

Abstract
Background
Histiocytic and dendritic cell neoplasms are a diverse group of tumors arising from monocytic or dendritic cell lineage. Whereas the genomic features for Langerhans cell histiocytosis and Erdheim‐Chester disease have been well described, other less common and often aggressive tumors in this broad category remain poorly characterized, and comparison studies across the World Health Organization diagnostic categories are lacking.
Methods
Tumor samples from a total of 102 patient cases within four major subtypes of malignant histiocytic and dendritic cell neoplasms, including 44 follicular dendritic cell sarcomas (FDCSs), 41 histiocytic sarcomas (HSs), 7 interdigitating dendritic cell sarcomas (IDCSs), and 10 Langerhans cell sarcomas (LCSs), underwent hybridization capture with analysis of up to 406 cancer‐related genes.
Results
Among the entire cohort of 102 patients, CDKN2A mutations were most frequent across subtypes and made up 32% of cases, followed by TP53 mutations (22%). Mitogen‐activated protein kinase (MAPK) pathway mutations were present and enriched among the malignant histiocytosis (M) group (HS, IDCS, and LCS) but absent in FDCS (72% vs. 0%; p < .0001). In contrast, NF‐κB pathway mutations were frequent in FDCSs but rare in M group histiocytoses (61% vs. 12%; p < .0001). Tumor mutational burden was significantly higher in M group histiocytoses as compared with FDCSs (median 4.0/Mb vs. 2.4/Mb; p = .012). We also describe a pediatric patient with recurrent secondary histiocytic sarcoma treated with targeted therapy and interrogated by molecular analysis to identify mechanisms of therapeutic resistance.
Conclusion
A total of 42 patient tumors (41%) harbored pathogenic mutations that were potentially targetable by approved and/or investigative therapies. Our findings highlight the potential value of molecular testing to enable precise tumor classification, identify candidate oncogenic drivers, and define personalized therapeutic options for patients with these aggressive tumors.
Implications for Practice
This study presents comprehensive genomic profiling results on 102 patient cases within four major subtypes of malignant histiocytic and dendritic cell neoplasms, including 44 follicular dendritic cell sarcomas (FDCSs), 41 histiocytic sarcomas (HSs), 7 interdigitating dendritic cell sarcomas (IDCSs), and 10 Langerhans cell sarcomas (LCSs). MAPK pathway mutations were present and enriched among the malignant histiocytosis (M) group (HS, IDCS, and LCS) but absent in FDCSs. In contrast, NF‐κB pathway mutations were frequent in FDCSs but rare in M group histiocytosis. A total of 42 patient tumors (41%) harbored pathogenic mutations that were potentially targetable by approved and/or investigative therapies.
Keywords: Malignant histiocytosis, Follicular dendritic cell sarcoma, Histiocytic sarcoma, Interdigitating dendritic cell sarcoma, Targeted therapy
Short abstract
Histiocytic and dendritic cell neoplasms are a diverse group of tumors arising from the monocytic or dendritic cell lineage. This article presents the molecular characteristics of the four major subtypes of malignant histiocytic and dendritic cell neoplasms, focusing on genomic alterations that could represent therapeutic targets.
Introduction
Histiocytic and dendritic cell neoplasms are a diverse group of tumors arising from the monocytic or dendritic cell lineage. They exhibit morphologic, phenotypic, and ultrastructural characteristics that parallel corresponding terminally differentiated hematopoietic cells, from which the World Health Organization (WHO) classification schema is derived [1]. These tumors arise de novo or following clonal evolution from another hematologic malignancy [1]. Although the genomic features for Langerhans cell histiocytosis and Erdheim‐Chester disease have been well described [2, 3, 4], other less common and often aggressive tumors in this broad category remain poorly characterized, and comparison studies are lacking.
We present the molecular characteristics of 102 patient cases within the four major subtypes of malignant histiocytic and dendritic cell neoplasms: follicular dendritic cell sarcoma (FDCS), histiocytic sarcoma (HS), interdigitating dendritic cell sarcoma (IDCS), and Langerhans cell sarcoma (LCS). Our findings demonstrate that the malignant histiocytosis (M) group (HS, IDCS, and LCS, as proposed by Emile et al. [5]), featuring a common hematopoietic origin, share reliance on MAPK pathway alterations. This contrasts with the frequent NF‐κB pathway alterations in FDCS, a tumor of mesenchymal origin. Collectively, nearly half of the M group malignant histiocytoses and FDCSs harbor genomic alterations that represent potential therapeutic targets.
Materials and Methods
Cohort and Genomic Analysis
We reviewed the Foundation Medicine, Inc. (Cambridge, MA), database cohort for specimens tested between January 2014 and December 2019. After review of submitted histology via hematoxylin and eosin digital slides by select authors (L.R.Z., J.A.F., R.P.H., Y.P.H., L.R.M., E.A.W.) and immunohistochemistry results from submitted pathology reports, we identified 88 patient tumors classified as FDCS, HS, IDCS, or LCS in accordance with the 2016 WHO classification [1].
Comprehensive genomic profiling (CGP) was performed in a Clinical Laboratory Improvement Amendments–certified, College of American Pathologists–accredited laboratory (Foundation Medicine, Inc.). Approval for this study, including a waiver of informed consent and a Health Insurance Portability and Accountability Act waiver of authorization, was obtained from Western Institutional Review Board (Protocol No. 20152817). Prior to nucleic acid extraction, hematoxylin and eosin–stained slides were reviewed to confirm the presence of tumor. DNA and RNA were extracted from 40‐micron sections of formalin‐fixed, paraffin‐embedded tissue. CGP was performed using FoundationOne Heme (Foundation Medicine, Inc., Cambridge, MA), a clinical‐grade, high‐throughput, hybridization capture–based next‐generation sequencing (NGS) assay that covers all exons of 406 genes with targeted RNA sequencing of 265 genes (supplemental online Table 1) [6, 7, 8, 9]. Data were analyzed for genomic alterations including short variant alterations, copy number alterations, and gene rearrangements [6, 7, 8, 9]. Tumor mutational burden (mutations/Mb) was determined based on 1.2 Mbp of sequenced DNA [8].
Additionally, 14 patient cases from the Massachusetts General Hospital (MGH) archives (9 HS, 2 IDCS, 3 LCS) from January 2007 to January 2021 underwent targeted DNA sequencing using MGH Heme SNaPshot Assays (V3 and V4; Thermo Fisher Scientific, Waltham, MA) that use ArcherDx platform (ArcherDx, Boulder, CO) and Illumina NextSeq (Illumina, San Diego, CA) to detect variants, insertion/deletions, and copy number alterations (supplemental online Table 2) [10]. Tumor histology and immunohistochemistry were reviewed in all 14 cases by select authors (L.R.Z., J.A.F., R.P.H., Y.P.H., L.R.M.). One patient with histiocytic sarcoma underwent serial biopsies and additional molecular testing, including the initial diagnostic specimen analyzed using MGH Solid Tumor SNaPshot [11], the first recurrence using the National Cancer Institute‐Children's Oncology Group Pediatric MATCH Clinical trial [12], and the second recurrence using the hybrid‐capture NGS panel OncoPanel POPv3 [13]. Approval for this portion of the study was obtained from Partners Institutional Review Board (Protocol No. 2011P001749). Human investigations were performed after approval by a local Human Investigations Committee and in accordance with an assurance filed with and approved by the Department of Health and Human Services, where appropriate.
Ultraviolet Mutational Signature
Ultraviolet (UV) mutational signature was evaluated in all samples containing at least 20 nondriver somatic missense alterations. Signatures were derived by analysis of the trinucleotide context and profiled using the Sanger COSMIC signatures of mutational processes in human cancer [14]. A positive signature was determined if a sample demonstrated at least 40% fit to a mutational process [14]. UV mutational signature is characterized by recurrent C > T and CC > TT base substitutions at dipyrimidine sites [15].
Results
Clinicopathologic and Molecular Characteristics
The combined cohorts of 102 patients included 44 FDCSs, 41 HSs, 7 IDCSs, and 10 LCSs (Fig. 1). Fifty‐three (52%) patients were women. The median age was 53 (range, <1 to >89) years. Sequenced tumor locations and CGP results are provided in supplemental online Table 3 and 4, respectively. Among the 58 patients with M group malignant histiocytoses (HS, IDCS, and LCS), 13 (22%) had a documented history of hematolymphoid neoplasm. Regarding the entire cohort of 102 patients, CDKN2A mutations were most frequent and were identified in 32% of cases, followed by TP53 mutations (22%). MAPK pathway mutations were present and enriched among the M group histiocytoses but absent in FDCS (72% vs. 0%; p < .0001). In contrast, NF‐κB pathway mutations were frequent in FDCSs but rare in M group histiocytoses (61% vs. 12%; p < .0001). The median tumor mutation burden (TMB) among 88 tested tumors was 3.2 mutations/Mb (range, 0–44; Q1–Q3, 1.6–5.7). TMB was significantly higher in M group histiocytoses as compared with FDCS (median 4.0/Mb vs. 2.4/Mb; p = .0043, Mann‐Whitney U test). A total of 42 (41%) patient tumors harbored 51 pathogenic mutations at initial sequencing that were potentially targetable by approved and/or investigative therapies. These included mutations in BRAF (n = 12), PTEN (n = 11), MAP2K1 (n = 10), ATM (n = 4), NRAS (n = 4), NF1 (n = 3), KIT (n = 3), BRCA2 (n = 2), FGFR3 (n = 1), and NTRK1 (n = 1).
Figure 1.
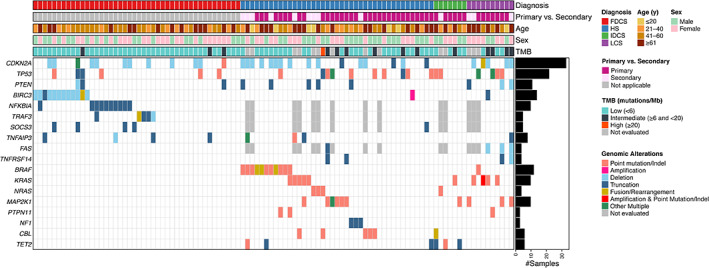
Clinicopathologic features and mutational landscape of histiocytic and dendritic cell sarcomas. Summary tile plot of pathogenic variants identified in FDCS, HS, IDCS, and LCS in 102 patients. Each column represents data (including age, sex, tumor type, and TMB) for an individual patient. Abbreviations: FDCS, follicular dendritic cell sarcoma; HS, histiocytic sarcoma; IDCS, interdigitating dendritic cell sarcoma; LCS, Langerhans cell sarcoma; TMB, tumor mutation burden.
Follicular Dendritic Cell Sarcoma
In the 44 patients (median age, 54; range, 22–77 years; 57% female) with FDCS, the most frequently mutated gene was CDKN2A, seen in 12 (27%) patients. TP53 alterations were present in 6 (14%) patients. Alterations in the NF‐κB signaling pathway were noted in 27 (61%) patients, including BIRC3 in 12 (27%), NFKBIA in 10 (23%), TRAF3 in 5 (11%), SOCS3 in 4 (9%), and TNFAIP3 in 4 (9%) patients. CCND2 amplification was present in 4 (9%) cases, whereas alterations in PTEN were detected in 3 (7%). Fusions detected included TYK2‐ATPAF2, MAP3K1‐GCOM1, and NTRK1‐PDIA3 in one FDCS each. One patient (2%; case 4) had a reported history of Castleman disease. This tumor contained the PDGFRB p.N666S alteration, a recurrent mutation in unicentric hyaline vascular Castleman disease, along with CDKN2A loss and frameshift alteration of ATM [16].
Histiocytic Sarcoma
In the 41 patients (median age, 47; range, <1 to >89 years; 44% female) with HS, a history of lymphoid neoplasm was noted in 9 (22%) patients. CDKN2A (16 patients, 39%) and TP53 (10 patients, 24%) were the most commonly altered genes. Mutations involving at least one gene in the MAPK pathway were identified in 29 (71%) patients, including five patients harboring more than one alteration. Recurrently mutated genes included BRAF in 11 (27%, including 4 canonical BRAF V600E mutations), KRAS in 5 (12%), MAP2K1 in 6 (15%), CBL in 5 (12%), NF1 in 3 (7%), NRAS in 3 (7%), and PTPN11 in 2 (5%) patients. Additionally, PTEN alterations were noted in five (12%) patients with HS who also harbored MAPK pathway alterations. PIK3CA amplification was present in one HS without an MAPK‐related alteration. Three (7%) patients had in‐frame insertion/deletion (indel) of CSF1R, all of which co‐occurred with MAPK pathway mutations. Fusions detected included MYC‐TRA (confirmed by cytogenetics and fluorescence in situ hybridization [FISH]), IGH‐BCL2, IGH‐BCL6, BRAF‐MBP, BRAF‐CLIP2, and BRAF‐NRF1 in one HS each.
Langerhans Cell Sarcoma
In the 10 patients (median age, 62.5; range, 11–78 years; 70% female) with LCS, a history of lymphoma was documented in 3 (30%) patients. Mutations in CDKN2A and TP53 were frequent, found in 5 (50%) and 4 (40%) patients, respectively, and appeared mutually exclusive of each other. Alterations in the MAPK pathway were noted in nine (90%) patients, including four with KRAS mutations. BRAF V600E mutation was present in one LCS. Three (30%) MAPK pathway–mutated LCSs harbored alterations in PTEN, and an additional LCS contained an in‐frame indel of CSF1R. IGH‐BCL2 fusion was present in one patient, while another contained BCL2 rearrangement identified by FISH.
Interdigitating Dendritic Cell Sarcoma
Of the seven patients (median age, 51; range, 22–71 years; 43% female) with IDCS, one had a history of lymphoid neoplasm (B‐lymphoblastic leukemia/lymphoma, B‐ALL). Four (57%) IDCSs harbored mutations in the MAPK pathway, including CBL‐USP2 fusion in one tumor. One IDCS without MAPK pathway alterations contained a loss‐of‐function mutation in FBXW7, a negative regulator of mTOR signaling. Three (43%) IDCS also harbored mutations in TET2, which were rare among other tumor types (7% in HS, 0% in LCS, 0% in FDCS). Alterations in CDKN2A were absent in IDCS herein.
Of note, three patient tumors from the FMI database with submitted diagnoses of IDCS were excluded from this study, as these tumors were of visceral location and contained UV mutational signatures, suggesting that they represented metastatic spindle‐cell melanoma rather than bona fide IDCS. UV mutational signature was absent in all remainder of patient tumors in which mutational signatures could be assessed.
Secondary Malignant Histiocytoses
Each of the 13 patients with M group histiocytoses and prior history of lymphoid neoplasm (Table 1) harbored genetic alterations that would be typical of the preceding lymphoid neoplasm. Additionally, all 13 secondary histiocytoses harbored mutations in the MAPK pathway, with 11 of the mutations being uncommon (<10% incidence) in their respective preceding lymphoid neoplasms. The remaining two patients both had a history of B‐ALL and both harbored variants in NRAS in their malignant histiocytosis, a gene also frequently mutated in B‐ALL [17]. Four patient cases underwent genetic testing that confirmed clonality with the preceding disease in all instances; in two, the preceding lymphoid neoplasm contained identical immunoglobulin heavy locus rearrangements to the secondary histiocytosis, although two others shared an identical genetic alteration in both neoplasms (Table 1). Lymphoma‐like genetic alterations were not restricted to patients with a documented history of lymphoma. Of the 45 primary M group histiocytoses, 13 (29%) harbored genetic alterations in at least one the following genes that may be present in small B‐cell lymphomas: CREBBP, MYD88, TNFRSF14, TNFAIP3, KMT2D, ARID1A, SETD2, EP300, SOCS1, CARD11, and/or NFKBIA.
Table 1.
M group malignant histiocytoses secondary to preceding lymphoid neoplasms
| Sequenced tumor diagnosis (case) | Age, yr (sex) | Reported prior malignancy | MAPK alteration | Identified mutations associated with prior malignancy | Mutations uncommon in prior malignancy | Confirmed clonality |
|---|---|---|---|---|---|---|
| Histiocytic sarcoma (69) | 4 (M) | T‐ALL | KRAS | ARID1A, PTEN, CDKN2A, CDKN2B | Prior not tested | |
| Histiocytic sarcoma (47) | 25 (M) | T‐ALL | BRAF | NOTCH1, PTEN, CDKN2A, CDKN2B | Prior not tested | |
| Histiocytic sarcoma (81) | 1 (M) | T‐ALL | BRAF | MYC (rearrangement), CDKN2A | Shared MYC rearrangement | |
| Histiocytic sarcoma (57) | 38 (M) | B‐ALL | NF1 | SETD2, PAX5, CDKN2A, CDKN2B | Prior not tested | |
| Histiocytic sarcoma (85) | 14 (M) | B‐ALL | NRAS | NRAS a | Shared NRAS variant | |
| Histiocytic sarcoma (80) | 78 (F) | Diffuse large B‐cell lymphoma | BRAF | TNFAIP3, TET2, CARD11, CDKN2A, CXCR4 | Prior not tested | |
| Histiocytic sarcoma (83) | 70 (F) | Diffuse large B‐cell lymphoma | NRAS | CREBBP, EP300, STAT3, CDKN2A, PTEN | CUX1 b | Prior not tested |
| Histiocytic sarcoma (60) | 62 (F) | Follicular lymphoma | BRAF | IGH/BCL2 (rearrangement), CREBBP, KMT2D, ARID1A | PTEN, SMARCA4, CDKN2A, CDKN2B, ASXL2, DNMT3A | Shared clonal IgH rearrangement |
| Histiocytic sarcoma (78) | 66 (F) | Follicular lymphoma | KRAS | TNFAIP3 | DNMT3A, CEBPA | Prior not tested |
| Interdigitating dendritic cell sarcoma (92) | 22 (M) | B‐ALL | NRAS | NRAS, a TP53 | Shared clonal IgH rearrangement | |
| Langerhans cell sarcoma (99) | 66 (F) | B‐cell lymphoma (NOS) | MAP2K1 | IGH/BCL2 (rearrangement), CARD11, SF3B1, TNFAIP3, HIST1HD, TNFRSF14, FAS | CDKN2A, CDKN2B, PTEN | Prior not tested |
| Langerhans cell sarcoma (100) | 71 (F) | Small B‐cell lymphoma c | KRAS | CREBBP, KLF2, KMT2D | CDKN2A, FBXW7 | Prior not tested |
| Langerhans cell sarcoma (102) | 65 (F) | Follicular lymphoma | MAP2K1 | KMT2D, CARD11, CREBBP, BCL2 (rearrangement) | PTEN | Prior not tested |
Variants of undetermined significance are not shown.
NRAS gain‐of‐function mutations are common in B‐ALL.
CUX1 loss; diffuse large B‐cell lymphoma may show CUX1 gain of copy alterations.
The patient's submitted history was lymphoplasmacytic lymphoma; however, the presence of a KLF2 mutation and absence of MYD88 mutation in the patient's LCS would favor a preceding splenic marginal zone lymphoma.
Abbreviations: B‐ALL, B‐lymphoblastic leukemia/lymphoma; F, female; M, male; T‐ALL, T‐acute lymphoblastic leukemia.
Illustrative Patient with Recurrent Secondary Histiocytic Sarcoma Treated by Targeted Therapy
One patient (case 81) in the HS group demonstrated response to two sequential targeted therapies in the course of tumor evolution (Fig. 2). An 18‐month‐old boy with a history of MYC‐rearranged T‐acute lymphoblastic leukemia (T‐ALL; Fig. 2A–2C) in remission presented with fevers and a rapidly enlarging right‐sided cranial HS (Fig. 2D). Sequencing studies identified the presence of BRAF V600E mutation (allele fraction [AF] 30%) that was absent in the prior T‐ALL by similar testing. MYC rearrangement was present by FISH, confirming clonality with his MYC‐rearranged T‐ALL (Fig. 2E). After tumor progression during a 5‐day treatment of clofarabine and dexamethasone, the patient received MAPK‐targeted therapy with oral dabrafenib and trametinib. Abrupt tumor regression was observed within 2–3 days, and the treatment was continued (aspects of this patient's initial care previously published) [18]. Partial tumor recurrence at 2 months led to craniotomy (recurrence 1; Fig. 2H–2K). Sequencing studies of the craniotomy specimen identified a pathogenic MTOR variant (p. T1977K; AF 8%) and persistence of BRAF V600E (AF 18%). Treatment by dabrafenib and trametinib was continued for 1 year after the resection, as the patient remained disease free. Fourteen months after the craniotomy and 2 months after cessation of dabrafenib and trametinib, a second recurrence was identified within the pleural cavity (recurrence 2; Fig. 2L–2O). Following poor response to reinstitution of MAPK‐targeted therapy, molecular testing revealed a persistent MTOR mutation with increased AF (25%) and persistence of the MYC rearrangement was identified by FISH; BRAF V600E alteration was absent, confirmed by next‐generation sequencing, polymerase chain reaction testing, and immunohistochemistry (Fig. 2O). mTOR‐directed therapy (one dose of temsirolimus and daily oral sirolimus) led to marked improvement of symptoms and radiologic abnormalities (supplemental online Fig. 1). The patient was continued on sirolimus and alive with no symptoms and improved imaging studies at 9 months after recurrence 2 (23 months after initial HS).
Figure 2.
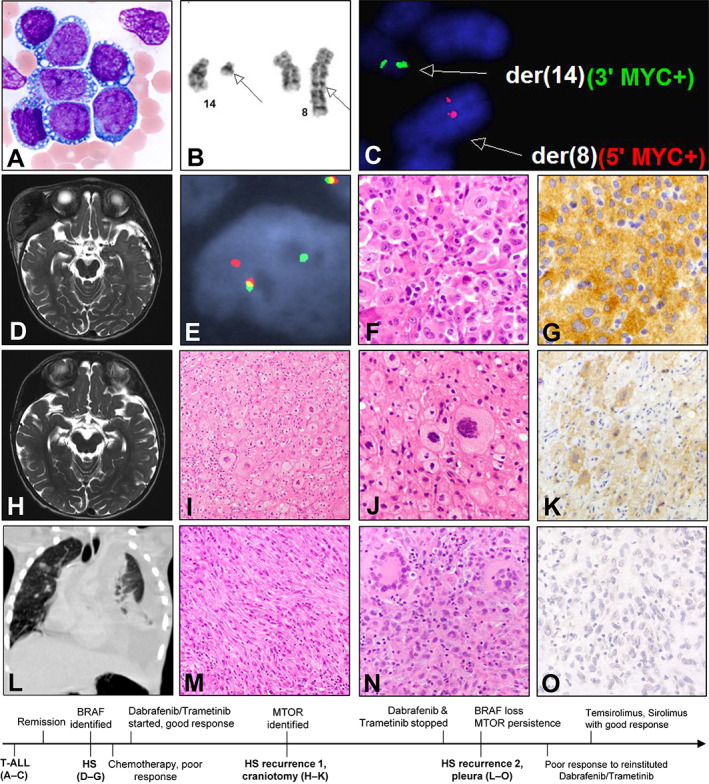
Illustrative patient case of secondary histiocytic sarcoma after T‐ALL. The patient's initial T‐ALL is characterized histologically by cytoplasmic vacuolization (A) and cytogenetically by t(8;14)(q24;q11) (B), indicative of TRA/MYC rearrangement as confirmed by MYC fluorescence in situ hybridization (FISH) (C). Magnetic resonance imaging (MRI) at initial diagnosis of histiocytic sarcoma shows a large mass destroying right temporal skull with intracranial extension (D). This histiocytic sarcoma harbors MYC rearrangement as confirmed by MYC FISH (E). Histopathologic examination shows large discohesive histiocytic cells with abundant eosinophilic cytoplasm (F). Immunohistochemistry (IHC) for BRAF V600E is strongly positive (G). MRI at recurrence 1 (H) shows partial regrowth that is amenable to surgical resection. The resected tumor displays extensive foamy changes, smaller nuclei, condensed chromatin, and multinucleated giant cell forms, with an appearance reminiscent of Erdheim‐Chester disease (I, J). BRAF V600E expression by IHC is reduced (K). Chest computed tomography at recurrence 2 shows marked consolidation of the left pleural cavity (L). Histopathologic examination shows prominent spindled morphology (M) and frequent multinucleated giant cells (N). IHC for BRAF V600E at recurrence 2 is negative (O). The history timeline below is not drawn to scale. Abbreviations: HS, histiocytic sarcoma; T‐ALL, T‐acute lymphoblastic leukemia.
Discussion
Our findings illustrated the genomic landscape of malignant histiocytic and dendritic cell neoplasms and identified potentially targetable mutations in 41% of patient cases. This study included a large number of patients affected by these rare tumors and compared the genetic findings across the WHO diagnostic categories of malignant histiocytic and dendritic cell neoplasms. HS, LCS, IDCS, and FDCS are all exceedingly rare, with limited data on optimal treatment. Localized disease is primarily managed by surgical resection, followed by adjuvant radiation and/or chemotherapy [19, 20, 21, 22]. Patients with multifocal or multisystem disease often receive intensive chemotherapy regimens, with consideration of allogenic stem cell transplantation [19, 22, 23]. Although isolated nodal disease shows higher survival rates, median overall survival for FDCS after surgical intervention is approximately 3 to 4 years [21, 24]. Outcomes for HS and LCS are dismal, with a median survival of several months for those with disseminated disease [19, 21, 25, 26, 27]. The illustrative HS patient case identified a putative genetic event driving transdifferentiation and demonstrated the potential for improved outcomes with targeted therapy.
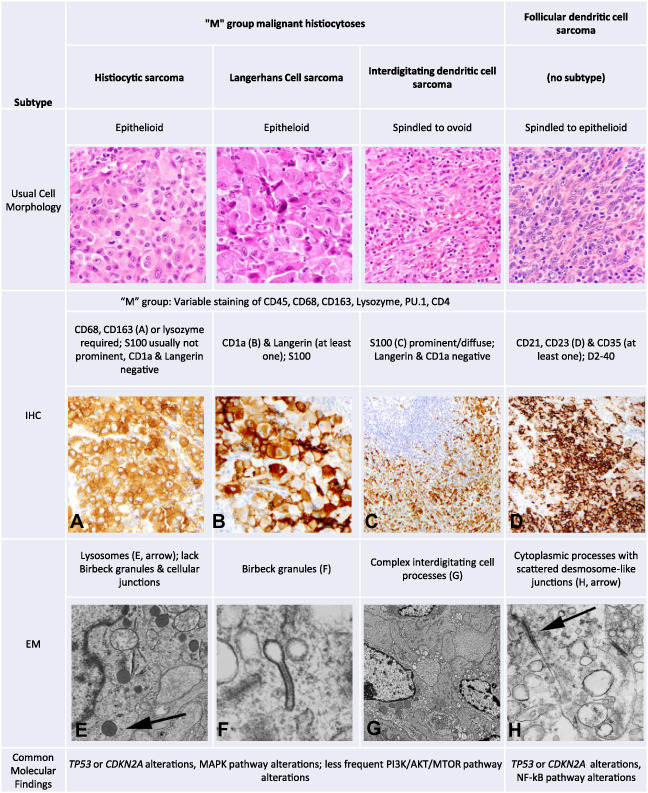
Malignant histiocytic and dendritic cell sarcomas differ from their benign phenotypic counterparts by the presence of malignant cytomorphology and more aggressive behavior [5]. As there are histologic similarities among tumor subtypes, immunohistochemistry plays a vital role in their classification (Table 2) [1]. In some patients, HS, IDCS, and LCS can share morphologic and immunophenotypic overlap, rendering precise classification challenging. Tumors with hybrid histomorphology or showing changes on subsequent biopsies that, in isolation, would be classified differently from the original tumor have been documented [28, 29, 30, 31, 32].
Table 2.
Pathologic characteristics of M group malignant histiocytoses and follicular dendritic cell sarcoma
|
Abbreviations: IHC, immunohistochemistry; EM, electron microscopy; M group, malignant histiocytosis group.
A recently revised classification schema for histiocytic and dendritic cell neoplasms addresses some of the diagnostic challenges aforementioned by grouping HS, LCS, and IDCS together as M group histiocytoses with additional subclassifiers [5]. Our molecular findings offer support for such classification, as HS, LCS, and IDCS harbor similar alterations with recurrent alterations in the MAPK pathway and frequent CDKN2A and TP53 mutations. Although MAPK pathway alterations are prevalent in M group histiocytoses and other more indolent histiocytic entities (including Langerhans cell histiocytosis, Erdheim‐Chester disease, and Rosai‐Dorfman disease/sinus histiocytosis with massive lymphadenopathy) [4, 33, 34, 35, 36, 37, 38], mutations in CDKN2A and TP53 appear rare in the latter group [34] and may help account for the more aggressive clinicopathologic features in M group histiocytoses.
This study demonstrates that FDCS harbors frequent alterations in the NF‐κB pathway, consistent with prior studies [39, 40], and genetically appears distinct from the M group histiocytoses. Although normal follicular dendritic cells play an integral role in hematolymphoid tissues, they arise from a mesenchymal stem cell precursor, in contrast to a common hematopoietic/myeloid progenitor among other macrophage/dendritic cell lineages [41, 42, 43, 44]. In FDCS, mutations in BIRC3, NFKBIA, and TRAF3 often occurred exclusive of one another. Alterations in the NF‐κB regulatory gene CYLD have been described in FDCS [39, 40] but were not examined in this study. Although BRAF V600E mutations have been rarely reported in FDCS [45], no recurrent alterations in MAPK pathway genes were identified in this large cohort. One FDCS patient case with a history of Castleman disease demonstrated PDGFRB N666S, a recurrent mutation in hyaline vascular Castleman disease [16]. To our knowledge, this is the first description of this variant in FDCS following Castleman disease.
Frequent alterations in the MAPK pathway were identified in the M group histiocytoses, consistent with recent reports on the molecular features of HS [13, 46, 47]. We identified activating mutations in the PI3K/AKT/mTOR pathway in six (15%) HSs, one (14%) IDCS, and three (30%) LCSs; these alterations were described in a subset of HS [13] and often co‐occurred with mutations in the MAPK pathway. In‐frame indels of CSF1R, a recurrent mutation in histiocytic neoplasms [4, 27], were present in four M group histiocytoses (3 HSs and 1 LCS; 9% of the 44 tumors with CSF1R coverage). They co‐occurred with MAPK alterations and appeared exclusive of PI3K/AKT/mTOR pathway alterations. CSF1R encodes the receptor tyrosine kinase CSF‐1R, a regulator of both the PI3K/AKT/mTOR and MAPK pathways. Activating CSF1R mutations in histiocytic neoplasms have also been shown to lead to ERK1/2 phosphorylation and augment MAPK pathway activity [4].
Mutations in PTPN11 (n = 2) or NF1 (n = 3) were noted in five (12%) patients with HS, with four representing primary HS. One study of primary HS showed predilection for the gastrointestinal tract in PTPN11‐ and/or NF1‐mutated tumors and frequent SETD2 alterations [46]. Only one (25%) such present case involved the abdomen; however, the possibility of subsequent gastrointestinal involvement cannot be excluded owing to limited follow‐up. In two patients with primary HS affecting the central nervous system, both contained PTPN11 alterations that co‐occurred with other mutations in the MAPK pathway aside from NF1. Although the NF1‐mutated secondary HS had a SETD2 alteration, the four primary HS cases lacked SETD2 mutations.
Both LCS and IDCS are rare and poorly characterized. Mutations in the MAPK pathway, as reported in case reports, were identified in a subset of tumors herein [48, 49, 50, 51, 52, 53, 54]. Our findings also highlight the diagnostic challenge of IDCS and its distinction from spindle‐cell melanoma, given their histologic and immunophenotypic overlap [55]. In difficult cases, analysis of mutation signature by NGS may be helpful in distinguishing metastatic melanoma from IDCS.
We explored the molecular mechanisms of the so‐called “transdifferentiation” phenomenon, in which the histiocytic/dendritic cell tumor arises from a shared progenitor or a subclone of a preceding/concurrent hematologic neoplasm [50, 53, 56, 57, 58]. Secondary M‐type histiocytoses harbored mutations that would be expected in the preceding lymphoid neoplasms, along with additional MAPK alterations that would be unusual for the preceding disease. In the illustrative HS patient case, acquisition of BRAF V600E mutation coincided with transformation from T‐ALL to histiocytic sarcoma. We also noted a second BRAF V600E‐mutated HS following T‐ALL likely arising in a similar fashion herein. Although some reports similarly show the absence of MAPK alterations in clonally related malignancies, others have noted their presence in the preceding neoplasms [47, 50, 59]. Similarly, we confirmed the presence of an NRAS mutation within the preceding B‐ALL of one secondary histiocytic sarcoma. Transdifferentiation may require multiple genetic events, with the number and sequence depending on the genomic state of the preceding neoplastic clone. Alterations in CDKN2A and TP53 have been implicated in the transformation of both follicular lymphoma and chronic lymphocytic leukemia into diffuse large B‐cell lymphoma [60, 61]; we hypothesize that temporally acquired mutations of the MAPK pathway may play a role in the diversion to histiocytic phenotype instead. Consistent with other descriptions [13], mutations seen in small B‐cell lymphomas could be identified in a subset of primary histiocytic and dendritic cell malignancies without a documented history of lymphoid neoplasm.
Our detailed clinical, histologic, and comprehensive molecular analysis of one pediatric patient with HS [18] demonstrated the potential for targeted therapy and uncovered genetic alterations that account for therapeutic resistance. The first HS recurrence coincided with the identification of a pathogenic MTOR mutation, followed by loss of BRAF V600E mutation in the second recurrence when it was unresponsive to MAPK‐directed therapy. Although NGS testing of the T‐ALL lacked MTOR coverage, MTOR gene mutations are rare in T‐ALL but have been described in HS; it is likely a small MTOR‐mutant subclone was selected for during targeted therapy [13]. Initiation of mTOR‐directed therapy led to prompt regression of this second recurrence, indicating reliance on aberrant mTOR signaling. The presence of frequent aberrations in both the MAPK and PI3K/AKT/mTOR pathway in a subset of M group histiocytoses (including the aforementioned patient) suggests that combination therapies to target these pathways may be valuable. Overall, the identification of targetable alterations in 41% of malignant histiocytic and dendritic cell tumors highlights the value of genomic profiling to identify potential targets in these rare aggressive tumors.
Limitations of this study include the possibility of incomplete medical history/follow‐up from the pathology reports in the FMI cohort. For example, one patient with HS with no documented history of lymphoid neoplasm contained an IGH‐BCL6 fusion, which may have arisen from a subclinical B‐cell lymphoma of germinal center origin. Additionally, immunohistochemistry results in the FMI cohort were available by pathology report only, and the database itself is enriched for aggressive and/or advanced diseases in which genetic testing is desired for targeted therapy.
Conclusion
We described the genomic features of a large cohort of malignant histiocytic and dendritic cell sarcomas, with identification of frequent alterations in the MAPK pathway among M group malignant histiocytoses (HS, IDCS, and LCS), alterations in the NF‐κB pathway in FDCS, and mutations in CDKN2A and TP53 in a subset of both. Our findings highlight the potential value of molecular testing to identify oncogenic drivers and treatment targets and define personalized therapeutic options for patients with these rare aggressive tumors.
Author Contributions
Conception/design: Lucas R. Massoth, Erik A. Williams
Provision of study material or patients: Lucas R. Massoth, Yin P. Hung, Judith A. Ferry, Robert P. Hasserjian, Valentina Nardi, Jeffrey S. Ross, Shakti H. Ramkissoon, Jo‐Anne Vergilio, Abner Louissaint, Lawrence R. Zukerberg, Erik A. Williams
Collection and/or assembly of data: Lucas R. Massoth, Yin P. Hung, Judith A. Ferry, Robert P. Hasserjian, Valentina Nardi, Martin Selig, Jonathan Keith Killian, Chelsea Marcus, Eric Severson, Daniel Duncan, Smruthy Sivakumar, Jeffrey S. Ross, Shakti H. Ramkissoon, Jo‐Anne Vergilio, Abner Louissaint, Lawrence R. Zukerberg, Erik A. Williams
Data analysis and interpretation: Lucas R. Massoth, Yin P. Hung, Judith A. Ferry, Robert P. Hasserjian, Valentina Nardi, G. Petur Nielsen, Sam Sadigh, Vinayak Venkataraman, Martin Selig, Alison M. Friedmann, Wesley Samore, Jonathan Keith Killian, Riza Milante, Joseph Giessinger, Kathleen Foley‐Peres, Chelsea Marcus, Eric Severson, Daniel Duncan, Julia A. Elvin, Smruthy Sivakumar, Jeffrey S. Ross, Vikram Desphande, Shakti H. Ramkissoon, Jo‐Anne Vergilio, Abner Louissaint, Lawrence R. Zukerberg, Erik A. Williams
Manuscript writing: Lucas R. Massoth, Yin P. Hung, Judith A. Ferry, Robert P. Hasserjian, Valentina Nardi, G. Petur Nielsen, Sam Sadigh, Vinayak Venkataraman, Martin Selig, Alison M. Friedmann, Wesley Samore, Jonathan Keith Killian, Riza Milante, Joseph Giessinger, Kathleen Foley‐Peres, Chelsea Marcus, Eric Severson, Daniel Duncan, Julia A. Elvin, Smruthy Sivakumar, Jeffrey S. Ross, Vikram Desphande, Shakti H. Ramkissoon, Jo‐Anne Vergilio, Abner Louissaint, Lawrence R Zukerberg, Erik A. Williams
Final approval of manuscript: Lucas R. Massoth, Yin P. Hung, Judith A. Ferry, Robert P. Hasserjian, Valentina Nardi, G. Petur Nielsen, Sam Sadigh, Vinayak Venkataraman, Martin Selig, Alison M. Friedmann, Wesley Samore, Jonathan Keith Killian, Riza Milante, Joseph Giessinger, Kathleen Foley‐Peres, Chelsea Marcus, Eric Severson, Daniel Duncan, Julia A. Elvin, Smruthy Sivakumar, Jeffrey S. Ross, Vikram Desphande, Shakti H. Ramkissoon, Jo‐Anne Vergilio, Abner Louissaint, Lawrence R. Zukerberg, Erik A. Williams
Disclosures
Jonathan Keith Killian: Foundation Medicine, Inc. (E), Foundation Medicine, Inc., Roche (OI); Chelsea Marcus: Foundation Medicine, Inc. (E), Foundation Medicine, Inc., Roche (OI); Eric Severson: Foundation Medicine, Inc. (E), Foundation Medicine, Inc., Roche (OI); Daniel Duncan: Foundation Medicine, Inc. (E), Foundation Medicine, Inc., Roche (OI); Smruthy Sivakumar: Foundation Medicine, Inc. (E), Foundation Medicine, Inc., Roche (OI); Jeffrey S. Ross: Foundation Medicine, Inc. (E), Foundation Medicine, Inc., Roche (OI); Shakti H. Ramkissoon: Foundation Medicine, Inc. (E), Foundation Medicine, Inc., Roche (OI); Jo‐Anne Vergilio: Foundation Medicine, Inc. (E), Foundation Medicine, Inc., Roche (OI); Erik A. Williams: Foundation Medicine, Inc. (E), Foundation Medicine, Inc., Roche (OI). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Supplemental Figure 1 Illustrative patient case radiologic studies prior to and one month after temsirolimus treatment. Chest X‐ray at second relapse, prior to initiation of temsirolimus, with consolidation of the left pleural cavity (A). After one month of targeted therapy, a sequential chest X‐ray shows marked improvement (B).
Supplemental Table 1 List of sequenced genes in the FoundationOne CDx and F1H platforms
Supplemental Table 2: MGH Heme Snapshot list of gene targets
Supplemental Table 3: Locations of sequenced tumor specimens
Supplemental Table 4 Known or likely pathogenic comprehensive genomic profiling results of all histiocytic and dendritic cell sarcomas
Acknowledgments
This study was supported in part by the MGH Vickery‐Colvin grant (L.R.M., L.R.Z.).
Disclosures of potential conflicts of interest may be found at the end of this article.
References
- 1. Swerdlow SH, Campo E, Harris NL et al., eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon: World Health Organization, 2017. [Google Scholar]
- 2. Papo M, Cohen‐Aubart F, Trefond L et al. Systemic histiocytosis (Langerhans cell histiocytosis, Erdheim–Chester disease, Destombes–Rosai–Dorfman disease): From oncogenic mutations to inflammatory disorders. Curr Oncol Rep 2019;21:62. [DOI] [PubMed] [Google Scholar]
- 3. Diamond EL, Durham BH, Haroche J et al. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discov 2016;6:154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Durham BH, Lopez Rodrigo E, Picarsic J et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat Med 2019;25:1839–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Emile JF, Abla O, Fraitag S et al. Revised classification of histiocytoses and neoplasms of the macrophage‐dendritic cell lineages. Blood 2016;127:2672–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Frampton GM, Fichtenholtz A, Otto GA et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31:1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sun JX, He Y, Sanford E et al. A computational approach to distinguish somatic vs. germline origin of genomic alterations from deep sequencing of cancer specimens without a matched normal. PLoS Comput Biol 2018;14:e1005965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chalmers ZR, Connelly CF, Fabrizio D et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 2017;9:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. He J, Abdel‐Wahab O, Nahas MK et al. Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood 2016;127:3004–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zheng Z, Liebers M, Zhelyazkova B et al. Anchored multiplex PCR for targeted next‐generation sequencing. Nat Med 2014;20:1479–1484. [DOI] [PubMed] [Google Scholar]
- 11. Massoth LR, Hung YP, Dias‐santagata D et al. Pan‐cancer landscape analysis reveals recurrent KMT2A‐MAML2 gene fusion in aggressive histologic subtypes of thymoma. JCO Precis Oncol 2020;4:109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lih CJ, Harrington RD, Sims DJ et al. Analytical validation of the next‐generation sequencing assay for a nationwide signal‐finding clinical trial: Molecular analysis for therapy choice clinical trial. J Mol Diagnostics 2017;19:313–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shanmugam V, Griffin GK, Jacobsen ED et al. Identification of diverse activating mutations of the RAS‐MAPK pathway in histiocytic sarcoma. Mod Pathol 2019;32:830–843. [DOI] [PubMed] [Google Scholar]
- 14. Alexandrov LB, Nik‐Zainal S, Wedge DC et al. Signatures of mutational processes in human cancer. Nature 2013;500:415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zehir A, Benayed R, Shah RH et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017;23:703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li Z, Lan X, Li C et al. Recurrent PDGFRB mutations in unicentric Castleman disease. Leukemia 2019;33:1035–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zaliova M, Stuchly J, Winkowska L et al. Genomic landscape of pediatric B‐other acute lymphoblastic leukemia in a consecutive European cohort. Haematologica 2019;104:1396–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Venkataraman V, Massoth LR, Sullivan RJ et al. Secondary histiocytic sarcoma with BRAFV600E mutation after T‐cell acute lymphoblastic leukemia in a very young child with dramatic response to dabrafenib and trametinib. Pediatr Blood Cancer 2020;67:3–5. [DOI] [PubMed] [Google Scholar]
- 19. Hornick JL, Jaffe ES, Fletcher CDM. Extranodal histiocytic sarcoma: Clinicopathologic analysis of 14 cases of a rare epithelioid malignancy. Am J Surg Pathol 2004;28:1133–1144. [DOI] [PubMed] [Google Scholar]
- 20. Saygin C, Uzunaslan D, Ozguroglu M et al. Dendritic cell sarcoma: A pooled analysis including 462 cases with presentation of our case series. Crit Rev Oncol Hematol 2013;88:253–271. [DOI] [PubMed] [Google Scholar]
- 21. Gounder M, Desai V, Kuk D et al. Impact of surgery, radiation and systemic therapy on the outcomes of patients with dendritic cell and histiocytic sarcomas. Eur J Cancer 2015;51:2413–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tella SH, Kommalapati A, Rech KL et al. Incidence, Clinical features, and outcomes of Langerhans cell sarcoma in the United States. Clin Lymphoma Myeloma Leuk 2019;19:441–446. [DOI] [PubMed] [Google Scholar]
- 23. Howard JEF, Dwivedi RC, Masterson L et al. Langerhans cell sarcoma: A systematic review. Cancer Treat Rev 2015;41:320–331. [DOI] [PubMed] [Google Scholar]
- 24. Jain P, Milgrom SA, Patel KP et al. Characteristics, management, and outcomes of patients with follicular dendritic cell sarcoma. Br J Haematol 2017;178:403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Skala SL, Lucas DR, Dewar R. Histiocytic sarcoma: Review, discussion of transformation from B‐cell lymphoma, and differential diagnosis. Arch Pathol Lab Med 2018;142:1322–1329. [DOI] [PubMed] [Google Scholar]
- 26. Shimono J, Miyoshi H, Arakawa F et al. Prognostic factors for histiocytic and dendritic cell neoplasms. Oncotarget 2017;8:98723–98732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hung YP, Lovitch SB, Qian X. Histiocytic sarcoma: New insights into FNA cytomorphology and molecular characteristics. Cancer Cytopathol 2017;125:604–614. [DOI] [PubMed] [Google Scholar]
- 28. Johnson RL, Boisot S, Ball ED et al. A case of interdigitating dendritic cell sarcoma/histiocytic sarcoma ‐ a diagnostic pitfall. Int J Clin Exp Pathol 2014;7:378–385. [PMC free article] [PubMed] [Google Scholar]
- 29. Porter DW, Gupte GL, Brown RM et al. Histiocytic sarcoma with interdigitating dendritic cell differentiation. J Pediatr Hematol Oncol 2004;26:827–830. [PubMed] [Google Scholar]
- 30. Vos JA, Abbondanzo SL, Barekman CL et al. Histiocytic sarcoma: A study of five cases including the histiocyte marker CD163. Mod Pathol 2005;18:693–704. [DOI] [PubMed] [Google Scholar]
- 31. Miettinen M, Fletcher CDM, Lasota J. True histiocytic lymphoma of small intestine: Analysis of two S‐100 protein‐positive cases with features of interdigitating reticulum cell sarcoma. Am J Clin Pathol 1993;100:285–292. [DOI] [PubMed] [Google Scholar]
- 32. Yang GCH, Besanceney CE, Tam W. Histiocytic sarcoma with interdigitating dendritic cell differentiation: A case report with fine needle aspiration cytology and review of literature. Diagn Cytopathol 2010;38:351–356. [DOI] [PubMed] [Google Scholar]
- 33. Baraban E, Sadigh S, Rosenbaum J et al. Cyclin D1 expression and novel mutational findings in Rosai‐Dorfman disease. Br J Haematol 2019;186:837–844. [DOI] [PubMed] [Google Scholar]
- 34. Lee LH, Gasilina A, Roychoudhury J et al. Real‐time genomic profiling of histiocytoses identifies early‐kinase domain BRAF alterations while improving treatment outcomes. JCI Insight 2017;2:e89473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Durham BH. Molecular characterization of the histiocytoses: Neoplasia of dendritic cells and macrophages. Semin Cell Dev Biol 2019;86:62–76. [DOI] [PubMed] [Google Scholar]
- 36. Chakraborty R, Burke TM, Hampton OA et al. Alternative genetic mechanisms of BRAF activation in Langerhans cell histiocytosis. Blood 2016;128:2533–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ozkaya N, Rosenblum MK, Durham BH et al. The histopathology of Erdheim‐Chester disease: A comprehensive review of a molecularly characterized cohort. Mod Pathol 2018;31:581–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Garces S, Medeiros LJ, Patel KP et al. Mutually exclusive recurrent KRAS and MAP2K1 mutations in Rosai‐Dorfman disease. Mod Pathol 2017;30:1367–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Andersen EF, Paxton CN, O'Malley DP et al. Genomic analysis of follicular dendritic cell sarcoma by molecular inversion probe array reveals tumor suppressor‐driven biology. Mod Pathol 2017;30:1321–1334. [DOI] [PubMed] [Google Scholar]
- 40. Griffin GK, Sholl LM, Lindeman NI et al. Targeted genomic sequencing of follicular dendritic cell sarcoma reveals recurrent alterations in NF‐κB regulatory genes. Mod Pathol 2016;29:67–74. [DOI] [PubMed] [Google Scholar]
- 41. Aguzzi A, Kranich J, Krautler NJ. Follicular dendritic cells: Origin, phenotype, and function in health and disease. Trends Immunol 2014;35:105–113. [DOI] [PubMed] [Google Scholar]
- 42. Fogg DK, Sibon C, Miled C et al. A clonogenic bone harrow progenitor specific for macrophages and dendritic cells. Science 2006;311:83–87. [DOI] [PubMed] [Google Scholar]
- 43. Collin M, Bigley V. Human dendritic cell subsets: An update. Immunology 2018;154:3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Collin M, Mcgovern N, Haniffa M. Human dendritic cell subsets. Immunology 2013;140:22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Go H, Jeon YK, Huh J et al. Frequent detection of BRAFV600E mutations in histiocytic and dendritic cell neoplasms. Histopathology 2014;65:261–272. [DOI] [PubMed] [Google Scholar]
- 46. Egan C, Nicolae A, Lack J et al. Genomic profiling of primary histiocytic sarcoma reveals two molecular subgroups. Haematologica 2020;105:951–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Egan C, Lack J, Skarshaug S et al. The mutational landscape of histiocytic sarcoma associated with lymphoid malignancy. Mod Pathol 2021;34:336–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lorillon G, Mourah S, Vercellino L et al. Sustained response to salvage therapy for dabrafenib‐resistant metastatic Langerhans cell sarcoma. Ann Oncol 2016;27:2305–2307. [DOI] [PubMed] [Google Scholar]
- 49. Mourah S, Lorillon G, Meignin V et al. Dramatic transient improvement of metastatic BRAFV600E‐mutated Langerhans cell sarcoma under treatment with dabrafenib. Blood 2015;126:2649–2652. [DOI] [PubMed] [Google Scholar]
- 50. Choi SM, Andea AA, Wang M et al. KRAS mutation in secondary malignant histiocytosis arising from low grade follicular lymphoma. Diagn Pathol 2018;13:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xerri L, Adélaïde J, Popovici C et al. CDKN2A/B deletion and double‐hit mutations of the MAPK pathway underlie the aggressive behavior of Langerhans cell tumors. Am J Surg Pathol 2018;42:150–159. [DOI] [PubMed] [Google Scholar]
- 52. Jouenne F, Reger De Moura C, Lorillon G et al. RASA1 loss in a BRAF‐mutated Langerhans cell sarcoma: A mechanism of resistance to BRAF inhibitor. Ann Oncol 2019;30:1170–1172. [DOI] [PubMed] [Google Scholar]
- 53. Chen W, Jaffe R, Zhang L et al. Langerhans cell sarcoma arising from chronic lymphocytic lymphoma/small lymphocytic leukemia: Lineage analysis and BRAF V600E mutation study. N Am J Med Sci 2013;5:386–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. O'Malley DP, Agrawal R, Grimm KE et al. Evidence of BRAF V600E in indeterminate cell tumor and interdigitating dendritic cell sarcoma. Ann Diagn Pathol 2015;19:113–116. [DOI] [PubMed] [Google Scholar]
- 55. Stowman AM, Mills SE, Wick MR. Spindle cell melanoma and interdigitating dendritic cell sarcoma: Do they represent the same process? Am J Surg Pathol 2016;40:1270–1279. [DOI] [PubMed] [Google Scholar]
- 56. Feldman AL, Arber DA, Pittaluga S et al. Clonally related follicular lymphomas and histiocytic/dendritic cell sarcomas: Evidence for transdifferentiation of the follicular lymphoma clone. Blood 2008;111:5433–5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ratei R, Hummel M, Anagnostopoulos I et al. Common clonal origin of an acute b‐lymphoblastic leukemia and a langerhans’ cell sarcoma: Evidence for hematopoietic plasticity. Haematologica 2010;95:1461–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shao H, Xi L, Raffeld M et al. Clonally related histiocytic/dendritic cell sarcoma and chronic lymphocytic leukemia/small lymphocytic lymphoma: A study of seven cases. Mod Pathol 2011;24:1421–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kemps PG, Hebeda KM, Pals ST et al. Spectrum of histiocytic neoplasms associated with diverse haematological malignancies bearing the same oncogenic mutation. J Pathol Clin Res 2021;7:10–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Casulo C, Burack WR, Friedberg JW. Transformed follicular non‐Hodgkin lymphoma. Blood 2015;125:40–47. [DOI] [PubMed] [Google Scholar]
- 61. Chigrinova E, Rinaldi A, Kwee I et al. Two main genetic pathways lead to the transformation of chronic lymphocytic leukemia to Richter syndrome. Blood 2013;122:2673–2682. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Supplemental Figure 1 Illustrative patient case radiologic studies prior to and one month after temsirolimus treatment. Chest X‐ray at second relapse, prior to initiation of temsirolimus, with consolidation of the left pleural cavity (A). After one month of targeted therapy, a sequential chest X‐ray shows marked improvement (B).
Supplemental Table 1 List of sequenced genes in the FoundationOne CDx and F1H platforms
Supplemental Table 2: MGH Heme Snapshot list of gene targets
Supplemental Table 3: Locations of sequenced tumor specimens
Supplemental Table 4 Known or likely pathogenic comprehensive genomic profiling results of all histiocytic and dendritic cell sarcomas