

Abstract
The physiological and pathophysiological relevance of the angiotensin II type 1 (AT1) G protein-coupled receptor no longer needs to be proven in the cardiovascular system. The renin–angiotensin system and the AT1 receptor are the targets of several classes of therapeutics (such as angiotensin converting enzyme inhibitors or angiotensin receptor blockers, ARBs) used as first-line treatments in cardiovascular diseases. The importance of AT1 in the regulation of the cerebrovascular system is also acknowledged. However, despite numerous beneficial effects in preclinical experiments, ARBs do not induce satisfactory curative results in clinical stroke studies. A better understanding of AT1 signaling and the development of biased AT1 agonists, able to selectively activate the β-arrestin transduction pathway rather than the Gq pathway, have led to new therapeutic strategies to target detrimental effects of AT1 activation. In this paper, we review the involvement of AT1 in cerebrovascular diseases as well as recent advances in the understanding of its molecular dynamics and biased or non-biased signaling. We also describe why these alternative signaling pathways induced by β-arrestin biased AT1 agonists could be considered as new therapeutic avenues for cerebrovascular diseases.
Keywords: AT1 receptor, Angiotensin II, cerebrovascular disease, biased agonism, beta-arrestin, RAS, TRV023, TRV027, Ang-(1–7)
1. Introduction
The systemic renin-angiotensin system (RAS) is a well-known hormonal cascade involved in many regulation processes such as the control of arterial pressure and vascular homeostasis. Angiotensinogen, a protein produced by the liver, is turned into angiotensin I, a decapeptide, by renin, an enzyme released by the kidney in response to various stimuli (blood pressure decrease, sympathetic stimulation). Angiotensin II (Ang II), an octapeptide, is generated by the cleavage of angiotensin I by the angiotensin-converting enzyme (ACE) mainly expressed at the endothelial surface. Ang II is known as the main effector of the RAS. Ang II binds with a similar affinity to two receptors: the Ang II type 1 receptor (AT1) and type 2 receptor (AT2), both belonging to the G protein-coupled receptor (GPCR) family and sharing 50% homology and 34% identity in their amino acid sequence [1,2,3].
Ang II activation of AT1 is acknowledged as triggering most of the known effects of RAS stimulation [4,5], such as vasoconstriction, water and sodium retention and aldosterone release by the adrenal glands. This leads to increases in blood pressure, cardiovascular remodeling and fibrosis [6,7,8]. In most physiological situations involving Ang II release, AT1 activation is predominant and will blunt the opposite effects induced by AT2 stimulation, such as vasodilation, apoptosis and antiproliferative effects [9,10,11,12]. Despite this AT1 predominance in Ang II mediated effects (mostly related to a more abundant expression of AT1 compared to AT2), the concept of a “protective arm” of RAS is a subject of intensive research: it is composed of AT2 and the Mas receptor (MasR), a GPCR activated by an endogenous truncated form of Ang II (Ang-(1–7)). For extensive reviews on the protective effect of AT2 activation, please refer to Matavelli’s or Carey’s works [13,14] and for the MasR, refer to Povlsen’s work [15].
Due to its wide physiological effects, AT1 plays a critical role in many pathological conditions and cardiovascular diseases, like cardiac hypertrophy, hypertension and heart failure. For decades, treatments preventing AT1 stimulation have been widely used as first line therapies for these cardiovascular diseases. ACE inhibitors, which prevent the cleavage of angiotensin I into Ang II, were developed in the 1980s. AT1 selective antagonists, called AT1 receptor blockers (ARBs or sartans), are a biphenyl-tetrazole based family of compounds first commercialized in the 1990s.
The involvement of AT1 in the regulation of cerebral circulation is also well established [16,17] and a cerebrovascular local production of Ang II has been shown [18]. AT1-dependent processes have been reported in cerebrovascular diseases including ischemic and hemorrhagic strokes, subarachnoid aneurysms, traumatic brain injury and might also be found in neurodegenerative processes, like Alzheimer’s and Parkinson’s diseases [19,20]. To date, etiological therapeutic treatments and/or therapeutic alternatives are scarce for all of these pathological conditions. This lack of efficient therapeutic options further justifies the growing interest for RAS and AT1 as a therapeutic target in brain disorders.
AT1 signaling pathways have been extensively studied over the past 25 years. AT1 is coupled to the heterotrimeric Gq/11 protein that represents the main AT1 signal transduction process involved in vasoconstriction of vascular smooth muscle cells (VSMC). However, recent advances suggest that AT1 activation also leads to β-arrestin-dependent signaling pathways that could exert protective effects against cardiovascular diseases.
In the present review, we first discuss the major role of AT1 in cerebrovascular diseases, before reviewing the preclinical and clinical studies showing the interest and limits of AT1 blockade with ARBs in such situations (with a particular emphasis on strokes). We analyze the structure/function relationship of AT1 with different agonists based on recent seminal articles that have led to the demonstration that a specific alternative active conformational state of AT1 almost exclusively activates β-arrestin signaling instead of G protein signaling. Finally, we show the therapeutic potential of promoting this exclusive AT1/β-arrestin biased signaling in pathophysiological situations where cerebral circulation is altered.
2. Implication of AT1 in Cerebrovascular Disease
2.1. Role of AT1 Activation in Strokes
Strokes are a major cause of death in industrialized countries and may lead to various physical disabilities consecutive to neurological deficit [21]. Strokes may result from two distinct etiologies: ischemia or hemorrhage. An ischemic stroke is the consequence of an obstruction or a vasospasm of a cerebral artery or arteriole [22], whereas a hemorrhagic stroke results from the rupture of the arterial, or arteriolar, wall. In both cases, the subsequent blood deprivation will lead to reduced oxygen levels, ischemia and necrosis of the downstream brain tissue.
Numerous preclinical data suggest that an overactivation of AT1 by Ang II might contribute to the severity of cerebral ischemia and its consequences, mainly through its vasoconstrictor effects on cerebral arteries [23]. Indeed, in a classic rodent model of ischemic stroke, the middle cerebral artery occlusion model (MCAo, in which the middle cerebral artery is temporarily occluded by a nylon monofilament), increases in the local production of Ang II and in the AT1-dependent vasoconstriction of cerebral arteries have been reported [24,25]. In the context of cerebral ischemia, Ang II and AT1 stimulation thus lead to a decreased perfusion of the penumbra and might expand the deleterious consequences of a stroke [26]. Besides these acute post-stroke effects, chronic AT1 stimulation is the main determinant of hypertension-induced inward remodeling of the cerebral arteriolar wall [17,27,28]. Indeed, hypertension, one of the major risk factors for strokes, induces a structural narrowing of the cerebral arterial lumen (thus reducing blood perfusion in case of a stroke) that only antihypertensive treatments targeting RAS and AT1 stimulation (ACE inhibitors and/or ARBs) are able to prevent [17,28,29].
Ang II and AT1 stimulation also induce a proinflammatory response [30] and increase oxidative stress through the production of ROS, thus promoting cellular damage and apoptosis [31,32]. These effects might also contribute to the increase in brain damage and the enlargement of the penumbra in case of ischemia and during reperfusion.
Numerous preclinical and clinical studies have investigated the effects of ARBs, confirming the involvement of AT1 in cerebral ischemia. These studies have been reviewed recently [33] and are discussed in Section 3.
2.2. Role of AT1 Activation in Cerebral Aneurysm
Cerebral aneurysms are an abnormal condition in which a weakness of the cerebral arterial wall causes a localized dilation or bulge. Cerebral aneurysms can thus lead to hemorrhagic strokes showing a high mortality rate [34]. Given their role as major determinants of the cerebral arterial and arteriolar wall structure, RAS and AT1 might be good candidates to participate in the development of cerebral aneurysms.
Regarding abdominal aortic aneurysms, the involvement of Ang II and RAS stimulation has been clearly demonstrated in animals, and strong evidence have been gathered in humans [35,36]. However, when considering cerebral aneurysms, the role of RAS components and AT1 seems less obvious. In a study comparing patients with unruptured or ruptured cerebral aneurysm to controls, a significant drop in the mRNA expressions of ACE and AT1 has been observed [37]. Immunohistochemistry also revealed a decrease in AT1 and Ang II expression in unruptured aneurysmal walls. This decrease was even more pronounced in the arterial walls of patients with ruptured cerebral aneurysm [37]. Given the hypertrophic and remodeling effect of AT1, the authors suggested that a decreased expression of RAS components could lead to a reduced wall thickness and vascular remodeling which may result in cerebral aneurysms [37].
Preclinical data do not completely support these observations. In an experimental model of aneurysm in rats combining a left internal carotid artery ligation with a left renal artery ligation and a high salt diet (to increase blood pressure), no change in AT1 and ACE protein expression could be observed in cerebral aneurysmal walls. The authors therefore excluded any implication of AT1 or ACE in cerebral aneurysm formation [38,39], as further supported by the lack of effect of an ARB treatment [38]. If not involved in the development of cerebral aneurysms, RAS has been proposed to play a pivotal role in their rupture. In a mouse model of cerebral aneurysm (obtained by combining induced systemic hypertension with a single injection of elastase into the cerebrospinal fluid at the right basal cistern), Tada et al. prevented the spontaneous aneurysmal rupture with either an ACE inhibitor or an ARB, in a pressure-independent manner [40]. It is noteworthy that, unlike the rat model evoked above, this particular model of intracranial aneurysm showed an increased expression of Ang II and AT1 in aneurysmal walls, as revealed by immunohistochemistry [40].
The role of AT1 in the pathogenesis of cerebral aneurysm still appears to be controversial. Even if some data suggest a less consistent role of AT1 compared to other cerebrovascular diseases, further investigations are required to clarify the role of AT1 and its alternative signaling pathways in cerebral aneurysm.
2.3. Role of AT1 Activation in Traumatic Brain Injury
The Ang II/AT1 axis also plays a pivotal role in traumatic brain injury (TBI), another major cause of death and disability. A direct lesion of the brain will result in neuronal damage, increased cell death, dysfunction of the blood-brain barrier and increased inflammatory response. TBI may thus lead to motor and cognitive disorders. In victims of severe TBI, the presence of the D allele of ACE genotype, leading to increased ACE activity, has been correlated with disease mortality [41]. The subsequent excessive AT1 activation might contribute to neuronal injury and vulnerability by inducing cerebrovascular remodeling and inflammation [42].
Two genes encode for AT1 in rodents, AT1A and AT1B, and both share 93–94% amino-acids identity with the unique human AT1 protein [43,44]. AT1A has been described as the most abundant AT1 isoform expressed in mouse brain [45]. A decrease in susceptibility to traumatic injury was observed in AT1A knockout mice [46].
2.4. Role of AT1 Activation in Neurodegenerative Diseases
AT1 has also been reported as key determinant in many neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases. However, the involvement of AT1 in these pathologies appears to be mostly located at the neuronal level. Recent reviews have been published on this matter [19,20,47] and this will not be further detailed here, as our present review addresses the cerebrovascular renin–angiotensin system.
2.5. Limitations
It is noteworthy that most of the studies cited above evaluated AT1 protein expression by means of antibodies targeting AT1 (Western blot and/or immunohistochemistry). These antibodies are notoriously unspecific [48,49,50] and the results obtained with these antibodies have therefore to be interpreted with caution. Nevertheless, the involvement of AT1 in cerebrovascular diseases has been proven by many other means.
The roles of AT1 in cerebrovascular diseases are summarized in Figure 1.
Figure 1.

Implication of AT1 in cerebrovascular diseases. ACE: angiotensin-converting enzyme; Ang II: angiotensin II; AT1: angiotensin II type 1 receptor; mRNA: messenger ribonucleic acid; ROS: reactive oxygen species. The figure was prepared using the Servier Medical Art website (Creative Commons Attribution 3.0 unported License).
3. Blocking AT1 by ARBs in Cerebrovascular Disease: Success and Limitations
AT1 thus appears to be a first-order therapeutic target against cerebrovascular diseases, as has previously been clearly established for cardiovascular diseases. Currently, eight ARBs are clinically available, among which the most studied are losartan, candesartan, telmisartan and valsartan (Figure 2). Often described as surmountable (losartan) or unsurmountable (candesartan, telmisartan, valsartan) antagonists on the basis of their impact on the Ang II concentration response curve [51], ARBs show an inverse agonistic action by stabilizing the inactive state of AT1 (see Section 4.2 below). ACE inhibitors and ARBs have been extensively studied to assess their potential preventive and curative effects in cerebrovascular pathologies.
Figure 2.

Chemical structures of the main angiotensin receptor blockers (ARB) used in therapeutics. The biphenyltetrazole (or the bioisosteric biphenyl carboxylic acid for telmisartan) is highlighted in blue.
Numerous preclinical studies show the beneficial effects of ARBs at the cerebrovascular level. ARBs regulate cerebral blood flow by blocking AT1 in cerebral arterial VSMC and endothelial cells [52]. ARBs improve blood perfusion, reduce apoptosis and decrease the infarct size after an ischemic stroke [53]. A 2-week pretreatment with candesartan before MCAo ischemic strokes in normotensive or hypertensive rats reduces the infarct volume and improves the lower and upper limits of cerebral blood flow autoregulation [54]. This chronic treatment also reduces vascular remodeling [55] and neuronal damage after cerebral ischemia [56,57]. Candesartan has also been reported to reduce neuronal damage in mice after a TBI [58] while valsartan could prevent these lesions [59]. Losartan has shown beneficial effects to prevent aneurysmal subarachnoid hemorrhage in a murine model [40]. Taken together, these preclinical data (which have recently been extensively reviewed [33,60]) provide evidence for the cerebrovascular potential of ARBs.
However, the transposition to clinic does not entirely support these preclinical data. Numerous clinical studies highlight the beneficial effect of long-term RAS blockades on stroke prevention. Early trials were conducted with ACE inhibitors. The stroke rate was reduced by 25% in patients receiving the ACE inhibitor captopril in the CAPPP (CAPtopril Prevention Project) trial [61] and this reduction reached 32% in the HOPE (Heart Outcome Prevention Evaluation) trial with the ACE inhibitor ramipril [62]. Similar drops in stroke recurrence were recorded in patients treated with perindopril, another ACE inhibitor, in the PROGRESS (perindopril PROtection aGainst REcurrent Stroke Study) study [63]. Interestingly, clinical studies using ARBs show identical results, suggesting that the beneficial effect of ACE inhibitors is mainly related to the prevention of AT1 stimulation. The LIFE (Losartan Intervention For Endpoint reduction in hypertension) study demonstrated that treatment with losartan prevented the risk of ischemia by 25% as compared to β-blockers that target β-adrenergic receptors [64]. The impact of candesartan was examined in the SCOPE (Study on COgnition and Prognosis in the Elderly) trial in which a risk reduction of 28% for non-fatal stroke was observed [65]. Similar results were found in the MOSES (MOrbidity and mortality after Stroke, Eprosartan compared with the calcium channel blocker nitrendipine for Secondary prevention) study with a 24% reduction in the stroke incidence in people treated with eprosartan [66]. Even if some studies failed to show such a dramatic effect (e.g., TRANSCEND [67] and PROFESS [68] trials), the prevention of strokes afforded by chronic ARBs treatment is well established. However, their efficacy in the acute and post-acute phases of strokes remains a matter of debate. The ACCESS (Acute Candesartan Cilexetil therapy in Stroke Survivors) study, in which candesartan therapy was initiated within 36 h after a stroke, revealed a lower mortality and a lower rate of vascular events after a one-year follow-up [69]. But a more recent trial failed to confirm these results. In the SCAST (Scandinavian Candesartan Acute Stroke Trial) study, candesartan treatment administered within 30 h after a stroke failed to show any beneficial effect when compared to placebo after 6 months [67,70]. In addition, ARBs act neither on cerebral lesions nor on the reduction in infarct volume, while this was observed in preclinical models. Thus, when considering acute stroke treatment, more in-depth studies should be carried out and the search for other therapeutic avenues that target AT1 should be considered.
4. Alternative Ligands to Target AT1 Signaling
AT1 has been a matter of intensive research over the last decades and new mechanisms for altering AT1 signaling have recently emerged, related to the particular properties of this GPCR (Figure 3).
Figure 3.

Overview of the cellular signaling pathways activated by AT1 after stimulation of the renin–angiotensin system (RAS). AA: arachidonic acid; ACE: angiotensin-converting enzyme; ACE2: angiotensin-converting enzyme 2; AKT: protein kinase B; Ang-(1–7): angiotensin (1–7); Ang I: angiotensin I; Ang II: angiotensin II; ARHGEF1: Rho guanine nucleotide exchange factor 1. AT1: angiotensin II type 1 receptor; AT2: angiotensin II type 2 receptor; ARBs: angiotensin receptor blockers; CaM: calmodulin; DAG: diacylglycerol; ER: endoplasmic reticulum; ERK1/2: extracellular signal regulated kinase ½; GRK: G protein-coupled receptor kinase; GSNO: S-nitrosoglutathione; IP3: inositol triphosphate; JAK2: Janus kinase 2; JNK: c-Jun N-terminal Kinase; MAPK: mitogen-activated protein kinases; MasR: Mas receptor; MLC: myosin light chain; MLCK: myosin light chain kinase; MLCP: myosin light chain phosphatase; mTOR: mammalian target of rapamycin; MYPT: myosin phosphatase target subunit; NFκB: nuclear factor kappa B; NOX: NADPH oxidase; PA: phosphatidic acid; PC: phosphatidylcholine; PI3K: phosphoinositide 3 kinase; PIP2: phosphatidylinositol-4, 5-diphosphate; PLA2: phospholipase A2; PLCβ: phospholipase Cβ; PLD: phospholipase D; PKC: protein kinase C; RhoA: RAS homolog family member A; ROCK: Rho associated protein kinase; ROS: reactive oxygen species; TRV023 and TRV027: β-arrestin biased AT1 agonists; TRV055 and TRV056: Gq biased AT1 agonists. The figure was prepared in part with the Servier Medical Art website (Creative Commons Attribution 3.0 unported License).
4.1. The Classical View of AT1 Signaling
After Ang II binding, AT1 adopts an active conformation that stimulates the Gq/11/phospholipase Cβ cascade [71] to induce VSMC contraction. Indeed, this pathway generates second messengers by phosphatidylinositol-4,5-diphosphate (PIP2) hydrolysis. The increase in inositol trisphosphate (IP3) will result in myosin light chain kinase (MLCK) activation through Ca2+/calmodulin complexes. The increase in intracellular Ca2+ has also been reported to activate the JAK2 kinase which phosphorylates the ARHGEF1 Rho guanine nucleotide exchange factor [72]. ARHGEF1 then activates the RhoA/Rho kinase–mediated phosphorylation of MYPT1 (myosin phosphatase target subunit 1) and inhibition of the myosin light chain phosphatase (MLCP) activity [72].
Recent studies have also reported that AT1 coupling to Gi/o, G12 and G13 [5,6,73,74,75,76]. Coupling of AT1 to Gi/o leads to an inhibition of adenylyl-cyclase and a decrease in cyclic adenosine monophosphate in kidneys, adrenal glomerula zone or liver [6]. AT1 activation, via its coupling to G12/13, may also trigger the RhoA/Rho kinase pathway evoked above and thus participate to vascular contraction, remodeling and inflammation induced by Ang II [77]. AT1 has also been reported to phosphorylate and activate the cytosolic phospholipase PLA2 [78], resulting in the release of arachidonic acid and its derivatives which are involved in vascular tone and in p22phox-based NADH/NADPH oxidase activation in VSMC [79]. Intracellular protein kinases, such as protein kinase C (PKC), mitogen-activated protein kinases (MAPK) and protein kinase B (AKT), may also stimulate NADPH oxidase and lead to the generation of reactive oxygen species (ROS) induced by Ang II [5,80].
After Ang II stimulation, AT1 also undergoes phosphorylation on its C-terminal serine and threonine residues by GPCR kinases (GRKs). This phosphorylation process is a common feature of GPCR and increases β-arrestin1 and β-arrestin2 affinities for the receptor [81,82]. The initial role of β-arrestins was described to arrest the response by interrupting the heterotrimeric G protein coupling and to induce receptor internalization through clathrin-coated pits. But a new role of β-arrestins has now emerged for GPCR [83,84] including AT1: once stably recruited by AT1, β-arrestins act as scaffolding proteins that are suggested to directly promote AT1 alternative signaling cascades with potential protective effects.
4.2. Targeting Different Active Conformations of AT1 with Biased Agonists
Many ligands of GPCR can selectively initiate one or more distinct signaling pathways when coupled to their receptor. This is the biased agonism theory, also known as functional selectivity [85], that was first described more than four decades ago but not explored until recently [86]. These biased agonists will preferentially stabilize one of the many conformations that GPCR can adopt, thus allowing only specific interactions between the receptor and specific downstream intracellular proteins. In addition to the receptor conformation, cellular background and simultaneous expression of other receptors may lead to phosphorylation changes at the C-terminus of activated receptors. This can induce the binding of different conformations of β-arrestins, different internalization routes and altered signaling. It is referred to as the barcode theory [87,88].
Because the biased signaling exploration held great promise for the development of more effective therapies, the concept became rapidly fashionable and over-simplified, stating that for each GPCR, in addition to the classical active state that couples to both G protein and beta-arrestin, one could find biased ligands triggering exclusive signaling either towards heterotrimeric G coupling or towards β-arrestin. This view of exclusive G proteins versus β-arrestins biased signaling has now been debated in a series of valuable articles [89,90,91,92] but still holds for AT1.
In the case of AT1, several biased agonists have been developed and pharmacologically characterized that allow specific activation of the G protein-dependent pathway without activating β-arrestins (TRV055 and TRV056, Figure 3) and vice versa (TRV023 and TRV027, Figure 3) [93]. Biased ligands that differentially activate AT1 coupling among the various G protein subtypes have also been described [75]. Combining very precise molecular and pharmacological characterizations of these biased AT1 agonists (allowed by the development of new tools, such as various biosensors based on bioluminescence resonance energy transfer BRET [75]) together with structural information [94,95,96] and molecular dynamic simulations of different categories of biased ligands/AT1 pairs [71,97,98], AT1 is now demonstrated to adopt at least three conformational states: the inactive one, the canonical active conformation that can couple to both Gq and β-arrestin pathways, but also an alternative active conformation coupling almost exclusively to β-arrestins. The following paragraph describes at the molecular level the results arising from these recent and highly relevant studies.
The very first X-ray crystallography study of AT1 consisted of the receptor co-crystallized with an ARB [99]. The resulting inactive form of the receptor showed an H-bond between N295 of the transmembrane domain TM7 (N2957.46) and N111 of the TM3 (N1113.35). This interaction ensures an inward positioning of the TM6, together with a slight outward movement of TM7, precluding any contact with intracellular effectors (Figure 4, in salmon). This conformation is stabilized by biphenyl-tetrazole ARBs, as they tightly interact with several residues, including N2957.46. They afford a stabilization of the N1113.35–N2957.46 H-bond and of the network of surrounding residues in the inactive conformation [51,99]. This stabilization of AT1 in an inactive state results in the pharmacologically observed robust inverse agonism.
Figure 4.
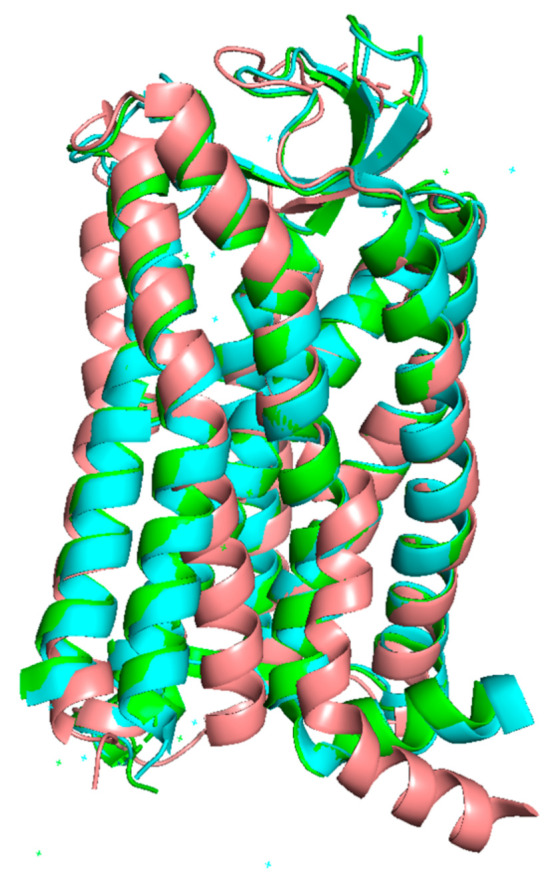
Superimposition of the three known structures of AT1. Inactive (salmon), canonical active (green) and alternative active (cyan). TM6 and TM7 are located in the front. In the inactive structure, TM6 is remarkably bent inside the receptor, while TM7 is slightly twisted outside. The reference inactive structure of AT1 has been chosen as PDB code 4YAY (from [99]). Canonical active and alternative active structures have been chosen as PDB code 6OS0 and 6OS1 respectively (from [96]). The figure was prepared with the PyMOL software (The PyMOL Molecular Graphics System, Version 2.5.0. Schrödinger, LLC).
In contrast, a more recent X-ray crystallography study [98] showed that in the presence of Ang II or of the biased agonist TRV 023 (Figure 4, in green and cyan, respectively), interaction with agonists induces an outward shift of TM6, which is a hallmark of GPCR activation [96]. In AT1, this shift in TM6 is due to a conformational change of N2957.46. Other events depend on the nature of the agonist, whether biased or not and have been well described by Suomivuori et al. and Wingler et al. in two recent and complementary articles published in 2020 [96,98]. The authors could decipher the following mechanisms by molecular dynamics and X-ray crystallography, both techniques providing converging results which are given below.
The natural agonist, Ang II, inserts itself in the ligand pocket of the receptor with its N-terminus oriented toward the extracellular side. The C-terminus with its hydrophobic phenylalanine will therefore position itself deeply into the core of the receptor, near the hydrophobic L112 of TM3 (L1123.36) and pushing it towards TM2. Consequently, the neighboring N1113.35 points outward from the receptor (Figure 5a in green). As a result, instead of pointing toward TM3, Y2927.43 points toward TM2, exchanging its position with F772.53. This movement of Y2927.43 induces a clockwise twist of TM7, such that the N461.50 below establishes an H-bond with N2957.46 instead of C2967.47 [98]. It also causes an outward shift of TM7 in the intracellular part of the receptor permitting Y3027.53 to point up (Figure 5b in green). Such a global phenomenon, described by Suomivuori et al. as a “long-range allosteric network”, drives the information from the ligand-binding pocket to the intracellular side of AT1, resulting in the “canonical active conformation”. The same pattern occurs with a greater extent when the ligand is a Gq biased agonist, allowing sufficient space for the α5 helix of Gq to accommodate with the receptor.
Figure 5.

Illustration of the long range allosteric network of AT1 inducing its switch to canonical active conformation. (a): Top view of the canonical (green) and alternative (cyan) active conformations of AT1. (b): Superimposition of the intracellular part of AT1 in its canonical (green) and alternative (cyan) active conformations. Canonical active and alternative active structures are simulation data available in supplementary material from [98]. The figure was prepared with the PyMOL software (The PyMOL Molecular Graphics System, Version 2.5.0. Schrödinger, LLC).
Alternatively, most arrestin-biased peptide agonists do not possess a phenylalanine on their C-terminus (Figure 6a) [94,100]. The consecutive lack of hydrophobic interaction with L1123.36 prevents the complete outward shift of the intracellular side of TM7, leading to the “alternative active conformation” (Figure 6b in cyan). It includes a downward rotamer of Y3027.53 which then points to the intracellular side. This has a repelling effect on R1263.50, thus also pointing down (Figure 5b, in cyan). R1263.50 belongs to the conserved DRY motif of GPCR that is mandatory for GPCR coupling to heterotrimeric G proteins. The models simulated by Suomivuori et al. have established that this downward position of R1263.50 induces a steric clash with the α5 helix of Gq, preventing its coupling to AT1 [98]. The same models have shown that both AT1 active conformations (canonical and alternative) can accommodate with the β-arrestin finger loop [98].
Figure 6.

β-arrestin biased agonists of AT1 favor its alternative active conformation. (a): Comparison of the amino acid sequences of angiotensin II (Ang II) and of the main β-arrestin biased agonists of AT1; Ang II: angiotensin II; SII: [Sar1,Ile4,Ile8] angiotensin II peptide analog; TRV023 and TRV027: β-arrestin biased AT1 agonists. (b): Superimposition of the structure of AT1 bound to Ang II (green, PDB code 6OS0; Ang II appears in orange) or to the β-arrestin biased agonist TRV023 (cyan, PDB code 6OS1; TRV023 appears in purple). The phenyl group of the C-terminal part of Ang II (highlighted in blue) goes deep into the ligand pocket, inducing a rotation of L1123.36 and of N1113.35 (yellow arrows). It yields the canonical structure of AT1 (appears in green). The C-terminal part of TRV023 comprises a less bulky alanine residue (highlighted in grey) which does not induce such motion, yielding the alternative structure (in cyan). This panel was prepared with the PyMOL software (The PyMOL Molecular Graphics System, Version 2.5.0. Schrödinger, LLC).
A very seductive hypothesis has been proposed to explain why AT1 can adopt such an alternative active conformation that binds almost exclusively to β-arrestin and not G proteins [96]. Wingler et al. have suggested that a biased signaling switch may predispose AT1 to adopt an exclusive β-arrestin conformation. Interestingly, Ang II binding to AT1 not only induces changes at the bottom of the ligand-binding pocket but also in residues constituting a key polar network in the transmembrane region of the receptor. In most class A GPCR, this polar network forms a sodium-binding site stabilizing the inactive conformation. This Na+/GPCR interaction is due to the bond established between Na+ and a highly conserved serine in TM7. However, one of the specificities of AT1 among GPCR family, is that this serine residue is replaced by the asparagine N2957.46 (involved in the initial outward shift of TM6 evoked above), thus alleviating Na+ binding and favoring the β-arrestin alternative active conformation. Similar residue substitutions in the conserved Na+-binding motif have been reported in several atypical chemokine receptors, such as the decoy receptor CXCR7 [101,102], that do not signal through G protein coupling, strengthening the hypothesis that the absence of the Na+ binding site could favor an exclusive β-arrestin active conformation.
4.3. Targeting AT1 by Covalent S-Nitrosation
S-nitrosation is defined as the covalent bond between nitric oxide (NO, a well-known gaseous transmitter playing an essential role in cardiovascular homeostasis) and the thiol moiety of cysteine residues of target proteins [103]. The so formed S-nitrosothiols are able to induce trans-nitrosation (the transfer of NO to another target protein) and/or to release NO, thus conferring on them potent vasodilator properties [104]. The S-nitrosation of proteins can induce dramatic changes in their functions [105] and recent papers suggest that AT1 functions could be regulated by S-nitrosation.
Studies carried out by Leclerc et al. in 2006 showed a slight decrease in the affinity of Ang II for the AT1 following exposure to sodium nitroprusside (SNP), a NO donor, in a cellular model of HEK293 cells overexpressing AT1 [106]. Interestingly, it was proposed that AT1 is the direct target of NO: using several AT1 cysteine mutants, the authors showed that mutation of the cysteine 289 (C2897.40) for a serine in human AT1 was no longer sensitive to NO, suggesting that S-nitrosation of the cysteine residue at position 289 in TM7 of AT1 was responsible for the slight loss of affinity for Ang II [106].
Based on these results, our group proposed that NO donors could directly reduce arterial contraction induced by Ang II activation of AT1. We investigated the physiological consequences of this phenomenon by studying the effect of NO in an ex-vivo model: isolated and perfused middle cerebral arteries of normotensive rats [107]. S-nitrosation was induced by pretreatment with S-nitrosoglutathione (GSNO), an endogenous S-nitrosothiol previously shown as protective in different models of cerebral ischemia (MCAo) [108,109] and autologous blood clots injected through the internal carotid artery [110]). GSNO pretreatment of rat middle cerebral arteries specifically abolished the vasoconstriction induced by Ang II. Other vasoconstrictors sharing the same Gq signaling pathway, such as phenylephrine, or targeting Gi/o coupled receptors, such as serotonin, still produced vasoconstriction after GSNO pre-treatment. GSNO therefore specifically inhibited the vasoconstriction induced by Ang II by acting upstream of the Gq proteins, i.e. probably on the receptor itself. In preliminary experiments, we could detect S-nitrosated AT1 after exposure of HEK293 cells overexpressing AT1 to GSNO, although we did not investigate which cysteine was targeted [107]. Similar results have just been published [111]. Using an ex-vivo model of rat aortic rings, Pinheiro et al. have observed a decrease in vascular contraction induced by Ang II when arteries were pretreated with nitrite or GSNO [111]. In vivo, a decrease in the blood pressure response to Ang II was also detected after oral administration of nitrite or GSNO [111]. Both treatments increased plasma concentrations of S-nitrosated species and enhanced total protein S-nitrosation in the vessels. The authors identified PKC as the target of S-nitrosation and as the underlying mechanisms of the decreased responses to Ang II. In Leclerc’s study, a decrease in the EC50 of Ang II-induced inositol phosphate production upon SNP treatment was reported, while Pinheiro et al. found, in HEK293T cells expressing AT1, a Bmax decrease in the Ang II-induced intracellular calcium mobilization after GSNO pre-treatment. Taken together, these studies suggest that S-nitrosation promoted by GSNO reduces the physiological responses induced by Ang II by acting directly on AT1 and on other proteins such as PKC.
Interestingly, the slight decrease in Ang II affinity described by Leclerc et al. after AT1 S-nitrosation might not be the only effect of NO on the receptor. Indeed, the AT1 dependent myogenic tone (the arterial contractile response consecutive to an increase in arterial transmural pressure and mechanical stretch [112]), which is independent of the binding of Ang II to AT1, decreases in isolated middle cerebral arteries pretreated with GSNO (Figure 3) [107]. In addition, in preliminary experiments, GSNO pre-treatment of HEK293 cells did not impair the kinetics and amplitude of activated AT1 internalization [107]. This suggests that the AT1/β-arrestin pathway could remain unaffected by the S-nitrosation of the receptor.
When looking at AT1 structure, it is noteworthy that C2897.40 is one of the residues involved in the “long-range allosteric network” (see Section 4.2). As shown in Figure 7, the thiol function of C2897.40 is oriented inside the receptor in the alternative active conformation (in cyan), whereas it points outward from the receptor in the inactive (in salmon) and canonical active (in green) states. This suggests that any modification on this particular residue could alter the process involved in conformational changes and may preclude or stabilize one or more conformations of AT1. This hypothesis is strengthened by several reports focusing on single nucleotide polymorphism of AT1 in humans. Among the few naturally occurring variants of AT1, one is related to C2897.40 where the cysteine is replaced by a tryptophan residue (C289W variant). This variant has not been associated with a pathology, but seems to be less expressed at the plasma membrane and decreases the potency of Ang II on phosphatidylinositol hydrolysis consecutive to Gq stimulation [113]. At least two other recent studies confirmed this reduced affinity of the C289W variant for Ang II but also showed that C289W is able to interact with β-arrestin2 at the plasma membrane upon Ang II stimulation [75,114]. However, C289W alters the β-arrestin2 conformation and reduces the β-arrestin-dependent activation of ERK1/2 [114]. Taken together, these data support the idea that a modification at C2897.40, such as S-nitrosation, may bias AT1 signaling, but further investigations are needed.
Figure 7.

Positions of the cysteine 289 residue in the three known AT1 structures. Superimposition of the three known AT1 structures: inactive (salmon), canonical active (green) and alternative active (cyan). TM6 and TM7 are located in the front. C289 residue is indicated by arrows. The reference inactive structure of AT1 has been chosen as PDB code 4YAY (from Zhang et al. [99]). Canonical active and alternative active structures are simulation data available in supplementary material from Suomivuori et al. [98]. The figure was prepared with the PyMOL software (The PyMOL Molecular Graphics System, Version 2.5.0. Schrödinger, LLC).
5. Promoting the β-Arrestin Pathway of AT1 to Treat Cerebrovascular Disease
Having established that AT1 is prone to biased agonism towards the β-arrestin pathway, we will now review the potential interests of promoting this AT1/β-arrestin signaling. AT1 recruits both β-arrestins equally and internalizes as a stable complex, inducing a slow recycling to the plasma membrane [115,116].
5.1. Biased Agonists towards AT1/β-Arrestins in Cardiovascular Diseases
In addition to the desensitization and internalization of AT1 which reduce Gq signaling, the activation of the β-arrestin pathway induces several signaling cascades involved in cellular processes, such as cell proliferation and growth, nutrient uptake and migration (Figure 3). Following stimulation either by Ang II or by the Ang II peptide analog SII, the first β-arrestin biased AT1 agonist identified (Figure 6a), the consecutive phosphoproteomes do not overlap, suggesting a Gq-independent phosphorylation process induced by β-arrestin biased agonists [117,118]. Moreover, in a very recent proteomic study, Pfeiffer et al. showed that the dynamic recruitment of proteins in close proximity to AT1 displays a completely different pattern as a function of time, depending on whether AT1 was stimulated by full agonists, Gq biased agonists, or β-arrestin biased agonists [119].
As previously mentioned, both β-arrestin1 and β-arrestin2 can be recruited after the phosphorylation of AT1 by GRKs. The use of β-arrestin1 and β-arrestin2 biosensors has shown that a given ligand/AT1 pair recruits both β-arrestins to the same extent but that they stabilize the receptor in distinct conformations [120]. Indeed, although the primary sequence and 3D structure of the two β-arrestin isoforms are very similar, conformational differences have been reported, suggesting functional differences [121]. This is illustrated at the cardiovascular level: on the one hand, β-arrestin1 exhibits harmful effects at the cardiac level, in particular by reducing cardiac function, promoting apoptosis and inflammation while, on the other hand, β-arrestin2 has been shown to have protective cardiac effects [122]. β-arrestin2 mediates signaling through Src, ERK1/2, AKT and PI3K activations and has been reported to be cytoprotective, anti-apoptotic and pro-survival [93,123,124,125,126]. β-arrestin2 has also been reported to inhibit the inflammation process after myocardial infarction [127]. However, one study has shown conflicting results, showing that β-arrestin2 is involved in cell death signaling after ischemic cardiac injury [128].
The importance of the exclusive AT1/β-arrestin pathway at the cardiovascular level has been studied through the use of various ligands biased towards β-arrestins, such as TRV027 and TRV023 (Figure 3 and Figure 6a) [129,130] and has been recently reviewed by Turu et al. [92]. Only TRV027 has been studied in clinical trials for heart failure (NCT01187836), kidney disease (NCT01444872) and currently for COVID-19/SARS-CoV-2 infection (NCT04419610; NCT04924660).
TRV027 increases cardiac contractility in vitro and reduces blood pressure in rats [131]. In addition, in a canine model of cardiac insufficiency, TRV027 reduces systemic and renovascular resistance and promotes cardiac unloading [132,133]. Based on these promising results, TRV027 has also been tested in humans, both in healthy subjects [134] and in patients with acute heart failure in the BLAST-AHF (Biased Ligand of the Angiotensin receptor STudy in Acute Heart Failure) study [135]. The beneficial effects of TRV027 were not confirmed by this first clinical study [136]. However, the interest of such β-arrestin-biased agonists of AT1 remains, since many other ligands with similar bias and convincing preclinical data are still to be tested.
Hence, TRV023 is another AT1 agonist that blocks G protein signaling and promotes the recruitment of β-arrestins (Figure 6a). In mice, TRV023 increases cardiac contractility in a β-arrestin2-dependent manner while it decreases blood pressure independently of β-arrestin2 (most likely through inhibition of the Gq pathway) [130]. TRV023 reduces cell death both ex vivo (intraventricular balloon-induced membrane stretch injury) and in vivo (left ventricular ischemia-reperfusion injury) by activating the MAPK and AKT pathways. These cytoprotective effects were not observed with losartan or in β-arrestin2 KO mice [130]. These results are strengthened by previous works showing that mechanical stretching in cardiac myocytes facilitates β-arrestin2 biased pro-survival signaling via EGFR transactivation mediated by AT1 independently of G protein or Ang II [137]. Moreover, TRV023 prevents Ang II-induced cardiac hypertrophy while preserving contractility more efficiently than losartan by improving the calcium response of myofilaments [138]. Finally, TRV023 also induces an increase in myocardiac contractility and in ventricular myosin light chain-2 phosphorylation in a transgenic mouse model of familial dilated cardiomyopathy [139].
Thus, the beneficial actions of these β-arrestin biased AT1 ligands appear promising, although not yet fully understood. These biased agonists also seem to bind to AT2, which could contribute to counteracting the cardiovascular harmful effects of AT1. Although AT2 does not couple to heterotrimeric G proteins and does not recruit β-arrestins [76,140], this receptor may play a role in the in vivo effects of these biased agonists, e.g., by modulating AT1/AT2 heterodimer signaling. However, to date no comprehensive study has been conducted to support this hypothesis.
5.2. Biased Agonists towards AT1/β-Arrestins in Cerebrovascular Diseases
To the best of our knowledge, no study has focused on the impact of specific β-arrestin biased agonists of AT1 on cerebrovascular diseases. The literature, however, supports the idea that these drugs might represent new hope for these clinical situations in which alternative treatments are lacking.
Interestingly, Ang-(1–7), a major component of the Ang-(1–7)/MasR/ACE2 protective axis of the renin–angiotensin system, has also been described as a β-arrestin biased ligand of AT1 [74,141]. Numerous studies report cardioprotective effects of Ang-(1–7) [141,142,143,144] as well as cerebroprotective effects in ischemic and hemorrhagic strokes [60,145,146]. Ang-(1–7) protects against central nervous system damage and neurological deficits induced by experimental ischemic strokes [145]. It reduces the infarct volume, oxidative stress, as well as the release of pro-inflammatory cytokines through Mas receptor stimulation [147]. However, even if MasR antagonists were used to prevent Ang-(1–7) effects, this does not completely rule out that its weak, exclusive β-arrestin biased agonist properties on AT1, together with its AT2 agonistic properties might contribute to the cerebrovascular beneficial effects of Ang-(1–7) [76,140,148,149]. The heterodimeric complexes between AT1/AT2 or AT1/MasR seem to inhibit AT1 signaling by Ang II, β-arrestin recruitment and receptor internalization [150,151,152,153]. It would be interesting to investigate how Ang-(1–7) and other β-arrestin biased ligands affect the signaling of these heterodimers.
Unlike reports at the cardiac level, both β-arrestin1 and β-arrestin2 seem to have neuroprotective effects on cerebrovascular diseases. The MCAo ischemic stroke model in mouse revealed an increase in protein expression of the two β-arrestins, suggesting their involvement in the pathophysiological process [154]. This study also reported that β-arrestin1 deletion worsens ischemic brain damage assessed by neuronal deficits, infarct size and mortality. These effects are correlated with the suppression of autophagy and induced neuronal apoptosis and necrosis in vitro and in vivo [154]. These results suggest that β-arrestin1 has neuroprotective effects related to autophagy. However, we should remain cautious, since β-arrestin1 deletion may affects a very wide variety of cellular functions, whether beneficial or deleterious.
Studies exploring the alterations of β-arrestin2 expression in strokes and neurovascular disease are scarce and the role of β-arrestin2 in neurological disorders is not yet clear. One study showed that β-arrestin2 plays a pivotal role in the immunodeficiency syndrome induced by strokes [155]. In particular, β-arrestin2 is believed to be a key intracellular mediator of adrenergic activity in monocytes after a stroke and may therefore be implicated in the stroke-induced adrenergic-mediated monocyte dysfunction [155]. However, these in vitro results concluding that there is a harmful role of β-arrestin2 after a stroke are not supported by in vivo experiments. Indeed, a more recent study suggests that β-arrestin2 signaling improves endothelial function and blood–brain barrier integrity after a stroke [156]. Using APC (activated protein C), a β-arrestin2 biased agonist of the PAR-1 receptor (protease-activated receptor 1), the authors showed that β-arrestin2 signaling has protective effects in mice under ischemic conditions (MCAo), notably as it increases angiogenesis. A high fat diet reduced β-arrestin2 expression in mice and the β-arrestin2 levels in the penumbra were inversely correlated with the severity of ischemic lesions after MCAo [156]. The authors identified PDFG-β as one of the key target molecules activated by β-arrestin2 to promote angiogenesis [156].
6. Conclusions
The pathophysiological role of AT1 in cerebrovascular diseases has been increasingly highlighted in recent years; however, therapeutic advances in this area have not followed. The development of new therapeutic approaches requires a better understanding of AT1 signaling pathways and its possible interactions with other components. Since β-arrestin-biased AT1 agonists have showed promising results in the cardiovascular disease context, it would be interesting to study them in models of cerebrovascular diseases such as strokes.
There is still much to learn about the regulation of AT1 signaling, whether through β-arrestin signaling or characterization of the potential beneficial effects of β-arrestin biased agonists.
Acknowledgments
We thank Frédéric Simonin, head of the GPCR, pain and inflammation team in Strasbourg for his support on the AT1 project and Jeremy Garwood for his critical reading of the manuscript.
Abbreviations
| ACE | Angiotensin-converting enzyme |
| AKT | protein kinase B |
| Ang II | angiotensin II |
| ARB | AT1 receptor blocker |
| AT1 | angiotensin II type 1 receptor |
| AT2 | angiotensin II type 2 receptor; |
| GPCR | G protein-coupled receptor |
| GRKs | G protein-coupled receptor kinases |
| GSNO | S-nitrosoglutathione |
| MAPK | mitogen-activated protein kinases |
| MCAo | middle cerebral artery occlusion |
| PKC | protein kinase C |
| RAS | renin–angiotensin system |
| ROS | reactive oxygen species |
| SNP | sodium nitroprusside |
| TBI | traumatic brain injury |
| TM (1 to 7) | transmembrane domain (1 to 7); |
| VSMC | vascular smooth muscle cells |
Funding
This work was funded by the French Ministry of Education, Research and Technology (Paris, France, EA3452). The authors acknowledge support of CITHEFOR by the “Impact Biomolecules” project of the “Lorraine Université d’Excellence” (Investissements d’avenir—ANR). Part of this work was supported by the IdEx Unistra (ANR-10-IDEX-0002) and by the SFRI-STRAT’US project (ANR-20-SFRI-0012) under the framework of the French Program “Investments for the Future” given to the Strasbourg drug discovery and development Institute (IMS), as part of the Interdisciplinary Thematic Institute (ITI) 2021-2028 program of the University of Strasbourg, CNRS and Inserm.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Mukoyama M., Nakajima M., Horiuchi M., Sasamura H., Pratt R.E., Dzau V.J. Expression cloning of type 2 angiotensin II receptor reveals a unique class of seven-transmembrane receptors. J. Biol. Chem. 1993;268:24539–24542. doi: 10.1016/S0021-9258(19)74498-6. [DOI] [PubMed] [Google Scholar]
- 2.Kambayashi Y., Bardhan S., Takahashi K., Tsuzuki S., Inui H., Hamakubo T., Inagami T. Molecular cloning of a novel angiotensin II receptor isoform involved in phosphotyrosine phosphatase inhibition. J. Biol. Chem. 1993;268:24543–24546. doi: 10.1016/S0021-9258(19)74499-8. [DOI] [PubMed] [Google Scholar]
- 3.Inagami T., Iwai N., Sasaki K., Yamano Y., Bardhan S., Chaki S., Guo D.F., Furuta H., Ohyama K., Kambayashi Y., et al. Cloning, Expression and Regulation of Angiotensin II Receptors. Eur. Hear. J. 1994;15:104–107. doi: 10.1093/eurheartj/15.suppl_D.104. [DOI] [PubMed] [Google Scholar]
- 4.Hunyady L., Catt K.J. Pleiotropic AT1 Receptor Signaling Pathways Mediating Physiological and Pathogenic Actions of Angiotensin II. Mol. Endocrinol. 2006;20:953–970. doi: 10.1210/me.2004-0536. [DOI] [PubMed] [Google Scholar]
- 5.Kawai T., Forrester S.J., O’Brien S., Baggett A., Rizzo V., Eguchi S. AT1 receptor signaling pathways in the cardiovascular system. Pharmacol. Res. 2017;125:4–13. doi: 10.1016/j.phrs.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Gasparo M., Catt K.J., Inagami T., Wright J.W., Unger T. International Union of Pharmacology. XXIII. The Angiotensin II Receptors. Pharmacol. Rev. 2000;52:415–472. [PubMed] [Google Scholar]
- 7.Kobori H., Nangaku M., Navar L.G., Nishiyama A. The Intrarenal Renin-Angiotensin System: From Physiology to the Pathobiology of Hypertension and Kidney Disease. Pharmacol. Rev. 2007;59:251–287. doi: 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- 8.Coble J.P., Grobe J.L., Johnson A.K., Sigmund C.D. Mechanisms of brain renin angiotensin system-induced drinking and blood pressure: Importance of the subfornical organ. Am. J. Physiol. Integr. Comp. Physiol. 2015;308:R238–R249. doi: 10.1152/ajpregu.00486.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsutsumi K., Saavedra J.M. Characterization of AT2 angiotensin II receptors in rat anterior cerebral arteries. Am. J. Physiol. Circ. Physiol. 1991;261:H667–H670. doi: 10.1152/ajpheart.1991.261.3.H667. [DOI] [PubMed] [Google Scholar]
- 10.Touyz R.M., Schiffrin E.L. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol. Rev. 2000;52:639–672. [PubMed] [Google Scholar]
- 11.Dimitropoulou C., White R.E., Fuchs L., Zhang H., Catravas J.D., Carrier G.O. Angiotensin II Relaxes Microvessels Via the AT 2 Receptor and Ca 2+ -Activated K + (BK Ca ) Channels. Hypertension. 2001;37:301–307. doi: 10.1161/01.HYP.37.2.301. [DOI] [PubMed] [Google Scholar]
- 12.Enzo R.P. The angiotensin II type 2 (AT2) receptor: An enigmatic seven transmembrane receptor. Front. Biosci. 2009;14:958–972. doi: 10.2741/3289. [DOI] [PubMed] [Google Scholar]
- 13.Matavelli L.C., Siragy H.M. AT2 Receptor Activities and Pathophysiological Implications. J. Cardiovasc. Pharmacol. 2015;65:226–232. doi: 10.1097/FJC.0000000000000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carey R.M. AT2Receptors: Potential Therapeutic Targets for Hypertension. Am. J. Hypertens. 2016;30:339–347. doi: 10.1093/ajh/hpw121. [DOI] [PubMed] [Google Scholar]
- 15.Povlsen A.L., Grimm D., Wehland M., Infanger M., Krüger M. The Vasoactive Mas Receptor in Essential Hypertension. J. Clin. Med. 2020;9:267. doi: 10.3390/jcm9010267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dupuis F., Atkinson J., Limiñana P., Chillon J.-M. Captopril improves cerebrovascular structure and function in old hypertensive rats. Br. J. Pharmacol. 2005;144:349–356. doi: 10.1038/sj.bjp.0706001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dupuis F., Atkinson J., Limiñana P., Chillon J.-M. Comparative effects of the angiotensin II receptor blocker, telmisartan, and the angiotensin-converting enzyme inhibitor, ramipril, on cerebrovascular structure in spontaneously hypertensive rats. J. Hypertens. 2005;23:1061–1066. doi: 10.1097/01.hjh.0000166848.95592.a5. [DOI] [PubMed] [Google Scholar]
- 18.Vincent J.-M., Kwan Y.W., Chan S.-L., Perrin-Sarrado C., Atkinson J., Chillon J.-M. Constrictor and Dilator Effects of Angiotensin II on Cerebral Arterioles. Stroke. 2005;36:2691–2695. doi: 10.1161/01.STR.0000190002.79052.bf. [DOI] [PubMed] [Google Scholar]
- 19.Kalra J., Prakash A., Kumar P., Majeed A.B.A. Cerebroprotective effects of RAS inhibitors: Beyond their cardio-renal actions. J. Renin-Angiotensin-Aldosterone Syst. 2015;16:459–468. doi: 10.1177/1470320315583582. [DOI] [PubMed] [Google Scholar]
- 20.Gironacci M., Vicario A., Cerezo G., Silva M.G. The depressor axis of the renin–angiotensin system and brain disorders: A translational approach. Clin. Sci. 2018;132:1021–1038. doi: 10.1042/CS20180189. [DOI] [PubMed] [Google Scholar]
- 21.Feigin V.L., Forouzanfar M.H., Krishnamurthi R., Mensah G.A., Connor M., Bennett D.A., Moran A.E., Sacco R.L., Anderson L., Truelsen T., et al. Global and regional burden of stroke during 1990–2010: Findings from the Global Burden of Disease Study 2010. Lancet. 2014;383:245–255. doi: 10.1016/S0140-6736(13)61953-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peisker T., Koznar B., Stetkarova I., Widimsky P. Acute stroke therapy: A review. Trends Cardiovasc. Med. 2017;27:59–66. doi: 10.1016/j.tcm.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 23.Näveri L., Strömberg C., Saavedra J.M. Angiotensin II AT, receptor mediated contraction of the perfused rat cerebral artery. NeuroReport. 1994;5:2278–2280. doi: 10.1097/00001756-199411000-00018. [DOI] [PubMed] [Google Scholar]
- 24.Stenman E., Edvinsson L. Cerebral Ischemia Enhances Vascular Angiotensin AT 1 Receptor–Mediated Contraction in Rats. Stroke. 2004;35:970–974. doi: 10.1161/01.STR.0000121642.53822.58. [DOI] [PubMed] [Google Scholar]
- 25.Kagiyama T., Kagiyama S., Phillips M.I. Expression of angiotensin type 1 and 2 receptors in brain after transient middle cerebral artery occlusion in rats. Regul. Pept. 2003;110:241–247. doi: 10.1016/S0167-0115(02)00223-9. [DOI] [PubMed] [Google Scholar]
- 26.Walther T., Olah L., Harms C., Maul B., Bader M., Hörtnagl H., Schultheiss H.-P., Mies G. Ischemic injury in experimental stroke depends on angiotensin II. FASEB J. 2001;16:169–176. doi: 10.1096/fj.01-0601com. [DOI] [PubMed] [Google Scholar]
- 27.Baumbach G.L., Sigmund C.D., Faraci F.M. Cerebral Arteriolar Structure in Mice Overexpressing Human Renin and Angiotensinogen. Hypertension. 2003;41:50–55. doi: 10.1161/01.HYP.0000042427.05390.5C. [DOI] [PubMed] [Google Scholar]
- 28.Dupuis F., Vincent J.-M., Limiñana P., Chillon J.-M., Capdeville-Atkinson C., Atkinson J. Effects of suboptimal doses of the AT1 receptor blocker, telmisartan, with the angiotensin-converting enzyme inhibitor, ramipril, on cerebral arterioles in spontaneously hypertensive rat. J. Hypertens. 2010;28:1566–1573. doi: 10.1097/HJH.0b013e328339f1f3. [DOI] [PubMed] [Google Scholar]
- 29.Chillon J.-M., Baumbach G. Effects of an Angiotensin-Converting Enzyme Inhibitor and a β-Blocker on Cerebral Arterioles in Rats. Hypertension. 1999;33:856–861. doi: 10.1161/01.HYP.33.3.856. [DOI] [PubMed] [Google Scholar]
- 30.Zhang M., Mao Y., Ramirez S., Tuma R., Chabrashvili T. Angiotensin II induced cerebral microvascular inflammation and increased blood–brain barrier permeability via oxidative stress. Neuroscience. 2010;171:852–858. doi: 10.1016/j.neuroscience.2010.09.029. [DOI] [PubMed] [Google Scholar]
- 31.Kazama K., Anrather J., Zhou P., Girouard H., Frys K., Milner T.A., Iadecola C. Angiotensin II Impairs Neurovascular Coupling in Neocortex Through NADPH Oxidase–Derived Radicals. Circ. Res. 2004;95:1019–1026. doi: 10.1161/01.RES.0000148637.85595.c5. [DOI] [PubMed] [Google Scholar]
- 32.De Silva T.M., Faraci F.M. Effects of angiotensin II on the cerebral circulation: Role of oxidative stress. Front. Physiol. 2013;3:484. doi: 10.3389/fphys.2012.00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wanderer S., Grüter B.E., Strange F., Sivanrupan S., Di Santo S., Widmer H.R., Fandino J., Marbacher S., Andereggen L. The Role of Sartans in the Treatment of Stroke and Subarachnoid Hemorrhage: A Narrative Review of Preclinical and Clinical Studies. Brain Sci. 2020;10:153. doi: 10.3390/brainsci10030153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Inci S., Spetzler R.F. Intracranial aneurysms and arterial hypertension: A review and hypothesis. Surg. Neurol. 2000;53:530–542. doi: 10.1016/S0090-3019(00)00244-5. [DOI] [PubMed] [Google Scholar]
- 35.Daugherty A., Cassis L. Angiotensin II and abdominal aortic aneurysms. Curr. Hypertens. Rep. 2004;6:442–446. doi: 10.1007/s11906-004-0038-0. [DOI] [PubMed] [Google Scholar]
- 36.Daugherty A., Rateri D.L., Cassis L.A. Role of the Renin-Angiotensin System in the Development of Abdominal Aortic Aneurysms in Animals and Humans. Ann. N. Y. Acad. Sci. 2006;1085:82–91. doi: 10.1196/annals.1383.035. [DOI] [PubMed] [Google Scholar]
- 37.Ohkuma H., Suzuki S., Fujita S., Nakamura W. Role of a Decreased Expression of the Local Renin-Angiotensin System in the Etiology of Cerebral Aneurysms. Circulation. 2003;108:785–787. doi: 10.1161/01.CIR.0000087339.31094.3C. [DOI] [PubMed] [Google Scholar]
- 38.Nishimura M., Aoki T., Kataoka H., Ishibashi R., Miyake T., Takagi Y., Morishita R., Hashimoto N. Role of angiotensin II type 1 receptor in cerebral aneurysm formation in rats. Int. J. Mol. Med. 2009;24:353–359. doi: 10.3892/ijmm_00000239. [DOI] [PubMed] [Google Scholar]
- 39.Ishibashi R., Aoki T., Nishimura M., Miyamoto S. Imidapril Inhibits Cerebral Aneurysm Formation in an Angiotensin-Converting Enzyme–Independent and Matrix Metalloproteinase-9–Dependent Manner. Neurosurgery. 2012;70:722–730. doi: 10.1227/NEU.0b013e3182326188. [DOI] [PubMed] [Google Scholar]
- 40.Tada Y., Wada K., Shimada K., Makino H., Liang E.I., Murakami S., Kudo M., Kitazato K.T., Nagahiro S., Hashimoto T. Roles of Hypertension in the Rupture of Intracranial Aneurysms. Stroke. 2014;45:579–586. doi: 10.1161/STROKEAHA.113.003072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kehoe A.D., Eleftheriou K.I., Heron M., Coats T.J., Montgomery H. Angiotensin-converting enzyme genotype may predict survival following major trauma. Emerg. Med. J. 2008;25:759–761. doi: 10.1136/emj.2006.045336. [DOI] [PubMed] [Google Scholar]
- 42.Ando H., Zhou J., Macova M., Imboden H., Saavedra J.M. Angiotensin II AT 1 Receptor Blockade Reverses Pathological Hypertrophy and Inflammation in Brain Microvessels of Spontaneously Hypertensive Rats. Stroke. 2004;35:1726–1731. doi: 10.1161/01.STR.0000129788.26346.18. [DOI] [PubMed] [Google Scholar]
- 43.Iwai N., Inagami T. Identification of two subtypes in the rat type I angiotensin II receptor. FEBS Lett. 1992;298:257–260. doi: 10.1016/0014-5793(92)80071-N. [DOI] [PubMed] [Google Scholar]
- 44.Yamasaki E., Thakore P., Krishnan V., Earley S. Differential expression of angiotensin II type 1 receptor subtypes within the cerebral microvasculature. Am. J. Physiol. Circ. Physiol. 2020;318:H461–H469. doi: 10.1152/ajpheart.00582.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johren O., Saavedra J.M. Expression of AT1A and AT1B angiotensin II receptor messenger RNA in forebrain of 2-wk-old rats. Am. J. Physiol. Metab. 1996;271:E104–E112. doi: 10.1152/ajpendo.1996.271.1.E104. [DOI] [PubMed] [Google Scholar]
- 46.Villapol S., Balarezo M.G., Affram K., Saavedra J.M., Symes A.J. Neurorestoration after traumatic brain injury through angiotensin II receptor blockage. Brain. 2015;138:3299–3315. doi: 10.1093/brain/awv172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Villapol S., Saavedra J.M. Neuroprotective Effects of Angiotensin Receptor Blockers. Am. J. Hypertens. 2015;28:289–299. doi: 10.1093/ajh/hpu197. [DOI] [PubMed] [Google Scholar]
- 48.Benicky J., Hafko R., Sanchez-Lemus E., Aguilera G., Saavedra J.M. Six Commercially Available Angiotensin II AT1 Receptor Antibodies are Non-specific. Cell. Mol. Neurobiol. 2012;32:1353–1365. doi: 10.1007/s10571-012-9862-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herrera M., Sparks M.A., Alfonso-Pecchio A.R., Harrison-Bernard L.M., Coffman T.M. Lack of Specificity of Commercial Antibodies Leads to Misidentification of Angiotensin Type 1 Receptor Protein. Hypertension. 2013;61:253–258. doi: 10.1161/HYPERTENSIONAHA.112.203679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bouressam M.-L., Lartaud I., Dupuis F., Lecat S. No answer to the lack of specificity: Mouse monoclonal antibody targeting the angiotensin II type 1 receptor AT1 fails to recognize its target. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2018;391:883–889. doi: 10.1007/s00210-018-1522-4. [DOI] [PubMed] [Google Scholar]
- 51.Takezako T., Unal H., Karnik S.S., Node K. Current topics in angiotensin II type 1 receptor research: Focus on inverse agonism, receptor dimerization and biased agonism. Pharmacol. Res. 2017;123:40–50. doi: 10.1016/j.phrs.2017.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou J., Pavel J., Macova M., Yu Z.-X., Imboden H., Ge L., Nishioku T., Dou J., Delgiacco E., Saavedra J.M. AT 1 Receptor Blockade Regulates the Local Angiotensin II System in Cerebral Microvessels From Spontaneously Hypertensive Rats. Stroke. 2006;37:1271–1276. doi: 10.1161/01.STR.0000217404.64352.d7. [DOI] [PubMed] [Google Scholar]
- 53.Lou M., Blume A., Zhao Y., Gohlke P., Deuschl G., Herdegen T., Culman J. Sustained Blockade of Brain AT1 Receptors before and after Focal Cerebral Ischemia Alleviates Neurologic Deficits and Reduces Neuronal Injury, Apoptosis, and Inflammatory Responses in the Rat. Br. J. Pharmacol. 2004;24:536–547. doi: 10.1097/00004647-200405000-00008. [DOI] [PubMed] [Google Scholar]
- 54.Nishimura Y., Ito T., Saavedra J.M. Angiotensin II AT1Blockade Normalizes Cerebrovascular Autoregulation and Reduces Cerebral Ischemia in Spontaneously Hypertensive Rats. Stroke. 2000;31:2478–2486. doi: 10.1161/01.STR.31.10.2478. [DOI] [PubMed] [Google Scholar]
- 55.Ito T., Yamakawa H., Bregonzio C., Terrón J.A., Falcón-Neri A., Saavedra J.M. Protection Against Ischemia and Improvement of Cerebral Blood Flow in Genetically Hypertensive Rats by Chronic Pretreatment With an Angiotensin II AT 1 Antagonist. Stroke. 2002;33:2297–2303. doi: 10.1161/01.STR.0000027274.03779.F3. [DOI] [PubMed] [Google Scholar]
- 56.Groth W., Blume A., Gohlke P., Unger T., Culman J. Chronic pretreatment with candesartan improves recovery from focal cerebral ischaemia in rats. J. Hypertens. 2003;21:2175–2182. doi: 10.1097/00004872-200311000-00028. [DOI] [PubMed] [Google Scholar]
- 57.Stier C.T., Adler L.A., Levine S., Chander P.N. Stroke prevention by losartan in stroke-prone spontaneously hypertensive rats. J. Hypertens. 1993;11:37–42. [PubMed] [Google Scholar]
- 58.Villapol S., Yaszemski A.K., Logan T.T., Sanchez-Lemus E., Saavedra J.M., Symes A.J. Candesartan, an Angiotensin II AT1-Receptor Blocker and PPAR-γ Agonist, Reduces Lesion Volume and Improves Motor and Memory Function After Traumatic Brain Injury in Mice. Neuropsychopharmacology. 2012;37:2817–2829. doi: 10.1038/npp.2012.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li J.-M., Mogi M., Iwanami J., Min L.-J., Tsukuda K., Sakata A., Fujita T., Iwai M., Horiuchi M. Temporary Pretreatment With the Angiotensin II Type 1 Receptor Blocker, Valsartan, Prevents Ischemic Brain Damage Through an Increase in Capillary Density. Stroke. 2008;39:2029–2036. doi: 10.1161/STROKEAHA.107.503458. [DOI] [PubMed] [Google Scholar]
- 60.Arroja M.M.C., Reid E., McCabe C. Therapeutic potential of the renin angiotensin system in ischaemic stroke. Exp. Transl. Stroke Med. 2016;8:1–14. doi: 10.1186/s13231-016-0022-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hansson L., Lindholm L.H., Niskanen L., Lanke J., Hedner T., Niklason A., Luomanmäki K., Dahlöf B., de Faire U., Mörlin C., et al. Effect of angiotensin-converting-enzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: The Captopril Prevention Project (CAPPP) randomised trial. Lancet. 1999;353:611–616. doi: 10.1016/S0140-6736(98)05012-0. [DOI] [PubMed] [Google Scholar]
- 62.Bosch J., Yusuf S., Pogue J., Sleight P., Lonn E., Rangoonwala B., Davies R., Ostergren J., Probstfield J. Use of ramipril in preventing stroke: Double blind randomised trial. BMJ. 2002;324:699. doi: 10.1136/bmj.324.7339.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.PROGRESS Collaborative Group Randomised trial of a perindopril-based blood-pressure-lowering regimen among 6105 individuals with previous stroke or transient ischaemic attack. Lancet. 2001;358:1033–1041. doi: 10.1016/S0140-6736(01)06178-5. [DOI] [PubMed] [Google Scholar]
- 64.Dahlöf B., Devereux R.B., Kjeldsen S.E., Julius S., Beevers G., de Faire U., Fyhrquist F., Ibsen H., Kristiansson K., Lederballe-Pedersen O., et al. Cardiovascular morbidity and mortality in the Losartan Intervention for Endpoint reduction in hypertension study (LIFE): A randomised trial against atenolol. Lancet. 2002;359:995–1003. doi: 10.1016/S0140-6736(02)08089-3. [DOI] [PubMed] [Google Scholar]
- 65.Lithell H., Hansson L., Skoog I., Elmfeldt D., Hofman A., Olofsson B., Trenkwalder P., Zanchetti A. The Study on Cognition and Prognosis in the Elderly (SCOPE) J. Hypertens. 2003;21:875–886. doi: 10.1097/00004872-200305000-00011. [DOI] [PubMed] [Google Scholar]
- 66.Schrader J., Lüders S., Kulschewski A., Hammersen F., Plate K., Berger J., Zidek W., Dominiak P., Diener H.C. The MOSES Study Group Morbidity and Mortality After Stroke, Eprosartan Compared with Nitrendipine for Secondary Prevention. Stroke. 2005;36:1218–1224. doi: 10.1161/01.STR.0000166048.35740.a9. [DOI] [PubMed] [Google Scholar]
- 67.Yusuf S., Teo K., Anderson C.S., Pogue J., Dyal L., Copland I., Schumacher H., Dagenais G., Sleight P. Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: A randomised controlled trial. Lancet. 2008;372:1174–1183. doi: 10.1016/s0140-6736(08)61242-8. [DOI] [PubMed] [Google Scholar]
- 68.Yusuf S., Diener H.-C., Sacco R.L., Cotton D., Ôunpuu S., Lawton W.A., Palesch Y., Martin R.H., Albers G.W., Bath P., et al. Telmisartan to Prevent Recurrent Stroke and Cardiovascular Events. N. Engl. J. Med. 2008;359:1225–1237. doi: 10.1056/NEJMoa0804593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schrader J., Lüders S., Kulschewski A., Berger J., Zidek W., Treib J., Einhäupl K., Diener H.C., Dominiak P. The ACCESS Study. Stroke. 2003;34:1699–1703. doi: 10.1161/01.STR.0000075777.18006.89. [DOI] [PubMed] [Google Scholar]
- 70.Sandset E.C., Bath P., Boysen G., Jatuzis D., Kõrv J., Lüders S., Murray G.D., Richter P.S., Roine R.O., Terént A., et al. The angiotensin-receptor blocker candesartan for treatment of acute stroke (SCAST): A randomised, placebo-controlled, double-blind trial. Lancet. 2011;377:741–750. doi: 10.1016/S0140-6736(11)60104-9. [DOI] [PubMed] [Google Scholar]
- 71.Cabana J., Holleran B., Leduc R., Escher E., Guillemette G., Lavigne P. Identification of Distinct Conformations of the Angiotensin-II Type 1 Receptor Associated with the Gq/11 Protein Pathway and the β-Arrestin Pathway Using Molecular Dynamics Simulations. J. Biol. Chem. 2015;290:15835–15854. doi: 10.1074/jbc.M114.627356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guilluy C., Brégeon J., Toumaniantz G., Rolli-Derkinderen M., Retailleau K., Loufrani L., Henrion D., Scalbert E., Bril A., Torres R.M., et al. The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat. Med. 2010;16:183–190. doi: 10.1038/nm.2079. [DOI] [PubMed] [Google Scholar]
- 73.Miura S.-I., Zhang J., Boros J., Karnik S.S. TM2-TM7 Interaction in Coupling Movement of Transmembrane Helices to Activation of the Angiotensin II Type-1 Receptor. J. Biol. Chem. 2003;278:3720–3725. doi: 10.1074/jbc.M211338200. [DOI] [PubMed] [Google Scholar]
- 74.Galandrin S., Denis C., Boularan C., Marie J., M’Kadmi C., Pilette C., Dubroca C., Nicaise Y., Seguelas M.-H., N’Guyen D., et al. Cardioprotective Angiotensin-(1–7) Peptide Acts as a Natural-Biased Ligand at the Angiotensin II Type 1 Receptor. Hypertension. 2016;68:1365–1374. doi: 10.1161/HYPERTENSIONAHA.116.08118. [DOI] [PubMed] [Google Scholar]
- 75.Namkung Y., LeGouill C., Kumar S., Cao Y., Teixeira L.B., Lukasheva V., Giubilaro J., Simões S.C., Longpré J.-M., Devost D., et al. Functional selectivity profiling of the angiotensin II type 1 receptor using pathway-wide BRET signaling sensors. Sci. Signal. 2018;11:eaat1631. doi: 10.1126/scisignal.aat1631. [DOI] [PubMed] [Google Scholar]
- 76.Connolly A., Holleran B.J., Simard É., Baillargeon J.-P., Lavigne P., Leduc R. Interplay between intracellular loop 1 and helix VIII of the angiotensin II type 2 receptor controls its activation. Biochem. Pharmacol. 2019;168:330–338. doi: 10.1016/j.bcp.2019.07.018. [DOI] [PubMed] [Google Scholar]
- 77.Wirth A., Benyó Z., Lukasova M., Leutgeb B., Wettschureck N., Gorbey S., Örsy P., Horváth B., Maser-Gluth C., Greiner E., et al. G12-G13–LARG–mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat. Med. 2008;14:64–68. doi: 10.1038/nm1666. [DOI] [PubMed] [Google Scholar]
- 78.Freeman E.J., Ruehr M.L., Dorman R.V. ANG II-induced translocation of cytosolic PLA2 to the nucleus in vascular smooth muscle cells. Am. J. Physiol. Content. 1998;274:C282–C288. doi: 10.1152/ajpcell.1998.274.1.C282. [DOI] [PubMed] [Google Scholar]
- 79.Zafari A.M., Ushio-Fukai M., Minieri C.A., Akers M., Lassègue B., Griendling K.K. Arachidonic Acid Metabolites Mediate Angiotensin II-Induced NADH/NADPH Oxidase Activity and Hypertrophy in Vascular Smooth Muscle Cells. Antioxid. Redox Signal. 1999;1:167–179. doi: 10.1089/ars.1999.1.2-167. [DOI] [PubMed] [Google Scholar]
- 80.Cat A.N.D., Montezano A.C., Burger D., Touyz R.M. Angiotensin II, NADPH Oxidase, and Redox Signaling in the Vasculature. Antioxid. Redox Signal. 2013;19:1110–1120. doi: 10.1089/ars.2012.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lohse M., Andexinger S., Pitcher J., Trukawinski S., Codina J., Faure J., Caron M., Lefkowitz R. Receptor-specific desensitization with purified proteins. Kinase dependence and receptor specificity of beta-arrestin and arrestin in the beta 2-adrenergic receptor and rhodopsin systems. J. Biol. Chem. 1992;267:8558–8564. doi: 10.1016/S0021-9258(18)42479-9. [DOI] [PubMed] [Google Scholar]
- 82.Gurevich V.V., Gurevich E.V. GPCR Signaling Regulation: The Role of GRKs and Arrestins. Front. Pharmacol. 2019;10:125. doi: 10.3389/fphar.2019.00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Reiter E., Ahn S., Shukla A., Lefkowitz R.J. Molecular Mechanism of β-Arrestin-Biased Agonism at Seven-Transmembrane Receptors. Annu. Rev. Pharmacol. Toxicol. 2012;52:179–197. doi: 10.1146/annurev.pharmtox.010909.105800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bologna Z., Teoh J.-P., Bayoumi A.S., Tang Y., Kim I.-M. Biased G Protein-Coupled Receptor Signaling: New Player in Modulating Physiology and Pathology. Biomol. Ther. 2017;25:12–25. doi: 10.4062/biomolther.2016.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Galandrin S., Oligny-Longpré G., Bouvier M. The evasive nature of drug efficacy: Implications for drug discovery. Trends Pharmacol. Sci. 2007;28:423–430. doi: 10.1016/j.tips.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 86.Roth B.L., Chuang D.-M. Multiple mechanisms of serotonergic signal transduction. Life Sci. 1987;41:1051–1064. doi: 10.1016/0024-3205(87)90621-7. [DOI] [PubMed] [Google Scholar]
- 87.Zhou X.E., He Y., de Waal P.W., Gao X., Kang Y., Van Eps N., Yin Y., Pal K., Goswami D., White T.A., et al. Identification of Phosphorylation Codes for Arrestin Recruitment by G Protein-Coupled Receptors. Cell. 2017;170:457–469.e13. doi: 10.1016/j.cell.2017.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Whalen E.J., Rajagopal S., Lefkowitz R.J. Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol. Med. 2011;17:126–139. doi: 10.1016/j.molmed.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Grundmann M., Merten N., Malfacini D., Inoue A., Preis P., Simon K., Rüttiger N., Ziegler N., Benkel T., Schmitt N.K., et al. Lack of beta-arrestin signaling in the absence of active G proteins. Nat. Commun. 2018;9:1–16. doi: 10.1038/s41467-017-02661-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Luttrell L.M., Wang J., Plouffe B., Smith J.S., Yamani L., Kaur S., Jean-Charles P.-Y., Gauthier C., Lee M.-H., Pani B., et al. Manifold roles of β-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci. Signal. 2018;11:eaat7650. doi: 10.1126/scisignal.aat7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gurevich V.V. Arrestin-mediated signaling: Is there a controversy? World J. Biol. Chem. 2018;9:25–35. doi: 10.4331/wjbc.v9.i3.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Turu G., Balla A., Hunyady L. The Role of β-Arrestin Proteins in Organization of Signaling and Regulation of the AT1 Angiotensin Receptor. Front. Endocrinol. 2019;10:519. doi: 10.3389/fendo.2019.00519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Violin J.D., Lefkowitz R.J. β-Arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol. Sci. 2007;28:416–422. doi: 10.1016/j.tips.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 94.Wingler L., Elgeti M., Hilger D., Latorraca N.R., Lerch M.T., Staus D.P., Dror R.O., Kobilka B.K., Hubbell W.L., Lefkowitz R.J. Angiotensin Analogs with Divergent Bias Stabilize Distinct Receptor Conformations. Cell. 2019;176:468–478.e11. doi: 10.1016/j.cell.2018.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wingler L., McMahon C., Staus D.P., Lefkowitz R.J., Kruse A.C. Distinctive Activation Mechanism for Angiotensin Receptor Revealed by a Synthetic Nanobody. Cell. 2019;176:479–490.e12. doi: 10.1016/j.cell.2018.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wingler L.M., Skiba M.A., McMahon C., Staus D.P., Kleinhenz A.L.W., Suomivuori C.-M., Latorraca N.R., Dror R.O., Lefkowitz R.J., Kruse A.C. Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science. 2020;367:888–892. doi: 10.1126/science.aay9813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Modestia S.M., De Sa M.M., Auger E., Trossini G.H.G., Krieger J.E., Rangel-Yagui C.D.O., Rangel-Yagui C.O. Biased Agonist TRV027 Determinants in AT1R by Molecular Dynamics Simulations. J. Chem. Inf. Model. 2018;59:797–808. doi: 10.1021/acs.jcim.8b00628. [DOI] [PubMed] [Google Scholar]
- 98.Suomivuori C.-M., Latorraca N.R., Wingler L.M., Eismann S., King M.C., Kleinhenz A.L.W., Skiba M.A., Staus D.P., Kruse A.C., Lefkowitz R.J., et al. Molecular mechanism of biased signaling in a prototypical G protein–coupled receptor. Science. 2020;367:881–887. doi: 10.1126/science.aaz0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang H., Unal H., Gati C., Han G.W., Liu W., Zatsepin N., James D., Wang D., Nelson G., Weierstall U., et al. Structure of the Angiotensin Receptor Revealed by Serial Femtosecond Crystallography. Cell. 2015;161:833–844. doi: 10.1016/j.cell.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Valero T.R., Sturchler E., Jafferjee M., Rengo G., Magafa V., Cordopatis P., McDonald P., Koch W.J., Lymperopoulos A. Structure–activity relationship study of angiotensin II analogs in terms of β -arrestin-dependent signaling to aldosterone production. Pharmacol. Res. Perspect. 2016;4:e00226. doi: 10.1002/prp2.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rajagopal S., Kim J., Ahn S., Craig S., Lam C.M., Gerard N.P., Gerard C., Lefkowitz R.J. β-arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc. Natl. Acad. Sci. USA. 2010;107:628–632. doi: 10.1073/pnas.0912852107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Galzi J.-L., Hachet-Haas M., Bonnet D., Daubeuf F., Lecat S., Hibert M., Haiech J., Frossard N. Neutralizing endogenous chemokines with small molecules: Principles and potential therapeutic applications. Pharmacol. Ther. 2010;126:39–55. doi: 10.1016/j.pharmthera.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stamler J.S., Simon D.I., Osborne J.A., Mullins M.E., Jaraki O., Michel T., Singel D.J., Loscalzo J. S-nitrosylation of proteins with nitric oxide: Synthesis and characterization of biologically active compounds. Proc. Natl. Acad. Sci. USA. 1992;89:444–448. doi: 10.1073/pnas.89.1.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ferro A. S-Nitrosothiols as Nitric Oxide-Donors: Chemistry, Biology and Possible Future Therapeutic Applications. Curr. Med. Chem. 2004;11:2679–2690. doi: 10.2174/0929867043364397. [DOI] [PubMed] [Google Scholar]
- 105.Zhang X., Huang B., Zhang L., Zhang Y., Zhao Y., Guo X., Qiao X., Chen C. SNObase, a database for S-nitrosation modification. Protein Cell. 2012;3:929–933. doi: 10.1007/s13238-012-2094-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Leclerc P.C., Lanctot P.M., Auger-Messier M., Escher E., LeDuc R., Guillemette G. S-nitrosylation of cysteine 289 of the AT1 receptor decreases its binding affinity for angiotensin II. Br. J. Pharmacol. 2006;148:306–313. doi: 10.1038/sj.bjp.0706725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bouressam M.-L., Lecat S., Raoul A., Gaucher C., Perrin-Sarrado C., Lartaud I., Dupuis F. S-nitrosoglutathione inhibits cerebrovascular angiotensin II-dependent and -independent AT 1 receptor responses: A possible role of S -nitrosation. Br. J. Pharmacol. 2019;176:2049–2062. doi: 10.1111/bph.14644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Khan M., Sekhon B., Giri S., Jatana M., Gilg A.G., Ayasolla K., Elango C., Singh A.K., Singh I. S-Nitrosoglutathione Reduces Inflammation and Protects Brain against Focal Cerebral Ischemia in a Rat Model of Experimental Stroke. Br. J. Pharmacol. 2005;25:177–192. doi: 10.1038/sj.jcbfm.9600012. [DOI] [PubMed] [Google Scholar]
- 109.Khan M., Dhammu T.S., Sakakima H., Shunmugavel A., Gilg A.G., Singh A.K., Singh I. The inhibitory effect of S-nitrosoglutathione on blood-brain barrier disruption and peroxynitrite formation in a rat model of experimental stroke. J. Neurochem. 2012;123:86–97. doi: 10.1111/j.1471-4159.2012.07947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Parent M., Boudier A., Perrin J., Vigneron C., Maincent P., Violle N., Bisson J.-F., Lartaud I., Dupuis F. In Situ Microparticles Loaded with S-Nitrosoglutathione Protect from Stroke. PLoS ONE. 2015;10:e0144659. doi: 10.1371/journal.pone.0144659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pinheiro L.C., Oliveira-Paula G.H., Ferreira G.C., de Paula T.D.-C., Duarte D.A., Costa-Neto C.M., Tanus-Santos J.E. Oral nitrite treatment increases S-nitrosylation of vascular protein kinase C and attenuates the responses to angiotensin II. Redox Biol. 2021;38:101769. doi: 10.1016/j.redox.2020.101769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Schnitzler M.M.Y., Storch U., Meibers S., Nurwakagari P., Breit A., Essin K., Gollasch M., Gudermann T. Gq-coupled receptors as mechanosensors mediating myogenic vasoconstriction. EMBO J. 2008;27:3092–3103. doi: 10.1038/emboj.2008.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hansen J.L., Haunsø S., Brann M.R., Sheikh S.P., Weiner D.M. Loss-of-Function Polymorphic Variants of the Human Angiotensin II Type 1 Receptor. Mol. Pharmacol. 2004;65:770–777. doi: 10.1124/mol.65.3.770. [DOI] [PubMed] [Google Scholar]
- 114.Cao Y., Kumar S., Namkung Y., Gagnon L., Cho A., Laporte S.A. Angiotensin II type 1 receptor variants alter endosomal receptor–β-arrestin complex stability and MAPK activation. J. Biol. Chem. 2020;295:13169–13180. doi: 10.1074/jbc.RA120.014330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Thomas W.G., Qian H. Arresting angiotensin type 1 receptors. Trends Endocrinol. Metab. 2003;14:130–136. doi: 10.1016/S1043-2760(03)00023-7. [DOI] [PubMed] [Google Scholar]
- 116.Gáborik Z., Hunyady L. Intracellular trafficking of hormone receptors. Trends Endocrinol. Metab. 2004;15:286–293. doi: 10.1016/j.tem.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 117.Christensen G.L., Kelstrup C.D., Lyngsø C., Sarwar U., Bøgebo R., Sheikh S.P., Gammeltoft S., Olsen J.V., Hansen J.L. Quantitative Phosphoproteomics Dissection of Seven-transmembrane Receptor Signaling Using Full and Biased Agonists. Mol. Cell. Proteom. 2010;9:1540–1553. doi: 10.1074/mcp.M900550-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kendall R.T., Strungs E.G., Rachidi S.M., Lee M.-H., El-Shewy H.M., Luttrell D.K., Janech M.G., Luttrell L.M. The β-Arrestin Pathway-selective Type 1A Angiotensin Receptor (AT1A) Agonist [Sar1,Ile4,Ile8]Angiotensin II Regulates a Robust G Protein-independent Signaling Network. J. Biol. Chem. 2011;286:19880–19891. doi: 10.1074/jbc.M111.233080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pfeiffer C.T., Wang J., Paulo J.A., Jiang X., Gygi S.P., Rockman H.A. Mapping Angiotensin II Type 1 Receptor-Biased Signaling Using Proximity Labeling and Proteomics Identifies Diverse Actions of Biased Agonists. J. Proteome Res. 2021;20:3256–3267. doi: 10.1021/acs.jproteome.1c00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sanni S., Hansen J., Bonde M., Speerschneider T., Christensen G.L., Munk S., Gammeltoft S., Hansen J. β-Arrestin 1 and 2 stabilize the angiotensin II type I receptor in distinct high-affinity conformations. Br. J. Pharmacol. 2010;161:150–161. doi: 10.1111/j.1476-5381.2010.00875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Srivastava A., Gupta B., Gupta C., Shukla A.K. Emerging Functional Divergence of β-Arrestin Isoforms in GPCR Function. Trends Endocrinol. Metab. 2015;26:628–642. doi: 10.1016/j.tem.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 122.McCrink K.A., Maning J., Vu A., Jafferjee M., Marrero C., Brill A., Bathgate-Siryk A., Dabul S., Koch W.J., Lymperopoulos A. β-Arrestin2 Improves Post–Myocardial Infarction Heart Failure via Sarco(endo)plasmic Reticulum Ca 2+ -ATPase–Dependent Positive Inotropy in Cardiomyocytes. Hypertension. 2017;70:972–981. doi: 10.1161/HYPERTENSIONAHA.117.09817. [DOI] [PubMed] [Google Scholar]
- 123.Revankar C.M., Vines C.M., Cimino D.F., Prossnitz E.R. Arrestins Block G Protein-coupled Receptor-mediated Apoptosis. J. Biol. Chem. 2004;279:24578–24584. doi: 10.1074/jbc.M402121200. [DOI] [PubMed] [Google Scholar]
- 124.Lefkowitz R.J., Rajagopal K., Whalen E.J. New Roles for β-Arrestins in Cell Signaling: Not Just for Seven-Transmembrane Receptors. Mol. Cell. 2006;24:643–652. doi: 10.1016/j.molcel.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 125.DeWire S., Ahn S., Lefkowitz R.J., Shenoy S.K. β-Arrestins and Cell Signaling. Annu. Rev. Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 126.Peterson Y.K., Luttrell L.M. The Diverse Roles of Arrestin Scaffolds in G Protein–Coupled Receptor Signaling. Pharmacol. Rev. 2017;69:256–297. doi: 10.1124/pr.116.013367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Watari K., Nakaya M., Nishida M., Kim K.-M., Kurose H. β-arrestin2 in Infiltrated Macrophages Inhibits Excessive Inflammation after Myocardial Infarction. PLoS ONE. 2013;8:e68351. doi: 10.1371/journal.pone.0068351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wang Y., Jin L., Song Y., Zhang M., Shan D., Liu Y., Fang M., Lv F., Xiao R.-P., Zhang Y. β-arrestin 2 mediates cardiac ischemia-reperfusion injury via inhibiting GPCR-independent cell survival signalling. Cardiovasc. Res. 2017;113:1615–1626. doi: 10.1093/cvr/cvx147. [DOI] [PubMed] [Google Scholar]
- 129.Rajagopal K., Whalen E.J., Violin J.D., Stiber J.A., Rosenberg P.B., Premont R., Coffman T.M., Rockman H.A., Lefkowitz R.J. β-Arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes. Proc. Natl. Acad. Sci. USA. 2006;103:16284–16289. doi: 10.1073/pnas.0607583103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kim K.-S., Abraham D., Williams B., Violin J.D., Mao L., Rockman H.A. β-Arrestin-biased AT1R stimulation promotes cell survival during acute cardiac injury. Am. J. Physiol. Circ. Physiol. 2012;303:H1001–H1010. doi: 10.1152/ajpheart.00475.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Violin J.D., DeWire S., Yamashita D., Rominger D.H., Nguyen L., Schiller K., Whalen E.J., Gowen M., Lark M.W. Selectively Engaging β-Arrestins at the Angiotensin II Type 1 Receptor Reduces Blood Pressure and Increases Cardiac Performance. J. Pharmacol. Exp. Ther. 2010;335:572–579. doi: 10.1124/jpet.110.173005. [DOI] [PubMed] [Google Scholar]
- 132.Boerrigter G., Lark M.W., Whalen E.J., Soergel D.G., Violin J.D., Burnett J.C., Jr J.C.B. Cardiorenal Actions of TRV120027, a Novel ß-Arrestin–Biased Ligand at the Angiotensin II Type I Receptor, in Healthy and Heart Failure Canines. Circ. Hear. Fail. 2011;4:770–778. doi: 10.1161/CIRCHEARTFAILURE.111.962571. [DOI] [PubMed] [Google Scholar]
- 133.Boerrigter G., Soergel D.G., Violin J.D., Lark M.W., Burnett J.C. TRV120027, a Novel β-Arrestin Biased Ligand at the Angiotensin II Type I Receptor, Unloads the Heart and Maintains Renal Function When Added to Furosemide in Experimental Heart Failure. Circ. Hear. Fail. 2012;5:627–634. doi: 10.1161/CIRCHEARTFAILURE.112.969220. [DOI] [PubMed] [Google Scholar]
- 134.Soergel D., Subach R.A., James I.E., Cowan C.L., Gowen M., Lark M. Trvo27, a beta-arrestin biased ligand at the angiotensin 2 type 1 receptor, produces rapid, reversible changes in hemodynamics in patients with stable systolic heart failure. J. Am. Coll. Cardiol. 2013;61:E683. doi: 10.1016/S0735-1097(13)60683-X. [DOI] [Google Scholar]
- 135.Felker G.M., Butler J., Collins S.P., Cotter G., Davison B.A., Ezekowitz J.A., Filippatos G., Levy P.D., Metra M., Ponikowski P., et al. Heart Failure Therapeutics on the Basis of a Biased Ligand of the Angiotensin-2 Type 1 Receptor. JACC Hear. Fail. 2015;3:193–201. doi: 10.1016/j.jchf.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 136.Pang P.S., Butler J., Collins S.P., Cotter G., Davison B.A., Ezekowitz J.A., Filippatos G., Levy P.D., Metra M., Ponikowski P., et al. Biased ligand of the angiotensin II type 1 receptor in patients with acute heart failure: A randomized, double-blind, placebo-controlled, phase IIB, dose ranging trial (BLAST-AHF) Eur. Hear. J. 2017;38:2364–2373. doi: 10.1093/eurheartj/ehx196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rakesh K., Yoo B., Kim I.-M., Salazar N.C., Kim K.-S., Rockman H.A. β-Arrestin-Biased Agonism of the Angiotensin Receptor Induced by Mechanical Stress. Sci. Signal. 2010;3:ra46. doi: 10.1126/scisignal.2000769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Monasky M.M., Taglieri D.M., Henze M., Warren C.M., Utter M.S., Soergel D.G., Violin J.D., Solaro R.J. The β-arrestin-biased ligand TRV120023 inhibits angiotensin II-induced cardiac hypertrophy while preserving enhanced myofilament response to calcium. Am. J. Physiol. Circ. Physiol. 2013;305:H856–H866. doi: 10.1152/ajpheart.00327.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tarigopula M., Davis R.T., Mungai P.T., Ryba D.M., Wieczorek D.F., Cowan C.L., Violin J.D., Wolska B.M., Solaro R.J. Cardiac myosin light chain phosphorylation and inotropic effects of a biased ligand, TRV120023, in a dilated cardiomyopathy model. Cardiovasc. Res. 2015;107:226–234. doi: 10.1093/cvr/cvv162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhang H., Han G.W., Batyuk A., Ishchenko A., White K.L., Patel N., Sadybekov A., Zamlynny B., Rudd M.T., Hollenstein K., et al. Structural basis for selectivity and diversity in angiotensin II receptors. Nat. Cell Biol. 2017;544:327–332. doi: 10.1038/nature22035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Teixeira L.B., Parreiras-E-Silva L.T., Bruder-Nascimento T., Duarte D.A., Simões S.C., Costa R.M., Rodríguez D.Y., Ferreira P.A.B., Silva C.A.A., Abrao E.P., et al. Ang-(1-7) is an endogenous β-arrestin-biased agonist of the AT1 receptor with protective action in cardiac hypertrophy. Sci. Rep. 2017;7:1–10. doi: 10.1038/s41598-017-12074-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ferreira A.J., Santos R.A., Almeida A.P. Angiotensin-(1-7): Cardioprotective Effect in Myocardial Ischemia/Reperfusion. Hypertension. 2001;38:665–668. doi: 10.1161/01.HYP.38.3.665. [DOI] [PubMed] [Google Scholar]
- 143.Grobe J.L., Mecca A.P., Lingis M., Shenoy V., Bolton T.A., Machado J.M., Speth R.C., Raizada M.K., Katovich M.J. Prevention of angiotensin II-induced cardiac remodeling by angiotensin-(1–7) Am. J. Physiol. Circ. Physiol. 2007;292:H736–H742. doi: 10.1152/ajpheart.00937.2006. [DOI] [PubMed] [Google Scholar]
- 144.Ferreira A.J., Jacoby B.A., Araújo C.A.A., Macedo F.A.F.F., Silva G.A.B., Almeida A.P., Caliari M., Santos R.A.S. The nonpeptide angiotensin-(1–7) receptor Mas agonist AVE-0991 attenuates heart failure induced by myocardial infarction. Am. J. Physiol. Circ. Physiol. 2007;292:H1113–H1119. doi: 10.1152/ajpheart.00828.2006. [DOI] [PubMed] [Google Scholar]
- 145.Mecca A.P., Regenhardt R., O’Connor T.E., Joseph J.P., Raizada M.K., Katovich M.J., Sumners C. Cerebroprotection by angiotensin-(1-7) in endothelin-1-induced ischaemic stroke. Exp. Physiol. 2011;96:1084–1096. doi: 10.1113/expphysiol.2011.058578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Regenhardt R., Bennion D., Sumners C. Cerebroprotective action of angiotensin peptides in stroke. Clin. Sci. 2014;126:195–205. doi: 10.1042/CS20130324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jiang T., Gao L., Guo J., Lu J., Wang Y., Zhang Y. Suppressing inflammation by inhibiting the NF-κB pathway contributes to the neuroprotective effect of angiotensin-(1-7) in rats with permanent cerebral ischaemia. Br. J. Pharmacol. 2012;167:1520–1532. doi: 10.1111/j.1476-5381.2012.02105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Turu G., Szidonya L., Gáborik Z., Buday L., Spät A., Clark A.J., Hunyady L. Differential β-arrestin binding of AT1and AT2angiotensin receptors. FEBS Lett. 2005;580:41–45. doi: 10.1016/j.febslet.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 149.Villela D., Leonhardt J., Patel N., Joseph J., Kirsch S., Hallberg A., Unger T., Bader M., Santos R.A., Sumners C., et al. Angiotensin type 2 receptor (AT2R) and receptor Mas: A complex liaison. Clin. Sci. 2014;128:227–234. doi: 10.1042/CS20130515. [DOI] [PubMed] [Google Scholar]
- 150.Lyngsø C., Erikstrup N., Hansen J.L. Functional interactions between 7TM receptors in the Renin-Angiotensin System—Dimerization or crosstalk? Mol. Cell. Endocrinol. 2009;302:203–212. doi: 10.1016/j.mce.2008.09.018. [DOI] [PubMed] [Google Scholar]
- 151.Porrello E.R., Pfleger K., Seeber R.M., Qian H., Oro C., Abogadie F., Delbridge L.M., Thomas W.G. Heteromerization of angiotensin receptors changes trafficking and arrestin recruitment profiles. Cell. Signal. 2011;23:1767–1776. doi: 10.1016/j.cellsig.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 152.Szalai B., Barkai L., Turu G., Szidonya L., Várnai P., Hunyady L. Allosteric interactions within the AT1 angiotensin receptor homodimer: Role of the conserved DRY motif. Biochem. Pharmacol. 2012;84:477–485. doi: 10.1016/j.bcp.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 153.Durdagi S., Erol I., Salmas R.E., Aksoydan B., Kantarcioglu I. Oligomerization and cooperativity in GPCRs from the perspective of the angiotensin AT1 and dopamine D2 receptors. Neurosci. Lett. 2019;700:30–37. doi: 10.1016/j.neulet.2018.04.028. [DOI] [PubMed] [Google Scholar]
- 154.Wang P., Xu T.-Y., Wei K., Guan Y.-F., Wang X., Xu H., Su D.-F., Pei G., Miao C.-Y. ARRB1/β-arrestin-1 mediates neuroprotection through coordination of BECN1-dependent autophagy in cerebral ischemia. Autophagy. 2014;10:1535–1548. doi: 10.4161/auto.29203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wang H., Deng Q.-W., Peng A.-N., Xing F.-L., Zuo L., Li S., Gu Z.-T., Yan F.-L. β-arrestin2 functions as a key regulator in the sympathetic-triggered immunodepression after stroke. J. Neuroinflamm. 2018;15:1–11. doi: 10.1186/s12974-018-1142-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kanki H., Sasaki T., Matsumura S., Yokawa S., Yukami T., Shimamura M., Sakaguchi M., Furuno T., Suzuki T., Mochizuki H. β-arrestin-2 in PAR-1-biased signaling has a crucial role in endothelial function via PDGF-β in stroke. Cell Death Dis. 2019;10:1–16. doi: 10.1038/s41419-019-1375-x. [DOI] [PMC free article] [PubMed] [Google Scholar]