

Abstract
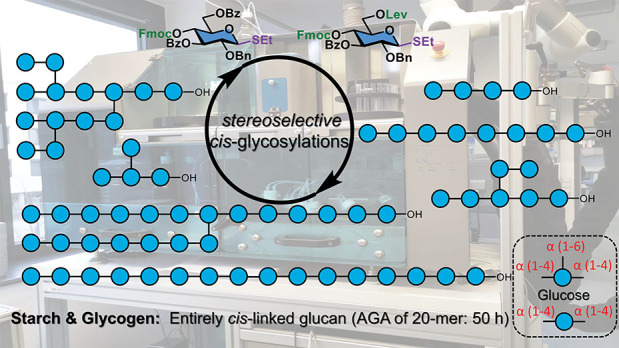
Polysaccharides are Nature’s most abundant biomaterials essential for plant cell wall construction and energy storage. Seemingly minor structural differences result in entirely different functions: cellulose, a β (1–4) linked glucose polymer, forms fibrils that can support large trees, while amylose, an α (1–4) linked glucose polymer forms soft hollow fibers used for energy storage. A detailed understanding of polysaccharide structures requires pure materials that cannot be isolated from natural sources. Automated Glycan Assembly provides quick access to trans-linked glycans analogues of cellulose, but the stereoselective installation of multiple cis-glycosidic linkages present in amylose has not been possible to date. Here, we identify thioglycoside building blocks with different protecting group patterns that, in concert with temperature and solvent control, achieve excellent stereoselectivity during the synthesis of linear and branched α-glucan polymers with up to 20 cis-glycosidic linkages. The molecules prepared with the new method will serve as probes to understand the biosynthesis and the structure of α-glucans.
Introduction
Polysaccharides are built from monosaccharide units connected through glycosidic bonds with different regio- and stereochemistry. Glucose-based polymers are the most abundant biomaterials on earth, but differ greatly in structure and function depending on whether they are β (1–4) linked as in cellulose, or α (1–4) linked as in starch and glycogen (Figure 1A). Cellulose (1) is a linear polymer that unlike starch does not coil or branch, but adopts an extended and stiff rodlike conformation.1,2 Hydrogen bonding between parallel chains form microfibrils with high tensile strength that are part of the polysaccharide matrix in the cell wall.1−5 Starch is a mixture of about one-quarter strictly linear amylose (2) and about three-quarters α (1–6) branched amylopectin (3).6,7 Glycogen (4), the polymer that stores energy in the form of glucose in animals, is highly branched.8 Amylose exists in a disordered amorphous conformation or can adopt two different helical conformations that can host other molecules.6,7,9−12 Amylopectin is linear and does not crystallize as well as the long linear chains of amylose.6,7,10,13
Figure 1.

Chemical structure, synthetic strategies, and Automated Glycan Assembly of starch and glycogen. (A) Chemical structure of β (1–4) linked polyglucoside cellulose (1), linear α (1–4) linked polyglucoside amylose (2), α (1–4) linked polyglucoside with α (1–6) branched α (1–4) linked polyglucoside side chains, amylopectin (3), and highly branched glycogen (4). (B) Participating neighboring protecting group ensures trans-glycoside construction, stereocontrolled cis-glycosidic bond formation is challenging: uncontrolled glycosylations result in anomeric mixtures. Typical solutions: Solvent assistance,19 bulky protecting group assistance,50 ester remote assistance,26 chiral-auxiliary-mediation,18 macrocyclic bis-thiourea catalysis.27 Strategy developed in this work for α (1–4) glucan AGA with simple chemistry. (C) Schematic view of Automated Glycan Assembly and the building blocks used to prepare cis-linked polyglucosides. (D) Synthetic amylose, and amylopectin oligo- and polysaccharides (5–11) prepared by AGA.
The molecular level understanding of cellulose structure formation has benefitted from synthetic glycans and single molecule glycan imaging.14,15 In order to understand the factors contributing to the structure of amylose, amylopectin, and glycogen, pure oligo- and polysaccharides containing exclusively cis-glycosidic linkages are needed. The stereocontrolled synthesis of linear or branched oligosaccharides has proven extremely challenging since glycosidic bond formation cannot rely on participating neighboring protecting groups as for trans-glycoside construction (Figure 1B). A host of methods to prepare cis-glycosides involving remote assistance, the use of chiral auxiliaries, or catalysts as well as substrate control combined with solvent and temperature effects has been developed (Figure 1B).16−34 Still, the synthesis of oligosaccharides with multiple cis-glycosidic linkages has been a major challenge to manual syntheses that relied on block couplings to prepare amylose and amylopectin sequences as long as 10-mers.7,35,36
Automated Glycan Assembly (AGA) has been used to prepare polysaccharides as large as 151-mers,37 a variety of biologically important complex glycans containing different monomers as well as natural and unnatural carbohydrate materials.5,38−45 AGA requires high yielding and completely selective glycosylation reactions as none of the intermediates, but only the products of the synthesis are purified.46 The separation of full-length oligosaccharide side-products that differ from the desired product only in the stereochemistry of one linkage is very difficult if not impossible. Therefore, very few oligosaccharides containing cis-glycosidic linkages have been prepared by AGA, exploiting building blocks with remote ester groups in the 3- and 6-positions.47−49
Here, we describe a simple strategy based on thioglycoside building blocks with protecting group patterns that, together with temperature and solvent control, allows for excellent cis-selectivity during AGA of α-glucans (Figure 1C). A series of glucans resembling linear or branched natural starch structures with an α (1–4) backbone and α (1–6) side chains were prepared to illustrate the power of the approach (Figure 1D). Branched 20-mer α-glucan 10, the largest full cis-linked branched glycan ever made, was obtained within 50 h AGA time.
Results and Discussion
Building Block Design
Thioglycosides with temporary fluorenylmethoxycarbonyl (Fmoc) protecting groups have proven as useful building blocks for AGA (Table 1A).40 Thus, Fmoc protection to mask the C4 hydroxyl group required for chain elongation was selected. The orientation of C3 and C6 functional groups on the glucopyranose ring makes them suitable remote assistant groups to block the β-face during glycosylation (Figure 1B). Remote assistance of a 6-acetyl (Ac) protecting group is not sufficient to prepare α (1–4) glucan oligosaccharides with high cis-selectivity and good isolated yield.49 In search for building blocks that maximize cis-selectivity and yield during AGA, a comprehensive evaluation of different C3 and C6 ester groups and their influence on α (1–4) glucan formation was undertaken (see Table S1). An electron rich, nonparticipating benzyl (Bn) ether group was selected as a permanent protecting group on the C2 hydroxyl group to allow for cis-glycoside formation with good reactivity. Thioglycosides 13–25 carry a C3 benzyl ether as well as different esters at C6, while building blocks 26–30 carry ester protection groups both on C3 and C6 (see Table 1A). Additional building blocks with ester at C3 but benzyl protection at C6-OH were also shown (Table S1, compound 110–112) as comparison. Thioglycosides 12 and 31–32, substituted with a fluorine or an ether at C6, respectively, were prepared for comparison. All building blocks were prepared from a single precursor using stratified methods in order to minimize experimental work (see Figure S1).
Table 1. Glucose Building Blocks and Solution Phase Glycosylationsa.

| entry | building block | acceptor | ratio (α/β) | yield (α + β) | entry | building block | acceptor | ratio (α/β) | yield (α + β) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 12 | i-PrOH | 1:2.9 | 93% | 15 | 12 | 33 | 1.5:1 | 83% |
| 2 | 13 | i-PrOH | 2.3:1 | 95% | 16 | 13 | 33 | 3.8:1 | 75% |
| 3 | 17 | i-PrOH | 2.5:1 | 92% | 17 | 17 | 33 | 5.5:1 | 69% |
| 4 | 20 | i-PrOH | 1.7:1 | 93% | 18 | 20 | 33 | 6.8:1 | 72% |
| 5 | 22 | i-PrOH | 2.2:1 | 90% | 19 | 22 | 33 | 5.6:1 | 68% |
| 6 | 23 | i-PrOH | 3.5:1 | 89% | 20 | 23 | 33 | 6.2:1 | 67% |
| 7 | 25 | i-PrOH | 1.9:1 | 90% | 21 | 25 | 33 | 7.5:1 | 44% |
| 8 | 26 | i-PrOH | 4.5:1 | 84% | 22 | 26 | 33 | 9.8:1 | 78% |
| 9 | 27 | i-PrOH | 6.4:1 | 88% | 23 | 27 | 33 | >10:1 | 62% |
| 10 | 28 | i-PrOH | 4.8:1 | 87% | 24 | 28 | 33 | >10:1 | 83% |
| 11 | 29 | i-PrOH | 7.1:1 | 74% | 25 | 29 | 33 | >10:1 | 64% |
| 12 | 30 | i-PrOH | 2.4:1 | 76% | 26 | 30 | 33 | >10:1 | 80% |
| 13 | 31 | i-PrOH | 1:1.1 | 96% | 27 | 31 | 33 | 3.9:1 | 75% |
| 14 | 32 | i-PrOH | 1:2.6 | 94% | 28 | 32 | 33 | 2.7:1 | 81% |
(A) The glucose thioglycoside building blocks were prepared from a common precursor as described in Figure S1. (B) Glycosylations using isopropanol and monosaccharide as nucleophiles. The ratio of α/β anomers were quantified by NMR, yield represented the isolated yield of α- and β-anomers (complete results in Table S1).
Solution-Phase Glycosylation
The differentially protected thioglycoside building blocks were used to glycosylate different nucleophiles to screen the influence of protecting groups on α-selectivity (Table 1B and Table S1). Initially, all building blocks were activated with N-iodosuccinimide (NIS)–triflic acid (TfOH) in DCM for 5 min at −15 °C followed by 60 min at 0 °C to glycosylate isopropanol (i-PrOH) as a model for a secondary hydroxyl group. Glycosylations involving 12–25 (entries 1–7 in Table 1B, and entries 1–14 in Table S1) as well as 31–32 (entries 13 and 14) suggest a relationship between electronic and steric properties of the C6 protecting groups and the α/β ratio of the glycoside products. An electron withdrawing group (EWG) on C6 improves cis-selectivity. Thioglycosides 12 (entry 1) and 32 (entry 14) that carry nonparticipating electron donating groups (EDGs), are less α-selective than building blocks 13–25 (entries 2–7, and entries 2–14 in Table S1) and 31 (entry 13). Esterification on 6-OH(entries 2–7, and entries 2–14 in Table S1) resulted in higher stereoselectivity than deoxyfluorination (entry 13), even though ester groups are less electron withdrawing than fluorine. These results further supported the ester remote assistance hypothesis during glycosylation.16,26,49 The argument whether the ester group really stabilized the intermediate by forming a covalent glycosyl dioxolenium ion during glycosylations has not been settled, although this intermediate can be detected under extreme conditions.51−54 We observed that 6-OH esterification alone does not ensure excellent selectivity during α (1–4) glycosylations. Selectivities for the glycosylation of isopropanol ranged from 1.5–3:1 (α/β) as changes in the electronic and steric properties of ester groups had little effect (entries 2–7, and entries 2–14 in Table S1). Additional experiments employing 3-OH esterified, 6-OH benzylated building blocks (entries 22–24 in Table S1) agreed with our previous findings that C3 esters are an EWG that can provide remote assistance, although usually less helpful than C6 esters.49 To increase the selectivity further, 3,6-disubstituted building blocks 26–30 were tested (entries 8–12). All of these compounds improved cis-glycoside formation.
With initial information concerning the selectivity of different thioglycoside building blocks in hand, we next investigated the influence of the reaction temperature on the product ratio (Table S2). Reactions at a constant temperature revealed that glycosylations at higher temperatures gave better stereoselectivity, as previously reported.28 However, activation temperatures above 0 °C may result in lower yields (Table S2, entry 3 vs 4). Solvent is another important factor to control the stereochemistry during glycosylation. Ethers are the most common alpha directing solvents.16,19,28,49 We tested our system in several different ethers (Table S3, entries 1–5). Although most of the selected ethers were helpful for producing cis-glycoside, the poor solubility of our building blocks and NIS makes these solvents not compatible with AGA. Dioxane is a good choice for use in the AGA system, but a balance between the solubility of the activator and the melting point of the reaction mixture needs to be maintained. For AGA we dissolved the activator in a DCM–dioxane mixture (2:1), and mixed it with the same volume of building block DCM solution (DCM–dioxane 5:1 during reaction).39 We tested several typical building blocks in a 5:1 DCM–dioxane mixture and proved that this condition can be used to increase the cis-product formation(Table S3, entries 1 and 6–14).
Next, the influence of the nucleophile on the outcome of the glycosylation was explored, by analyzing the glycosylation of the secondary hydroxyl group at the C4 position of monosaccharide 33 (entries 15–28).55 EWG and ester remote assistance contribute both to the formation of cis-linked disaccharide. Higher cis-selectivity was obtained with the more sterically hindered monosaccharide-acceptor compared to isopropanol, in particular when building blocks with bulky pivaloyl (Piv) 17 (entry 3 vs 17), triphenylacetyl 25 (entry 7 vs 21) protecting groups or disubstituted thioglycosides 27–30 (entries 9–12 vs 23–26) were used. For sterically more demanding nucleophiles, the protecting group pattern on the glycosylating agent might have an important impact on the stereochemical outcome of the glycosylation. Bulky protecting groups had been used in the solution phase synthesis to introduce cis-linked glucose residues, and an α-glucan decamer was prepared recently.50,56 Overall, selectivities exceeding 10:1 (α/β) were achieved when all factors favoring cis-glycoside formation were combined using building blocks 27–30 carrying 3,6-diesters for AGA.
Glycosylations on Solid Support
Using the optimized solution-phase conditions, glycosylations were carried out on a Merrifield resin functionalized with the 5-aminopentanol photolabile linker (35) (Table 2). The coupling cycle involved an acidic wash with trimethylsilyl trifluoromethanesulfonate (TMSOTf), followed by NIS-TfOH induced glycosylation (−20 °C for 5 min → 0 °C for 60 min), capping of any unreacted nucleophile with acetic anhydride (Ac2O) and methanesulfonic acid (MsOH), and cleavage of the temporary Fmoc protective group with triethylamine (TEA). After completion of each synthesis, the product was released from the resin by irradiation with UV light. The α (1–4) glucose pentasaccharide containing a β-glycosidic linkage between the first unit and the amino pentanol linker served as a model to study the cis-selectivity on both C4 secondary and C6 primary hydroxyl groups.49
Table 2. Screening of Glycosylation Conditions by AGA.

| entry | building block | R1 | R2 | product | selectivity | yield |
|---|---|---|---|---|---|---|
| 1 | 13 | OAc | OBn | 36 | 0.35 | 40% |
| 2 | 17 | OPiv | OBn | 37 | 0.93 | 4% |
| 3 | 23 | O(p-NO2Bz) | OBn | 38 | 0.68a | 14% |
| 4 | 27 | OPiv | OPiv | 39 | >0.95 | 3% |
| 5 | 28 | OBz | OBz | 40 | 0.74 | 17% |
| 6 | 29 | O(p-NO2Bz) | O(p-NO2Bz) | 41 | 0.45a | 19% |
The p-nitrobenzoyl group present in building blocks 23 and 29 is not stable to UV irradiation. Therefore, the data was calculated based on deprotected products.
The products 36–41 were characterized by analytical HPLC and were purified in protected form to remove any deletion sequences. All pentamer products were collected. The isolated yield of the combined pentamers is reported independently of the stereochemistry of the linkages. The selectivity is calculated as the ratio of the amount of desired product containing four α-linkages to all pentamer products, as measured by NMR spectroscopy (for details, see Supporting Information). For protected glucans in this study, the chemical shift of α-linked anomeric proton is usually > 4.8 ppm and anomeric carbon < 100 ppm; for β-linked anomeric proton, the chemical shift is usually < 4.7 ppm and anomeric carbon > 100 ppm.
In search for a suitable building block for α-glucan AGA, both yield and cis-selectivity were taken into consideration. The results for thioglycoside 13 were in good agreement with our earlier findings (Table 2, entry 1).49 Despite the promising yield obtained with monoacetylated 13, only 35% of the pentamer products had the desired stereochemistry (entry 1). The pivaloyl group at 6-OH (17 and 27) resulted in good cis-selectivity on solid phase (entries 2 and 4). However, bulky esters at C3 or C6 lowered the reactivity of 17 and 27 both as glycosylating agents and nucleophiles to render them unsuitable for the assembly of longer oligosaccharides. Building blocks 23 and 28 showed similar yields and cis-selectivity (entries 3 and 5). The trans-linked side-products arose mainly from the glycosylation of primary hydroxyl group of 34. Di(p-nitrobenzoyl) protected building block 29 showed a slightly lower cis-selectivity (entry 6). Additionally, the p-nitrobenzoyl group was not stable under UV irradiation and complicated purification and characterization of the protected oligomers. 3,6-Dibenzoylated thioglycoside building block 28 emerged as the best choice considering selectivity and yield (entry 5). Similar good cis-selectivity was observed recently with galactose building blocks bearing multiple benzoyl (Bz) groups.57
AGA of Starch and Glycogen α-Glucans
On the basis of the preliminary AGA results described above, the reaction conditions were optimized in the context of the synthesis of a series of α-glucans (Figure 2). The 3,6-di-O-benzyolated thioglycoside 28 carrying a temporary 4-OFmoc protecting group was employed to assemble the α (1–4) glycan backbone. α (1–6) Glycan branching was installed using building block 30 with benzoate ester at C3 and a 4-OFmoc and a 6-OLev temporary protecting groups. A double glycosylation cycle (−20 °C for 5 min → 0 °C for 60 min, twice) with thioglycoside 28 was used for each elongation to increase yields and minimize the formation of deletion sequences. Traceless linker (42)42 was cleaved by photolysis and removed after global deprotection to release the natural starch/glycogen α-glucan. Linear amylose oligomer α (1–4) glucan, including tetramer 5, octamer 6, and 16-mer 7, were prepared with good yield and selectivity (Figure 2A, see Supporting Information for full characterization). The HPLC analysis of the crude protected 16-mer 45 after AGA displayed the (partially capped) deletion sequences as the record of glycan elongation (Figure 3A). No trans-glycosylic linkages were observed by NMR spectroscopy, indicating that even the first glycosylation between 28 and linker 42 proceeded with excellent cis-selectivity.
Figure 2.

AGA of amylose, amylopectin, and glycogen α-glucans. (A) AGA of amylose oligosaccharides 5–7. (B) AGA of amylopectin polysaccharides 8–10. (C) AGA of glycogen oligosaccharide 11.
Figure 3.

HPLC analysis of protected 16-mer 45, 20-mer 48, and 14-mer 49. Left side, crude mixture after AGA; right side, target compound after purification. (A) 16-mer 45; (B) 20-mer 48; (C) 14-mer 49. Signal was collected through ELSD.
To extend the AGA scope to natural amylopectin and glycogen structures, the selective installation of α (1–6) branching points within the α (1–4) backbone had to be developed. Glycosylation of the C6 primary hydroxyl group of 34 using building block 28 did not result in satisfactory cis-selectivity (Table 2). Two synthetic strategies were tested in the context of the syntheses of 8–11, as the order of Fmoc and Lev cleavage may influence yield and selectivity. In Strategy A, after incorporation of building block 30, the Fmoc group was cleaved and one glycosylation was performed in the presence of the levaloyl group. Then, Lev was cleaved and the C6 hydroxyl group was glycosylated (Figure S2A). After the branching point was introduced, α (1–4) elongation of backbone and side chain was achieved simultaneously. Tetramer 8 and heptamer 9 were prepared using this strategy (Figure 2B). Protected heptamer 47 was fully analyzed using HPLC and HSQC NMR to evaluate the cis-selectivity of Strategy A. After AGA, the crude product was analyzed with HPLC and all the detectable side-products were assigned according to MALDI-TOF MS (Figure S2B). Interestingly, most of the deletion sequences were capped by benzoyl groups that may result from the acetylated deletion sequences (capping step) through the ester exchange reaction. The MS signal of the desired heptamer was only observed for the main peak (red square) and its surrounding area, together with the signal for some deletion sequences (black square). The main product proved to be the desired fully cis-linked heptamer 47 by HSQC NMR (Figure S2C). The mixture of deletion sequences was also analyzed by HSQC NMR showing no evidence of β linkage (Figure S2D). These results strongly suggest that Strategy A is a highly selective method for the installation of α (1–6) linkages in our synthesis. Glycosylation at C4-OH may increase the glycan bulk for the subsequent glycosylation of the C6 hydroxyl group, thereby promoting cis-selectivity.
Strategy B was tested for trimer 50 (Figure S3) which not only allowed for the formation of different patterns during glycan chain elongation, but also gave a much higher yield compared to Strategy A and greatly simplified the synthesis of longer amylopectins. After glycosylation with building block 30, cleavage of the levulinic ester was followed by glycosylation on C6-OH in the presence of 4-OFmoc (Figure S3A). After Fmoc cleavage, the C4 hydroxyl groups were glycosylated. Only one main fraction was observed by HPLC analysis of the crude product (Figure S3B) that proved to be the desired cis-linked trimer by HSQC NMR analysis (Figure S3C). The higher selectivity observed for this reaction, compared to the glycosylation of 34, could result from the electron withdrawing 4-OFmoc in 30, instead of an electron donating benzyl ether in 34 that renders it less nucleophilic.
Amylopectin 20-mer 10 was assembled using Strategy B (Figure 2B). After HPLC purification (Figure 3B), we obtained the protected 20-mer in 24% yield. The major side products are (partially capped) deletion sequences formed during chain extension after the α (1–6) branching point was installed. The simultaneous elongation of backbone and side chain on large glycan may not be as efficient using the standard glycosylation condition.
The assembly of a more challenging, hyperbranched glycogen model 11 was undertaken. Up to the secondary branching points, the assembly followed the procedures established above. Two secondary branching points was introduced with one double glycosylation, but in order to add four glucose units in one pot at the end, two double glycosylation cycles were required. The HPLC analysis of the crude product after AGA indicated that the deletion sequences were mainly caused by incomplete multiple-point glycosylation (Figure 3C).
Since the end-stage deletion sequences partially overlap with the product peak, three HPLC purifications were required for 48 and 49 (see Supporting Information, Method B3 in AGA part). The first purification yielded pure fraction (I) (purity >90% indicated by MS) and fraction (II) (purity >50%). The second purification of fraction (II) yielded fraction (III) (purity >90%), before the third purification of combined fractions (I) and (III) gave the product in high purity. After deprotection, both 10 and 11 were obtained in good yield and stereochemical purity, as confirmed by HSQC NMR (see Supporting Information for full characterization). The entire AGA process to access the protected 20-mer 48 took about 50 h and for protected 14-mer 49 took only 30 h.
Comparison of Synthetic α-Glucans and Natural Starch/Glycogen
The properties of the synthetic α-glucan were compared to natural potato starch and glycogen from bovine liver. All characteristic signals in the NMR spectra of the synthetic glucans exactly match those of the natural compounds (Figure 4). Except for glucose, all compounds show signals from 5.30 to 5.50 ppm that represent the α (1–4) anomeric protons of the essential starch/glycogen linkages. Signals at 5.24, 4.67, and 3.29 ppm represent the α, β anomeric protons and the β H2 at the reducing end that are not observed in natural starch and glycogen. The signals at 4.99–4.96 ppm in starch and glycogen represent the α (1–6) anomeric protons of the glycan branching points. The signals at 3.45–3.39 ppm represent the C4 protons of the nonreducing end. These signals are indicative of the “degree of branching” of starch/glycogen polysaccharides with strong signals (compared to the signals of reducing end protons) for glycogen and 14-mer 11. In contrast to the broad signals for heterogeneous natural polysaccharides, all homogeneous synthetic compounds show sharp NMR signals. Our data highlight the importance of synthetic compounds as standard to characterize naturally sourced compounds.
Figure 4.

Comparison of 1H NMR spectra of natural starch (a), glycogen (b), glucose (c), and synthetic α-glucans (d–j).
The iodine–starch test is a simple, but characteristic detection method for α (1–4) glucans.9 Although the details of this interaction was not entirely understood, an iodine–iodide complex is known to embed into the helix formed by α (1–4) glucan.58,59 With increasing glucan chain length, more complex is bound, causing a color change from yellow, orange, red-brown, to purple and dark blue. Amylose tetramer 5, amylopectin tetramer 8, and heptamer 9 show a light yellow color similar to glucose. Amylose octamer 6, amylopectin 20-mer 10, and glycogen 14-mer 11 result in a deep yellow color, indicative of weak interactions between iodide complex and glucan. Amylose 16-mer 7 gave a red-brown color similar to glycogen indicating that the α (1–4) glucan chain has the required length to strongly interact with the iodine–iodide complex. The long α (1–4) glucan backbone of natural starch is poorly soluble at room temperature and has a very strong bonding with the complex, indicated by the dark blue color (Figure 5). Our results match literature reports that six continuous α (1–4) glucose residues are needed to form the repeating unit of helical structure, which can assemble with iodine complex nicely.60,61
Figure 5.

I2–KI staining of glucose, natural starch/glycogen and synthetic α-glucans.
Conclusions
On the basis of a comprehensive study of 21 differentially protected thioglycoside building blocks, glycosylation conditions that result in high yielding cis-selective glycosidic bond formation were developed. With amylose and amylopectin oligo- and polysaccharides as targets, 3,6-dibenzoylated glucose thioglycoside 28 provided the perfect balance between cis-selectivity and reactivity during AGA required to prepare long α (1–4) glycans. Differentially protected thioglycoside 30 was the key to α (1–6) branching with excellent cis-selectivity and good yield using AGA. The 20-mer amylopectin polysaccharide 10 is the largest entirely cis-linked branched carbohydrate assembled by chemical synthesis to date. NMR studies and iodine-stain tests, confirmed that the synthetic glucans share common structural properties with natural starch and glycogen. This simple and efficient AGA method provides convenient access to large, well-defined cis-linked starch and glycogen polysaccharides for biological and material science investigations. Future improvements on the instrumentation hardware and the chemistry will shorten assembly times. Increased glycosylation efficiencies will save building blocks, reduce deletion sequences, and facilitate HPLC purification.
Acknowledgments
We thank the Max Planck Society financial support. We thank Sabrina Leichnitz and Giulio Fittolani for the help in collection of HRMS data, Theodore Tyrikos-Ergas and Zhouxiang Zhao for the help during resin preparation, and Elena Shanina for the help in collection of NMR spectrum for amylopectin 20-mer.
Glossary
ABBREVIATIONS
- AGA
automated glycan assembly
- Ac
acetyl
- Fmoc
fluorenylmethoxycarbonyl
- Bn
benzyl
- Bz
benzoyl
- NIS
N-iodosuccinimide
- TfOH
triflic acid
- DCM
dichloromethane
- i-PrOH
isopropanol
- EWG
electron withdrawing group
- EDG
electron donating group
- Piv
pivaloyl
- TMSOTf
trimethylsilyl trifluoromethanesulfonate
- Ac2O
acetic anhydride
- MsOH
methanesulfonic acid
- TEA
trimethylamine
- Lev
levaloyl
- HPLC
high-performance liquid chromatography
- HRMS
high-resolution mass spectrometry
- MALDI-TOF MS
matrix-assisted laser desorption ionization–time-of-flight mass spectrometry
- ELSD
vaporative light scattering detector
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c02188.
Experimental procedures, additional data, and spectral data for all reactions and compounds (PDF)
This work is supported by the Max Planck Society.
The authors declare no competing financial interest.
Supplementary Material
References
- O’Sullivan A. C. Cellulose: the structure slowly unravels. Cellulose 1997, 4 (3), 173–207. 10.1023/A:1018431705579. [DOI] [Google Scholar]
- Habibi Y.; Lucia L. A.; Rojas O. J. Cellulose nanocrystals: chemistry, self-assembly, and applications. Chem. Rev. 2010, 110 (6), 3479–3500. 10.1021/cr900339w. [DOI] [PubMed] [Google Scholar]
- Nishiyama Y.; Langan P.; Chanzy H. Crystal structure and hydrogen-bonding system in cellulose Iβ from synchrotron X-ray and neutron fiber diffraction. J. Am. Chem. Soc. 2002, 124 (31), 9074–9082. 10.1021/ja0257319. [DOI] [PubMed] [Google Scholar]
- Cosgrove D. J. Growth of the plant cell wall. Nat. Rev. Mol. Cell Biol. 2005, 6 (11), 850. 10.1038/nrm1746. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Tyrikos-Ergas T.; Zhu Y.; Fittolani G.; Bordoni V.; Singhal A.; Fair R. J.; Grafmüller A.; Seeberger P. H.; Delbianco M. Systematic hydrogen-bond manipulations to establish polysaccharide structure–property correlations. Angew. Chem. 2019, 131 (37), 13261–13266. 10.1002/ange.201906577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buleon A.; Colonna P.; Planchot V.; Ball S. Starch granules: structure and biosynthesis. Int. J. Biol. Macromol. 1998, 23 (2), 85–112. 10.1016/S0141-8130(98)00040-3. [DOI] [PubMed] [Google Scholar]
- Damager I.; Engelsen S. B.; Blennow A.; Lindberg Møller B.; Motawia M. S. First principles insight into the α-glucan structures of starch: their synthesis, conformation, and hydration. Chem. Rev. 2010, 110 (4), 2049–2080. 10.1021/cr900227t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manners D. J. Recent developments in our understanding of glycogen structure. Carbohydr. Polym. 1991, 16 (1), 37–82. 10.1016/0144-8617(91)90071-J. [DOI] [Google Scholar]
- Bates F. L.; French D.; Rundle R. Amylose and amylopectin content of starches determined by their iodine complex formation. J. Am. Chem. Soc. 1943, 65 (2), 142–148. 10.1021/ja01242a003. [DOI] [Google Scholar]
- Tester R. F.; Karkalas J.; Qi X. Starch—composition, fine structure and architecture. J. Cereal Sci. 2004, 39 (2), 151–165. 10.1016/j.jcs.2003.12.001. [DOI] [Google Scholar]
- Popov D.; Buléon A.; Burghammer M.; Chanzy H.; Montesanti N.; Putaux J.-L.; Potocki-Veronese G.; Riekel C. Crystal structure of A-amylose: A revisit from synchrotron microdiffraction analysis of single crystals. Macromolecules 2009, 42 (4), 1167–1174. 10.1021/ma801789j. [DOI] [Google Scholar]
- Putseys J.; Lamberts L.; Delcour J. Amylose-inclusion complexes: Formation, identity and physico-chemical properties. J. Cereal Sci. 2010, 51 (3), 238–247. 10.1016/j.jcs.2010.01.011. [DOI] [Google Scholar]
- Jane J.; Chen Y.; Lee L.; McPherson A.; Wong K.; Radosavljevic M.; Kasemsuwan T. Effects of amylopectin branch chain length and amylose content on the gelatinization and pasting properties of starch. Cereal Chem. 1999, 76 (5), 629–637. 10.1094/CCHEM.1999.76.5.629. [DOI] [Google Scholar]
- Wu X.; Delbianco M.; Anggara K.; Michnowicz T.; Pardo-Vargas A.; Bharate P.; Sen S.; Pristl M.; Rauschenbach S.; Schlickum U.; Abb S.; Seeberger P. H.; Kern K. Imaging single glycans. Nature 2020, 582 (7812), 375–378. 10.1038/s41586-020-2362-1. [DOI] [PubMed] [Google Scholar]
- Anggara K.; Zhu Y.; Delbianco M.; Rauschenbach S.; Abb S.; Seeberger P. H.; Kern K. Exploring the molecular conformation space by soft molecule–surface collision. J. Am. Chem. Soc. 2020, 142 (51), 21420–21427. 10.1021/jacs.0c09933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demchenko A. V.; Rousson E.; Boons G.-J. Stereoselective 1, 2-cis-galactosylation assisted by remote neighboring group participation and solvent effects. Tetrahedron Lett. 1999, 40 (36), 6523–6526. 10.1016/S0040-4039(99)01203-4. [DOI] [Google Scholar]
- Demchenko A. V. 1, 2-cis-O-Glycosylation: methods, strategies, principles. Curr. Org. Chem. 2003, 7 (1), 35–79. 10.2174/1385272033373175. [DOI] [Google Scholar]
- Boltje T. J.; Kim J.-H.; Park J.; Boons G.-J. Chiral-auxiliary-mediated 1, 2-cis-glycosylations for the solid-supported synthesis of a biologically important branched α-glucan. Nat. Chem. 2010, 2 (7), 552–557. 10.1038/nchem.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh H.; Hansen H. S.; Manabe S.; van Gunsteren W. F.; Hünenberger P. H. Theoretical investigation of solvent effects on glycosylation reactions: stereoselectivity controlled by preferential conformations of the intermediate oxacarbenium-counterion complex. J. Chem. Theory Comput. 2010, 6 (6), 1783–1797. 10.1021/ct1001347. [DOI] [PubMed] [Google Scholar]
- Aubry S.; Sasaki K.; Sharma I.; Crich D.. Influence of protecting groups on the reactivity and selectivity of glycosylation: chemistry of the 4,6-O-benzylidene protected mannopyranosyl donors and related species. In Reactivity Tuning in Oligosaccharide Assembly; Springer, 2010; pp 141–188. [DOI] [PubMed] [Google Scholar]
- Lu S. R.; Lai Y. H.; Chen J. H.; Liu C. Y.; Mong K. K. T. Dimethylformamide: an unusual glycosylation modulator. Angew. Chem., Int. Ed. 2011, 50 (32), 7315–7320. 10.1002/anie.201100076. [DOI] [PubMed] [Google Scholar]
- Yasomanee J. P.; Demchenko A. V. Effect of remote picolinyl and picoloyl substituents on the stereoselectivity of chemical glycosylation. J. Am. Chem. Soc. 2012, 134 (49), 20097–20102. 10.1021/ja307355n. [DOI] [PubMed] [Google Scholar]
- Yasomanee J. P.; Demchenko A. V. Hydrogen bond mediated aglycone delivery: Synthesis of linear and branched α-glucans. Angew. Chem. 2014, 126 (39), 10621–10624. 10.1002/ange.201405084. [DOI] [PubMed] [Google Scholar]
- Baek J. Y.; Kwon H.-W.; Myung S. J.; Park J. J.; Kim M. Y.; Rathwell D. C.; Jeon H. B.; Seeberger P. H.; Kim K. S. Directing effect by remote electron-withdrawing protecting groups at O-3 or O-4 position of donors in glucosylations and galactosylations. Tetrahedron 2015, 71 (33), 5315–5320. 10.1016/j.tet.2015.06.014. [DOI] [Google Scholar]
- Nigudkar S. S.; Demchenko A. V. Stereocontrolled 1, 2-cis glycosylation as the driving force of progress in synthetic carbohydrate chemistry. Chem. Sci. 2015, 6 (5), 2687–2704. 10.1039/C5SC00280J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarova B. S.; Tsvetkov Y. E.; Nifantiev N. E. Design of α-selective glycopyranosyl donors relying on remote anchimeric assistance. Chem. Rec. 2016, 16 (1), 488–506. 10.1002/tcr.201500245. [DOI] [PubMed] [Google Scholar]
- Park Y.; Harper K. C.; Kuhl N.; Kwan E. E.; Liu R. Y.; Jacobsen E. N. Macrocyclic bis-thioureas catalyze stereospecific glycosylation reactions. Science 2017, 355 (6321), 162–166. 10.1126/science.aal1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S.; Moon S.; Hentschel F.; Gilmore K.; Seeberger P. H. An empirical understanding of the glycosylation reaction. J. Am. Chem. Soc. 2018, 140 (38), 11942–11953. 10.1021/jacs.8b04525. [DOI] [PubMed] [Google Scholar]
- Wang L.; Overkleeft H. S.; van der Marel G. A.; Codée J. D. Reagent controlled stereoselective synthesis of α-glucans. J. Am. Chem. Soc. 2018, 140 (13), 4632–4638. 10.1021/jacs.8b00669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vorm S.; van Hengst J. M.; Bakker M.; Overkleeft H. S.; van der Marel G. A.; Codée J. D. Mapping the relationship between glycosyl acceptor reactivity and glycosylation stereoselectivity. Angew. Chem. 2018, 130 (27), 8372–8376. 10.1002/ange.201802899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H.; Vu M. D.; Liu X.-W. Recent advances in reagent-controlled stereoselective/stereospecific glycosylation. Carbohydr. Res. 2019, 473, 72–81. 10.1016/j.carres.2018.10.006. [DOI] [PubMed] [Google Scholar]
- Hettikankanamalage A. A.; Lassfolk R.; Ekholm F. S.; Leino R.; Crich D. Mechanisms of stereodirecting participation and ester migration from near and far in glycosylation and related reactions. Chem. Rev. 2020, 120 (15), 7104–7151. 10.1021/acs.chemrev.0c00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.; Levi S. M.; Jacobsen E. N. Highly selective β-mannosylations and β-rhamnosylations catalyzed by bis-thiourea. J. Am. Chem. Soc. 2020, 142 (27), 11865–11872. 10.1021/jacs.0c04255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayfield A. B.; Metternich J. B.; Trotta A. H.; Jacobsen E. N. Stereospecific furanosylations catalyzed by bis-thiourea hydrogen-bond donors. J. Am. Chem. Soc. 2020, 142 (8), 4061–4069. 10.1021/jacs.0c00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damager I.; Olsen C. E.; Møller B. L.; Motawia M. S. Chemical synthesis of 6‴-α-maltotriosyl-maltohexaose as substrate for enzymes in starch biosynthesis and degradation. Carbohydr. Res. 1999, 320 (1–2), 19–30. 10.1016/S0008-6215(99)00131-7. [DOI] [PubMed] [Google Scholar]
- Damager I.; Jensen M. T.; Olsen C. E.; Blennow A.; Møller B. L.; Svensson B.; Motawia M. S. Chemical synthesis of a dual branched malto-decaose: A potential substrate for α-amylases. ChemBioChem 2005, 6 (7), 1224–1233. 10.1002/cbic.200400449. [DOI] [PubMed] [Google Scholar]
- Joseph A. A.; Pardo-Vargas A.; Seeberger P. H. Total synthesis of polysaccharides by automated glycan assembly. J. Am. Chem. Soc. 2020, 142 (19), 8561–8564. 10.1021/jacs.0c00751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plante O. J.; Palmacci E. R.; Seeberger P. H. Automated Solid-Phase Synthesis of Oligosaccharides. Science 2001, 291 (5508), 1523–1527. 10.1126/science.1057324. [DOI] [PubMed] [Google Scholar]
- Delbianco M.; Kononov A.; Poveda A.; Yu Y.; Diercks T.; Jiménez-Barbero J.; Seeberger P. H. Well-defined oligo-and polysaccharides as ideal probes for structural studies. J. Am. Chem. Soc. 2018, 140 (16), 5421–5426. 10.1021/jacs.8b00254. [DOI] [PubMed] [Google Scholar]
- Guberman M. n.; Seeberger P. H. Automated glycan assembly: A perspective. J. Am. Chem. Soc. 2019, 141 (14), 5581–5592. 10.1021/jacs.9b00638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guberman M.; Bräutigam M.; Seeberger P. H. Automated glycan assembly of Lewis type I and II oligosaccharide antigens. Chem. Sci. 2019, 10 (21), 5634–5640. 10.1039/C9SC00768G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Mai Hoang K.; Pardo-Vargas A.; Zhu Y.; Yu Y.; Loria M.; Delbianco M.; Seeberger P. H. Traceless photolabile linker expedites the chemical synthesis of complex oligosaccharides by automated glycan assembly. J. Am. Chem. Soc. 2019, 141 (22), 9079–9086. 10.1021/jacs.9b03769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo-Vargas A.; Bharate P.; Delbianco M.; Seeberger P. H. Automated glycan assembly of arabinomannan oligosaccharides from Mycobacterium tuberculosis. Beilstein J. Org. Chem. 2019, 15 (1), 2936–2940. 10.3762/bjoc.15.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Tyrikos-Ergas T.; Schiefelbein K.; Grafmüller A.; Seeberger P. H.; Delbianco M. Automated access to well-defined ionic oligosaccharides. Org. Biomol. Chem. 2020, 18 (7), 1349–1353. 10.1039/D0OB00137F. [DOI] [PubMed] [Google Scholar]
- Tyrikos-Ergas T.; Bordoni V.; Fittolani G.; Chaube M.; Grafmüller A.; Seeberger P. H.; Delbianco M. Systematic structural characterization of chitooligosaccharides enabled by Automated Glycan Assembly. Chem. - Eur. J. 2021, 27 (7), 2321–2325. 10.1002/chem.202005228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeberger P. H. The logic of automated glycan assembly. Acc. Chem. Res. 2015, 48 (5), 1450–1463. 10.1021/ar5004362. [DOI] [PubMed] [Google Scholar]
- Werz D. B.; Castagner B.; Seeberger P. H. Automated synthesis of the tumor-associated carbohydrate antigens Gb-3 and Globo-H: incorporation of α-galactosidic linkages. J. Am. Chem. Soc. 2007, 129 (10), 2770–2771. 10.1021/ja069218x. [DOI] [PubMed] [Google Scholar]
- Walvoort M. T.; van den Elst H.; Plante O. J.; Kröck L.; Seeberger P. H.; Overkleeft H. S.; van der Marel G. A.; Codée J. D. Automated solid-phase synthesis of β-mannuronic acid alginates. Angew. Chem., Int. Ed. 2012, 51 (18), 4393–4396. 10.1002/anie.201108744. [DOI] [PubMed] [Google Scholar]
- Hahm H. S.; Hurevich M.; Seeberger P. H. Automated assembly of oligosaccharides containing multiple cis-glycosidic linkages. Nat. Commun. 2016, 7 (1), 1–8. 10.1038/ncomms12482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boons G.-J.; Bowers S.; Coe D. M. Trityl ethers in oligosaccharide synthesis: A novel strategy for the convergent assembly of oligosaccharides. Tetrahedron Lett. 1997, 38 (21), 3773–3776. 10.1016/S0040-4039(97)00732-6. [DOI] [Google Scholar]
- Elferink H.; Severijnen M. E.; Martens J.; Mensink R. A.; Berden G.; Oomens J.; Rutjes F. P.; Rijs A. M.; Boltje T. J. Direct experimental characterization of glycosyl cations by infrared ion spectroscopy. J. Am. Chem. Soc. 2018, 140 (19), 6034–6038. 10.1021/jacs.8b01236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marianski M.; Mucha E.; Greis K.; Moon S.; Pardo A.; Kirschbaum C.; Thomas D. A.; Meijer G.; Helden G.; Gilmore K.; Seeberger P. H.; Pagel K. Remote participation during glycosylation reactions of galactose building blocks: direct evidence from cryogenic vibrational spectroscopy. Angew. Chem., Int. Ed. 2020, 59 (15), 6166–6171. 10.1002/anie.201916245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen T.; Elferink H.; van Hengst J. M. A.; Houthuijs K. J.; Remmerswaal W. A.; Kromm A.; Berden G.; van der Vorm S.; Rijs A. M.; Overkleeft H. S.; Filippov D. V.; Rutjes F. P. J. T.; van der Marel G. A.; Martens J.; Oomens J.; Codee J. D. C.; Boltje T. J. Characterization of glycosyl dioxolenium ions and their role in glycosylation reactions. Nat. Commun. 2020, 11 (1), 1–9. 10.1038/s41467-020-16362-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crich D. En route to the transformation of glycoscience: A chemist’s perspective on internal and external crossroads in glycochemistry. J. Am. Chem. Soc. 2021, 143 (1), 17–34. 10.1021/jacs.0c11106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.; Gim S.; Kim D.; Arnon Z. A.; Gazit E.; Seeberger P. H.; Delbianco M. Oligosaccharides Self-Assemble and Show Intrinsic Optical Properties. J. Am. Chem. Soc. 2019, 141 (12), 4833–4838. 10.1021/jacs.8b11882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q.; Zhang H.; Zhang Y.; Zhou S.; Gao J. Stereoselective synthesis of a branched α-decaglucan. Org. Biomol. Chem. 2020, 18 (33), 6549–6557. 10.1039/D0OB01402H. [DOI] [PubMed] [Google Scholar]
- Shadrick M.; Singh Y.; Demchenko A. V. Stereocontrolled α-galactosylation under cooperative catalysis. J. Org. Chem. 2020, 85 (24), 15936–15944. 10.1021/acs.joc.0c01279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teitelbaum R. C.; Ruby S. L.; Marks T. J. On the structure of starch-iodine. J. Am. Chem. Soc. 1978, 100 (10), 3215–3217. 10.1021/ja00478a045. [DOI] [Google Scholar]
- Madhu S.; Evans H. A.; Doan-Nguyen V. V.; Labram J. G.; Wu G.; Chabinyc M. L.; Seshadri R.; Wudl F. Infinite polyiodide chains in the pyrroloperylene–iodine complex: insights into the starch–iodine and perylene–iodine complexes. Angew. Chem., Int. Ed. 2016, 55 (28), 8032–8035. 10.1002/anie.201601585. [DOI] [PubMed] [Google Scholar]
- Hinrichs W.; Büttner G.; Steifa M.; Betzel C.; Zabel V.; Pfannemüller B.; Saenger W. An amylose antiparallel double helix at atomic resolution. Science 1987, 238 (4824), 205–208. 10.1126/science.238.4824.205. [DOI] [PubMed] [Google Scholar]
- Fittolani G.; Seeberger P. H.; Delbianco M. Helical polysaccharides. Pept. Sci. 2020, 112 (1), e24124. 10.1002/pep2.24124. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.