

Abstract
Alzheimer’s disease (AD) is a chronic disabling disease that affects the central nervous system. The main consequences of AD include the decline of cognitive functions and language disorders. One of the causes leading to AD is the decrease of neurotransmitter acetylcholine (ACh) levels in the brain, in part due to a higher activity of acetylcholinesterase (AChE), the enzyme responsible for its degradation. Many acetylcholinesterase inhibitors (AChEIs), both natural and synthetic, have been developed and used through the years to counteract the progression of the disease. The first of such drugs approved for a therapeutic use was tacrine, that binds through a reversible bond to the enzyme. However, tacrine has since been withdrawn because of its adverse effects. Currently, donepezil and galantamine are very promising AChEIs with clinical benefits. Moreover, rivastigmine is considered a pseudo-irreversible compound with anti-AChE action, providing similar effects at the clinical level. The purpose of this review is to provide an overview of what has been published over the last decade on the effectiveness of AChEIs in AD, analysing the most relevant issues under the clinical and methodological profiles and the consequent possible welfare effects for the whole world. Furthermore, novel drugs and possible therapeutic approaches are also discussed.
Keywords: Alzheimer’s disease, behavioral/cognitive syndromes, acetylcholinesterase’s inhibitors, Alzheimer’s treatment, disease modifying therapeutics, phase 3 Alzheimer’s clinical trials
Introduction
Alzheimer’s disease (AD) was first discovered in 1901 by Dr. Alois Alzheimer, a German psychiatrist who described its typical symptoms in one of his patients. Nowadays, AD is one of the most studied neurodegenerative disorders, due to its commonness worldwide. Indeed, about 30 million people are currently affected by the disease, 1 and the number is expected to double in the coming decades. 2 AD is one of the most common neurodegenerative diseases in the older population, with a typical onset after age 65, and it is characterized by a slow, gradual and irreversible deterioration of cognitive and mental functions. In addition, the disease entails considerable memory loss and inability to form new memories, ultimately leading to behavioural disorders. Disease progression is usually different for each patient, also due to pre-existing or concomitant illnesses that may be responsible for the severity of observed symptoms. However, AD presents a number of common clinical manifestations and, as a rule, is often anticipated, even decades before its actual onset, by the so-called mild cognitive impairment (MCI), a very slow decline of many and varied cognitive functions related to memory, orientation and verbal skills.
The exact causes of AD occurrence and progression are not fully understood yet. From the knowledge we possess so far, AD can be considered as a multifactorial pathology, that depends on a combination of both genetic and environmental factors. 1 Currently, there are 7 competing hypotheses associated with AD onset:
Deposition of beta-amyloid aggregates, by the improper cleavage of the precursor protein of amyloid (APP)4,5;
Precipitation of intracellular neurofibrillary tangles, due to hyperphosphorylation of tau proteins6,7;
Chronic peripheral8,10 and neuro-inflammation 11 by microglial activation;
Metabolic disorders such as those provoked by dysregulation of cholesterol homeostasis, 14 type 2 diabetes and obesity.15,16
For the sake of clarity, only the implications of cholinergic system alteration will be discussed in detail in the present review. According to such hypothesis, the typical slowing down of learning and memory processes found in AD is mostly caused by a decrease of acetylcholine (ACh) neuronal levels, leading to a loss of cholinergic transmission at pre-synaptic level. Therefore, many pharmacological strategies have been designed with the aim to slow down AD symptoms and restore ACh levels in the synaptic cleft, for example, the use of ACh precursors or cholinergic agonists to ameliorate ACh synthesis and effects, respectively. Unfortunately, such strategies were mostly ineffective in AD treatment, due to severe side-effects.
The decrease in ACh levels can be due, on one hand, to a limited activity of the enzyme choline O-acetyltransferase (ChAt), responsible for its synthesis, and, on the other, to an increased catalytic functioning of acetylcholinesterase (AChE), the enzyme responsible for its degradation. 3 The latter enzyme was often chosen as a target of therapeutic approaches aiming at its inhibition. 17 The mechanism of action of AChE inhibitors (AChEIs) is particularly effective since it leads to an increase in ACh concentrations at synaptic level, with the consequent improvement of cholinergic neurotransmission and recovery of cognitive functions in AD patients. However, AChEIs do not lack adverse reactions or toxicity; in some cases, they also presented a short half-life and non-selectivity in enzymatic inhibition. Indeed, AChE is not the only cholinesterase (ChE) present in the human body. In fact, while AChE can be found in blood and neuronal synapses, butyrylcholinesterase (BuChE) is the main ChE in liver, glia, neurons and in tangles and neuritic plaques. 18 The principal difference between the two enzymes is represented by their physiological substrates: ACh and butyrylcholine (BuCh), respectively.19,20 Many ChE inhibitors (ChEIs) are unable to distinguish between AChE and BuChE, thus lacking proper selectivity. In such context, much interest has been focused through the years on the design and development of novel specific AChE inhibitors.
Acetylcholinesterase
AChE (EC 3.1.1.7) 21 is a pivotal enzyme involved in the cholinergic nervous system, that includes both the peripheral and central nervous systems. Its main activity is the catalysis of ACh hydrolysis, thus yielding choline and acetate ions.
AChE can exist in two different molecular forms: simple homomeric oligomers of catalytic subunits (monomers, dimers and tetramers) and heteromeric associations of catalytic and structural subunits. 22 Homomeric oligomers are usually found in soluble form inside cells, presumably intended to be secreted or associated with the external membrane through attachment to a glycol-phospholipid. Heteromeric AChEs are instead frequently found in neuronal synapses, usually consisting of a tetramer of catalytic subunits bound through disulphide bridges to a 20 kDa structural subunit, associated with a lipid and localized on the outer surface of the cell membrane.
Molecular cloning has shown that one single gene encodes for all AChE forms found in vertebrates. In fact, alternative splicing results in distinct gene products that differ in their C-terminal sequence, while presenting the same conserved catalytic core. Thus, AChEs of different species usually share substrate selectivity and specificity of inhibition.
From a kinetic point of view, AChE is one of the most efficient enzymes ever studied: indeed, a single molecule of AChE is able to hydrolyse 6 × 105 molecules of ACh per minute, resulting in a turnover time of 100 µsec. AChEs located in the post-synaptic membrane of the cholinergic junctions at the nerve level are responsible for most of ACh degradation. Additional hydrolysis by BuChE also occurs, however to a much lesser extent and with a significantly lower affinity for ACh.
The 3-dimensional structure of the dimeric form of AChE from Torpedo californica (TcAChE, Figure 1) was solved by X-ray crystallography and deposited in the Protein Data Bank23,24 with entry code 2ACE. Such structure shows the active site of the enzyme in a symmetrical central position in each subunit and at the base of a narrow 20 Å deep gorge. 22 The active site consists of two subsites, an anionic and an esterase ones. In the latter, three amino acids in particular (Ser200, Glu327 and His440) are essential for catalytic activity. From a functional point of view, substrate binding and recognition occurs in the anionic pocket, where the carbonyl oxygen of the acetyl group in ACh forms a hydrogen bond with the hydroxyl group of Ser200. A tetrahedral intermediate is thus formed and, within few microseconds, a water molecule breaks the new bond releasing choline and acetic acid. In such way, Ser200 can revert to its original free -OH form (Figure 2).
Figure 1.
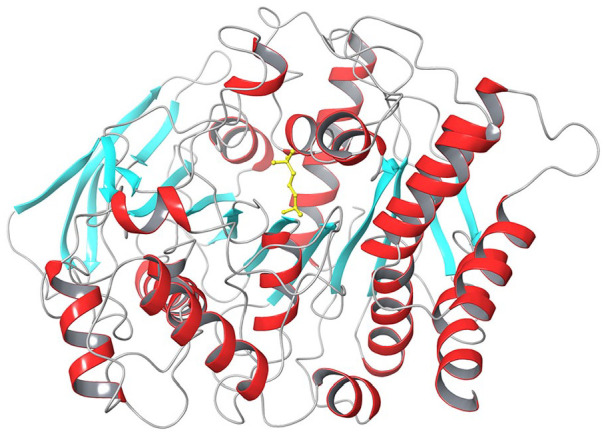
Crystal structure of TcAChE (PDB: 2ACE), shown in cartoons coloured by secondary structure elements (α-helices in red, β-sheets in light blue and loops in grey). ACh bound in the active site is shown as yellow sticks.
Figure 2.

Mechanism of ACh hydrolysis catalysed by AChE.
Acetylcholinesterase Inhibitors
Inhibitors (either drugs or toxins) that show an effect on ChEs’ functioning can be divided into two classes according to their structure and mechanism of action: (i) molecules that interact with the enzyme by covalent bonds, such as organophosphates and carbamates; (ii) molecules that are able to establish reversible bonds with the enzyme and usually contain aminic groups. The resolution of the crystallographic structure of AChE in complex with different inhibitors was particularly useful for the elucidation of inhibition mechanisms. 22
The cholinergic deficit in AD has led to the therapeutic use of reversible AChE inhibitors, drugs with indirect parasympathetic mimic action. Such inhibitors, by blocking the activity of the enzyme, maintain the cerebral availability of ACh and can thus compensate, but not stop, cells death caused by the disease. They can also lead to improvement of some cognitive (memory, attention) and behavioural (apathy, agitation, hallucinations) symptoms during the mild phase of the disease. 25 Unfortunately, such capacity decreases with the progression of the neurodegenerative disorder. AChE inhibitors are not free from side effects, with the most common being nausea and, in some cases, cardiac arrhythmia.26,27 Nonetheless, they are still the first choice in AD treatment, even though only few patients respond to therapy. This is probably due to vascular system health in AD patients, that could lead to blood-brain barrier (BBB) dysfunctions28,29 and to lower permeability for anticholinesterase drugs.30,31 It is thus necessary to adapt the dosage to the patient’s individual characteristics during treatment. Classic AChEIs still in use are donepezil, galantamine and rivastigmine 32 all approved by both Food and Drug Administration and European Medicine Agency. Tacrine (1,2,3,4-tetrahydroacridin-9-amine; Figure 4), the first commercialised AChE/BuChE inhibitor (1993) 33 with a non-competitive and rapidly reversible inhibition mechanism, was instead withdrawn in 2013 because of its severe hepatotoxicity 34 and is used nowadays only as a reference due to its impressive IC50 of 77 nM.
Figure 4.

Structure of natural AChE inhibitors.
Physostigmine, tacrine and cardanol are mostly used as chemical scaffolds for the design of derivatives.
The State of the Art
Most clinical studies were designed to test the possible effects of new drugs with respect to placebo-treated controls. Unfortunately, only few studies entailed direct comparison of different inhibitors. This disparity can be mainly attributed to pharmaceutical governing laws, that consider placebo-controlled studies sufficient for the assessment of the effectiveness and safety of new drugs. However, such features would be determined more reliably with comparison of head-to-head inhibitors, also belonging to different chemical classes.
In addition, some of the known ChEs inhibitors have additional effects on other biochemical processes involved in the complex etiopathogenetic mechanisms of AD. 35 For this reason, cholinesterase inhibitors can be divided into two general classes: (i) ‘single target’ ligands that specifically inhibit AChE/BuChE; (ii) ‘multi-target’ cholinesterase inhibitors that exert effects on other enzymatic routes (generally mediated by monoamine oxidases) and modulate other biological pathways (eg, inflammation, free radical production) responsible for the pathogenesis of AD (Table 1).
Table 1.
Main features of cholinesterase inhibitors.
| Classic/derivatives/analogues/hybrid natural and synthetic | Enzyme specificity (AChE/BuChE) | Additional targets | Type of experimentation | Pre-clinical/clinical studies/in use |
|---|---|---|---|---|
| Single target | ||||
| Tacrine | AChE/BuChE | In use 1993-2013 | ||
| Donepezil | AChE | In use since 1996 | ||
| Rivastigmine | AChE (G1 isoform) and BuChE | In use since 2000 | ||
| Galantamine | AChE | In use since 2001 | ||
| Huperzine-A | AChE | ‘In vitro’ and ‘In vivo’ studies | Phase IV in China | |
| Tolserine (Physostigmine derivate) | AChE, especially human AChE | Theoretical chemistry studies | ||
| Eseroline (Physostigmine derivate) | AChE | Opioids agonist | Theoretical chemistry studies | |
| Tacrine analogues (N-alkyl-7-methoxytacrine hydrochloride) | AChE better than tacrine | Theoretical chemistry studies | ||
| Cardanol (Anacardium occidentale nutshell) derivatives [N, N-dimethycarbamoyl, N, N-dimethylamine, and pyrrolidine substitutions)] | AChE | Theoretical chemistry studies | ||
| Multi-target | ||||
| Phenserine (physostigmine derivate) | AChE | APP cleavage | Phase II | |
| Ladostigil N-propargyl-(3R) aminoindan-5yl)-ethyl methyl carbamate) | AChE | APP cleavage; MAO | Phase II | |
| Galangin (flavonoid extracted from Alpiniae officinarum sp.) | AChE | Free radical scavenger | ‘In vitro’ studies | |
| Liquiritigenin (chalcone isoliquiritigenin derivate) | AChE | MAO; BDNF; ERK; CREB and free radicals’ production | ‘In vivo’ studies | |
| Hybrid compounds (tacrine/donepezil; tacrine/ferulic acid; tacrine-8-hydroxyquinoline; donepezil-AP2238) | AChE; BuChE | Aβ mediated toxicity; MAO; antioxidant properties | ‘In vitro’ studies | |
| Fucoxanthin (Brown seaweed) | AChE | Oxidative stress; inflammation via MAPK pathway | Enzymatic assay; ‘in vitro’ and ‘in vivo’ studies | |
| Phloroglucinol (Ecklonia maxima) | AChE | Aβ 1-42-induced neuritic loss | Enzymatic assay; ‘in vitro’ and ‘in vivo’ studies | |
| Dibenzodioxine-2,4,7,9-tetraol (Ecklonia maxima) | AChE | Aβ mediated toxicity | Enzymatic assay; ‘in vitro’ and ‘in vivo’ studies | |
| Eckol (Ecklonia maxima) | AChE | Aβ 25-35-induced toxicity | Enzymatic assay; ‘in vitro’ and ‘in vivo’ studies | |
| Fucosterol (Ecklonia stolonifera) | BuChE | Aβ 1-42-induced memory impairment; inflammation via NFkB pathway | Enzymatic assay; ‘in vitro’ and ‘in vivo’ studies | |
| Fucosterol (Panida australis) | AChE/BuChE | Enzymatic assay; ‘in vitro’ and ‘in vivo’ studies | ||
| Fucosterol (Sargassum horridum) | AChE | Enzymatic assay; ‘in vitro’ and ‘in vivo’ studies | ||
Single-Target ChEs Inhibitors
Inhibitors currently in use
Donepezil
Donepezil [2-(1-Benzylpiperidin-4-yl)methyl-5,6-dimethoxy-2,3-dihydro-1H-inden-1-one] (Figure 3) was approved in 1996 and is generally administered during the first month of treatment at a daily dose of 5 mg, that can later be increased up to 10 mg for mild to moderate AD treatment. In 2010, it was also approved for moderate to severe AD with a 23 mg/day posology. 36 The most frequently observed adverse reactions comprise diarrhoea, muscle cramps and fatigue. The effects of donepezil in patients with mild or moderate AD were well described by double-blind clinical trials lasting from 12 to 16 weeks. 37 In such context, the range of doses used went from 1 to 10 mg per day with the highest doses providing the greatest benefits. Behavioural observations and analyses were conducted following the official neurological scale ADAS (Alzheimer’s Disease Assessment Scale) and the MMSE (Mini-Mental State Examination), the fastest and most commonly used test both in clinic and research. Moreover, in 2003, a meta-analysis pointed out that improvements in MMSE score were maintained up to 52 weeks with 10 mg daily doses of donepezil with respect to placebo controls. 38 AD2000,39,40 a randomised clinical study on 565 community residents, also showed that MMSE scores were maintained also more than 52 weeks with 5 or 10 mg daily doses of donepezil. Thirty studies, involving 8257 participants with mild, moderate or severe AD, have recently demonstrated that prolonged treatments with donepezil (12-24 weeks) provide some benefits in cognitive functions and in the recovery of daily activities. 38
Figure 3.
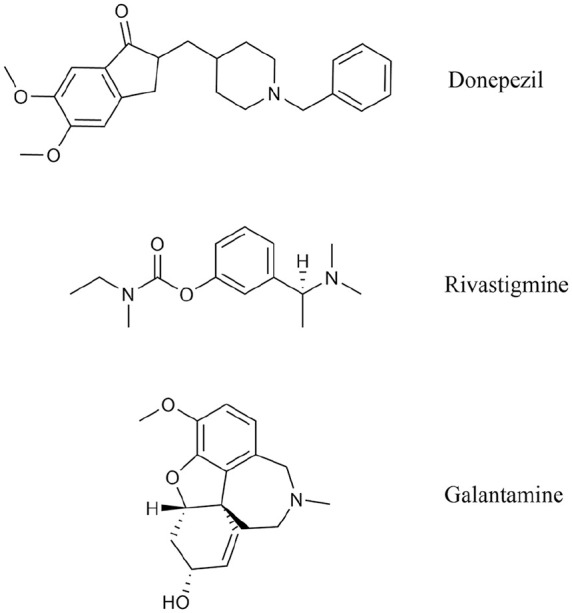
Structure of AChE inhibitors currently in use.
Rivastigmine
Approved for AD treatment in 2000, rivastigmine [3-[1-(Dimethylamino)ethyl]phenyl ethyl(methyl)carbamate] (Figure 3) is a non-competitive, pseudo-irreversible inhibitor of both AChE and BuChE. It is used in the treatment of mild to moderate AD at a starting daily dose of 3 mg that can be increased up to 4-fold. Treatment with rivastigmine usually leads to a functional and global rehabilitation of the cognitive, behavioural skills to a greater extent with respect to donepezil.41,42 Good tolerability was observed in clinical trials 43 and the most frequent adverse reactions were generally found at the gastrointestinal level, for example, nausea, vomiting and, less frequently, diarrhoea. For such reasons, the route of administration is often transdermal 44 by patches that slowly release a progressive amount of the drug and can be replaced every 24 hours. Usually, the treatment begins with a dose of 4.6 mg per day and can be increased until 9.5 mg per day after the first 4 weeks of treatment. Such rivastigmine doses proved to improve cognitive (eg, short- and long-term memory, verbal expression, language comprehension, orientation skills) and non-cognitive (eg, agitation, aggressiveness, hallucinations and delusion) symptoms in several clinical studies on patients with mild AD. 45 Rivastigmine, in contrast to donepezil and galantamine, has a specific effect on the G1 isoform of AChE localised in cortex, hippocampus and neuritic plaques, the areas mostly affected by ACh deficit in AD.
Galantamine
Approved for AD treatment in 2001, galantamine [(4aS,6R,8aS)-5,6,9,10,11,12-Hexahydro-3-methoxy-11-methyl-4aH-[1]benzofuro [3a,3,2-ef][2]benzazepin-6-ol] (Figure 3) is a competitive, rapidly reversible, potent AChE inhibitor. It is generally used at a dose between 16 and 24 mg per day. Several randomised placebo-controlled clinical studies showed that the therapeutic effects of galantamine were reached with doses from 16 up to 32 mg per day, administered for 21-26 weeks. Moreover, galantamine benefits on sleep quality of both patients with mild to moderate AD and their caregivers were pointed out in a double-blind, head-to-head, randomised pilot study. 46 The most frequently observed adverse reactions comprise nausea, vomiting, diarrhoea, gastric pain, asthenia and insomnia.
Natural inhibitors
Huperzine A
Huperzine A (HupA; Figure 4) is a natural alkaloid extracted from Huperzia serrata, an herb used in traditional Chinese medicine. With respect to classic inhibitors, HupA presents a better permeability through the blood-brain barrier. It is a potent, highly specific and reversible AChE inhibitor and its low toxicity was assessed in several studies.47,48 In China, HupA reached phase IV clinical trials with appreciable improvements in cognitive deficit. 47
Physostigmine derivatives
Classic ChE inhibitors were used as a model for the design of new possible ligands, 20 for example, physostigmine derivatives. 49 Physostigmine (Figure 4) is an alkaloid, extracted from Calabar bean, that proved to modulate ChE functioning. However, it was never used in therapy because of its short half-life and numerous side effects. 50 Physostigmine derivatives include tolserine, eseroline and phenserine (see section 7.3.2). Of these, only the latter was tested in clinical studies, 49 mainly thanks to its ability to counteract APP production.
Tolserine
Tolserine (Figure 4) was experimentally characterized as an efficient inhibitor of human erythrocytic AChE, presenting a higher potency than its structural analogues physostigmine and phenserine. 51 In fact, its IC50 value was determined as 8.13 nM with a partial non-competitive inhibition mechanism. However, to date, no data are available regarding preclinical studies on this compound.
Eseroline
Unlike physostigmine, eseroline (Figure 4) is a competitive and reversible AChE inhibitor. Moreover, its cyclic alkyl carbamate derivative was also characterized as a highly selective and very promising inhibitor of AChE. 49
Cardanol derivatives
Cardanol (Figure 4) is a non-isoprenoid phenolic lipid obtained from Anacardium occidentale nutshell. Fifteen cardanol derivatives were produced by functionalization with either of the following chemical groups: methyl; acetyl; N, N-dimethylcarbamoyl; N, N-dimethylamine; N, N-diethylamine; piperidine; pyrrolidine; N, N-methylbenzylamine. Theoretical studies showed that the N, N-dimethycarbamoyl, N, N-dimethylamine and pyrrolidine substitutions presented similarities with rivastigmine and could thus represent future AChEIs to test in AD treatment. 52
Tacrine analogues
Several synthetic analogues incorporating the main functional moieties derived from diverse chemotypes (eg, from acridine, quinoline, carbamates and other heterocyclic analogues) showed the desired pharmacological effects. Among them, many tacrine analogues proved to possess marked inhibitory activity, like for example N-alkyl-7-methoxytacrine hydrochloride. 53
Multi-Target Cholinesterase Inhibitors
Ladostigil
Recent advances in organic chemistry and pharmacology allowed the synthesis of multifunctional compounds which, acting at different levels, may allow better control over AD progression.
Ladostigil [(N-propargyl-(3R) aminoindan-5yl)-ethyl methyl carbamate] (Figure 5) is both a MAO-A/-B and ChE inhibitor. Such dual action made this compound particularly promising for clinical trials. 54
Figure 5.

Structure of ladostigil and of hybrid multi-target AChE inhibitors.
Hybrid molecular structures
In the search for effective AChE inhibitors, researchers decided to investigate the possibility of synthetizing hybrid molecules. These ligands were developed with the aim to either bind to both catalytic and peripheral sites in AChE or have an additional action on β-amyloid (Aβ) aggregation.
The first of such compounds, donepezil-AP2238 (Figure 5), besides being able to bind to both anionic sites in AChE, proved to inhibit Aβ-mediated toxicity to a greater extent with respect to donepezil.55,56
Other two hybrid inhibitors, tacrine-ferulic acid 57 (T6FA, Figure 5) and tacrine-8-hydroxyquinoline 58 (Figure 5), were more effective than their ancestor in inhibiting AChE in vitro while possessing weak toxicity. Moreover, T6FA showed potent activity in inhibiting Aβ aggregation both in vitro and in vivo. 57
Donepezil-tacrine (Figure 5) hybrids were also developed and were found to inhibit AChE, BChE and Aβ-aggregation. 59
Particularly interesting data were obtained on a donepezil-chromone-melatonin hybrid, that showed good inhibition on ChEs and monoaminoxidases (both MAO-A and MAO-B) and also antioxidant properties. 60
Furthermore, tacrine-acridine hybrids are currently under study as multi-target drugs in AD treatment. 61
Natural organic compounds
Flavonoids
Flavonoids are a group of natural compounds with a well-known free-radical-scavenging capacity. They can be extracted from plants and were extensively used in traditional Chinese medicine. Among them, galangin, a compound isolated from Alpiniae officinarum rhizomes, showed an AChE inhibitory activity by over 55% with an IC50 of 120 µM in vitro. 62 However, the safety of such compounds has yet to be tested in preclinical and clinical studies.
Phenserine
Phenserine, already mentioned as a physostigmine derivative, is a non-competitive, selective AChE inhibitor with reduced adverse effects if compared to traditional anti-ChEs. It can be considered as a multi-target drug thanks to its ability to inhibit Aβ-aggregation and was used for the treatment of cognitive impairments induced by traumatic brain injury in mice.63,64 Phenserine was also tested in Phase II studies with moderate success. 65 Indeed, patients treated with phenserine (10 and 15 mg) versus placebo for 12 weeks showed a significative improvement of cognitive functions. 66 These results were in agreement with phenserine potential effectiveness in AD symptomatic therapy. Further investigations are still in progress to understand its mechanism of action and potential therapeutic use. 67
Chalcone derivatives
Chalcone is an aromatic ketone that forms the central core for a variety of important biological compounds, collectively known as chalcones or chalconoids. Such compounds are selective AChE inhibitors and also possess anti-Aβ aggregation properties, 68 thus they were selected as promising scaffolds for the development of new drugs for AD treatment.
Among them, the flavanone liquiritigenin, isolated from Glycyrrhizae uralensis, was already well known in traditional Chinese medicine as a life enhancer, used for the treatment of cough and influenza and for detoxification. 69 In vivo studies demonstrated that liquiritigenin might have potential learning and memory enhancement effects in mice.70,71 Its mechanism of action has also been elucidated and involves the inhibition of several enzymatic pathways, including those mediated by AChE and MAOs. 72 Moreover, liquiritigenin proved to have a regulatory effect on free radicals production and on the expression of factors involved in the regulation of cognitive functions (eg, BDNF, ERK, CREB). 70
Metabolites from marine algae
During the past decade, many researchers worldwide focused their attention on various algal metabolites, 73 including phenolic compounds, alkaloids, terpenes, phytosterols, polysaccharides, tannins and carotenoids. Several preclinical studies have confirmed the neuroprotective activity of such compounds in a range of neurodegenerative and traumatic events (eg, stroke) as well as in metabolic disorders (diabetes, obesity). Moreover, algal metabolites possess promising antioxidant and anti-inflammatory properties and can participate in defence mechanisms.74,75
Several algal metabolites, such as fucoxanthin, phloroglucinol, dibenzodioxine-2,4,7,9-tetraol, eckol and fucosterol (all reported in Figure 6) showed encouraging inhibitory effects on ChEs with considerable IC50 values.
Figure 6.
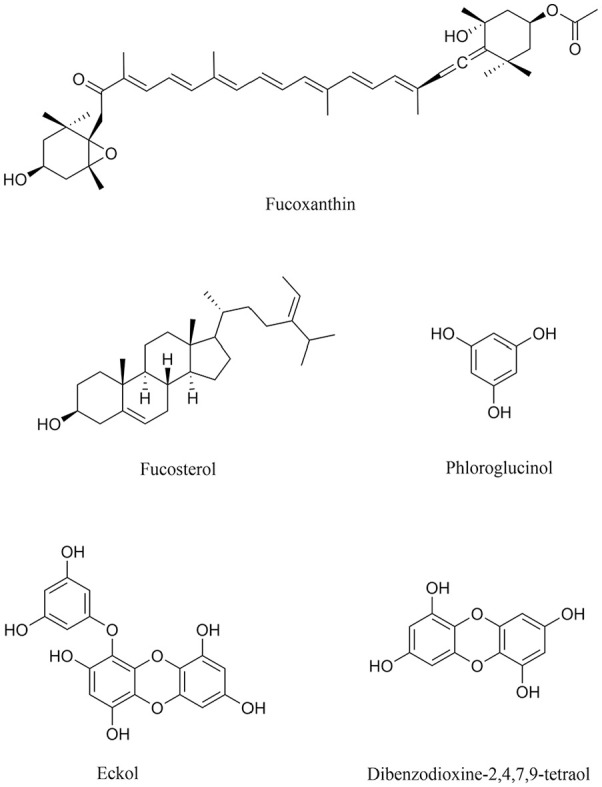
Structure of algal metabolites as promising novel drugs to treat AD.
In particular, fucoxanthin, a carotenoid extracted from Brown seaweed binds in a non-competitive fashion to the AChE anionic site with an IC50 of 81.2 µM. 76 Fucosterols extracted from distinct algal species inhibit different forms of ChEs, with the one from Ecklonia stolonifera 78 being the most effective, with an IC50 around 422 µM for BuChE. When tested in in vitro and in vivo experimental models, fucoxanthin and fucosterol led to an improvement of oxidative stress and inflammation via MAPK- and NF-kB-mediated pathways.79,80 Fucosterol also led to a decreased Aβ-induced neurotoxicity in hippocampal neurons 81 and SH-SY5Y neuroblastoma cells. 82
Phloroglucinol, dibenzodioxine-2,4,7,9-tetraol and eckol from Ecklonia maxima 77 are potent and specific AChE ligands with IC50 values between 76.7 and 579.3 µM. Biological assays also showed that phloroglucinol is able to revert the Aβ 1-42 slowing down of dendritic spine density and of synaptic protein (synaptophysin) in primary cultures of rat hippocampal cells. 84 The phlorotannin eckol instead protects PC12 cells against the toxicity induced by Aβ25-35 fragment. 83
The chemical features and effects of algal metabolites suggest a possible use in drug design studies and make them, once safety has been verified, also evaluable in clinical trials.
Novel Therapeutics by Marine Algae
The continuing failure of therapies aimed at reducing β-amyloid deposits and/or tau protein aggregates is guiding research efforts towards the discovery of new and more effective therapeutic strategies, that take into account the involvement of alternative pathways in the pathogenesis and progression of AD. In such context, numerous studies, both on animal models and humans, have shown a correlation between gut microbiota dysbiosis and neuroinflammation in AD. 85 In particular, gut microbiota alterations in AD mouse models led to the accumulation of phenylalanine and isoleucine in peripheral blood, prompting the activation, proliferation and differentiation of type 1 (Th1) T helper cells. Once in the brain, such cells stimulate and support the neuroinflammatory process through the activation of microglia cells (M1). 86 The same study reported that the administration of the algal oligosaccharide sodium oligomannate (GV-971) led to a restored homeostasis of the gut environment, thus inhibiting the onset of the neuroinflammatory process. In fact, the amount of compound reaching the brain through the blood-brain barrier (BBB) inhibited the formation of novel amyloid fibrils and led to the degradation of pre-formed ones into non-toxic monomers, while simultaneously restoring gut microbiota and reducing immune response. 86 Taken together, such effects led to a general improvement of cognitive processes.
Sodium oligomannate also showed neuroprotective effects against Aβ toxicity in neuroblastoma cells 87 and was able to revert memory disorders in the 5XFAD transgenic animal model. 86 GV-971 was developed by Shanghai Green Valley Pharmaceuticals and approved for first clinical use in patients with mild to moderate AD by National Medical Products Administration (NMPA; Chinese equivalent of the FDA) in November 2019. 88 The 9-month trial showed a clear recovery of cognitive functions in participants compared to baseline (ADAS scale) but no disease-specific biomarker was monitored in the trial. However, thanks to such studies, neuroinflammation triggered by gut dysbiosis is now recognised as a target for the development of future therapies. 89
Currently, sodium oligomannate has completed the first phase 3 clinical trial 90 (recommended dosage: 450 mg twice a day) and represents the first new drug approved for therapeutic use since 2003.91,92
New Approaches in Clinical Trials
As already mentioned, neurodegenerative diseases have a multifactorial aetiology and involve different physiological processes and pathogenetic mechanisms that are usually difficult to characterise in detail. Over the past 5 years, such multifactorial features led to the design of novel therapeutics.
In fact, unlike cholinesterase inhibitors that have specific targets and mechanisms of action, many compounds currently under preclinical and/or clinical study have multiple biological targets and can be considered as disease-modifying therapeutics (DMTs), able to act on AD onset and progression.92,93 According to the federal government’s database, that collects most data from clinical trials around the world, DMTs represent the 59% of compounds currently in phase 3 trials, with most of them including amyloids as a target. 93 The remaining 41% comprises symptomatic drugs, that lead to the improvement of cognitive and memory functions as well as neuropsychiatric symptoms in advanced stages of AD without acting on the actual biological causes of the disease. However, some compounds, for example, sodium oligomannate, may also act both as symptomatic drugs and DMTs.
From a methodological point of view, symptomatic therapy is cost-effective and easier to study, with no need for a large number of samples and usually completed in a relatively short time (3-6 months). On the other hand, the development of DMTs is way more expensive and time-consuming (12-24 months), also due to the absence of precise pathways and mechanisms of action.
However, symptomatic treatments and DMTs are both important for successful therapies. In fact, many clinical trials also entail the combination of both approaches, 91 leading to slowdown of disease progression, reduced local toxicity and increased neuroprotection.94,95
AD phase 3 trials
Despite DMTs being the compounds of choice in clinical trials, the number of anti-amyloid agents studied in advanced AD declined in 2019 and 2020. 96 Such compounds are mostly evaluated in stages of the disease preceding the clinical manifestations of dementia, taking as reference the Research Framework 97 to define the AD stages and the FDA guidance98,99 for the design and development of cognitive scales appropriated for early AD. According to Research Framework, AD stages preceding dementia are the preclinical (β-amyloid and tau biomarkers are present without cognitive impairment), and the prodromal (presence of brain changes and not disabling mild cognitive impairment MCI). 97 DMTs are particularly effective in preclinical AD trials, leading to a slowdown of disease progression towards MCI and dementia.100-102
Currently, there are 29 agents under study in 36 phase 3 AD clinical trials. Of such drugs, 17 are DMTs (5 biological agents, 12 small molecules) and 12 are symptomatic (4 cognitive enhancers, 8 targeting behavioural symptoms). Recognised targets include amyloid (n = 6 agents, including monoclonal, vaccine and anti-aggregation antibodies), synaptic plasticity/neuroprotection (n = 4), inflammation/infection/immunity (n = 3), metabolism/bionergetics (n = 2), tau and vascularisation (n = 1). Participants in the 36 trials are in different stages of AD: cognitively normal (4 trials), prodromal/MCI or prodromal/mild (11 trials), cognitively normal and MCI/mild (1 trial), mild/moderate (11 trials) and mild/severe (9 trials). 103
Many DMTs (including Aβ-targeting agents and β-secretase 1 inhibitors) failed in phase 3 clinical trials due to enrolment of patients with symptomatic AD, toxicity or lack of significant effects. 104 Aducanumab, a monoclonal antibody tested in two phase 3 clinical trials in 2019, had been declared ineffective by preliminary statistical results. The antibody, developed by the companies Biogen and Eisai, then showed good effects in better designed studies (ENGAGE and EMERGE), 105 showing reduction in beta-amyloid plaques and stabilisation of cognitive decline in early-stage patients. These data led the two pharmaceutical companies to submit the antibody for approval by FDA. 106
Symptomatic drugs also had their share of failure, also due to the inability to confirm with PET the presence of amyloid plaques in the brains of selected subjects. Fortunately, research on biomarkers has recently improved with the development of ultra-sensitive methods capable of measuring analytes present even at low concentrations in the blood, reflecting pathological changes at a central level. 91
Considerations on preclinical AD phase 3 trials
To date, no drug was approved for preclinical AD but 7 phase 3 trials are currently in progress. 107 Of these, 3 involve compounds that act on amyloid deposits (reducing or preventing their formation) and 4 involve neuroprotective drugs (Table 2). 107 Scale–cognitive subscale (ADAS-cog), clinical dementia rating (CDR) score, and Mini-Mental Status Examination (MMSE) assess both cognitive and functional outcomes 98 in trials concerning early-stage AD with absent or limited cognitive deficits. 108 Participants in such trials, all with normal scored in cognitive tests but at high risk of developing AD, were selected on the basis of the following criteria: (i) family history of dementia; (ii) positivity for the apolipoprotein E4 gene; (iii) presence of β-amyloid in the brain, as detected by PET. Patients with pacemaker or artificial heart valves were not included, to avoid interferences during diagnostic tests and cerebrospinal fluid sampling. Patients with a history of cancer, mental illnesses, brain traumas, drugs or alcohol abuse were also excluded from the trials.
Table 2.
Preclinical AD phase 3 trials.
| Drugs | Type and mechanism of action | Age (y) and estimated number of enrolled | Assessment test a | Trial length |
|---|---|---|---|---|
| Solanezumab | DMT Monoclonal antibody directed at b-amyloid monomers | 65-85 (n = 1 150) | Cognition, function and biomarker evaluations | Start February 2014/estimated end July 2022 |
| Gantenerumab + Solanezumab | DMT Monoclonal antibody directed at plaques and b-amyloid oligomers | 18-80 (n = 490) | Cognition, function and biomarker evaluations | Start December 2012/estimated end March 2021 |
| CAD106 | Amyloid vaccine | 60-75 (n = 481) | Cognition, function and biomarker evaluations | Start November 2015/estimated end March 2025 |
| Icosapent ethyl | Purified form of omega-3 fatty acid EPA with neuroprotection action | 50-75 (n = 150) | Assessment test + regional cerebral blood flow measurement | Start June 2017/estimated end November 2021 |
| Losartan + Amlodipine + Atorvastatin | Angiotensin II receptor blocker + Calcium channel blocker + Cholesterol agent, each of them with neuroprotection action by cardiovascular system improving | 60-85 (n = 640) | Neurocognitive skills assessment | Start September 2016/estimated end March 2022 |
Food and Drug Administration. 99
If such trials succeed, many patients will benefit of the slowdown of disease progression and dementia onset but this would represent a huge challenge for global health in terms of economic commitment, especially due to the management of a large number of patients. The continuation and further development of new experimental approaches will also require a greater effort from sponsors, manufacturers and governments to carefully assess the cost/benefit ratios before new therapies are marketed.107,109
Discussion
AD is one of the best studied neurodegenerative disorders and its progression to dementia was found in more than 60% patients. Recent studies provided the elucidation of the molecular mechanisms underlying the pathology, of reliable methods of diagnosis and of effective therapies. Much effort was dedicated to the design and development of AChE inhibitors, since alterations in the cholinergic system are among the most accredited causes of AD. Anti-AChE drugs currently in use comprise donepezil, rivastigmine and galantamine. Such compounds showed an interesting ability in reducing AD characteristic clinical symptoms, like cognitive and behavioural disorders, and in improving quality of life. For what regards safety, apart from few side effects, anti-AChE drugs are usually well tolerated by patients even for long periods of treatment. However, therapies with AChE inhibitors proved to be most effective during the early stages of the disease, when symptoms range from mild to moderate. As dementia and more severe symptoms occur, other molecular pathways are involved in AD progression, thus requiring the use of additional drugs. In this regard, new generation ChE inhibitors of natural and/or synthetic origin were designed and studied but, so far, only few of them have reached clinical trials. Many of these inhibitors behave as multi-targeted drugs not only by inhibiting ChE but also by modulating other enzymatic and biosynthetic pathways that are involved in dementia and AD (eg, Aβ aggregation). Multi-targeted ChE inhibitors are currently being studied also as possible disease-modifying drugs, that will alter the course of the disease rather than act on clinical symptoms. 110 The starting point for the development of these drugs is the amyloid cascade hypothesis, which considers Aβ accumulation in senile plaques as one of the main causes of Alzheimer’s dementia. So far, disease-modifying drugs have been designed to act on secretase modulation, immune system, amyloids, metal-chelation, inflammation, oxidative stress and neuroprotection. However, results obtained are pretty controversial and non-conclusive. To date, such drugs seem to be more effective in the preclinical stages of the disease, when Aβ and tau tangles are not compromised, with a normal quality of life for patients. As many phase 3 clinical trials in symptomatic patients have failed, it was considered more cost-effective to use DMTs in the pre-clinical phase. Such lack of effectiveness underlines the difficulty in developing therapeutic agents that are truly capable of counterbalancing the devastating effects of such a complex neurodegenerative disease.
Pending answers from ongoing preclinical trials, new scenarios are being considered involving different pathways and approaches. In fact, AD can no longer be considered a disease triggered by the cascade of events following amyloid deposition. Its development also depends on a disruption of the brain-gut connection, when the latter undergoes dysbiosis and inflammatory processes. 86
Recent data have shown that poly-oligosaccharides are able to restore gut microbiota, for example, sodium oligomannate (GV-971) that was able to improve cognitive ability in patients with mild to moderate AD in phase 3 clinical trials with no safety issues. The implications of these results can be easily transferred from the diagnosis to the therapy of AD. The identification of the specific bacteria associated with the immune response, the amino acids produced, and the type of immune effectors present in the brain may be an additional weapon in early diagnosis and deserves further large-scale evaluation. On the other hand, having identified intestinal dysbiosis as a triggering event will serve to direct therapy towards finding compounds to be used in prevention. The study of new molecules to be used for the treatment of AD is aimed also in this perspective. 111
Conclusions
Further investigations are still needed to understand the relationship at the molecular level among the many factors involved in AD pathogenesis and progression. Early diagnosis seems, so far, the best strategy that could really facilitate AD treatment before clinical signs of the disease become evident. Studies on preclinical AD could also finally provide effective therapies to treat the very early stages of the disease and hamper its progression. However, it is necessary to set up an accurate, rigorous, possibly not expensive screening to be carried out worldwide, with the help of specialists in Alzheimer’s disease and neurologists, in order to evaluate disease parameters and clearly identify patients with preclinical AD. Moreover, since many drugs currently in clinical trials are of the biological kind and need to be administered parenterally, a sufficient number of infusion clinics centres could also be necessary.
Are healthcare facilities and specialists ready to embrace this forthcoming therapeutic revolution? This is the question to which a research carried out by the RAND Corporation, 112 an important and authoritative US think tank, attempted to provide an answer. The research investigated six European countries (France, Germany, Spain, Sweden, Great Britain and Italy), most of which lacked the resources and facilities needed to welcome the arrival of innovative therapies against AD. One of the main issues would be the lack of specialists able to diagnose the disease at a very early stage and therefore correctly recommend preclinical drugs. Furthermore, early diagnosis requires genetic and neuroimaging investigations that are often very expensive and difficult to perform on a large number of potential future AD patients.
On the other hand, it is important to carry out clinical trials that aim at precision in the choice of drug, target, biomarker, participants and disease staging for a successful outcome of the study. 113 The molecular and neurochemical mechanisms underlying the processes of vulnerability and resilience to cognitive, emotional and affective disorders must also be considered since they could represent and issue in finding molecules that selectively interact with one or more individual targets (neurotransmitters, receptors, enzymes) capable of modulating altered cognitive functions.
The challenge is therefore wide open both for researchers, companies and global health organizations in the development of new successful therapies to treat different stages of AD.
Acknowledgments
The authors thanks Francesco Frustaci, Antonio Macrì and Domenico Saturnino for technical support.
Footnotes
Funding: The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: MA coordinated the working team. IV developed and wrote the draft. LS was responsible for editing and figure preparation. AP and RM contributed in the final revision before submission. All the authors read and approved the final manuscript.
ORCID iDs: Immacolata Vecchio  https://orcid.org/0000-0001-7188-9861
https://orcid.org/0000-0001-7188-9861
Annamaria Paoletti
https://orcid.org/0000-0002-0967-6635
References
- 1. Prince M, Guerchet M, Prina M. Policy brief for heads of government: the global impact of dementia 2013–2050. 2013. https://www.alz.co.uk/research/GlobalImpactDementia2013.pdf
- 2. Shah H, Albanese E, Duggan C, et al. Research priorities to reduce the global burden of dementia by 2025. Lancet Neurol. 2016;15:1285-1294. [DOI] [PubMed] [Google Scholar]
- 3. Stanciu GD, Luca A, Rusu RN, et al. Alzheimer’s disease pharmacotherapy in relation to cholinergic system involvement. Biomolecules. 2019;10:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ferreira-Vieira TH, Guimaraes IM, Silva FR, Ribeiro FM. Alzheimer’s disease: targeting the cholinergic system. Curr Neuropharmacol. 2016;14:101-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ferreira S, Raimundo A, Menezes R, Martins I. Islet amyloid polypeptide & amyloid beta peptide roles in Alzheimer’s disease: two triggers, one disease. Neural Regen Res. 2021;16:1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1:a006189-a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chong FP, Ng KY, Koh RY, Chye SM. Tau proteins and tauopathies in Alzheimer’s disease. Cell Mol Neurobiol. 2018;38:965-980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Leblhuber F, Ehrlich D, Steiner K, et al. The immunopathogenesis of Alzheimer’s disease is related to the composition of gut microbiota. Nutrients. 2021;13:361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Devi S, Kumar V, Singh SK, Dubey AK, Kim J-J. Flavonoids: potential candidates for the treatment of neurodegenerative disorders. Biomedicines. 2021;9:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Suβ P, Lana A, Schlachetzki JM. Chronic peripheral inflammation: a possible contributor to neurodegenerative diseases. Neural Regen Res. 2021;16:1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guzman-Martinez L, Maccioni RB, Andrade V, Navarrete LP, Pastor MG, Ramos-Escobar N. Neuroinflammation as a common feature of neurodegenerative disorders. Front Pharmacol. 2019;10:1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huat TJ, Camats-Perna J, Newcombe EA, Valmas N, Kitazawa M, Medeiros R. Metal toxicity links to Alzheimer’s disease and neuroinflammation. J Mol Biol. 2019;431:1843-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang N, Yu X, Xie J, Xu H. New insights into the role of ferritin in iron homeostasis and neurodegenerative diseases. Mol Neurobiol. 2021;58:2812-2823. [DOI] [PubMed] [Google Scholar]
- 14. Mouzat K, Chudinova A, Polge A, et al. Regulation of brain cholesterol: what role do liver X receptors play in neurodegenerative diseases? Int J Mol Sci. 2019;20:3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Frohlich J, Chaldakov GN, Vinciguerra M. Cardio-and neurometabolic adipobiology: consequences and implications for therapy. Int J Mol Sci. 2021;22:4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vinuesa A, Pomilio C, Gregosa A, Bentivegna M. Inflammation and insulin resistance as risk factors and potential therapeutic targets for Alzheimer’s disease. 2021;15:1-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martinez A, Castro A. Novel cholinesterase inhibitors as future effective drugs for the treatment of Alzheimer’s disease. Expert Opin Investig Drugs. 2006;15:1-12. [DOI] [PubMed] [Google Scholar]
- 18. DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener. 2019;14:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mehta M, Adem A, Sabbagh M. New acetylcholinesterase inhibitors for Alzheimer’s disease. Int J Alzheimers Dis. 2012;2012:1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sharma K. Cholinesterase inhibitors as Alzheimer’s therapeutics (Review). Mol Med Rep. 2019;20:1479-1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thapa S, Lv M, Xu H. Acetylcholinesterase: a primary target for drugs and insecticides. Mini Rev Med Chem. 2017;17:1665-1676. [DOI] [PubMed] [Google Scholar]
- 22. Sussman JL, Harel M, Silman I. Three-dimensional structure of acetylcholinesterase and of its complexes with anticholinesterase drugs. Chem Biol Interact. 1993;87:187-197. [DOI] [PubMed] [Google Scholar]
- 23. Axelsen PH, Harel M, Silman I, Sussman JL. Structure and dynamics of the active site gorge of acetylcholinesterase: synergistic use of molecular dynamics simulation and X-ray crystallography. Protein Sci. 1994;3:188-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Silman I, Millard CB, Ordentlich A, et al. A preliminary comparison of structural models for catalytic intermediates of acetylcholinesterase. Chem Biol Interact. 1999;119-120:43-52. [DOI] [PubMed] [Google Scholar]
- 25. Dou K-X, Tan M-S, Tan C-C, et al. Comparative safety and effectiveness of cholinesterase inhibitors and memantine for Alzheimer’s disease: a network meta-analysis of 41 randomized controlled trials. Alzheimers Res Ther. 2018;10:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nordberg A, Svensson A-L. Cholinesterase inhibitors in the treatment of Alzheimer’s disease. Drug Saf. 1998;19:465-480. [DOI] [PubMed] [Google Scholar]
- 27. Kho J, Ioannou A, Mandal AKJ, Missouris CG. Donepezil induces ventricular arrhythmias by delayed repolarisation. Naunyn Schmiedebergs Arch Pharmacol. 2021;394:559-560. [DOI] [PubMed] [Google Scholar]
- 28. Zenaro E, Piacentino G, Constantin G. The blood-brain barrier in Alzheimer’s disease. Neurobiol Dis. 2017;107:41-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cai Z, Qiao P-F, Wan C-Q, Cai M, Zhou N-K, Li Q. Role of blood-brain barrier in Alzheimer’s disease. J Alzheimers Dis. 2018;63:1223-1234. [DOI] [PubMed] [Google Scholar]
- 30. Banks WA. Drug delivery to the brain in Alzheimer’s disease: consideration of the blood–brain barrier. Adv Drug Deliv Rev. 2012;64:629-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chakraborty A, de Wit NM, van der Flier WM, de Vries HE. The blood brain barrier in Alzheimer’s disease. Vascul Pharmacol. 2017;89:12-18. [DOI] [PubMed] [Google Scholar]
- 32. Bond M, Rogers G, Peters J, et al. The effectiveness and cost-effectiveness of donepezil, galantamine, rivastigmine and memantine for the treatment of Alzheimer’s disease (review of Technology Appraisal No. 111): a systematic review and economic model. Health Technol Assess. 2012;16:1-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Crismon ML. Tacrine: first drug approved for Alzheimer’s disease. Ann Pharmacother. 1994;28:744-751. [DOI] [PubMed] [Google Scholar]
- 34. Watkins PB, Zimmerman HJ, Knapp MJ, Gracon SI, Lewis KW. Hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. JAMA. 1994;271:992-998. [PubMed] [Google Scholar]
- 35. Anand P, Singh B. A review on cholinesterase inhibitors for Alzheimer’s disease. Arch Pharm Res. 2013;36:375-399. [DOI] [PubMed] [Google Scholar]
- 36. English C. Donepezil 23 mg: is it more advantageous compared to the original? Ment Health Clin. 2012;1:272-273. [Google Scholar]
- 37. Knowles J. Donepezil in Alzheimer’s disease: an evidence-based review of its impact on clinical and economic outcomes. Core Evid. 2006;1:195-219. [PMC free article] [PubMed] [Google Scholar]
- 38. Birks JS, Harvey RJ. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst Rev. 2018;6:CD001190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schneider LS. AD2000: donepezil in Alzheimer’s disease. Lancet. 2004;363:2100-2101. [DOI] [PubMed] [Google Scholar]
- 40. Holmes C, Burns A, Passmore P, Forsyth D, Wilkinson D. AD2000: design and conclusions. Lancet. 2004;364:1213-1214. [DOI] [PubMed] [Google Scholar]
- 41. Cumbo E, Ligori LD. Differential effects of current specific treatments on behavioral and psychological symptoms in patients with Alzheimer’s disease: a 12-month, randomized, open-label trial. J Alzheimers Dis. 2014;39:477-485. [DOI] [PubMed] [Google Scholar]
- 42. Bullock R. Treatment of behavioural and psychiatric symptoms in dementia: implications of recent safety warnings. Curr Med Res Opin. 2005;21:1-10. [DOI] [PubMed] [Google Scholar]
- 43. Nguyen K, Hoffman H, Chakkamparambil B, Grossberg GT. Evaluation of rivastigmine in Alzheimer’s disease. Neurodegener Dis Manag. 2021;11:35-48. [DOI] [PubMed] [Google Scholar]
- 44. Winblad B, Machado JC. Use of rivastigmine transdermal patch in the treatment of Alzheimer’s disease. Expert Opin Drug Deliv. 2008;5:1377-1386. [DOI] [PubMed] [Google Scholar]
- 45. Birks JS, Grimley Evans J. Rivastigmine for Alzheimer’s disease. Cochrane Database Syst Rev. 2015;4:CD001191. [DOI] [PubMed] [Google Scholar]
- 46. Ancoli-Israel S, Amatniek J, Ascher S, Sadik K, Ramaswamy K. Effects of galantamine versus donepezil on sleep in patients with mild to moderate Alzheimer disease and their caregivers. Alzheimer Dis Assoc Disord. 2005;19:240-245. [DOI] [PubMed] [Google Scholar]
- 47. Wang R, Yan H, Tang X. Progress in studies of huperzine A, a natural cholinesterase inhibitor from Chinese herbal medicine. Acta Pharmacol Sin. 2006;27:1-26. [DOI] [PubMed] [Google Scholar]
- 48. Ha GT, Wong RK, Zhang Y. Huperzine A as potential treatment of Alzheimer’s disease: an assessment on chemistry, pharmacology, and clinical studies. Chem Biodivers. 2011;8:1189-1204. [DOI] [PubMed] [Google Scholar]
- 49. Zhan Z-J, Bian H-L, Wang J-W, Shan W-G. Synthesis of physostigmine analogues and evaluation of their anticholinesterase activities. Bioorg Med Chem Lett. 2010;20:1532-1534. [DOI] [PubMed] [Google Scholar]
- 50. Coelho Filho JMJ, Birks J. Physostigmine for dementia due to Alzheimer’s disease. Cochrane Database Syst Rev. 2001;2:CD001499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kamal MA, Greig NH, Alhomida AS, Al-Jafari AA. Kinetics of human acetylcholinesterase inhibition by the novel experimental Alzheimer therapeutic agent, tolserine. Biochem Pharmacol. 2000;60:561-570. [DOI] [PubMed] [Google Scholar]
- 52. de Paula AAN, Martins JBL, dos Santos ML, et al. New potential AChE inhibitor candidates. Eur J Med Chem. 2009;44:3754-3759. [DOI] [PubMed] [Google Scholar]
- 53. Torre P, Saavedra L, Caballero J, et al. A novel class of selective acetylcholinesterase inhibitors: synthesis and evaluation of (E)-2-(Benzo[d]thiazol-2-yl)-3-heteroarylacrylonitriles. Molecules. 2012;17:12072-12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Weinreb O, Amit T, Bar-Am O, Youdim MBH. A novel anti-Alzheimer’s disease drug, ladostigil neuroprotective, multimodal brain-selective monoamine oxidase and cholinesterase inhibitor. Int Rev Neurobiol. 2011;100:191-215. [DOI] [PubMed] [Google Scholar]
- 55. Piazzi L, Rampa A, Bisi A, et al. 3-(4-{[Benzyl(methyl)amino]methyl}phenyl)-6,7-dimethoxy-2H-2-chromenone (AP2238) inhibits both acetylcholinesterase and acetylcholinesterase-induced β-amyloid aggregation: a dual function lead for Alzheimer’s disease therapy. J Med Chem. 2003;46:2279-2282. [DOI] [PubMed] [Google Scholar]
- 56. Rizzo S, Bartolini M, Ceccarini L, et al. Targeting Alzheimer’s disease: novel indanone hybrids bearing a pharmacophoric fragment of AP2238. Bioorg Med Chem. 2010;18:1749-1760. [DOI] [PubMed] [Google Scholar]
- 57. Pi R, Mao X, Chao X, et al. Tacrine-6-ferulic acid, a novel multifunctional dimer, inhibits amyloid-β-mediated Alzheimer’s disease-associated pathogenesis in vitro and in vivo. PLoS One. 2012;7:e31921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fernández-Bachiller MI, Pérez C, González-Muñoz GC, et al. Novel tacrine−8-hydroxyquinoline hybrids as multifunctional agents for the treatment of Alzheimer’s disease, with neuroprotective, cholinergic, antioxidant, and copper-complexing properties. J Med Chem. 2010;53:4927-4937. [DOI] [PubMed] [Google Scholar]
- 59. Camps P, Formosa X, Galdeano C, et al. Pyrano[3,2-c]quinoline−6-chlorotacrine hybrids as a novel family of acetylcholinesterase- and β-amyloid-directed anti-Alzheimer compounds. J Med Chem. 2009;52:5365-5379. [DOI] [PubMed] [Google Scholar]
- 60. Pachón-Angona I, Refouvelet B, Andrýs R, et al. Donepezil + chromone + melatonin hybrids as promising agents for Alzheimer’s disease therapy. J Enzyme Inhib Med Chem. 2019;34:479-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chufarova N, Czarnecka K, Skibiński R, Cuchra M, Majsterek I, Szymański P. New tacrine-acridine hybrids as promising multifunctional drugs for potential treatment of Alzheimer’s disease. Arch Pharm (Weinheim). 2018;351:1800050. [DOI] [PubMed] [Google Scholar]
- 62. Guo AJY, Xie HQ, Choi RCY, et al. Galangin, a flavonol derived from Rhizoma Alpiniae Officinarum, inhibits acetylcholinesterase activity in vitro. Chem Biol Interact. 2010;187:246-248. [DOI] [PubMed] [Google Scholar]
- 63. Tweedie D, Fukui K, Li Y, et al. Cognitive impairments induced by concussive mild traumatic brain injury in mouse are ameliorated by treatment with phenserine via multiple non-cholinergic and cholinergic mechanisms. PLoS One. 2016;11:e0156493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Greig NH, Lecca D, Hsueh S, et al. (−)-Phenserine tartrate (PhenT) as a treatment for traumatic brain injury. CNS Neurosci Ther. 2020;26:636-649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Klein J. Phenserine. Expert Opin Investig Drugs. 2007;16:1087-1097. [DOI] [PubMed] [Google Scholar]
- 66. Winblad B, Giacobini E, Frölich L, et al. Phenserine efficacy in Alzheimer’s disease. J Alzheimer’s Dis. 2011;22:1201-1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Becker RE, Greig NH, Lahiri DK, et al. (-)-Phenserine and inhibiting pre-programmed cell death: in pursuit of a novel intervention for Alzheimer’s disease. Curr Alzheimer Res. 2018;15:883-891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhao F-C, Wu Y, Song X-J. Design and development of a novel chalcone derivative as an anticholinesterase inhibitor for possible treatment of dementia. Med Sci Monit. 2017;23:3311-3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ramalingam M, Kim H, Lee Y, Lee Y-I. Phytochemical and pharmacological role of liquiritigenin and isoliquiritigenin from radix glycyrrhizae in human health and disease models. Front Aging Neurosci. 2018;10:348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ko Y-H, Kwon S-H, Lee S-Y, Jang C-G. Liquiritigenin ameliorates memory and cognitive impairment through cholinergic and BDNF pathways in the mouse hippocampus. Arch Pharm Res. 2017;40:1209-1217. [DOI] [PubMed] [Google Scholar]
- 71. Du Y, Luo M, Du Y, et al. Liquiritigenin decreases Aβ levels and ameliorates cognitive decline by regulating microglia M1/M2 transformation in AD mice. Neurotox Res. 2021;39:349-358. [DOI] [PubMed] [Google Scholar]
- 72. Liu RT, Tang JT, Zou LB, Fu JY, Lu QJ. Liquiritigenin attenuates the learning and memory deficits in an amyloid protein precursor transgenic mouse model and the underlying mechanisms. Eur J Pharmacol. 2011;669:76-83. [DOI] [PubMed] [Google Scholar]
- 73. Hannan MA, Dash R, Haque MN, et al. Neuroprotective potentials of marine algae and their bioactive metabolites: pharmacological insights and therapeutic advances. Mar Drugs. 2020;18:347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Alghazwi M, Kan YQ, Zhang W, Gai WP, Garson MJ, Smid S. Neuroprotective activities of natural products from marine macroalgae during 1999–2015. J Appl Phycol. 2016;28:3599-3616. [Google Scholar]
- 75. Barbalace MC, Malaguti M, Giusti L, Lucacchini A, Hrelia S, Angeloni C. Anti-inflammatory activities of marine algae in neurodegenerative diseases. Int J Mol Sci. 2019;20:3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lin J, Huang L, Yu J, et al. Fucoxanthin, a marine carotenoid, reverses scopolamine-induced cognitive impairments in mice and inhibits acetylcholinesterase in vitro. Mar Drugs. 2016;14:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kannan RRR, Aderogba MA, Ndhlala AR, Stirk WA, Van Staden J. Acetylcholinesterase inhibitory activity of phlorotannins isolated from the brown alga, Ecklonia maxima (Osbeck) Papenfuss. Food Res Int. 2013;54:1250-1254. [Google Scholar]
- 78. Yoon NY, Chung HY, Kim HR, Choi JS. Acetyl- and butyrylcholinesterase inhibitory activities of sterols and phlorotannins from Ecklonia stolonifera. Fish Sci. 2008;74:200-207. [Google Scholar]
- 79. Zhao D, Kwon S-H, Chun YS, Gu M-Y, Yang HO. Anti-neuroinflammatory effects of fucoxanthin via inhibition of Akt/NF-κB and MAPKs/AP-1 pathways and activation of PKA/CREB pathway in lipopolysaccharide-activated BV-2 microglial cells. Neurochem Res. 2017;42:667-677. [DOI] [PubMed] [Google Scholar]
- 80. Jung HA, Jin SE, Ahn BR, Lee CM, Choi JS. Anti-inflammatory activity of edible brown alga Eisenia bicyclis and its constituents fucosterol and phlorotannins in LPS-stimulated RAW264.7 macrophages. Food Chem Toxicol. 2013;59:199-206. [DOI] [PubMed] [Google Scholar]
- 81. Oh J, Choi J, Nam T-J. Fucosterol from an edible brown alga Ecklonia stolonifera prevents soluble amyloid beta-induced cognitive dysfunction in aging rats. Mar Drugs. 2018;16:368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gan SY, Wong LZ, Wong JW, Tan EL. Fucosterol exerts protection against amyloid β-induced neurotoxicity, reduces intracellular levels of amyloid β and enhances the mRNA expression of neuroglobin in amyloid β-induced SH-SY5Y cells. Int J Biol Macromol. 2019;121:207-213. [DOI] [PubMed] [Google Scholar]
- 83. Liu X, Liu D, Lin G, et al. Anti-ageing and antioxidant effects of sulfate oligosaccharides from green algae Ulva lactuca and Enteromorpha prolifera in SAMP8 mice. Int J Biol Macromol. 2019;139:342-351. [DOI] [PubMed] [Google Scholar]
- 84. Yang E-J, Ahn S, Ryu J, et al. Phloroglucinol attenuates the cognitive deficits of the 5XFAD mouse model of Alzheimer’s disease. PLoS One. 2015;10:e0135686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zhuang Z-Q, Shen L-L, Li W-W, et al. Gut microbiota is altered in patients with Alzheimer’s disease. J Alzheimers Dis. 2018;63:1337-1346. [DOI] [PubMed] [Google Scholar]
- 86. Wang X, Sun G, Feng T, et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019;29:787-803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jiang R, Du X, Zhang X, et al. Synthesis and bioassay of β-(1,4)-D-mannans as potential agents against Alzheimer’s disease. Acta Pharmacol Sin. 2013;34:1585-1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Syed YY. Sodium oligomannate: first approval. Drugs. 2020;80:441-444. [DOI] [PubMed] [Google Scholar]
- 89. Sochocka M, Donskow-Łysoniewska K, Diniz BS, Kurpas D, Brzozowska E, Leszek J. The gut microbiome alterations and inflammation-driven pathogenesis of Alzheimer’s disease—a critical review. Mol Neurobiol. 2019;56:1841-1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Xiao S, Chan P, Wang T, et al. A 36-week multicenter, randomized, double-blind, placebo-controlled, parallel-group, phase 3 clinical trial of sodium oligomannate for mild-to-moderate Alzheimer’s dementia. Alzheimers Res Ther. 2021;13:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Cummings J. New approaches to symptomatic treatments for Alzheimer’s disease. Mol Neurodegener. 2021;16:1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement (N Y). 2019;5:272-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement (N Y). 2020;6:1-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Cheng Y, Chen Z, Liao T, et al. An intranasally delivered peptide drug ameliorates cognitive decline in Alzheimer transgenic mice. EMBO Mol Med. 2017;9:703-715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Abraham CR, Mullen PC, Tucker-Zhou T, Chen CD, Zeldich E. Klotho is a neuroprotective and cognition-enhancing protein. Vitam Horm. 2016;101:215-238. [DOI] [PubMed] [Google Scholar]
- 96. Huang LK, Chao SP, Hu CJ. Clinical trials of new drugs for Alzheimer disease. J Biomed Sci. 2020;27:1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Jack CR, Bennett DA, Blennow K, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14:535-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Sabbagh MN, Hendrix S, Harrison JE. FDA position statement “Early Alzheimer’s disease: developing drugs for treatment, Guidance for Industry.” Alzheimers Dement (N Y). 2019;5:13-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Food and Drug Administration. Federal Register. Early Alzheimer’s disease: developing drugs for treatment; draft guidance for industry; availability. 2018:7060-7061. https://www.federalregister.gov/documents/2018/02/16/2018-03226/early-alzheimers-disease-developing-drugs-for-treatment-draft-guidance-for-industry-availability [Google Scholar]
- 100. Vos SJ, Xiong C, Visser PJ, et al. Preclinical Alzheimer’s disease and its outcome: a longitudinal cohort study. Lancet Neurol. 2013;12:957-965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Amariglio RE, Buckley RF, Mormino EC, et al. Amyloid-associated increases in longitudinal report of subjective cognitive complaints. Alzheimers Dement (N Y). 2018;4:444-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Insel PS, Hansson O, Mackin RS, Weiner M, Mattsson N. Amyloid pathology in the progression to mild cognitive impairment. Neurobiol Aging. 2018;64:76-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Yiannopoulou KG, Papageorgiou SG. Current and future treatments in Alzheimer disease: an update. J Cent Nerv Syst Dis. 2020;12:117957352090739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Marasco RA. Current and evolving treatment strategies for the Alzheimer disease continuum. Am J Manag Care. 2020;26:S167-S176. [DOI] [PubMed] [Google Scholar]
- 105. Budd Haeberlein S, O’Gorman J, Chiao P, et al. Clinical development of aducanumab, an anti-Aβ human monoclonal antibody being investigated for the treatment of early Alzheimer’s disease. J Prev Alzheimers Dis. 2017;4:255-263. [DOI] [PubMed] [Google Scholar]
- 106. Biogen. Biogen completes submission of Biologics License Application to FDA for aducanumab as a treatment for Alzheimer’s disease. 2020. https://investors.biogen.com/news-releases/news-release-details/biogen-completes-submission-biologics-license-application-fda
- 107. Hung A, Schneider M, Lopez MH, McClellan M. Preclinical Alzheimer disease drug development: early considerations based on phase 3 clinical trials. J Manag Care Spec Pharm. 2020;26:888-900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Wang J, Logovinsky V, Hendrix SB, et al. ADCOMS: a composite clinical outcome for prodromal Alzheimer’s disease trials. J Neurol Neurosurg Psychiatry. 2016;87:993-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Langa KM, Burke JF. Preclinical Alzheimer disease—early diagnosis or overdiagnosis? JAMA Intern Med. 2019;179:1161. [DOI] [PubMed] [Google Scholar]
- 110. Ghezzi L, Scarpini E, Galimberti D. Disease-modifying drugs in Alzheimer’s disease. Drug Des Devel Ther. 2013;7:1471-1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ewen ST, Fauzi A, Quan TY, Chamyuang S, Yin ACY. A review on advances of treatment modalities for Alzheimer’s disease. Life Sci. 2021;276:119129. [DOI] [PubMed] [Google Scholar]
- 112. Hlávka JP, Mattke S, Liu JL. Assessing the preparedness of the health care system infrastructure in six European countries for an Alzheimer’s treatment. Rand Health Q. 2019;8:2. [PMC free article] [PubMed] [Google Scholar]
- 113. Cummings J, Feldman HH, Scheltens P. The “rights” of precision drug development for Alzheimer’s disease. Alzheimers Res Ther. 2019;11:76. [DOI] [PMC free article] [PubMed] [Google Scholar]