sequence. The AarI, BsaI, BsmBI restriction modification sites are filtered by default, whereas other sites can be manually supplied (e.g. a Shine-Dalgarno motif or terminator U-tract). The advanced settings allows users to tweak the random seed and sampling options (i.e., quick or deep, which uses different numbers of iterations and parallel processes). Here users can also customise the region for optimisation or disable the terminator checks.
Once the input sequence passes a sanity check, the optimisation task is rapid [O(1) time using RNAplfold v2.4.11 (using parameters -W 210 -u 210)] with our simulated annealing algorithm. A list of optimized sequences are returned after checking for terminators using cmsearch (Infernal v1.1.2) (66) with RMfam models (67,68). If terminators are found, an option to use the full-length sequence for optimisation will be prompted to users. In a default case (E. coli T7 lac promoter system), the optimised sequence closest to the chosen expression level is selected as the first solution (Figure 2). For other expression hosts and/or promoters, the optimised sequence with the minimum changes in nucleotides is selected as the first solution. The altered nucleotides are highlighted (Figure 2). The accessibility of translation initiation sites for both the input and optimised sequences is shown as opening energy (kcal/mol). The results can be exported as a PDF or CSV file. When the default settings are used, the opening energy for each sequence is indicated on the distributions of the opening energy of 8780 ‘success’ and 2650 ‘failure’ groups of the PSI:Biology target genes. Furthermore, options for solubility and SP analyses using SoDoPE and Razor, respectively, are available for each sequence on the same results page (Figure 2).
Figure 2.

The results of TIsigner shows a protein expression optimised nucleotide sequence. The highlighted nucleotides show changes made to the input sequence. The opening energy of the input sequence before and after optimisation is annotated over the distributions of the opening energy for 8780 ‘success’ and 2650 ‘failure’ experiments from PSI:Biology. Further optimised sequences, if found, are also displayed. The results can be downloaded in either CSV or PDF format using the download icon on the bottom right. Each resulting sequence can be analysed for solubility or signal peptide.
SoDoPE
SoDoPE is our interactive solubility analysis and optimisation tool based on the SWI (32). SoDoPE accepts either a nucleotide or protein sequence (Figure 1). Upon submission, a query is sent to the HMMER web service for domain annotation (69). Successful annotations are displayed as interactive graphics, in which the annotated domains are represented as discorectangles, above a grey band that represents the input protein sequence (Figure 3). Information about a protein domain is shown upon a mouse hover. The domains can be selected for solubility analysis. For a complete domain annotation report, a link to the HMMER results page is also provided.
Figure 3.

Exploring and optimizing protein solubility using SoDoPE interactive graphics. Upon clicking a protein domain or selecting a region of interest, its solubility is optimised in real-time, and a list of regions with extended boundaries and higher probabilities of solubility is returned as green buttons (clickable). The probabilities of solubility of the selected region with and without fusion tags can be visualized in a barplot. The flexibility and hydrophobicity profile plots for the selected region can also be selectively viewed. The sequence can also be checked for the presence of a signal peptide or optimized for protein expression.
In addition, a two-way slider is available for navigation through any region of interest (Figure 3). The probability of solubility, flexibility and GRAVY (grand average of hydropathicity) is shown in real-time according to the user-selected region. The selected region is optimised for higher solubility using simulated annealing. Only the regions with extended boundaries and also higher probability of solubility is returned. SP analysis can also be done using Razor (see below).
A profile plot of flexibility and/or hydrophilicity corresponding to the user selected region is generated (Figure 3). This allows an estimation of rigid/flexible regions and possible helices, that may be helpful for mutagenesis experiments. The sequence of the selected region is shown, with the option of sequence conversion between nucleotide and amino acid sequence format. In particular, the nucleotide sequence can be redirected to TIsigner for optimising protein expression (Figures 1 and 3, through the ‘view DNA | optimise expression’ button).
The contributions of several solubility-enhancing tags to user selected regions can be compared and shown in a bar plot, including thioredoxin (TRX), maltose binding protein (MBP), small ubiquitin-related modifier (SUMO) and glutathione-S-transferase (GST) tags (Figure 3). Users can also input a fusion sequence of interest either in a nucleotide or protein sequence format.
Razor
Razor is our SP prediction tool which is based upon random forest models of protein features from the eukaryotic SP sequences of the SignalP 5.0 dataset and the animal toxin annotation project (52,60,70). Razor accepts either a protein or a nucleotide sequence (Figure 1). After validation, the N-terminal region is checked for the presence of a SP using five random forest models. This gives five SP scores (S-scores) for a given sequence. For detecting the cleavage site, we use a sliding window of 30 residues and our optimised weight matrix for residues around the cleavage site. The scored subsequences are scored by additional five random forest models to give the cleavage site scores (C-scores) along the sequence, which is displayed as a step plot (Figure 4). The Y-score, which is the geometric mean of S-scores and the max of C-scores, is used to infer whether the given sequence has a SP or not. The median of these five Y-scores is displayed as the final score. The cleavage site from the model with the median of max of C-scores is used to annotate the predicted region.
Figure 4.

Detection of signal peptides using Razor. The dotted annotation in the step plot for the cleavage site scores (C-scores) shows the most likely position for proteolytic cleavage. The sequence can also be checked and optimised for protein solubility and expression.
If any of the models detect a SP in the input sequence, we further check whether the SP belongs to toxins, using five random forests trained on toxin SPs. The final toxin score is the median of scores from those random forest models. Furthermore, since we noticed a lack of tools specialising in predicting SPs from fungi, any detected signal peptide is checked for such origin. Similarly, we use five random forests for detecting fungal SPs, with the final fungal score being the median score of these models. Since we have five random forest models in each step (eukaryotic, toxin and fungal SP detection steps), stars are displayed as an indication of the number of models agreeing on the sequence falling on either category (Figure 4).
Razor is linked with SoDoPE for checking and optimising protein solubility (Figure 4). If a nucleotide sequence was submitted, this sequence can also be optimised for protein expression using TIsigner (Figure 1).
DISCUSSION
Low protein expression and solubility are the major hindrances to a successful recombinant protein production. Based on our comprehensive studies on these two problems, we have developed novel tools to optimise protein expression (TIsigner) and solubility (SoDoPE), and assessed their predictive performance using independent datasets (Supplementary Table S1). Our tools offer some unique features in an interactive way. TIsigner allows tuning of protein expression from low to high levels, whereas SoDoPE allows easy navigation of protein sequence/domains with real-time solubility prediction. Based on our assessment of similar tools, none of the publicly available tools provides these features.
Our third tool, Razor, is designed to check the presence of SPs. Compared to other related tools, Razor also predicts toxin and fungal SPs (Supplementary Table S2). These would be helpful for users in choosing the expression and purification systems that prevent the harmful intracellular accumulation of recombinant secretory proteins/toxins.
Our tools are interactive, fast, and accurate. Importantly, our tools are highly integrated, allowing a seamless transition between the optimisation tools. To make such transition intuitive, our web services limits one input sequence at a time and we aim to remove this input sequence limitation in the future. For optimising a large number of sequence, we provide the command-line version of each of our tools (see below).
GENERAL INFORMATION
Demo input and results are available for new users to get started. A list of frequently asked questions is also available for each tool. The frontend is written in React and uses responsive web design principles. The backend is written in Flask and Python v3.6. The website is hosted on a virtual machine (Red Hat Enterprise Linux 8) running on Intel Xeon (8 × 2.60 GHz) with 4GiB RAM, by the Information Technology Services at the University of Otago.
DATA AVAILABILITY
The web server is available at https://tisigner.com. This website is free and open to all users and there is no login required. All our tools, and the website are open-sourced (https://github.com/Gardner-BinfLab/TISIGNER-ReactJS; https://github.com/Gardner-BinfLab/TIsigner/tree/master/TIsigner_cmd; https://github.com/Gardner-BinfLab/SoDoPE_paper_2020/tree/master/SWI; https://github.com/Gardner-BinfLab/Razor) and privacy friendly (no data stored).
Supplementary Material
Contributor Information
Bikash K Bhandari, Department of Biochemistry, School of Biomedical Sciences, University of Otago, Dunedin 9054, New Zealand.
Chun Shen Lim, Department of Biochemistry, School of Biomedical Sciences, University of Otago, Dunedin 9054, New Zealand.
Paul P Gardner, Department of Biochemistry, School of Biomedical Sciences, University of Otago, Dunedin 9054, New Zealand; Biomolecular Interaction Centre, University of Canterbury, Christchurch 8140, New Zealand.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ministry of Business, Innovation and Employment Smart Idea [UOOX1709 to P.P.G]; MBIE Data Science Programmes [UOAX1932 to P.P.G]; Royal Society of New Zealand Te Apārangi Marsden Fund [19-UOO-040 to P.P.G]. Funding for open access charge: Ministry of Business, Innovation and Employment Smart Idea [UOOX1709].
Conflict of interest statement. None declared.
REFERENCES
- 1. Berlec A., Strukelj B.. Current state and recent advances in biopharmaceutical production in Escherichia coli, yeasts and mammalian cells. J. Ind. Microbiol. Biotechnol. 2013; 40:257–274. [DOI] [PubMed] [Google Scholar]
- 2. Esposito D., Chatterjee D.K.. Enhancement of soluble protein expression through the use of fusion tags. Curr. Opin. Biotechnol. 2006; 17:353–358. [DOI] [PubMed] [Google Scholar]
- 3. Hou Q., Bourgeas R., Pucci F., Rooman M.. Computational analysis of the amino acid interactions that promote or decrease protein solubility. Scientific Rep. 2018; 8:14661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kramer R.M., Shende V.R., Motl N., Pace C.N., Scholtz J.M.. Toward a molecular understanding of protein solubility: increased negative surface charge correlates with increased solubility. Biophys. J. 2012; 102:1907–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mazurenko S. Predicting protein stability and solubility changes upon mutations: data perspective. ChemCatChem. 2020; 12:5590–5598. [Google Scholar]
- 6. Rosano G.L., Ceccarelli E.A.. Recombinant protein expression in Escherichia coli: advances and challenges. Front. Microbiol. 2014; 5:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vihinen M. Solubility of proteins. ADMET DMPK. 2020; 8:391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bernstein J.A., Khodursky A.B., Lin P.-H., Lin-Chao S., Cohen S.N.. Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:9697–9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. de Sousa Abreu R., Penalva L.O., Marcotte E.M., Vogel C.. Global signatures of protein and mRNA expression levels. Mol. Biosyst. 2009; 5:1512–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lim C.S., T. Wardell S.J., Kleffmann T., Brown C.M.. The exon–intron gene structure upstream of the initiation codon predicts translation efficiency. Nucleic Acids Res. 2018; 46:4575–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nieuwkoop T., Finger-Bou M., van der Oost J., Claassens N.J.. The ongoing quest to crack the genetic code for protein production. Mol. Cell. 2020; 80:193–209. [DOI] [PubMed] [Google Scholar]
- 12. Taniguchi Y., Choi P.J., Li G.-W., Chen H., Babu M., Hearn J., Emili A., Xie X.S.. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science. 2010; 329:533–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brule C.E., Grayhack E.J.. Synonymous codons: choose wisely for expression. Trends Genet. 2017; 33:283–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. dos Reis M., Savva R., Wernisch L.. Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res. 2004; 32:5036–5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gutman G.A., Hatfield G.W.. Nonrandom utilization of codon pairs in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1989; 86:3699–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sabi R., Tuller T.. Modelling the efficiency of codon–tRNA interactions based on codon usage bias. DNA Res. 2014; 21:511–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sharp P.M., Li W.H.. The codon Adaptation Index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987; 15:1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. de Smit M.H., van Duin J.. Secondary structure of the ribosome binding site determines translational efficiency: a quantitative analysis. Proc. Natl. Acad. Sci. U.S.A. 1990; 87:7668–7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dvir S., Velten L., Sharon E., Zeevi D., Carey L.B., Weinberger A., Segal E.. Deciphering the rules by which 5′-UTR sequences affect protein expression in yeast. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:E2792–E2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kudla G., Murray A.W., Tollervey D., Plotkin J.B.. Coding-sequence determinants of gene expression in Escherichia coli. Science. 2009; 324:255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Plotkin J.B., Kudla G.. Synonymous but not the same: the causes and consequences of codon bias. Nat. Rev. Genet. 2011; 12:32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tuller T., Zur H.. Multiple roles of the coding sequence 5′ end in gene expression regulation. Nucleic Acids Res. 2015; 43:13–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Umu S.U., Poole A.M., Dobson R.C., Gardner P.P.. Avoidance of stochastic RNA interactions can be harnessed to control protein expression levels in bacteria and archaea. Elife. 2016; 5:e13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mauger D.M., Cabral B.J., Presnyak V., Su S.V., Reid D.W., Goodman B., Link K., Khatwani N., Reynders J., Moore M.J.et al.. mRNA structure regulates protein expression through changes in functional half-life. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:24075–24083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cambray G., Guimaraes J.C., Arkin A.P.. Evaluation of 244,000 synthetic sequences reveals design principles to optimize translation in Escherichia coli. Nat. Biotechnol. 2018; 36:1005–1015. [DOI] [PubMed] [Google Scholar]
- 26. Bhandari B.K., Lim C.S., Gardner P.P.. Protein yield is tunable by synonymous codon changes of translation initiation sites. 2021; bioRxiv doi:22 February 2021, preprint: not peer reviewed 10.1101/726752. [DOI] [PMC free article] [PubMed]
- 27. Terai G., Asai K.. Improving the prediction accuracy of protein abundance in Escherichia coli using mRNA accessibility. Nucleic Acids Res. 2020; 48:e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bernhart S.H., Hofacker I.L., Stadler P.F.. Local RNA base pairing probabilities in large sequences. Bioinformatics. 2006; 22:614–615. [DOI] [PubMed] [Google Scholar]
- 29. Chan W.-C., Liang P.-H., Shih Y.-P., Yang U.-C., Lin W.-C., Hsu C.-N.. Learning to predict expression efficacy of vectors in recombinant protein production. BMC Bioinformatics. 2010; 11:S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Costa S., Almeida A., Castro A., Domingues L.. Fusion tags for protein solubility, purification and immunogenicity in Escherichia coli: the novel Fh8 system. Front. Microbiol. 2014; 5:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Waldo G.S. Genetic screens and directed evolution for protein solubility. Curr. Opin. Chem. Biol. 2003; 7:33–38. [DOI] [PubMed] [Google Scholar]
- 32. Bhandari B.K., Gardner P.P., Lim C.S.. Solubility-weighted index: fast and accurate prediction of protein solubility. Bioinformatics. 2020; 36:4691–4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Luirink J., Dobberstein B.. Mammalian and Escherichia coli signal recognition particles. Mol. Microbiol. 1994; 11:9–13. [DOI] [PubMed] [Google Scholar]
- 34. Palmer T., Berks B.C.. The twin-arginine translocation (Tat) protein export pathway. Nat. Rev. Microbiol. 2012; 10:483–496. [DOI] [PubMed] [Google Scholar]
- 35. Rusch S.L., Kendall D.A.. Interactions that drive Sec-dependent bacterial protein transport. Biochemistry. 2007; 46:9665–9673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. von Heijne G. The signal peptide. J. Membr. Biol. 1990; 115:195–201. [DOI] [PubMed] [Google Scholar]
- 37. Freudl R. Signal peptides for recombinant protein secretion in bacterial expression systems. Microb. Cell Fact. 2018; 17:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Karyolaimos A., Dolata K.M., Antelo-Varela M., Borras A.M., Elfageih R., Sievers S., Becher D., Riedel K., de Gier J.-W.. Escherichia coli can adapt its protein translocation machinery for enhanced periplasmic recombinant protein production. Front. Bioeng. Biotechnol. 2020; 7:465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rosano G.L., Morales E.S., Ceccarelli E.A.. New tools for recombinant protein production in Escherichia coli: A 5-year update. Protein Sci. 2019; 28:1412–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zamani M., Nezafat N., Negahdaripour M., Dabbagh F., Ghasemi Y.. In Silico evaluation of different signal peptides for the secretory production of human growth hormone in E. coli. Int. J. Peptide Res. Ther. 2015; 21:261–268. [Google Scholar]
- 41. Owji H., Nezafat N., Negahdaripour M., Hajiebrahimi A., Ghasemi Y.. A comprehensive review of signal peptides: structure, roles, and applications. Eur. J. Cell Biol. 2018; 97:422–441. [DOI] [PubMed] [Google Scholar]
- 42. Ma R.J., Wang Y.H., Liu L., Bai L.L., Ban R.. Production enhancement of the extracellular lipase LipA in Bacillus subtilis: effects of expression system and Sec pathway components. Protein Expression Purif. 2018; 142:81–87. [DOI] [PubMed] [Google Scholar]
- 43. Agostini F., Cirillo D., Livi C.M., Delli Ponti R., Tartaglia G.G.. ccSOL omics: a webserver for solubility prediction of endogenous and heterologous expression in Escherichia coli. Bioinformatics. 2014; 30:2975–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chin J.X., Chung B. K.-S., Lee D.-Y.. Codon optimization OnLine (COOL): a web-based multi-objective optimization platform for synthetic gene design. Bioinformatics. 2014; 30:2210–2212. [DOI] [PubMed] [Google Scholar]
- 45. Grote A., Hiller K., Scheer M., Münch R., Nörtemann B., Hempel D.C., Jahn D.. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005; 33:W526–W531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Puigbò P., Guzmán E., Romeu A., Garcia-Vallvé S.. OPTIMIZER: a web server for optimizing the codon usage of DNA sequences. Nucleic Acids Res. 2007; 35:W126–W131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hebditch M., Carballo-Amador M.A., Charonis S., Curtis R., Warwicker J.. Protein-Sol: a web tool for predicting protein solubility from sequence. Bioinformatics. 2017; 33:3098–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hon J., Marusiak M., Martinek T., Kunka A., Zendulka J., Bednar D., Damborsky J.. SoluProt: prediction of soluble protein expression in Escherichia coli. Bioinformatics. 2021; doi:10.1093/bioinformatics/btaa1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Smialowski P., Doose G., Torkler P., Kaufmann S., Frishman D.. PROSO II–a new method for protein solubility prediction. FEBS J. 2012; 279:2192–2200. [DOI] [PubMed] [Google Scholar]
- 50. Sormanni P., Aprile F.A., Vendruscolo M.. The CamSol method of rational design of protein mutants with enhanced solubility. J. Mol. Biol. 2015; 427:478–490. [DOI] [PubMed] [Google Scholar]
- 51. Zayni S., Damiati S., Moreno-Flores S., Amman F., Hofacker I., Ehmoser E.-K.. Enhancing the cell-free expression of native membrane proteins by in-silico optimization of the coding sequence – an experimental study of the human voltage-dependent anion channel. 2018; bioRxiv doi:07 September 2018, preprint: not peer reviewed 10.1101/411694. [DOI] [PMC free article] [PubMed]
- 52. Almagro Armenteros J.J., Tsirigos K.D., Sønderby C.K., Petersen T.N., Winther O., Brunak S., von Heijne G., Nielsen H.. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019; 37:420–423. [DOI] [PubMed] [Google Scholar]
- 53. Bagos P.G., Tsirigos K.D., Plessas S.K., Liakopoulos T.D., Hamodrakas S.J.. Prediction of signal peptides in archaea. Protein Eng. Des. Sel. 2009; 22:27–35. [DOI] [PubMed] [Google Scholar]
- 54. Hiller K., Grote A., Scheer M., Münch R., Jahn D.. PrediSi: prediction of signal peptides and their cleavage positions. Nucleic Acids Res. 2004; 32:W375–W379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Käll L., Krogh A., Sonnhammer E. L.L.. A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 2004; 338:1027–1036. [DOI] [PubMed] [Google Scholar]
- 56. Savojardo C., Martelli P.L., Fariselli P., Casadio R.. DeepSig: deep learning improves signal peptide detection in proteins. Bioinformatics. 2017; 34:1690–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gupta S., Kapoor P., Chaudhary K., Gautam A., Kumar R.Open Source Drug Discovery Consortium and Raghava G. P.S. . In silico approach for predicting toxicity of peptides and proteins. PLoS One. 2013; 8:e73957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Naamati G., Askenazi M., Linial M.. ClanTox: a classifier of short animal toxins. Nucleic Acids Res. 2009; 37:W363–W368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wong E. S.W., Hardy M.C., Wood D., Bailey T., King G.F.. SVM-based prediction of propeptide cleavage sites in spider toxins identifies toxin innovation in an Australian tarantula. PLoS One. 2013; 8:e66279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bhandari B.K., Gardner P.P., Lim C.S.. Razor: annotation of signal peptides from toxins. 2021; bioRxiv doi:07 March 2021, preprint: not peer reviewed 10.1101/2020.11.30.405613. [DOI]
- 61. Bernhart S.H., Mückstein U., Hofacker I.L.. RNA accessibility in cubic time. Algorithms Mol. Biol. 2011; 6:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lorenz R., Bernhart S.H., Höner Zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shilling P.J., Mirzadeh K., Cumming A.J., Widesheim M., Köck Z., Daley D.O.. Improved designs for pET expression plasmids increase protein production yield in Escherichia coli. Commun. Biol. 2020; 3:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chen L., Oughtred R., Berman H.M., Westbrook J.. TargetDB: a target registration database for structural genomics projects. Bioinformatics. 2004; 20:2860–2862. [DOI] [PubMed] [Google Scholar]
- 65. Seiler C.Y., Park J.G., Sharma A., Hunter P., Surapaneni P., Sedillo C., Field J., Algar R., Price A., Steel J.et al.. DNASU plasmid and PSI:Biology-Materials repositories: resources to accelerate biological research. Nucleic Acids Res. 2014; 42:D1253–D1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nawrocki E.P., Eddy S.R.. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013; 29:2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gardner P.P., Eldai H.. Annotating RNA motifs in sequences and alignments. Nucleic Acids Res. 2015; 43:691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kalvari I., Nawrocki E.P., Ontiveros-Palacios N., Argasinska J., Lamkiewicz K., Marz M., Griffiths-Jones S., Toffano-Nioche C., Gautheret D., Weinberg Z.et al.. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2021; 49:D192–D200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Potter S.C., Luciani A., Eddy S.R., Park Y., Lopez R., Finn R.D.. HMMER web server: 2018 update. Nucleic Acids Res. 2018; 46:W200–W204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jungo F., Bougueleret L., Xenarios I., Poux S.. The UniProtKB/Swiss-Prot Tox-Prot program: a central hub of integrated venom protein data. Toxicon. 2012; 60:551–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The web server is available at https://tisigner.com. This website is free and open to all users and there is no login required. All our tools, and the website are open-sourced (https://github.com/Gardner-BinfLab/TISIGNER-ReactJS; https://github.com/Gardner-BinfLab/TIsigner/tree/master/TIsigner_cmd; https://github.com/Gardner-BinfLab/SoDoPE_paper_2020/tree/master/SWI; https://github.com/Gardner-BinfLab/Razor) and privacy friendly (no data stored).