

Abstract
Oncostatin M (OSM) is a pleiotropic cytokine elevated in a number of inflammatory conditions including periodontal disease. OSM is produced by a variety of immune cells and has diverse functionality such as regulation of metabolic processes, cell differentiation and the inflammatory response to bacterial pathogens. The oral cavity is under constant immune surveillance including complementary neutrophil and macrophage populations, due to a persistent symbiotic bacterial presence. Periodontal disease is characterized by a dysbiotic bacterial community, with an abundance of Treponema denticola. Despite strong associations with severe periodontal disease, the source and mechanism of release of OSM has not been defined in the oral cavity. We show that OSM protein is elevated in the gingival epithelium and immune cell infiltrate during periodontal disease. Furthermore, salivary and oral neutrophil OSM is elevated in correlation with the presence of T. denticola. In an air pouch infection model, T. denticola stimulated higher levels of OSM than the oral pathogen Porphorymonas gingivalis despite differential recruitment of innate immune cells suggesting T. denticola has distinct properties to elevate OSM levels. OSM release and transcription was increased in isolated human blood, oral neutrophils or macrophages exposed to T. denticola in vitro as measured by ELISA, qPCR and microscopy. Using transcription, translation, and actin polymerization inhibition, we found that T. denticola stimulates both OSM release through degranulation and de novo synthesis in neutrophils and also OSM release and synthesis in macrophages. Differential induction of OSM by T. denticola may promote clinical periodontal disease.
Keywords: periodontitis, oral bacteria, spirochete, inflammation, periodontal disease
Summary Sentence:
Innate immune cells which encounter T. denticola differentially induce OSM to promote gingival inflammation during periodontal disease.
Introduction
Periodontal disease is a chronic inflammatory condition that affects 47% of the population in the United States caused by dysbiosis of the oral polymicrobial biofilm community [1, 2]. The spirochete Treponema denticola is an understudied pathogen associated with severe periodontal disease, classically in association with the oral bacteria Tannerella forsythia and Porphorymonas gingivalis [3, 4]. Spirochetes are minor components of the healthy subgingival biofilm community but are highly abundant in the dysbiotic community of deep periodontal pockets, where they preferentially colonize at the biofilm-tissue interface [4]. Through this close localization with both immune cells and resident tissue cells [5, 6], T. denticola induces inflammation and bone alveolar bone loss [7, 8] and can also invade the underlying connective tissue to disseminate throughout the host [9, 10]. Notably, immune cell recruitment and cytokine responses to T. denticola are markedly different than other well-studied oral pathogens such as P. gingivalis [11–13]. Despite the clear role in periodontitis and extra-oral infections [14], T. denticola and its interaction with host cells remains poorly understood and virulence mechanisms are not well characterized.
Neutrophils and macrophages coordinate to form the innate immune response in the oral cavity, a site under constant immune surveillance with active production of cytokines and chemokines, in response to both the homeostatic bacterial community and dysbiotic pathogenic changes [15]. The environment of the oral cavity and the complex periodontal milieu is a unique reservoir of cytokines, host cells and bacterial components crucial to manipulating the immune response. Neutrophils (PMNs), are the primary immune cell recruited to the oral cavity comprising greater than 95% of leukocytes in the gingival crevice and 60% of the immune cells populating the surrounding gingival tissues [16, 17]. PMN dysregulation is a hallmark of periodontal disease and a direct cause of the prolonged and damaging inflammatory response [18, 19]. While traditionally thought to be static cells only releasing preformed granule mediators, PMNs also contain extensive transcriptional capabilities with many identified transcription factors that respond differentially to specific stimuli or conditions [20]. T. denticola disrupts PMN homeostasis through an uncharacterized mechanism at the plasma membrane, altering signaling pathways and important antimicrobial functions such as chemotaxis [21–23].
Resident macrophages are present in low numbers in healthy gingiva, but migratory monocytes and recruited tissue macrophages are elevated in periodontal disease, where they have important roles in phagocytosis killing, antigen presentation and resolution of inflammation (reviewed in [2]). T. denticola modifies cytokine responses, phagocytosis and also induces activation of caspases and the inflammasome complex in macrophages [24–26]. Despite the importance of PMN and macrophage action and signaling in maintaining periodontal health, the knowledge of cytokine profile and cellular responses to T. denticola in driving inflammation leading to subsequent gingival tissue destruction are lacking.
Oncostatin M (OSM) is a pleiotropic, multifunctional cytokine of the gp130/IL-6 family involved in hematopoietic, metabolic, cell differentiation and inflammatory processes and is produced by a variety of cell types including neutrophils, monocytes/macrophages, T-cells and dendritic cells (reviewed in [27]) and can activate a number of signaling pathways including the JAK/STAT pathway following receptor engagement [27–33]. OSM is elevated in many inflammatory conditions; both with sterile origins such as rheumatoid arthritis, inflammatory skin conditions and lung injury (reviewed in [27]) as well as bacterial induced- inflammatory conditions including bacterial peritonitis, pneumonia, tuberculosis and periodontal disease [32–36]. Despite reports of local elevation in periodontal disease, its cellular sources and biological role to influence tissue destruction or bone loss characteristic of periodontal disease is largely undefined. Within the oral cavity the abundant PMN population likely serves as the source of OSM, but macrophages responding to the dysbiotic bacterial community represent another important source [32, 33, 37]. OSM is associated with multiple granule subtypes in PMNs [31, 38] and is also synthesized de novo upon PMN or macrophage stimulation by bacteria [36, 39–41] or by other sterile stimuli such as IFN-y, GM-CSF and PMA [36, 42]. Despite the strong implication for OSM to initiate cross talk between immune cells and resident matrix cells to trigger tissue destructive elements like matrix- metalloprotease activity and to shape the inflammatory response, very little is known about OSM regulation in the context of periodontal disease.
There is a crucial need to understand the biological role of unique cytokine signatures of immune cells in response to T. denticola and to define the sources and stimulation for OSM within the oral cavity. We hypothesized that oral pathogens may be an important trigger for immune cells to release and synthesize OSM in vitro and in vivo. We found that T. denticola induces a disproportionate production of OSM from neutrophils and macrophages through degranulation and de novo synthesis. Furthermore, elevated OSM protein localized in gingival tissue specimens and oral neutrophils and in a murine air pouch model of inflammation, correlated with the presence of T. denticola. This is a previously uncharacterized cytokine response by immune cells to T. denticola and increases our understanding of the impact this understudied bacterium has on modulating cell signaling in the oral cavity to promote disease.
Material and Methods
Subject population and study criteria
Clinical samples were collected from a total of 43 subjects by AM, who is a licensed periodontist. This study population was grouped as non-periodontitis subjects (NP or healthy, N = 14) and subjects with periodontal disease (PD, N = 29). The general and clinical characteristics of these populations are described in Table 1. Periodontal disease classification was performed according to the 2017 guidelines provided by the American Academy of Periodontology (AAP) [43]. Briefly, subjects with at least two periodontal pockets with clinical attachment loss (CAL) >2–3 mm on two or more non-adjacent teeth were diagnosed as periodontitis patients. Subjects in this study with periodontal disease were classified as Stage III periodontitis, Grade B with observable inflammation, bleeding on probing and required surgical periodontal treatment. Some of the periodontitis patients also had underlying systemic conditions such as diabetes and habit history of smoking. Subjects with absence of clinical signs of inflammation and no periodontal pockets were considered as non-periodontitis patients. Non-periodontitis subjects were recruited from within the UB School of Dental Medicine community or were undergoing procedures such as crown lengthening or non-periodontal related tooth extraction at the UB Dental Clinics. Collection protocols were approved by The University at Buffalo Institutional Review Board and written informed consent was obtained from subjects by authorized personnel prior to collection of samples. Due to sample availability, the exact number of samples analyzed for each type of experimental assay varies and is denoted as N values in each methods section below and within the results section and figure legends.
Table 1:
Characteristics of the Study population
| Patient Characteristics | Non-Periodontitis (NP) | Periodontal Disease (PD) |
|---|---|---|
| Subjects (N) | 14 | 29 |
| Sex (Male) | 5 | 6 |
| Sex (Female) | 9 | 9 |
| Mean Age | 41.7 ± 18.5 years | 60.9 ± 11.5 years |
| Subjects with diabetes | 0 | 3 |
| Subjects with history of smoking | 0 | 9 |
| Subjects with other underlying non-oral conditions | 0 | 12 |
Collection of whole saliva
Unstimulated whole saliva samples were collected by expectoration clinic chair side (PD, N = 29, NP, N = 11). Briefly, patients were provided a sterile tube to expectorate 3 ml of saliva. The collected saliva was stored on ice and transported to the laboratory where it was aliquoted into volumes of 100 μl and stored at −80 °C until experimental usage.
Isolation of oral wash PMNs
Isolation of neutrophils from oral washes were collected as described [44] from NP subjects (N =7) or subjects with PD (N =9). Briefly the oral cavity of each participant was rinsed for 30 seconds with 5ml of sterile 0.9% NaCl and collected in to a 50ml tube on ice. This process was repeated 10 to 30 times (30 for healthy, 10 for periodontal disease) with 3 minutes between rinses to allow for repopulation of cells to the oral cavity. The pooled washes were sequentially filtered through 40 μm, 20 μm and 10 μm nylon mesh filters attached to Swinnex 25mm filter holders (Millipore, Burlington, MA) to remove epithelial cells and other debris. Filtrate was centrifuged (500 x g, 10 min at 4°C) followed by resuspension in HBSS (without calcium or magnesium) and use in assay. Purity of oral wash neutrophils were ~ 98% by morphological staining and visual observation as previously reported [44].
Collection and processing of tissue samples
Gingival tissue specimens of periodontal pockets (N = 12) were obtained from patients with periodontal disease who were treated by surgical periodontal therapy by AM. Non-periodontitis gingival tissue (N = 7) was obtained from patients undergoing procedures such as crown lengthening or non-periodontal tooth extraction. Resected tissues which would be normally discarded following surgical procedures were immediately fixed in 10% buffered formalin for 24 hours. Specimens were then paraffin embedded (Leica EG1150H Paraffin Embedding Station) and 5 μm sections were processed (Leica RM2245 - Semi-automated Rotary Microtome) onto charged slides. Tissue sections were stained with H&E for general histological analysis and by immunohistochemistry for OSM protein expression and localization.
Detection of T. denticola by PCR in clinical samples
Saliva aliquots (100 μl) from periodontal disease (N = 15) or non-periodontal disease patients (N = 5) were centrifuged at 10,000 rpm for 3 min. Pellets were resuspended in 50 mM NaOH (100 μl) and boiled for 10 minutes at 95 °C. The solution was neutralized with 100 mM Tris (10 μl) and centrifuged at 10,000 rpm for 5 min. The resulting supernatant containing DNA was stored at −20 °C until use [45]. PCR was performed with T. denticola 16S rRNA primers TAATACCGTATGTGCTCATTTACAT and TCAAAGAAGCATTCCCTCTTCTTCTTA and Universal 16S rRNA primers AGAGTTTGATCMTGGCTCAG and ACCGCGGCTGCTGGCAC. Primers (0.4 μM final concentration) and saliva template DNA (0.5 ul) were combined with dH2O and Hot Start Taq 2X Master Mix (NEB, Ipswitch, MA) in a 25 ul final volume. PCR program was as follows: 95 °C for 2 minutes followed by 40X replications at 95 °C for 30 seconds, 55 °C for 30 seconds, 72 °C for 1 minutes with a final extension at 72 °C for 5 min. Results were visualized by ethidium bromide staining of a 1% agarose gel.
Bacterial growth conditions and preparation of bacterial products
T. denticola strain ATTCC 35405 was routinely grown anaerobically at 37°C in NOS media. Purity and viability of spirochetes were confirmed by darkfield microscopy. P. gingivalis strain A7436 (gift of T. Van Dyke, Forsyth Institute, Boston, MA) was routinely grown anaerobically at 37°C on Blood Agar plates or Trypticase- Yeast Extract Broth supplemented with hemin and menadione [46]. To prepare heat-killed bacteria, cultures were incubated at 95 °C for 10 minutes and cooled to room temperature before use. In some experiments, T. denticola conditioned media was collected following 3-day anaerobic growth, filtered sterilized and added to neutrophils to examine the effect of secreted bacterial products on OSM release. T. denticola outer membrane vesicles (OMVs) were prepared from large scale cultures as described [23].
Isolation of human peripheral blood leukocytes
Blood (40 – 50 ml) was drawn from healthy donors (N = 5 donors) in a vacutainer containing sodium citrate as an anticoagulant (BD Vacutainer Citrate tubes) under protocols approved by The University at Buffalo Institutional Review Board. Equal volumes of blood were layered over separation gradient media (1-Step Polymorphs, Accurate Chemicals, Westbury, NY) and centrifuged according to manufacturer’s directions. Bands corresponding to peripheral blood mononuclear cells (PBMCs) and polymorphonuclear neutrophils (PMNs) were aspirated and washed twice with Hank’s Buffered Saline Solution (HBSS without calcium or magnesium, Corning, 21–022-CV). Remaining red blood cells were lysed with 1ml RBC lysis buffer, 1X (BioLegend, San Diego, CA) on ice for 5 minutes with gentle agitation followed by HBSS wash and resuspended with HBSS. Cells were counted using a hemocytometer. On average, PBMC yield ~3 × 107 and PMN yield 3 – 4 × 107 was obtained. Neutrophil viability was assessed by trypan blue staining and neutrophil purity was ~ 95% as assessed by morphological staining and visual observation similar to reported literature [44]. PMNs were used immediately for experiments and PBMCs were differentiated to macrophages prior to experimentation.
Human PBMCs differentiation into macrophages
PBMCs (3 × 106 cells/well) were transferred to a 6-well plate in RPMI 1640 with L-glutamine (Corning 10–040-CV) containing 10% fetal bovine serum (Qualified USDA approved, Gibco) and incubated overnight to allow attachment. Unattached cells were removed and complete RPMI-1640 media containing human macrophage-colony stimulating factor (15 ng/ml M-CSF) (Shenandoah Biotechnologies, Warwick, PA) was added. Media was exchanged every 3–4 days until fully differentiated, on average 6 days. Viability of macrophage cells was examined by MTT assay (Thermo Fisher, Waltham, MA).
Mouse bone marrow immune cell isolation
Murine bone marrow was isolated as previously described [23]. All procedures were approved by the University at Buffalo Institutional Animal Care and Use Committee. Briefly, C57BL/6J wild-type mice (male, 6 weeks old) were purchased from Jackson Laboratory (Bar Harbor, ME). Mice were euthanized by CO2 inhalation, femurs and tibias removed, and cells were isolated from bone marrow by fractionation into discontinuous Percoll (Sigma) gradients (80%, 65%, and 55%). Mature neutrophils were isolated from the 80%–65% interface and monocytes isolated from 65%−55% interface. Neutrophils were washed with HBSS (without calcium or magnesium) and used immediately.
Bacterial - immune cell co-incubation assays
Bacteria were grown to mid-log phase (3 days), centrifuged (2000 rpm, 15 minutes) and washed with sterile, anaerobic PBS. PMNs (5 × 106) or differentiated macrophages (2 × 106 cells) were exposed to T. denticola (MOI of 100), bacterial conditioned media or OMVs (30 μg/ml) in 6-well tissue culture plates for the appropriate incubation period (15 min or 3 hours) at 5% CO2, 37°C. For experiments with oral neutrophils, equal numbers of oral neutrophils (~ 5 ×106 per condition, depending on donor) were exposed to sham tissue culture media or T. denticola (MOI of 100) for 3 hours at 5% CO2, 37°C. In some experiments, Actinomycin D (AcD 10 μg/ml, Abcam, Cambridge, MA), cycloheximide (CHX, 10 μg/ml, Sigma, St. Louis, MO), or Latrunculin A (1 μM, Fisher, Hampton, NH) were added for 30 minutes prior to addition of T. denticola. After co-incubation, conditioned cell culture media was collected, centrifuged and frozen at −80 °C. Immune cells were collected for RNA isolation or protein lysate preparation. Cells were washed gently with DPBS (Corning) and lysed for RNA isolation (E.Z.N.A. HP Total RNA kit, Omega Bio-Tek, Norocross, GA) with quantification using a Nanodrop 1000. For protein lysates, cells were gently washed with ice cold PBS and 400μl of ice cold RIPA buffer (50mM Tris pH 8.0, 150mM NaCl, 1% Triton X-1000, 0.5% sodium deoxycholate, 0.1% SDS) with 1X protease inhibitor cocktail (Halt Protease Inhibitor Cocktail, Thermo Fisher) was added to the well. Cells were gently scraped, incubated at 4°C with gentle agitation for 30 minutes then centrifuged (10,000 × g, 10 min, 4°C). A BCA assay was used to quantify protein.
Oncostatin M ELISA
100μl of RIPA lysate (normalized across each individual experiment), conditioned cell culture media, murine lavage fluid or human saliva samples (NP = 11, PD = 21) were added to a high binding 96 well microplate (Greiner Bio-one) that was incubated overnight with OSM capture antibody. ELISAs were performed following the manufacturer’s instructions (Human Oncostatin M DuoSet or Mouse OSM DuoSet ELISA, R&D Systems)
Quantitative reverse transcriptase PCR (qPCR)
RNA (100 ng) was converted to cDNA using iScript (Bio-Rad, Hercules, CA) and qPCR performed (10 ng template cDNA) using SsoAdvanced Universal SYBR Green Mix (Bio-Rad, Hercules, CA) with an Applied Biosystems 7500 machine. Relative fold change (ΔΔCt) of OSM was determined against the housekeeping gene GAPDH in the untreated samples (control). The primers used are as follows: OSM (Forward (F) primer: GAAAGAGTACCGCGTGCTCCTT, Reverse (R) primer: CTCTCAGTTTAGGAACATCCAGG) GAPDH (F primer: GGAGCGAGATCCCTCCAAAAT, R primer: GGCTGTTGTCATACTTCTCATGG).
Immune cell recruitment in vivo using the air pouch model
All animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee at the University at Buffalo. Murine air pouch model of inflammation was performed as described [47]. Briefly, air pouches were formed on the dorsal region of anesthetized male 6-week-old BLC57 mice (Jackson Laboratories, Bar Harbor, ME. 5 mice per treatment group) by subcutaneous injection of 3 ml of sterilized air on day 1 followed by re-inflation with 2ml on day 3. On day 5, bacteria (1 × 109 T. denticola or P. gingivalis) in 1 ml PBS were injected into the air pouch while sham mice were injected with PBS alone. After 6-hour exposure, mice were euthanized by CO2 inhalation and air pouches were washed with 2 ml of PBS. Lavage fluid was centrifuged, the cell pellet resuspended in 200 μl of PBS and used to quantify cell recruitment using a Cellometer T4 Bright Field Counter (Nexcelom, Lawrence, MA) and by flow cytometry. Lavage fluid supernatant was stored at −80 °C for ELISA analysis. Bacterial exposure for 6 hours was selected in order to examine early immune cell recruitment in the airpouch model. This time point is within the timeframe reported to examine neutrophil recruitment to the air pouch in response to P. gingivalis or T. denticola derived peptides [46, 47].
Flow cytometry
Characterization of immune cell types recruited to the airpouch following bacterial exposure was determined by flow cytometry. Lavage cell pellets were resuspended in PBS/ 1% BSA with Fc block (TruStain Fc, Biolegend) followed by a 30-minute incubation with fluorescent labeled primary antibodies for neutrophil (Ly6G-V450, clone 1A8. BD Biosciences, San Jose,CA) or monocyte/macrophage lineage (F4/80-PE, clone T45–2342. BD Biosciences, San Jose, CA.). Unstained cells and isotype antibodies were also included as controls. Cells were washed with PBS/1% BSA, fixed with 2% paraformaldehyde, washed again and analyzed using a LSRFortessa Flow cytometer (BD Biosciences, San Jose, CA). Data was analyzed using FlowJo Software (Version 10.6.2, Ashland, OR).
OSM Immunofluorescence and Immunohistochemistry
Localization of OSM in mouse immune cells was visualized using indirect immunofluorescence as described [23]. Briefly, macrophages grown on coverslips were exposed to T. denticola while PMNs treated with T. denticola as above were allowed to attach to BSA-coated coverslips. Cells were fixed, permeabilized and stained with a mouse anti-mouse OSM antibody (A9, sc374039, 1:100, Santa Cruz Biotechnology, Dallas, TX) and visualized by fluorescence microscopy (Nikon Eclipse TE2000-U). OSM localization in human gingival tissue samples was determined by immunohistochemistry. Tissue sections were deparaffinized and hydrated (3 × 5 min Histoclear, 2 × 5 min 100% ETOH, 2 × 5 min 95% ETOH, 2 × 5 min dH20) followed by antigen unmasking (10 mM sodium citrate buffer pH 6 with 0.05% Tween), 10 min in boiling bath). Blocking of endogenous peroxide activity was performed followed by blocking with 5% goat serum (1 hr) and primary antibody incubation overnight at 4°C (1:100, mouse anti- human OSM antibody (clone 17001, MAB295), R&D Systems, Minneapolis, MN). Reactions were developed using the SignalStain Boost mouse IHC Detection Reagent (Cell Signaling Technology, Danvers, MA), nuclei counterstained with Hematoxylin, followed by dehydration (2 × 10 min 95% ETOH, 2 × 10 min 100% ETOH, 2 × 10 min Histoclear) and mounting (SignalStain Mounting media, Cell Signaling Technology, Danvers, MA).
Statistical Analysis
Comparisons between two groups were performed by t test, paired and unpaired as appropriate. For some experiments, significance among groups was analyzed by ANOVA with post-hoc Tukey HSD or Dunn’s multi-comparison tests as appropriate. All statistical analysis was performed using PRISM software (GraphPad, San Diego, CA). Results are based on at least 3 independent experiments as indicated by the n-value within the figure legends. Statistical significance was defined as P < 0.05. Error bars represent the standard error of the mean (SEM) and statistical information is presented in figure legends.
Results
OSM is elevated in the saliva, oral neutrophils and gingival tissues of periodontal patients.
OSM is elevated during periodontal disease in gingival crevicular fluid [34, 35, 48] and in tissue specimens [39, 49], however there remains limited knowledge on localization and cellular source of OSM in the gingival tissues of periodontal patients. While diffuse cytoplasmic and nuclear staining for OSM was observed in healthy gingival epithelium, increased OSM protein was observed in the gingival epithelium and connective tissues of subjects with periodontal disease (Fig 1A, B, D, E). Furthermore, infiltration of immune cells is observed in the connective tissue of periodontal disease subjects, with clear localization of OSM within the cellular infiltrate (Fig 1C, F). The amount of OSM present in the saliva of periodontal patients averaged 62 pg/ml of OSM compared to 23 pg/ml in saliva from healthy individuals (Fig 2A). Upon closer stratification of the periodontal population in patients with factors such as smoking or underlying systemic conditions, no significant increase in the amount of salivary OSM levels was observed compared to subjects with only periodontal disease (Fig 2A). Periodontal patients typically have higher numbers of PMNs in their oral cavity compared to healthy individuals [50]. Our oral PMN isolations yielded similar results with healthy individuals averaging 1.8 × 106 PMNs compared to 3.1 × 106 PMNs from periodontal patients. Significantly more OSM, 82 pg/ml, was also present in isolated oral PMNs isolated from periodontal patients compared to the 24 pg/ml in healthy controls, suggesting neutrophils are an important source of OSM in the oral cavity (Fig 2C). Given the high amount of OSM present in diseased PMNs, we also assessed OSM gene transcript levels in the oral PMNs. PMNs from patients with periodontal disease have 0.24-fold more OSM transcript than healthy individuals (Fig 2D). While not significant given the low number of samples tested, this increase is on trend with the rest of the data reported herein. Finally, DNA was isolated from saliva samples to investigate the carriage rate of T. denticola in these patients, as it is routinely associated with chronic periodontal disease [3]. As expected, T. denticola was detected in 60% (12 out of 20 total) periodontal saliva samples by PCR, while none was found in any of the healthy samples tested (Fig 2E).
Figure 1. OSM protein is elevated in periodontitis gingival tissue.
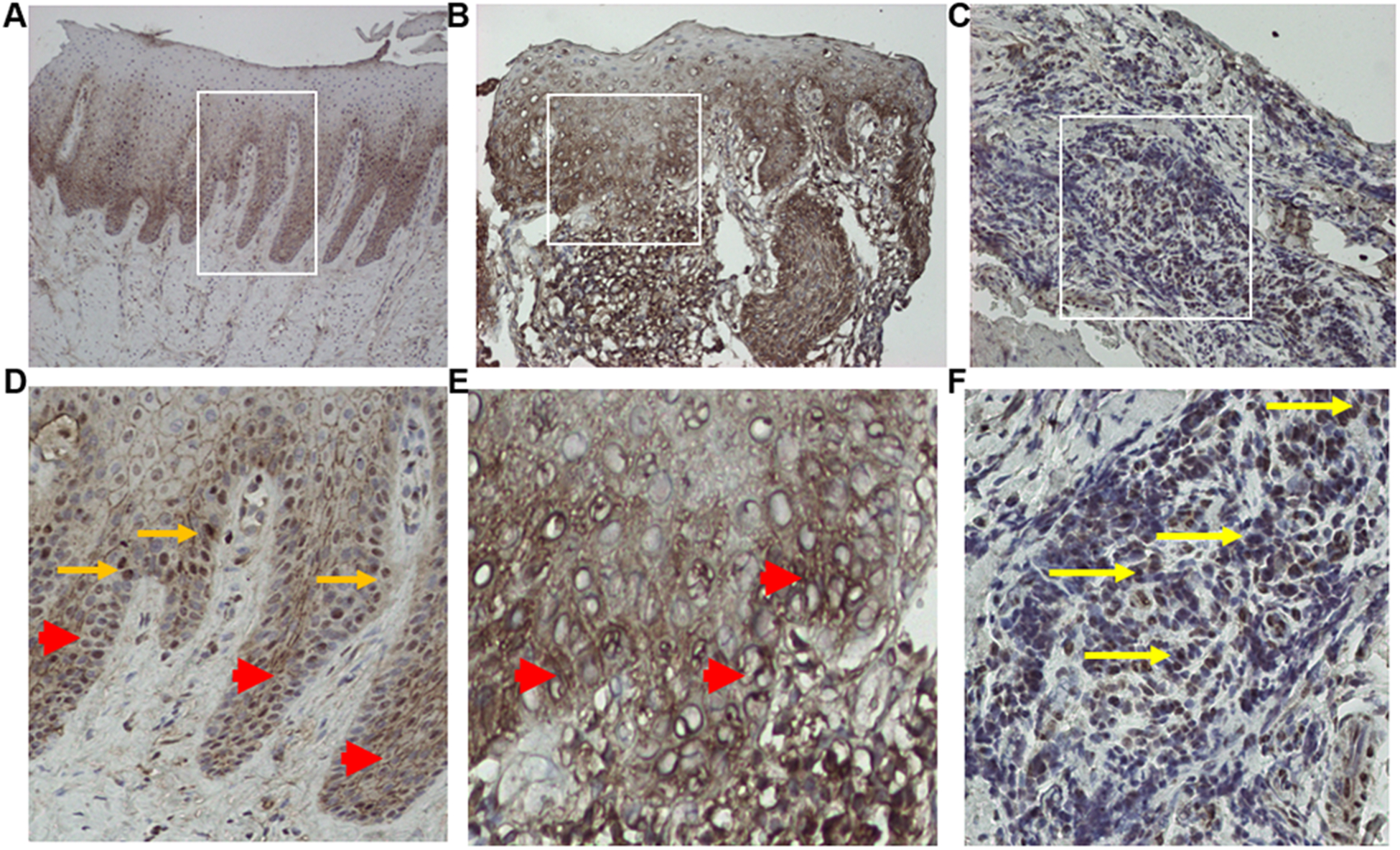
OSM protein localizes to the nucleus (arrow) and cytoplasm (arrowhead) of gingival epithelium in healthy tissue (Panel A, D) while more intense cytoplasmic staining and edema is noted in the epithelium and connective tissue from subjects with periodontal disease (panel B, E). OSM protein is also seen in the immune cell infiltrate (yellow arrow) in the gingival connective tissue in periodontitis (Panel C, F). Representative images of individual tissue specimens are shown of immunohistochemistry staining with an OSM antibody. Panels D- F are higher magnification images of the inset areas of Panels A -C.
Figure 2. OSM protein and gene expression is elevated concurrently with T. denticola in periodontal disease.

A) Patients with periodontal disease (PD, N= 29) have increased OSM protein in saliva measured by ELISA compared to oral healthy controls (N= 11). ULC= underlying non-oral condition. Grey symbols indicate subjects with ULC that are also a smoker. B) Number of oral PMNs isolated (Healthy: N= 7, PD: N = 9). C) OSM protein release (Healthy: N =6, PD: N = 7) and D) OSM gene expression (N = 5 each) in equal numbers of oral PMNs is elevated in periodontal disease. E) PD patients have detectable T. denticola in saliva. Representative image of 16s rRNA PCR detection of bacteria in a subset of subjects. T.d = T. denticola primers, univ =universal 16s primer, gDNA = T. d genomic DNA control. Graphs represent mean ± SEM * p< 0.05, **** p<0.001 by unpaired t-test.
T. denticola stimulates OSM release in vivo.
Since many periodontal subjects with elevated OSM levels also carried T. denticola, we speculated that T. denticola may play an important role in OSM elevation during disease. To assess the initial PMN and OSM response to bacterial infection, T. denticola or P. gingivalis were injected into preformed air pouches in mice. After 6 hours, infiltrating cells were collected and counted, and secreted OSM present in lavage fluid measured by ELISA. P. gingivalis is a potent immune stimulator, while T. denticola stimulates a much weaker immune response [13, 51]. Consistent with these previous studies, this experiment showed a similar immune response, with P. gingivalis recruiting 1.7 × 107 PMNs to the site of infection compared to 1.2 × 107 immune cells recruited by T. denticola (Fig 3A). Both bacterial species recruit monocytes/macrophages and neutrophils, however the neutrophil population is greater than the monocyte population as determined by flow cytometry (Fig 3B). Interestingly, while T. denticola recruited fewer overall cells and similar neutrophils compared than P. gingivalis, there was on average 17 pg/ml OSM present in the T. denticola air pouch lavage fluid compared to 8 pg/ml present with P. gingivalis (Fig 3C). These results suggest T. denticola elicits a unique OSM response compared to other oral pathogens.
Figure 3. T. denticola provokes differential OSM release and neutrophil recruitment in vivo.
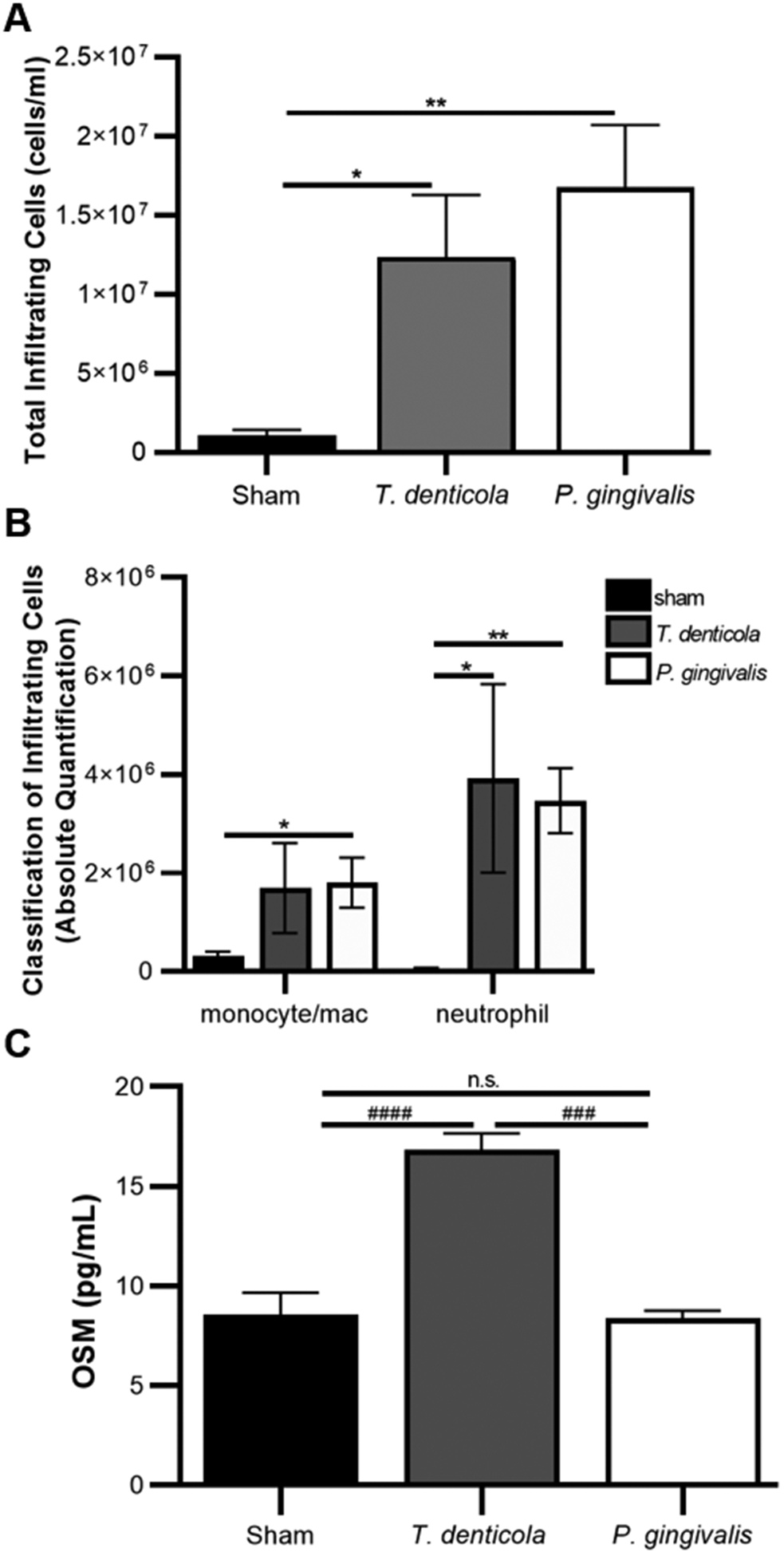
A) Both T. denticola and P. gingivalis recruit immune cells as quantified using air pouch lavage fluid analyzed for immune cell numbers using a cellometer. B) Flow cytometry classification of cells recruited to the air pouch reveal abundant neutrophil populations and moderate monocyte/macrophage populations. C) Lavage fluid from T. denticola animals had higher OSM levels than those exposed to P. gingivalis as quantified with ELISA. Graphs represent mean ± SEM, n=5, * p< 0.05, ** p<0.01 by unpaired t-test. #### p< 0.0001, ### p= 0.0005 by ANOVA with the Dunn’s multiple comparison test.
T. denticola stimulates OSM release from oral and blood PMNs.
Transcriptomic analysis indicates oral PMNs are identifiably different from circulating blood PMNs [50] and have a unique CD profile [52], suggesting they may respond to stimuli in a differential manner compared to circulating PMNs. To test if PMNs produced a similar OSM response to T. denticola independent of the tissue isolation site, oral and blood PMNs from healthy donors were exposed to T. denticola for 3 hours and OSM released into the surrounding media was measured by ELISA. Both populations of PMNs released similar amounts of OSM in response to T. denticola, with blood PMNs producing 783 pg/ml and oral PMNs producing 676 pg/ml of OSM (Fig 4A). Unstimulated PMNs released 114 and 109 pg/ml of OSM from blood and oral isolation sites, respectively. GM-CSF is a known simulator of OSM release in PMNs and was used as a positive control in blood PMNs [31]. These results suggested the PMN-OSM response to T. denticola is not site specific due to cellular differentiation as neutrophils migrate from blood to tissues and comparable OSM response has implications for cytokine release in the local gingival environment.
Figure 4. T. denticola stimulates OSM release from PMNs and macrophages in vitro.

A) OSM was released from human PMNs following exposure to T. denticola (MOI 100) for 3 hours as quantified by ELISA from peripheral blood or oral wash PMNs with GMCSF serving as a positive control for stimulating PMNs. B) Macrophages differentiated from human peripheral blood monocytes also secrete OSM following incubation with T. denticola. Increased OSM release is due to cellular action following T. denticola treatment, not cell death and membrane rupture to release cellular contents as no cell death was observed by C) trypan blue staining of PMNs and D) MTT assay of macrophages. Graphs represent mean ± SEM, N = 4, * P < 0.05, *** P < 0.01, **** P < 0.001 by paired t-test.
Macrophages release OSM in response to T. denticola.
Macrophages are crucial in immune surveillance in the oral cavity and as they also produce OSM [37], the ability of T. denticola to stimulate OSM release from macrophages was also investigated. Cultured primary human macrophages exposed to T. denticola for 3 hours released 103 pg/ml OSM compared to 68 pg/ml in untreated macrophages (Fig 4B), suggesting a similar recognition pathway in both PMNs and macrophages in response to T. denticola. To confirm the increased levels of OSM observed in the T. denticola-treated cells was a direct cellular secretion response and not due to cell death and membrane rupture to release any existing cytoplasmic pools, viability assays were conducted with both PMNs and macrophages. Following 3 hours of exposure to T. denticola, no differences were observed in cell viability, suggesting the elevated OSM levels observed is a cellular response to the bacteria (Fig 4C and D).
T. denticola stimulates granule release from PMNs.
To confirm the involvement of PMN granules in the release of OSM, the actin cytoskeleton of bone marrow PMNs was disrupted by treatment with Latrunculin A. Latrunculin A is documented to induce granule exocytosis into the extracellular media, through sequestration of globular actin leading to disruption of the cortical F-actin network [53]. T. denticola disrupts actin rearrangement in neutrophils including by preventing actin uncapping and free-barbed end formation [21]. As expected, cells treated with Latrunculin A alone released 13.5 pg/ml OSM compared to 9.4 pg/ml in control cells, which was very similar to the amount released by T. denticola at 12.9 pg/ml. However, the combined effect of Latrunculin A and T. denticola resulted in the release of 26.3 pg/ml of OSM compared 12.9 pg/ml of cells exposed to only T. denticola (Fig 5). The similar amount of OSM released in response to T. denticola and Latrunculin A, as well as the additive effect of both treatments, strongly suggest that T. denticola is stimulating degranulation events in PMNs to release OSM.
Figure 5. T. denticola stimulates OSM release from granules.

Mouse PMNs were treated with or without Latrunculin A to disrupt the actin cytoskeleton and release granules for 30 minutes followed by 3 hours of exposure to T. denticola. Media was collected and OSM levels assessed by ELISA. Graphs represent mean ± SEM, n=3, # P < 0.05 by ANOVA with Tukey’s multiple comparisons test.
T. denticola stimulates OSM gene transcription in innate immune cells.
Since both PMNs and macrophages release OSM in response to T. denticola, we next assessed if the bacteria-cell interaction also stimulated OSM gene transcription. Human blood PMNs and macrophages were pretreated with Actinomycin D (AcD) to inhibit RNA transcription followed by incubation with T. denticola. RNA was isolated and the relative quantity of OSM gene transcript compared to the housekeeping gene GAPDH was determine by qPCR. OSM gene transcription increased 0.4-fold in T. denticola treated cells and this increase was blocked by AcD treatment (Fig 6A). Macrophages had a 13-fold increase in OSM gene transcription following T. denticola incubation which was also blocked by AcD pretreatment (Fig 6B). These results suggest direct bacteria-cell interaction stimulates the gene transcription machinery to increase OSM cytokine levels. To see if gene transcription correlated with possible protein translation and OSM release, OSM present in the media was measured by ELISA. For both PMNs (Fig 6C) and macrophages (Fig 6D), AcD treatment reduced the amount of OSM in the media to levels below untreated cells. AcD treatment completely blocked any OSM secretion from macrophages, suggesting active transcription and protein translation was required for macrophages to release OSM in response to T. denticola (Fig 6D). In AcD-treated PMNs however, T. denticola still elicited a significantly higher amount of OSM release compared to AcD treatment alone (Fig 6C). While the OSM release is much lower than the T. denticola treated cells with no AcD, this difference suggests PMNs may release granules containing OSM, which is an independent process that does not rely on transcription and de novo protein synthesis. Additionally, it was noted that despite greatly increased gene transcription in macrophages compared to PMNs, T. denticola triggers 3 times more OSM release into media in PMNs and only 1.3 times as much in macrophages compared to untreated cells. This also suggested PMNs release stores of OSM, likely in granules, in response to T. denticola.
Figure 6. T. denticola stimulates OSM gene transcription in innate immune cells.

Human blood PMNs and macrophages were treated with Actinomycin D (AcD) to inhibit transcription for 30 minutes prior to 3-hour incubation with T. denticola (MOI 100). Both A) PMNs and B) macrophages increased OSM gene expression as determined by qPCR which was inhibited by AcD. Conditioned media was collected and OSM protein quantified by ELISA. C) PMNs treated with AcD showed reduced OSM release and PMNs exposed to AcD and T. denticola released more OSM than control AcD treated cells. D) Macrophages treated with AcD released less OSM and was not influenced by T. denticola treatment. Graphs represent mean ± SEM, N = 4, * P < 0.05, ** P < 0.01, *** P < 0.01, **** P < 0.001 by paired t-test.
T. denticola stimulates de novo synthesis of OSM in innate immune cells.
OSM is located in a number of granule subtypes including primary and secondary granules within PMNs [31]. To further differentiate between a degranulation event and de novo synthesis in human blood PMNs, cells were pretreated with cycloheximide (CHX) to prevent protein translation prior to analysis of secreted OSM and OSM remaining in cell lysates. Increased OSM protein was detected in both conditioned media and cell lysates on PMNs and treatment with CHX reduced the amount of OSM found in both the media (Fig 7A) and the lysate (Fig 7B) of PMNs in response to T. denticola, however, similar to the previous AcD experiments, T. denticola-treated cells were still capable of release some OSM into the media and had some remaining in the cell lysate. These results indicate T. denticola stimulates de novo OSM protein synthesis in PMNs within 3 hours of exposure, but also that there is OSM within the cell made prior to CHX treatment, likely stored in granules, that is released in response to T. denticola. Following exposure to T. denticola, increased protein levels are observed in macrophage conditioned media and cell lysates (Fig 7C and D). As macrophages do not have a comparable cytokine storage mechanism to the PMN granules, treatment with CHX prevented any de novo synthesis which was reflected in the lack of OSM secreted into the media (Fig 7C) or found in the lysate (Fig 7D). These results are in line with the AcD experiments, suggesting the OSM released from macrophages treated with T. denticola is due to active transcription and translation to produce new OSM protein. Active synthesis of OSM in response to T. denticola is also supported by the immunofluorescence data (Fig 7E), in which increased cellular fluorescence intensity is observed in both PMNs and macrophages exposed to bacteria. As observed above (Fig 6), PMNs continue to release and produce more OSM than macrophages, supporting the hypothesis that PMNs are the primary source of increased OSM in the oral cavity during periodontal disease.
Figure 7. T. denticola stimulates OSM translation in innate immune cells. Human blood.

PMNs (A and B) and macrophages (C and D) were treated with cyclohexamide (CHX) to inhibit protein translation for 30 minutes prior to 3 hours incubation with T. denticola (MOI 100). OSM was quantified by ELISA in media (A and C) and cellular lysates (B and D). A) PMNs released less OSM followed CHX treatment and B) had less OSM present in the cell lysates, but CHX was not able to block all OSM release or production in the cell. C) CHX treatment blocked OSM release from macrophages and D) there was no difference in OSM levels in the macrophage lysates. E) Increased OSM protein was observed in immune cells by immunofluorescence. Representative images are shown. Graphs represent mean ± SEM, N = 4, * P < 0.05, ** P < 0.01, *** P < 0.01, **** P < 0.001 by paired t-test.
T. denticola secretes a component to trigger OSM release from PMNs.
Similar to other gram-negative bacteria of the red complex, T. denticola secretes OMVs [24]. To investigate if OMVs or perhaps an uncharacterized secreted protein from T. denticola could also stimulate OSM release, human blood PMNs were exposed to sterile bacterial growth bacteria (NOS) or to conditioned NOS media collected after growing T. denticola for 3 days containing both OMVs and other possible bacterial components. This T. denticola-NOS media was filter sterilized to remove any possible bacteria. As NOS is a complex medium that contains rabbit sera, it was not surprising to see PMNs responded to this media alone. There was 39 pg/ml OSM released in response to NOS media compared to the 25 pg/ml from untreated cells (Fig 8A). However, PMNs released 73 pg/ml OSM in response to the T. denticola-NOS media, which was significantly higher than the NOS media alone. Furthermore, exposure of PMNs to purified OMVs is also able to increase the amount of OSM released from PMNs from 13 to 24 pg/ml (Fig 8B). These results suggest the T. denticola OMVs and possibly another other secreted component activate PMNs and trigger the release of OSM.
Figure 8. T. denticola secreted components stimulate OSM release from PMNs.

A) Human blood PMNs were exposed to bacterial growth media (NOS) or T. denticola conditioned NOS media containing secreted components. T. denticola – NOS was filtered sterilized prior to addition to PMNs and contained no bacteria (N = 3). B) PMNs exposed to purified OMVs have elevated OSM release compared to control PMNs (N = 2). C) Mouse PMNs exposed to T. denticola, heat-killed (HK) T. denticola, or purified OMVs release OSM (n=3). Graphs represent mean ± SEM, ** P < 0.01, *** P < 0.01 by paired t-test, ## P < 0.0001, ## P = 0.01 by ANOVA with Tukey’s multiple comparisons test.
To further investigate the aspect of T. denticola that is triggering release of OSM from PMNs, mouse bone marrow PMNs were exposed to heat-killed T. denticola. Live and heat-killed bacteria released similar amounts of OSM (Fig 8C), suggesting the element of T. denticola to stimulate release from PMNs is not a heat-labile protein. Similar to human PMNs, mouse PMNs also released significantly more OSM in response to OMVs at 16 pg/ml compared to 9 pg/ml from untreated control cells (Fig 8C).
T. denticola does not stimulate immediate OSM release from immune cells.
Finally, to further investigate the hypothesized granule release from human blood PMNs and to confirm the de novo synthesis requirement of OSM in macrophages, OSM release was quantified after 15 minutes incubation with T. denticola along with CHX treatment. No increase in OSM was observed following exposure to T. denticola in PMNs (Fig 9A), supporting the theory that the largest pools of OSM in granules are in primary or secondary granules, which take longer to release their contents upon stimulation than tertiary granules. Moreover, no difference was observed in macrophages (Fig 9B), further supporting the conclusion that macrophages begin de novo OSM synthesis in response to T. denticola. More experiments must be performed to further characterize the mechanism and type of PMN granules released due to interaction with T. denticola.
Figure 9. T. denticola does not cause immediate OSM release from PMNs or macrophages.

A) Human blood PMNs and B) human macrophages were treated with or without CHX for 30 minutes followed by a 15-minute exposure to T. denticola (MOI 100, N = 4). Media was collected and OSM levels assessed by ELISA. Graphs represent mean ± SEM.
Discussion
The periodontal environment is complex with a multitude of bacterial populations, host cells and inflammatory mediators. The goal of this study was to examine the presence of a novel cytokine, OSM, in periodontal disease and to define how the oral spirochete T. denticola modulates differential OSM response from innate immune cells. OSM is increased in the tissue and gingival crevicular fluid during periodontal disease [34, 35, 48, 54], yet knowledge of OSM regulation or specific cellular sources is lacking. While OSM is well documented to be produced by hematopoietic cells [27], there is more limited reports of OSM in resident cells in health and disease. In addition to infiltrating leukocytes, elevated OSM is localized in the marginal keratinocytes of regenerating skin wounds, the parenchyma tissue of pathological brain conditions and in odontoblasts and fibroblasts within inflamed dental pulp [55–57]. In line with these reports, our data indicates that OSM is elevated in keratinocytes of the gingival epithelium and immune cell infiltrate in clinical periodontal disease (Fig 1).
Moreover, we also report elevated OSM levels from isolated oral neutrophils and in saliva during clinical periodontal disease (Fig 2). Increased OSM secretion from infiltrating neutrophils has been observed in conditions such as rheumatoid arthritis, acute lung injury and mucosal airway disease [29, 30, 58], but to our knowledge, this is the first report of a cellular source of oral OSM in periodontal disease. Oral neutrophils as a local source of OSM in the gingival environment has important implications for orchestrating tissue destruction characteristic of periodontal disease [59, 60], as OSM is able to regulate matrix metalloproteinases (MMPs) in a number of cell types [61, 62], which are also significantly elevated in periodontal disease and are responsible for much of the associated tissue damage.
Oral PMNs are elevated in periodontal disease, including in our cohort of subjects, and evidence suggests a heterogenous population with differential phenotypic and functional properties [50, 52]. Furthermore, different functional capacity and transcriptomic signatures exist between oral and blood neutrophils likely dependent on local environmental cues and cellular differentiation states due to transit to the oral cavity. In our study, PMNs isolated from oral washes or blood of healthy individuals yielded a comparable OSM response to T. denticola (Fig 4), suggesting this effect is due to specific bacteria-cell interaction independent of the tissue isolation site and although oral PMNs can show exhausted capacity in some functions [63], their ability to actively respond to T. denticola and release cytokine remains. Additionally, elevated release of OSM by oral PMNs periodontal disease subjects further supports the notion that oral neutrophils still display some functionality even in disease states.
Our clinical data showing correlation of salivary T. denticola presence with elevated oral PMN counts and OSM levels (Fig 1 and 2) lends support to a putative relationship between these elements. T. denticola is typically less immunostimulatory than other oral pathogens such as P. gingivalis due in part to its ability to degrade inflammatory cytokines or differentially regulate cell signaling pathways [64], although in dual species models of infection, a clear synergistic effect exists between these species [51]. To examine a direct role for bacteria in orchestrating PMN derived-OSM we utilized a murine air pouch model of inflammation. Both T. denticola and P. gingivalis were able to recruit immune cells to the air pouch, yet fewer were recruited in response to T. denticola compared to P. gingivalis. Despite overall similarity in the amount of monocyte/ macrophage or PMN recruitment between the bacterial species, more OSM was detected in air pouch lavage fluid following exposure to T. denticola compared to P. gingivalis (Fig 3). This suggests that T. denticola differentially modulates cellular responses in vivo to release OSM, and this is likely from PMNs as these are abundantly recruited during the first 6 hours of exposure to T. denticola compared to cells of the monocytes/ macrophage lineage. Furthermore, OSM release is increased by T. denticola more robustly from neutrophils than from differentiated macrophages in vitro (Fig 4), supporting a novel role for neutrophils in production of OSM. Variable induction of OSM release or gene expression by different bacteria has been reported in which OSM was increased following intratracheal instillation of E. coli but not Klebsiella pneumoniae [36] or only in response to pathogenic Yersinia strains [40]. Similarly, we saw increased OSM gene expression in both PMNs and macrophages in response to T. denticola (Fig 6). Evidence in the literature also suggests that P. gingivalis has limited ability to induce OSM gene or protein expression in monocytes [65]. While P. gingivalis is recognized as a keystone pathogen to promote a dysbiotic community and inflammatory state, differential cytokine responses by T. denticola also highlight a novel role for this organism.
Inappropriate PMN function is a significant source of inflammation in periodontal disease [18, 19, 66, 67], yet there is still limited knowledge of how oral pathogens such as T. denticola contribute to this dysregulation. Our previous work suggests that T. denticola modulates neutrophil function by disruption of the PIP3 signaling pathway [22, 23], therefore our data presented herein extend knowledge of how T. denticola also regulates OSM release from PMNs. As macrophages are abundant in periodontal disease and synthesize OSM [33], we also wished to examine their response to T. denticola in vitro. Both neutrophils and macrophages responded to T. denticola exposure with increased protein release, gene expression and synthesis (Fig 6). Unsurprisingly, the amount of OSM produced from macrophages was much lower than that of PMNs, but as OSM is a known component of granules, we hypothesized the high levels observed could be due to degranulation events, while the OSM released from macrophages would require de novo synthesis. This hypothesis was supported by the relatively minimal changes in OSM gene transcription in PMNs compared to macrophages following T. denticola exposure (Fig 6) and the blocking of OSM production by CHX in macrophages (Fig 7). Furthermore, as PMNs typically make fewer molecules of a given cytokine compared to other leukocytes [68], we were not surprised to see overall low changes in gene transcription. Interestingly, there was more OSM in PMN lysates following exposure to T. denticola as well as in the media (Fig 7), suggesting PMNs are undergoing some de novo synthesis. The increase in OSM as visualized by microscopy (Fig 7) also suggests an increase in de novo synthesis. PMNs were still able to secrete OSM into the media, with more remaining in the lysate as well following CHX treatment, supporting our hypothesis that T. denticola triggers at least some degranulation events to release OSM.
PMNs combat pathogens by undergoing degranulation events to rapidly release preformed stores of cytokines and there is a hierarchy in terms of time to PMN granule release, which contain both similar and dissimilar cytokines [38, 69, 70]. The actin cytoskeleton network controls neutrophil granule access to the plasma membrane and the exocytosis of granule contents. Disruption of the actin cytoskeleton following treatment with Latrunculin A decreases basal actin turnover and prevents elongation of actin filaments upon stimulation, which modifies release of a subset of granules [53]. In this study, exposure of control neutrophils to Latrunculin A modestly increases OSM release from granules due to disruption of the basal cortical network, which could be akin to priming the neutrophils. An additive effect of OSM release was seen following pre-treatment with Latrunculin A and T. denticola exposure (Fig 5), which suggests that these neutrophils may be sensitized to respond to the secondary bacterial challenge with further granule release. The major outer sheath protein of T. denticola is able to impair free-barbed end formation and inhibit actin uncapping in neutrophils in response to fMLP stimulation [21] which prevents actin elongation and turnover. If the actin network normally prevents release of granules containing OSM, this extended actin alteration could also further allow elevated granule exocytosis. OSM is present in at least primary and secondary granules [31], which take more time and stronger stimulation to release than tertiary granules. To further support the evidence that tertiary granules are likely not responsible for the OSM-T. denticola response, no increase in OSM was observed following 15 minutes of exposure to the bacteria (Fig 9). It is possible that degranulation events in neutrophils exposed to T. denticola are due to signaling mechanisms unrelated or in parallel with cytoskeleton rearrangement, however PMN degranulation in response to T. denticola is poorly characterized and experiments are currently underway to investigate which granule subtype containing OSM is released in response to T. denticola. These experiments will also shed further light onto which ligand on the cell surface T. denticola may be interacting with, as this is currently poorly understood for both PMNs and macrophages, although TLR2 has been implicated in monocytes [71].
We were particularly interested in how macrophages responded to T. denticola, because while macrophages appear capable of phagocytosing T. denticola [26] and T. denticola membrane components can activate macrophages [72], very little is known about this interaction or the cellular responses. The polarization of macrophages is of particular interest in the oral cavity, as M1, or classically activated macrophages, are usually higher in periodontal patients and contribute to inflammation. However, M2, or alternatively activated macrophages, generally associated with wound healing and tissue repair, are also found within periodontal tissues [73, 74]. P. gingivalis is associated with M1 polarization and hyperinflammation [75], but the lipopolysaccharide has been shown to support M2 polarization [74]. T. forsythia supports an M2 phenotype and has been associated with a delayed macrophage response [76, 77]. T. denticola has also been suggested to drive the M2 polarization [77]. M2 macrophages may play an important role in supporting the dysbiotic polymicrobial biofilm associated with periodontal disease, as P. gingivalis can survive inside of M2 macrophages [78]. Our data showing T. denticola manipulating OSM levels further suggests T. denticola would encourage an M2 phenotype as OSM promotes an M2 polarization in lung fibrosis [79]. To fully appreciate the macrophage response to T. denticola, additional experiments will need to be performed at later time points.
Outer membrane vesicles (OMVs) are an important virulence factor for many bacteria including T. denticola and we have previously shown that OMVs can impair migration and PIP3 signaling of PMNs [23]. Other studies suggest that the T. denticola OMVs are less immunostimulatory than those of the oral pathogens P. gingivalis and Tannerella forsythia [13, 80], yet they can all prime and activate the macrophage inflammasome complex [24]. Our data herein showing increased OSM release from PMNs following exposure to T. denticola conditioned media or OMVs (Fig 8), is in line with a report that secreted molecules from Helicobacter pylori induce OSM secretion from macrophages [41]. The notion of secreted products including OMVs regulating neutrophil response supports how T. denticola could influence host cells without requiring physical contact which has significance for local effects in the oral cavity as well as in extra-oral systemic conditions which are associated with periodontal pathogens, such as cardiovascular disease [9, 14].
In conclusion, this work describes the novel finding that T. denticola stimulates release and de novo synthesis of the cytokine OSM from innate immune cells. These results have important implications on how T. denticola both manipulates the innate immune response and supports the polymicrobial environment within the oral cavity. To our knowledge this is the first in depth assessment of OSM in clinical oral disease and highlight oral neutrophils as a significant cellular source. A greater understanding of how cytokines and inflammation are modulated by oral pathogens is a necessary step to develop appropriate treatment strategies for periodontal disease.
Acknowledgments
The authors thank Sarah Metcalfe for assistance with flow cytometry and Natalie Anselmi for assistance with statistical analysis. This work was supported by NIH/NICDR R01DE027073 and R03DE024769 (MBV) and the Innovative Micro-Programs Accelerating Collaboration in Themes (IMPACT) program of the University at Buffalo (MBV and AM). Graphical abstract was created with BioRender.com.
Abbreviations Page
- OSM
Oncostatin M
- PMN
Polymorphonuclear leukocyte
- PBMC
Peripheral blood mononuclear cell
- AcD
Actinomycin D
- CHX
Cycloheximide
- T. d
Treponema denticola
- OMV
outer membrane vesicle
Footnotes
Conflict of Interest Disclosure
The authors declare no conflicts of interest.
References
- 1.Eke PI, Dye BA, Wei L, Slade GD, Thornton-Evans GO, Borgnakke WS, Taylor GW, Page RC, Beck JD, Genco RJ (2015) Update on prevalence of periodontitis in adults in the United States: NHANES 2009 to 2012. Journal of periodontology 86, 611–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hajishengallis G and Korostoff JM (2017) Revisiting the Page & Schroeder model: the good, the bad and the unknowns in the periodontal host response 40 years later. Periodontology 2000 75, 116–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holt SC and Ebersole JL (2005) Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia: the “red complex”, a prototype polybacterial pathogenic consortium in periodontitis. Periodontology 2000 38, 72–122. [DOI] [PubMed] [Google Scholar]
- 4.Ellen RP and Galimanas VB (2005) Spirochetes at the forefront of periodontal infections. Periodontology 2000 38, 13–32. [DOI] [PubMed] [Google Scholar]
- 5.Chukkapalli SS, Rivera MF, Velsko IM, Lee J-Y, Chen H, Zheng D, Bhattacharyya I, Gangula PR, Lucas AR, Kesavalu L (2014) Invasion of Oral and Aortic Tissues by Oral Spirochete Treponema denticola in ApoE(−/−) Mice Causally Links Periodontal Disease and Atherosclerosis. Infection and immunity 82, 1959–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frank RM (1980) Bacterial penetration in the apical pocket wall of advanced human periodontitis. J Periodontal Res 15, 563–73. [DOI] [PubMed] [Google Scholar]
- 7.Izard J, Sasaki H, Kent R (2012) Pathogenicity of Treponema denticola Wild-Type and Mutant Strain Tested by an Active Mode of Periodontal Infection Using Microinjection. International journal of dentistry 2012, 549169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee SF, Andrian E, Rowland E, Marquez IC (2009) Immune Response and Alveolar Bone Resorption in a Mouse Model of Treponema denticola Infection. Infection and immunity 77, 694–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Foschi F, Izard J, Sasaki H, Sambri V, Prati C, Müller R, Stashenko P (2006) Treponema denticola in Disseminating Endodontic Infections. Journal of dental research 85, 761–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Velsko IM, Chukkapalli SS, Rivera-Kweh MF, Zheng D, Aukhil I, Lucas AR, Larjava H, Kesavalu L (2015) Periodontal pathogens invade gingiva and aortic adventitia and elicit inflammasome activation in αvβ6 integrin-deficient mice. Infection and immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shin J, Ji S, Choi Y (2008) Ability of oral bacteria to induce tissue-destructive molecules from human neutrophils. Oral diseases 14, 327–34. [DOI] [PubMed] [Google Scholar]
- 12.Ji S and Choi Y (2013) Innate immune response to oral bacteria and the immune evasive characteristics of periodontal pathogens. Journal of periodontal & implant science 43, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cecil JD, O?Brien-Simpson NM, Lenzo JC, Holden JA, Chen Y-Y, Singleton W, Gause KT, Yan Y, Caruso F, Reynolds EC (2016) Differential Responses of Pattern Recognition Receptors to Outer Membrane Vesicles of Three Periodontal Pathogens. PLoS ONE 11, e0151967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chistiakov DA, Orekhov AN, Bobryshev YV (2016) Links between atherosclerotic and periodontal disease. Experimental and Molecular Pathology 100, 220–235. [DOI] [PubMed] [Google Scholar]
- 15.Moutsopoulos NM and Konkel JE (2018) Tissue-Specific Immunity at the Oral Mucosal Barrier. Trends in Immunology 39, 276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ryder MI (2010) Comparison of neutrophil functions in aggressive and chronic periodontitis. Periodontology 2000 53, 124–37. [DOI] [PubMed] [Google Scholar]
- 17.Delima AJ and Van Dyke TE (2003) Origin and function of the cellular components in gingival crevice fluid. Periodontology 2000 31, 55–76. [DOI] [PubMed] [Google Scholar]
- 18.Cortes-Vieyra R, Rosales C, Uribe-Querol E (2016) Neutrophil Functions in Periodontal Homeostasis. Journal of immunology research 2016, 1396106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scott DA and Krauss J (2012) Neutrophils in periodontal inflammation. Frontiers of oral biology 15, 56–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X, Kluger Y, Nakayama Y, Poddar R, Whitney C, DeTora A, Weissman SM, Newburger PE (2004) Gene expression in mature neutrophils: early responses to inflammatory stimuli. Journal of leukocyte biology 75, 358–72. [DOI] [PubMed] [Google Scholar]
- 21.Visser MB, Sun CX, Koh A, Ellen RP, Glogauer M (2013) Treponema denticola major outer sheath protein impairs the cellular phosphoinositide balance that regulates neutrophil chemotaxis. PLoS One 8, e66209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones MM, Vanyo ST, Visser MB (2017) The C- terminal region of the Major Outer Sheath Protein (Msp) of Treponema denticola inhibits neutrophil chemotaxis. Molecular oral microbiology, n/a-n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones MM, Vanyo ST, Visser MB (2019) The Msp protein of Treponema denticola interrupts activity of phosphoinositide processing in neutrophils. Infection and immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cecil JD, O’Brien-Simpson NM, Lenzo JC, Holden JA, Singleton W, Perez-Gonzalez A, Mansell A, Reynolds EC (2017) Outer Membrane Vesicles Prime and Activate Macrophage Inflammasomes and Cytokine Secretion In Vitro and In Vivo. Frontiers in immunology 8, 1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abiko Y, Nagano K, Yoshida Y, Yoshimura F (2014) Characterization of Treponema denticola mutants defective in the major antigenic proteins, Msp and TmpC. PLoS One 9, e113565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gaibani P, Vocale C, Ambretti S, Cavrini F, Izard J, Miragliotta L, Pellegrino MT, Sambri V (2010) Killing of Treponema denticola by mouse peritoneal macrophages. Journal of dental research 89, 521–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richards CD (2013) The enigmatic cytokine oncostatin m and roles in disease. ISRN Inflamm 2013, 512103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boniface K, Diveu C, Morel F, Pedretti N, Froger J, Ravon E, Garcia M, Venereau E, Preisser L, Guignouard E, Guillet G, Dagregorio G, Pene J, Moles JP, Yssel H, Chevalier S, Bernard FX, Gascan H, Lecron JC (2007) Oncostatin M secreted by skin infiltrating T lymphocytes is a potent keratinocyte activator involved in skin inflammation. Journal of immunology (Baltimore, Md. : 1950) 178, 4615–22. [DOI] [PubMed] [Google Scholar]
- 29.Cross A, Edwards SW, Bucknall RC, Moots RJ (2004) Secretion of oncostatin M by neutrophils in rheumatoid arthritis. Arthritis and rheumatism 50, 1430–6. [DOI] [PubMed] [Google Scholar]
- 30.Grenier A, Combaux D, Chastre J, Gougerot-Pocidalo MA, Gibert C, Dehoux M, Chollet-Martin S (2001) Oncostatin M production by blood and alveolar neutrophils during acute lung injury. Lab Invest 81, 133–41. [DOI] [PubMed] [Google Scholar]
- 31.Grenier A, Dehoux M, Boutten A, Arce-Vicioso M, Durand G, Gougerot-Pocidalo MA, Chollet-Martin S (1999) Oncostatin M production and regulation by human polymorphonuclear neutrophils. Blood 93, 1413–21. [PubMed] [Google Scholar]
- 32.Hurst SM, McLoughlin RM, Monslow J, Owens S, Morgan L, Fuller GM, Topley N, Jones SA (2002) Secretion of oncostatin M by infiltrating neutrophils: regulation of IL-6 and chemokine expression in human mesothelial cells. Journal of immunology (Baltimore, Md. : 1950) 169, 5244–51. [DOI] [PubMed] [Google Scholar]
- 33.O’Kane CM, Elkington PT, Friedland JS (2008) Monocyte-dependent oncostatin M and TNF-alpha synergize to stimulate unopposed matrix metalloproteinase-1/3 secretion from human lung fibroblasts in tuberculosis. European journal of immunology 38, 1321–30. [DOI] [PubMed] [Google Scholar]
- 34.Lin SJ, Chen YL, Kuo MY, Li CL, Lu HK (2005) Measurement of gp130 cytokines oncostatin M and IL-6 in gingival crevicular fluid of patients with chronic periodontitis. Cytokine 30, 160–7. [DOI] [PubMed] [Google Scholar]
- 35.Sakai A, Ohshima M, Sugano N, Otsuka K, Ito K (2006) Profiling the cytokines in gingival crevicular fluid using a cytokine antibody array. Journal of periodontology 77, 856–64. [DOI] [PubMed] [Google Scholar]
- 36.Traber KE, Hilliard KL, Allen E, Wasserman GA, Yamamoto K, Jones MR, Mizgerd JP, Quinton LJ (2015) Induction of STAT3-Dependent CXCL5 Expression and Neutrophil Recruitment by Oncostatin-M during Pneumonia. Am J Respir Cell Mol Biol 53, 479–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Torossian F, Guerton B, Anginot A, Alexander KA, Desterke C, Soave S, Tseng HW, Arouche N, Boutin L, Kulina I, Salga M, Jose B, Pettit AR, Clay D, Rochet N, Vlachos E, Genet G, Debaud C, Denormandie P, Genet F, Sims NA, Banzet S, Levesque JP, Lataillade JJ, Le Bousse-Kerdiles MC (2017) Macrophage-derived oncostatin M contributes to human and mouse neurogenic heterotopic ossifications. JCI Insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rorvig S, Ostergaard O, Heegaard NH, Borregaard N (2013) Proteome profiling of human neutrophil granule subsets, secretory vesicles, and cell membrane: correlation with transcriptome profiling of neutrophil precursors. Journal of leukocyte biology 94, 711–21. [DOI] [PubMed] [Google Scholar]
- 39.Demmer RT, Behle JH, Wolf DL, Handfield M, Kebschull M, Celenti R, Pavlidis P, Papapanou PN (2008) Transcriptomes in healthy and diseased gingival tissues. Journal of periodontology 79, 2112–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Subrahmanyam YV, Yamaga S, Prashar Y, Lee HH, Hoe NP, Kluger Y, Gerstein M, Goguen JD, Newburger PE, Weissman SM (2001) RNA expression patterns change dramatically in human neutrophils exposed to bacteria. Blood 97, 2457–68. [DOI] [PubMed] [Google Scholar]
- 41.Zeaiter Z, Diaz H, Stein M, Huynh HQ (2011) Helicobacter pylori Induces Expression and Secretion of Oncostatin M in Macrophages In Vitro. Digestive Diseases and Sciences 56, 689–697. [DOI] [PubMed] [Google Scholar]
- 42.Setiadi H, Yago T, Liu Z, McEver RP (2019) Endothelial signaling by neutrophil-released oncostatin M enhances P-selectin-dependent inflammation and thrombosis. Blood Adv 3, 168–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Papapanou PN, Sanz M, Buduneli N, Dietrich T, Feres M, Fine DH, Flemmig TF, Garcia R, Giannobile WV, Graziani F, Greenwell H, Herrera D, Kao RT, Kebschull M, Kinane DF, Kirkwood KL, Kocher T, Kornman KS, Kumar PS, Loos BG, Machtei E, Meng H, Mombelli A, Needleman I, Offenbacher S, Seymour GJ, Teles R, Tonetti MS (2018) Periodontitis: Consensus report of workgroup 2 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. Journal of Clinical Periodontology 45, S162–S170. [DOI] [PubMed] [Google Scholar]
- 44.Lakschevitz FS and Glogauer M (2014) High-purity neutrophil isolation from human peripheral blood and saliva for transcriptome analysis. Methods Mol Biol 1124, 469–83. [DOI] [PubMed] [Google Scholar]
- 45.Kim J, Kim M, Lee D, Baik B, Yang Y, Kim J (2014) Rapid Detection of Pathogens Associated with Dental Caries and Periodontitis by PCR Using a Modified DNA Extraction Method. J Korean Acad Pediatr Dent 41, 292–297. [Google Scholar]
- 46.Herrera BS, Hasturk H, Kantarci A, Freire MO, Nguyen O, Kansal S, Van Dyke TE (2015) Impact of resolvin E1 on murine neutrophil phagocytosis in type 2 diabetes. Infection and immunity 83, 792–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koh A, Amin Mohsen, Visser Michelle B., Sima Corneliu, Ellen Richard P., Glogauer Michael (2015) Inhibition of neutrophil chemotaxis by a multivalent reagent based on a short peptide sequence in Treponema denticola major outer sheath protein. J Pharmaceut Biol 5, 56–65. [Google Scholar]
- 48.Thorat Manojkumar S, Pradeep AR, Garg G, Raju A (2010) Gingival crevicular fluid levels of oncostatin M in periodontal conditions. Cytokine 50, 248–52. [DOI] [PubMed] [Google Scholar]
- 49.Papapanou PN, Behle JH, Kebschull M, Celenti R, Wolf DL, Handfield M, Pavlidis P, Demmer RT (2009) Subgingival bacterial colonization profiles correlate with gingival tissue gene expression. BMC microbiology 9, 221–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lakschevitz FS, Aboodi GM, Glogauer M (2013) Oral Neutrophil Transcriptome Changes Result in a Pro-Survival Phenotype in Periodontal Diseases. PLOS ONE 8, e68983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Orth RK, O’Brien-Simpson NM, Dashper SG, Reynolds EC (2011) Synergistic virulence of Porphyromonas gingivalis and Treponema denticola in a murine periodontitis model. Molecular oral microbiology 26, 229–40. [DOI] [PubMed] [Google Scholar]
- 52.Fine N, Hassanpour S, Borenstein A, Sima C, Oveisi M, Scholey J, Cherney D, Glogauer M (2016) Distinct Oral Neutrophil Subsets Define Health and Periodontal Disease States. Journal of dental research 95, 931–8. [DOI] [PubMed] [Google Scholar]
- 53.Jog NR, Rane MJ, Lominadze G, Luerman GC, Ward RA, McLeish KR (2007) The actin cytoskeleton regulates exocytosis of all neutrophil granule subsets. American Journal of Physiology-Cell Physiology 292, C1690–C1700. [DOI] [PubMed] [Google Scholar]
- 54.Lu HK, Chen YL, Chang HC, Li CL, Kuo MY (2006) Identification of the osteoprotegerin/receptor activator of nuclear factor-kappa B ligand system in gingival crevicular fluid and tissue of patients with chronic periodontitis. J Periodontal Res 41, 354–60. [DOI] [PubMed] [Google Scholar]
- 55.Ruprecht K, Kuhlmann T, Seif F, Hummel V, Kruse N, Bruck W, Rieckmann P (2001) Effects of oncostatin M on human cerebral endothelial cells and expression in inflammatory brain lesions. J Neuropathol Exp Neurol 60, 1087–98. [DOI] [PubMed] [Google Scholar]
- 56.Goren I, Kampfer H, Muller E, Schiefelbein D, Pfeilschifter J, Frank S (2006) Oncostatin M expression is functionally connected to neutrophils in the early inflammatory phase of skin repair: implications for normal and diabetes-impaired wounds. J Invest Dermatol 126, 628–37. [DOI] [PubMed] [Google Scholar]
- 57.Huang F-M, Tsai C-H, Yang S-F, Chang Y-C (2009) The upregulation of oncostatin M in inflamed human dental pulps. International endodontic journal 42, 627–631. [DOI] [PubMed] [Google Scholar]
- 58.Pothoven KL, Norton JE, Suh LA, Carter RG, Harris KE, Biyasheva A, Welch K, Shintani-Smith S, Conley DB, Liu MC, Kato A, Avila PC, Hamid Q, Grammer LC 3rd, Peters AT, Kern RC, Tan BK, Schleimer RP (2017) Neutrophils are a major source of the epithelial barrier disrupting cytokine oncostatin M in patients with mucosal airways disease. The Journal of allergy and clinical immunology 139, 1966–1978.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Franco C, Patricia H-R, Timo S, Claudia B, Marcela H (2017) Matrix Metalloproteinases as Regulators of Periodontal Inflammation. International journal of molecular sciences 18, 440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gursoy UK, Kononen E, Tervahartiala T, Gursoy M, Pitkanen J, Torvi P, Suominen AL, Pussinen P, Sorsa T (2018) Molecular Forms and Fragments of Salivary MMP-8 in Relation to Periodontitis. J Clin Periodontol. [DOI] [PubMed] [Google Scholar]
- 61.Fan X, Hughes BG, Ali MAM, Chan BYH, Launier K, Schulz R (2016) Matrix metalloproteinase-2 in oncostatin M-induced sarcomere degeneration in cardiomyocytes. American Journal of Physiology-Heart and Circulatory Physiology 311, H183–H189. [DOI] [PubMed] [Google Scholar]
- 62.Langdon C, Leith J, Smith F, Richards CD (1997) Oncostatin M stimulates monocyte chemoattractant protein-1- and interleukin-1-induced matrix metalloproteinase-1 production by human synovial fibroblasts in vitro. Arthritis and rheumatism 40, 2139–46. [DOI] [PubMed] [Google Scholar]
- 63.Moonen CGJ, Hirschfeld J, Cheng L, Chapple ILC, Loos BG, Nicu EA (2019) Oral Neutrophils Characterized: Chemotactic, Phagocytic, and Neutrophil Extracellular Trap (NET) Formation Properties. Frontiers in immunology 10, 635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miyamoto M, Ishihara K, Okuda K (2006) The Treponema denticola Surface Protease Dentilisin Degrades Interleukin-1β (IL-1β), IL-6, and Tumor Necrosis Factor Alpha. Infection and immunity 74, 2462–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barksby HE, Nile CJ, Jaedicke KM, Taylor JJ, Preshaw PM (2009) Differential expression of immunoregulatory genes in monocytes in response to Porphyromonas gingivalis and Escherichia coli lipopolysaccharide. Clin Exp Immunol 156, 479–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nicu EA and Loos BG (2016) Polymorphonuclear neutrophils in periodontitis and their possible modulation as a therapeutic approach. Periodontology 2000 71, 140–163. [DOI] [PubMed] [Google Scholar]
- 67.Rijkschroeff P, Jansen ID, van der Weijden FA, Keijser BJ, Loos BG, Nicu EA (2016) Oral polymorphonuclear neutrophil characteristics in relation to oral health: a cross-sectional, observational clinical study. International journal of oral science 8, 191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tsuyoshi K, Yusuke M, Takeo I, Tsuyoshi O, Mitsuru A, Steven LK (2005) Neutrophil-Derived Cytokines: Potential Therapeutic Targets in Inflammation. Current Drug Targets - Inflammation & Allergy 4, 273–279. [DOI] [PubMed] [Google Scholar]
- 69.Lacy P (2006) Mechanisms of degranulation in neutrophils. Allergy Asthma Clin Immunol 2, 98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cassatella MA, Ostberg NK, Tamassia N, Soehnlein O (2019) Biological Roles of Neutrophil-Derived Granule Proteins and Cytokines. Trends Immunol 40, 648–664. [DOI] [PubMed] [Google Scholar]
- 71.Ruby J, Martin M, Passineau MJ, Godovikova V, Fenno JC, Wu H (2018) Activation of the Innate Immune System by Treponema denticola Periplasmic Flagella through Toll-Like Receptor 2. Infection and immunity 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rosen G, Sela MN, Naor R, Halabi A, Barak V, Shapira L (1999) Activation of murine macrophages by lipoprotein and lipooligosaccharide of Treponema denticola. Infection and immunity 67, 1180–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou L-N, Bi C-S, Gao L-N, An Y, Chen F, Chen F-M (2019) Macrophage polarization in human gingival tissue in response to periodontal disease. Oral diseases 25, 265–273. [DOI] [PubMed] [Google Scholar]
- 74.Yang J, Zhu Y, Duan D, Wang P, Xin Y, Bai L, Liu Y, Xu Y (2018) Enhanced activity of macrophage M1/M2 phenotypes in periodontitis. Archives of oral biology 96, 234–242. [DOI] [PubMed] [Google Scholar]
- 75.Yu S, Ding L, Liang D, Luo L (2018) Porphyromonas gingivalis inhibits M2 activation of macrophages by suppressing α-ketoglutarate production in mice. Molecular oral microbiology 33, 388–395. [DOI] [PubMed] [Google Scholar]
- 76.Sekot G, Posch G, Messner P, Matejka M, Rausch-Fan X, Andrukhov O, Schäffer C (2011) Potential of the Tannerella forsythia S-layer to delay the immune response. Journal of dental research 90, 109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Parisi L, Gini E, Baci D, Tremolati M, Fanuli M, Bassani B, Farronato G, Bruno A, Mortara L (2018) Macrophage Polarization in Chronic Inflammatory Diseases: Killers or Builders? Journal of immunology research 2018, 8917804–8917804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lam RS, O’Brien-Simpson NM, Holden JA, Lenzo JC, Fong SB, Reynolds EC (2016) Unprimed, M1 and M2 Macrophages Differentially Interact with Porphyromonas gingivalis. PloS one 11, e0158629–e0158629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ayaub EA, Dubey A, Imani J, Botelho F, Kolb MRJ, Richards CD, Ask K (2017) Overexpression of OSM and IL-6 impacts the polarization of pro-fibrotic macrophages and the development of bleomycin-induced lung fibrosis. Scientific Reports 7, 13281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhu Y, Dashper SG, Chen Y-Y, Crawford S, Slakeski N, Reynolds EC (2013) Porphyromonas gingivalis and Treponema denticola Synergistic Polymicrobial Biofilm Development. PLOS ONE 8, e71727. [DOI] [PMC free article] [PubMed] [Google Scholar]