

Abstract
Single-cell RNA-sequencing (scRNA-seq) has become an essential tool for characterizing gene expression in eukaryotes but current methods are incompatible with bacteria. Here, we introduce microSPLiT, a high-throughput scRNA-seq method for gram-negative and gram-positive bacteria that can resolve heterogeneous transcriptional states. We applied microSPLiT to >25,000 Bacillus subtilis cells sampled at different growth stages, creating an atlas of changes in metabolism and lifestyle. We retrieved detailed gene expression profiles associated with known, but rare, states such as competence and prophage induction, and also identified novel and unexpected gene expression states including the heterogeneous activation of a niche metabolic pathway in a subpopulation of cells. MicroSPLiT paves the way to high-throughput analysis of gene expression in bacterial communities otherwise not amenable to single-cell analysis such as natural microbiota.
One Sentence Summary:
A high-throughput microbial single-cell RNA sequencing method reveals gene expression states in bacteria.
Gene expression in bacteria is highly heterogeneous even in isogenic populations grown under the same lab conditions. Bacteria can randomly differentiate into subpopulations that assume different roles for the survival of the community; a strategy known as bet hedging (1, 2). For example, gene expression programs governing developmental and stress-response states such as competence or antibiotic resistance may switch on stochastically in a small number of single cells (3–5). Population level gene expression measurements are insufficient to resolve such rare states which, to date, have been characterized only in tractable model systems and through methods such as fluorescence microscopy that can only measure a limited set of reporter genes at a time (6).
Single-cell RNA-seq (scRNA-seq) methods developed for eukaryotic cells can provide comprehensive gene expression profiles for tens of thousands of cells (7–11). Although the need for microbial scRNA-seq has been recognized (12), technical challenges have long prevented adapting scRNA-seq technology to microbes. Specifically, bacteria have low mRNA content, about two orders of magnitude less than human cells (14) and bacterial mRNA is not polyadenylated which makes separation from rRNA challenging. Bacteria have diverse cell walls and membranes which can interfere with the lysis or permeabilization steps required for scRNA-seq. Finally, their small size can hinder microfluidic single-cell isolation. Recent work has begun to address these issues and demonstrated that scRNA-seq methods can be adapted to bacteria. However, in spite of rapid progress from sequencing just a few cells (13, 14) to performing experiments in a 96-well format (15), these prior approaches remain relatively low-throughput compared to the state-of-the-art in eukaryotic scRNA-seq.
We have managed to overcome the difficulties of performing high-throughput scRNA-seq with bacterial cells with a technique we have named microSPLiT (Microbial Split-Pool Ligation Transcriptomics). We applied microSPLiT to profile gene expression states in >25,000 single B. subtilis cells, uncovering both rare and unexpected states present in as little as 0.1% of the population. A technically similar and concurrently developed approach termed PETRI-seq also supports the use of single-cell transcriptomics for gene expression analysis in prokaryotes (16).
Developing microSPLiT.
MicroSPLiT builds on SPLiT-seq, a eukaryotic scRNA-seq approach, which labels the cellular origin of RNA through combinatorial barcoding (7). In SPLiT-seq, cells are fixed, permeabilized and mRNA is converted to cDNA through in-cell reverse transcription (RT) with barcoded poly-T and random hexamer primers in a multi-well format. Cells are then pooled, randomly split into a new 96-well plate, and a well-specific barcode is appended to the cDNA through ligation. This split-ligation-pool cycle is repeated and a fourth, optional barcode is added during sequencing library preparation to ensure that each cell acquires a unique barcode combination with high likelihood (Figs. 1A and S1A–B).
Fig. 1. MicroSPLiT development and validation.
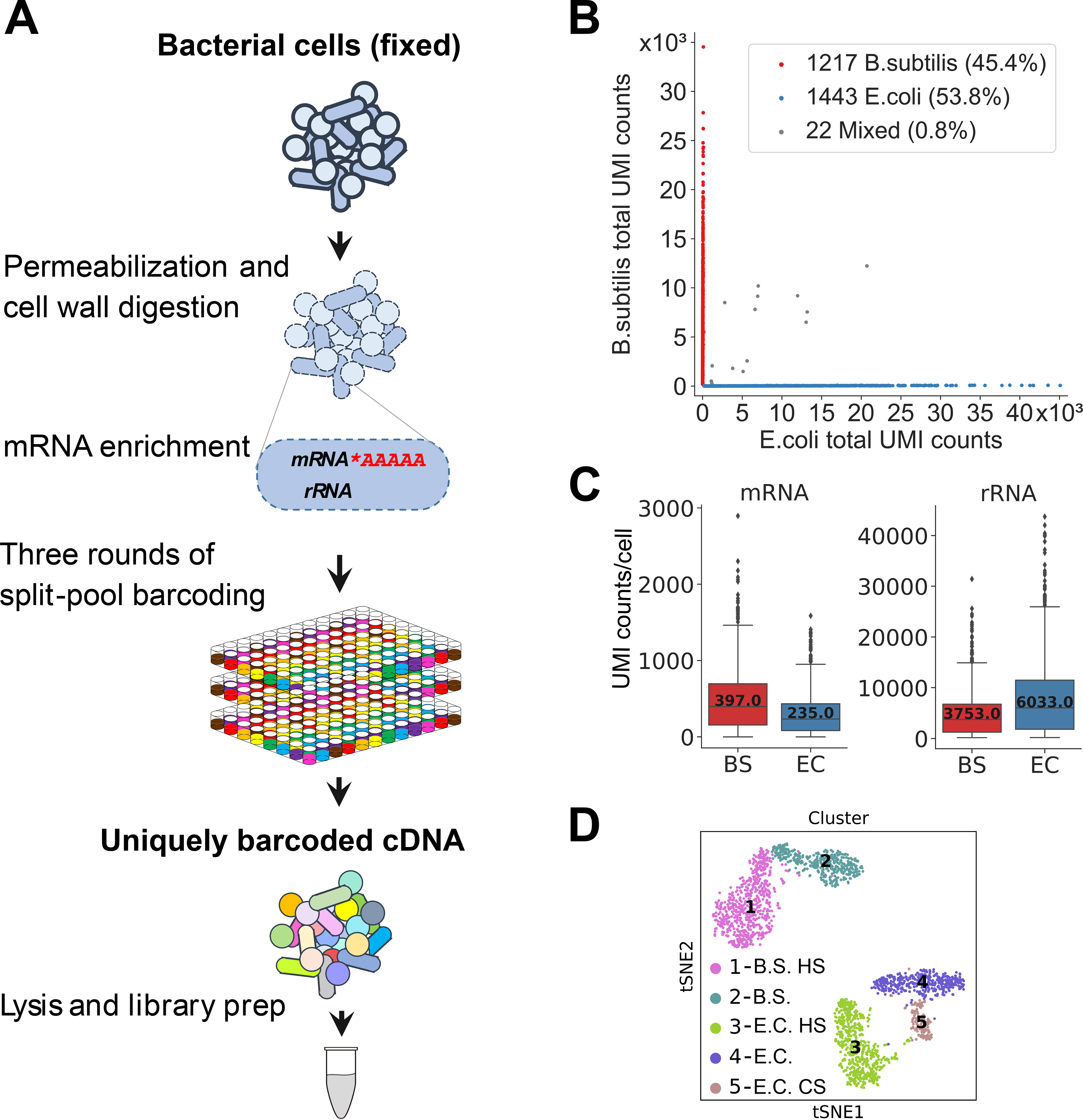
(A) MicroSPLiT method summary. Fixed bacterial cells are permeabilized with Tween-20 and lysozyme. The mRNA is then polyadenylated in-cell with E. coli Poly(A) Polymerase I (PAP). The cellular RNA then undergoes three rounds of combinatorial barcoding including in-cell reverse transcription (RT) and two in-cell ligation reactions, followed by lysis and library preparation. (B) Barnyard plot for the E. coli and B. subtilis species-mixing experiment. Each dot corresponds to a putative single-cell transcriptome. Total UMI (unique molecular identifier) counts for all types of RNA are plotted. (C) mRNA and rRNA UMI counts per cell for both species. Error bars represent 95% confidence intervals. (D) t-stochastic neighbor embedding (t-SNE) of the data from heat shock experiment showing distinct clusters. “HS” – heat shock, “CS” – cold shock (see (20)).
Because SPLiT-seq does not require cell isolation, it is compatible with a wide range of cell shapes and sizes. Moreover, because SPLiT-seq already uses random hexamer primers, in addition to poly-T primers for RT, we reasoned that it might be suitable for detecting bacterial mRNA. However, a direct application of the mammalian SPLiT-seq protocol to bacteria, not surprisingly, resulted in low total UMI (unique molecular identifier) counts (<100 max UMIs/cell, median 0 mRNA reads/cell) and a bias toward gram-negative over gram-positive bacteria (Fig. S1C).
We thus set out to optimize sample processing steps for bacteria. With SPLiT-seq’s multiplexing capabilities we tested a range of approaches to cell wall removal and membrane permeabilization. A protocol utilizing a combination of a mild detergent, Tween-20, and lysozyme resulted in the best capture efficiency for both gram-positive Bacillus subtilis and gram-negative Escherichia coli bacteria. (Figure 1A, inset, and Table S1). For the mRNA enrichment, we then tested polyadenylation with E. coli Poly(A) Polymerase I (PAP) which preferentially polyadenylates mRNA (17, 18), 5’-phosphate-dependent exonuclease (“Terminator”, Epicentre) (19) and reverse transcription with ribosomal RNA-specific probes followed by RNaseH-mediated degradation (Fig. S2 and Table S1). We found that the treatment of fixed and permeabilized cells with PAP resulted in the highest (about 2.5-fold, or approximately 7% of total RNA) enrichment of mRNA reads (Figs. 1A, inset, and S2). We also optimized the fixation protocol as well as the downstream enzymatic reaction conditions (Table S1 and (20)). Notably, we found that RT resulted in cell clumping and that mild sonication after this step was necessary to reliably obtain single cell suspensions (Fig. S3).
microSPLiT generates high-quality single-cell RNA-seq data.
In order to validate microSPLiT performance on a mixture of gram-positive and gram-negative organisms, we grew E. coli MW1255 and B. subtilis PY79 cells to OD600=0.5 and subjected half of each culture to a brief 47°C heat-shock. We performed a microSPLiT experiment on both samples, using the first barcode as a sample identifier. We prepared and sequenced a cDNA library from 2682 total bacteria from heat-shocked and control treatments and aligned the reads to a combined B. subtilis-E. coli genome. 99.2% of the putative single cell transcriptomes were unambiguously assigned to a single species (Fig. 1B). The rest is attributed to multi-species cellular aggregates, with true aggregate frequency including same-species aggregates expected to be double the multi-species one (1.6%). We sampled a median of 235 mRNA transcripts per cell for E. coli and 397 for B. subtilis (Fig. 1C), or approximately 5–10% of the estimated total mRNA (21), and 3.7% and 9.5% of all detected RNA molecules per cell for each respective species (Fig. S4A–B). We also detected a median count of 3753 and 6033 rRNA per cell for B. subtilis and E. coli (Fig. 1C) as well as a median number of 18 and 20 tRNA molecules per cell (Fig. S4C). We observed 230 median unique genes per cell for B. subtilis and 138 for E. coli (Fig. S4D). The majority of detected genes for both species had on average between 0 and 5 UMIs per gene (Fig. S4E–F). The summed E. coli expression data was correlated with independently published bulk transcriptomic data (r=0.736, Fig. S5A) (16). The most highly expressed genes detected only in the bulk assay but not with microSPLiT encode tRNA species (Fig. S5B). When we discarded multiply aligned reads, the proportion of mRNA reads increased to 90.5% for B. subtilis and 28.2% for E. coli (Fig. S6A–B), while mRNA UMI counts per cell were not strongly affected (Fig. S6C–D).
Even with in-cell polyadenylation, the majority of rRNA and mRNA molecules were captured by random hexamer primers, while tRNA were predominantly retrieved by poly-T primers (Fig. S7). The mRNA and rRNA UMI counts in each cell were highly correlated (r=0.97 and 0.94), as were UMI counts of each detected mRNA captured by poly-T and random hexamer primers (r=0.87 and 0.94) (Fig. S8). tRNA UMI counts captured by poly-T and random hexamer primers displayed lower correlation (Fig. S9) which could be due to the transient native polyadenylation of some tRNA species (22). We found that the 23S and 16S rRNA abundances were highly correlated with each other, while correlation with 5S rRNA was lower (Fig. S10). We also quantified the effect of sequencing depth on gene and UMI detection by subsampling analysis, suggesting that additional sequencing would only modestly increase UMI counts (Fig. S11).
Next, we tested whether microSPLiT could detect transcriptional responses to heat shock (Fig. S12A). Unsupervised clustering identified five distinct clusters which were visualized by t-distributed stochastic neighbor embedding (t-SNE) (Fig. 1D). The first barcode identified two pairs of clusters corresponding to the heat treated and control cultures, and gene expression analysis within each pair further labeled them as corresponding to B. subtilis and E. coli cells (Fig. S12B). Enriched within each heat shock cluster were classical heat shock genes differentially expressed in each of the E. coli and B. subtilis heat treated clusters as compared to the control clusters (Figs. S12C–D and S13A–B) (23, 24).
Unexpectedly, we found an additional small cluster, representing E. coli cells from both control and heat treated samples that expressed a different signature of DEAD-box helicase deaD induction as well as cold shock genes cspA-G, consistent with a transcriptional response to cold (Figs. S12C and S13C) (25). This subpopulation of E. coli might be displaying a very rapid response to cold from a brief cold centrifugation step performed as the first step in sample preparation before formaldehyde fixation and is thus likely an artifact of the workflow.
Transcriptional patterns during B. subtilis growth in rich medium.
Next, we applied microSPLiT to capture transcriptional states across the B. subtilis growth curve in a rich medium (LB). In total, we sampled ten optical density (OD) points along the growth curve of the laboratory strain PY79 ranging from OD0.5 (early exponential phase) to OD6.0 (early stationary phase), with one replicate of 4 OD points ((20), Fig. 2A). The first barcode was used to record sample identity (i.e. OD) and the replicates are consistent and produced a combined dataset of 25,214 cells (Figs. 2B–C and S14). Unsupervised clustering of the combined datasets revealed 14 clusters (Fig. 2B), most of which overlapped with a single OD (Fig. 2C). The most notable exceptions are two smaller clusters that contain cells from multiple ODs: cluster 9 with cells from OD2–3.2 that differentially express myo-inositol metabolism pathway genes, and a very small cluster 13 containing only 36 cells from 5 different OD points uniquely expressing genes associated with the defective PBSX prophage (Figs. 2B and S15–S18).
Fig. 2. MicroSPLiT detects global transcriptional states during B. subtilis growth.
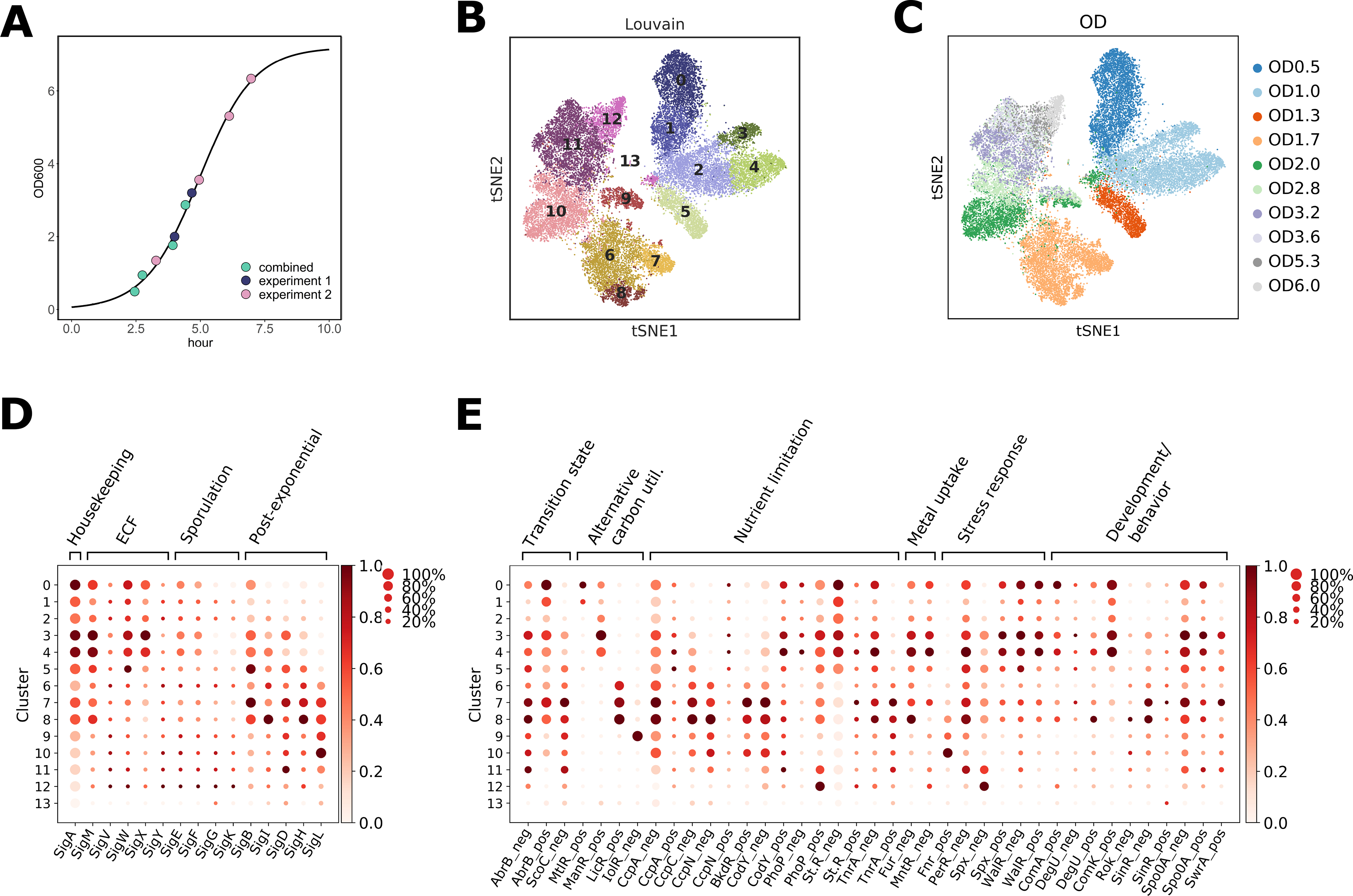
(A) Optical density (OD) points sampled in two experiments overlaid on the growth curve from experiment 2 (20). (B) and (C) t-SNE embedding of the combined growth curve data colored by (B) cluster or (C) OD. (D) Inferred normalized sigma factor activity for each cluster, calculated from averaged expression of genes regulated by each sigma factor. The size of each dot indicates the proportion of cells in the cluster in which the sigma factor is active, while the color indicates the average activity normalized from 0 to 1 across all clusters for each sigma factor. (E) Inferred activity of select transcriptional regulators per cluster, calculated and normalized for all clusters as above, plotted as in (D). “Neg” indicates that activity was calculated for the genes known to be negatively regulated by this TR, and “pos” indicates the activity was calculated for the genes positively regulated by the given TR.
We then turned to an analysis of alternative sigma factors which are the primary regulators of prokaryotic RNA polymerase specificity and thus directly shape transcriptional changes in response to environmental conditions (26, 27). To understand whether microSPLiT could capture variation in sigma factor utilization across different growth stages, we averaged expression of genes regulated by each sigma factor, recording for each cluster, both the percentage of cells expressing at least one gene regulated by a given sigma factor and the average intensity of gene expression (Figs. 2D and (20)). The patterns of sigma factor utilization are largely consistent with the literature, with housekeeping σA activity highest at the early growth stage relative to other time points, and the activity of general stress response sigma factor σB rising as cells begin to exit from exponential phase (clusters 3–4, OD1.3–1.7) and then declining as cells approach stationary phase (Fig. 2D). Sporulation sigma factors were more active at later ODs, but in a small fraction of cells (clusters 10–12, Fig. 2D) and the extracellular function (ECF) sigma factors were divided into two groups with different patterns of activities (Fig. 2D and (20)). Additionally, correlations between the sigma factor regulons largely agreed with the concept of molecular time sharing, i.e. the idea that sigma factors compete for RNA polymerases (Fig. S19) (28).
To obtain an even finer-grained picture, we inferred the activity profiles of select transcriptional regulators from expression of the genes in their respective regulons (Figs. 2E and S20) (28). This revealed changes in regulation of carbon utilization, stress responses, metal uptake, developmental decisions and more (Fig. 2E). For example, we observe cellular response to a variety of intrinsic and cell-envelope stresses, as well as temporal activation patterns of a battery of developmental regulators (Fig. 2E). These data indicate that microSPLiT captures known regulatory programs and reveals heterogeneity in a wide range of cellular pathways.
Central carbon metabolism changes and transient activation of alternative carbon utilization pathways.
Given the changes in regulation of carbon utilization observed in our analysis of transcriptional regulators, we turned to a more comprehensive examination of carbon metabolism genes enriched in each cluster (Figs. 3A and S15–S18). When glucose and other preferred sugars are present, they are converted to pyruvate during glycolysis; the primary metabolic route when these sugars are abundant. In these conditions promoting rapid growth, pyruvate is then converted to lactate, acetate, acetoin, and other by-products through fermentation. Upon depletion of preferred sugars, cells redirect the fermentation by-products to be metabolized in the TCA cycle generating additional adenosine triphosphate (ATP) and carbon dioxide (29).
Fig. 3. Central carbon metabolism changes and alternative carbon sources utilization during B. subtilis growth.
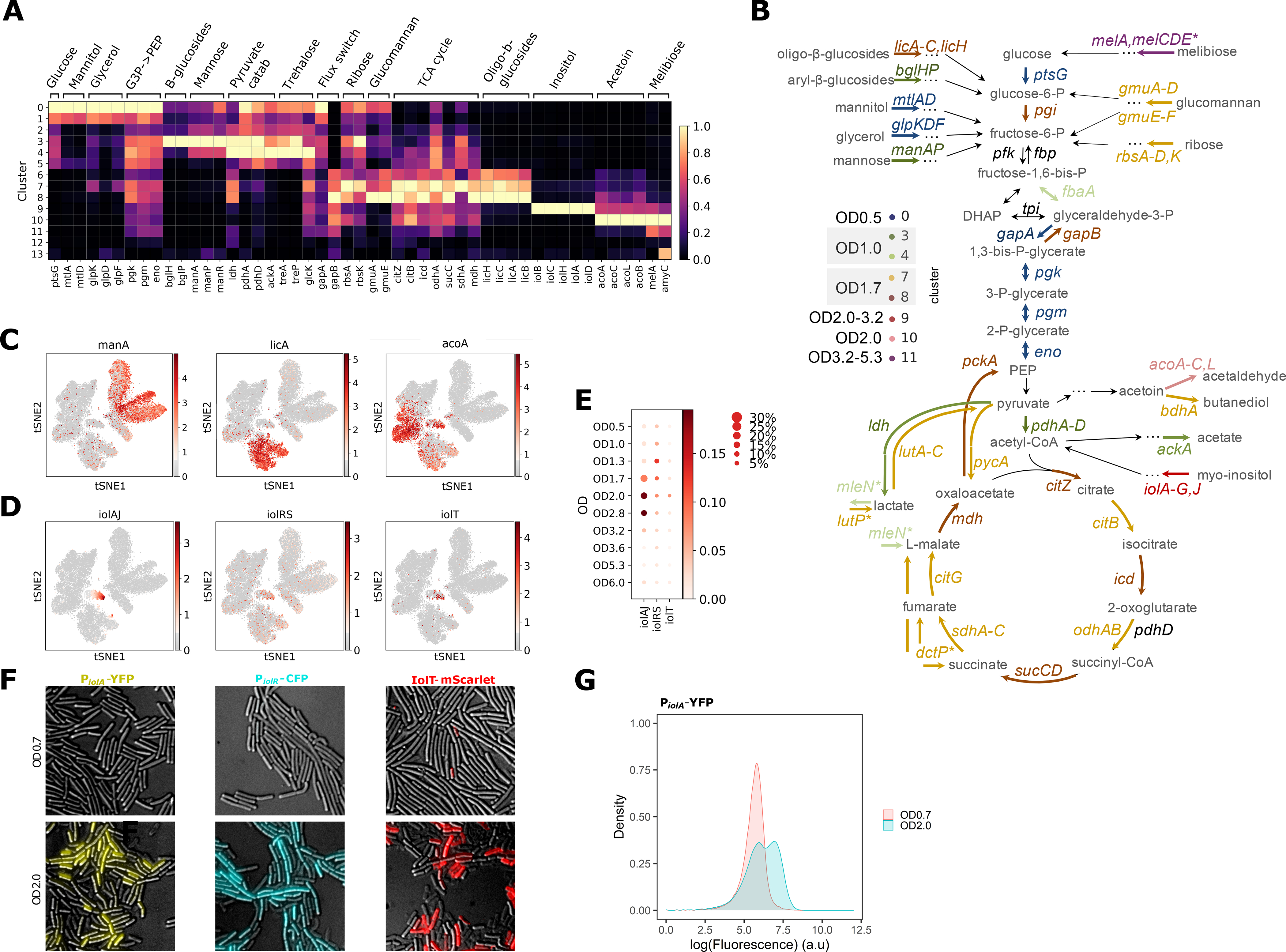
(A) Normalized expression of genes from select metabolic pathways and central carbon metabolism shown per cluster. Gene expression shows distinct carbon utilization programs associated with different clusters and growth states. (B) Schematic of the central carbon metabolism pathway showing alternative carbon sources, metabolic products and genes in the pathway. The genes are color-coded according to the cluster they are highest expressed in. (C) Expression of select genes from (A) overlaid on the t-SNE plot to illustrate the differential patterns of activation. (D) Expression of each of the three inositol utilization operons, averaged across all genes in a given operon, and overlaid on the t-SNE plot. (E) Activities of the three inositol utilization operons across ODs. The size of each dot indicates the proportion of cells in each OD sample expressing any of the genes in the selected operon, while the color shows the average expression of the genes in a given operon. (F) Fluorescence and DIC microscopy overlays of B. subtilis expressing PiolA-YFP (left), PiolR-CFP (middle) or IolT-mScarlet-I (right) grown in LB to OD0.7 (top row) or OD2.0 (bottom row). (G) Flow cytometry of PiolA-YFP strain grown to OD0.7 or 2.0.
As expected, in the clusters corresponding to early ODs (clusters 0–4) we observe peak expression of genes involved in glycolysis such as glucose permease (ptsG), and genes involved in rapid growth and fermentation such as lactate dehydrogenase (ldh), pyruvate dehydrogenase (pdhA) and acetate kinase (ackA) (Fig. 3A–B and (20)). At OD1.7 cells appear to undergo a dramatic transition from glycolysis to gluconeogenesis with multiple genes from the gluconeogenetic pathway activated in clusters 7 and 8 ((20) and Figs. 3A–C, S16–S17). We also find a different pattern of pyruvate production and utilization, together with catabolism of acetoin, another fermentation product, and additional nutrient fluxes into the TCA cycle ((20) and Figs. 3A–C, S16–S17).
Additionally, we observe expression of pathways responsible for uptake and utilization of different carbon sources. As the preferred sources of carbon are depleted, the major repressor of alternative carbon utilization pathways CcpA becomes inactive, permitting the cells to catabolize a variety of carbohydrates (30) (Fig. 3A–C). We find that the activation and suppression of these pathways happen in varying proportions of the cells in each OD sample and appear to follow a temporal order (Figs 3A–C, S15–S18, S21, and (20)).
Heterogeneous activation of myo-inositol catabolism pathway at intermediate growth stages.
Inositol is an abundant resource in soil, and B. subtilis is able to subsist on inositol as its sole carbon source (31). While LB medium is not typically expected to contain myo-inositol (further “inositol”), heterogeneous inositol utilization pathway activation is observed in a small (3–15% across OD1.7–3.2) subpopulation in both of our independent LB growth experiments (cluster 9, see Figs. 3A, 3D–E and S22). The inositol catabolism intermediate, 2-deoxy-5-keto-D-gluconic acid 6-phosphate (DKGP), is responsible for the pathway induction (31). We hypothesize that these results arise from trace amounts of inositol present in the LB medium, potentially from the yeast extract since yeast is capable of inositol production as a precursor to the essential membrane component, phosphatidylinositol.
There are three operons involved in inositol utilization, iolT (main transporter), iolRS (the first gene is a repressor and the second is a likely dehydrogenase), and iolA through J (metabolic enzymes, further “iolAJ”), with IolC producing and IolJ cleaving the pathway-activating DKGP intermediate. iolRS and iolAJ are normally transcribed by σA through divergent transcription (32). In the absence of the inducer, IolR suppresses transcription of all three operons (33). In addition, CcpA represses the iolAJ operon in the presence of glucose (34) ( Fig. S23A). Interestingly, we observe that the pathway suppressor iolR gene is more broadly expressed both outside and inside of cluster 9 (Figs. 3D–E and S22A).
To validate our findings, we constructed fluorescent reporters of all three operons in the inositol metabolism pathway (transcriptional reporters PiolA-YFP and PiolR-CFP and protein fusion IolT-mScarlet-I, (20)). As expected, we observed widespread expression of all three operons in the presence of inositol as a sole carbon source (Fig. S23B,D). In agreement with our clustering analysis (Fig. 3E), B. subtilis cells grown in LB show heterogeneous expression of PiolA-YFP in 22.7% of cells at OD2 and in 44% of cells at OD4, as opposed to cells grown in LB to OD0.7 (threshold set to 1% positive cells in the “off” state OD0.7, Fig. 3F–G and S23C,E). While microSPLiT data shows that the proportion of cells expressing inositol metabolism genes (belonging to cluster 9) drops from 5% at OD3.2 to 0.3% at OD5.3, the accumulation of YFP expressing cells at OD4 is consistent with the delay in fluorescent protein maturation and with the high stability of fluorescent proteins which mainly get cleared from cells by dilution during cell division.
Altogether, our transcriptomics and fluorescent reporter data indicate that the transcription of genes in the inositol metabolism pathway is activated in a heterogeneous fashion in a subpopulation of cells grown in LB between logarithmic and early stationary phases. The source of the activating molecule, as well as the underlying gene regulatory network architecture behind this behavior remain to be determined.
Motility, antimicrobials production, stress response, and metal ion import.
Next, we turned to examine a variety of B. subtilis behaviors thought to enhance survival in adverse conditions (35). Bacteria universally produce peptide and small molecule antimicrobials that are meant to target both closely and distantly related organisms (Fig. 4A) (36). We observe the expression of at least three broad spectrum antimicrobials – subtilosin, bacillaene, and plipastatin (37–39) - in various fractions of cells across ODs (Fig. 4A and S24). We also see a rise in spore killing factor (SKF) and spore delay protein (SDP) in the last three ODs (Fig. S25).
Fig. 4. Intrinsic stress responses and developmental gene expression.
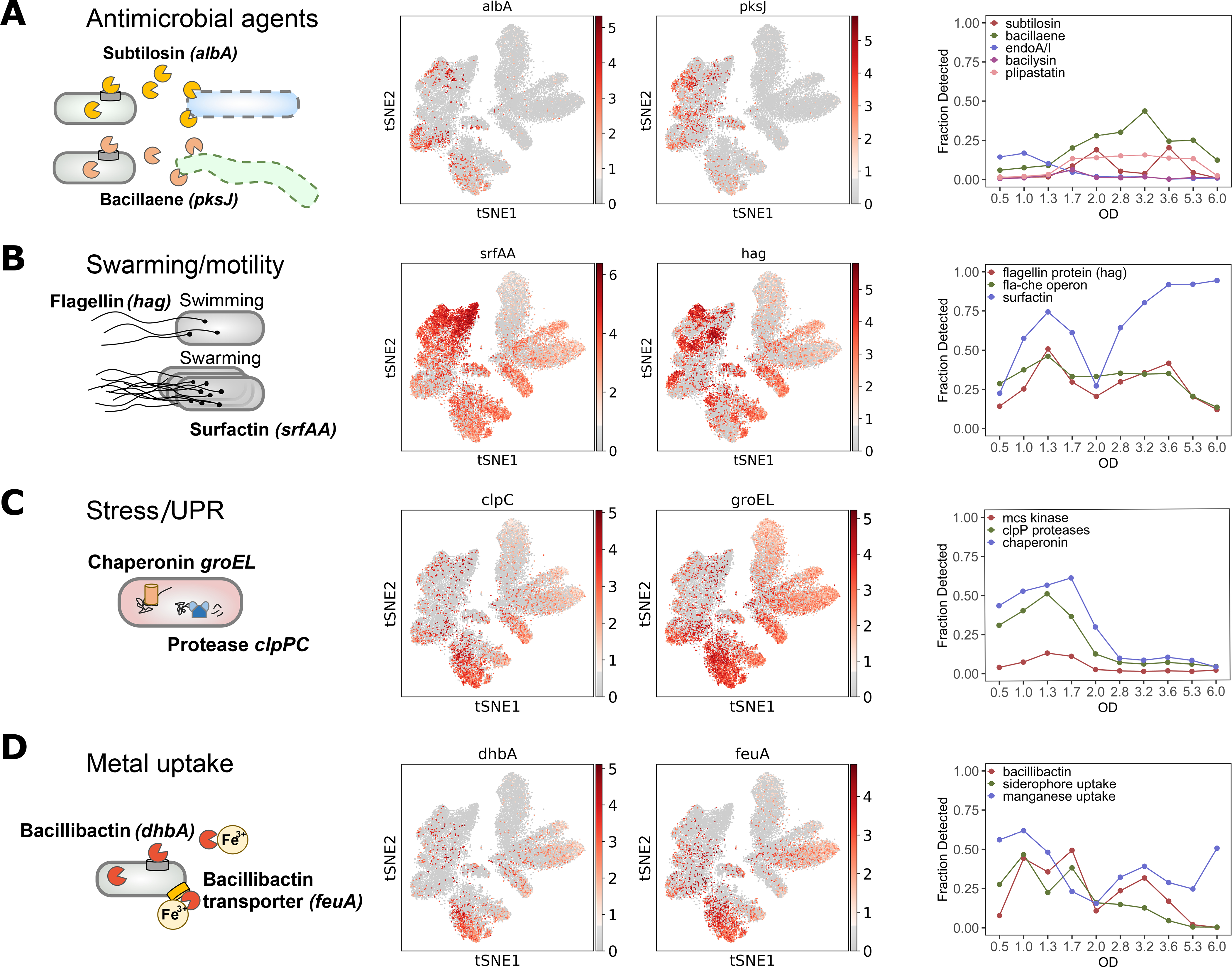
Left to right: Pathway diagram. Overlays of expression of genes representative of each pathway on the t-SNE. Fraction of cells expressing at least one of the genes in the indicated operon as a function of OD. (A) Antimicrobial agents (subtilosin (albA) and bacillaene (pksJ)) and endoA toxin-antitoxin system. (B) Swarming and motility (surfactin (srfAA) and flagellin (hag)). (C) Intrinsic stress and unfolded protein response (UPR) (GroEL chaperonin (groEL) and ClpCP protease (clpC)). (D) Iron (bacillibactin (dhbA) and siderophore transporter (feuA)) and manganese uptake. The genes used for fractional plots are listed in Table S2.
During active growth, B. subtilis can morphologically present as filamentous sessile cells or smaller motile cells (40, 41). Similarly, B. subtilis populations are expected to be differentiated into surfactin-producing and extracellular matrix-producing bacteria as cell density increases (42). We profiled the fraction of cells expressing motility genes (fla-che operon and flagellin encoded by hag), which noticeably declines at OD6.0. Meanwhile, surfactin (srfA-D) reaches almost 100% detection at OD6.0, consistent with the PY79 strain having defective matrix production genes that cannot negatively regulate srfA-D expression ((43), Fig. 4B).
We also found that genes involved in the unfolded protein response such as ClpP associated proteases (clpP,C,X,E), McsA and McsB kinases (mcsA,B), and chaperonins (groEL,ES) peak at OD1.7, the same time as the cells switch from glycolysis to gluconeogenesis (Fig. 4C). Expression of these genes is induced by the transient increase of regulatory sigma factor σB activity during normal exponential growth (see Fig. 2D) which is attributed to intrinsic cellular stresses (44). Additionally, we profiled expression of genes associated with metal uptake such as siderophore bacillibactin (dhbA) with associated transporter (feuA) and manganese transporter (mntA-D) (Fig. 4D and (20)).
microSPLiT quantifies a rare stress response.
Cluster 13 (36 cells, or 0.142% of total cells, representing ODs between 0.5 and 2.8) contains a rare subpopulation of cells expressing PBSX prophage genes (Fig. 5A). The PBSX element is a defective prophage that is non-infectious but upon induction causes the release of phage-like particles (45) containing 13 kb of random fragmented chromosomal DNA (46, 47). Prophage gene expression is induced by DNA damage (45, 48) and is known to activate in a small fraction of cells during exponential growth (49, 50) (Fig. 5B). The majority of genes (25) differentially expressed in cluster 13 represent known PBSX prophage genes with functions in PBSX prophage-mediated lysis (xlyA,B, xhlA,B), phage release (xepA), and phage replication (xtmA,B), and many PBSX-associated genes of unknown function (Fig. 5C). Thus, we not only identify a rare subpopulation of cells in the state of prophage induction, but also capture the expression of major phage-associated operons. We also identified eleven host genes with known or putative functions expressed in the PBSX prophage cluster (Fig. 5C). Five of these genes have previously been shown to be induced only in PBSX-harboring strains of B. subtilis after DNA damage (46). The rest, including a chemoreceptor (mcpC), an ATP-binding cassette transporter (liaL), a cell wall binding protein (ykuG), an ammonium transporter (amtB), a sucrose-6-phospate hydrolase (sacA), and a regulatory protein of homologous recombination (recX), have not previously been linked to prophage induction (20).
Fig. 5. Rare developmental states induced by cellular stress.
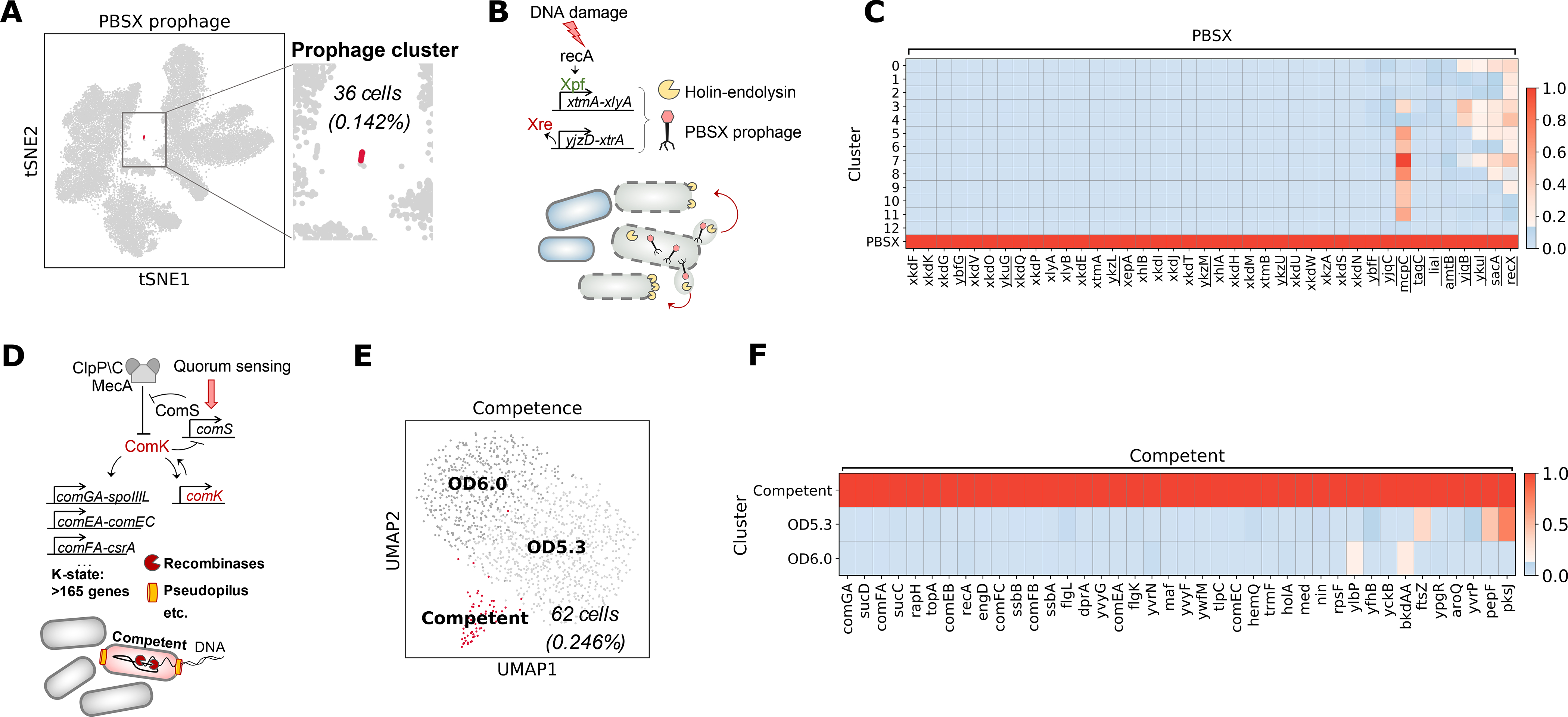
(A) Overview of PBSX prophage induction. (B) PBSX prophage cluster (36 cells) shown on the t-SNE plot. (C) Normalized averaged expression of genes enriched in the PBSX prophage cluster, including both prophage and host genes (underscored). (D) Overview of competence development. (E) UMAP embedding of the subclustered OD5.3 and 6.0 samples, showing the competence cluster (62 cells) in red. (F) Normalized averaged expression of genes enriched in the discovered competence cluster (shown in red) relative to the rest of the cells in OD5.3 and 6.0 samples.
microSPLiT captures a rare stochastically induced developmental state.
Under stress or nutrient limitation, a small fraction (2–5%) of B. subtilis cells undergoes stochastic transient differentiation into a state of natural competence, characterized by the ability to uptake extracellular DNA and integrate it into the chromosome (Fig. 5D) (49). The master transcriptional regulator of competence ComK is activated via a positive feedback loop, inducing expression of a suite of >165 genes involved in a variety of cellular processes in addition to DNA uptake (51). Competence is expected to naturally occur under nutrient limitation. We thus separately subclustered the last two OD points (OD5.3 and 6.0). UMAP embedding revealed a small cluster (62 cells, or 4.6% of cells at OD 5.3 and 6) expressing a distinct transcriptional signature of the competent state, or K-state (Fig. 5E–F). The observed frequency of competence is comparable to previous reports (3–10%, (51, 52)). The most enriched gene was comGA, as expected from prior transcriptomic data (53, 54), followed by the succinyl-CoA synthetase (sucCD) operon which is induced in competent cells (51, 53, 55). We also see enrichment of genes encoding the DNA uptake machinery: comF and comE operons, the response regulator (rapH) which represses sporulation development in competent cells (54), genes necessary for processing of internalized ssDNA such as recA along with genes for single-strand DNA binding proteins SsbA and SsbB (51), and other genes related to DNA processing (Fig. 5F, (20)). Overall, we capture the majority of genes associated with the state of competence as defined in two previous microarray studies (53, 54) as well as other approaches (56). In addition, we found four genes not previously linked to the competent state (Fig. 5F, (20)).
Discussion
We applied microSPLiT to B. subtilis cells growing in liquid rich medium, which is not associated with cellular heterogeneity. Nevertheless, we found a variety of subpopulations displaying differential gene expression of select metabolic, stress response or developmental pathways. In particular, we identified a myo-inositol catabolism pathway, which was activated only in a fraction of cells at later OD points in a distinct temporal fashion. We anticipate microSPLiT to be broadly useful in identifying heterogeneous cell states in more varied environments, such as in multi-species biofilms and natural microbiota.
We were able to detect subpopulations of cells as rare as 0.142% (Fig. 5A), pointing to microSPLiT’s potential to uncover physiologically relevant rare cell states, such as persistence, that are hard to study by bulk or low-throughput methods. The regulators for many such states are not well known and currently reporters or mutants producing the desired state at a higher frequency cannot be engineered. Even for better understood and inducible states, such as prophage induction by UV irradiation, microSPLiTcan produce state-specific transcriptional signatures free of artifacts introduced by the perturbation.
In order to use microSPLiT on complex natural communities, the protocol will likely need to be further optimized, particularly the permeabilization and mRNA enrichment steps as cell wall and membrane composition vary among bacteria. However, alternate treatment for different subsamples may still provide optimal results. In addition, we experienced lower mRNA counts from bacteria in stationary phase as opposed to logarithmic growth phase (Figs. S26 and S27), consistent with their slower growth rate and smaller cell size at this stage. Although the resulting data still reliably identified rare cell states, such as the K-state, further improvement of the protocol should increase sensitivity for applications to slower dividing bacteria or challenging environmental conditions. Finally, it may become desirable to increase cell retention, currently about 25% (20) between the RT step and sub-library preparation, as the method is applied to sparse natural communities rather than lab-grown cultures. Still, we expect microSPLiT to provide an exciting new dimension to studies of bacterial gene expression heterogeneity and community behavior facilitated by the potential scalability to millions of bacterial cells and single-cell resolution without the need for constructing reporters.
Materials and Methods Summary
Experimental Methods
Bacterial Culture
Escherichia coli MW1255, a derivative of MG1655, and Bacillus subtilis PY79 overnight cultures were inoculated into fresh 1.25x LB Lennox medium (Sigma-Aldrich) at 1:1000 or 1:250, respectively, and grown at 37C with shaking (200rpm). For the heat shock experiment, upon reaching the OD600 = 0.5, half of each culture was transferred to the separate 37C incubator where the temperature was increased to 47C, and kept there with shaking (200rpm) for 8 minutes from the time the temperature stabilized. Both control and heat-treated samples then were immediately centrifuged at 4C, 5000 rcf for 5 minutes, and resuspended in cold formaldehyde. Since we found a cluster in these data that may represent an artifact of the cold centrifugation conditions, the B. subtilis growth curve samples were instead centrifuged at room temperature, 10000 rpm for 1 minute before fixation.
Fixation and permeabilization
For the steps below, centrifugation was performed at 4C, 5000 rcf for 5 minutes. Following centrifugation, the bacterial pellet was resuspended in 1mL of fresh, cold, 4% formaldehyde solution (in 1x PBS) and incubated at 4C overnight. The next morning, cells were centrifuged and resuspended in 1ml cold 100mM Tris-HCL+RI (‘RI’ indicates that SUPERase-In RNase Inhibitor, ThermoFisher, was added to a final concentration of 0.1 U/uL). Cells were centrifuged and resuspended in 250ul of 0.04% Tween-20 in 1x PBS, then permeabilized for 3 min on ice. We then added 1ml of cold PBS+RI, centrifuged the cells and resuspended in 200ul lysozyme mix per sample on ice as follows: 0.1M Tris-HCL pH7, 0.05M EDTA, 2.5mg/ml lysozyme, 0.25U/ml SUPERase-In. We incubated the samples at 37C in the thermocycler for 15 minutes as we found that precise timing of lysozyme incubation is critical to maintaining cell integrity at later stages of the protocol. Following the cell wall digestion step, we immediately added 1ml of cold PBS+RI, centrifuged the cells and counted the cells stained with SYTO9 using Accuri C6 flow cytometer. For the species-mixing experiment, we mixed the E. coli and B. subtilis cells at equal proportions and took 0.6M cells for each of heat shocked and control samples. For the B. subtilis growth curve experiments, we took 0.25M cells for each OD sample.
In-cell Polyadenylation
In order to enrich for mRNA capture, we performed in situ polyadenylation with E. coli Poly(A) Polymerase I (PAP) from NEB. For 0.25M cell samples, the reaction proceeded in 50μl volume, and in 100μl volume for the 0.6M cell samples. For each 0.6M cells bacterial pellet, we added 66μl of water, 4μl of SUPERase-In, 10μl of 10x PAP Buffer, 10μl of 10mM ATP, and 10μl of PAP. The reaction mixture was incubated at 37C for 30min, then centrifuged upon addition of 1ml cold PBS+RI. We also added 1μL of 10% Tween-20 in order to make the cells easier to pellet. The cells then were resuspended in 0.5ml of cold PBS+RI.
Reducing aggregate formation
Two steps were crucial to break down cell aggregates and reduce the doublet rate: first, we vortexed and double-filtered the bacterial cells prior to reverse transcription; and second, we performed sonication with double filtration right after the reverse transcription.
Following the polyadenylation step, cells in 500ul of PBS-RI were vortexed for 1 minute on the highest setting, filtered through 10μm pluriStrainer (pluriSelect) by pipetting through the membrane, then filtered again through 1μm pluriStrainer with gentle suction, and finally, right before adding to the reverse transcription wells, vortexed again on the highest setting for 1 minute.
Following the reverse transcription step and after resuspension in 2 mL of cold PBS+RI, cells were vortexed for 1 minute on the highest setting, filtered through 10μm and 1μm pluriStrainer as above, and briefly sonicated at 10% power for 5s on ice for 1 pulse (Sharpertek Ultrasonic Cell Crusher) followed by immediate distribution to the ligation plate. We found that the sonication step can be replaced by a second vigorous vortexing step, with the roughly 2-fold increase in resulting detected doublet rate (0.7% to 1.3%).
In-cell Reverse Transcription
Like in SPLiT-seq, the first round of barcoding occurs through an in situ reverse transcription (RT) reaction. Cells are split into up to 48 wells, each containing barcoded well-specific reverse transcription primers. We used both random hexamer and anchored poly(dT)15 barcoded RT primers in each well at the ratio of 1:2 (2.5μM ramdom hexamer + 5μM poly(dT)15). In addition to primers, each RT well had a mix of 1x RT Buffer, 0.25U/μL RNase Inhibitor (Enzymatics), 0.25U/μL SUPERase-In RNase Inhibitor, 500μM dNTPs each (ThermoFisher), 7.5% of PEG8000, 20U/μL of Maxima H Minus Reverse Transcriptase (ThermoFisher). We pipetted 4 μL of cells at about 1M cells per mL in PBS-RI into every well, in the total resulting RT reaction volume of 20ul. The plate was incubated in a thermocycler for 10 min at 23˚C followed by 50˚C for 50 min. RT reactions were pooled back together and after adding 9.6 μL of 10% Triton X-100, cells were centrifuged for 5 min at 3000g at 4˚C in a swinging bucket rotor centrifuge. The supernatant was removed and cells were resuspended in 2 mL of cold 1X PBS-RI. The cells then underwent two rounds of filtration and sonication as described above.
In-cell Ligations
The oligonucleotide plates for the second and third barcoding round were prepared as previously described (7). We prepared a 2.04 mL ligation mix containing 727.5 μL of RNase-free water, 500 μL 10X T4 Ligase buffer (NEB), 20 μL T4 DNA Ligase (2000 U/μL, NEB), 30 μL RNase inhibitor (40 U/μL, Enzymatics), 12.5 μL SuperaseIn RNase Inhibitor (20 U/μL), and 750 μL of 50% PEG8000. This ligation mix and the 2 mL of sonicated and filtered cells in 1X PBS were added to a basin and mixed thoroughly to make a total of 4.04 mL. The ligation steps were performed as in the SPLiT-seq protocol (7), except we did vigorous vortexing combined with the double filtration technique as described above where the protocol called for a filtration step.
Lysis and Sublibrary Generation
After the third round of barcoding, we performed a final vigorous vortexing and double filtration step as described above. Then 70 μL of 10% Triton-X100 was added to the cell solution before spinning it down for 5 min at 3000G and 4˚C. We carefully aspirated the supernatant, leaving about 30 μL to avoid removing the pellet. We then resuspended the cells in 4 mL of wash buffer (4 mL of 1X PBS, 40 μL of 10% Triton X-100 and 10 μL of SUPERase-In RNase Inhibitor) and spun down for 5 min at 3000G and 4˚C. We then aspirated the supernatant and resuspended in 50 μL of PBS+RI. After counting cells, we aliquoted them into sublibraries (in 1.7 mL tubes). After adding the desired number of cells to each sublibrary, we brought the volume of each to 50 μL by adding 1x PBS and froze the cells at −80C overnight. Next morning, we flash-thawed the cells and added 50 μL of 2X lysis buffer (20 mM Tris (pH 8.0), 400 mM NaCl, 100 mM EDTA (pH 8.0), and 4.4% SDS) and 10 μL of proteinase K solution (20mg/mL). We incubated cells at 55˚C for 2 hours with shaking at 200 rpm to lyse the cells and reverse the formaldehyde crosslinks.
Computational Methods
Alignment and Generation of Cell-gene Matrices
The data preprocessing and alignment was performed using a modified SPLiT-seq pipeline (7), where the cDNA reads were mapped to either a combined B. subtilis – E. coli genome (ASM904v1.45 and ASM80076v1.37 from EnsemblBacteria (57)) or only the B. subtilis 168 genome (ASM904v1.45 from EnsemblBacteria) using STAR with the splicing isoform detection switched off (58). In addition, we kept the highest-scored multimapping reads, assigning a fractional count based on the number of equally-good alignments, since bacterial genomes are known to contain overlapping CDSs. We then generated a matrix of gene counts for each cell (N x K matrix, with N cells and K genes).
Processing of Data from the Heat Shock Experiment
Clustering and data analysis for the species-mixing experiment with heat shock treatment was performed using Scanpy (59). We only kept transcriptomes with the number of total reads higher than 200. Then, we removed the ribosomal and tRNA reads from the data, retaining only reads representing the mRNA counts for both species. We further filtered cells based on the mRNA counts, retaining cells expressing >100 reads and >100 genes, and additionally filtered the genes retaining the genes expressed in >5 cells. We then applied standard Scanpy normalization and scaling, dimensionality reduction, and clustering as described in the Scanpy tutorial (59, 60). The clusters were produced by Louvain graph-clustering method and manually inspected for the top differentially expressed genes. After inspection, three pairs of transcriptionally similar clusters with fewer differentially expressed genes were merged, resulting in clusters 1, 2 and 3 in Fig. 1D.
Processing of B. subtilis Data from the Growth in Rich Media Experiment
Clustering and data analysis for the combined 10 samples of B.S. grown in rich medium was performed using Scanpy (59) and verified with Seurat v3 (61) and UNCURL (62). Experiment 1 sampled OD points 0.5, 1.0, 1.7, 2.0, 2.8, and 3.2, while in experiment 2 we collected OD points 0.5, 1.0, 1.3, 1.7, 2.8, 3.5, 5.3, and 6.0. For the data from both experiments separately, we discarded any transcriptomes with the number of total reads fewer than 200. Then, we filtered the data and retained only reads representing the mRNA counts (excluding the ribosomal and tRNA reads). Finally, we combined the data matrices together. Since the read depth decreased for the higher OD samples (Fig. S26), for selecting the highest quality data, we implemented differential thresholds for each OD in the combined matrix, retaining top 75% of the cells by read counts for each OD sample. This resulted in retention of 25,214 transcriptomes from both experiments. Finally, we performed batch correction through Scanpy, using a python implementation of ComBat (63, 64). Cells that passed the QC were clustered using a pipeline described in previous studies (7, 60).
Supplementary Material
Acknowledgments:
Microscopy was performed at the University of Washington - W. M. Keck Microscopy Center.
Funding:
This work was supported by NIH grants NIH R01HG009136 and 580 R01HG009892 to G.S. and NIH R01DK104908–01 to W.D. L.P. was supported by Mary Gates Undergraduate Research Scholarship.
Footnotes
Competing interests: A.K., L.B. R.W.D. and G.S. are inventors on a patent application for microSPLiT filed by the University of Washington. C.R., A.B.R and G.S. are cofounders and shareholders of Split Biosciences, a single-cell RNA sequencing company.
Data and materials availability: All relevant sequencing files were deposited to Sequence Read Archive (SRA): GSM4594094, GSM4594095 and GSM4594096. Processed data was submitted to GEO, accession number GSE151940.
References and Notes:
- 1.Raj A, van Oudenaarden A, Nature, Nurture, or Chance: Stochastic Gene Expression and Its Consequences. Cell. 135, 216–226 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eldar A, Elowitz MB, Functional roles for noise in genetic circuits. Nature. 467, 167–173 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuchina A, Espinar L, Cagatay T, Balbin AO, Zhang F, Alvarado A, Garcia-Ojalvo J, Suel GM, Temporal competition between differentiation programs determines cell fate choice. Molecular Systems Biology. 7, 557–557 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elowitz MB, Stochastic Gene Expression in a Single Cell. Science. 297, 1183–1186 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Russell JR, Cabeen MT, Wiggins PA, Paulsson J, Losick R, Noise in a phosphorelay drives stochastic entry into sporulation in Bacillus subtilis. EMBO J. 36, 2856–2869 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Locke JCW, Elowitz MB, Using movies to analyse gene circuit dynamics in single cells. Nat. Rev. Microbiol 7, 383–392 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenberg AB, Roco CM, Muscat RA, Kuchina A, Sample P, Yao Z, Gray L, Peeler DJ, Mukherjee S, Chen W, Pun SH, Sellers DL, Tasic B, Seelig G, Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science, eaam8999 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao J, Packer JS, Ramani V, Cusanovich DA, Huynh C, Daza R, Qiu X, Lee C, Furlan SN, Steemers FJ, Adey A, Waterston RH, Trapnell C, Shendure J, Comprehensive single-cell transcriptional profiling of a multicellular organism. Science. 357, 661–667 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, Kirschner MW, Droplet Barcoding for Single-Cell Transcriptomics Applied to Embryonic Stem Cells. Cell. 161, 1187–1201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan HC, Fu GK, Fodor SPA, Combinatorial labeling of single cells for gene expression cytometry. Science. 347, 1258367–1258367 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, Trombetta JJ, Weitz DA, Sanes JR, Shalek AK, Regev A, McCarroll SA, Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 161, 1202–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sampaio NMV, Dunlop MJ, Functional roles of microbial cell-to-cell heterogeneity and emerging technologies for analysis and control. Current Opinion in Microbiology. 57, 87–94 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang J, Chen L, Chen Z, Zhang W, RNA-seq based transcriptomic analysis of single bacterial cells. Integr Biol (Camb). 7, 1466–1476 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Kang Y, Norris MH, Zarzycki-Siek J, Nierman WC, Donachie SP, Hoang TT, Transcript amplification from single bacterium for transcriptome analysis. Genome Res. 21, 925–935 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Imdahl F, Vafadarnejad E, Homberger C, Saliba A-E, Vogel J, Single-cell RNA-sequencing reports growth-condition-specific global transcriptomes of individual bacteria. Nature Microbiology. 5, 1202–1206 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Blattman SB, Jiang W, Oikonomou P, Tavazoie S, Prokaryotic single-cell RNA sequencing by in situ combinatorial indexing. Nature Microbiology. 5, 1192–1201 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wendisch VF, Zimmer DP, Khodursky A, Peter B, Cozzarelli N, Kustu S, Isolation of Escherichia coli mRNA and comparison of expression using mRNA and total RNA on DNA microarrays. Anal. Biochem 290, 205–213 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Wangsanuwat C, Heom KA, Liu E, O’Malley MA, Dey SS, “Efficient and cost-effective bacterial mRNA sequencing from low input samples through ribosomal RNA depletion” (preprint, Genomics, 2020), doi: 10.1101/2020.06.19.162412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He S, Wurtzel O, Singh K, Froula JL, Yilmaz S, Tringe SG, Wang Z, Chen F, Lindquist EA, Sorek R, Hugenholtz P, Validation of two ribosomal RNA removal methods for microbial metatranscriptomics. Nature Methods. 7, 807–812 (2010). [DOI] [PubMed] [Google Scholar]
- 20. Supplementary Materials.
- 21.Bartholomäus A, Fedyunin I, Feist P, Sin C, Zhang G, Valleriani A, Ignatova Z, Bacteria differently regulate mRNA abundance to specifically respond to various stresses. Philos Trans A Math Phys Eng Sci. 374 (2016), doi: 10.1098/rsta.2015.0069. [DOI] [PubMed] [Google Scholar]
- 22.Mohanty BK, Kushner SR, New Insights into the Relationship between tRNA Processing and Polyadenylation in Escherichia coli. Trends in Genetics. 35, 434–445 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arsène F, Tomoyasu T, Bukau B, The heat shock response of Escherichia coli. International Journal of Food Microbiology. 55, 3–9 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Schumann W, The Bacillus subtilis heat shock stimulon. Cell Stress Chaperones. 8, 207–217 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thieringer HA, Jones PG, Inouye M, Cold shock and adaptation. BioEssays. 20, 49–57 (1998). [DOI] [PubMed] [Google Scholar]
- 26.Haldenwang WG, The sigma factors of Bacillus subtilis. Microbiol. Mol. Biol. Rev. 59, 1–30 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Helmann JD, Bacillus subtilis extracytoplasmic function (ECF) sigma factors and defense of the cell envelope. Current Opinion in Microbiology. 30, 122–132 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park J, Dies M, Lin Y, Hormoz S, Smith-Unna SE, Quinodoz S, Hernández-Jiménez MJ, Garcia-Ojalvo J, Locke JCW, Elowitz MB, Molecular Time Sharing through Dynamic Pulsing in Single Cells. Cell Syst. 6, 216–229.e15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stülke J, Hillen W, Regulation of Carbon Catabolism in Bacillus Species. Annual Review of Microbiology. 54, 849–880 (2000). [DOI] [PubMed] [Google Scholar]
- 30.Pechter KB, Meyer FM, Serio AW, Stulke J, Sonenshein AL, Two Roles for Aconitase in the Regulation of Tricarboxylic Acid Branch Gene Expression in Bacillus subtilis. Journal of Bacteriology. 195, 1525–1537 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh KD, Schmalisch MH, Stulke J, Gorke B, Carbon Catabolite Repression in Bacillus subtilis: Quantitative Analysis of Repression Exerted by Different Carbon Sources. Journal of Bacteriology. 190, 7275–7284 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshida K, Yamaguchi M, Morinaga T, Kinehara M, Ikeuchi M, Ashida H, Fujita Y, myo-Inositol Catabolism in Bacillus subtilis. J. Biol. Chem 283, 10415–10424 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Yoshida KI, Aoyama D, Ishio I, Shibayama T, Fujita Y, Organization and transcription of the myo-inositol operon, iol, of Bacillus subtilis. Journal of Bacteriology. 179, 4591–4598 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoshida K-I, Shibayama T, Aoyama D, Fujita Y, Interaction of a Repressor and its Binding Sites for Regulation of the Bacillussubtilis iol Divergon. Journal of Molecular Biology. 285, 917–929 (1999). [DOI] [PubMed] [Google Scholar]
- 35.Marciniak BC, Pabijaniak M, de Jong A, Dűhring R, Seidel G, Hillen W, Kuipers OP, High- and low-affinity cre boxes for CcpA binding in Bacillus subtilis revealed by genome-wide analysis. BMC Genomics. 13, 401 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ben-Jacob E, Schultz D, Bacteria determine fate by playing dice with controlled odds. Proceedings of the National Academy of Sciences. 107, 13197–13198 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Granato ET, Meiller-Legrand TA, Foster KR, The Evolution and Ecology of Bacterial Warfare. Current Biology. 29, R521–R537 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Shelburne CE, An FY, Dholpe V, Ramamoorthy A, Lopatin DE, Lantz MS, The spectrum of antimicrobial activity of the bacteriocin subtilosin A. J Antimicrob Chemother. 59, 297–300 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Patel PS, Huang S, Fisher S, Pirnik D, Aklonis C, Dean L, Meyers E, Fernandes P, Mayerl F, Bacillaene, a novel inhibitor of procaryotic protein synthesis produced by Bacillus subtilis: production, taxonomy, isolation, physico-chemical characterization and biological activity. J. Antibiot 48, 997–1003 (1995). [DOI] [PubMed] [Google Scholar]
- 40.Gao L, Han J, Liu H, Qu X, Lu Z, Bie X, Plipastatin and surfactin coproduction by Bacillus subtilis pB2-L and their effects on microorganisms. Antonie van Leeuwenhoek. 110, 1007–1018 (2017). [DOI] [PubMed] [Google Scholar]
- 41.van Gestel J, Vlamakis H, Kolter R, From Cell Differentiation to Cell Collectives: Bacillus subtilis Uses Division of Labor to Migrate. PLOS Biology. 13, e1002141 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kearns DB, Losick R, Cell population heterogeneity during growth of Bacillus subtilis. Genes Dev. 19, 3083–3094 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.López D, Vlamakis H, Losick R, Kolter R, Paracrine signaling in a bacterium. Genes Dev. 23, 1631–1638 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krüger E, Witt E, Ohlmeier S, Hanschke R, Hecker M, The clp proteases of Bacillus subtilis are directly involved in degradation of misfolded proteins. J. Bacteriol 182, 3259–3265 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mhatre E, Troszok A, Gallegos-Monterrosa R, Lindstädt S, Hölscher T, Kuipers OP, Kovács ÁT, The impact of manganese on biofilm development of Bacillus subtilis. Microbiology (Reading, Engl.). 162, 1468–1478 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Wood HE, Dawson MT, Devine KM, McConnell DJ, Characterization of PBSX, a defective prophage of Bacillus subtilis. J. Bacteriol 172, 2667–2674 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goranov AI, Kuester-Schoeck E, Wang JD, Grossman AD, Characterization of the Global Transcriptional Responses to Different Types of DNA Damage and Disruption of Replication in Bacillus subtilis. Journal of Bacteriology. 188, 5595–5605 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anderson LM, Bott KF, DNA packaging by the Bacillus subtilis defective bacteriophage PBSX. J. Virol 54, 773–780 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shingaki R, Kasahara Y, Inoue T, Kokeguchi S, Fukui K, Chromosome DNA fragmentation and excretion caused by defective prophage gene expression in the early-exponential-phase culture of Bacillus subtilis. Canadian Journal of Microbiology. 49, 313–325 (2003). [DOI] [PubMed] [Google Scholar]
- 50.Nanda AM, Thormann K, Frunzke J, Impact of Spontaneous Prophage Induction on the Fitness of Bacterial Populations and Host-Microbe Interactions. Journal of Bacteriology. 197, 410–419 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Süel GM, Garcia-Ojalvo J, Liberman LM, Elowitz MB, An excitable gene regulatory circuit induces transient cellular differentiation. Nature. 440, 545–550 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Hamoen LW, Venema G, Kuipers OP, Controlling competence in Bacillus subtilis: shared use of regulators. Microbiology,. 149, 9–17 (2003). [DOI] [PubMed] [Google Scholar]
- 53.Berka RM, Hahn J, Albano M, Draskovic I, Persuh M, Cui X, Sloma A, Widner W, Dubnau D, Microarray analysis of the Bacillus subtilis K-state: genome-wide expression changes dependent on ComK. Molecular Microbiology. 43, 1331–1345 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Ogura M, Yamaguchi H, Kobayashi K, Ogasawara N, Fujita Y, Tanaka T, Whole-genome analysis of genes regulated by the Bacillus subtilis competence transcription factor ComK. J. Bacteriol 184, 2344–2351 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grundy FJ, Waters DA, Allen SH, Henkin TM, Regulation of the Bacillus subtilis acetate kinase gene by CcpA. Journal of Bacteriology. 175, 7348–7355 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rosenthal AZ, Qi Y, Hormoz S, Park J, Li SH-J, Elowitz MB, Metabolic interactions between dynamic bacterial subpopulations. Elife 7 (2018), doi: 10.7554/eLife.33099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Howe KL, Contreras-Moreira B, De Silva N, Maslen G, Akanni W, Allen J, Alvarez-Jarreta J, Barba M, Bolser DM, Cambell L, Carbajo M, Chakiachvili M, Christensen M, Cummins C, Cuzick A, Davis P, Fexova S, Gall A, George N, Gil L, Gupta P, Hammond-Kosack KE, Haskell E, Hunt SE, Jaiswal P, Janacek SH, Kersey PJ, Langridge N, Maheswari U, Maurel T, McDowall MD, Moore B, Muffato M, Naamati G, Naithani S, Olson A, Papatheodorou I, Patricio M, Paulini M, Pedro H, Perry E, Preece J, Rosello M, Russell M, Sitnik V, Staines DM, Stein J, Tello-Ruiz MK, Trevanion SJ, Urban M, Wei S, Ware D, Williams G, Yates AD, Flicek P, Ensembl Genomes 2020—enabling non-vertebrate genomic research. Nucleic Acids Research. 48, D689–D695 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wolf FA, Angerer P, Theis FJ, SCANPY: large-scale single-cell gene expression data analysis. Genome Biology. 19, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luecken MD, Theis FJ, Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol 15, e8746 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, Satija R, Comprehensive Integration of Single-Cell Data. Cell. 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mukherjee S, Zhang Y, Fan J, Seelig G, Kannan S, Scalable preprocessing for sparse scRNA-seq data exploiting prior knowledge. Bioinformatics. 34, i124–i132 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Johnson WE, Li C, Rabinovic A, Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 8, 118–127 (2007). [DOI] [PubMed] [Google Scholar]
- 64.Pederson Brent, patsy version of ComBat for removing batch effects. (GitHub, 2012; https://github.com/brentp/combat.py).
- 65.Sastalla I, Chim K, Cheung GYC, Pomerantsev AP, Leppla SH, Codon-Optimized Fluorescent Proteins Designed for Expression in Low-GC Gram-Positive Bacteria. Appl Environ Microbiol. 75, 2099–2110 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ellis B, Haaland P, Hahne F, Le Meur N, Gopalakrishnan N, Spidlen J, Jiang M, Finak G, flowCore: Basic structures for flow cytometry data (2020).
- 67.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, Vanderplas J, Passos A, Cournapeau D, Brucher M, Perrot M, Duchesnay É, Scikit-learn: Machine Learning in Python. Journal of Machine Learning Research. 12, 2825–2830 (2011). [Google Scholar]
- 68.van der Maaten L, Accelerating t-SNE using Tree-Based Algorithms. Journal of Machine Learning Research. 15, 3221–3245 (2014). [Google Scholar]
- 69.McInnes L, Healy J, Melville J, UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv:1802.03426 [cs, stat] (2018) (available at http://arxiv.org/abs/1802.03426). [Google Scholar]
- 70.Zhu B, Stülke J, SubtiWiki in 2018: from genes and proteins to functional network annotation of the model organism Bacillus subtilis. Nucleic Acids Res. 46, D743–D748 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chastanet A, Vitkup D, Yuan G-C, Norman TM, Liu JS, Losick RM, Broadly heterogeneous activation of the master regulator for sporulation in Bacillus subtilis. Proceedings of the National Academy of Sciences. 107, 8486–8491 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Luo Y, Asai K, Sadaie Y, Helmann JD, Transcriptomic and Phenotypic Characterization of a Bacillus subtilis Strain without Extracytoplasmic Function Factors. Journal of Bacteriology. 192, 5736–5745 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ludwig H, Homuth G, Schmalisch M, Dyka FM, Hecker M, Stülke J, Transcription of glycolytic genes and operons in Bacillus subtilis: evidence for the presence of multiple levels of control of the gapA operon: Regulation of glycolysis in Bacillus subtilis. Molecular Microbiology. 41, 409–422 (2001). [DOI] [PubMed] [Google Scholar]
- 74.Larsson JT, Coordinated patterns of cytochrome bd and lactate dehydrogenase expression in Bacillus subtilis. Microbiology. 151, 3323–3335 (2005). [DOI] [PubMed] [Google Scholar]
- 75.Wei Y, Guffanti AA, Ito M, Krulwich TA, Bacillus subtilis YqkI Is a Novel Malic/Na + -Lactate Antiporter That Enhances Growth on Malate at Low Protonmotive Force. Journal of Biological Chemistry. 275, 30287–30292 (2000). [DOI] [PubMed] [Google Scholar]
- 76.Lowe PN, Hodgson JA, Perham RN, Dual role of a single multienzyme complex in the oxidative decarboxylation of pyruvate and branched-chain 2-oxo acids in Bacillus subtilis. Biochemical Journal. 215, 133–140 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chai Y, Kolter R, Losick R, A Widely Conserved Gene Cluster Required for Lactate Utilization in Bacillus subtilis and Its Involvement in Biofilm Formation. Journal of Bacteriology. 191, 2423–2430 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tojo S, Kumamoto K, Hirooka K, Fujita Y, Heavy Involvement of Stringent Transcription Control Depending on the Adenine or Guanine Species of the Transcription Initiation Site in Glucose and Pyruvate Metabolism in Bacillus subtilis. Journal of Bacteriology. 192, 1573–1585 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nicholson WL, The Bacillus subtilis ydjL (bdhA) Gene Encodes Acetoin Reductase/2,3-Butanediol Dehydrogenase. Applied and Environmental Microbiology. 74, 6832–6838 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Baik S-H, Ogasawara N, Asai K, Kasahara Y, Moriya S, Regulation of the transport system for C4-dicarboxylic acids in Bacillus subtilis. Microbiology. 146, 263–271 (2000). [DOI] [PubMed] [Google Scholar]
- 81.Blencke H-M, Homuth G, Ludwig H, Mäder U, Hecker M, Stülke J, Transcriptional profiling of gene expression in response to glucose in Bacillus subtilis: regulation of the central metabolic pathways. Metabolic Engineering. 5, 133–149 (2003). [DOI] [PubMed] [Google Scholar]
- 82.Kormelink T, Koenders E, Hagemeijer Y, Overmars L, Siezen RJ, de Vos WM, Francke C, Comparative genome analysis of central nitrogen metabolism and its control by GlnR in the class Bacilli. BMC Genomics. 13, 191 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Randazzo P, Aucouturier A, Delumeau O, Auger S, Revisiting the in vivo GlnR-binding sites at the genome scale in Bacillus subtilis. BMC Research Notes. 10 (2017), doi: 10.1186/s13104-017-2703-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Morabbi Heravi K, Altenbuchner J, Cross Talk among Transporters of the Phosphoenolpyruvate-Dependent Phosphotransferase System in Bacillus subtilis. Journal of Bacteriology. 200 (2018), doi: 10.1128/JB.00213-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Garrity LF, Schiel SL, Merrill R, Reizer J, Saier MH, Ordal GW, Unique regulation of carbohydrate chemotaxis in Bacillus subtilis by the phosphoenolpyruvate-dependent phosphotransferase system and the methyl-accepting chemotaxis protein McpC. J. Bacteriol. 180, 4475–4480 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jordan S, Junker A, Helmann JD, Mascher T, Regulation of LiaRS-Dependent Gene Expression in Bacillus subtilis: Identification of Inhibitor Proteins, Regulator Binding Sites, and Target Genes of a Conserved Cell Envelope Stress-Sensing Two-Component System. Journal of Bacteriology. 188, 5153–5166 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ye B-C, Zhang Y, Yu H, Yu W-B, Liu B-H, Yin B-C, Yin C-Y, Li Y-Y, Chu J, Zhang S-L, Time-Resolved Transcriptome Analysis of Bacillus subtilis Responding to Valine, Glutamate, and Glutamine. PLoS ONE. 4, e7073 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cárdenas PP, Carrasco B, Defeu Soufo C, César CE, Herr K, Kaufenstein M, Graumann PL, Alonso JC, RecX Facilitates Homologous Recombination by Modulating RecA Activities. PLoS Genetics. 8, e1003126 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Smits WK, Bongiorni C, Veening J-W, Hamoen LW, Kuipers OP, Perego M, Temporal separation of distinct differentiation pathways by a dual specificity Rap-Phr system in Bacillus subtilis. Molecular Microbiology. 65, 103–120 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.