

Abstract
Purpose
Genotoxic chemotherapy and radiotherapy can cause DNA double stranded breaks (DSBs) in primordial follicle (PMF) oocytes, which then undergo apoptosis. The development of effective new fertility preservation agents has been hampered, in part, by a limited understanding of DNA repair in PMF oocytes. This study investigated the induction of classical DSB repair pathways in the follicles of wild type (WT) and apoptosis-deficient Puma-/- mice in response to DSBs caused by the chemotherapy agent cisplatin.
Methods
Adult C57BL/6 WT and Puma-/- mice were injected i.p. with saline or cisplatin (5 mg/kg); ovaries were harvested at 8 or 24 h. Follicles were counted, and H2A histone family member (γH2AX) immunofluorescence used to demonstrate DSBs. DNA repair protein RAD51 homolog 1 (RAD51) and DNA-dependent protein kinase, catalytic subunit (DNA-PKcs) immunofluorescence were used to identify DNA repair pathways utilised.
Results
Puma-/- mice retained 100% of follicles 24 h after cisplatin treatment. Eight hours post-treatment, γH2AX immunofluorescence showed DSBs across follicular stages in Puma-/- mice; staining returned to control levels in PMFs within 5 days, suggesting repair of PMF oocytes in this window. RAD51 immunofluorescence eight hours post-cisplatin was positive in damaged cell types in both WT and Puma-/- mice, demonstrating induction of the homologous recombination pathway. In contrast, DNA-PKcs staining were rarely observed in PMFs, indicating non-homologous end joining plays an insignificant role.
Conclusion
PMF oocytes are able to conduct high-fidelity repair of DNA damage accumulated during chemotherapy. Therefore, apoptosis inhibition presents a viable strategy for fertility preservation in women undergoing treatment.
Supplementary Information
The online version contains supplementary material available at 10.1007/s10815-021-02184-3.
Keywords: Oocyte, Follicle, Fertility, Chemotherapy, DNA repair, Apoptosis
Introduction
Whilst modern cancer treatment regimens have resulted in a rapid rise in survivorship rates, curative treatments have well-documented off-target effects resulting in unwanted effects for survivors [1, 2]. In women, DNA-damaging treatments such as conventional chemotherapy and radiotherapy for common cancers deplete the ovarian reserve of primordial follicles, reducing fertility and increasing premature or early menopause with multisystem adverse effects beyond reproductive capacity [3]. Cancer treatment is now the commonest cause of acquired premature ovarian insufficiency [4, 5]. Accordingly, oncofertility has emerged as a new field of clinical practice and research, with the goals of improving reproductive and fertility outcomes following cancer treatment. Despite this, there are currently no effective non-invasive approaches to prevent treatment-induced ovarian damage.
Female fertile potential is dependent on the pool of primordial follicles. These structures comprise one immature oocyte, encased within a single layer of pre-granulosa cells. The population of primordial follicles established during prenatal life forms the pool from which all mature hormone-producing follicles are drawn and from which mature oocytes are ovulated [6, 7]. During the female reproductive lifespan, the number of these primordial follicles declines as they are activated to commence growth, resume meiosis, mature, and are eventually ovulated—or as is more common, undergo atresia and die. Exhaustion of the primordial follicle pool results in age-related infertility and, subsequently, menopause [8, 9]. Thus, the number and quality of primordial follicles present is critically important for female fertility and ovarian endocrine function.
Primordial follicle oocytes are unique in that they exist in a state of meiotic arrest and can remain quiescent for many years in women prior to recruitment into the growing follicle pool. This extreme longevity combined with their arrested state results in particular sensitivity to genotoxic stress, endogenous or exogenous. It is therefore crucial that genomic integrity is maintained, with rigorous surveillance leading to the detection and repair of DNA damage, or the elimination of damaged oocytes by apoptosis [10]. Because these primordial follicles are irreplaceable, interventions to prevent damage and loss and/or enhancement of DNA repair have been the subject of intense interest.
Many conventional chemotherapies commonly used in females for cancers such as breast, haematological, and solid organ malignancies exert their anti-cancer actions through interference with DNA, causing both a therapeutic effect in cancer cells and off-target effects in healthy tissues. Of the several types of DNA damage, double-stranded breaks (DSBs) are the most deleterious, and undetected or unrepaired DSBs can result in chromosome breaks, deletions, translocations, and point mutations [11]. The quality of DSB repair is also critical to the outcome of the damaged cell; poor quality repair can lead to genomic instability or cell death. The extreme longevity of primordial follicle oocytes makes them vulnerable to the accumulation of DSBs, both as a result of the normal ageing process [12, 13] and due to exogenous stressors such as cancer treatment [10, 14–16]. Indeed, karyotypic studies of human oocytes [12, 17], and karyotypic and microarray analyses of early miscarriages [18–21], have shown significant rates of chromosomal aberrations, supporting this notion of vulnerability over extended periods of meiotic arrest. However, it remains unclear whether sensitivity of primordial follicles to DNA damage is due to a reduced threshold for triggering apoptosis, insufficient DNA repair capacity, or both.
The repair of DNA DSBs in somatic cells has been well-characterised, with homologous recombination (HR) and non-homologous end joining (NHEJ) the two main pathways involved. Identification of which pathway(s) is activated following DNA DSBs in oocytes is particularly important because of the risk of transmitting germline mutations to future generations of offspring. In somatic cells, the initial event after the generation of DNA DSBs is the phosphorylation of γH2AX by ATM kinase, leading to the recruitment and targeting of repair factors [22]. From here, the HR pathway utilises the sister chromatid as a DNA template for accurate error-free repair, involving the action of DNA polymerases, nucleases, helicases and ligases. One protein critical to this process is RAD51, which is involved in locating and invading the homologous intact chromatid, DNA pairing, and then strand exchange [23]. This requirement of a sister chromatid to act as template, however, restricts HR to the S and G2 phases of the cell cycle. By contrast, NHEJ culminates in the two broken ends of the DNA DSB being ligated after the removal of ssDNA (single stranded DNA) overhangs at the break site and is thus error-prone. However, it is functional in any phase of the cell cycle. The catalytic sub-unit of DNA-dependent protein kinase (DNA-PKcs) is an essential protein involved in binding the broken ends prior to ligation and provides a marker for detecting the induction of NHEJ [24]. Whilst the “decision-making” mechanisms involved in directing damaged somatic cells toward HR or NHEJ are still under investigation, it is likely that HR constitutes the predominating pathway in oocytes, given that sister chromatids are present [25]. Indeed, HR repair factors, RAD51, BRCA1, and BRCA2 have been localised within PMF oocytes in mice [13, 26], and heterozygous deletion of BRCA1 has been found to impair fertility in mice [13].
In recent years, prevention of oocyte apoptosis has emerged as a promising strategy to ameliorate cancer treatment-induced primordial follicle loss and has been studied in mice following both γ-irradiation and chemotherapy [27–31]. We have previously shown that elimination of the powerful pro-apoptotic BH3-only protein, PUMA, results in complete preservation of the ovarian reserve in mice following single-dose treatment with cisplatin or cyclophosphamide, two chemotherapeutic agents which exert cell damage by causing DNA DSBs. Moreover, long-term fertility trials conducted in that study demonstrated that Puma-/- females treated with either drug had normal fertility outcomes and apparently normal offspring [30]. Subsequently, we have demonstrated that it is indeed the primordial follicle oocyte which is the primary target of DNA damage induced by these drugs [32]. Critically, it has recently been shown that PMF oocytes are indeed highly capable of functionally efficient DNA repair in response to DNA damage caused by γ-irradiation and that genetic fidelity is not compromised [33]. However, the question of whether oocytes rescued from apoptosis after chemotherapy are capable of similarly error-free repair has not been examined; given that chemotherapy is used far more widely in clinical practice, this question must be answered prior to the pursuit of apoptosis prevention as a means of fertility preservation in a clinical oncology setting.
The current study aimed to address these knowledge gaps by examining DNA repair in primordial follicle oocytes following cisplatin treatment in both wild type and Puma-/- mice. Specifically, we aimed to determine which of the classical DNA DSB repair pathways, HR and/or NHEJ, is preferentially utilised in the PMF oocyte after the generation of chemotherapy-induced DSBs. We have previously shown that elimination of PUMA results in survival of the ovarian reserve in its entirety and preserves fertility [30]. Therefore, it was essential to elucidate whether DNA repair conducted within PMF oocytes of Puma-/- mice would be of sufficient quality to prevent the transmission of mutations to future generations—a critical question in determining whether apoptosis inhibition is indeed a feasible means of ovarian protection. In this study we demonstrate that oocytes of Puma-/- mice, like those of wild-type mice, incur DNA DSBs in response to cisplatin. Thus, this genetic knock-out mouse model provides the ideal backdrop for the investigation of DNA repair in PMF oocytes, demonstrating that inhibition of PUMA represents a viable approach to female fertility preservation during cancer treatment.
Materials and methods
Mice
C57BL/6 mice were housed in a photo-controlled animal facility with a 12-h light/dark cycle and given free access to commercial feed (Barastoc, Melbourne, Australia) and water. The generation and genotyping of Puma-/- mice on a C57BL/6 background has been described in detail previously [34]. All animal experiments and procedures were carried out in accordance with the NHMRC Australian Code of Practice for the Care and Use of Animals and approved by the Monash University School of Biomedical Sciences (MARP/2017/077) and the Monash Medical Centre Animal Ethics (MMCB/2014/13) Committees.
Injection of mice
Post-natal day 50 wild type (WT) and Puma-/- female mice received a single intraperitoneal (i.p.) injection of saline or cisplatin (5 mg/kg) (Pfizer, New York, NY, USA) (N = 3/treatment/genotype/timepoint); the cisplatin dose was based on our previous work and on doses commonly used in for the treatment of tumours in mouse studies [32, 35, 36]. Mice were culled by cervical dislocation 8 h, 24 h or 5 days after treatment; one ovary from each mouse was fixed in Bouin’s solution and the contralateral ovary in 10% neutral buffered formalin.
Follicular quantification
Bouin’s-fixed ovaries were processed through ethanol and embedded in glycomethacrylate, after which they were cut into 20 μm sections, stained with periodic acid-Schiff, and counterstained with Mayer’s haematoxylin. Stereology was used to quantify primordial, transitional and primary follicles, using the 100× oil immersion objective on an Olympus BX50 microscope (Tokyo, Japan) equipped with an Autoscan stage (Autoscan Systems Pty Ltd, Melbourne, Victoria, Australia) in conjunction with the StereoInvestigator stereological system (Version 11.06.02, MBF Bioscience 2015, MicroBrightField Inc, Williston, VT, USA). Every 6th section was evaluated and the stereological methods used have previously been described in detail [37]. Secondary, antral, and atretic follicles were quantified using light microscopy to evaluate every 9th section, and then the number was multiplied by a factor of 9 to obtain an estimated total count per ovary, and then again by 2 to obtain an estimation per animal. Follicles were classified histologically by developmental stage as previously described [30].
Immunofluorescence
γH2AX staining was used to mark DNA double-stranded breaks; RAD51 was used to the detect induction of homologous recombination DNA repair; DNA-PKcs was used to identify the induction of non-homologous end-joining DNA repair; c-Kit was used to stain oocytes of primordial, transitional, and primary follicles to aid in their identification. Ovaries fixed in 10% neutral buffered formalin overnight, were processed in ethanol, embedded in paraffin and serially sectioned at 5–μm intervals. Three to five slides (4 sections per slide) were selected to represent the middle and edges of the ovary, and a total of 6–8 sections per ovary were examined. Sections were deparaffinised and rehydrated and then subjected to microwave antigen retrieval in sodium citrate buffer (pH 6) for 10 min. Following cooling to room temperature, sections were blocked with 10% donkey serum at room temperature for 1 h and then incubated at 4 °C with primary antibody. Primary antibodies were diluted in 10% donkey serum, and concentrations used were as follows: rabbit γH2AX antibody (ab22551, Abcam, Cambridge, UK; and 97185, Cell Signalling Technology, Danvers, MA, USA) at 1:500; rabbit antibody RAD51 (ab133534, Abcam, Cambridge, UK) at 1:500; and rabbit DNA-PKcs antibody (ab18192, Abcam, Cambridge, UK) at 1:500; all sections were co-stained with goat c-Kit antibody (af1356, Novus Biologicals, Centennial, CO, USA) at 1:500. After washing with Tris-NaCl-Tween (TNT) buffer (all washes performed at room temperature), sections were incubated with donkey anti-rabbit 568 at 1:500 (A10042, Life Technologies, Carlsbad, CA, USA) and donkey anti-goat Alexa 488 at 1:500 (A11055, Life Technologies, Carlsbad, CA, USA) for 1 h at room temperature, washed again in TNT buffer, incubated with DAPI (SK203, Agilent DAKO, Santa Clara, CA, USA) for 10 min at room temperature, washed again in TNT buffer, and then mounted with FluorSave Reagent (345789, EMD Millipore Corp, Burlington, MA, USA). Slides were visualized with confocal microscopy, using the 40x objective on a Nikon Eclipse 90i microscope (Nikon Corp, Tokyo, Japan); images were processed using FIJI software (National Institutes of Health, New York, NY, USA). Oocytes were considered to be γH2AX-positive if three or more punctate foci were present (Suh et al., 2006); positive granulosa and/or theca cells were also recorded, and the follicle as a whole was considered to be positive if any cells within it were positive; follicles were considered negative if no positive cells were seen (Fig. S1). Percentages of follicles staining positively for γH2AX were calculated by dividing the number of γH2AX-positive follicles by the total number of follicles (in a given follicular stage) and multiplying by 100 for each individual animal. Subanalyses of γH2AX-positive follicles to identify cell targets were conducted by pooling absolute counts of follicles, stratified by cell type affected and expressing this as a percentage of the total number of γH2AX-positive follicles of that follicular stage. The same methods were applied for the quantification and subanalyses of RAD51- and DNA-PKcs-positive follicles.
Statistical analysis
Data are shown as mean ± SEM, and statistical analysis was undertaken using GraphPad Prism 8.0 (GraphPad Software Inc, La Jolla, CA, USA). Comparisons were made using unpaired Student’s t test with statistical significance set at P ≤ 0.05.
Results
The ovarian reserve is maintained 24 hours after cisplatin treatment in PUMA-deficient mice
It has previously been shown that the cisplatin administration protocol used in this study results in an 83% reduction in PMF numbers of WT mice and a net decrease in follicle numbers across all stages, 24 h after injection [32], but that Puma-/- mice retain all of their PMFs when assessed 5 days after treatment [30]. To confirm that elimination of PUMA indeed protects follicles from cisplatin treatment, stereologic enumeration of primordial follicles was undertaken in Puma-/- females, 24 h following cisplatin treatment. In keeping with previous studies of Puma-/- mice given this dose of cisplatin, no depletion of primordial follicles was seen (Fig. 1a). A statistically significant increase in the number of transitional follicles was observed after cisplatin exposure (Fig. 1b, P < 0.05), however no difference was detected in follicles of the primary (Fig. 1c), secondary (Fig. 1d) or antral (Fig. 1e) classes; likewise, there was no change in the number of atretic secondary and antral follicles (Fig. 1f).
Fig. 1.

Follicular quantification in Puma-/- mice by follicular stage and treatment group. Primordial (a), transitional (b), primary (c), secondary (d), antral (e), and atretic (f) follicles were counted in ovaries harvested 24 h after Puma-/- mice were treated with either saline or 5 mg/kg cisplatin (cisplat) (N = 3/treatment group). Data are expressed as mean follicles per animal ± SEM; statistical comparisons were made using Student’s unpaired t test
Double-stranded DNA breaks are detected in follicles of all stages within 8 h of cisplatin treatment in Puma-/- mice
γH2AX immunofluorescence in formalin-fixed sections was used to detect DNA DSBs in ovarian follicles of Puma-/- mice, 8 h, 24 h, and 5 days following cisplatin treatment. Firstly, the magnitude of DNA damage sustained in each follicular class was assessed by identifying and counting γH2AX-positive follicles and expressing this count as a percentage of the total number of follicles of that class. In cisplatin-treated Puma-/- mice at 8 h, 53.4% of primordial follicles stained positively for γH2AX (vs 8 h-saline, 3.3%, P < 0.01; Fig. 2a). The percentage of γH2AX-positive follicles increased with advancing follicular stage, with cisplatin treatment inducing γH2AX positivity in 100% of primary (vs 8 h-saline, 76.7%, P < 0.001; Fig. 2a) and secondary follicles (vs 8 h-saline, 51.9%, P < 0.01; Fig. 2a).Trends towards increased γH2AX positivity in Puma-/- mice treated with cisplatin were also observed in transitional and antral follicles, although these did not reach statistical significance (Fig. 2a). Notably, whilst a proportion of γH2AX-positive secondary and antral follicles were identified in saline controls, these appeared qualitatively different from cisplatin-treated γH2AX-positive follicles of the same class; those from cisplatin-treated mice displayed visibly higher proportions of positively staining granulosa cells (Fig. S2a and b).
Fig. 2.
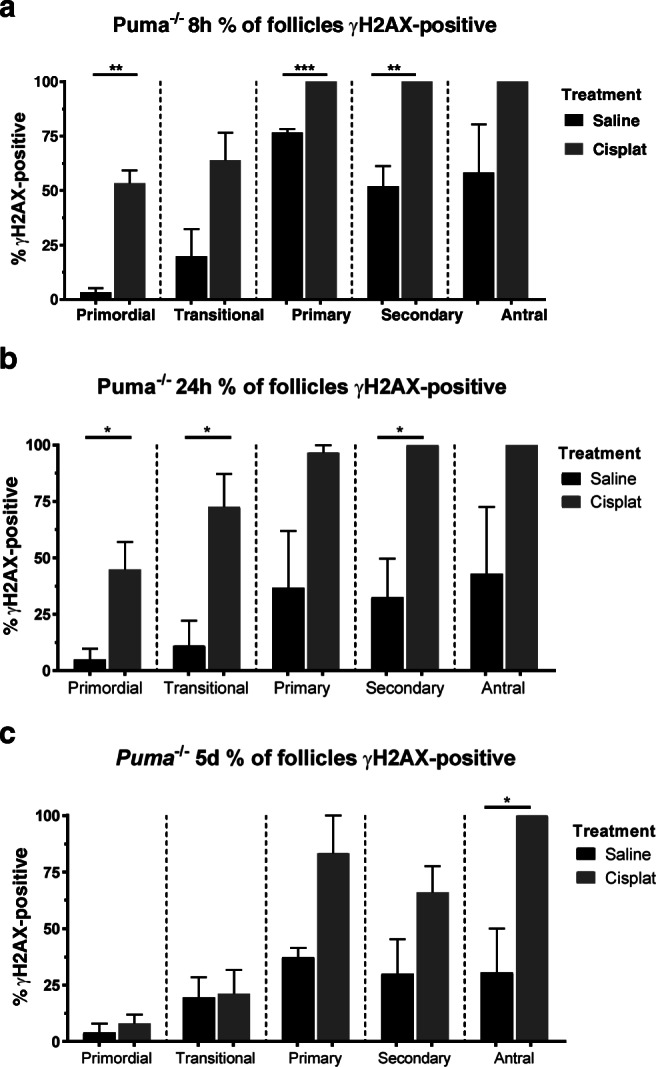
γH2AX-positive follicles by follicular stage and treatment group in Puma-/- mice. Following treatment with saline or cisplatin (cisplat), γH2AX-positive follicles were counted, and expressed as a percentage of the total follicles seen in each respective follicular stage. This was performed at three time-points post-treatment: 8 h (a), 24 h (b), and 5 days (c). Data are expressed as mean percentage ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001 (Student’s unpaired t test)
24 hours following cisplatin treatment, 44.9% of primordial follicles were γH2AX positive, significantly higher than saline controls (vs 24 h-saline, 4.9%, P < 0.05; Fig. 2b). Higher proportions of γH2AX-positive follicles were also observed in the growing follicle pool, including 72.6% of transitional (vs 24 h-saline, 11.1%, P < 0.05; Fig. 2b) and 100% of secondary (vs. 24 h-saline, 32.6%, P < 0.05; Fig. 2b) follicles. Trends towards increased γH2AX-positivity were also observed in primary and antral classes, although these did not reach statistical significance.
At 5 days after cisplatin treatment, the proportion of γH2AX-positive primordial and transitional follicles was observed to fall to control levels (vs 5d-saline, Fig. 2c). Whilst a trend toward persistently increased γH2AX positivity was still observed in primary and secondary follicles, these did not reach statistical significance; a significantly higher proportion of antral follicles in cisplatin-treated mice persisted at this timepoint (5d-cisplatin, 100% vs 5d-saline, 30.5%, P < 0.05; Fig. 2c).
Cisplatin induces double-stranded DNA breaks in oocytes of Puma-/- small follicles, but predominantly within somatic cells of Puma-/- large follicles
γH2AX-positive follicles in Puma-/- mice were then further examined to determine the cell type primarily affected within the follicle. Whilst statistical analysis was not possible due to smaller numbers of follicles in transitional, primary, secondary, and antral classes, visualisation by confocal microscopy revealed that cisplatin treatment resulted in punctate γH2AX staining indicative of double-stranded breaks within only the oocytes of primordial and transitional follicles, but this shifted towards a predominance of somatic cell staining (granulosa and theca cells) in secondary and antral follicles (Fig. 3a). These observations were quantified across all treatment timepoints, by classifying visualised follicles as oocyte-positive, somatic-cell positive, or both oocyte- and somatic-cell positive, then expressing this number as a proportion of the total number of γH2AX-positive follicles in each respective follicular class. Eight hours following treatment, primordial and transitional follicles of both saline (Fig. 3b, i) and cisplatin (Fig. 3b, ii) groups showed γH2AX staining in oocytes only. Oocyte staining remained predominant in primary follicles (Fig. 3b), but the staining pattern underwent a shift at this stage, when somatic cell staining rose in both 8-h saline and 8-h cisplatin groups, with staining of both oocyte and somatic cells universally seen in secondary and antral follicles of 8-h cisplatin mice (Fig. 3b, ii).
Fig. 3.

γH2AX staining in follicles by follicular stage and treatment group in Puma-/- mice. γH2AX-stained follicles of all stages were visualised by confocal microscopy at 8 hours, 24 hours, and 5 days following treatment with saline or cisplatin (cisplat). Red = γH2AX; green = c-Kit; blue = DAPI. Representative images of positively staining primordial, transitional, primary, secondary, and antral follicles at 8 hours following cisplatin treatment (a). Scale bars = 10 μm (primordial, transitional, primary follicles); 50 μm (secondary); 100 μm (antral). Follicles staining positively for γH2AX were further analysed to assess the cell types affected at each follicular stage, and expressed as a total percentage of γH2AX-positive follicles for that stage. Analysis was conducted at 8 hours following treatment (b) with saline (i) or cisplatin (ii); 24 h following treatment (c) with saline (i) or cisplatin (ii); 5 days following treatment (D) with saline (i) or cisplatin (ii)
24 hours after treatment with either saline or cisplatin, oocyte-only staining continued to predominate in primordial, transitional, and primary follicles of both treatment groups (Fig. 3c, i and ii). In secondary follicles 24 hours after saline treatment, somatic cell-only staining was mostly seen, whilst in the antral class, approximately even proportions of oocyte-only, somatic cell-only and both oocyte- and somatic-cell staining were observed (Fig. 3c, i). By contrast, mice injected with cisplatin 24 h prior, in which higher proportions of γH2AX-positive secondary and antral follicles had been seen (Fig. 2b), showed predominance of somatic cell-only staining in these follicle classes (Fig. 3c, ii).
Similar γH2AX staining patterns persisted in the follicles of mice evaluated 5 days after injection with saline or cisplatin (Fig. 3d). Again, oocyte-only staining predominated in primordial, transitional, and primary follicles in both saline- (Fig. 3d, i) and cisplatin-treated (Fig. 3d, ii) groups, whilst cisplatin-treated mice, in which larger numbers of positively staining antral follicles had been seen (Fig. 2c) had higher proportions of somatic cell-only γH2AX staining observed in secondary and antral follicles (Fig. 3d, ii).
Cisplatin treatment leads to the induction of the classical homologous recombination pathway in follicles of all stages
Next, RAD51 immunofluorescence was used to assess the degree to which the homologous recombination DNA repair pathway is employed following DNA double-stranded breaks, in follicles of all stages. Since the highest proportion of γH2AX-positive primordial follicles was observed in Puma-/- mice at the 8 h time-point (Fig. 2a), this time-point was also used to examine RAD51 staining patterns.
Because the induction of DNA repair pathways in normal primordial follicle oocytes has not been characterised before, WT mice were included in all experiments examining these pathways and compared with the response in Puma-/mice. Firstly, in order to examine the role of homologous recombination in PMF oocyte repair, RAD51-positive follicles were quantified in WT mice, 8 h after saline or cisplatin injection. Following cisplatin administration, a significant increase was observed in the percentage of RAD51-positive primordial (WT-cisplatin, 25.0% vs WT-saline, 0%, P < 0.01; Fig. 4a), primary (WT-cisplatin, 49.8.0% vs WT-saline, 6.3%, P < 0.05; Fig. 4a), secondary (WT-cisplatin, 100.0% vs WT-saline, 16.7%, P < 0.01; Fig. 4a), and antral follicles (WT-cisplatin, 100.0% vs WT-saline, 12.5%, P < 0.01; Fig. 4a). No significant difference was seen in the transitional follicle class.
Fig. 4.
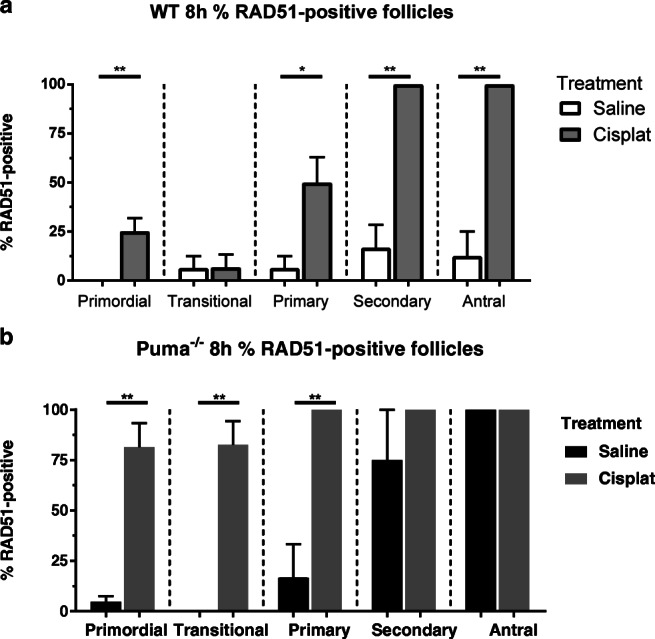
RAD51-positive follicles by follicular stage and treatment group in WT and Puma-/- mice. Eight hours following treatment with saline or cisplatin (cisplat), RAD51-positive follicles were counted, and expressed as a percentage of the total follicles seen in each respective follicular stage. This was performed in WT mice (a) and Puma-/- mice (b). Data are expressed as mean percentage ± SEM; P < 0.05, **P < 0.01 (Student’s unpaired t test)
In comparison, very high proportions of RAD51-positive follicles were seen across all stages in Puma-/- mice 8 h after cisplatin therapy, in which all follicles were rescued (Fig. 1); 81.4% of primordial follicles (vs Puma-/--saline, 4.6%, P <0.01, Fig. 4b), 82.5% of transitional follicles (vs Puma-/--saline, 0%, P < 0.01) and 100% of primary follicles (vs Puma-/--saline, 16.7%, P<0.01). Interestingly, high proportions of secondary and antral follicles were RAD51-positive in the saline-treated Puma-/- mice, with no statistically significant difference seen between cisplatin- and saline-treated animals in follicles of these classes (Puma-/--cisplatin secondary follicles, 100%, vs Puma-/--saline secondary follicles, 75%; Puma-/--cisplatin antral follicles, 100% vs Puma-/--saline antral follicles, 100%). This observation is consistent with higher proportions of γH2AX-positive follicles of these classes in the saline-treated Puma-/- animals at the same timepoint (Fig. 2a).
The homologous recombination DNA repair pathway is activated at sites of DNA double-strand breaks in follicles of all stages following cisplatin treatment
RAD51-positive follicles were further analysed to assess the cell types predominantly affected at each follicular stage. Similar to the γH2AX staining pattern described earlier, confocal microscopy demonstrated punctate RAD51 staining in the oocytes of primordial, transitional and primary follicles, but predominantly within the somatic cells of secondary and antral follicles (Fig. 5a). When this observation was quantified, in RAD51-positive follicles of saline-treated WT mice, only oocytes stained positively in primordial, transitional, and primary follicles, and only somatic cell staining was seen in secondary and antral follicles (Fig. 5b, i). A similar pattern was seen in RAD51-positive follicles of WT mice treated with cisplatin, with only oocytes staining positively in primordial and transitional follicles, mixed staining in a majority of primary follicles, and somatic cell-only staining predominating at the secondary and antral follicle stages (Fig. 5b, ii).
Fig. 5.

RAD51 staining in follicles by follicular stage and treatment group in WT and Puma-/- mice. RAD51-stained follicles of all stages were visualised by confocal microscopy at 8 hours following treatment with saline or cisplatin (cisplat). Red = RAD51; green = c-Kit; blue = DAPI. Representative images of positively staining primordial, transitional, primary, secondary, and antral follicles of Puma-/- mice, 8 hours following treatment with cisplatin (A). Scale bars = 10 μm (primordial, transitional, primary follicles); 50 μm (secondary); 100 μm (antral). Follicles staining positively for RAD51 were further analysed to assess the cell types affected at each follicular stage, and expressed as a total percentage of RAD51-positive follicles for that stage. Analysis was conducted in WT mice (B) 8 hours following treatment with saline (i) or cisplatin (ii); and in Puma-/- mice (C) 8 hours following treatment with saline (i) or cisplatin (ii)
Similarly, in Puma-/- mice, RAD51 staining was seen only in the oocytes of primordial and transitional follicles of saline-treated animals; both oocyte and somatic cell staining was observed in primary follicles, and then somatic cell-only staining predominated in secondary and antral follicles (Fig. 5c, i). Similarly, in Puma-/- cisplatin-treated mice, only oocytes of primordial and transitional follicles stained positively for RAD51, with oocyte-only staining predominating at the primary stage, then somatic cell-only staining predominating in secondary and antral follicles (Fig. 5c, ii).
Overall, the patterns of RAD51 staining seen in both treatment groups of both genotypes suggest that homologous recombination is activated at sites of DNA DSBs.
The non-homologous end-joining pathway is seldom employed in primordial follicles following cisplatin treatment
In order to assess the degree to which NHEJ is activated following cisplatin-induced DNA DSBs, DNA-PKcs immunofluorescence was performed in ovaries harvested 8 h after treatment with saline or cisplatin. Visualisation and quantification of DNA-PKcs-positive follicles revealed only very small numbers of positively staining primordial and transitional follicles (Fig. S3a; Figure 6a and b). In WT animals treated with saline, no positive follicles were seen; in contrast, in cisplatin-treated WT mice, whilst only 7.3% (vs WT-saline, 0%, P < 0.0001) and 16.7% (vs WT-saline, 0%, P < 0.0001) of primordial and transitional follicles, respectively, stained positively for DNA-PKcs, higher proportions were seen in the primary (WT-cisplatin, 67.5% vs WT-saline, 0%, P <0.0001), secondary (WT-cisplatin, 77.8%, vs WT-saline, P < 0.0001), and antral (WT-cisplatin 100% vs WT-saline, 0%) follicle classes.
Fig. 6.
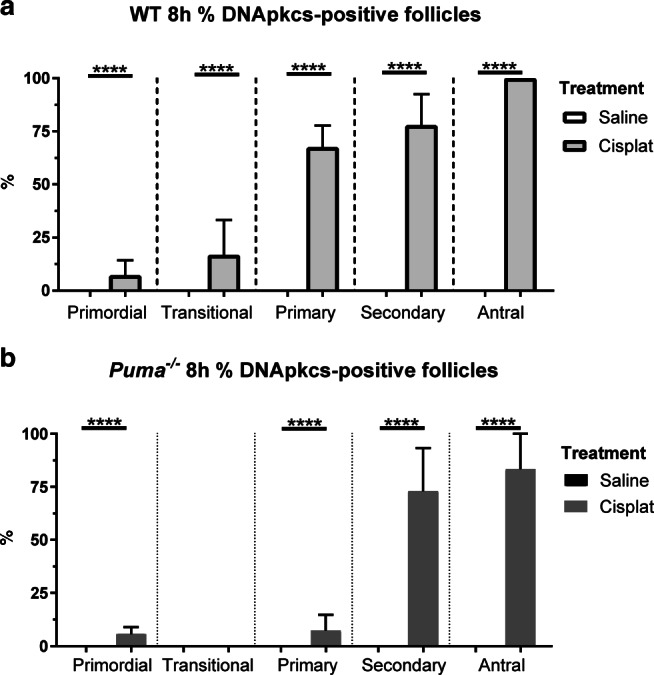
DNA-PKcs-positive follicles by follicular stage and treatment group in WT and Puma-/- mice. Eight hours following treatment with saline or cisplatin (cisplat), DNA-PKcs-positive follicles were counted, and expressed as a percentage of the total follicles seen in each respective follicular stage. This was performed in WT mice (a) and Puma-/- mice (b). Data are expressed as mean percentage ± SEM; P < 0.05, **P < 0.01 (Student’s unpaired t test)
Similarly, in Puma-/- mice treated with saline, no DNA-PKcs staining was seen in any follicle class (Fig. 6b). In the cisplatin-treated Puma-/- group, a very small number of positively staining primordial (Puma-/--cisplatin, 5.7% vs Puma-/--saline, 0%, P < 0.0001) and primary follicles (Puma-/--cisplatin, 7.4% vs Puma-/--saline, 0%, P < 0.0001) was seen; no staining was seen in transitional follicles; in contrast, large proportions of secondary (Puma-/--cisplatin, 73.0% vs Puma-/--saline, 0%, P < 0.0001) and antral (Puma-/--cisplatin, 83.3% vs Puma-/--saline, 0%, P < 0.0001) stained positively for DNA-PKcs (Fig. 6b).
Confocal visualisation showed that in larger follicles staining positively for DNA-PKcs, only somatic cells stained positively (Fig. S3a); numbers of positive primordial and transitional follicles were too small for meaningful analysis (Fig. 6a, b; Fig. S3b, c).
Overall, these results show that the NHEJ pathway is seldom employed by primordial follicles after DNA DSBs induced by cisplatin treatment.
Discussion
It is well established that primordial follicle oocytes incur DNA DSBs during genotoxic cancer treatment (Gonfloni et al., 2009, Livera et al., 2008, Nguyen et al., 2019, Oktem and Oktay, 2007, Suh et al., 2006). We have previously demonstrated that this direct damage and subsequent apoptosis is the primary mechanism by which the ovarian reserve is depleted following genotoxic chemotherapy (Nguyen et al., 2019). Therefore, novel pharmacological strategies should be aimed either at preventing apoptosis or enhancing DNA repair in PMF oocytes. Accordingly, proposed avenues of exploration for future fertility preservation have included inhibition of apoptosis [27, 29–31, 38–50], and enhancement of DNA repair pathways or prevention of damage [51]. A recent study by conducted by Stringer et al. used Tap63-/- mice to demonstrate that in the absence of apoptosis, PMF oocytes utilise the homologous recombination pathway to conduct highly effective DNA repair in response to DSBs induced by irradiation, and that this repair does not give rise to higher rates of mutations [33]. Building on these findings, in this study we show that RAD51 is localised to sites of DNA DSBs induced by the chemotherapeutic agent, cisplatin, suggesting that repair within primordial follicle oocytes in this context also occurs predominantly via the homologous recombination pathway. Because homologous recombination results in high fidelity repair, our findings suggest that elimination of apoptosis is unlikely to result in the propagation of mutations to future generations of offspring following chemotherapy, and further strengthens the argument for this approach to prevent ovarian damage due to chemoradiation.
In keeping with our previous findings, these data confirm that the prevention of apoptosis by elimination of PUMA results in complete preservation of the ovarian reserve [30]. However, the formation of DNA DSBs in response to cisplatin was unaffected, indicating that PUMA loss, by preventing apoptosis, redirects damaged oocytes to repair their DNA. Most studies examining PMF apoptosis following either γ-irradiation or various genotoxic chemotherapies have demonstrated that apoptosis prevention rescues all or part of the ovarian reserve, but the presence or absence of DSBs in apoptosis-deficient oocytes has not previously been reported [27, 31, 42, 43, 50, 52]. We have previously reported that in response to γ-irradiation, PMF oocytes of Puma-/- and Puma-/-/Noxa-/- female mice sustain levels of DSBs comparable to WT mice, as demonstrated by the appearance of γH2AX foci in PMF oocyte nuclei [29]. Similarly, we now demonstrate that the survival of oocytes in Puma-/- mice after DSBs induced by cisplatin is linked to their ability to avoid apoptosis and conduct effective DNA repair, rather than prevention of the damage itself. Collectively, our data therefore suggest that integrity of DNA repair within PMF oocytes is a critical factor determining whether apoptosis prevention forms a viable means of ovarian protection from chemotherapy or radiation for cancer treatment.
Interestingly, we observed an increase in the number of transitional follicles in cisplatin compared with saline-treated Puma-/- mice. Whilst we cannot be certain of the underlying reason, it could be because the cell cycle was halted in the granulosa cells of transitional follicles in order to complete repair of cisplatin induced damage, temporarily slowing follicular progression to the primary stage. This idea is consistent with the small but non-significant decrease in primary follicles observed. Increased activation of primordial follicles is a less likely explanation, as primordial follicles were not depleted, nor were secondary follicles, both of which are prerequisites for chemotherapy induced follicle depletion via “burnout”. Our previous study using the same chemotherapy protocol also showed no net increase in the growing follicle pool 5 days after a single dose of cisplatin, in support of there being no significant activation effect due to cisplatin treatment [30].
Previously, we have studied the induction of DNA DSBs in WT mice in response to treatment with cisplatin (using the same treatment protocol as the current study) and cyclophosphamide. In WT mice treated with saline, no significant γH2AX staining was detected at 8 or 24 h following injection. In contrast, we now report that saline-treated Puma-/- mice had much a higher proportion of γH2AX-positive follicles at all stages of development although still significantly less than seen after cisplatin administration. Accordingly, when compared with WT mice in this study, saline-treated Puma-/- mice demonstrated higher proportions of RAD51-positive follicles than their WT counterparts. The reasons for the increased levels of γH2AX staining at baseline in Puma-/- mice are unclear. It may be related to the inability of these follicles to undergo apoptosis at normal DNA damage thresholds when compared with WT mice, thus necessitating the initiation of repair as demonstrated by the increased presence of RAD51 foci. Interestingly, this observation is consistent with that made previously in a study of Puma-/- and Puma-/-/Noxa-/- mice receiving γ-irradiation, in which apoptosis-deficient mice were found to have a residual level of positive γH2AX staining within some PMF oocytes even 5 days following treatment [29].
Our demonstration of punctate RAD51 staining within PMF oocytes—likely localising to the same site where DNA DSBs occur, as indicated by γH2AX staining—suggests that homologous recombination is the predominant pathway induced after cisplatin-induced DNA damage. Whilst it is known that HR is a critical mechanism of DSB break repair during meiotic recombination in mammalian oocytes, this has not been previously shown to occur in prophase-arrested oocytes. During normal meiotic recombination oocytes sustain many DSBs, promoted by the action of Spo11, which are essential to allow DNA exchange between homologous sister chromatids [53, 54]; these DSBs are tolerated and repaired by homologous recombination, in a pathway involving many factors such as MRN11, RAD51, NBS1, BRCA1 and BRCA2. However, under normal circumstances, arrested PMF oocytes do not demonstrate this same tolerance for DSBs, and indeed it has been shown that the threshold for a PMF oocyte to undergo apoptosis may be a single unrepaired break [27]. This low threshold for apoptosis may represent a crucial safeguard for germline integrity [10, 55]. However, it is unclear why oocytes undergoing meiotic recombination can tolerate and indeed repair hundreds of DSBs, whereas arrested PMF oocytes cannot. The results of our study suggest that in the absence of effective apoptotic mechanisms, PMF oocytes are able to “redirect” their critical DNA damage response pathways from one of apoptosis, to one favouring high-fidelity repair. This is supported by our finding that Puma-/- mice have higher levels of RAD51 within PMF oocyte nuclei after cisplatin treatment than their WT counterparts, which instead commit to apoptosis in high proportions, as evidenced by reduced follicle counts 24 h after treatment. Given the ability of oocytes to commit to this pathway earlier in development, the known presence of HR factors within oocytes, and the presence of a sister chromatid to provide the template for faithful repair, the finding that apoptosis-deficient oocytes utilise HR as the predominant repair pathway is perhaps unsurprising. Interestingly, RAD51-positive follicles were seen in higher proportions than γH2AX at our earliest treatment timepoint, 8 h after injection. However, this may indicate that peak γH2AX positivity occurs at an earlier time than captured by our study, which is unsurprising given that in studies of γ-irradiation, γH2AX foci are seen in high proportions by 3 h post-treatment [10, 27, 29].
This study demonstrated a relative paucity of DNA-PKcs staining within PMF oocytes after cisplatin-induced DNA DSBs, in both WT and Puma-/- mice. Given that DNA-PKcs is an essential part of the NHEJ repair pathway, this indicates that NHEJ does not form a major component of the DSB repair response within PMF oocytes after cisplatin. NHEJ pathway components have previously been shown to be expressed within oocytes. However, it appears that NHEJ may be utilised by oocytes at much later stages of development; in Xenopus laevis, it has been shown that oocytes at the later, germinal vesicle stage do not express NHEJ factors until metaphase II, at which time NHEJ becomes the predominant repair mechanism [56, 57]. However, in somatic cells, it has previously been shown that HR and NHEJ may compete or cooperate to repair DSB. Notably, unlike HR, NHEJ is able to operate throughout the cell cycle, and has been shown to be the main DSB repair pathway in higher eukaryotes, particularly in phases of the cell cycle where a sister chromatid is absent [58, 59]. Our data showed DNA-PKcs in very few primordial follicles, meaning that we cannot draw accurate conclusions about the cell types employing NHEJ within PMFs after cisplatin-induced DSBs. However, high numbers of both RAD51 and DNA-PKcs-positive follicles were seen in later follicular stages, particularly secondary and antral follicles, in which DNA-PKcs foci were observed only within somatic cells. This is consistent with our findings that in larger growing follicles, high proportions of somatic (granulosa and theca) cells stained positively for both RAD51 and DNA-PKcs, suggesting that both HR and NHEJ are used within these cells to repair DSBs sustained in these circumstances. A much smaller proportion of oocytes were shown to sustain DSBs in growing follicles of Puma-/- mice; this is consistent with our previous studies examining chemotherapy-induced DSBs in WT mice [32]. In keeping with the previously noted observations that NHEJ plays a major role in the oocyte only from the MII stage [56, 57, 60], we found only RAD51 staining within secondary and antral follicle oocytes.
In this in vivo study of DNA repair in mouse ovarian follicles, we have demonstrated the induction of DNA DSBs within the primordial follicle oocytes of apoptosis-deficient mice and shown that when the opportunity for apoptosis is removed, primordial follicle oocytes are capable of initiating the high-fidelity DSB repair pathway, homologous recombination. Additionally, we have shown that this pathway likely predominates over the more error-prone repair pathway, non-homologous end-joining. Given the promise shown by numerous studies of apoptosis inhibition for fertility preservation in cancer, our findings provide strong evidence that this approach is likely to be safe and effective in preventing ovarian damage during chemotherapy without presenting significant risks of transmitting germline mutations to future generations.
Supplementary Information
(DOCX 128 kb)
(DOCX 4033 kb)
(PDF 1065 kb)
Acknowledgements
The authors acknowledge the facilities, scientific and technical assistance of Monash Micro Imaging, the Monash Animal Research Platform, and the Monash Histology Platform, Monash University, Victoria, Australia.
Authors’ contributions
QNN planned experiments, obtained ethics approvals, conducted experiments, analysed data, and prepared the manuscript. NZ provided laboratory support and assisted with experiments. MH and JKF provided supervision, assisted in planning experiments, and revised the manuscript. KJH provided supervision, gave assistance with ethics applications, project funding, laboratory resources and infrastructure, planned experiments, and prepared the manuscript.
Funding
This work was supported by fellowships and grants from the National Health and Medical Research Council Australia (Project Grant #1007027, Program Grant #1016701, and Fellowship KJH #1050130); National Breast Cancer Foundation (#NC14-001). This work was made possible through Victorian State Government Operational Infrastructure Support and Australian Government NHMRC IRIISS.
Data Availability
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
Declarations
Conflicts of interest/competing interests
None to declare.
Ethics approval
All animal experiments and procedures were carried out in accordance with the NHMRC Australian Code of Practice for the Care and Use of Animals and approved by the Monash University School of Biomedical Sciences (MARP/2017/077) and the Monash Medical Centre Animal Ethics (MMCB/2014/13) Committees.
Consent to participate
Not applicable
Consent for publication
Not applicable
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bath LE, Wallace WH, Critchley HO. Late effects of the treatment of childhood cancer on the female reproductive system and the potential for fertility preservation. BJOG. 2002;109(2):107–114. doi: 10.1111/j.1471-0528.2002.t01-1-01007.x. [DOI] [PubMed] [Google Scholar]
- 2.Hudson MM. Reproductive outcomes for survivors of childhood cancer. Obstetrics and gynecology. 2010;116(5):1171–1183. doi: 10.1097/AOG.0b013e3181f87c4b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Faubion SS, Kuhle CL, Shuster LT, Rocca WA. Long-term health consequences of premature or early menopause and considerations for management. Climacteric. 2015;18(4):483–491. doi: 10.3109/13697137.2015.1020484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meirow D, Biederman H, Anderson RA, Wallace WH. Toxicity of chemotherapy and radiation on female reproduction. Clinical obstetrics and gynecology. 2010;53(4):727–739. doi: 10.1097/GRF.0b013e3181f96b54. [DOI] [PubMed] [Google Scholar]
- 5.Byrne J, Fears TR, Gail MH, Pee D, Connelly RR, Austin DF, Holmes GF, Holmes FF, Latourette HB, Meigs JW, Strong LC, Myers MH, Mulvihill JJ. Early menopause in long-term survivors of cancer during adolescence. American journal of obstetrics and gynecology. 1992;166(3):788–793. doi: 10.1016/0002-9378(92)91335-8. [DOI] [PubMed] [Google Scholar]
- 6.Beaumont HM. The radiosensitivity of germ-cells at various stages of ovarian development. International journal of radiation biology and related studies in physics, chemistry, and medicine. 1962;4:581–590. doi: 10.1080/09553006214550391. [DOI] [PubMed] [Google Scholar]
- 7.Bristol-Gould SK, Kreeger PK, Selkirk CG, Kilen SM, Mayo KE, Shea LD, Woodruff TK. Fate of the initial follicle pool: empirical and mathematical evidence supporting its sufficiency for adult fertility. Developmental biology. 2006;298(1):149–154. doi: 10.1016/j.ydbio.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 8.de Bruin JPV, E.R. Female reproductive aging: concepts and consequences. Preservation of Fertility. London: Taylor and Francis; 2004. [Google Scholar]
- 9.Findlay JK, Hutt KJ, Hickey M, Anderson RA. How Is the Number of Primordial Follicles in the Ovarian Reserve Established? Biology of reproduction. 2015;93(5):111. doi: 10.1095/biolreprod.115.133652. [DOI] [PubMed] [Google Scholar]
- 10.Hanoux V, Pairault C, Bakalska M, Habert R, Livera G. Caspase-2 involvement during ionizing radiation-induced oocyte death in the mouse ovary. Cell death and differentiation. 2007;14(4):671–681. doi: 10.1038/sj.cdd.4402052. [DOI] [PubMed] [Google Scholar]
- 11.Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25(5):409–433. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim AS, Tsakok MF. Age-related decline in fertility: a link to degenerative oocytes? Fertility and sterility. 1997;68(2):265–271. doi: 10.1016/s0015-0282(97)81513-0. [DOI] [PubMed] [Google Scholar]
- 13.Titus S, Li F, Stobezki R, Akula K, Unsal E, Jeong K, et al. Impairment of BRCA1-related DNA double-strand break repair leads to ovarian aging in mice and humans. Science translational medicine. 2013;5(172):172ra21. doi: 10.1126/scitranslmed.3004925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kerr JB, Brogan L, Myers M, Hutt KJ, Mladenovska T, Ricardo S, Hamza K, Scott CL, Strasser A, Findlay JK. The primordial follicle reserve is not renewed after chemical or gamma-irradiation mediated depletion. Reproduction. 2012;143(4):469–476. doi: 10.1530/REP-11-0430. [DOI] [PubMed] [Google Scholar]
- 15.Tatone C, Amicarelli F, Carbone MC, Monteleone P, Caserta D, Marci R, Artini PG, Piomboni P, Focarelli R. Cellular and molecular aspects of ovarian follicle ageing. Human reproduction update. 2008;14(2):131–142. doi: 10.1093/humupd/dmm048. [DOI] [PubMed] [Google Scholar]
- 16.Kerr JB, Hutt KJ, Cook M, Speed TP, Strasser A, Findlay JK, Scott CL. Cisplatin-induced primordial follicle oocyte killing and loss of fertility are not prevented by imatinib. Nature medicine. 2012;18(8):1170–1172. doi: 10.1038/nm.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ravel C, Berthaut I, Bresson JL, Siffroi JP. Genetics Commission of the French Federation of C. Prevalence of chromosomal abnormalities in phenotypically normal and fertile adult males: large-scale survey of over 10,000 sperm donor karyotypes. Hum Reprod. 2006;21(6):1484–1489. doi: 10.1093/humrep/del024. [DOI] [PubMed] [Google Scholar]
- 18.van den Berg MM, van Maarle MC, van Wely M, Goddijn M. Genetics of early miscarriage. Biochimica et biophysica acta. 2012;1822(12):1951–1959. doi: 10.1016/j.bbadis.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 19.McFadden DE, Friedman JM. Chromosome abnormalities in human beings. Mutation research. 1997;396(1-2):129–140. doi: 10.1016/s0027-5107(97)00179-6. [DOI] [PubMed] [Google Scholar]
- 20.Pellicer A, Rubio C, Vidal F, Minguez Y, Gimenez C, Egozcue J, et al. In vitro fertilization plus preimplantation genetic diagnosis in patients with recurrent miscarriage: an analysis of chromosome abnormalities in human preimplantation embryos. Fertility and sterility. 1999;71(6):1033–1039. doi: 10.1016/s0015-0282(99)00143-0. [DOI] [PubMed] [Google Scholar]
- 21.Zhang YX, Zhang YP, Gu Y, Guan FJ, Li SL, Xie JS, Shen Y, Wu BL, Ju W, Jenkins EC, Brown WT, Zhong N. Genetic analysis of first-trimester miscarriages with a combination of cytogenetic karyotyping, microsatellite genotyping and arrayCGH. Clin Genet. 2009;75(2):133–140. doi: 10.1111/j.1399-0004.2008.01131.x. [DOI] [PubMed] [Google Scholar]
- 22.Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27(3):247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 23.Baumann P, West SC. Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem Sci. 1998;23(7):247–251. doi: 10.1016/s0968-0004(98)01232-8. [DOI] [PubMed] [Google Scholar]
- 24.Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J. 2009;417(3):639–650. doi: 10.1042/BJ20080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Winship AL, Stringer JM, Liew SH, Hutt KJ. The importance of DNA repair for maintaining oocyte quality in response to anti-cancer treatments, environmental toxins and maternal ageing. Human reproduction update. 2018;24:119–134. doi: 10.1093/humupd/dmy002. [DOI] [PubMed] [Google Scholar]
- 26.Kujjo LL, Laine T, Pereira RJ, Kagawa W, Kurumizaka H, Yokoyama S, et al. Enhancing survival of mouse oocytes following chemotherapy or aging by targeting Bax and Rad51. PloS one. 2010;5(2):e9204. doi: 10.1371/journal.pone.0009204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suh EK, Yang A, Kettenbach A, Bamberger C, Michaelis AH, Zhu Z, Elvin JA, Bronson RT, Crum CP, McKeon F. p63 protects the female germ line during meiotic arrest. Nature. 2006;444(7119):624–628. doi: 10.1038/nature05337. [DOI] [PubMed] [Google Scholar]
- 28.Gonfloni S, Di Tella L, Caldarola S, Cannata SM, Klinger FG, Di Bartolomeo C, et al. Inhibition of the c-Abl-TAp63 pathway protects mouse oocytes from chemotherapy-induced death. Nature medicine. 2009;15(10):1179–1185. doi: 10.1038/nm.2033. [DOI] [PubMed] [Google Scholar]
- 29.Kerr JB, Hutt KJ, Michalak EM, Cook M, Vandenberg CJ, Liew SH, Bouillet P, Mills A, Scott CL, Findlay JK, Strasser A. DNA damage-induced primordial follicle oocyte apoptosis and loss of fertility require TAp63-mediated induction of Puma and Noxa. Molecular cell. 2012;48(3):343–352. doi: 10.1016/j.molcel.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen QN, Zerafa N, Liew SH, Morgan FH, Strasser A, Scott CL, Findlay JK, Hickey M, Hutt KJ. Loss of PUMA protects the ovarian reserve during DNA-damaging chemotherapy and preserves fertility. Cell Death Dis. 2018;9(6):618. doi: 10.1038/s41419-018-0633-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Livera G, Petre-Lazar B, Guerquin MJ, Trautmann E, Coffigny H, Habert R. p63 null mutation protects mouse oocytes from radio-induced apoptosis. Reproduction. 2008;135(1):3–12. doi: 10.1530/REP-07-0054. [DOI] [PubMed] [Google Scholar]
- 32.Nguyen QN, Zerafa N, Liew SH, Findlay JK, Hickey M, Hutt KJ. Cisplatin- and cyclophosphamide-induced primordial follicle depletion is caused by direct damage to oocytes. Molecular human reproduction. 2019;25(8):433–444. doi: 10.1093/molehr/gaz020. [DOI] [PubMed] [Google Scholar]
- 33.Stringer JM, Winship A, Zerafa N, Wakefield M, Hutt K. Oocytes can efficiently repair DNA double-strand breaks to restore genetic integrity and protect offspring health. Proceedings of the National Academy of Sciences of the United States of America. 2020;117(21):11513–11522. doi: 10.1073/pnas.2001124117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302(5647):1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 35.Mabuchi S, Ohmichi M, Nishio Y, Hayasaka T, Kimura A, Ohta T, Saito M, Kawagoe J, Takahashi K, Yada-Hashimoto N, Sakata M, Motoyama T, Kurachi H, Tasaka K, Murata Y. Inhibition of NFkappaB increases the efficacy of cisplatin in in vitro and in vivo ovarian cancer models. J Biol Chem. 2004;279(22):23477–23485. doi: 10.1074/jbc.M313709200. [DOI] [PubMed] [Google Scholar]
- 36.Spanos WC, Nowicki P, Lee DW, Hoover A, Hostager B, Gupta A, Anderson ME, Lee JH. Immune response during therapy with cisplatin or radiation for human papillomavirus-related head and neck cancer. Arch Otolaryngol Head Neck Surg. 2009;135(11):1137–1146. doi: 10.1001/archoto.2009.159. [DOI] [PubMed] [Google Scholar]
- 37.Myers M, Britt KL, Wreford NG, Ebling FJ, Kerr JB. Methods for quantifying follicular numbers within the mouse ovary. Reproduction. 2004;127(5):569–580. doi: 10.1530/rep.1.00095. [DOI] [PubMed] [Google Scholar]
- 38.Bolcun-Filas E, Rinaldi VD, White ME, Schimenti JC. Reversal of female infertility by Chk2 ablation reveals the oocyte DNA damage checkpoint pathway. Science. 2014;343(6170):533–536. doi: 10.1126/science.1247671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hancke K, Strauch O, Kissel C, Gobel H, Schafer W, Denschlag D. Sphingosine 1-phosphate protects ovaries from chemotherapy-induced damage in vivo. Fertility and sterility. 2007;87(1):172–177. doi: 10.1016/j.fertnstert.2006.06.020. [DOI] [PubMed] [Google Scholar]
- 40.Hancke K, Walker E, Strauch O, Gobel H, Hanjalic-Beck A, Denschlag D. Ovarian transplantation for fertility preservation in a sheep model: can follicle loss be prevented by antiapoptotic sphingosine-1-phosphate administration? Gynecological endocrinology : the official journal of the International Society of Gynecological Endocrinology. 2009;25(12):839–843. doi: 10.3109/09513590903159524. [DOI] [PubMed] [Google Scholar]
- 41.Kaya H, Desdicioglu R, Sezik M, Ulukaya E, Ozkaya O, Yilmaztepe A, Demirci M. Does sphingosine-1-phosphate have a protective effect on cyclophosphamide- and irradiation-induced ovarian damage in the rat model? Fertility and sterility. 2008;89(3):732–735. doi: 10.1016/j.fertnstert.2007.03.065. [DOI] [PubMed] [Google Scholar]
- 42.Kim SY, Cordeiro MH, Serna VA, Ebbert K, Butler LM, Sinha S, Mills AA, Woodruff TK, Kurita T. Rescue of platinum-damaged oocytes from programmed cell death through inactivation of the p53 family signaling network. Cell death and differentiation. 2013;20(8):987–997. doi: 10.1038/cdd.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim SY, Nair DM, Romero M, Serna VA, Koleske AJ, Woodruff TK, Kurita T. Transient inhibition of p53 homologs protects ovarian function from two distinct apoptotic pathways triggered by anticancer therapies. Cell death and differentiation. 2019;26(3):502–515. doi: 10.1038/s41418-018-0151-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luan Y, Edmonds ME, Woodruff TK, Kim SY. Inhibitors of apoptosis protect the ovarian reserve from cyclophosphamide. The Journal of endocrinology. 2019;240(2):243–256. doi: 10.1530/JOE-18-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meng Y, Xu Z, Wu F, Chen W, Xie S, Liu J, Huang X, Zhou Y. Sphingosine-1-phosphate suppresses cyclophosphamide induced follicle apoptosis in human fetal ovarian xenografts in nude mice. Fertility and sterility. 2014;102(3):871–877. doi: 10.1016/j.fertnstert.2014.05.040. [DOI] [PubMed] [Google Scholar]
- 46.Paris F, Perez GI, Fuks Z, Haimovitz-Friedman A, Nguyen H, Bose M, Ilagan A, Hunt PA, Morgan WF, Tilly JL, Kolesnick R. Sphingosine 1-phosphate preserves fertility in irradiated female mice without propagating genomic damage in offspring. Nature medicine. 2002;8(9):901–902. doi: 10.1038/nm0902-901. [DOI] [PubMed] [Google Scholar]
- 47.Pascuali N, Scotti L, Di Pietro M, Oubina G, Bas D, May M, et al. Ceramide-1-phosphate has protective properties against cyclophosphamide-induced ovarian damage in a mice model of premature ovarian failure. Hum Reprod. 2018;33(5):844–859. doi: 10.1093/humrep/dey045. [DOI] [PubMed] [Google Scholar]
- 48.Rinaldi VD, Bolcun-Filas E, Kogo H, Kurahashi H, Schimenti JC. The DNA Damage Checkpoint Eliminates Mouse Oocytes with Chromosome Synapsis Failure. Molecular cell. 2017;67(6):1026–1036. doi: 10.1016/j.molcel.2017.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spears N, Lopes F, Stefansdottir A, Rossi V, De Felici M, Anderson RA, et al. Ovarian damage from chemotherapy and current approaches to its protection. Human reproduction update. 2019;25(6):673–693. doi: 10.1093/humupd/dmz027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tuppi M, Kehrloesser S, Coutandin DW, Rossi V, Luh LM, Strubel A, Hötte K, Hoffmeister M, Schäfer B, de Oliveira T, Greten F, Stelzer EHK, Knapp S, de Felici M, Behrends C, Klinger FG, Dötsch V. Oocyte DNA damage quality control requires consecutive interplay of CHK2 and CK1 to activate p63. Nat Struct Mol Biol. 2018;25(3):261–269. doi: 10.1038/s41594-018-0035-7. [DOI] [PubMed] [Google Scholar]
- 51.Stringer J, Groenewegen E, Liew SH, Hutt KJ. Nicotinamide mononucleotide does not protect the ovarian reserve from cancer treatments. Reproduction. 2019. 10.1530/REP-19-0337. [DOI] [PubMed]
- 52.Rinaldi VD, Hsieh K, Munroe R, Bolcun-Filas E, Schimenti JC. Pharmacological Inhibition of the DNA Damage Checkpoint Prevents Radiation-Induced Oocyte Death. Genetics. 2017;206(4):1823–1828. doi: 10.1534/genetics.117.203455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McDougall A, Elliott DJ, Hunter N. Pairing, connecting, exchanging, pausing and pulling chromosomes. EMBO Rep. 2005;6(2):120–125. doi: 10.1038/sj.embor.7400331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carroll J, Marangos P. The DNA damage response in mammalian oocytes. Front Genet. 2013;4:117. doi: 10.3389/fgene.2013.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Levine AJ, Tomasini R, McKeon FD, Mak TW, Melino G. The p53 family: guardians of maternal reproduction. Nature reviews Molecular cell biology. 2011;12(4):259–265. doi: 10.1038/nrm3086. [DOI] [PubMed] [Google Scholar]
- 56.Goedecke W, Vielmetter W, Pfeiffer P. Activation of a system for the joining of nonhomologous DNA ends during Xenopus egg maturation. Mol Cell Biol. 1992;12(2):811–816. doi: 10.1128/mcb.12.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hagmann M, Adlkofer K, Pfeiffer P, Bruggmann R, Georgiev O, Rungger D, Schaffner W. Dramatic changes in the ratio of homologous recombination to nonhomologous DNA-end joining in oocytes and early embryos of Xenopus laevis. Biol Chem Hoppe Seyler. 1996;377(4):239–250. doi: 10.1515/bchm3.1996.377.4.239. [DOI] [PubMed] [Google Scholar]
- 58.Her J, Bunting SF. How cells ensure correct repair of DNA double-strand breaks. J Biol Chem. 2018;293(27):10502–10511. doi: 10.1074/jbc.TM118.000371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annual review of biochemistry. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martin JH, Bromfield EG, Aitken RJ, Lord T, Nixon B. Double Strand Break DNA Repair occurs via Non-Homologous End-Joining in Mouse MII Oocytes. Sci Rep. 2018;8(1):9685. doi: 10.1038/s41598-018-27892-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 128 kb)
(DOCX 4033 kb)
(PDF 1065 kb)
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.