

Abstract
Simple Summary
The tumor suppressor protein p53 is frequently mutated in human cancers, and its reactivation represents an encouraging hope for precision anticancer therapy. Despite this, the clinical use of compounds capable of restoring the wild-type-like function to mutant p53 (mutp53) is not yet a reality. A more detailed understanding of the structural features of p53 mutations and the molecular mechanisms of mutp53 reactivators is critical to drive an efficient translation of these compounds into the clinic. In addition, this knowledge will provide insights into wild-type and mutp53 pharmacology at molecular levels, fostering the design and development of improved mutp53-targeted therapies to halt cancer.
Abstract
p53 is a transcription factor with a pivotal role in cell homeostasis and fate. Its impairment is a major event in tumor onset and development. In fact, about half of human cancers bear TP53 mutations that not only halt the normal function of p53, but also may acquire oncogenic gain of functions that favor tumorigenesis. Although considered undruggable for a long time, evidence has proven the capability of many compounds to restore a wild-type (wt)-like function to mutant p53 (mutp53). However, they have not reached the clinic to date. Structural studies have strongly contributed to the knowledge about p53 structure, stability, dynamics, function, and regulation. Importantly, they have afforded relevant insights into wt and mutp53 pharmacology at molecular levels, fostering the design and development of p53-targeted anticancer therapies. Herein, we provide an integrated view of mutp53 regulation, particularly focusing on mutp53 structural traits and on targeting agents capable of its reactivation, including their biological, biochemical and biophysical features. With this, we expect to pave the way for the development of improved small molecules that may advance precision cancer therapy by targeting p53.
Keywords: mutant p53, reactivators, targeted anticancer drugs, transcription factor
1. Introduction
The p53 protein belongs to a family of transcription factors, alongside p63 and p73, whose full activity is essential for the prevention of tumor onset and development. p53 is the central hub of a complex and intricate network, regulating major signaling and cell fate decision pathways. It senses diverse physical or chemical distress cellular signals, such as DNA damage, that eventually lead to its activation, stabilization, and accumulation in the cell [1] (Figure 1). Once active, p53 transcriptionally regulates many genes involved in major cellular processes, including cell cycle arrest, senescence, DNA damage repair, metabolic adaptation, and apoptosis [2] (Figure 1). Importantly, p53 activity is tightly controlled by a complex feedback-regulated network involving endogenous negative regulators (e.g., Murine Double Minute 2, MDM2) as well as by post-translational modifications (PTMs) and by interaction with distinct signaling proteins [3,4,5] (Figure 1).
Figure 1.

p53 signaling pathway. In unstressed conditions, p53 has a short half-life, being normally kept at low levels by MDM2, an E3 ubiquitin ligase that is itself a transcriptional target of p53, inhibiting p53 transcriptional activity and targeting it for proteasomal degradation via polyubiquitination (Ub). p53 is activated by stress stimuli, such as DNA damage, which activate the upstream kinases ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related protein (ATR), leading to p53 phosphorylation by checkpoint kinase (CHK)1 and CHK2. Other cellular stresses include hypoxia, nutrient deprivation, or oncogene activation, which dislocate p53 from the regulation of inhibitors (e.g., MDM2) through post-translational modifications (PTM), like phosphorylation (P) and acetylation (Ac), inducing stabilization, rapid accumulation, and activation of p53. Molecular chaperones, including Heat Shock Proteins (HSPs), through transient cyclic bind-unwind-release reactions, assist p53 to adopt its proper tertiary and quaternary structure and carry out its tumor suppressor activities. Depending on the stimulus, cell context, or PTM pattern, active p53 transcriptionally regulates the expression of several target genes involved in the control of major cellular processes, including cell cycle, apoptosis, DNA repair, autophagy, senescence, metabolism, metastasis, and angiogenesis. p53 also plays transcription-independent mitochondrial roles, as it can directly bind to BAX/BAK, resulting in their activation and initiation of apoptosis; p53 can also bind to BCL-2/BCL-xL, inhibiting their anti-apoptotic functions, through the disruption of inhibitory complexes between BCL-xL/BCL-2 with BAK/BAX, which results in mitochondrial outer membrane permeabilization (MOMP), cytochrome c (cyt c) release, and subsequent apoptosis.
The cell fate specified by p53 activation is context- and tissue-dependent, being mainly related to the nature of cellular stress [1]. In case of mild DNA damage, p53 drives cell cycle arrest for DNA repair, while under severe and irreparable DNA damage, p53 stimulates apoptotic or senescence programs. Other crucial factors, including the expression levels and PTM status of p53, cellular localization, co-factor enrollment, and the architecture of the promoters of target genes, also dictate the type of p53-mediated cellular responses based on the transcription of different subsets of p53 effectors [6,7].
The impairment of p53 tumor suppressor functions, either by mutations in the TP53 gene (which encodes the p53 protein) or by other abnormalities in the p53 pathway, is a common event among human cancers. Therefore, targeting p53 constitutes an appealing anticancer therapeutic strategy. However, since its discovery, p53 has represented a challenging target for drug discovery, being frequently considered as “undruggable”. In fact, wild-type (wt) p53 and most p53 mutant (mutp53) forms lack binding pockets or allosteric sites, other than the DNA-binding groove itself, hindering rational drug design [8]. Importantly, over 2000 mutp53 forms can be found in human tumors, with differences in their structure, stability, and biological functions, making it virtually impossible to target all of them with a single drug [9,10]. Nonetheless, in vitro and in vivo evidence has proven that restoration of mutp53, with re-establishment of wt-like activity, elicits impressive tumor regression and cell death [11,12,13,14].
This review addresses the most relevant achievements in the functional and structural elucidation of mutp53 as well as of the mutp53-targeting agents reported to date. With this systematization, we expect to bring new insights into mutp53 pharmacology, aiming at the development of improved molecules capable of restoring wt-like properties to mutp53.
2. p53 Structure and DNA Recognition
Structural studies with full-length p53 are challenging. Indeed, its flexibility, labile stability, and proneness to aggregation have hampered crystallographic studies. Its molecular weight represents a limitation for studies using conventional nuclear magnetic resonance (NMR) spectroscopy. On the other hand, p53 size is at the limits for cryo-electron microscopy (EM) methodology [15]. To overcome these limitations, a combined multi-technique approach was followed to construct a jigsaw from individual domain data. For this, specific recombinant p53 domains were produced by bioengineering techniques and analyzed by X-ray crystallography and NMR spectroscopy. In addition, some studies to check the spatial 3D arrangement of folded domains were pursued with the full-length protein, using suitable methodologies such as small angle X-ray scattering (SAXS), cryo-EM, and fluorescence resonance energy transfer (FRET) [15,16]. NMR spectroscopy proved to be important for understanding protein-protein and protein-DNA interactions in solution [17]. To complement these studies, electrophoretic mobility shift assay [18], protein microarrays, transactivation assays, fluorescence anisotropy, and isothermal calorimetric [18] titrations, among others, have also been used to unveil binding features and to characterize affinities for DNA and other proteins, peptides, or small molecules [17,19,20,21,22]. Recombinant p53 proteins with specific mutations (naturally occurring alone or combined with second-site mutations that can rescue DNA binding ability) have also contributed to the study of the relevance of specific amino acid residues in p53 stability and activity [23,24,25,26]. Furthermore, computational studies, such as molecular superposition and dynamics, have been used for predictive and comparison purposes in the structure-function relationship [15].
Active p53 is a homotetramer of four identical chains of 393 amino acids. Each monomer presents a modular structure divided into different domains: an acidic disordered N-terminal region, comprising the transactivation domain (TAD) and the proline-rich region (PRR); a central core sequence-specific DNA-binding domain (DBD); a C-terminal region, encompassing an oligomerization domain (OD) and a disordered regulatory domain (CRD) [15] (Figure 2).
Figure 2.

p53 structural domains with overview of post-translational modifications, interacting proteins, and most frequent TP53 missense mutations in human cancers. (A) Each p53 domain (transactivation domain, TAD; proline-rich region, PRR; DNA-binding domain, DBD; oligomerization domain, OD; C-terminal regulatory domain, CRD) is target of different post-translational modifications (PTMs; alone or concomitant) that regulate p53 function. p53 interacts directly with different PTM enzymes, transcription co-factors, and other nuclear or cytoplasmic proteins (some examples are depicted in the white boxes, being associated with a p53 interacting domain) that modulate p53 function. Image based on [27,28,29,30]. (B) Graphic depicting TP53 missense mutations relative frequency per codon (DBD encoding codons are highlighted in fading blue box, residues 94–292). Highlighted in green, yellow, orange, red, and dark-red are codons 175, 245, 248, 249, 273, and 282, hotspots for the highest incidence, and codons 280 and 220. These codons code for critical amino acid residues for p53 structure and function. (Graph constructed with GraphPad 7 and data from IARC TP53 Database, R20, July 2019 [31]).
In solution, using SAXS, NMR, and EM techniques, it was possible to understand that full-length p53 forms a tetramer with an opened, cross-shaped structure, having the ODs at its center and a pair of loosely coupled DBD dimers at the ends, which are accessible to bind to cognate DNA and partner proteins. Upon DNA binding, the structure closes around DNA and becomes more compact [17,32,33] (Figure 3A). p53 is a biologically active transcription factor as a homotetramer (assembled as a dimer of dimers; p53 dimers are formed co-translationally [34]), and DNA recognition occurs upon a cooperative binding process [35]. This means that the formation of DBD-DBD dimers interaction followed by the tetramerization through the ODs is favored in the presence of DNA, and both protein-protein and protein-DNA interactions contribute to the overall complex stability [36].
Figure 3.

Representation of p53 quaternary structure in solution, structural insights on p53 DBD binding to DNA, and p53 specific DNA-binding modes. (A) Spatial conformations of open unbound and closed/compacted bound p53 tetramer to DNA response element (RE) sequence. Pink and orange protein-like cartoons represent the DBDs (each color for one dimer), and sticks represent the respective ODs. The disordered N-terminal and C-terminal are depicted in yellow and blue, respectively. Representation is based on NMR, SAXS, and EM experimental studies [17,32,33]. (B) The 3D model of the DBD binding to cognate DNA sequence (PDB ID code: 1TUP, [42]) highlights the structural organization in helixes, loops, and strands in the β-sandwich. Furthermore, it depicts the amino acid residues involved in DNA recognition, establishing interactions with DNA, specifically K120 (L1), S241 and R248 (L3), R273 (S10), A276, C277, and R280 (H2). Other residues are also key partners for the correct binding positioning or structural stability, such as R175 (L2), Y220 (S7–S8 turn), G245 (L3), R249 (L3), and R282 (H2). Other regions of the DBD that are important for the assembly in dimers, namely H1, L2, and L3, are highlighted by a gray ellipse. Image created using PyMOL software [44]. H—helix; L—loop; S—strand. Image based on [15]. (C) p53 binding to three pentameric DNA sequences belonging to decameric half-sites of the consensus region of p53 target genes. The side chain of residue R280 is a steady point of contact between p53 and DNA, making two invariant hydrogen bonds to the conserved guanine base (G7) in the DNA major groove. The residue R273 anchors p53 to DNA backbone phosphates (grey pipe) in the central region of each half-site. The side chain of residue S241 also makes invariant contacts to DNA backbone phosphates. The specific interactions established by residues K120, A276, and C277 dramatically change when the base pair, at positions 3 and 8 of the DNA half-site, is changed from G/C to A/T. When in presence of the G/C base pair, the residue K120 makes three (I) or two (II) hydrogen-bonds with successive guanine bases (G2 and G3); when in presence of A/T base pair (III), it establishes interactions with G2 and T8. Regarding positions 3 and 8, the residue C277 interacts with C8 in the case of the G/C base pair (I,II); in the case of the A/T base pair (III), it establishes van der Waals interactions with T8. The most marked binding difference is observed with the residue A276: the interaction between its side chain and the DNA base only occurs with the A/T base pair (III), with a hydrophobic interaction occurring among the methyl groups of A276 and T8. The doublet central DNA half-site A/T base pairs (positions 5 and 6) are crucial in water-mediated interactions, playing an essential role in the cooperative binding of the core dimer to its DNA half-site through minor groove hydration. This interaction is directly mediated by the side chain of residue R248 (I) or indirectly via water molecules (II,III). The base pairs at positions 1 and 10 do not interfere with protein-DNA complex stability. Image based on [36,45].
p53 exerts its transcriptional activity through recognition of a short specific DNA sequence in the target genes’ promoters, called the response element (RE) [36,37]. The REs enable a coordinated and flexible response, given the existence of many variations of the consensus sequence. Thus, upon stress stimuli, p53 can bind to multiple scattered REs in the genome, eliciting the transcription of sets of genes and related cellular responses [38]. The p53 target RE binding site consists of two decameric motifs (half-sites) with the consensus sequence RRRCWWGYYY (R = A, G; W = A, T; Y = C, T); C4 and G7 positions should be conserved among the variant sequences [39,40]. The degenerate nature of the consensus already exemplifies why there are many variant sequences of REs among the different p53 target genes. In addition, mismatches from the consensus are frequent among established p53 REs at target gene promoters. Thus, there is a wide range in sequence-specific binding affinities, and this is correlated with variations in the protein-DNA contact geometry, providing insights into the mechanism of p53 function and regulation and the potential to elicit gene expression changes that are tailored to specific stress conditions through modulation of nuclear p53 protein levels or by specific PTMs or protein cofactors [36,41].
In 1994, Cho et al. reported for the first time the crystallographic model of p53 DBD with DNA at 2.2 Å [42]. The p53 DBD is an immunoglobulin-like central β-sandwich of two antiparallel β-sheets, providing the basic scaffold for the DNA-binding surface (Figure 3B). This surface is constituted by two large loops, L2 and L3, the latter intercalated by a short helix, H1, stabilized by a tetrahedral coordination of two residues of each loop to a zinc atom (L2–C176, H179; L3–C238, C242) and by a loop-sheet-helix (LSH) motif constituted by L1, β-strands S2 and S2′, the end of β-strand S10 and C-terminal of H2 (Figure 3B).
The LSH motif makes specific contacts with the major groove of the target DNA, through hydrogen bonds, salt-bridges, and structural water molecules in a tight network of hydrogen bonds that ensures the correct orientation of each amino acid residue. In the DNA major groove, the side chains of residues K120 (L1), A276, C277, and R280 (H2) interact with DNA bases via hydrogen bonds, and the side chain of residue R273 (S10) and the backbone amide of residues K120 and A276 establish interactions with the DNA backbone phosphates via salt bridge interactions (Figure 3B,C). The L3 contains the residues S241 and R248, which have contact with DNA backbone phosphates in the DNA minor groove; R248 is essential for DNA recognition (Figure 3B,C) [42,43]. Other residues also play an important role in stabilizing the p53 DNA-binding surface, such as R175 (L2), G245 (L3), R249 (L3), and R282 (H2) [42,43] (Figure 3B,C). The finer details of DNA-contact geometry show an interesting variation that depends on the DNA sequence [36]. A description of the known modes of binding of each p53 residue to the DNA consensus region was provided by Kytainer et al. [36] (Figure 3C).
Each half-site is recognized by a symmetrical dimer of two p53 DBDs with a relatively small, self-complementary protein-protein interface with around 600 Å2 of buried surface area [26]. The interface is stabilized by hydrophobic, water-mediated polar interactions and hydrogen bonds between the zinc regions, involving H1, L2, and L3, of the two p53 DBDs (Figure 3B). There are two buried water molecules that provide an internal hydrogen-bonding network as a central anchor, linking the two zinc regions, which support H1 and L3 configuration. A nonpolar interaction shell is formed by surface residues from H1 and L3 hairpin via residues P177, H178, M243, and G244. Next to this hydrophobic area, there are two hydrophilic networks that contribute to its stability, one far from DNA, involving charged residues from the two monomers (specifically R174, E180, R181, and several water molecules), and the second one near to DNA, with a first-shell hydration of the protein surface facing DNA, via N239, S241, and R248 side chains [42].
The interaction of protein-DNA is the ultimate contribution to dimer stabilization. Dimer protein-protein interactions may also contribute to DBD integrity, considering the bidentated salt bridges between the polar and charged surface residues D184 with R175, and R249 with E171 on opposite sides of each monomer [36,46]. Kantarci et al., in addition to acknowledging that both p53 DBD monomers in the dimer establish the same interactions with the DNA RE, advanced the hypothesis that, in a dimer, the p53 DBD interactions with DNA were stronger for one monomer than for the other [47]. In fact, this has also been supported by the spatial rearrangement of the tetramer complexed with DNA verified in cryo-EM works [17,32,33].
3. Dynamics and Regulation of p53
Although intrinsic domain disorder hampers p53 studies, it allows a structural plasticity, reflected in binding promiscuity observed by p53 interaction with a wide range of target DNA sequences and interacting proteins [45] (Figure 2A and Figure 3C). Additionally, intrinsic disorder is directly correlated with low thermodynamic and kinetic stability: in fact, p53 half-life time at body temperature is about 9 min, with rapid cycles between folded and unfolded states [19,48,49]. These features have been postulated as an evolutionary advantage, providing a tight regulation of functionally active p53 cellular levels [45]. Additionally, in normal cells, under no stress conditions, p53 levels are low, as it is negatively regulated by different proteins, including its major endogenous inhibitor MDM2 (Figure 1). This protein not only prevents DNA recognition by directly binding to p53, but also targets p53 for proteasomal degradation via polyubiquitination or nuclear export via monoubiquitination by its E3 ligase activity. Under stress stimuli, p53 is displaced from MDM2 negative regulation, with subsequent stabilization and activation of its transcriptional functions [50] (Figure 1). Furthermore, heat shock proteins (HSPs), such as HSP40, HSP70, and HSP90, act like chaperones, transiently binding to free p53 and promoting its correct folding upon their dissociation for DNA RE binding [28,51,52,53] (Figure 1).
Besides p53 protein stabilization, several other factors seem to reinforce the fine tuning of stimuli-dependent p53 regulation and cell fate decision, including the protein level dynamics, cellular localization, and PTMs [54] (Figure 1 and Figure 2A). Methylated or ubiquitinated p53 correlates with unstressed cells. In contrast, phosphorylation, acetylation, neddylation, or sumoylation are more frequent in cells upon DNA damage, increasing p53 DNA-binding affinity to target genes [29,55,56]. One PTM may influence the occurrence of another, unlocking additional layers of regulation, affecting protein stability, function, protein-protein interaction, and the recruitment of transcriptional co-factors and machinery, favoring DNA-binding toward specific target genes [57,58].
4. Mutant p53 Functions in Tumorigenesis
TP53 mutations occur in more than half of human cancers, with colorectal, head and neck, esophagus, female genital organs, and lung cancers exhibiting the highest prevalence (37–43%; International Agency for Research on Cancer (IARC) TP53 Database, R20, July 2019) [31]. TP53 mutations occur in both germline (associated with Li-Fraumeni syndrome) and sporadic contexts and can be found throughout the whole gene [31] (Figure 2B). Missense mutations are the most frequent alterations in TP53 (over 75%) and mainly occur in the DBD region (over 80%) [59]. In fact, in the p53 DBD, there are six hotspot missense mutations in codons 175, 245, 248, 249, 273, and 282, with high clinical significance [31] (Figure 2B). As previously mentioned, these codons code for residues with important roles in p53 structure and function.
TP53 mutations in the TAD are associated with the loss of transactivation of specific genes, such as CDKN1A, hence blocking the capacity of inducing cell cycle arrest without compromising apoptosis. TP53 mutations in the OD often halt tetramerization, translating into loss of function (LOF). In addition, TP53 mutations in the DBD exhibit diverse degrees of functionality and, consequently, different pathological relevance (reviewed in [60]). Besides being primarily associated with LOF [60] (Figure 4A), TP53 mutations in the DBD commonly occur in a single allele. As such, first-stage tumors are heterozygous, expressing both wt and mutp53. Although wtp53 is still expressed, a dominant negative effect (DNE) [18] of mutp53 over wtp53 is observable and can be explained by the formation of heterotetramers (wtp53 dimer plus mutp53 dimer) devoid of transcriptional activity [60] (Figure 4B). During tumor progression, the loss of heterozygosity is commonly observed and is associated with gain of function (GOF) of sporadic or inherited TP53 mutations [50] (Figure 4C). GOF can be manifested through mutp53 interaction with diverse transcriptional factors or co-factors. For instance, mutp53 heterooligomerizes with p63 or p73, blocking their tumor suppressor activity by inhibiting their transcriptional activity, or stimulating the transcription of non-canonical genes [61,62]. Mutp53 GOF may also occur through association of mutp53 with C-ets-1/2 (Ets1/2), yes-associated protein (YAP1), peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 (Pin1), or promyelocytic leukemia protein (PML), with transcriptional enhancement of their set of target genes [61,62]. Both scenarios result in altered gene expression patterns that contribute to survival, tumor progression, and more aggressive phenotypes [62]. GOF has been strongly associated with some specific mutp53 forms, such as R175H, R248W, R273H, and R280K (reviewed in [60,62,63]).
Figure 4.

Functional activities of mutp53 in cancer. (A) In general, p53 mutations lead to loss of DNA-binding ability and impairment of the p53 response (loss of function, LOF). (B) A dominant negative effect [18] of mutp53 over wtp53 occurs through the formation of heterotetramers and supramolecular aggregates with wtp53. (C) Mutp53 gain of function (GOF) activities impact multiple hallmarks of cancer cell biology, affecting the chromatin structure, transcriptional regulation, and microRNA biogenesis, shaping the proteome, and rewiring tumor cell metabolic pathways. It also encompasses cytoplasmic functions and cell-extrinsic effects, namely affecting tumor microenvironment and the inflammatory response. Oncogenic GOF of mutp53, driving tumor development and dissemination, relies on the direct interaction with transcription factors (TFs, grey boxes) or co-factors and other protein effectors (yellow boxes), altering their activity, or on the transcriptional modulation of target genes (green boxes). Data retrieved from and based on [8,62,72].
It is worth noting that mutp53 is expressed in cancer cells at higher levels than wtp53 in normal cells, indicating that mutp53 is somehow more stable than wtp53 and accumulates in tumor cells [64]. Initially, it was thought that the MDM2 regulatory axis would not work on mutp53 once canonical genes transcription was halted [62]. Nevertheless, MDM2 may be expressed by other pathways [65], and evidence has suggested that mutp53 levels can be controlled by MDM2 in normal tissues, but not in tumor tissues [66]. This raised the possibility of additional events in tumorigenesis responsible for mutp53 accumulation. Indeed, in cancer cells, the occurrence of mutp53 is also associated with increased levels of HSP70 or HSP90, which often bind to and stabilize mutp53 and/or participate in aggregosomes [67]. This impairs the activity of MDM2 or other E3 ligase regulatory proteins, like the carboxy terminus of HSP70-interacting protein [68], culminating in mutp53 accumulation and potentiation of its GOF [53]. Indeed, distinct mutp53 forms have been described as aggregation-prone and are localized in the cytoplasm and perinuclear region rather than in the nucleus as functional wtp53 [69]. These aggregosomes may have a prion-like amyloid behavior [70] and have also been associated with DNE and GOF events by sequestration of wtp53, TAp63, and TAp73. Co-aggregation is possible, since these isoforms share highly conserved aggregating sequences in the same structural motif [69,71].
Deleterious Effects of Mutant p53 on DNA Binding and Protein Stability
Through the crystallographic model of wtp53 DBD bound to DNA, Cho et al. unveiled the deleterious effects of common cancer mutations [42,45]. Subsequent studies have been pursued to further understand the impact of mutations on p53 structure, stability, and function [45]. In this context, based on the known p53 intrinsic instability, a superstable quadruple mutp53 DBD was developed (M133L, V120A, N239Y, N268D) and used for structural studies (Table 1), enabling its handling without compromising its function [25,73]. These mutations maintain the basic structural scaffold and turn p53 DBD structural framework more rigid, increasing its thermodynamic stability by 2.6 kcal/mol (compared to wtp53 DBD) [25]. The most frequent TP53 mutations occur in highly conserved sequence regions and coincide with key amino acid residues for DNA recognition and structural stability [74,75] (Figure 2B and Figure 3B). Indeed, single amino acid mutations in the p53 DBD can result in the removal of DNA-contact residues (contact mutp53; e.g., R248Q, R248W, R273H, R273C, R280K) or conformational changes in different parts of the DBD, including structural rearrangements on the DNA-binding surface, creation of internal cavities, or formation of surface crevices in regions remote from the DNA-binding site (structural mutp53; e.g., G245S, G245D, R249S, R175H, Y220C, R282W) [45].
Table 1.
Impact of mutations on p53 DBD structural features and DNA recognition. Description of main physicochemical amino acid changes that influence the surrounding chemical environment, affecting interactions and structural rearrangements; consequences for mutp53 thermal stability, folding, and DNA-binding ability.
| Variable (DBD) |
Amino Acid Location and Biochemical Mutation Type | Mutp53 Structural Modifications | Thermo-Dynamic Stability & | Estimated Folded Protein at 37 °C # | DNA-Binding Affinity at 20 °C # | Structural Elucidation of p53 DBD (PDB ID Code Eample) | Ref. |
|---|---|---|---|---|---|---|---|
| Wtp53 | |||||||
| with DNA | NA | NA | NA | NA | +++ | 1TUP *, 1TSR *, 2AC0, 2ADY, 2AHI, 2ATA | [20,36,42] |
| w/o DNA | NA | NA | + | +++ | NA | 2OCJ, 1UOL * | [20,87] |
| Oncogenic mutp53 | |||||||
| DNA contact region | |||||||
| S241F | L3; uncharged polar to hydrophobic aromatic/small to large residue | Increase of hydrophobicity and steric clash halts DNA contacts (theoretical interpretation) | ND | ND | ND | ND | [45] |
| R248Q | L3; cationic to uncharged polar residue | Alteration of L3 conformation (NMR data) | +/− | +++ | − | ND | [20,76] |
| R248W | L3; cationic to hydrophobic aromatic bulky residue | Increase of hydrophobicity and steric clash halts DNA contacts (theoretical interpretation) | ND | ND | ND | ND | [45] |
| R273C | S10; cationic to uncharged polar/large to small residue | Alteration of hydrogen bonds network in DNA-binding surface affecting buttressing residues and DNA contacts | ND | ND | ND | 2J20 *, 4IBQ | [24,79] |
| R273H | S10; aliphatic to aromatic/large to bulky-small residue | Alteration of hydrogen bonds network in DNA-binding surface affecting buttressing residues and DNA contacts | + | +++ | − | 2BIM *, 4IBS, 4IJT | [20,24,48,78] |
| C277F | S10-H2 turn; uncharged polar to hydrophobic aromatic/small to large residue | Increase of hydrophobicity and steric clash halts DNA contacts (theoretical interpretation) | ND | ND | ND | ND | [45] |
| R280K | H2; large to small residue | Alteration of hydrogen bonds network in DNA-binding surface affecting buttressing residues and DNA contacts | ND | ND | − | 6FF9 | [45,80,81] |
| Structural—DNA region | |||||||
| F134L | S2′; aromatic to aliphatic residue | ND | − | − | − | ND | [20] |
| H168R | L2; aromatic to aliphatic/bulky-small to large residue | Alteration of L2 conformation | ND | ND | ND | 2BIN * | [78] |
| G245S | L3; small to large uncharged polar residue | Small distortion of L3/dimerization interface | +/− | +++ | + | 2J1Y * | [20,48,79] |
| G245D | L3; small to large anionic residue | Small distortion of L3 and LSH (in silico data) | ND | ND | ND | ND | [88] |
| R249S | L3; cationic to uncharged polar/large to small residue | Alteration of L3 conformation affecting R248-mediated DNA anchoring and of dimer interface; increased flexibility of the β-sandwich | +/− | +++ | − | 2BIO *, 3D05, 3DO6, 3DO7 | [20,48,78,82] |
| R282Q | H2; cationic to uncharged polar residue | Flexibility is decreased in L1 and increased in L3 | ND | ND | ND | 2PCX | [89] |
| R282W | H2; aliphatic to aromatic/cationic to hydorphobic bulky residue | Impaired LSH anchoring to β-sandwich, increase of L1 flexibility | − | − | ++ | 2J21 * | [20,48,79] |
| Structural—Zinc region | |||||||
| R175A | L2; cationic to hydrophobic/large to small residue | Interference with zinc-binding (smaller effect than R175H) (theoretical interpretation) | +/− | +++ | + | ND | [20,45] |
| R175H | L2; aliphatic to aromatic/large to bulky-small residue | Alteration of L2 and L3 conformation, loss of zinc-binding (theoretical interpretation) | − | − | − | ND | [20,45,48] |
| M237I | L3; large to small residue/decrease in atom electro-negativity | ND | − | +/− | − | ND | [20] |
| C242S | L3; decrease in atom electro-negativity | Zinc ligand substitution with loss of zinc coordination (theoretical interpretation) | − | +/− | − | ND | [20,45] |
| Structural—-sandwich | |||||||
| V143A | S3; large to small residue | Internal hydrophobic cavity | − | − | + | 2J1W * | [20,48,79] |
| L145Q | S3; hydrophobic to uncharged polar/small to large residue | -sheet and loop-sheet-helix motif destabilization (in silico data) | − | +/− | +/− | ND | [20,90] |
| P151S | S3/S4 turn; hydrophobic to uncharged polar residue | ND | − | − | +/− | ND | [20] |
| V157F | S4; aliphatic to aromatic/small to large-bulky residue | Internal hydrophobic cavity | − | − | ++ | 4KVP | [91] |
| I195T | S5; hydrophobic to uncharged polar/large to small residue | ND | − | − | − | ND | [20] |
| Y220C | S7–S8 turn; large to small residue | Hydrophobic crevice in the -sandwich surface at S7–S8 turn | − | − | +/− | 2JIX *, 6SHZ * | [20,79,84] |
| Y220H | S7–S8 turn; hydrophobic to cationic residue | Mild alteration of intermolecular interactions on -sandwich surface at S7–S8 turn, no crevice observed | ND | ND | ND | 6SI1 * | [84] |
| Y220N | S7–S8 turn; aromatic to aliphatic/bulky hydrophobic to uncharged polar residue | Hydrophobic crevice in the -sandwich surface at S7–S8 turn | ND | ND | ND | ND | [84] |
| Y220S | S7–S8 turn; aromatic to aliphatic/bulky hydrophobic to small uncharged polar residue | Hydrophobic crevice in the -sandwich surface at S7–S8 turn | ND | ND | ND | 6SI2 * | [84] |
| I232T | S8; hydrophobic to uncharged polar/large to small residue | ND | − | +/− | + | ND | [20] |
| I255F | S9; aliphatic to aromatic residue | ND | − | − | +/− | ND | [20] |
| F270C | S10; hydrophobic aromatic to uncharged polar/large to small residue | Internal hydrophobic cavity (theoretical interpretation) | − | − | +/− | ND | [20] |
| F270L | S10; aromatic to aliphatic/large to small residue | Internal hydrophobic cavity | ND | ND | ND | 2J1Z * | [79] |
* The protein has four stabilizing mutations (M133L, V203A, N239Y, N268D). & Difference in free energy of unfolding between wt- and mutp53 DBD at 10 °C (), which is extrapolated to 37 °C; free energy of wtp53 used as reference (0 kcal/mol); (+) stable protein ≤ 0.5 kcal/mol, (+/−) weakly destabilized protein 0.6–2.5 kcal/mol, (−) strongly destabilized protein ≥ 2.6 kcal/mol. # Relative folded protein and DNA-binding affinity: (+++) 100–85%, (++) 84–70%, (+) 69–55%, (+/−) 54–40%, (−) ≤ 39%. NA—not applicable; ND—not determined; w/o—without.
Mutp53 studies by X-ray crystallography and NMR have enlightened local structural changes and their impact on DNA recognition, which are summarized in Table 1. In particular, NMR and in silico simulation data have provided insights into structural changes of L3, with L2 rearrangements, caused by the change of arginine to glutamine in the contact mutp53 R248Q [76,77]. Although structural data is lacking, it is speculated that the substitution of the arginine for a tryptophan in contact mutp53 R248W will abolish the anchoring of p53 to DNA minor grooves due to the hydrophobicity and steric clash of tryptophan, preventing the establishment of hydrogen bonds with DNA [45]. The same is thought to occur in other contact mutp53 forms, such as S241F and C277F [45]. In contact mutp53 R273H and R273C, the histidine and cysteine residues, respectively, alter the hydrogen-bonding network in the DNA-binding surface, compromising the direct contacts to DNA backbone phosphates due to the shorter lateral chains, compared to arginine [24,78,79]. The contact mutp53 R280K exhibits loss of DNA-binding ability with impairment of canonical target genes transcription [80]. Although the switch from arginine to lysine is not markedly different, the lysine is shorter than arginine, and the absence of a guanidinium group affects the p53 DNA-binding ability. It is observed that lysine 280 halts the establishment of essential hydrogen-bonds to the DNA major groove and the contribution of the extended hydrogen-bonding network with other residues [81]. Conformational changes observed in the DNA-binding region of structural mutp53 G245S are small, with the overall conformation conserved [79]. Nevertheless, the serine side chain displaces a structural water molecule, leading to small structural shifts in the proximal residues. This is enough to disturb the protein-protein interface of the dimer, reducing DNA-binding affinity [79]. Conversely, the structural mutp53 R249S leads to a large conformational change of L3, affecting p53 DBD anchoring to the DNA minor groove via R248 contacts [78]. The guanidinium group of the R249 is responsible for stabilizing the L3 hairpin conformation by establishing one salt bridge interaction with D171 and hydrogen bonds with G245 and M246. When the arginine residue changes to serine (R249S), these interactions are abolished. Consequently, L3 acquires more flexibility along with M246 displacement elicited by M243 from its buried location within the hydrophobic zinc-cluster region, leading to the formation of a short α-helix. This significant conformational change ultimately displaces R248, halting mutp53 DNA interaction [78]. Additionally, NMR data on mutp53 R249S evidenced an increased flexibility of the β-sandwich close to the DNA-binding surface [82].
The residue R282 (H2 C-terminal) is responsible for maintaining the structural integrity of the LSH motif by packing H2 to the S2-S2′ β-hairpin, anchoring it to the protein core. Through the crystallographic model of structural mutp53 R282W, it was observed that the inclusion of a hydrophobic and bulky tryptophan residue compromised those anchoring interactions. Consequently, the L1 flexibility increases with displacement of the DNA-contact residue K120, affecting DNA recognition [83]. Structural mutp53 Y220C, Y220H, Y220N, and Y220S, although positioned away from the DNA-binding surface, impart deleterious effects on the overall protein stability [19,84]. The mutations occur in the periphery of the β-sandwich hydrophobic core, at the beginning of the loop that connects S7 and S8, leading to a mutation-induced crevice on the p53 surface in Y220C, Y220N, and Y220S [79,84]. As the imidazole group of mutant histidine 220 roughly superimposes with the phenol of wt tyrosine, the interactions are partially conserved without observation of crevice formation when comparing with the other Y220 mutants [84]. Other mutations in the β-sandwich may occur within its hydrophobic core, such as V143A and F270L. Valine and phenylalanine side chains form an integral part of the hydrophobic core network, and these mutations create internal cavities. Of note, these cavities occur without collapse of the surrounding structure guaranteed by other hydrophobic residues, but they cause a strong destabilization of the core domain [79].
Although no structural data of zinc region mutp53 R175H are available, it is possible to speculate why it abrogates p53 activity. Residue R175 (L2) is embedded between L2 and L3, next to the zinc cluster, and its guanidinium group is responsible for stabilizing this region by establishing hydrogen bonds with P191 and M237 and one salt bridge with D184. If the arginine is replaced by a shorter and bulkier histidine (R175H), structural distortions and interference with zinc binding are expected [45]. Such effects may justify the observed complete loss of DNA-binding ability by R175H [20,85]. In fact, for substitutions bearing smaller side chains, such in the case of R175A (not occurring in cancer), R175C, or R175L, residual binding [20] or wt-like activity [86] can be observed. Conversely, the introduction of large, bulky side chains (e.g., R175W and R175Y) abrogated the wt-like functions in cancer cells [86].
Regarding mutp53 stability, Bullock et al. have classified mutp53 DBD in accordance with its thermodynamic and kinetic stability and DNA-binding affinity, relating it to the mutation-specific local structural changes [19,20] (Table 1). The mutp53 stability will dictate the relative amount of folded and potentially functional protein under physiological conditions. It was observed that mutp53 has higher propensity to aggregate than wtp53, with at least 50% of mutp53 denaturated at physiological temperature [20]. For example, the contact mutp53 R273H does not significantly destabilize the DBD, but it halts DNA-binding ability (Table 1), since it is a crucial residue for DNA recognition [20]. Although considered a contact mutp53, R248Q decreases the thermodynamic stability by inducing a structural distortion [20,76,77] (Table 1). The structural distortion induced by mutp53 G245S and R249S slightly reduces protein thermal stability and halts DNA contacts, although G245S retains partial DNA-binding ability at sub-physiological conditions (20 °C) (Table 1). In addition, mutp53 R282W exhibits increased protein destabilization, but it retains partial DNA-binding ability at 20 °C (Table 1), given that most contact residues are undamaged for interaction [20] (Table 1). Similarly, in structural mutp53 Y220C or other β-sandwich mutants, despite the conservation of some DNA-binding affinity at 20 °C, the mutations dramatically decrease p53 stability (Table 1) with extensive protein unfolding at physiological temperature [20]. Among the recently described Y220 mutants, Y220H was the least destabilizing; nevertheless, all showed lower melting temperatures than wtp53, similar to Y220C, which suggests that these cancer mutp53 forms are also largely unfolded under physiological conditions [84]. This makes β-sandwich structural mutp53 temperature-dependent with respect to its DNA-binding ability [20]. Nevertheless, the same is not verified for the zinc region structural mutp53 R175H, C242S, or M237I, which are extensively denaturated independently of the temperature, thus reinforcing the importance of the zinc ion tethering for the DBD correct folding [20] (Table 1).
It was demonstrated that mutp53 DBD plus OD can lead to misfolding of the tetramer, but not of the monomer. Misfolding appears to involve the intramolecular association of DBD-DBD within the tetramer, promoted by destabilizing mutation in the DBD [21]. In addition, in the case of DNE, although there is a wtp53 dimer, the p53 DBD mutation in mutp53 dimer affects the overall tetramer stability, leading to the loss of binding cooperativity to DNA and subsequent impairment of transcriptional activity [34,35,45,92]. Moreover, mutations in the DBD not only affect DNA recognition, but may also interfere with binding to wtp53 target proteins. For instance, mutp53 forms bearing mutations that do not interfere with L2 and L3 conformation are still capable of interacting with TP53-binding protein 2 (53BP2); however mutp53 G245S impairs this interaction [45]. Importantly, mutp53 cellular half-life is not correlated with thermodynamic stability, as mutp53 tends to accumulate as previously mentioned [53,93].
Despite all the knowledge that has been generated around p53 structure and related topics, the elucidation of how several mutations impact on p53 conformation, stability, and function is still missing.
5. Targeting Mutant p53
The restoration of the activity to target proteins poses intrinsic difficulties as a therapeutic strategy when compared to target inhibition [94]. Indeed, regarding mutp53, it could be arguable whether the reestablishment of its tumor suppressor function would be sufficient to counteract a context of multiple oncogenic alterations, including expression of c-Myc, GTPase HRas (RAS), or phosphoinositide 3-kinase (PI3K) cancer drivers. However, multiple in vitro and in vivo studies have proven the concept that the reactivation of wt-like function to mutp53 can elicit cell death and halt tumor progression [50,94]. Although mutp53 has long been regarded as an undruggable target [95], some studies have shown that the insertion of artificial second-site mutations can alleviate the effect of inactivating TP53 mutations [78,96]. The purpose of these rescue mutations is to enable the establishment of new interactions with the DNA, favoring protein stability and correct folding or compensating for missing contacts caused by the original mutated residue [24,26,78,91,96,97,98]. Indeed, this provided the conceptual basis for the feasibility of mutp53 reactivation, by enhancing specific-sequence DNA-binding and transcription of wtp53 target genes. Another positive aspect of mutp53 as a therapeutic target is its high expression levels in cancer cells [99,100].
As formerly evidenced, p53 mutants are not all equal, differing on their structure, stability, and function. Contact mutants, for their structural similarity to wtp53, do not have well-defined hydrophobic pockets to be docked by small molecules [42,95]. Therefore, reactivation of contact mutp53 seems to be a daunting task, since the therapeutic strategy must rely on the introduction of extra interaction points to compensate for the missing DNA contacts [95]. Conversely, structural mutants are found to be kinetically and thermodynamically destabilized in a temperature-dependent (e.g., β-sandwich mutations) or temperature-independent (e.g., zinc region mutations) manner. For both structural mutant forms, small molecules acting as chaperones, increasing the level of correctly folded protein at physiological temperature, may be a feasible therapeutic strategy due to the conservation of the DNA-binding residues [20,95].
In the past two decades, consistent efforts from academic and industry research groups have led to the identification of several small molecules (and peptides) that stabilize p53 native conformation and restore sequence-specific DNA binding, rescuing wt-like transcriptional functions and ultimately resulting in cell death and tumor suppression (reviewed in [94,101]) (Table 2). Among these mutp53-targeting agents, two small molecule reactivators have entered clinical trials: PRIMA-1MET (APR-246; Phase III trials; NCT03745716 [102]) and COTI-2 (Phase I trial; NCT02433626 [103]).
Table 2.
Summary of reported mutp53-targeting agents. Agents are organized by their chemical class, discovery strategy, targeted mutp53 forms, mechanism of action, and main biophysical and cellular outcomes.
| Chemical Name (Class) | Discovery Strategy | Targeted * Mutp53 | Mechanism of Action | Observations | Ref. |
|---|---|---|---|---|---|
| Reactivators—Cysteine-targeting alkylation | |||||
|
CP-31398 (Styrylquinazoline) 
|
Chemical library; protein-based screening assay | V173A; S241F; R249S; R273H | Michael addition # Binding: ND |
Transcription of p53 target genes; p53-dependent and -independent in vitro antitumor activity | [104,105,106,107] |
|
MIRA-1 (Maleimide) 
|
Chemical library; cellular screening assay | R175H; P176Y/R248W; R248Q; R248W; R273H; R273H/P309S; R280K; R282W | Michael addition # Binding: ND |
Rescue of wt conformation to mutp53 R248W, R175H; restoration of DNA binding to mutp53 R175H, R248Q, P176Y/R248W, R280K, R282W; restoration of transcriptional activity to mutp53 R175H, R273H, R273H/P309S; p53-dependent and -independent in vitro and in vivo antitumor activity | [108] |
|
STIMA-1 (Styrylquinazoline) 
|
Chemical library; cellular screening assay | R175H; R273H | Michael addition # Binding: ND |
Restoration of DNA binding to mutp53 R175H; restoration of transcriptional activity to mutp53 R175H, R273H; p53-dependent and -independent in vitro antitumor activity | [107] |
|
KSS-9 (Piperlongumine derivative) 
|
Rational design | R175H | Michael addition #
Binding: ND |
Rescue of wt conformation; restoration of DNA binding to mutp53; transcription of p53 target genes; p53-dependent and -independent in vitro antitumor activity | [109] |
|
PRIMA-1 PRIMA-1MET (Quinuclidinone) 
|
Chemical library; cellular screening assay | R175H; R273H; D259Y/K286E; K286E; S241F; R273C; P223L/V274F | Metabolized to methylene quinuclidinone (active metabolite), Michael addition Binding: mutp53 R175H, R273H |
Enhanced thermal stability of wtp53, mutp53 R175H, R273H; rescue of wt conformation to mutp53 R175H; restoration of transcriptional activity to mutp53 R175H, R273H, D259Y/K286E, K286E, S241F, R273C, P223L/V274F; p53-dependent and -independent in vitro and in vivo antitumor activity | [110,111,112,113,114,115,116,117,118,119] |
|
PK11007 (Sulfonylpyrimidine) 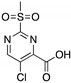
|
Chemical library; protein thermal stability-based screening assay | Y220C; V143A | Binds p53 by nucleophilic aromatic substitution Binding: mutp53 Y220C |
Enhanced thermal stability of mutp53; transcription of p53 target genes; p53-dependent and -independent in vitro antitumor activity | [120] |
|
HO-3867 (Diarylidenyl piperidone curcumin analogue) 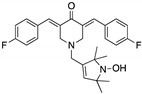
|
Chemical library; cellular screening assay | K132Q; R156P; Y163H; R175H; H193R; L194F; Y205F; P223L/V274F; C238Y; N239D; S241F; G245S; G245V; M246I; R248Q; R248W; R249S; R273H; C277F; R280K; E285K | Binds p53 by Michael addition Binding: mutp53 Y220C |
Rescue of wt conformation to mutp53 P223L/V274F, R273H, R280K; restoration of transcriptional activity to mutp53 K132Q, R156P, Y163H, R175H, H193R, L194F, Y205F, C238Y, N239D, S241F, G245S, G245V, M246I, R248W, R248Q, R249S, R273H, C277F, R280K, E285K; p53-dependent in vitro and in vivo antitumor activity | [121,122] |
| Thermal stabilizers | |||||
|
3-Benzoylacrylic acid (Benzoylacrylate) 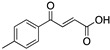
|
Chemical library; protein thermal stability-based screening assay | Y220C; R175H; G245D; R249S; R282W | Michael addition Binding: mutp53 Y220C |
Enhanced thermal stability of mutp53 Y220C, R175H, G245D, R249S, R282W; absence of in vitro antitumor activity evaluation | [123] |
| Reactivators—Zinc chelators | |||||
|
ZMC1/NSC319726 (Thiosemicarbazone) 
|
Database analysis; cellular screening assay | R175H; C176F; C238S; C242S; C242F; G245S | Zn2+ chelator Binding: mutp53 R175H |
Increased cellular zinc concentration; rescue of wt conformation to mutp53 R175H, C176F, C238S, C242S; restoration of transcriptional activity to mutp53 R175H, C176F, C238S, C242S, G245S; p53-dependent and -independent in vitro and in vivo antitumor activity | [124,125] |
|
COTI-2 (Thiosemicarbazone) 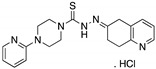
|
Rational drug design; virtual drug screening | R175H | Zn2+ chelator Binding: ND |
p53-dependent and -independent in vitro and in vivo antitumor activity | [126,127,128] |
|
Benzothiazolyl, Benzoxazolyl Hydrazones (C85) 
|
Chemical library; biophysical and cellular screening assay | R175H | Zn2+ chelator Binding: ND |
Increased cellular zinc concentration; rescue of wt conformation to mutp53 R175H, zinc-deficient p53-dependent in vitro and in vivo antitumor activity | [129] |
| Reactivators—Non-covalent binding | |||||
|
PK083 and analogs (Carbazole) 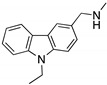
|
Rational drug design; virtual drug screening | Y220C, Y220N, Y220S | Binds to a cleft in C-terminal of mutp53 Y220C, Y220N, and Y220S DBD | Enhanced thermal stability (Y220C, Y220N, Y200S); rescue of wt conformation to mutp53 (PK083-Y220C); inhibition of aggregation (PK9318 analog-Y220C); transcription of p53 target genes (PK9318 analog-Y220C); p53-dependent in vitro antitumor activity | [84,130,131,132] |
|
PK7088 (Pyrazole) 
|
Rational drug design; NMR protein-based screening assay | Y220C | Binds to a cleft in C-terminal of mutp53 Y220C DBD | Enhanced thermal stability; rescue of wt conformation to mutp53; transcription of p53 target genes; weak in vitro antitumor activity | [131,133] |
|
PK5196 (Halogen-phenol derivative) 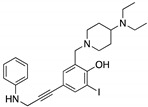
|
Rational drug design; NMR protein-based screening assay | Y220C | Binds to a cleft in C-terminal of mutp53 Y220C DBD | Enhanced thermal stability; in vitro antitumor activity | [131,134] |
|
MB725 (Aminobenzothiazole) 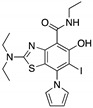
|
Rational drug design; NMR protein-based screening assay | Y220C | Binds to a cleft in C-terminal of mutp53 Y220C DBD | Enhanced thermal stability; transcription of p53 target genes; p53-dependent in vitro antitumor activity | [131] |
|
SLMP53-1 (Tryptophan-derived isoindolinone) 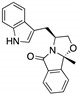
|
Chemical library; yeast-targeted screening assay | R175H, G245D, R248Q, R248W, R273H, R280K, R282W | Binds to mutp53 R280K DBD; in silico proposes that SLMP53-1 bridges DNA-binding surface of mutp53 R280K to DNA minor groove | Enhanced thermal stability of wt- and mutp53 R280K; restoration of DNA binding to mutp53 R280K; restoration of transcriptional activity to mutp53 R280K; p53-dependent in vitro and in vivo antitumor activity | [135,136] |
|
HBAP (2-[(4-hydroxybenzyl) amino]phenol) 
|
Chemical library; cellular screening assay |
R280K; R273H | Binds to mutp53 R280K and R273H DBD | Transcription of p53 target genes; in vitro and in vivo antitumor activity | [137] |
|
MANIO (Thiazole derivative) 
|
Chemical library; cellular screening assay |
Y126C; R175H; G245D; G245S; R248Q; R248W; R280K; R282W; R273C; R273H | Binds to wt- and mutp53 R248W DBD; in silico proposes that MANIO fits between the DNA molecule and the protein pocket at the dimer interface | Enhanced thermal stability of wt- and mutp53 R248W, Y126C, and R273H; increase of protein DNA-binding ability; transcription of p53 target genes; p53-dependent in vitro and in vivo antitumor activity | [138] |
|
SCH529074 (Piperazinylquinazoline) 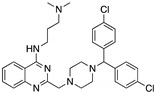
|
Chemical library; DNA-binding assay | R175H; S241F; R248W; R249S; R273H | Binds to wtp53 nearby DNA binding surface | Protected wtp53 conformation from thermal denaturation; rescue of wt conformation to mutp53 S241F, R248W, R273H; restoration of DNA binding to mutp53 R175H, R249S, R273H; transcription of p53 target genes by mutp53s R175H, S241F R248W, R249S, R273H; blocked MDM2-mediated ubiquitination of p53; p53-dependent in vitro and in vivo antitumor activity | [139] |
Stictic acid
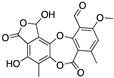
|
Chemical library; virtual screening | R175H; G245S | In silico binding to wtp53 and mutp53 R175H, R273H, G245S to a transiently open pocket (L1/S3) | Enhanced thermal stability of mutp53 R175H, G245S; transcription of p53 target genes by mutp53 R175H, G245S; p53-dependent in vitro antitumor activity | [140] |
Curcumin, Flavokawain B, Alpinetin
Curcumin 
Flavokawain B 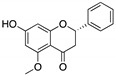
Alpinetin |
Crude extracts; cellular screening assay | R273H | In silico binding to mutp53 R273H bridging DNA-binding surface to DNA sequence | p53-dependent and -independent in vitro antitumor activity | [141] |
|
pCAPs (Peptides) pCAP221 sequence: RRKHNKHRPEPDSDER pCAP242 sequence: RRLIVRILKLPNPPER pCAP250 sequence: RRHSTPHPD |
Phage peptide display-protein screening assay | V135A; S241F; R249S; R280K | Binding to unknown local of mutp53 R175H and R249S | Rescue of wt conformation to mutp53 R175H, R249S; restoration of DNA binding to mutp53 R175H, R249S; transcription of p53 target genes by mutp53 V135A, S241F, R280K; p53-dependent in vitro and in vivo antitumor activity | [142] |
|
CDB3 (Peptide) Sequence: REDEDEIEW |
Rational drug design; NMR protein-based screening assay | R175H; I195T; R249S; R273H | Binding to wt and mutp53 R249S, R273H, nearby DNA binding surface and G245S, R175H unknown local | Enhanced thermal stability of wt and mutp53s R175H, R249S, R273H; rescue of wt conformation to mutp53 R175H, R249S, R273H; enhanced DNA binding to mutp53 I195T; transcription of p53 target genes by mutp53 R175H, R273H; p53-dependent in vitro antitumor activity | [82,143,144] |
| Reactivators—Chaperone-mediated effect | |||||
|
Chetomin (Epidithiodioxopiperazine) 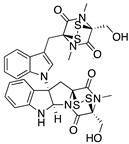
|
Natural products database; cellular luciferase reporter screening assay |
R175H | Binds to HSP40 | Rescue of wt conformation to mutp53; transcription of p53 target genes; MDM2 negative regulation; p53-dependent and -independent in vitro and in vivo antitumor activity | [145] |
|
SLMP53-2 (Tryptophanol-derived oxazoloisoindolinone) 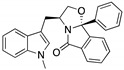
|
Chemical library; cellular screening assay | Y220C | Enhances the mutp53 Y220C interaction with HSP70 | Rescue of wt conformation to mutp53; transcription of p53 target genes; p53-dependent in vitro and in vivo antitumor activity | [146] |
| Reactivators—Unknown binding | |||||
|
Ellipticine (Alkaloid) 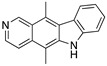
|
Chemical library; cellular screening assay | R175H; L194F; S241F; R249S; R273C; R273H | ND | Restoration of DNA binding to mutp53 R175H, S241F; rescue of wt conformation to mutp53 S241F; restoration of transcriptional activity to mup53 R175H; L194F, S241F, R249S, R273C, R273H; p53-dependent in vitro antitumor activity | [147] |
|
P53R3 (Piperazinylquinazoline) 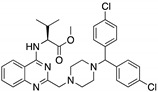
|
Chemical library; DNA-binding assay | M237I; R175H; R273H R248W | ND | Restoration of DNA binding to mutp53 R175H, M237I, R273H; restoration of transcriptional activity to mup53 M237I; p53-dependent in vitro antitumor activity | [148] |
|
PEITC (Phenethyl isothiocyanate) 
|
Cellular screening assay | R175H | ND | Rescue of wtp53 conformation to mutp53; transcription of p53 target genes; p53-dependent in vitro and in vivo antitumor activity | [149] |
|
WR-1065 (Aminothiol) 
|
Active metabolite of amifostine; cellular screening assay | V272M | ND | Restoration of DNA binding; rescue of wt conformation to mutp53; transcription of p53 target genes; p53-dependent and -independent in vitro antitumor activity | [150] |
| Disruptors of protein-protein interaction | |||||
|
RETRA (Thiazolthiophenyl ethanone) 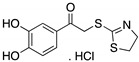
|
Chemical library; cellular screening assay | R273H | Disrupts mutp53-TAp73 complexes | Increased TAp73 expression; transcription of p53-shared target genes; TAp73-dependent in vitro and in vivo antitumor activity | [151] |
|
Prodigiosin (Pyrrolyl pyrromethane) 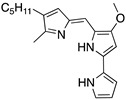
|
Chemical library; cellular screening assay | R273H; S241F; R248Q | Disrupts mutp53-p73 complexes | Transcription of p53-shared target genes; p73-dependent in vitro antitumor activity | [152] |
|
LEM2 (Xanthone) 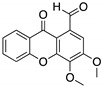
|
Chemical library; yeast-targeted screening assay | R273H | Disrupts mutp53-TAp73 and MDM2-TAp73 complexes | Enhanced thermal stability of TAp73; transcription of TAp73- and p53-shared target genes; TAp73-dependent in vitro antitumor activity | [153] |
|
Statins (Lovastatin) 
|
Cellular screening assay | R156P; V157F; R175H; Y220C; R248W | Inhibition of the mevalonate pathway, with CHIP-mediated mutp53 degradation | In vitro and in vivo suppression of mutp53-expressing cancer cell growth | [154] |
|
ATRA (Retinoic acid; tretinoin) 
|
Chemical library; mechanism-based screening assay (protein active site) | R273H; R280K | Disrupts mutp53-Pin1 interaction (via Pin1 inhibition and degradation) | In vitro and in vivo antitumor activity | [155] |
|
ReACp53 (Peptide) Sequence: RRRRRRRRRRPILTRITLE |
Structure-based rational design; cellular screening assay | R175H; R248Q | Binds to mutp53 aggregation prone region (S9) | Inhibits mutp53 aggregates; shifts the folding equilibrium toward the wt conformation; transcription of p53 target genes; p53-dependent in vitro and in vivo antitumor activity | [156] |
| Inducers of mutp53 degradation | |||||
|
17-AAG; 17-DMAG (Demethoxygeldanamycin derivatives)  17-AAG  17-DMAG |
HSP90 inhibitor; cellular evaluation | L194F; R273H; R273H/P309S; R280K | Inhibits HSP90 with increase of MDM2 and CHIP function | In vivo antitumor activity in synergism with SAHA | [157,158] |
|
Geldanamycin (Benzoquinone ansanamycin) 
|
HSP90 inhibitor; cellular evaluation | R175H; L194F; R248Q; R273H; R280K; R172H (mouse) |
Inhibits HSP90 with increase of MDM2 and CHIP function | In vitro antitumor activity | [159,160] |
|
Ganetespib (Phenylindolyl triazolone) 
|
HSP90 inhibitor; cellular evaluation | C124R; R172H; L194F; S241F; R248Q; R273H; C275F | Inhibits HSP90 with mutp53 degradation | In vivo and in vitro antitumor activity | [158] |
|
SAHA (Suberoyl-anilide hydroxamic acid) 
|
HDAC inhibitor; cellular evaluation | L194F; P223L/V274F; R249S; R273H; R273H/P309S; R280K | Inhibits HDAC6/8 (HSP90 machinery) with mutp53 CHIP-ubiquitin/proteasome-mediated degradation; decreases association with YY-1 transcription factor halting GOF | In vivo and in vitro antitumor activity | [158,161,162,163] |
Sodium butyrate
|
HDAC inhibitor; cellular evaluation | R249S; R280K | Inhibits HDAC8 (HSP90 machinery) with decreased association with YY-1 transcription factor halting GOF | In vitro antitumor activity | [163] |
Arsenic trioxide

|
Cellular evaluation | R175H; H179Y/R282W; R248W; R270H; R273H; R273H/P309S | Induces mutp53 nuclear proteasome-mediated degradation | Besides inducing mutp53 degradation, arsenic compounds stabilized wtp53 levels in cancer cells; in vitro antitumor activity | [164] |
|
Gambogic acid (Xanthone) 
|
Cellular evaluation | R175H; G266E; R273H; R280K | Depletes mutp53 via HSP90-CHIP ubiquitin/proteasome-mediated degradation | In vitro antitumor activity | [165] |
|
Spautin-1 (Fluorobenzylquinazolin amine) 
|
USP inhibitor; cellular evaluation |
P98S; P151H; S158inF; A161T; R175C/D/H; L194F; S227K/R; S241F; G245C; R248L/Q/W; E258K; G266E; R273H/L; R280K; R282W | Inhibits deubiquitinating enzymes leading to mutp53 lysosome-mediated degradation | In vitro antitumor activity | [166] |
|
YK-3-237 (Chalcone) 
|
Chemical library; cellular evaluation | V157F; M237I; R249S; R273H; R280K | Activates deacetylase SIRT1 reducing p53 levels | Transcription of p53 target genes; p53-dependent in vitro antitumor activity | [167] |
|
NSC59984 (Methylpiperazinylnitrofuranyl propenone) 
|
Chemical library; p53-reporter gene cellular screen | R175H/L; S241F; R273H/P309F | Induces MDM2-ubiquitin-proteasome-mediated degradation | Transcription of p53 target genes; p73-dependent in vitro and in vivo antitumor activity | [168] |
|
Disulfiram (Tetraethylthiuram disulfide)  |
Cellular evaluation | R273H | Thiol-conjugation with mutp53 proteasome-mediated degradation | In vivo and in vitro antitumor activity | [169] |
# The compound is a Michael acceptor, suggesting that the mechanism of action occurs by Michael addition to p53 cysteine-thiol groups; nevertheless, the reaction was not determined. ND—not determined. CHIP—E3 ubiquitin-protein ligase CHIP; HDAC—histone deacetylase; HSP—heat shock protein; Pin1—peptidyl-prolyl cis-trans isomerase NIMA-interacting 1; SIRT-1—NAD-dependent protein deacetylase sirtuin-1; USP—ubiquitin carboxyl-terminal hydrolase; YY-1—transcriptional repressor protein YY1. * Targeted mutp53 by small molecules through mechanisms of reactivation of wt-like p53 activities, disruption of mutp53 interaction with other proteins, or protein degradation.
Although, some mutp53-targeting agents have shown to bind and stabilize mutp53 (Table 2), the exact mechanism of mutp53 reactivation is still far from being fully understood [94]. The understanding of the precise molecular mechanism underlying the p53 activation of a mutp53-targeting agent, as well as of its off-target effects, is important data for the design of new, more efficient, and safe therapies.
Some of the first molecules reported to activate p53 are Michael acceptors that form adducts by covalently binding to p53 cysteines, causing its stabilization and reactivation [113,119,120,121]. On the other hand, the hydrophobic crevice suitable for binding exhibited by mutp53 Y220C prompted the design of several molecules able to reactivate its wt-like function [130,131,133]. Notably, the affinity of Y220C-cavity binders has progressively improved since PhiKan083 was developed, and further optimizations may be predicted [132]. Furthermore, in silico studies have identified new druggable p53 regions, namely a transiently open putative binding pocket in L1/S3 of the p53 DBD, in its folded conformation. This pocket is suitable to accommodate small molecules, thermodynamically stabilizing p53 in its native conformation and reactivating its transcriptional function [140]. The targeted therapy using zinc metallochaperones represents a unique approach, which is not based on a direct protein-ligand interaction, but instead on increasing the intracellular levels of zinc that are appropriate for remetallating and refolding of conformational mutp53 DBD [125]. A different approach targeting the DNA-binding surface of mutp53 DBD describes a chaperone mechanism (through non-covalent binding) by peptides or small molecules for the rescuing of the unfolded mutp53, shifting the equilibrium towards the native state and then being displaced in the presence of DNA [142,144]. In this regard, in silico results denoted a distinct molecular mechanism of mutp53 reactivation by SLMP53-1 and MANIO, not dependent on covalent bonds or mutation-created hydrophobic pockets, as described for other mutp53 reactivators. SLMP53-1 and MANIO reactivates mutp53 R280K and R248W, respectively, through binding to a hydrophobic pocket of the protein homodimer, establishing an interface with the DNA minor groove, and compensating for the loss of direct contacts between the K280/W248 residues and the DNA. Interestingly, the superposition of wt- and mutp53 models showed that the compounds have a similar binding pattern to wtp53, which is consistent with the reported activation of wtp53 by SLMP53-1 and MANIO [135,138]. It is also interesting to note that these compounds can reactivate other mutp53 forms, both contact and structural, which further supports a molecular mechanism unrelated to the formation of pockets derived from specific mutation sites.
An alternative mutp53-targeting approach includes disruptors of protein-protein interaction, which inhibit the interaction of mutp53 with other proteins, thereby suppressing mutp53 GOF activities (Table 2). The small molecule LEM2, besides inducing thermal stability to p73, also halted the interaction of mutp53 and p73, leading to tumor suppression by the transcription of p53-shared target genes [153]. Moreover, ReACp53, a mutp53 targeting peptide, has shown the capability to inhibit mutp53 containing aggregosomes by binding to S9 (an aggregation prone region) and favoring wt-like folding to structural mutp53 forms [156]. In silico studies disclosed that mutp53 forms exhibit an “open” state of the S6-S7 turn, exposing S9, and therefore representing a putative binding pocket to be explored in drug design [170].
Mutp53 protein degradation by small molecules, rather than its reactivation, with observed cellular growth inhibitory effect, constitutes another mutp53 targeting strategy [67] (Table 2). This strategy could also be explored through a novel pharmacological approach, such as PROteolysis TArgeting Chimeras (PROTACs) [171]. For this, the combined knowledge of mutp53 structure and putative binding pockets or surfaces, known mutp53-targeting agents, and small molecules that bind to E3 ligases may boost the discovery of effective therapeutic options.
Although none of the reported mutp53-targeting compounds have reached the clinic, the structural and mechanistic diversity among them is encouraging. It is reasonable to expect that some of them will prove effective in clinical applications in the near future [94]. Furthermore, the pharmacological reactivation of mutp53 also poses a great opportunity in combination therapy with anticancer drugs known to trigger p53-dependent cancer cell death or to inhibit mutp53 downstream pathways [94].
However, it has to be considered that although the potential of reactivating wt-like functions of mutp53 with canonical gene transcription has been proven, this does not always translate into cell death or halt tumor growth, as this also depends on the cancer cell context [58,94].
6. Conclusions
Structural studies have strongly contributed to knowledge about p53 stability, its behavior in the presence of inhibitors or co-factors, and how the protein recognizes and binds to target DNA sequences, thereby inducing coordinated changes in gene transcription within its vast and complex regulatory network. Furthermore, they have provided insights into wt and mutp53 pharmacology at molecular levels, fostering the design and development of p53-based targeted therapies to halt cancer. However, despite all the strategies detailed above to target mutp53, drugging mutp53 still prompts researchers and clinicians to investigate new possibilities in the universe of mutp53 pharmacology, structure, and biology.
Concerning the structural elucidation of mutp53, it would be valuable to further understand the structural and functional differences of other mutp53 forms. In particular, the classical categorization of mutp53 into contact and structural forms, and the possibility of reactivating each class by establishing extra contacts to DNA or by increasing thermal stability [95,120] seem to be inadequate based on the reported data for mutp53 reactivators. In fact, many mutp53 reactivators are capable of reactivating both contact and structural mutp53 with restoration of DNA-binding and transcriptional activity. Some of them have even induced the thermal stability of both mutp53 classes to increase the fraction of wt-like conformation [49,108,111,121,139,143]. Other important issues to be further explored are the mechanisms underlying the mutp53 aggregation-prone ability [83,156,172,173], the study of mutp53 interactomes, the mapping of potential PTMs on mutp53, and the impact of small molecules in these events. All such studies would greatly contribute towards a better understanding of mutp53 pathobiology and pharmacology, galvanizing the discovery and development of new mutp53 targeting agents for personalized cancer therapy.
Acknowledgments
The authors acknowledge the financial support from PT national funds (FCT/MCTES, Fundação para a Ciência e Tecnologia and Ministério da Ciência, Tecnologia e Ensino Superior) through UID/QUI/50006/2020 (LAQV/REQUIMTE) and UIDB/04423/2020, UIDP/04423/2020 (Group of Natural Products and Medicinal Chemistry CIIMAR). A.I. wishes to acknowledge the financial support from the Italian Foundation for Cancer Research (AIRC IG grant # 18985).
Author Contributions
Conceptualization, A.S.G. and L.S.; writing—original draft preparation, A.S.G. and L.S.; investigation, writing—review and editing, A.S.G., H.R., A.I., E.S. and L.S.; project administration, L.S.; funding acquisition, A.I., E.S. and L.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Funds through Fundação para a Ciência e Tecnologia, I.P. via the project UID/QUI/50006/2020.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Riley T., Sontag E., Chen P.A., Levine A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008;9:402–412. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 2.Menendez D., Inga A., Resnick M. The expanding universe of p53 targets. Nat. Rev. Cancer. 2009;9:724–737. doi: 10.1038/nrc2730. [DOI] [PubMed] [Google Scholar]
- 3.Joerger A.C., Fersht A.R. Structural Biology of the Tumor Suppressor p53. Annu. Rev. Biochem. 2008;77:557–582. doi: 10.1146/annurev.biochem.77.060806.091238. [DOI] [PubMed] [Google Scholar]
- 4.Li M., He Y., Dubois W., Wu X., Shi J., Huang J. Distinct Regulatory Mechanisms and Functions for p53-Activated and p53-Repressed DNA Damage Response Genes in Embryonic Stem Cells. Mol. Cell. 2012;46:30–42. doi: 10.1016/j.molcel.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fischer M. Census and evaluation of p53 target genes. Oncogene. 2017;36:3943–3956. doi: 10.1038/onc.2016.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brady C.A., Attardi L.D. p53 at a glance. J. Cell Sci. 2010;123:2527–2532. doi: 10.1242/jcs.064501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vousden K.H., Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 8.Blandino G., Di Agostino S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J. Exp. Clin. Cancer Res. 2018;37:1–13. doi: 10.1186/s13046-018-0705-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chillemi G., Kehrloesser S., Bernassola F., Desideri A., Dötsch V., Levine A.J., Melino G. Structural Evolution and Dynamics of the p53 Proteins. Cold Spring Harb. Perspect. Med. 2017;7:a028308. doi: 10.1101/cshperspect.a028308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y., Wang Z., Chen Y., Petersen R.B., Zheng L., Huang K. Salvation of the fallen angel: Reactivating mutant p53. Br. J. Pharmacol. 2019;176:817–831. doi: 10.1111/bph.14572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Selivanova G. Wild type p53 reactivation: From lab bench to clinic. FEBS Lett. 2014;588:2628–2638. doi: 10.1016/j.febslet.2014.03.049. [DOI] [PubMed] [Google Scholar]
- 12.Christophorou M.A., Martin-Zanca D., Soucek L., Lawlor E.R., Brown-Swigart L., Verschuren E., Evan G.I. Temporal dissection of p53 function In Vitro and In Vivo. Nat. Genet. 2005;37:718–726. doi: 10.1038/ng1572. [DOI] [PubMed] [Google Scholar]
- 13.Ventura A., Kirsch D.G., McLaughlin M.E., Tuveson D.A., Grimm J., Lintault L., Newman J., Reczek E.E., Weissleder R., Jacks T. Restoration of p53 function leads to tumour regression In Vivo. Nat. Cell Biol. 2007;445:661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 14.Xue W., Zender L., Miething C., Dickins R.A., Hernando E., Krizhanovsky V., Cordon-Cardo C., Lowe S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joerger A.C., Fersht A.R. The Tumor Suppressor p53: From Structures to Drug Discovery. Cold Spring Harb. Perspect. Biol. 2010;2:a000919. doi: 10.1101/cshperspect.a000919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang F., Rajagopalan S., Settanni G., Marsh R., Armoogum D.A., Nicolaou N., Bain A.J., Lerner E., Haas E., Ying L., et al. Multiple conformations of full-length p53 detected with single-molecule fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA. 2009;106:20758–20763. doi: 10.1073/pnas.0909644106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tidow H., Melero R., Mylonas E., Freund S.M.V., Grossmann J.G., Carazo J.M., Svergun D.I., Valle M., Fersht A.R. Quaternary structures of tumor suppressor p53 and a specific p53 DNA complex. Proc. Natl. Acad. Sci. USA. 2007;104:12324–12329. doi: 10.1073/pnas.0705069104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.James S.L., Abate D., Abate K.H., Abay S.M., Abbafati C., Abbasi N., Abbastabar H., Abd-Allah F., Abdela J., Abdelalim A., et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392:1789–1858. doi: 10.1016/S0140-6736(18)32279-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bullock A.N., Henckel J., DeDecker B.S., Johnson C.M., Nikolova P.V., Proctor M.R., Lane D., Fersht A.R. Thermodynamic stability of wild-type and mutant p53 core domain. Proc. Natl. Acad. Sci. USA. 1997;94:14338–14342. doi: 10.1073/pnas.94.26.14338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bullock A.N., Henckel J., Fersht A.R. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: Definition of mutant states for rescue in cancer therapy. Oncogene. 2000;19:1245–1256. doi: 10.1038/sj.onc.1203434. [DOI] [PubMed] [Google Scholar]
- 21.Lubin D.J., Butler J.S., Loh S.N. Folding of Tetrameric p53: Oligomerization and Tumorigenic Mutations Induce Misfolding and Loss of Function. J. Mol. Biol. 2010;395:705–716. doi: 10.1016/j.jmb.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 22.Tidow H., Veprintsev D., Freund S.M.V., Fersht A.R. Effects of Oncogenic Mutations and DNA Response Elements on the Binding of p53 to p53-binding Protein 2 (53BP2) J. Biol. Chem. 2006;281:32526–32533. doi: 10.1074/jbc.M604725200. [DOI] [PubMed] [Google Scholar]
- 23.Ahn J.-H., Kim T.J., Lee J.H., Choi J.-H. Mutant p53 stimulates cell invasion through an interaction with Rad21 in human ovarian cancer cells. Sci. Rep. 2017;7:1–11. doi: 10.1038/s41598-017-08880-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eldar A., Rozenberg H., Diskin-Posner Y., Rohs R., Shakked Z. Structural studies of p53 inactivation by DNA-contact mutations and its rescue by suppressor mutations via alternative protein-DNA interactions. Nucleic Acids Res. 2013;41:8748–8759. doi: 10.1093/nar/gkt630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nikolova P.V., Henckel J., Lane D., Fersht A.R. Semirational design of active tumor suppressor p53 DNA binding domain with enhanced stability. Proc. Natl. Acad. Sci. USA. 1998;95:14675–14680. doi: 10.1073/pnas.95.25.14675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suad O., Rozenberg H., Brosh R., Diskin-Posner Y., Kessler N., Shimon L., Frolow F., Liran A., Rotter V., Shakked Z. Structural Basis of Restoring Sequence-Specific DNA Binding and Transactivation to Mutant p53 by Suppressor Mutations. J. Mol. Biol. 2009;385:249–265. doi: 10.1016/j.jmb.2008.10.063. [DOI] [PubMed] [Google Scholar]
- 27.Collavin L., Lunardi A., Del Sal G. p53-family proteins and their regulators: Hubs and spokes in tumor suppression. Cell Death Differ. 2010;17:901–911. doi: 10.1038/cdd.2010.35. [DOI] [PubMed] [Google Scholar]
- 28.Fernandez-Fernandez M.R., Sot B. The relevance of protein-protein interactions for p53 function: The CPE contribution. Protein Eng. Des. Sel. 2010;24:41–51. doi: 10.1093/protein/gzq074. [DOI] [PubMed] [Google Scholar]
- 29.Kruse J.-P., Gu W. Modes of p53 Regulation. Cell. 2009;137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maclaine N.J., Hupp T.R. The regulation of p53 by phosphorylation: A model for how distinct signals integrate into the p53 pathway. Aging. 2009;1:490–502. doi: 10.18632/aging.100047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bouaoun L., Sonkin D., Ardin M., Hollstein M., Byrnes G., Zavadil J., Olivier M. TP53Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016;37:865–876. doi: 10.1002/humu.23035. [DOI] [PubMed] [Google Scholar]
- 32.Melero R., Rajagopalan S., Lázaro M., Joerger A., Brandt T., Veprintsev D., Lasso G., Gil D., Scheres S., Carazo J.M., et al. Electron microscopy studies on the quaternary structure of p53 reveal different binding modes for p53 tetramers in complex with DNA. Proc. Natl. Acad. Sci. USA. 2011;108:557–562. doi: 10.1073/pnas.1015520107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wells M., Tidow H., Rutherford T.J., Markwick P., Jensen M.R., Mylonas E., Svergun D.I., Blackledge M., Fersht A.R. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc. Natl. Acad. Sci. USA. 2008;105:5762–5767. doi: 10.1073/pnas.0801353105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nicholls C.D., McLure K.G., Shields M.A., Lee P.W.K. Biogenesis of p53 Involves Cotranslational Dimerization of Monomers and Posttranslational Dimerization of Dimers. J. Biol. Chem. 2002;277:12937–12945. doi: 10.1074/jbc.M108815200. [DOI] [PubMed] [Google Scholar]
- 35.Weinberg L.R., Veprintsev D.B., Fersht A.R. Cooperative binding of tetrameric p53 to DNA. J. Mol. Biol. 2004;341:1145–1159. doi: 10.1016/j.jmb.2004.06.071. [DOI] [PubMed] [Google Scholar]
- 36.Kitayner M., Rozenberg H., Kessler N., Rabinovich D., Shaulov L., Haran T.E., Shakked Z. Structural Basis of DNA Recognition by p53 Tetramers. Mol. Cell. 2006;22:741–753. doi: 10.1016/j.molcel.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 37.Lane D.P. Cancer. p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 38.Tebaldi T., Zaccara S., Alessandrini F., Bisio A., Ciribilli Y., Inga A. Whole-genome cartography of p53 response elements ranked on transactivation potential. BMC Genom. 2015;16:464. doi: 10.1186/s12864-015-1643-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.El-Deiry W.S. Definition of a consensus binding site for p53. Nat. Genet. 1992;1:45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- 40.Funk W.D., Pak D.T., Karas R.H., Wright W.E., Shay J.W. A transcriptionally active DNA-binding site for human p53 protein complexes. Mol. Cell. Biol. 1992;12:2866–2871. doi: 10.1128/MCB.12.6.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jordan J.J. Noncanonical DNA motifs as transactivation targets by wild type and mutant p53. PLoS Genet. 2008;4:e1000104. doi: 10.1371/annotation/f7fc9c28-14ae-480d-a69e-ee9cc4fba9a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cho Y., Gorina S., Jeffrey P., Pavletich N. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science. 1994;265:346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 43.Wright J.D., Noskov S.Y., Lim C. Factors governing loss and rescue of DNA binding upon single and double mutations in the p53 core domain. Nucleic Acids Res. 2002;30:1563–1574. doi: 10.1093/nar/30.7.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DeLano W. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002;40:82–92. [Google Scholar]
- 45.Joerger A.C., Fersht A.R. Structure–function–rescue: The diverse nature of common p53 cancer mutants. Oncogene. 2007;26:2226–2242. doi: 10.1038/sj.onc.1210291. [DOI] [PubMed] [Google Scholar]
- 46.Ho W.C., Fitzgerald M.X., Marmorstein R. Structure of the p53 Core Domain Dimer Bound to DNA. J. Biol. Chem. 2006;281:20494–20502. doi: 10.1074/jbc.M603634200. [DOI] [PubMed] [Google Scholar]
- 47.Kantarci N., Doruker P., Haliloglu T. Cooperative Fluctuations Point to the Dimerization Interface of P53 Core Domain. Biophys. J. 2006;91:421–432. doi: 10.1529/biophysj.106.077800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ang H.C., Joerger A., Mayer S., Fersht A.R. Effects of Common Cancer Mutations on Stability and DNA Binding of Full-length p53 Compared with Isolated Core Domains. J. Biol. Chem. 2006;281:21934–21941. doi: 10.1074/jbc.M604209200. [DOI] [PubMed] [Google Scholar]
- 49.Friedler A., Veprintsev D.B., Hansson L.O., Fersht A.R. Kinetic instability of p53 core domain mutants: Implications for rescue by small molecules. J. Biol. Chem. 2003;278:24108–24112. doi: 10.1074/jbc.M302458200. [DOI] [PubMed] [Google Scholar]
- 50.Bieging K.T., Mello S.S., Attardi L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer. 2014;14:359–370. doi: 10.1038/nrc3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walerych D., Gutkowska M., Klejman M.P., Wawrzynow B., Tracz-Gaszewska Z., Wiech M., Zylicz M., Zylicz A. ATP Binding to Hsp90 Is Sufficient for Effective Chaperoning of p53 Protein. J. Biol. Chem. 2010;285:32020–32028. doi: 10.1074/jbc.M110.112110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walerych D., Olszewski M.B., Gutkowska M., Helwak A., Zylicz M., Zylicz A. Hsp70 molecular chaperones are required to support p53 tumor suppressor activity under stress conditions. Oncogene. 2009;28:4284–4294. doi: 10.1038/onc.2009.281. [DOI] [PubMed] [Google Scholar]
- 53.Wawrzynow B., Zylicz A., Zylicz M. Chaperoning the guardian of the genome. The two-faced role of molecular chaperones in p53 tumor suppressor action. Biochim. Biophys. Acta (BBA) Bioenerg. 2018;1869:161–174. doi: 10.1016/j.bbcan.2017.12.004. [DOI] [PubMed] [Google Scholar]
- 54.Luo Q., Beaver J.M., Liu Y., Zhang Z. Dynamics of p53: A Master Decider of Cell Fate. Genes. 2017;8:66. doi: 10.3390/genes8020066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kumari R., Kohli S., Das S. p53 regulation upon genotoxic stress: Intricacies and complexities. Mol. Cell. Oncol. 2014;1:e969653. doi: 10.4161/23723548.2014.969653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reed S.M., Quelle D.E. p53 Acetylation: Regulation and Consequences. Cancers. 2014;7:30–69. doi: 10.3390/cancers7010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gu B., Zhu W.-G. Surf the Post-translational Modification Network of p53 Regulation. Int. J. Biol. Sci. 2012;8:672–684. doi: 10.7150/ijbs.4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kastenhuber E.R., Lowe S.W. Putting p53 in Context. Cell. 2017;170:1062–1078. doi: 10.1016/j.cell.2017.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Petitjean A., Mathe E., Kato S., Ishioka C., Tavtigian S.V., Hainaut P., Olivier M. Impact of mutant p53 functional properties onTP53mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007;28:622–629. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 60.Sabapathy K., Lane D. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 2018;15:13–30. doi: 10.1038/nrclinonc.2017.151. [DOI] [PubMed] [Google Scholar]
- 61.Kim M.P., Lozano G. Mutant p53 partners in crime. Cell Death Differ. 2018;25:161–168. doi: 10.1038/cdd.2017.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Freed-Pastor W.A., Prives C. Mutant p53: One name, many proteins. Genes Dev. 2012;26:1268–1286. doi: 10.1101/gad.190678.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Muller P.A., Vousden K.H. Mutant p53 in Cancer: New Functions and Therapeutic Opportunities. Cancer Cell. 2014;25:304–317. doi: 10.1016/j.ccr.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Prives C., White E. Does control of mutant p53 by Mdm2 complicate cancer therapy? Genes Dev. 2008;22:1259–1264. doi: 10.1101/gad.1680508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Manfredi J.J. The Mdm2-p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev. 2010;24:1580–1589. doi: 10.1101/gad.1941710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lang G.A., Iwakuma T., Suh Y.-A., Liu G., Rao V., Parant J.M., Valentin-Vega Y.A., Terzian T., Caldwell L.C., Strong L.C., et al. Gain of Function of a p53 Hot Spot Mutation in a Mouse Model of Li-Fraumeni Syndrome. Cell. 2004;119:861–872. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 67.Yue X., Zhao Y., Xu Y., Zheng M., Feng Z., Hu W. Mutant p53 in Cancer: Accumulation, Gain-of-Function, and Therapy. J. Mol. Biol. 2017;429:1595–1606. doi: 10.1016/j.jmb.2017.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chipuk J.E., Maurer U., Green D.R., Schuler M. Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription. Cancer Cell. 2003;4:371–381. doi: 10.1016/S1535-6108(03)00272-1. [DOI] [PubMed] [Google Scholar]
- 69.Xu J., Reumers J., Couceiro J., De Smet F., Gallardo R., Rudyak S., Cornelis A., Rozenski J., Zwolinska A., Marine J.-C., et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011;7:285–295. doi: 10.1038/nchembio.546. [DOI] [PubMed] [Google Scholar]
- 70.Ano Bom A.P. Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: Implications for cancer. J. Biol. Chem. 2012;287:28152–28162. doi: 10.1074/jbc.M112.340638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang G., Fersht A.R. Multisite aggregation of p53 and implications for drug rescue. Proc. Natl. Acad. Sci. USA. 2017;114:E2634–E2643. doi: 10.1073/pnas.1700308114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mantovani F., Walerych D., Del Sal G. Targeting mutant p53 in cancer: A long road to precision therapy. FEBS J. 2016;284:837–850. doi: 10.1111/febs.13948. [DOI] [PubMed] [Google Scholar]
- 73.Joerger A., Allen M.D., Fersht A.R. Crystal Structure of a Superstable Mutant of Human p53 Core Domain. J. Biol. Chem. 2004;279:1291–1296. doi: 10.1074/jbc.M309732200. [DOI] [PubMed] [Google Scholar]
- 74.Soussi T., May P. Structural aspects of the p53 protein in relation to gene evolution: A second look. J. Mol. Biol. 1996;260:623–637. doi: 10.1006/jmbi.1996.0425. [DOI] [PubMed] [Google Scholar]
- 75.Walker D.R., Bond J.P., Tarone R.E., Harris C.C., Makalowski W., Boguski M.S., Greenblatt M.S. Evolutionary conservation and somatic mutation hotspot maps of p53: Correlation with p53 protein structural and functional features. Oncogene. 1999;18:211–218. doi: 10.1038/sj.onc.1202298. [DOI] [PubMed] [Google Scholar]
- 76.Wong K.-B., DeDecker B.S., Freund S.M.V., Proctor M.R., Bycroft M., Fersht A.R. Hot-spot mutants of p53 core domain evince characteristic local structural changes. Proc. Natl. Acad. Sci. USA. 1999;96:8438–8442. doi: 10.1073/pnas.96.15.8438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ng J.W.K., Lama D., Lukman S., Lane D., Verma C.S., Sim A.Y.L. R248Q mutation-Beyond p53-DNA binding. Proteins Struct. Funct. Bioinform. 2015;83:2240–2250. doi: 10.1002/prot.24940. [DOI] [PubMed] [Google Scholar]
- 78.Joerger A., Ang H.C., Veprintsev D., Blair C.M., Fersht A.R. Structures of p53 Cancer Mutants and Mechanism of Rescue by Second-site Suppressor Mutations. J. Biol. Chem. 2005;280:16030–16037. doi: 10.1074/jbc.M500179200. [DOI] [PubMed] [Google Scholar]
- 79.Joerger A., Ang H.C., Fersht A.R. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl. Acad. Sci. USA. 2006;103:15056–15061. doi: 10.1073/pnas.0607286103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Malcikova J., Tichy B., Damborsky J., Kabathova J., Trbusek M., Mayer J., Pospisilova S. Analysis of the DNA-binding activity of p53 mutants using functional protein microarrays and its relationship to transcriptional activation. Biol. Chem. 2010;391:197–205. doi: 10.1515/bc.2010.027. [DOI] [PubMed] [Google Scholar]
- 81.Gomes A.S., Trovão F., Pinheiro B.A., Freire F., Gomes S., Oliveira C., Domingues L., Romão M.J., Saraiva L., Carvalho A.L. The Crystal Structure of the R280K Mutant of Human p53 Explains the Loss of DNA Binding. Int. J. Mol. Sci. 2018;19:1184. doi: 10.3390/ijms19041184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Friedler A., DeDecker B.S., Freund S.M., Blair C., Rüdiger S., Fersht A.R. Structural Distortion of p53 by the Mutation R249S and its Rescue by a Designed Peptide: Implications for “Mutant Conformation”. J. Mol. Biol. 2004;336:187–196. doi: 10.1016/j.jmb.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 83.Zhang Y., Coillie S.V., Fang J.-Y., Xu J. Gain of function of mutant p53: R282W on the peak? Oncogenesis. 2016;5:e196. doi: 10.1038/oncsis.2016.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bauer M.R., Krämer A., Settanni G., Jones R.N., Ni X., Tareque R.K., Fersht A.R., Spencer J., Joerger A.C. Targeting Cavity-Creating p53 Cancer Mutations with Small-Molecule Stabilizers: The Y220X Paradigm. ACS Chem. Biol. 2020;15:657–668. doi: 10.1021/acschembio.9b00748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dearth L.R., Qian H., Wang T., Baroni T.E., Zeng J., Chen S.W., Yi S.Y., Brachmann R.K. Inactive full-length p53 mutants lacking dominant wild-type p53 inhibition highlight loss of heterozygosity as an important aspect of p53 status in human cancers. Carcinogenesis. 2007;28:289–298. doi: 10.1093/carcin/bgl132. [DOI] [PubMed] [Google Scholar]
- 86.Ryan K.M., Vousden K.H. Characterization of Structural p53 Mutants Which Show Selective Defects in Apoptosis but Not Cell Cycle Arrest. Mol. Cell. Biol. 1998;18:3692–3698. doi: 10.1128/MCB.18.7.3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang Y., Rosengarth A., Luecke H. Structure of the human p53 core domain in the absence of DNA. Acta Crystallogr. Sect. D Biol. Crystallogr. 2007;63:276–281. doi: 10.1107/S0907444906048499. [DOI] [PubMed] [Google Scholar]
- 88.Pintus S., Ivanisenko N., Demenkov P., Ramachandran S., Kolchanov N., Ivanisenko V. The substitutions G245C and G245D in the Zn2+-binding pocket of the p53 protein result in differences of conformational flexibility of the DNA-binding domain. J. Biomol. Struct. Dyn. 2013;31:78–86. doi: 10.1080/07391102.2012.691364. [DOI] [PubMed] [Google Scholar]
- 89.Tu C., Tan Y.-H., Shaw G., Zhou Z., Bai Y., Luo R., Ji X. Impact of low-frequency hotspot mutation R282Q on the structure of p53 DNA-binding domain as revealed by crystallography at 1.54 Å resolution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2008;64:471–477. doi: 10.1107/S0907444908003338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Calhoun S., Daggett V. Structural Effects of the L145Q, V157F, and R282W Cancer-Associated Mutations in the p53 DNA-Binding Core Domain. Biochemistry. 2011;50:5345–5353. doi: 10.1021/bi200192j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wallentine B.D., Wang Y., Tretyachenko-Ladokhina V., Tan M., Senear D.F., Luecke H. Structures of oncogenic, suppressor and rescued p53 core-domain variants: Mechanisms of mutant p53 rescue. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013;69:2146–2156. doi: 10.1107/S0907444913020830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Aramayo R., Sherman M.B., Brownless K., Lurz R., Okorokov A., Orlova E.V. Quaternary structure of the specific p53–DNA complex reveals the mechanism of p53 mutant dominance. Nucleic Acids Res. 2011;39:8960–8971. doi: 10.1093/nar/gkr386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yamamoto S., Iwakuma T. Regulators of Oncogenic Mutant TP53 Gain of Function. Cancers. 2018;11:4. doi: 10.3390/cancers11010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bykov V.J.N., Eriksson S.E., Bianchi J., Wiman K. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer. 2018;18:89–102. doi: 10.1038/nrc.2017.109. [DOI] [PubMed] [Google Scholar]
- 95.Joerger A.C., Fersht A.R. The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu. Rev. Biochem. 2016;85:375–404. doi: 10.1146/annurev-biochem-060815-014710. [DOI] [PubMed] [Google Scholar]
- 96.Nikolova P.V., Wong K.-B., Dedecker B., Henckel J., Fersht A.R. Mechanism of rescue of common p53 cancer mutations by second-site suppressor mutations. EMBO J. 2000;19:370–378. doi: 10.1093/emboj/19.3.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Baroni T.E., Wang T., Qian H., Dearth L.R., Truong L.N., Zeng J., Denes A.E., Chen S.W., Brachmann R.K. A global suppressor motif for p53 cancer mutants. Proc. Natl. Acad. Sci. USA. 2004;101:4930–4935. doi: 10.1073/pnas.0401162101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brachmann R.K., Yu K., Eby Y., Pavletich N.P., Boeke J.D. Genetic selection of intragenic suppressor mutations that reverse the effect of common p53 cancer mutations. EMBO J. 1998;17:1847–1859. doi: 10.1093/emboj/17.7.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Brosh R., Rotter V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer. 2009;9:701–713. doi: 10.1038/nrc2693. [DOI] [PubMed] [Google Scholar]
- 100.Terzian T., Suh Y.-A., Iwakuma T., Post S.M., Neumann M., Lang G.A., Van Pelt C.S., Lozano G. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008;22:1337–1344. doi: 10.1101/gad.1662908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Parrales A., Iwakuma T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Front. Oncol. 2015;5:288. doi: 10.3389/fonc.2015.00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.NIH-ClinicalTrials APR-246 & Azacitidine for the Treatment of TP53 Mutant Myelodysplastic Syndromes (MDS) [(accessed on 11 April 2021)];2018 Available online: https://clinicaltrials.gov/ct2/show/NCT03745716.
- 103.NIH-ClinicalTrials Study of COTI-2 as Monotherapy or Combination Therapy for the Treatment of Malignancies (COTI2-101) [(accessed on 11 April 2019)];2019 Available online: https://www.clinicaltrials.gov/ct2/show/NCT02433626.
- 104.Foster B.A. Pharmacological Rescue of Mutant p53 Conformation and Function. Science. 1999;286:2507–2510. doi: 10.1126/science.286.5449.2507. [DOI] [PubMed] [Google Scholar]
- 105.Madka V., Zhang Y., Li Q., Mohammed A., Sindhwani P., Lightfoot S., Wu X.-R., Kopelovich L., Rao C.V. p53-stabilizing Agent CP-31398 Prevents Growth and Invasion of Urothelial Cancer of the Bladder in Transgenic UPII-SV40T Mice. Neoplasia. 2013;15:966–974. doi: 10.1593/neo.13704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rippin T.M., Bykov V.J.N., Freund S.M.V., Selivanova G., Wiman K., Fersht A.R. Characterization of the p53-rescue drug CP-31398 in vitro and in living cells. Oncogene. 2002;21:2119–2129. doi: 10.1038/sj.onc.1205362. [DOI] [PubMed] [Google Scholar]
- 107.Zache N., Lambert J.M., Rökaeus N., Shen J., Hainaut P., Bergman J., Wiman K.G., Bykov V.J. Mutant p53 targeting by the low molecular weight compound STIMA-1. Mol. Oncol. 2008;2:70–80. doi: 10.1016/j.molonc.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bykov V.J.N., Issaeva N., Zache N., Shilov A., Hultcrantz M., Bergman J., Selivanova G., Wiman K. Reactivation of Mutant p53 and Induction of Apoptosis in Human Tumor Cells by Maleimide Analogs. J. Biol. Chem. 2005;280:30384–30391. doi: 10.1074/jbc.M501664200. [DOI] [PubMed] [Google Scholar]
- 109.Punganuru S.R., Madala H.R., Venugopal S.N., Samala R., Mikelis C., Srivenugopal K.S. Design and synthesis of a C7-aryl piperlongumine derivative with potent antimicrotubule and mutant p53-reactivating properties. Eur. J. Med. Chem. 2016;107:233–244. doi: 10.1016/j.ejmech.2015.10.052. [DOI] [PubMed] [Google Scholar]
- 110.Bykov V.J., Issaeva N., Selivanova G., Wiman K.G. Mutant p53-dependent growth suppression distinguishes PRIMA-1 from known anticancer drugs: A statistical analysis of information in the National Cancer Institute database. Carcinogenesis. 2002;23:2011–2018. doi: 10.1093/carcin/23.12.2011. [DOI] [PubMed] [Google Scholar]
- 111.Bykov V.J., Issaeva N., Shilov A., Hultcrantz M., Pugacheva E., Chumakov P., Bergman J., Wiman K., Selivanova G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002;8:282–288. doi: 10.1038/nm0302-282. [DOI] [PubMed] [Google Scholar]
- 112.Bykov V.J.N., Zache N., Stridh H., Westman J., Bergman J., Selivanova G., Wiman K.G. PRIMA-1MET synergizes with cisplatin to induce tumor cell apoptosis. Oncogene. 2005;24:3484–3491. doi: 10.1038/sj.onc.1208419. [DOI] [PubMed] [Google Scholar]
- 113.Lambert J.M., Gorzov P., Veprintsev D., Söderqvist M., Segerbäck D., Bergman J., Fersht A.R., Hainaut P., Wiman K.G., Bykov V.J. PRIMA-1 Reactivates Mutant p53 by Covalent Binding to the Core Domain. Cancer Cell. 2009;15:376–388. doi: 10.1016/j.ccr.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 114.Lambert J.M.R., Moshfegh A., Hainaut P., Wiman K.G., Bykov V.J.N. Mutant p53 reactivation by PRIMA-1MET induces multiple signaling pathways converging on apoptosis. Oncogene. 2009;29:1329–1338. doi: 10.1038/onc.2009.425. [DOI] [PubMed] [Google Scholar]
- 115.Messina R.L. Reactivation of p53 mutants by prima-1 [corrected] in thyroid cancer cells. Int. J. Cancer. 2012;130:2259–2270. doi: 10.1002/ijc.26228. [DOI] [PubMed] [Google Scholar]
- 116.Aryee D.N.T., Niedan S., Ban J., Schwentner R., Muehlbacher K., Kauer M., Kofler R., Kovar H. Variability in functional p53 reactivation by PRIMA-1Met/APR-246 in Ewing sarcoma. Br. J. Cancer. 2013;109:2696–2704. doi: 10.1038/bjc.2013.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li X.-L., Zhou J., Chan Z.-L., Chooi J.-Y., Chen Z.-R., Chng W.-J. PRIMA-1met (APR-246) inhibits growth of colorectal cancer cells with different p53 status through distinct mechanisms. Oncotarget. 2015;6:36689–36699. doi: 10.18632/oncotarget.5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang W., Yi B., Wang C., Chen D., Bae S., Wei S., Guo R.-J., Lu C., Nguyen L., Yang W.-H., et al. Silencing of CD24 Enhances the PRIMA-1–Induced Restoration of Mutant p53 in Prostate Cancer Cells. Clin. Cancer Res. 2016;22:2545–2554. doi: 10.1158/1078-0432.CCR-15-1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhang Q., Bykov V.J.N., Wiman K.G., Zawacka-Pankau J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018;9:1–12. doi: 10.1038/s41419-018-0463-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bauer M., Joerger A., Fersht A.R. 2-Sulfonylpyrimidines: Mild alkylating agents with anticancer activity toward p53-compromised cells. Proc. Natl. Acad. Sci. USA. 2016;113:E5271–E5280. doi: 10.1073/pnas.1610421113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Madan E., Parker T.M., Bauer M., Dhiman A., Pelham C.J., Nagane M., Kuppusamy M.L., Holmes M., Holmes T.R., Shaik K., et al. The curcumin analog HO-3867 selectively kills cancer cells by converting mutant p53 protein to transcriptionally active wildtype p53. J. Biol. Chem. 2018;293:4262–4276. doi: 10.1074/jbc.RA117.000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Selvendiran K., Ahmed S., Dayton A., Kuppusamy M.L., Tazi M., Bratasz A., Tong L., Rivera B.K., Kálai T., Hideg K. Safe and targeted anticancer efficacy of a novel class of antioxidant-conjugated difluorodiarylidenyl piperidones: Differential cytotoxicity in healthy and cancer cells. Free Radic. Biol. Med. 2010;48:1228–1235. doi: 10.1016/j.freeradbiomed.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kaar J.L., Basse N., Joerger A., Stephens E., Rutherford T.J., Fersht A.R. Stabilization of mutant p53 via alkylation of cysteines and effects on DNA binding. Protein Sci. 2010;19:2267–2278. doi: 10.1002/pro.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yu X., Vazquez A., Levine A.J., Carpizo D.R. Allele-Specific p53 Mutant Reactivation. Cancer Cell. 2012;21:614–625. doi: 10.1016/j.ccr.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yu X., Blanden A.R., Narayanan S., Jayakumar L., Lubin D., Augeri D., Kimball S.D., Loh S.N., Carpizo D.R. Small molecule restoration of wildtype structure and function of mutant p53 using a novel zinc-metallochaperone based mechanism. Oncotarget. 2014;5:8879–8892. doi: 10.18632/oncotarget.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Silver N., Osman A., Patel A., Tanaka N., Tang L., Zhou G., Myers J. A Novel Third Generation Thiosemicarbazone, COTI-2, Is Highly Effective in Killing Head and Neck Squamous Cell Carcinomas (HNSCC) Bearing a Variety of TP53 Mutations. Int. J. Radiat. Oncol. 2016;94:942. doi: 10.1016/j.ijrobp.2015.12.272. [DOI] [Google Scholar]
- 127.Salim K., Vareki S.M., Danter W., Koropatnick J. COTI-2, a new anticancer drug currently under clinical investigation, targets mutant p53 and negatively modulates the PI3K/AKT/mTOR pathway. Eur. J. Cancer. 2016;69:S19. doi: 10.1016/S0959-8049(16)32638-7. [DOI] [Google Scholar]
- 128.Salim K.Y., Vareki S.M., Danter W.R., San-Marina S., Koropatnick J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines In Vitro and In Vivo. Oncotarget. 2016;7:41363–41379. doi: 10.18632/oncotarget.9133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gilleran J.A., Yu X., Blayney A.J., Bencivenga A.F., Na B., Augeri D.J., Blanden A.R., Kimball S.D., Loh S.N., Roberge J.Y., et al. Benzothiazolyl and Benzoxazolyl Hydrazones Function as Zinc Metallochaperones to Reactivate Mutant p53. J. Med. Chem. 2021;64:2024–2045. doi: 10.1021/acs.jmedchem.0c01360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Boeckler F., Joerger A., Jaggi G., Rutherford T.J., Veprintsev D., Fersht A.R. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. USA. 2008;105:10360–10365. doi: 10.1073/pnas.0805326105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Baud M.G., Bauer M., Verduci L., Dingler F.A., Patel K.J., Roy D.H., Joerger A., Fersht A.R. Aminobenzothiazole derivatives stabilize the thermolabile p53 cancer mutant Y220C and show anticancer activity in p53-Y220C cell lines. Eur. J. Med. Chem. 2018;152:101–114. doi: 10.1016/j.ejmech.2018.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bauer M., Jones R.N., Tareque R.K., Springett B., Dingler F.A., Verduci L., Patel K.J., Fersht A.R., Joerger A.C., Spencer J. A structure-guided molecular chaperone approach for restoring the transcriptional activity of the p53 cancer mutant Y220C. Futur. Med. Chem. 2019;11:2491–2504. doi: 10.4155/fmc-2019-0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Liu X., Wilcken R., Joerger A., Chuckowree I.S., Amin J., Spencer J., Fersht A.R. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 2013;41:6034–6044. doi: 10.1093/nar/gkt305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wilcken R., Liu X., Zimmermann M.O., Rutherford T.J., Fersht A.R., Joerger A., Boeckler F.M. Halogen-Enriched Fragment Libraries as Leads for Drug Rescue of Mutant p53. J. Am. Chem. Soc. 2012;134:6810–6818. doi: 10.1021/ja301056a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Soares J., Raimundo L., Pereira N.A., Monteiro Â., Gomes S., Bessa C., Pereira C., Queiroz G., Bisio A., Fernandes J., et al. Reactivation of wild-type and mutant p53 by tryptophanolderived oxazoloisoindolinone SLMP53-1, a novel anticancer small-molecule. Oncotarget. 2016;7:4326–4343. doi: 10.18632/oncotarget.6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gomes A.S., Ramos H., Gomes S., Loureiro J., Soares J., Barcherini V., Monti P., Fronza G., Oliveira C., Domingues L., et al. SLMP53-1 interacts with wild-type and mutant p53 DNA-binding domain and reactivates multiple hotspot mutations. Biochim. Biophys. Acta Gen. Subj. 2020;1864:129440. doi: 10.1016/j.bbagen.2019.129440. [DOI] [PubMed] [Google Scholar]
- 137.Xu C., Zhuang J., Zhang X. 2-[(4-Hydroxybenzyl) Amino] Phenol (HBAP) Restores the Mutated p53 to the Level Similar to That of Wild-Type p53 Protein and Inhibits Breast Cancer Growth in vivo to by Inducing Tumor Cells Apoptosis. Front. Cell Dev. Biol. 2020;8:574799. doi: 10.3389/fcell.2020.574799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ramos H., Soares M.I., Silva J., Raimundo L., Calheiros J., Gomes C., Reis F., Monteiro F.A., Nunes C., Reis S., et al. A selective p53 activator and anticancer agent to improve colorectal cancer therapy. Cell Rep. 2021;35:108982. doi: 10.1016/j.celrep.2021.108982. [DOI] [PubMed] [Google Scholar]
- 139.Demma M., Maxwell E., Ramos R., Liang L., Li C., Hesk D., Rossman R., Mallams A., Doll R., Liu M., et al. SCH529074, a Small Molecule Activator of Mutant p53, Which Binds p53 DNA Binding Domain (DBD), Restores Growth-suppressive Function to Mutant p53 and Interrupts HDM2-mediated Ubiquitination of Wild Type p53. J. Biol. Chem. 2010;285:10198–10212. doi: 10.1074/jbc.M109.083469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wassman C.D., Baronio R., Demir Ö., Wallentine B.D., Chen C.-K., Hall L.V., Salehi F., Lin D.-W., Chung B.P., Hatfield G.W., et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 2013;4:1407. doi: 10.1038/ncomms2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Malami I., Muhammad A., Etti I.C., Waziri P.M., Alhassan A.M. An In Silico approach in predicting the possible mechanism involving restoration of wild-type p53 functions by small molecular weight compounds in tumor cells expressing R273H mutant p53. EXCLI J. 2017;16:1276–1287. doi: 10.17179/excli2017-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tal P., Eizenberger S., Cohen E., Goldfinger N., Pietrokovski S., Oren M., Rotter V. Cancer therapeutic approach based on conformational stabilization of mutant p53 protein by small peptides. Oncotarget. 2016;7:11817–11837. doi: 10.18632/oncotarget.7857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Friedler A., Hansson L.O., Veprintsev D., Freund S.M.V., Rippin T.M., Nikolova P.V., Proctor M.R., Rüdiger S., Fersht A.R. A peptide that binds and stabilizes p53 core domain: Chaperone strategy for rescue of oncogenic mutants. Proc. Natl. Acad. Sci. USA. 2002;99:937–942. doi: 10.1073/pnas.241629998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Issaeva N., Friedler A., Bozko P., Wiman K., Fersht A.R., Selivanova G. Rescue of mutants of the tumor suppressor p53 in cancer cells by a designed peptide. Proc. Natl. Acad. Sci. USA. 2003;100:13303–13307. doi: 10.1073/pnas.1835733100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hiraki M., Hwang S.-Y., Cao S., Ramadhar T., Byun S., Yoon K.W., Lee J.H., Chu K., Gurkar A., Kolev V., et al. Small-Molecule Reactivation of Mutant p53 to Wild-Type-like p53 through the p53-Hsp40 Regulatory Axis. Chem. Biol. 2015;22:1206–1216. doi: 10.1016/j.chembiol.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gomes S., Bosco B., Loureiro J.B., Ramos H., Raimundo L., Soares J., Nazareth N., Barcherini V., Domingues L., Oliveira C., et al. SLMP53-2 Restores Wild-Type-Like Function to Mutant p53 through Hsp70: Promising Activity in Hepatocellular Carcinoma. Cancers. 2019;11:1151. doi: 10.3390/cancers11081151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Peng Y., Li C., Chen L., Sebti S., Chen J. Rescue of mutant p53 transcription function by ellipticine. Oncogene. 2003;22:4478–4487. doi: 10.1038/sj.onc.1206777. [DOI] [PubMed] [Google Scholar]
- 148.Weinmann L., Wischhusen J., Demma M.J., Naumann U., Roth P., DasMahapatra B., Weller M. A novel p53 rescue compound induces p53-dependent growth arrest and sensitises glioma cells to Apo2L/TRAIL-induced apoptosis. Cell Death Differ. 2008;15:718–729. doi: 10.1038/sj.cdd.4402301. [DOI] [PubMed] [Google Scholar]
- 149.Aggarwal M., Saxena R., Sinclair E., Fu Y., Jacobs A., Dyba M., Wang X., Cruz I., Berry D., Kallakury B., et al. Reactivation of mutant p53 by a dietary-related compound phenethyl isothiocyanate inhibits tumor growth. Cell Death Differ. 2016;23:1615–1627. doi: 10.1038/cdd.2016.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.North S., Pluquet O., Maurici D., El-Ghissassi F., Hainaut P. Restoration of wild-type conformation and activity of a temperature-sensitive mutant of p53 (p53(V272M)) by the cytoprotective aminothiol WR1065 in the esophageal cancer cell line TE-1. Mol. Carcinog. 2002;33:181–188. doi: 10.1002/mc.10038. [DOI] [PubMed] [Google Scholar]
- 151.Kravchenko J.E., Ilyinskaya G.V., Komarov P.G., Agapova L.S., Kochetkov D.V., Strom E., Frolova E.I., Kovriga I., Gudkov A., Feinstein E., et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc. Natl. Acad. Sci. USA. 2008;105:6302–6307. doi: 10.1073/pnas.0802091105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hong B., Prabhu V.V., Zhang S., Heuvel A.P.J.V.D., Dicker D.T., Kopelovich L., El-Deiry W.S. Prodigiosin Rescues Deficient p53 Signaling and Antitumor Effects via Upregulating p73 and Disrupting Its Interaction with Mutant p53. Cancer Res. 2014;74:1153–1165. doi: 10.1158/0008-5472.CAN-13-0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gomes S., Raimundo L., Soares J., Loureiro J.B., Leão M., Ramos H., Monteiro M.N., Lemos A., Moreira J., Pinto M., et al. New inhibitor of the TAp73 interaction with MDM2 and mutant p53 with promising antitumor activity against neuroblastoma. Cancer Lett. 2019;446:90–102. doi: 10.1016/j.canlet.2019.01.014. [DOI] [PubMed] [Google Scholar]
- 154.Parrales A., Ranjan A., Iyer S.V., Padhye S., Weir S.J., Roy A., Iwakuma A.P.A.R.S.V.I.T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016;18:1233–1243. doi: 10.1038/ncb3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wei S., Kozono S., Kats L., Nechama M., Li W., Guarnerio J., Luo M., You M.-H., Yao Y., Kondo A., et al. Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nat. Med. 2015;21:457–466. doi: 10.1038/nm.3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Soragni A., Janzen D.M., Johnson L.M., Lindgren A.G., Nguyen T.Q.A., Tiourin E., Soriaga A.B., Lu J., Jiang L., Faull K.F., et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell. 2016;29:90–103. doi: 10.1016/j.ccell.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Li D., Marchenko N.D., Schulz R., Fischer V., Velasco-Hernandez T., Talos F., Moll U.M. Functional Inactivation of Endogenous MDM2 and CHIP by HSP90 Causes Aberrant Stabilization of Mutant p53 in Human Cancer Cells. Mol. Cancer Res. 2011;9:577–588. doi: 10.1158/1541-7786.MCR-10-0534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Alexandrova E.M., Yallowitz A.R., Li D., Xu S., Schulz R., Proia D.A., Lozano G., Dobbelstein M., Moll U.M. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nat. Cell Biol. 2015;523:352–356. doi: 10.1038/nature14430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Blagosklonny M.V., Toretsky J., Neckers L. Geldanamycin selectively destabilizes and conformationally alters mutated p53. Oncogene. 1995;11:933–939. [PubMed] [Google Scholar]
- 160.Wang C., Chen J. Phosphorylation and hsp90 Binding Mediate Heat Shock Stabilization of p53. J. Biol. Chem. 2003;278:2066–2071. doi: 10.1074/jbc.M206697200. [DOI] [PubMed] [Google Scholar]
- 161.Marks P.A. Discovery and development of SAHA as an anticancer agent. Oncogene. 2007;26:1351–1356. doi: 10.1038/sj.onc.1210204. [DOI] [PubMed] [Google Scholar]
- 162.Li D., Marchenko N., Moll U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011;18:1904–1913. doi: 10.1038/cdd.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wang Z.-T., Chen Z.-J., Jiang G.-M., Wu Y.-M., Liu T., Yi Y.-M., Zeng J., Du J., Wang H.-S. Histone deacetylase inhibitors suppress mutant p53 transcription via HDAC8/YY1 signals in triple negative breast cancer cells. Cell. Signal. 2016;28:506–515. doi: 10.1016/j.cellsig.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 164.Yan W., Zhang Y., Zhang J., Liu S., Cho S.J., Chen X. Mutant p53 Protein Is Targeted by Arsenic for Degradation and Plays a Role in Arsenic-mediated Growth Suppression. J. Biol. Chem. 2011;286:17478–17486. doi: 10.1074/jbc.M111.231639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wang J., Zhao Q., Qi Q., Gu H.-Y., Rong J.-J., Mu R., Zou M.-J., Tao L., You Q.-D., Guo Q.-L. Gambogic acid-induced degradation of mutant p53 is mediated by proteasome and related to CHIP. J. Cell. Biochem. 2011;112:509–519. doi: 10.1002/jcb.22941. [DOI] [PubMed] [Google Scholar]
- 166.Vakifahmetoglu-Norberg H. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013;27:718–730. doi: 10.1101/gad.220897.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Yi Y.W., Kang H.J., Kim H.J., Kong Y., Brown M.L., Bae I. Targeting Mutant p53 by a SIRT1 Activator YK-3-237 Inhibits the Proliferation of Triple-Negative Breast Cancer Cells. Oncotarget. 2013;4:984–994. doi: 10.18632/oncotarget.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zhang S., Zhou L., Hong B., Heuvel A.P.J.V.D., Prabhu V.V., Warfel N.A., Kline C.L.B., Dicker D.T., Kopelovich L., El-Deiry W.S. Small-Molecule NSC59984 Restores p53 Pathway Signaling and Antitumor Effects against Colorectal Cancer via p73 Activation and Degradation of Mutant p53. Cancer Res. 2015;75:3842–3852. doi: 10.1158/0008-5472.CAN-13-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Paranjpe A., Srivenugopal K.S. Degradation of NF-κB, p53 and other regulatory redox-sensitive proteins by thiol-conjugating and -nitrosylating drugs in human tumor cells. Carcinogenesis. 2013;34:990–1000. doi: 10.1093/carcin/bgt032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Pradhan M.R., Siau J.W., Kannan S., Nguyen M.N., Ouaray Z., Kwoh C.K., Lane D.P., Ghadessy F., Verma C.S. Simulations of mutant p53 DNA binding domains reveal a novel druggable pocket. Nucleic Acids Res. 2019;47:1637–1652. doi: 10.1093/nar/gky1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Demir Ö., Barros E.P., Offutt T.L., Rosenfeld M., Amaro R.E. An integrated view of p53 dynamics, function, and reactivation. Curr. Opin. Struct. Biol. 2021;67:187–194. doi: 10.1016/j.sbi.2020.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Silva J.L., Cino E.A., Soares I.N., Ferreira V., De Oliveira G.A.P. Targeting the Prion-like Aggregation of Mutant p53 to Combat Cancer. Acc. Chem. Res. 2017;51:181–190. doi: 10.1021/acs.accounts.7b00473. [DOI] [PubMed] [Google Scholar]
- 173.Silva J.L., Gallo C.V.D.M., Costa D.C., Rangel L.P. Prion-like aggregation of mutant p53 in cancer. Trends Biochem. Sci. 2014;39:260–267. doi: 10.1016/j.tibs.2014.04.001. [DOI] [PubMed] [Google Scholar]