

Abstract
Acquired PARP inhibitor (PARPi) resistance in BRCA1- or BRCA2-mutant ovarian cancer often results from secondary mutations that restore expression of functional protein. RAD51C is a less commonly studied ovarian cancer susceptibility gene whose promoter is sometimes methylated, leading to homologous recombination (HR) deficiency and PARPi sensitivity. For this study, the PARPi-sensitive patient-derived ovarian cancer xenograft PH039, which lacks HR gene mutations but harbors RAD51C promoter methylation, was selected for PARPi resistance by cyclical niraparib treatment in vivo. PH039 acquired PARPi resistance by the third treatment cycle and grew through subsequent treatment with either niraparib or rucaparib. Transcriptional profiling throughout the course of resistance development showed widespread pathway level changes along with a marked increase in RAD51C mRNA, which reflected loss of RAD51C promoter methylation. Analysis of ovarian cancer samples from the ARIEL2 Part 1 clinical trial of rucaparib monotherapy likewise indicated an association between loss of RAD51C methylation prior to on-study biopsy and limited response. Interestingly, the PARPi resistant PH039 model remained platinum sensitive. Collectively, these results not only indicate that PARPi treatment pressure can reverse RAD51C methylation and restore RAD51C expression, but also provide a model for studying the clinical observation that PARPi and platinum sensitivity are sometimes dissociated.
Graphical Abstract
Graphical Abstract.

During PARP inhibitor treatment, an ovarian cancer PDX exhibits loss of RAD51C promoter methylation and altered expression of >750 genes in association with PARP inhibitor resistance and persistent carboplatin sensitivity.
INTRODUCTION
Up to 50% of high-grade serous ovarian carcinomas (HGSOC) display homologous recombination (HR) deficiency (1,2). The lesions most commonly implicated are mutations in BRCA1 and BRCA2, which are present in 17–25% of HGSOCs (1–6). An additional 10–15% of HGSOC cases have lost BRCA1 expression as a consequence of CDK12 inactivation (1,7–8) or BRCA1 promoter methylation (1–2,9–12), which often reflects loss of the zinc finger protein ZC3H18 (13). Whatever the cause, the resulting loss of BRCA1 or BRCA2 is associated with increased sensitivity to PARP inhibitors (PARPis) (14–16), which are approved for ovarian cancer both as maintenance therapy and as monotherapy for relapsed, platinum-sensitive disease (17–19).
The lesions contributing to BRCA1 or BRCA2 loss are often reversed when HGSOCs lacking these proteins become therapy resistant (20). For example, secondary mutations that restore the BRCA1 or BRCA2 open reading frame have been observed in DNA from platinum- (21) and PARPi-resistant (16,22–23) HGSOC. Additionally, loss of promoter methylation in a single copy of the BRCA1 gene was shown to be sufficient to restore HR and cause PARPi resistance (12,24).
Another 6–10% of HGSOC cases harbor mutations in other genes that contribute to HR (1,3–6,25–27). These inactivated genes include RAD51C and RAD51D, which encode RAD51 paralogs that participate in HR repair by modulating the action of the RAD51 recombinase (28). Previous studies have established that HGSOCs with RAD51C and RAD51D mutations are sensitive to the PARPi rucaparib (26). Conversely, secondary mutations that restore the RAD51C or RAD51D open reading frame and reestablish HR are observed when RAD51C- or RAD51D-mutant HGSOCs become rucaparib resistant (16,26). Promoter hypermethylation resulting in silencing of RAD51C has also been reported in 1–2% of ovarian cancers (1–2,9–11), but the number of reported cases has been too small to definitively determine whether RAD51C methylation is associated with increased platinum sensitivity (10,11). Even less is currently known about the changes that lead to PARPi resistance in RAD51C methylated tumors.
HR alterations that modulate PARPi sensitivity generally affect sensitivity to platinating agents as well. In preclinical models, loss of HR components sensitizes to both drug classes and restoration of HR produces resistance to both (24,26,29–30). Moreover, the degree of ovarian cancer shrinkage with the PARPi olaparib in the clinical setting has been correlated with platinum-free interval (an index of platinum sensitivity) (31). Despite this general correlation, olaparib responses at any particular platinum-free interval are quite variable (31), raising the possibility that factors other than HR deficiency might also play important roles in platinum sensitivity. Consistent with this notion, defects in nucleotide excision repair have been reported to sensitize HGSOCs to cisplatin without affecting PARPi sensitivity (32). It has also been observed that 40% of ovarian cancers progressing on PARPi treatment will respond to subsequent platinum-based therapy (33), although the basis for these divergent responses remains incompletely understood.
Patient-derived xenograft (PDX) models can provide a unique view into acquired resistance. In particular, intraperitoneal HGSOC PDX models retain critical features of the original cancers, including the pathogenic mutations, copy number alterations, gene expression profiles, degree of stromal infiltration, propensity to metastasize to bowel or induce ascites, sensitivity or resistance to drug treatment, and tumor heterogeneity (34–36), providing an opportunity for studying drug resistance under conditions somewhat similar to the source tumors. While acquired drug resistance studies in solid tumor PDX models typically rely on endpoint analyses, losing much of the time-course resolution of changes, serial passaging of PDX models can also allow for time resolution of signaling changes while simultaneously controlling for tumor evolution in a matched untreated control.
At the present time little is known about the stability of RAD51C methylation under PARPi treatment pressure. In the clinical setting, it is also difficult to determine whether alterations that affect PARPi sensitivity concomitantly affect platinum sensitivity. Because sister tumors can be simultaneously assayed for response to multiple agents, PDXs are a potentially important tool for addressing these questions. Accordingly, the present study was undertaken to (i) determine whether changes in RAD51C methylation would occur in a RAD51C-methylated HGSOC under PARPi selection, (ii) determine whether other genes exhibited altered methylation and expression during selection for stable PARPi resistance and (iii) assess whether resistance to carboplatin accompanied PARPi selection. Here, we report the loss of RAD51C methylation over a time course of acquired PARPi resistance in a PDX model and its differential impact on PARPi versus platinum sensitivity.
MATERIALS AND METHODS
Reagents
Rucaparib was provided by Clovis Oncology. Antibodies for immunoblotting were purchased as follows: Murine anti-RAD51C (NB100-177) from Novus (Centennial, CO), murine anti-RAD51 (MS-988-P0) from Thermo (Fremont, CA) and murine anti-LMNB (sc-377000) from Santa Cruz Biotechnology. Chicken polyclonal anti-B23/nucleophosmin was raised as previously described (37).
Resistance development in vivo
All animal studies were performed under protocols approved by the Mayo Clinic Animal Care and Use Committee. Utilizing previously described methods (34), PDX PH039 was established intraperitoneally in five female SCID Beige mice (C.B-17/IcrHsd-PrkdcscidLystbg–J; Envigo, Indianapolis, IN). When tumor size (largest cross-sectional area) as monitored by transabdominal ultrasound (SonoSite S-Series Ultrasound, Fujifilm SonoSite, Bothell, WA) reached 0.5–0.8 cm2, mice were randomized to the niraparib arm (n = 3) or source tumor arm (n = 2) as indicated in Figure 1A. On the source tumor arm, tumors were monitored for growth twice weekly by ultrasound, harvested when they reached predefined size criteria, and re-established in two mice. Tumor was passaged five times to control for any changes unrelated to niraparib treatment. On the niraparib arm, mice treated with 100 mg/kg niraparib (dissolved in 0.5% methylcellulose) daily for 21 days via oral gavage were monitored for regrowth of tumors, which were harvested and re-established in three mice. Once established, the tumor was again treated with niraparib for 21 days. This cyclic drug exposure continued until the tumor grew through treatment in two sequential passages. Additional details of the animal studies are described in the Supplementary Methods.
Figure 1.

Development of PARPi resistance in PH039. (A) To develop PARP inhibitor resistance, PH039, an intraperitoneal ovarian cancer PDX with no known mutations in HR, was treated with niraparib (100 mg/kg) daily via oral gavage for 21 days. Tumors were subsequently allowed to regrow and were then re-established in three new mice. This process was repeated for a total of four treatment cycles of niraparib. Tumor was also passaged five times without treatment to account for any genetic drift with passaging alone. (B) Representative ultrasounds showing shrinkage of PH039 with first treatment cycle and lack of shrinkage with fourth treatment cycle. (C) Tumor cross-sectional area relative to treatment day 0 as monitored in each animal by ultrasound. Shaded area, period of niraparib treatment. Error bars, mean ± sd of 3–5 individual PDXs at each timepoint.
RNA sequencing
RNA was harvested from snap-frozen xenograft tissue using an RNeasy Plus Minikit (Qiagen, Germany) or a Direct-zol RNA MiniPrep (Zymo Research, Cat# R2050). RNA sample quality was assessed by RNA integration number (RIN) on the RNA ScreenTape System (Agilent) according to manufacturer’s protocol. cDNA was generated with an Illumina TruSeq mRNA kit. The cDNAs were then denatured and polymerase chain reaction (PCR) enriched. The resulting genomic library was analyzed on an Illumina HiSeq 4000.
Bioinformatic analysis of RNAseq data
The bioinformatic pipeline is outlined in Supplementary Figure S1. Tophat (38,39) was used for sequence alignment to the UCSC HG38 (human) or UCSC MM10 (mouse) in conjunction with Picard (http://broadinstitute.github.io/picard) (see Supplementary Material for description and validation of method for addressing multi-mapping of sequences). Aligned reads were quantified using Subread v1.4.6 (40,41). Differential expression analysis was performed with EdgeR (42) and Bioconductor v3.4 (43) in R (44). For pairwise comparisons, gene filtering was set to include any gene with a total of more than two or three counts per million (two if two was the lowest number of samples for a given condition in the comparison and three if three was the lowest number of samples for a given condition.). IPA pathway analysis (45) (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuitypathway-analysis) was performed for genes with |log2 FC| > 1 and P-value< 0.01 unless otherwise noted. Figures were generated in R using ComplexHeatmap (46), pvclust (47), dendextend (48), MASS (49), circlize (50), colorspace (51), GetoptLong (https://github.com/jokergoo/GetoptLong) and ggplot2 (52). Heatmap colors based on www.ColorBrewer.org by Cynthia A. Brewer, Penn State. Genes corresponding to the ontology tag DNA Repair (GO:0006281) in Homo Sapiens were downloaded from AmiGO 2 (53,54) and intersected with differentially expressed genes.
SNV calling
SAMtools (v1.3.1) (55) was applied to RNAseq mouse depleted files to generate mpileups against GRCh38.86 human reference genome. VarScan2 (v2.4.1) (56) was used for baseline calling (S0 samples #1 and #2 against the human genome). Somatic calling using S0 as reference was performed for two samples passaged without selection (S2 #1, S2 #2) and two niraparib-resistant samples (C3 #1 and C3 #2). Reads were filtered for min-coverage of 10 and min-base-qual of 20. This allowed us to identify common variants at each time point that may contribute to the observed cisplatin sensitive phenotype. Variants were annotated with SNPEFFECT (57) and dbSNP15 was used for SNP annotations. For SNV selection, we filtered high confidence missense somatic variants and indels (P-value somatic <0.05). We inspected variants for 281 DNA damage repair genes (58), and all ATP transporters (*ATP) and solute carriers (*SLC), including all SLC31 family copper transporters (CTR1, CTR2) as well as ATOX1 and CCS previously described to impact cisplatin sensitivity (59).
Transcription factor analysis
Transcription factor analysis was performed focusing on transcription factors and targets that are expressed in the PDX samples. Log2 average fold change across all samples was compared to S0, and C3 was compared to S2 for each gene. Transcription factors, targets, and their mode of regulation (‘Activation’, ‘Repression’, ‘Unknown’) were extracted from the TRRUST v2 database (60). Entries with ‘Unknown’ modes of regulation and transcription factors with less than five annotation entries were excluded from the analysis. A transcription factor activity score was calculated for each transcription factor in the comparisons described above using the following formula:
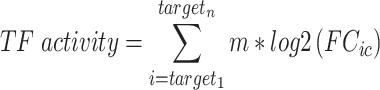 |
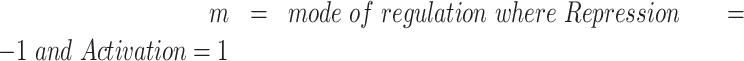 |
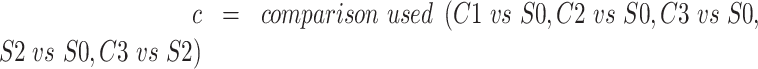 |
This analysis was performed using R 3.6.2; and heatmaps were generated with the ComplexHeatmap package.
Immunoblotting
Nuclei and cytosol were prepared from snap frozen tumors using a Thermo nuclear isolation kit according to the supplier’s instructions. Aliquots containing 30 μg (nuclei) or 50 μg protein (cytosol) (assayed by the bicinchoninic acid method) were separated on SDS-polyacrylamide gels containing 5–20% (wt/vol) acrylamide, transferred to nitrocellulose and probed with antibodies as previously described (61).
Immunohistochemistry, quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) and DNA methylation analysis
Immunohistochemistry (62), qRT-PCR (63) and assays for DNA methylation (64–66) were performed as previously reported and are described in the Supplementary Methods.
Clinical samples
ARIEL2 Part 1 was a multi-center international phase II clinical trial of single-agent rucaparib in patients with platinum-sensitive relapsed ovarian cancer (16). This study was conducted in accordance the Declaration of Helsinki and Good Clinical Practice Guidelines of the International Conference on Harmonisation. All patients signed informed written consent before participation. Of 196 patients enrolled, four had documented RAD51C promoter methylation at some point during the course of their disease. Pretreatment biospsies from three of these patients were adequate for RAD51C methylation by methylation-sensitive high resolution melting (MS-HRM) analysis (66). Tumor shrinkage is presented as the greatest % change at any timepoint in the sum of the longest dimensions of up to five prespecified target lesions relative to baseline (67). Time on therapy was defined as the number of days from initiation to cessation of rucaparib.
RESULTS
Generation of PARPi resistance in vivo
To systematically study genomic changes that occur during selection for PARPi resistance in vivo, mice bearing PH039, a HGSOC PDX that is extremely sensitive to PARPi but lacks demonstrable mutations in DNA repair genes (65), were treated with multiple cycles of niraparib (Figure 1A). At each step, tumors that regrew were transplanted into additional mice and selected for further resistance.
With the first cycle of PARPi exposure, PH039 shrank dramatically (Figure 1B and C) and remained below baseline for >50 days after the end of the treatment (Figure 1C). With the second cycle, shrinkage was again observed, but the tumor regrew more quickly. By the third and fourth cycles, the xenograft grew through niraparib treatment. Histological examination indicated that the tumor recurring after treatment was an adenocarcinoma that was indistinguishable from the source tumor (Supplementary Figure S2A). Moreover, the signature TP53 mutation in this PDX (65) was conserved throughout (Supplementary Figure S2B). Gene expression profiling (see below) also confirmed that the resistant tumor was of human origin.
Transcriptome changes associated with mouse passaging and with niraparib resistance
RNA sequencing (RNAseq) was performed on the original passage of PH039 (denoted S0), PH039 passaged without drug treatment (denoted S2 to indicate two passages in untreated mice) and PH039 selected for niraparib resistance (denoted C1-C3 to indicate the number of prior cycles of therapy). The strategy for distinguishing mouse (stroma) versus human (tumor cell) contributions to levels of each transcript is described in the Supplementary Results, Supplementary Tables S1–S4 and Supplementary Figures S1 and S3–S6. This gene-by-gene analysis not only demonstrated the need to identify and address the nonuniform homologies between human and mouse genes even for PDX tissue with very low mouse tissue infiltration, but also showed that this approach provided a robust pipeline for quality control in identifying genes disproportionately affected by mouse tissue contamination.
Further analysis demonstrated that certain transcripts changed with repeated passaging in the absence of drug treatment, possibly reflecting adaptation of the tumor to the mouse microenvironment and resulting in diverging and converging patterns of evolution (Supplementary Results and Figures S7 and S8). In addition to these transcript level changes, diverging and converging patterns of upstream pathway regulation were seen when comparing untreated tumor evolution with treated tumor evolution (Figure 2A).
Figure 2.

Changes in gene expression during PARPi resistance development in PH039. (A) Evolution of PH039 upstream signaling pathways through selective passaging and drug treatment. Differential expression results (absolute value of log2 fold change >1 and P-value < 0.01) for the pairwise comparisons of samples listed above the heatmap were analyzed through the use of IPA (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuitypathway-analysis) (45). Upstream regulators changed and detected in all five conditions are shown in the heatmap and colored by z-score. Clustering (rows) represents unsupervised UPGMA clustering on the displayed upstream regulators. (B) Changes in transcription factor activity scores of the top 5 differentially activated transcription factors between S2 and C3. Each circle represents a target gene that was analyzed. The Y values for each target gene indicate the change in activity, respectively for in the indicated comparison. The colors correspond to the average change in transcription factor activity calculated as described in the Methods. (C) Differential expression results (absolute value of log2 fold change >1 and P-value < 0.01) for the pairwise comparisons (bottom) were analyzed through the use of IPA (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuitypathway-analysis (45) and changes in upstream regulators detected in every condition are shown in the heatmap. Color represents the z-score for each upstream regulator in each condition. (D) Genes differentially expressed between S2 and C3 (absolute value of log2 fold change >1 and FDR < 0.01) are shown as mean-subtracted log counts per million. Kernel density plots (bottom) are represented for the differentially expressed genes. Hierarchical clustering (columns) was performed using UPGMA on all 13 900 genes passing the filtering threshold. Hierarchical clustering (rows) was performed using UPGMA on the differentially expressed genes shown. Genes with GO terms matching DNA repair are indicated.
To determine whether specific transcription factor activities might contribute to these changes, the TRRUST v2 database was used to examine combined target expression as a measure of putative transcription factor activity for those transcription factors that had at least five identified target genes (Figure 2B and Supplementary Figure S9). This analysis indicated a higher activity score for CEBPD, which had several target genes upregulated in the resistant PH039 samples, and a lower activity score for REST, which had several target genes downregulated (Figure 2B). In addition, the interferon regulatory factor (IRF) family members IRF9 and IRF1 had higher activity scores in the resistant models when compared to both the parental cell line (S0) and passaged control (S2) (Figure 2B and Supplementary Figure S9B).
Predicted upstream regulators of the transcriptional changes seen in the resistant tumors were also analyzed by Ingenuity Pathway Analysis (Figure 2C) and showed activation of multiple inflammation-related pathways, including INFG, INFL1, INFA2, TNF, INFB1, INFL4, TLR7 and TLR9, as well as downregulation of the SOCS1 pathway, which antagonizes these inflammatory pathways. Interestingly, this activation of inflammatory signaling was identified in two separate analyses, was demonstrated after controlling for mouse tissue contamination in samples that were >50 days from the last PARPi treatment (C1), and was observed in the absence of a functional immune response.
Unsupervised clustering revealed that the untreated tumor tissue, regardless of passage number, clustered with the source PDX (Figure 2D). Additionally, the PDXs that regrew after drug treatment also clustered together, with samples harvested at regrowth after Cycle 1 showing an intermediate gene expression pattern (Figure 2D, columns C1) relative to tumors that regrew after two and three cycles of treatment, which showed a more distinct RNA expression pattern (Figure 2D). Of the >700 differentially expressed transcripts identified in pairwise comparison of S2 and C3, seven genes (RAD51C, ISG15, PARP9, DTX3L, UBE2L6, PARP10 and RAD21L1) were associated by gene ontology with DNA repair and are highlighted in Figure 2D. Among these, the top differentially expressed gene was RAD51C (log2 fold change 8.5).
Altered methylation and re-expression of RAD51C associates with PARPi resistance
Consistent with the RNAseq analysis, qRT-PCR failed to detect RAD51C mRNA in untreated tumor at passage one (S0) and passage six (S5) but revealed readily detectable RAD51C message in the PDX that regrew after Cycle 1 (C1) of niraparib, which increased further after additional treatment (C2-C4, Figure 3A). This expression of RAD51C mRNA was accompanied by readily detectable RAD51C protein (Figure 3B), which was localized to the nuclei of tumor cells (Figure 3C). In accord with these results, niraparib induced formation of phospho-H2AX foci (Figure 3D), which are consistent with PARPi-induced DNA damage (68), and increased RAD51 foci (Figure 3E), which suggest restoration of HR.
Figure 3.

Re-expression of RAD51C in PARPi-selected PH039. (A) qRT-PCR results for RAD51C mRNA normalized to GAPDH mRNA. (B) Immunoblot of RAD51C protein across the samples. B23/nucleophosmin served as a loading control. (C) Immunohistochemical RAD51C staining of parental tumor after passaging (S5) versus resistant tumor (C4). (D and E) phospho-Ser139-histone H2AX (D) and RAD51C (E) staining of PH039 (S3) or niraparib-selected PH039R (C4) untreated (-) or after 8 days of niraparib (+).
Analysis of whole exome sequencing failed to provide any evidence of RAD51C gene amplification. Instead, as previously reported (65), untreated PH039 showed >80% methylation of the RAD51C promoter (Figure 4A, lanes 1–4) that persisted during serial passaging (Figure 4A, lanes 5–12). Methylation-sensitive high-resolution melting (MS-HRM) analysis demonstrated that this reflected heterogeneous methylation of the promoter (Supplementary Figure S10A). At the end of Cycle 1, the methylation was not detectably different (Figure 4A, lanes 13–16; Supplementary Figure S10A). On the other hand, at the end of Cycle 2 when the regrowing PDX was no longer PARPi responsive (Figure 1C), the locus was 95% unmethylated (Figure 4A, lanes 17–20) and was maintained in this unmethylated state through Cycles 3 and 4 of treatment (Figure 4A, lanes 21–32; Supplementary Figure S10A). This was further confirmed through allele-specific reduced representation bisulfite sequencing (Figure 4B and C), which showed that the diluent-treated PDX contained multiple distinct patterns of RAD51C promoter methylation, virtually all of which were lost in the niraparib-selected PDX.
Figure 4.

Loss of RAD51C methylation in PARPi-selected PH039. (A) RAD51C promoter methylation assessed by bisulfite modification followed by methylation-sensitive PCR in individual PDX samples. Controls were methylated (m) and unmethylated (um) RAD51C promoter sequence. (B) Allele-specific RRBS results for representative samples chosen from four untreated PH039 tumors (S0-S5), two tumors analyzed at C1 and two tumors analyzed at C2. Shown for each sample are allele-specific methylation patterns that occurred with >1% frequency in the sample (left) and the frequency of reads with that pattern (right). (C) Frequency of methylation of specific CpGs determined by allele-specific RRBS on DNA from each of the indicated samples. Numbers below panel indicate location relative to the transcription start site. (D) Frequency of completely unmethylated RAD51C reads as determined by allele-specific RBBS on DNA from each of the indicated samples. A total of 210 000–350 000 reads/sample were examined.
In further analysis, the allele-specific reduced representation bisulfite sequencing was searched for the presence of the fully unmethylated alleles that ultimately predominated after drug selection. Of 210 000–350 000 reads per sample, an average of 0.016% were fully unmethylated in the untreated PDXs (Figure 4D). The frequency of unmethylated reads increased 90-fold when the PDX grew back after the first cycle of treatment and another 60-fold (to over 80% of all reads) when the PDX grew back after the second cycle of treatment (Figure 4C and D).
Additional analysis of DNA methylation on Illumina EPIC arrays not only confirmed that the RAD51C promoter became unmethylated during the course of selection (Supplementary Figure S11A and S11B), but also revealed that the change in methylation was greater for one of the RAD51C promoter probes than for any other probe (Supplementary Figure S11A). Moreover, changes in methylation were extensive, with over 1100 probes showing a 4-fold change in methylation between S2 and C3 (Supplementary Figure S11A, S11C and S11D). Among the 700+ genes with altered expression during the course of resistance development (Figure 2D), 64 had a probe with a ≥4-fold change in methylation during resistance development (Supplementary Figure S11C).
Further xenograft studies revealed that the niraparib-selected PH039 PDX was cross-resistant to the PARPi rucaparib (Figure 5A). Based on these results, we examined the response of ovarian cancers with RAD51C promotor methylation identified in ARIEL2 Part 1, a phase II study of the PARPi rucaparib in patients with platinum-sensitive relapsed ovarian cancer (16). Of four cases with RAD51C methylation, three had biopsies with sufficient material for MS-HRM analysis harvested immediately prior to rucaparib. Of these three, two cancers showed retention of RAD51C methylation at the time of study entry (Supplementary Figure S10B, patients 1 and 2) and had 33–83% decreases in target lesions by CT on rucaparib treatment, which lasted 339–618 days before progression (Figure 5B). In contrast, the third ovarian cancer had lost RAD51C promoter methylation by the time of study entry and had limited shrinkage in response to rucaparib when removed from the study on day 50.
Figure 5.

Resistance of RAD51C unmethylated ovarian cancer to rucaparib. (A) Growth of parental (top, S3) or niraparib-selected PH039 (bottom, C4) without treatment or during treatment with the PARPi rucaparib at 300 or 450 mg/kg/d for 56 days. (B) Three ovarian carcinomas with previous RAD51C methylation and platinum-free intervals of 10.8, 32.7 and 24.2 months (patients 1–3, respectively) were treated with rucaparib monotherapy on the ARIEL2 Part 1 clinical trial after screening biopsies were obtained. MS-HRM analysis on those screening biopsies (Supplementary Figure S10B, summarized above bars) was compared to days on rucaparib treatment (top bars) and relative change in target lesion size as assessed by CT scan using RESIST 1.1 guidelines (bottom bars). Error bars in (A), ± sd derived from 6–8 mice in each cohort.
Dissociation of PARPi and platinum response in PH039
Additional PDX studies demonstrated that PH039R, despite being resistant to niraparib and rucaparib, was still sensitive to carboplatin (Figure 6A). This is in marked contrast to the BRCA2-mutated HGSOC PDX PH077, a second PDX model that was also selected by multiple cycles of niraparib treatment. When PH077 became resistant to niraparib (Figure 6B, squares), it harbored a secondary mutation that restored the BRCA2 open reading frame (Supplementary Figure S12A) and was carboplatin resistant (Figure 6B, triangles). Likewise, in tissue culture we observed that PARPi and platinum sensitivity varied in parallel when RAD51C status was altered (Figure 6C and Supplementary Figure S12B), in agreement with previous studies (26).
Figure 6.

Impact of niraparib selection on platinum sensitivity. (A) Response of PH039S (left) and PH039R (right) to carboplatin 51 mg/kg/week for 4 weeks. (B) Response of PH077S (left) and PH077R (right) to 100 mg/kg/day niraparib for 28 days or 51 mg/kg/week carboplatin for 4 weeks. Error bars in A and B, ± sd derived from 6–8 mice in each cohort. (C) Response of parental and RAD51C–/– Ovcar8 to niraparib and carboplatin in colony forming assays. Results are representative of three independent experiments. Error bars, ± sd derived from three replicate plates at each concentration. (D) qRT-PCR for selected transcripts that have been associated with cisplatin or carboplatin sensitivity. Error bars, ± sd derived from the 2–3 independent cryopreserved PDX samples shown at each indicated passage. Expression levels in untreated (S0, S2, S5) and niraparib-resistant (C2, C3, C4) xenografts were compared by unpaired two-sided t test. C1 was omitted because of its intermediate gene expression pattern as depicted in Figure 2D. (E) Homozygous repair protein genetic polymorphisms observed in PH039 that are associated with high platinum sensitivity (Supplementary Figure S12E and Supplementary Table S6).
In order to better understand the unexpected platinum sensitivity of PH039R, we examined DNA sequencing and RNAseq data for mutations and gene expression changes that might be associated with selective platinum sensitivity. Targeted capture and massively parallel sequencing [BROCA analysis (25,69), Supplementary Table S5] failed to identify mutations in any of the HR/ovarian cancer susceptibility genes sequenced. Moreover, whole exome sequencing failed to identify mutations of 53BP1, PTIP or other genes known to be involved in PARPi resistance in the PH039R xenograft. Instead, further examination of the RNAseq results revealed several changes that might contribute to platinum sensitivity (Figures 2D and 6D; Supplementary Figure S12C). Among the top 100 differentially expressed genes between PH039S and PH039R, 21 were previously associated with platinum response, and 18 of the changes in expression were reported to convey platinum sensitivity (Supplementary Figure S12C). In contrast, none of these changes occurred with repeated passaging alone. These changes included upregulation of mRNA encoding the ubiquitin-like protein ISG15 and the XIAP neutralizer XAF1 as well as downregulation of mRNA encoding the glutathione transferase isoform GSTμ, the small G protein RAB18 and the forkhead transcription factor FOXA1 (Figure 6D), all of which have been previously associated with increased platinum sensitivity (70–77). On the other hand, when PH077 became resistant to niraparib and carboplatin, none of these transcripts were significantly altered in the same direction (Supplementary Figure S12D).
In addition to these changes in gene expression, analysis of the whole exome sequencing revealed SNVs in critical components of the nucleotide excision repair (XPC G939K) and base excision pathways (XRCC1 Q391R) that were previously associated with increased platinum sensitivity (78–81). These SNVs were present at >90% variant allele frequency in the earliest passage PH039 (S0), persisted in the niraparib-selected PDX (Figure 6E and Supplementary Figure S12E) and were also confirmed in >90% of RNA reads. Although the potential role of the XPC SNV is controversial (see Supplementary Table S6), XRCC1 Q391R reportedly exhibits diminished ability to support repair after ultraviolet and ionizing radiation compared to wild-type XRCC1. Moreover, XRCC1 Q391R has been associated with enhanced platinum sensitivity in other neoplasms (Supplementary Table S6), raising the possibility that this SNV could also potentially contribute to platinum sensitivity of the PH039R xenograft.
DISCUSSION
Results of the present study demonstrated that selection for PARP inhibitor resistance in a RAD51C promoter-methylated ovarian cancer PDX model is associated loss of RAD51C methylation and with development of resistance to multiple PARPis in vivo. Similar results were observed in clinical samples from ovarian cancer patients treated on ARIEL2 Part 1, a single-agent phase II clinical trial of the PARPi rucaparib. Interestingly, in the PH039 PDX characterized here, development of PARPi resistance was not accompanied by carboplatin resistance. These observations have potentially important implications for improved understanding of the relationship between PARPi and platinum resistance.
In the present study, we have examined changes in the transcriptome and methylome over the time-course of PARPi resistance development in a rare but informative genomic subtype of high-grade serous ovarian cancer. Use of a PDX model is ideal for this study, as this number of sequential samples cannot be acquired during the course of clinical treatment. The RNAseq data obtained over the course of resistance development provide a unique look into tumor evolution through sequential passaging with and without drug. Importantly, there are a number of changes during serial passaging even without drug (Figure 2D; Supplementary Figures S7 and S8), possibly reflecting evolution of this TP53-mutant ovarian cancer as it adapts to growth in mice. These observations highlight the potential importance of including both the PDX passaged without drug (S2) and the starting PDX (S0) as controls when examining changes that accompany drug selection.
Comparing the transcriptome of niraparib-selected PH039 to the untreated PDX passaged an equal number of times, the most prominent transcriptional pathway changes reflect increased inflammatory signaling (Figure 2A and B). These results are consistent with previous reports that acute PARPi treatment, which is thought to cause release of DNA fragments into the cytoplasm, activates interferon-induced signaling through the cGAS/STING pathway in xenografts and cell culture systems (82–84). Interestingly, however, we observed activation of this signaling over 50 days after PARPi withdrawal (C1 samples, Figures 1C and 2), suggesting that the DNA damage persists long after drug treatment or that PARPis also activate inflammatory signaling through some other persistent process.
The PDX on which these studies were performed, PH039, is among the most PARPi sensitive ovarian cancer PDXs we have examined (34,65). In the therapy naïve model, the RAD51C promoter is extensively methylated, a modification that is known to disrupt HR and contribute to PARPi sensitivity (2). Our further analysis demonstrated that this RAD51C methylation displays extensive heterogeneity of CpG methylation patterns when analyzed in an allele-by-allele basis (Figure 4B). Upon repeated treatment with niraparib, we observed loss of RAD51C promoter methylation accompanied by reappearance of RAD51C mRNA and protein (Figures 3, 4 and Supplementary Figure S10). Conceptually this could represent either a PARPi-induced dynamic change in methylation or selection for a subpopulation of cells with preexisting loss of RAD51C promoter methylation. Even though the untreated PDX did not contain a discernible subpopulation of RAD51C expressing cells (Figure 3C), examination of allele-specific reduced representation bisulfite sequencing indicated that approximately 1 in 6000 reads was fully unmethylated in the untreated PDX (Figure 4D). With each of the first two cycles of niraparib treatment, the frequency of these unmethylated alleles increased 60- to 90-fold. These results are consistent with possibility that niraparib might select for a small population of preexisting cells harboring unmethylated RAD51C loci, but we cannot rule out a contribution of dynamic demethylation as well. Moreover, because the number of unmethylated bisulfite reads in aliquots of the untreated PDX was small (16–131 reads), we cannot completely rule out the possibility that these reads are of technical rather than biological origin. Accordingly, this proposed explanation for the emergence of resistance must be viewed as tentative.
The change in RAD51C expression was accompanied by changes in expression of over 700 other transcripts. Because over 90% of these changes occurred without alterations in methylation of the corresponding gene promoter (Supplementary Figure S11C), we examined changes in transcription factor networks that could account for the altered gene expression. As indicated in Figure 2B and Supplementary Figure S9, multiple transcription factors—particularly members of the IRF family—are activated in PARPi resistant PH039.
PARPi sensitivity and platinum sensitivity are generally thought to parallel each other. In PH077, the second ovarian cancer PDX characterized in the present study, this was clearly the case (Figure 6B). This PDX contained a BRCA2 frameshift mutation at the outset; and selection for niraparib resistance was accompanied by a secondary mutation that restored the BRCA2 reading frame (Supplementary Figure S12A), resulting in carboplatin resistance (Figure 6B). In contrast, PH039R was resistant to two different PARPis (Figures 1C and 5A) but sensitive to carboplatin (Figure 6A). Because changes in RAD51C expression clearly affect both PARPi and platinum sensitivity (Figure 6C and Supplementary Figure S12B), the carboplatin sensitivity of PH039R was unexpected.
Further examination revealed multiple factors that might, in aggregate, contribute to the platinum sensitivity of PH039R. Among the changes in expression between PH039S and PH039R are many that have been reported to convey platinum sensitivity (Supplementary Figure S12C), including upregulation of mRNA encoding XAF1 as well as downregulation of FOXA1, RAB18, and GSTM, all of which have been confirmed by qRT-PCR (Figure 6D). Among the differentially expressed genes, ISG15 is particularly interesting because its increased expression is (i) tightly linked to the increased inflammatory signaling seen after PARPi treatment (Figure 2A–C) and (ii) tied to increased replication stress (85), which would enhance platinum sensitivity. In contrast, the RNAseq analysis did not identify upregulation of any of the ATP binding cassette transporters previously implicated in PARPi resistance (data not shown); and the observation that phospho-H2AX foci form when the resistant PDXs are treated with niraparib (Figure 3D) shows that niraparib still induces DNA damage, further arguing against transport-mediated resistance. On the other hand, previous studies have also shown that cells with nucleotide excision repair (NER) defects are particularly sensitive to cisplatin, which forms the same DNA lesions as carboplatin (86), and resistant to PARPis (32). Whole exome sequencing failed to reveal any new mutations in DNA repair genes, including NER genes, in PH039R compared to the parental PDX. Instead, we identified SNVs in repair genes (Figure 6E) that have previously been associated with impaired repair in preclinical studies (Supplementary Table S6) and cisplatin sensitivity in the clinical setting (78–81). Especially noteworthy in this regard is the homozygous XRCC1 Q399R allele because the homogeneous G allele seen in PH039 is associated with impaired base excision repair and a higher response rate of several cancers to platinum-containing therapy (Supplementary Table S6). Accordingly, it is possible that the persistent platinum sensitivity of the PH039R PDX reflects the combined effects of several sensitizing features, including gene expression changes that counterbalance RAD51C re-expression by increasing platinum sensitivity, concomitant presence of one or more single nucleotide polymorphisms that also convey platinum sensitivity, and possibly other factors yet to be identified.
Several observations have previously suggested that the relationship between platinum resistance and PARPi resistance in the clinic might also be more nuanced than generally recognized. First, a large randomized study demonstrated that 67% of suboptimally debulked ovarian cancers exhibit objective responses to cisplatin (87), yet the fraction of ovarian cancers with well-established HR defects is only 40–50% (1,2). These observations raise the possibility that a subset of HR proficient ovarian cancers is platinum sensitive. Second, while the correlation between platinum-free interval (a measure of platinum sensitivity) and % tumor shrinkage on PARPi (a measure of PARPi sensitivity) is statistically significant (31), the correlation is somewhat modest (R2 = 0.26), again suggesting that factors beyond HR pathway integrity also might play a role platinum sensitivity. Finally, it has been reported that 40% of ovarian cancers progressing on the PARPi olaparib still respond to subsequent platinum therapy (33), again suggesting that PARPi sensitivity and platinum sensitivity might be dissociated under certain circumstances.
Although ovarian cancer PDXs have several advantages for studying the emergence of drug resistance, they also have several limitations. First, unlike continuously growing cell lines, tumor cells from ovarian cancer PDXs survive for only a few days in tissue culture, making it difficult to conduct gene knockdown, knockout, or re-expression studies and assess drug response ex vivo. Second, we have not yet been successful in introducing transgenes into these ovarian cancer PDXs for long-term expression, limiting our ability to definitively assess the role of the gene expression changes shown in Figure 6D and Supplementary Figure S12C in platinum sensitivity. Finally, the ability to follow PDX growth is limited by the lifespan of mice. Although human ovarian cancers sometimes recur as late as 8–10 years after treatment, PDXs must recur in 12–18 months if they are to be studied in murine hosts, a consideration that led us to use intermittent rather than continuous niraparib dosing in selecting for resistance (Figure 1). Accordingly, we cannot definitively state that the mechanism of PARPi resistance observed in PH039R reflects a mechanism that would be observed with continuous dosing. However, the fact that similar results are seen in clinical material (Figure 5), coupled with the observation that the resulting PDXs are resistant to continuous dosing (Figure 6A and B), suggests that the observed mechanism is in fact germane to the clinical setting.
In summary, we have shown that changes accompanying the development of PARPi resistance in an ovarian cancer PDX with initial RAD51C silencing can potentially contribute to retained platinum sensitivity in vivo. Whether similar events occur in other ovarian or breast PDXs with RAD51C methylation remains to be further investigated. As the present study illustrates, the ability to examine sister tumors for sensitivity to a variety of agents represents one of the clear-cut benefits of PDXs for understanding patterns of cross resistance.
DATA AVAILABILITY
Data have been deposited into the GEO database as data super series GSE165052 and are available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE165052.
Supplementary Material
ACKNOWLEDGEMENTS
We thank David Toft for his kind gift of H90-10 antibody, Alan D’Andrea for helpful discussions, and the three anonymous reviewers for their insightful suggestions.
Author Contributions:
Conceived and designed project: R.M.H., C.D.M., P.H., S.H.K.
Conducted experiments: R.M.H., K.N., R.L.K., A.V., X.H., N.M.P., X.W.M., M.R.R., P.A.S., K.S.F., K.L.P., M.A.B., E.M.W., A.D., J.M.W., S.H.K.
Analyzed data: R.M.H., C.D.M., K.N., C.C., T.M.W., O.K., C.A.R., M.W., H.L.
Provided critical reagents: K.K.L., T.C.H, I.A.M.
Project supervision: M.S.S., M.W., C.L.S., A.E.W.H., E.M.S., H.L., S.J.W. and S.H.K.
Initial manuscript preparation: R.M.H., C.D.M. and S.H.K.
Manuscript editing: All
Contributor Information
Rachel M Hurley, Department of Molecular Pharmacology and Experimental Therapeutics, Mayo Clinic, Rochester, MN 55905 USA.
Cordelia D McGehee, Department of Molecular Pharmacology and Experimental Therapeutics, Mayo Clinic, Rochester, MN 55905 USA.
Ksenija Nesic, The Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria 3052, Australia; Department of Medical Biology, University of Melbourne, Parkville, Victoria 3010, Australia.
Cristina Correia, Department of Molecular Pharmacology and Experimental Therapeutics, Mayo Clinic, Rochester, MN 55905 USA; Division of Oncology Research, Mayo Clinic, Rochester, MN 55905 USA.
Taylor M Weiskittel, Department of Molecular Pharmacology and Experimental Therapeutics, Mayo Clinic, Rochester, MN 55905 USA.
Rebecca L Kelly, Department of Molecular Pharmacology and Experimental Therapeutics, Mayo Clinic, Rochester, MN 55905 USA.
Annapoorna Venkatachalam, Department of Molecular Pharmacology and Experimental Therapeutics, Mayo Clinic, Rochester, MN 55905 USA.
Xiaonan Hou, Division of Medical Oncology, Mayo Clinic, Rochester, MN 55905 USA.
Nicholas M Pathoulas, Division of Oncology Research, Mayo Clinic, Rochester, MN 55905 USA.
X Wei Meng, Department of Molecular Pharmacology and Experimental Therapeutics, Mayo Clinic, Rochester, MN 55905 USA; Division of Oncology Research, Mayo Clinic, Rochester, MN 55905 USA.
Olga Kondrashova, The Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria 3052, Australia; Department of Medical Biology, University of Melbourne, Parkville, Victoria 3010, Australia.
Marc R Radke, Department of Obstetrics & Gynecology, University of Washington, Seattle, WA 98195, USA.
Paula A Schneider, Division of Oncology Research, Mayo Clinic, Rochester, MN 55905 USA.
Karen S Flatten, Division of Oncology Research, Mayo Clinic, Rochester, MN 55905 USA.
Kevin L Peterson, Division of Oncology Research, Mayo Clinic, Rochester, MN 55905 USA.
Marc A Becker, Division of Medical Oncology, Mayo Clinic, Rochester, MN 55905 USA.
Ee Ming Wong, Precision Medicine, School of Clinical Sciences at Monash Health, Monash University, Victoria 3800, Australia; Department of Clinical Pathology, Melbourne Medical School, The University of Melbourne, Parkville, Victoria 3000, Australia.
Melissa S Southey, Precision Medicine, School of Clinical Sciences at Monash Health, Monash University, Victoria 3800, Australia; Department of Clinical Pathology, Melbourne Medical School, The University of Melbourne, Parkville, Victoria 3000, Australia; Cancer Epidemiology Division, Cancer Council Victoria, Melbourne, Victoria 3004, Australia.
Alexander Dobrovic, University of Melbourne Department of Surgery, Austin Hospital, Heidelberg, Victoria 3084, Australia.
Kevin K Lin, Clovis Oncology, San Francisco, CA 94158, USA.
Thomas C Harding, Clovis Oncology, San Francisco, CA 94158, USA.
Iain McNeish, Division of Cancer, Department of Surgery and Cancer, Imperial College London, Hammersmith Campus, London, W12 0NN United Kingdom.
Christian A Ross, Division of Information Technology, Mayo Clinic, Rochester, MN 55905, USA.
Jill M Wagner, Division of Medical Oncology, Mayo Clinic, Rochester, MN 55905 USA.
Matthew J Wakefield, The Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria 3052, Australia; Department of Obstetrics and Gynaecology, University of Melbourne, Parkville, Victoria 3052, Australia.
Clare L Scott, The Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria 3052, Australia; Department of Medical Biology, University of Melbourne, Parkville, Victoria 3010, Australia; Department of Obstetrics and Gynaecology, University of Melbourne, Parkville, Victoria 3052, Australia.
Paul Haluska, Division of Medical Oncology, Mayo Clinic, Rochester, MN 55905 USA.
Andrea E Wahner Hendrickson, Division of Medical Oncology, Mayo Clinic, Rochester, MN 55905 USA.
Larry M Karnitz, Department of Molecular Pharmacology and Experimental Therapeutics, Mayo Clinic, Rochester, MN 55905 USA; Division of Oncology Research, Mayo Clinic, Rochester, MN 55905 USA.
Elizabeth M Swisher, Department of Obstetrics & Gynecology, University of Washington, Seattle, WA 98195, USA.
Hu Li, Department of Molecular Pharmacology and Experimental Therapeutics, Mayo Clinic, Rochester, MN 55905 USA; Center for Individualized Medicine, Mayo Clinic, Rochester, MN 55905 USA.
S John Weroha, Division of Medical Oncology, Mayo Clinic, Rochester, MN 55905 USA.
Scott H Kaufmann, Department of Molecular Pharmacology and Experimental Therapeutics, Mayo Clinic, Rochester, MN 55905 USA; Division of Oncology Research, Mayo Clinic, Rochester, MN 55905 USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Cancer Online.
FUNDING
National Institutes of Health [P50 CA136393, F30 CA213737, T32 GM072474, T32 GM085641]; Ovarian Cancer Research Alliance (to E.M.S., P.H., S.H.K.); Stand Up to Cancer [SU2C-AACR-DT16-15] (to A.E.W.H., E.M.S., S.J.W., S.H.K.); Stafford Fox Medical Research Foundation (to C.L.S., O.K., M.W., K.N.); National Breast Cancer Foundation of Australia (to A.D.); Mayo Foundation for Education and Research (to R.M.H., C.D.M., T.M., R.L.K., A.V.)
Conflict of interest statement. K.K.L. and T.C.H. are employees of Clovis Oncology, which supported the ARIEL2 clinical trial. C.L.S. declares Advisory Boards for Clovis Oncology, AstraZeneca, Roche, Eisai Inc, Sierra Oncology, Takeda, MSD and Grant/Research support from Clovis Oncology, Eisai Inc, Sierra Oncology, Roche and Beigene. The authors have no other conflicts of interest to disclose.
REFERENCES
- 1. Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474:609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Konstantinopoulos P.A., Ceccaldi R., Shapiro G.I., D’Andrea A.D. Homologous recombination deficiency: exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 2015; 5:1137–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alsop K., Fereday S., Meldrum C., deFazio A., Emmanuel C., George J., Dobrovic A., Birrer M.J., Webb P.M., Stewart C. et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012; 30:2654–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pennington K.P., Walsh T., Harrell M.I., Lee M.K., Pennil C.C., Rendi M.H., Thornton A., Norquist B.M., Casadei S., Nord A.S. et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014; 20:764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Norquist B.M., Harrell M.I., Brady M.F., Walsh T., Lee M.K., Gulsuner S., Bernards S.S., Casadei S., Yi Q., Burger R.A. et al. Inherited mutations in women with ovarian carcinoma. JAMA Oncol. 2016; 2:482–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lilyquist J., LaDuca H., Polley E., Davis B.T., Shimelis H., Hu C., Hart S.N., Dolinsky J.S., Couch F.J., Goldgar D.E. Frequency of mutations in a large series of clinically ascertained ovarian cancer cases tested on multi-gene panels compared to reference controls. Gynecol. Oncol. 2017; 147:375–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blazek D., Kohoutek J., Bartholomeeusen K., Johansen E., Hulinkova P., Luo Z., Cimermancic P., Ule J., Peterlin B.M. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 2011; 25:2158–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Joshi P.M., Sutor S.L., Huntoon C.J., Karnitz L.M. Ovarian cancer-associated mutations disable catalytic activity of CDK12, a kinase that promotes homologous recombination repair and resistance to cisplatin and poly(ADP-ribose) polymerase inhibitors. J. Biol. Chem. 2014; 289:9247–9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Swisher E.M., Gonzalez R.M., Taniguchi T., Garcia R.L., Walsh T., Goff B.A., Welcsh P. Methylation and protein expression of DNA repair genes: association with chemotherapy exposure and survival in sporadic ovarian and peritoneal carcinomas. Mol. Cancer. 2009; 8:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cunningham J.M., Cicek M.S., Larson N.B., Davila J., Wang C., Larson M.C., Song H., Dicks E.M., Harrington P., Wick M. et al. Clinical characteristics of ovarian cancer classified by BRCA1, BRCA2, and RAD51C status. Sci. Rep. 2014; 4:4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bernards S.S., Pennington K.P., Harrell M.I., Agnew K.J., Garcia R.L., Norquist B.M., Swisher E.M. Clinical characteristics and outcomes of patients with BRCA1 or RAD51C methylated versus mutated ovarian carcinoma. Gynecol. Oncol. 2018; 148:281–285. [DOI] [PubMed] [Google Scholar]
- 12. Swisher E.M., Kwan T.T., Oza A.M., Tinker A.V., Ray-Coquard I., Oaknin A., Coleman R.L., Aghajanian C., Konecny G.E., O’Malley D.M. et al. Molecular and clinical determinants of response and resistance to rucaparib for recurrent ovarian cancer treatment in ARIEL2 (Parts 1 and 2). Nat. Commun. 2021; 12:2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kanakkanthara A., Huntoon C.J., Hou X., Heinzen E.P., Oberg A.L., Weroha S.J., Kaufmann S.H., Karnitz L.M. ZC3H18 deficiency impairs BRCA1 transcription and causes homologous recombination DNA repair defect in ovarian cancer. Nat. Commun. 2019; 10:4632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ledermann J., Harter P., Gourley C., Friedlander M., Vergote I., Rustin G., Scott C.L., Meier W., Shapira-Frommer R., Safra T. et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014; 15:852–861. [DOI] [PubMed] [Google Scholar]
- 15. Mirza M.R., Monk B.J., Herrstedt J., Oza A.M., Mahner S., Redondo A., Fabbro M., Ledermann J.A., Lorusso D., Vergote I. et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N. Engl. J. Med. 2016; 375:2154–2164. [DOI] [PubMed] [Google Scholar]
- 16. Swisher E.M., Lin K.K., Oza A.M., Scott C.L., Giordano H., Sun J., Konecny G.E., Coleman R.L., Tinker A.V., O’Malley D.M. et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017; 18:75–87. [DOI] [PubMed] [Google Scholar]
- 17. Scott C.L., Swisher E.M., Kaufmann S.H. Poly (ADP-Ribose) polymerase inhibitors: recent advances and future development. J. Clin. Oncol. 2015; 33:1397–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Konstantinopoulos P.A., Matulonis U.A. PARP inhibitors in ovarian cancer: a trailblazing and transformative journey. Clin. Cancer Res. 2018; 24:4062–4065. [DOI] [PubMed] [Google Scholar]
- 19. Curtin N.J., Szabo C. Poly(ADP-ribose) polymerase inhibition: past, present and future. Nat. Rev. Drug Discov. 2020; 19:711–736. [DOI] [PubMed] [Google Scholar]
- 20. Lord C.J., Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat. Med. 2013; 19:1381–1388. [DOI] [PubMed] [Google Scholar]
- 21. Norquist B., Wurz K., Pennil C., Garcia R., Gross J., Sakai W., Karlan B., Taniguchi T., Swisher E.M. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J. Clin. Oncol. 2011; 29:3008–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barber L.J., Sandhu S., Chen L., Campbell J., Kozarewa I., Fenwick K., Assiotis I., Rodrigues D.N., Reis Filho J.S., Moreno V. et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J. Pathol. 2013; 229:422–429. [DOI] [PubMed] [Google Scholar]
- 23. Lin K.K., Harrell M.I., Oza A.M., Oaknin A., Ray-Coquard I., Tinker A.V., Helman E., Radke M.R., Say C., Vo L.T. et al. BRCA reversion mutations in circulating tumor DNA predict primary and acquired resistance to the PARP inhibitor Rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2019; 9:210–219. [DOI] [PubMed] [Google Scholar]
- 24. Kondrashova O., Topp M., Nesic K., Lieschke E., Ho G.Y., Harrell M.I., Zapparoli G.V., Hadley A., Holian R., Boehm E. et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018; 9:3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Walsh T., Casadei S., Lee M.K., Pennil C.C., Nord A.S., Thornton A.M., Roeb W., Agnew K.J., Stray S.M., Wickramanayake A. et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. U. S. A. 2011; 108:18032–18037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kondrashova O., Nguyen M., Shield-Artin K., Tinker A.V., Teng N.N.H., Harrell M.I., Kuiper M.J., Ho G.Y., Barker H., Jasin M. et al. Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor Rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2017; 7:984–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Norquist B.M., Brady M.F., Harrell M.I., Walsh T., Lee M.K., Gulsuner S., Bernards S.S., Casadei S., Burger R.A., Tewari K.S. et al. Mutations in homologous recombination genes and outcomes in ovarian carcinoma patients in GOG 218: an NRG Oncology/Gynecologic Oncology Group Study. Clin. Cancer Res. 2018; 24:777–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Suwaki N., Klare K., Tarsounas M. RAD51 paralogs: roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin. Cell Dev. Biol. 2011; 22:898–905. [DOI] [PubMed] [Google Scholar]
- 29. Edwards S.L., Brough R., Lord C.J., Natrajan R., Vatcheva R., Levine D.A., Boyd J., Reis-Filho J.S., Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008; 451:1111–1115. [DOI] [PubMed] [Google Scholar]
- 30. Sakai W., Swisher E.M., Karlan B.Y., Agarwal M.K., Higgins J., Friedman C., Villegas E., Jacquemont C., Farrugia D.J., Couch F.J. et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008; 451:1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fong P.C., Yap T.A., Boss D.S., Carden C.P., Mergui-Roelvink M., Gourley C., De Greve J., Lubinski J., Shanley S., Messiou C. et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol. 2010; 28:2512–2519. [DOI] [PubMed] [Google Scholar]
- 32. Ceccaldi R., O’Connor K.W., Mouw K.W., Li A.Y., Matulonis U.A., D’Andrea A.D., Konstantinopoulos P.A. A unique subset of epithelial ovarian cancers with platinum sensitivity and PARP inhibitor resistance. Cancer Res. 2015; 75:628–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ang J.E., Gourley C., Powell C.B., High H., Shapira-Frommer R., Castonguay V., De Greve J., Atkinson T., Yap T.A., Sandhu S. et al. Efficacy of chemotherapy in BRCA1/2 mutation carrier ovarian cancer in the setting of PARP inhibitor resistance: a multi-institutional study. Clin. Cancer Res. 2013; 19:5485–5493. [DOI] [PubMed] [Google Scholar]
- 34. Weroha S.J., Becker M.A., Enderica-Gonzalez S., Harrington S.C., Oberg A.L., Maurer M.J., Perkins S., Al Hilli M., Butler K., McKinstry S. et al. Tumorgrafts as in vivo surrogates for women with ovarian cancer. Clin. Cancer Res. 2014; 20:1288–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Scott C.L., Mackay H.J., Haluska P. Jr Patient-derived xenograft models in gynecologic malignancies. Am. Soc. Clin. Oncol. Educ. Book. 2014; e258–e266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu Y., Chanana P., Davila J.I., Hou X., Zanfagnin V., McGehee C.D., Goode E.L., Polley E.C., Haluska P., Weroha S.J. et al. Gene expression differences between matched pairs of ovarian cancer patient tumors and patient-derived xenografts. Sci. Rep. 2019; 9:6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fields A.P., Kaufmann S.H., Shaper J.H. Analysis of the internal nuclear matrix. Oligomers of a 38 kD nucleolar polypeptide stabilized by disulfide bonds. Exp. Cell Res. 1986; 164:139–153. [DOI] [PubMed] [Google Scholar]
- 38. Trapnell C., Pachter L., Salzberg S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009; 25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013; 14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014; 30:923–930. [DOI] [PubMed] [Google Scholar]
- 41. Liao Y., Smyth G.K., Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013; 41:e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huber W., Carey V.J., Gentleman R., Anders S., Carlson M., Carvalho B.S., Bravo H.C., Davis S., Gatto L., Girke T. et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods. 2015; 12:115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. R Core Team R: A language and environment for statistical computing. R Foundation for Statistical Computing. 2016; Vienna, Austria. [Google Scholar]
- 45. Kramer A., Green J., Pollard J. Jr, Tugendreich S Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014; 30:523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016; 32:2847–2849. [DOI] [PubMed] [Google Scholar]
- 47. Suzuki R., Shimodaira H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics. 2006; 22:1540–1542. [DOI] [PubMed] [Google Scholar]
- 48. Galili T. dendextend: an R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics. 2015; 31:3718–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Venables W.N., Ripley B.D. Modern Applied Statistics with S. 2002; 4th ednSpringer. [Google Scholar]
- 50. Gu Z., Gu L., Eils R., Schlesner M., Brors B. circlize Implements and enhances circular visualization in R. Bioinformatics. 2014; 30:2811–2812. [DOI] [PubMed] [Google Scholar]
- 51. Zeileis A., Fischer J.C., Hornik K., Ihaka R., McWhite C.D., Murrell P., Stauffer R., Wilke C.O. colorspace: A Toolbox for Manipulating and Assessing Colors and Palettes. J. Stat. Soft. 2020; 96: 10.18637/jss.v096.i01. [DOI] [Google Scholar]
- 52. Ito K., Murphy D. Application of ggplot2 to pharmacometric graphics. CPT Pharmacometrics Syst. Pharmacol. 2013; 2:e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. The Gene Ontology, C. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019; 47:D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Carbon S., Ireland A., Mungall C.J., Shu S., Marshall B., Lewis S., Ami G.O.H.Web Presence Working Group AmiGO: online access to ontology and annotation data. Bioinformatics. 2009; 25:288–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Koboldt D.C., Larson D.E., Wilson R.K. Using VarScan 2 for germline variant calling and somatic mutation detection. Curr. Protoc. Bioinformatics. 2013; 44:15.4.1–15.4.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. De Baets G., Van Durme J., Reumers J., Maurer-Stroh S., Vanhee P., Dopazo J., Schymkowitz J., Rousseau F. SNPeffect 4.0: on-line prediction of molecular and structural effects of protein-coding variants. Nucleic Acids Res. 2012; 40:D935–D939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Knijnenburg T.A., Wang L., Zimmermann M.T., Chambwe N., Gao G.F., Cherniack A.D., Fan H., Shen H., Way G.P., Greene C.S. et al. Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Rep. 2018; 23:239–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bompiani K.M., Tsai C.Y., Achatz F.P., Liebig J.K., Howell S.B. Copper transporters and chaperones CTR1, CTR2, ATOX1, and CCS as determinants of cisplatin sensitivity. Metallomics. 2016; 8:951–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Han H., Cho J.W., Lee S., Yun A., Kim H., Bae D., Yang S., Kim C.Y., Lee M., Kim E. et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018; 46:D380–D386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kaufmann S.H. Reutilization of immunoblots after chemiluminescent detection. Anal. Biochem. 2001; 296:283–286. [DOI] [PubMed] [Google Scholar]
- 62. Hurley R.M., Wahner Hendrickson A.E., Visscher D.W., Ansell P., Harrell M.I., Wagner J.M., Negron V., Goergen K.M., Maurer M.J., Oberg A.L. et al. 53BP1 as a potential predictor of response in PARP inhibitor-treated homologous recombination-deficient ovarian cancer. Gynecol. Oncol. 2019; 153:127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yun S., Vincelette N.D., Knorr K.L., Almada L.L., Schneider P.A., Peterson K.L., Flatten K.S., Dai H., Pratz K.W., Hess A.D. et al. 4EBP1/c-MYC/PUMA and NF-kappaB/EGR1/BIM pathways underlie cytotoxicity of mTOR dual inhibitors in malignant lymphoid cells. Blood. 2016; 127:2711–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Esteller M., Silva J.M., Dominguez G., Bonilla F., Matias-Guiu X., Lerma E., Bussaglia E., Prat J., Harkes I.C., Repasky E.A. et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J. Natl. Cancer Inst. 2000; 92:564–569. [DOI] [PubMed] [Google Scholar]
- 65. Al Hilli M.M., Becker M.A., Weroha S.J., Flatten K.S., Hurley R.M., Harrell M.I., Oberg A.L., Maurer M.J., Hawthorne K.M., Hou X. et al. In vivo anti-tumor activity of the PARP inhibitor niraparibin homologous recombination deficient and proficient ovarian carcinoma. Gynecol. Oncol. 2016; 143:379–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Snell C., Krypuy M., Wong E.M., Loughrey M.B., Dobrovic A. BRCA1 promoter methylation in peripheral blood DNA of mutation negative familial breast cancer patients with a BRCA1 tumour phenotype. Breast Cancer Res. 2008; 10:R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Eisenhauer E.A., Therasse P., Bogaerts J., Schwartz L.H., Sargent D., Ford R., Dancey J., Arbuck S., Gwyther S., Mooney M. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer. 2009; 45:228–247. [DOI] [PubMed] [Google Scholar]
- 68. Ito S., Murphy C.G., Doubrovina E., Jasin M., Moynahan M.E. PARP inhibitors in clinical use induce genomic instability in normal human cells. PLoS One. 2016; 11:e0159341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Swisher E.M., Harrell M.I., Norquist B.M., Walsh T., Brady M., Lee M., Hershberg R., Kalli K.R., Lankes H., Konnick E.Q. et al. Somatic mosaic mutations in PPM1D and TP53 in the blood of women with ovarian carcinoma. JAMA Oncol. 2016; 2:370–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Huo Y., Zong Z., Wang Q., Zhang Z., Deng H. ISG15 silencing increases cisplatin resistance via activating p53-mediated cell DNA repair. Oncotarget. 2017; 8:107452–107461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ju W.C., Huang G.B., Luo X.Y., Ren W.H., Zheng D.Q., Chen P.J., Lou Y.F., Li B. X-linked inhibitor of apoptosis-associated factor l (XAFl) enhances the sensitivity of colorectal cancer cells to cisplatin. Med. Oncol. 2014; 31:273. [DOI] [PubMed] [Google Scholar]
- 72. Zhao W.J., Deng B.Y., Wang X.M., Miao Y., Wang J.N. XIAP associated factor 1 (XAF1) represses expression of X-linked inhibitor of apoptosis protein (XIAP) and regulates invasion, cell cycle, apoptosis, and cisplatin sensitivity of ovarian carcinoma cells. Asian Pac. J. Cancer Prev. 2015; 16:2453–2458. [DOI] [PubMed] [Google Scholar]
- 73. Matsumoto T., Hayase R., Kodama J., Kamimura S., Yoshinouchi M., Kudo T. Immunohistochemical analysis of glutathione S-transferase mu expression in ovarian tumors. Eur. J. Obstet. Gynecol. Reprod. Biol. 1997; 73:171–176. [DOI] [PubMed] [Google Scholar]
- 74. Bader P., Fuchs J., Wenderoth M., von Schweinitz D., Niethammer D., Beck J.F. Altered expression of resistance associated genes in hepatoblastoma xenografts incorporated into mice following treatment with adriamycin or cisplatin. Anticancer Res. 1998; 18:3127–3132. [PubMed] [Google Scholar]
- 75. Wu B., Qi R., Liu X., Qian L., Wu Z. Rab18 overexpression promotes proliferation and chemoresistance through regulation of mitochondrial function in human gastric cancer. Onco Targets Ther. 2018; 11:7805–7820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ji X., Guo X., Wang Y., Li X., Li H. Rab18 Regulates Proliferation, Invasion and Cisplatin Sensitivity Through STAT3 Signaling in Head and Neck Squamous Cell Carcinoma. Onco Targets Ther. 2020; 13:4123–4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Li Y.L., Zhao Y.G., Chen B., Li X.F. MicroRNA-132 sensitizes nasopharyngeal carcinoma cells to cisplatin through regulation of forkhead box A1 protein. Pharmazie. 2016; 71:715–718. [DOI] [PubMed] [Google Scholar]
- 78. Zhu X.L., Sun X.C., Chen B.A., Sun N., Cheng H.Y., Li F., Zhang H.M., Feng J.F., Qin S.K., Cheng L. et al. XPC Lys939Gln polymorphism is associated with the decreased response to platinum based chemotherapy in advanced non-small-cell lung cancer. Chin. Med. J. (Engl.). 2010; 123:3427–3432. [PubMed] [Google Scholar]
- 79. Giachino D.F., Ghio P., Regazzoni S., Mandrile G., Novello S., Selvaggi G., Gregori D., DeMarchi M., Scagliotti G.V. Prospective assessment of XPD Lys751Gln and XRCC1 Arg399Gln single nucleotide polymorphisms in lung cancer. Clin. Cancer Res. 2007; 13:2876–2881. [DOI] [PubMed] [Google Scholar]
- 80. Jalal S., Earley J.N., Turchi J.J. DNA repair: from genome maintenance to biomarker and therapeutic target. Clin. Cancer Res. 2011; 17:6973–6984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hildebrandt M.A., Gu J., Wu X. Pharmacogenomics of platinum-based chemotherapy in NSCLC. Expert Opin. Drug Metab. Toxicol. 2009; 5:745–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ding L., Kim H.J., Wang Q., Kearns M., Jiang T., Ohlson C.E., Li B.B., Xie S., Liu J.F., Stover E.H. et al. PARP inhibition Elicits STING-dependent antitumor immunity in Brca1-deficient ovarian cancer. Cell Rep. 2018; 25:2972–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Shen J., Zhao W., Ju Z., Wang L., Peng Y., Labrie M., Yap T.A., Mills G.B., Peng G. PARPi triggers the sting-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res. 2019; 79:311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wang Z., Sun K., Xiao Y., Feng B., Mikule K., Ma X., Feng N., Vellano C.P., Federico L., Marszalek J.R. et al. Niraparib activates interferon signaling and potentiates anti-PD-1 antibody efficacy in tumor models. Sci. Rep. 2019; 9:1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Raso M.C., Djoric N., Walser F., Hess S., Schmid F.M., Burger S., Knobeloch K.P., Penengo L. Interferon-stimulated gene 15 accelerates replication fork progression inducing chromosomal breakage. J. Cell Biol. 2020; 219:e202002175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer. 2007; 7:573–584. [DOI] [PubMed] [Google Scholar]
- 87. Muggia F.M., Braly P.S., Brady M.F., Sutton G., Niemann T.H., Lentz S.L., Alvarez R.D., Kucera P.R., Small J.M. Phase III randomized study of cisplatin versus paclitaxel versus cisplatin and paclitaxel in patients with suboptimal stage III or IV ovarian cancer: a gynecologic oncology group study. J. Clin. Oncol. 2000; 18:106–115. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data have been deposited into the GEO database as data super series GSE165052 and are available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE165052.