

Abstract
Given their primacy in governing the action potential (AP) of excitable cells, voltage-gated Na+ (Nav) channels are important pharmacological targets of therapeutics for a diverse array of clinical indications. Despite historically being a traditional drug target, therapeutics targeting Nav channels lack isoform selectivity, giving rise to off-target side effects. To develop isoform-selective modulators of Nav channels with improved target-specificity, the identification and pharmacological targeting of allosteric sites that display structural divergence among Nav channel isoforms represents an attractive approach. Despite the high homology among Nav channel α subunit isoforms (Nav1.1–Nav1.9), there is considerable amino acid sequence divergence among their constituent C-terminal domains (CTD), which enables structurally and functionally specific protein: protein interactions (PPI) with auxiliary proteins. Although pharmacological targeting of such PPI interfaces between the CTDs of Nav channels and auxiliary proteins represents an innovate approach for developing isoform-selective modulators of Nav channels, appreciable modulation of PPIs using small molecules has conventionally been difficult to achieve. After briefly discussing the challenges of modulating PPIs using small molecules, this current frontier review that follows subsequently expounds on approaches for circumventing such difficulties in the context of developing small molecule modulators of PPIs between transmembrane ion channels and their auxiliary proteins. In addition to broadly discussing such approaches, the implementation of such approaches is specifically discussed in the context of developing small molecule modulators between the CTD of Nav channels and auxiliary proteins. Developing allosteric modulators of ion channels by targeting their PPI interfaces with auxiliary proteins represents an innovative and promising strategy in ion channel drug discovery that could expand the “druggable genome” and usher in first-in-class PPI-targeting therapeutics for a multitude of channelopathies.
Keywords: Sodium channels, Selectivity, Protein:protein interaction, Allosteric modulators, Channelopathies, Drug discovery
1. INTRODUCTION
In neurons, voltage-gated Na+ (Nav) channels are a primary molecular determinant of the action potential (AP) [1]. Despite the central role of the pore-forming α subunit in conferring this functionality, protein: protein interactions (PPI) between the α subunit and auxiliary proteins are essential for enabling the full physiological function of the Nav channel, which ultimately enables the repetitive firing of APs [2–11]. As these PPIs enable specificity of the intermolecular regulation of different Nav channel isoforms [12–15], pharmacological targeting of PPI interfaces between Nav channels and their auxiliary proteins represents a novel approach for developing highly target-selective therapeutics. Despite holding promise, identification of compounds that modulate such PPIs is vexingly difficult for a multitude of reasons, chief among which is that PPI interfaces are structurally divergent from the targets of most conventional small molecules. This challenge is further complicated in the context of developing modulators of PPIs involving transmembrane ion channels and auxiliary proteins, as adequate screening platforms to support early-phase drug discovery initiatives targeting these surfaces are lacking in both the academic and industry sectors. As such, integration of complementary approaches ranging from in-cell and biophysical assays to functional and behavioral studies is necessary to elucidate the potential of PPIs as druggable targets for the treatment of a diverse array of disease states, especially neurologic and neuropsychiatric disorders. In addition to broadly discussing such approaches, the mini-review that follows discusses a targeted approach for developing allosteric modulators of Nav channels by targeting their PPI interfaces with auxiliary proteins. As such efforts to expand the “druggable genome” by targeting PPIs represent a largely unexplored area in central and peripheral nervous system drug discovery, implementation of such approaches has the potential of identifying and establishing novel classes of PPI-targeting neurotherapeutics, which, in the specific case of Nav channels and auxiliary proteins, would result in a first-in-class family of allosteric modulators.
2. BARRIERS TO DEVELOPING SMALL MOLECULE MODULATORS OF PPIS
The approaches expounded upon below to identify small molecule modulators of PPIs between transmembrane ion channels and their auxiliary proteins are born of necessity to circumvent, broadly speaking, two pharmacological difficulties. The first of these difficulties arise due to the sheer size and unorthodox geometry of the target. PPI interfaces generally range in size from 1000 to 4000 Å2, whereas conventional small molecule drugs are generally designed against targets that are only 300 to 1000 Å2 [16, 17]. Furthermore, conventional small molecule drugs are ordinarily designed against well-defined binding pockets of enzymes and receptors, whereas PPI interfaces tend to be devoid of such clearly demarcated topographical regions [18]. Given these discrepancies between the conventional surfaces that small molecules are designed to target and the structural properties of PPI interfaces, it is, therefore, unsurprising that high-throughput screenings (HTS) of small-molecule libraries against PPI complexes often yield few viable hits that could serve as PPI modulators [18].
The second, and intimately related, pharmacological challenge of developing small molecule modulators of PPIs stems from ill-defined characterization of druggable motifs along vast PPI interfaces. In particular, the discovery that there are residues of PPI interfaces that are crucial to the assembly of two proteins, conventionally identified using alanine scanning mutagenesis [19], has led to the viewingof some “hot spot” residues as druggable motifs of PPI interfaces [20]. This notion that a PPI interface “hot spot” implies a druggable target for small molecules is, however, oftentimes uninformative from a drug discovery perspective. For example, alanine scanning can identify a residue as a “hot spot” due to the amino acid’s contribution to protein stability, which is unrelated to developing modulators of a PPI [21]. Furthermore, interactions of a small molecule with residues beyond a single “hot spot” are necessary to enhance specificity and modulatory effects on the assembly of two proteins [21], underscoring the need to reconceptualize druggable pockets of PPI interfaces.
3. DEVELOPMENT OF SMALL MOLECULES TARGETING “HOT SEGMENTS” OF PPIS
Although developing a viable small-molecule PPI modulator by targeting “hot spot” residues of PPI interfaces is hindered by the difficulties described above, targeting “hot segments” of PPIs could potentially circumvent many of those obstacles. “Hot segments” [22, 23], or the related “small-molecule inhibitor starting points (SMISP)” [21, 24], are clusters of amino acids at a PPI interface. These clusters of amino acids, unlike conventional “hot spots,” are identified through methods that depart from alanine scanning and determine the primacy of clusters of amino acids in governing a PPI by directly assessing the PPI interface structure [21–24]. Using these methods, between 15% and 40% of all PPIs have been shown to exhibit “hot segments” and that such clusters of amino acids confer more than half of the protein:protein binding energy involved in a given PPI [22, 23]. Furthermore, the “druggability” of such “hot segments” or “SMISPs” can be determined using some of these methods [21, 24], enabling target tractability to be rapidly assessed. Given the increased amenability of such clusters of amino acids at PPI interfaces to serve as druggable pockets, such pharmacological targeting of “hot segments” represents a promising approach for developing PPI-targeting therapeutics.
4. WORKFLOW FOR DEVELOPING COMPOUNDS TARGETING “HOT SEGMENTS” OF PPI INTERFACES
To develop small molecules targeting a “hot segment” of a PPI, the first phase of the workflow, as in Fig. (1), is to access the structure of a functionally relevant PPI interface. If structural data of the PPI interface suggests that the interaction between the two proteins is disproportionately mediated by a specific cluster of amino acids at a key domain of one of the binding partners, putative “hot segments” of the PPI can be investigated using a variety of previously reported in silico prediction methods, such as PocketQuery [24] and RosettaDock [25]. If in silico interrogation of a PPI is suggestive of a well-defined “hot segment,” development of small molecule modulators of the PPI can proceed by screening chemical libraries or compounds rationally designed against the “hot segment” using targeted platforms for HTS that are tailored to the specific target. Using a robust and scalable in-cell HTS platform is essential for this early-phase discovery. A desirable platform should include cost-effective primary screenings that enable reconstitution of the PPI interface in a controllable yet physiologically relevant environment to enable reliable initial validation of hits. Especially in the context of developing modulators of PPIs involving transmembrane ion channels and intracellular auxiliary proteins, an in-cell primary assay is desirable as it ensures that identified lead compounds have the necessary drug-like properties, especially favorable molecular weights and cLogP values, to reach their intracellular sites of action both ex vivo and in vivo.
Fig. (1). Workflow for developing small molecules targeting “hot segments” of PPIs in the central and peripheral nervous systems.
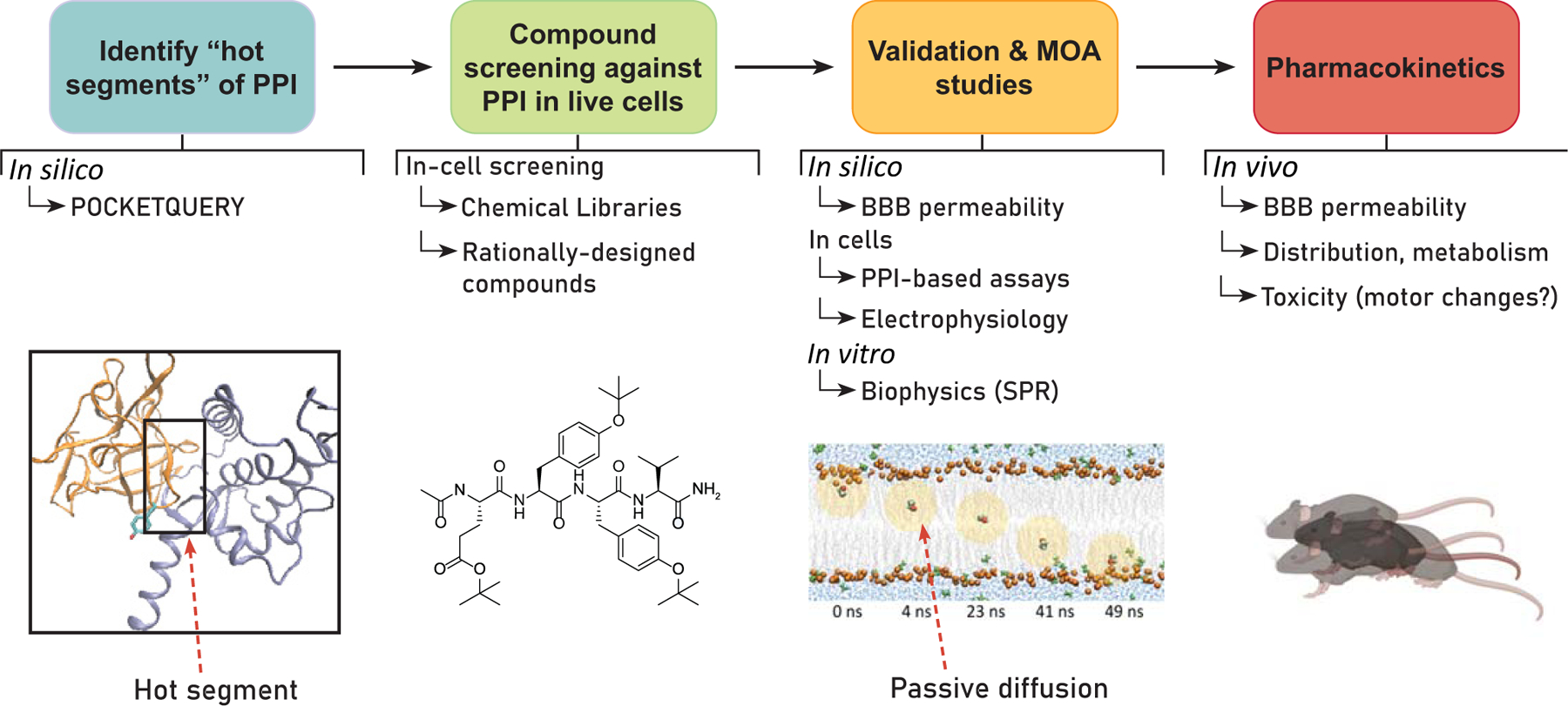
Phase 1) Based on existing structural data, potential “hot segments” of a PPI interface can be identified using in silico prediction methods, such as PocketQuery [24]. Phase 2) Screening of compounds against PPI interfaces with predicted “hot segments” using in-cell HTS platforms. Phase 3) After preliminary screening, hits are selected for orthogonal validation and mechanism of action (MOA) studies. These investigations can include in silico prediction of blood-brain barrier (BBB) permeability [26], in-cell assays to characterize PPI selectivity, electrophysiology to assess functional activity, and protein: ligand binding assays to determine binding affinities. Phase 4) Lead compounds identified are selected for in vivo pharmacokinetic (PK) and behavioral studies. Some in vivo studies of interest for the development of these modulators include single-unit electrophysiological recordings, assessments of mechanical hypersensitivity, and operant behaviors for the development of anti-epileptics, local anesthetics, and neuropsychopharmacological agents, respectively. Of particular interest for central nervous system (CNS) drug discovery, BBB permeability and deleterious effects on locomotor activity are assessed.
After completion of screening, identified hits with favorable drug-like properties are further pharmacodynamically evaluated using complementary techniques. At this juncture, employing an amalgam of complementary and orthogonal approaches, including in silico prediction of blood-brain barrier (BBB) permeability [26], in-cell toxicity assays, protein: ligand binding studies using surface plasmon resonance (SPR), and functional studies using electrophysiology is crucial to robustly characterize lead compounds in silico to ex vivo prior to advancing into in vivo studies. Having characterized lead compounds displaying favorable in silico to ex vivo performance, the in vivo pharmacokinetic (PK) profile of leads should be assessed prior to advancing into behavioral paradigms that seek to characterize the organismal effects of modulation of a targeted PPI.
5. “HOT SEGMENTS” OF PPI INTERFACES BETWEEN NAV CHANNELS AND THEIR AUXILIARY PROTEINS AS NOVEL SITES FOR ALLOSTERIC MODULATION
Given their role as the fundamental molecular determinant of neuronal excitability, it is unsurprising that Nav channels have been the target of many central nervous system (CNS) drug discovery campaigns [1]. However, given that three pharmacologically relevant Nav channel isoforms are abundantly expressed in the CNS (Nav1.1, Nav1.2, and Nav1.6) [1], which have greater than 85% overall homology in terms of their respective pore-forming α-subunits [27], such efforts have been of limited success on account of developed lead compounds often failing to demonstrate isoform selectively [28].
To progress toward achieving isoform-selectivity, some conceptual delineation between orthosteric and allosteric sites is necessary to identify novel druggable sites of Nav channels that could confer functionally relevant and targeted modulation. Structurally, the pore-forming α subunit of Nav channels is comprised of four transmembrane domains, denoted as DI-DIV. Each of these four transmembrane domains is comprised of six segments, denoted as S1–S6. In addition, the pore-forming α subunit also features an intracellular N-terminal domain and intracellular C-terminal domain (CTD) [29, 30]. Crucially, the orthosteric modulator of Nav channels, which is voltage itself, exerts its effects through positive gating charges at intervals of three amino acid residues in S4, leading this transmembrane segment to conventionally be considered the “orthosteric site” of these channels [31]. Although this definition is technically correct, it is somewhat uninformative in the context of developing isoform-selective, allosteric modulators of Nav channels. For example, small molecules targeting Nav channels for epilepsy and local anesthesia bind to regions that, despite being outside of S4 are still, however, highly homologous among Nav channel isoforms [32, 33], resulting in such molecules being classified as allosteric modulators despite lacking selectively.
To arrive at a more restrictive and informative conceptualization of allosteric sites of Nav channels in the context of developing isoform-selective modulators, a criterion worthy of consideration is the homology of specific structural domains of the Nav channel across isoforms. Considering this criterion, one particular structural domain of Nav channels that represents a promising and truly allosteric site to target for the development of isoform-selective modulators is the intracellular CTD, as CTDs display appreciable amino acid sequence divergence across Nav channels isoforms that enables functionally specific PPIs with distinct auxiliary proteins [2–4, 6–15]. As such, identification of compounds targeting “hot segments” of PPIs between the CTD of specific Nav channels and specific auxiliary proteins could serve as a novel approach for the development of isoform-selective modulators of Nav channels. In the case of the CNS, such modulators would have broad applicability for neurologic and neuropsychiatric disorders, such as epilepsy and bipolar disorder, respectively, whereas such modulators targeting peripheral nervous system isoforms of Nav channels, such as Nav1.7, would have applicability for the treatment of neuropathic pain and as local anesthetics [29, 34]. Compounds targeting these PPIs would gain the definition of allosteric modulators by two separate and complementary criteria: i) they target a domain of the Nav channel, the CTD, that is highly divergent across isoforms and is not directly related to the orthosteric site (i.e. the voltage sensor); and ii) they involve a binding partner, the auxiliary protein, that is distinct from the pore-forming α subunit.
Of particular interest, PPIs between intracellular fibroblast growth factors (iFGF) and CTDs of different Nav channel isoforms have been shown to modulate Nav channel activity in an isoform-dependent fashion [35–37], and these changes were further shown to be distinct from those mediated by other iFGF isoforms and splice variants [12, 14]. Given this isoform-dependent modulation of distinct Nav channels by distinct iFGFs, in addition to iFGF dysfunction increasingly being associated with neurologic and neuropsychiatric disorders [34, 38, 39], PPI interfaces between different Nav:iFGF pairs may serve as promising targets for pharmacological probe and drug development of allosteric modulators of Nav channels (Fig. 2A–C).
Fig. (2). Diagrammatic representation of how “hot segments” of PPI interfaces between the CTD of Nav channels and intracellular fibroblast growth factors (iFGFs) function as allosteric sites for Nav channel modulation.
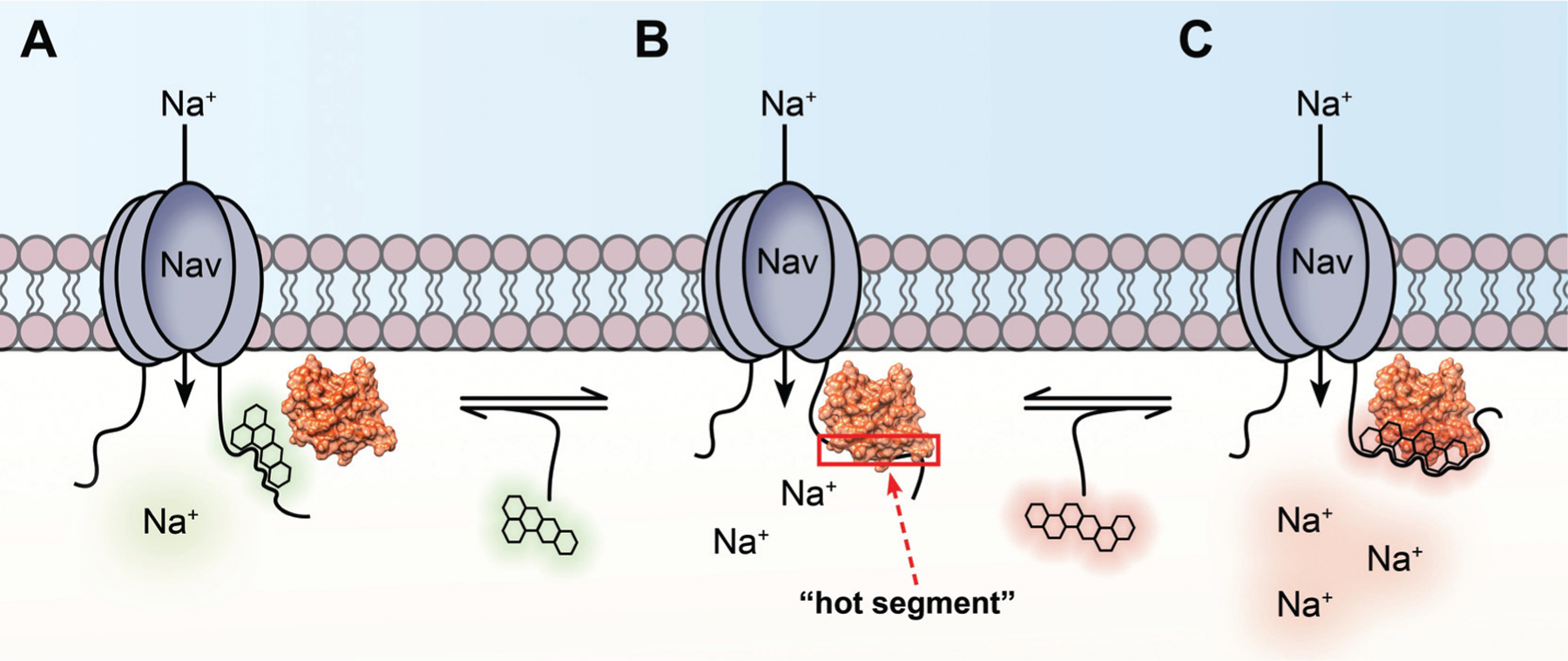
A) Small molecule targeting the iFGF:Nav PPI interface that inhibits association of the complex. Given that such PPIs are necessary for full physiological activity of Nav channels, such inhibition of complex assembly is anticipated to decrease Nav channel activity, resulting in molecules that inhibit the association of the complex functioning as negative allosteric modulators (NAM). B) Representation of a “hot segment” (red box) along an iFGF:Nav PPI interface that could serve as a druggable pocket for novel allosteric modulators of Nav channels. CTDs of Nav channel isoforms show less sequence homology relative to other structural components of the pore-forming α subunit. This amino acid sequence divergence among CTDs of Nav channel isoforms gives rise to structurally divergent PPI interfaces with auxiliary proteins, intermolecular interactions with functionally selective effects on different Nav channel isoforms. C) Small molecule targeting the iFGF:Nav PPI interface that stabilizes and inhibits dissociation of the complex. Inhibiting dissociation of the complex is anticipated to exacerbate iFGF-mediated regulatory effects on the channel, thereby leading to increased Nav channel activity. As such, molecules that inhibit dissociation of the complex are anticipated to function as positive allosteric modulators (PAM) of Nav channels.
As depicted in Fig. (2), pharmacological targeting of “hot segments” of iFGF:Nav PPI interfaces not only represents an approach for developing isoform-selective modulators of Nav channels but also, through bidirectional manipulation of complex assembly, presents opportunities for negative versus positive allosteric modulation of Nav channel activity. For example, small molecules that inhibit the association of iFGF:Nav complex assembly (Fig. 2A) are anticipated to abrogate iFGF-mediated regulation of Nav channels, thereby preventing the full physiological function of Nav channels and decreasing their activity. Such effects, consistent with studies in which iFGFs have been genetically deleted [34, 40], could classify such molecules as negative allosteric modulators (NAM) of Nav channels. In contrast, small molecules that stabilize and inhibit dissociation of iFGF:Nav complexes (Fig. 2C) are anticipated to exacerbate iFGF-mediated regulation of Nav channels. Potentiation of iFGF-mediated effects on electrophysiological parameters of Nav channels including inactivation and resurgent INa+ [41] would increase Nav channel activity. Such effects, which would manifest as increased firing rates of neurons, could classify such molecules as positive allosteric modulators (PAM) of Nav channels.
Through targeting “hot segments” of the PPI between the CTD of the Nav1.6 channel and its auxiliary protein fibroblast growth factor 14 (FGF14), isoform-selective probes targeting the Nav1.6 channel macromolecular complex have been developed to inform efforts to usher in first-in-class allosteric modulators of Nav channels targeting PPI interfaces. To begin, molecular modeling of the PPI interface between the CTD of Nav1.6 and FGF14 was performed, which identified clusters of amino acids on the β12 sheet of FGF14, namely FLPK (1, Fig. 3) and PLEV (2, Fig. 3), and β8–β9 loop of FGF14, namely EYYV (3, Fig. 3), that were putative “hot segments” at the FGF14:Nav1.6 PPI interface [42, 43]. In subsequent works, these clusters of amino acids were used as scaffolds for the development of rationally designed peptidomimetics selectively targeting the Nav1.6 channel macromolecular complex. Reported by Ali et al. [40], a benzyl chloroformate (Cbz) functional group was added to the N-terminus of the FLPK tetrapeptide, and a Boc functional group was added to the constituent lysine residue of the tetrapeptide, which produced ZL181 (4, Fig. 3) [40]. Functional studies using 4 revealed that the peptidomimetic functioned as an isoform-selective modulator of the Nav1.6 channel macromolecular complex, as the compound suppressed Nav1.6-mediated peak transient INa+ density and suppressed maximal and instantaneous firing frequencies of medium spiny neurons (MSN) of the nucleus accumbens (NAc) [40]. Subsequently, Liu et al. [44] optimized the scaffold of compound 4 by replacing the N-terminal Cbz functional group with an acetyl functional group, replacing the Fmoc function group added to the constituent lysine residue with a Boc functional group, and adding a methoxy group to the C-terminus of the tetrapeptide, thereby producing ZL0177 (5, Fig. 3), which displayed a roughly five-fold improvement in terms of its inhibitory potency toward the FGF14:Nav1.6 complex assembly with an IC50 value of 11 μM [44].
Fig. (3).
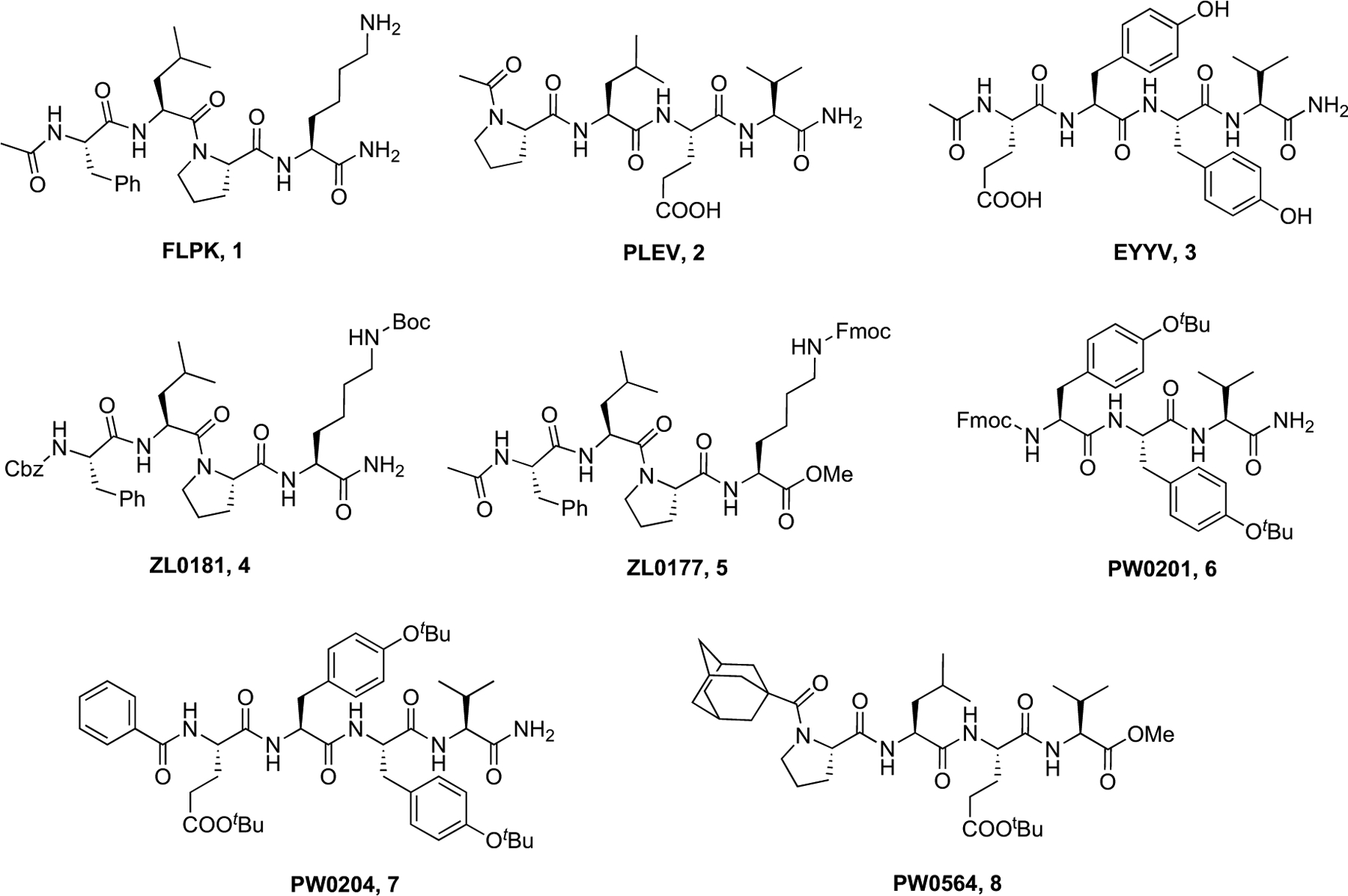
Reported representative FGF14:Nav1.6 complex assembly modulators.
In more recent works, we [45] optimized the parental EYYV peptide via two synthetic routes. Firstly, the tetrapeptide was truncated to a tripeptide (YYV), the hydroxyl groups of constituent tyrosine residues were protected by tert-butyl functional groups, and a Fmoc functional group was added to the N-terminus, which produced compound PW0201 (6, Fig. 3). Secondly, hydroxyl groups of residues comprising the tetrapeptide scaffold were replaced with tert-butyl functional groups and a benzoyl substituent was added to the N-terminus of the tetrapeptide, which produced compound PW0204 (7, Fig. 3). Interestingly, the compounds displayed opposite effects in vitro, with compound 6 and compound 7 negatively and positively modulating the Nav1.6 channel macromolecular, respectively [45]. Relatedly, Wang et al. [46] optimized the PLEV tetrapeptide through the addition of N-terminal 1-adamantyl carbonyl pharmacophore and a C-terminal methoxy group, which produced compound PW0564 (8, Fig. 3). In vitro, compound 8 was shown to have functional and selective effects on the Nav1.6 channel macromolecular complex.
CONCLUSION
Overall, this review introduces novel approaches and design strategies that could lead to the development of isoform-selective, allosteric modulators of transmembrane domain ion channels by targeting “hot segments” of their PPI interfaces with regulatory accessory proteins. As PPI interfaces represent hundreds of unexplored pharmacological targets, implementation of such approaches could considerably expand the concept of the “druggable genome” and usher in the development of novel classes of PPI-targeting neurotherapeutics.
ACKNOWLEDGEMENTS
Declared none.
FUNDING
This work was supported by the National Institutes of Health (NIH) Grants R01 MH12435101 (F.L.), R01 MH111107 (F.L. and J.Z.), UTMB Technology Commercialization Program (F.L. and J.Z.), the Houston Area Molecular Biophysics Program Grant No. T32 GM008280 (N.M.D.), NIA T32 Fellowship Grant No. T32 AG051131 (P.A.W.), and the Pharmaceutical Research and Manufacturers of America (PhRMA) Foundation Pre-doctoral Fellowship in Pharmacology/Toxicology (P.A.W.), USA.
LIST OF ABBREVIATIONS
- AP
Action Potential
- BBB
Blood-Brain Barrier
- Cbz
Benzyl Chloroformate
- CNS
Central Nervous System
- CTD
C-Terminal Domain
- FGF14
Fibroblast Growth Factor 14
- HTS
High-Throughput Screening
- iFGF
Intracellular Fibroblast Growth Factor
- MOA
Mechanism of Action
- MSN
Medium Spiny Neuron
- NAc
Nucleus Accumbens
- NAM
Negative Allosteric Modulator
- Nav
Voltage-gated Na+ Channel
- PAM
Positive Allosteric Modulator
- PK
Pharmacokinetic
- PPI
Protein:Protein Interaction
- SMISP
Small-Molecule Inhibitor Starting Point
- SPR
Surface Plasmon Resonance
Footnotes
CONFLICT OF INTEREST
The authors have no conflicts of interest, financial or otherwise.
REFERENCES
- [1].Catterall WA Forty years of sodium channels: Structure, function, pharmacology, and epilepsy. Neurochem. Res, 2017, 42(9), 2495–2504. 10.1007/s11064-017-2314-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pitt GS; Lee S-Y Current view on regulation of voltage-gated sodium channels by calcium and auxiliary proteins. Protein Sci, 2016, 25(9), 1573–1584. 10.1002/pro.2960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].White HV; Brown ST; Bozza TC; Raman IM Effects of FGF14 and NaVβ4 deletion on transient and resurgent Na current in cerebellar purkinje neurons. J. Gen. Physiol, 2019, 151(11), 1300–1318. 10.1085/jgp.201912390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lewis AH; Raman IM Resurgent current of voltage-gated Na(+) channels. J. Physiol, 2014, 592(22), 4825–4838. 10.1113/jphysiol.2014.277582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wang Q; McEwen DG; Ornitz DM Subcellular and developmental expression of alternatively spliced forms of fibroblast growth factor 14. Mech. Dev, 2000, 90(2), 283–287. 10.1016/S0925-4773(99)00241-5 [DOI] [PubMed] [Google Scholar]
- [6].Gade AR; Marx SO; Pitt GS An interaction between the III-IV linker and CTD in NaV1.5 confers regulation of inactivation by CaM and FHF. J. Gen. Physiol, 2020, 152(2), e201912434. 10.1085/jgp.201912434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tseng T-T; McMahon AM; Johnson VT; Mangubat EZ; Zahm RJ; Pacold ME; Jakobsson E Sodium channel auxiliary subunits. J. Mol. Microbiol. Biotechnol, 2007, 12(3–4), 249–262. 10.1159/000099646 [DOI] [PubMed] [Google Scholar]
- [8].Goetz R; Dover K; Laezza F; Shtraizent N; Huang X; Tchetchik D; Eliseenkova AV; Xu C-F; Neubert TA; Ornitz DM; Goldfarb M; Mohammadi M Crystal structure of a fibroblast growth factor homologous factor (FHF) defines a conserved surface on FHFs for binding and modulation of voltage-gated sodium channels. J. Biol. Chem, 2009, 284(26), 17883–17896. 10.1074/jbc.M109.001842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Goldfarb M; Schoorlemmer J; Williams A; Diwakar S; Wang Q; Huang X; Giza J; Tchetchik D; Kelley K; Vega A; Matthews G; Rossi P; Ornitz DM; D’Angelo E Fibroblast growth factor homologous factors control neuronal excitability through modulation of voltage-gated sodium channels. Neuron, 2007, 55(3), 449–463. 10.1016/j.neuron.2007.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang C; Wang C; Hoch EG; Pitt GS Identification of novel interaction sites that determine specificity between fibroblast growth factor homologous factors and voltage-gated sodium channels. J. Biol. Chem, 2011, 286(27), 24253–24263. 10.1074/jbc.M111.245803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yan H; Wang C; Marx SO; Pitt GS Calmodulin limits pathogenic Na+ channel persistent current. J. Gen. Physiol, 2017, 149(2), 277–293. 10.1085/jgp.201611721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Laezza F; Lampert A; Kozel MA; Gerber BR; Rush AM; Nerbonne JM; Waxman SG; Dib-Hajj SD; Ornitz DM FGF14 N-terminal splice variants differentially modulate Nav1.2 and Nav1.6-encoded sodium channels. Mol. Cell. Neurosci, 2009, 42(2), 90–101. 10.1016/j.mcn.2009.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Laezza F; Gerber BR; Lou J-Y; Kozel MA; Hartman H; Craig AM; Ornitz DM; Nerbonne JM The FGF14(F145S) mutation disrupts the interaction of FGF14 with voltage-gated Na+ channels and impairs neuronal excitability. J. Neurosci, 2007, 27(44), 12033–12044. 10.1523/JNEUROSCI.2282-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lou J-Y; Laezza F; Gerber BR; Xiao M; Yamada KA; Hartmann H; Craig AM; Nerbonne JM; Ornitz DM Fibroblast growth factor 14 is an intracellular modulator of voltage--gated sodium channels. J. Physiol, 2005, 569(Pt 1), 179–193. 10.1113/jphysiol.2005.097220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Effraim PR; Huang J; Lampert A; Stamboulian S; Zhao P; Black JA; Dib-Hajj SD; Waxman SG Fibroblast growth factor homologous factor 2 (FGF-13) associates with Nav1.7 in DRG neurons and alters its current properties in an isoform-dependent manner. Neurobiol Pain, 2019, 6, 100029. 10.1016/j.ynpai.2019.100029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jones S; Thornton JM Principles of protein-protein interactions. Proc. Natl. Acad. Sci. USA, 1996, 93(1), 13–20. 10.1073/pnas.93.1.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pelay-Gimeno M; Glas A; Koch O; Grossmann TN Structure-based design of inhibitors of protein-protein interactions: Mimicking peptide binding epitopes. Angew. Chem. Int. Ed. Engl, 2015, 54(31), 8896–8927. 10.1002/anie.201412070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Arkin MR; Wells JA Small-molecule inhibitors of protein-protein interactions: Progressing towards the dream. Nat. Rev. Drug Discov, 2004, 3(4), 301–317. 10.1038/nrd1343 [DOI] [PubMed] [Google Scholar]
- [19].Wells JA Systematic mutational analyses of protein-protein interfaces. Methods Enzymol, 1991, 202, 390–411. 10.1016/0076-6879(91)02020-A [DOI] [PubMed] [Google Scholar]
- [20].Whitty A; Kumaravel G Between a rock and a hard place? Nat. Chem. Biol, 2006, 2(3), 112–118. 10.1038/nchembio0306-112 [DOI] [PubMed] [Google Scholar]
- [21].Koes DR; Camacho CJ Small-molecule inhibitor starting points learned from protein-protein interaction inhibitor structure. Bioinformatics, 2012, 28(6), 784–791. 10.1093/bioinformatics/btr717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].London N; Raveh B; Schueler-Furman O Druggable protein-protein interactions-from hot spots to hot segments. Curr. Opin. Chem. Biol, 2013, 17(6), 952–959. 10.1016/j.cbpa.2013.10.011 [DOI] [PubMed] [Google Scholar]
- [23].London N; Raveh B; Movshovitz-Attias D; Schueler-Furman O Can self-inhibitory peptides be derived from the interfaces of globular protein-protein interactions? Proteins, 2010, 78(15), 3140–3149. 10.1002/prot.22785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Koes DR; Camacho CJ PocketQuery: Protein-protein interaction inhibitor starting points from protein-protein interaction structure. Nucleic Acids Res, 2012, 40) (Web Server issue), W387–W392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lyskov S; Gray JJ The rosettadock server for local protein-protein docking. Nucleic Acids Res, 2008, 36(Web Server issue), W233–W238. 10.1093/nar/gkn216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang Y; Gallagher E; Jorgensen C; Troendle EP; Hu D; Searson PC; Ulmschneider MB An experimentally validated approach to calculate the blood-brain barrier permeability of small molecules. Sci. Rep, 2019, 9(1), 6117. 10.1038/s41598-019-42272-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li T; Chen J Voltage-gated sodium channels in drug discovery. IntechOpen, 2018. 10.5772/intechopen.78256 [DOI] [Google Scholar]
- [28].Focken T; Burford K; Grimwood ME; Zenova A; Andrez J-C; Gong W; Wilson M; Taron M; Decker S; Lofstrand V; Chowdhury S; Shuart N; Lin S; Goodchild SJ; Young C; Soriano M; Tari PK; Waldbrook M; Nelkenbrecher K; Kwan R; Lindgren A; de Boer G; Lee S; Sojo L; DeVita RJ; Cohen CJ; Wesolowski SS; Johnson JP Jr; Dehnhardt CM; Empfield JR Identification of cns-penetrant aryl sulfonamides as isoform-selective Nav1.6 inhibitors with efficacy in mouse models of epilepsy. J. Med. Chem, 2019, 62(21), 9618–9641. 10.1021/acs.jmedchem.9b01032 [DOI] [PubMed] [Google Scholar]
- [29].Catterall WA; Swanson TM Structural basis for pharmacology of voltage-gated sodium and calcium channels. Mol. Pharmacol, 2015, 88(1), 141–150. 10.1124/mol.114.097659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Catterall WA From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron, 2000, 26(1), 13–25. 10.1016/S0896-6273(00)81133-2 [DOI] [PubMed] [Google Scholar]
- [31].Changeux J-P; Christopoulos A Allosteric modulation as a unifying mechanism for receptor function and regulation. Cell, 2016, 166(5), 1084–1102. 10.1016/j.cell.2016.08.015 [DOI] [PubMed] [Google Scholar]
- [32].Fozzard HA; Sheets MF; Hanck DA The sodium channel as a target for local anesthetic drugs. Front. Pharmacol, 2011, 2, 68–68. 10.3389/fphar.2011.00068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lipkind GM; Fozzard HA Molecular model of anticonvulsant drug binding to the voltage-gated sodium channel inner pore. Mol. Pharmacol, 2010, 78(4), 631–638. 10.1124/mol.110.064683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yang L; Dong F; Yang Q; Yang P-F; Wu R; Wu Q-F; Wu D; Li C-L; Zhong Y-Q; Lu Y-J; Cheng X; Xu FQ; Chen L; Bao L; Zhang X FGF13 selectively regulates heat nociception by interacting with Nav1.7. Neuron, 2017, 93(4), 806–821.e9. 10.1016/j.neuron.2017.01.009 [DOI] [PubMed] [Google Scholar]
- [35].Liu Cj; Dib-Hajj SD; Waxman SG Fibroblast growth factor homologous factor 1B binds to the C terminus of the tetrodotoxin-resistant sodium channel rNav1.9a (NaN). J. Biol. Chem, 2001, 276(22), 18925–18933. 10.1074/jbc.M101606200 [DOI] [PubMed] [Google Scholar]
- [36].Liu CJ; Dib-Hajj SD; Renganathan M; Cummins TR; Waxman SG Modulation of the cardiac sodium channel Nav1.5 by fibroblast growth factor homologous factor 1B. J. Biol. Chem, 2003, 278(2), 1029–1036. 10.1074/jbc.M207074200 [DOI] [PubMed] [Google Scholar]
- [37].Wittmack EK; Rush AM; Craner MJ; Goldfarb M; Waxman SG; Dib-Hajj SD Fibroblast growth factor homologous factor 2B: Association with Nav1.6 and selective colocalization at nodes of Ranvier of dorsal root axons. J. Neurosci, 2004, 24(30), 6765–6775. 10.1523/JNEUROSCI.1628-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Di Re J; Wadsworth PA; Laezza F Intracellular fibroblast growth factor 14: Emerging risk factor for brain disorders. Front. Cell. Neurosci, 2017, 11, 103. 10.3389/fncel.2017.00103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Paucar M; Lundin J; Alshammari T; Bergendal Å; Lindefeldt M; Alshammari M; Solders G; Di Re J; Savitcheva I; Granberg T; Laezza F; Iwarsson E; Svenningsson P Broader phenotypic traits and widespread brain hypometabolism in spinocerebellar ataxia 27. J. Intern. Med, 2020, 288(1), 103–115. 10.1111/joim.13052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ali SR; Liu Z; Nenov MN; Folorunso O; Singh A; Scala F; Chen H; James TF; Alshammari M; Panova-Elektronova NI; White MA; Zhou J; Laezza F Functional modulation of voltage-gated sodium channels by a fgf14-based peptidomimetic. ACS Chem. Neurosci, 2018, 9(5), 976–987. 10.1021/acschemneuro.7b00399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yan H; Pablo JL; Wang C; Pitt GS FGF14 modulates resurgent sodium current in mouse cerebellar Purkinje neurons. eLife, 2014, 3, e04193. 10.7554/eLife.04193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ali SR; Singh AK; Laezza F Identification of amino acid residues in fibroblast growth factor 14 (fgf14) required for structure--function interactions with voltage-gated sodium channel nav1.6. J. Biol. Chem, 2016, 291(21), 11268–11284. 10.1074/jbc.M115.703868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ali S; Shavkunov A; Panova N; Stoilova-McPhie S; Laezza F Modulation of the FGF14:FGF14 homodimer interaction through short peptide fragments. CNS Neurol. Disord. Drug Targets, 2014, 13(9), 1559–1570. 10.2174/1871527313666141126103309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Liu Z; Wadsworth P; Singh AK; Chen H; Wang P; Folorunso O; Scaduto P; Ali SR; Laezza F; Zhou J Identification of peptidomimetics as novel chemical probes modulating fibroblast growth factor 14 (FGF14) and voltage-gated sodium channel 1.6 (Nav1.6) protein-protein interactions. Bioorg. Med. Chem. Lett, 2019, 29(3), 413–419. 10.1016/j.bmcl.2018.12.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Dvorak NM; Wadsworth PA; Wang P; Chen H; Zhou J; Laezza F Bidirectional modulation of the voltage-gated sodium (Nav1.6) channel by rationally designed peptidomimetics. Molecules, 2020, 25(15), 3365. 10.3390/molecules25153365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wang P; Wadsworth PA; Dvorak NM; Singh AK; Chen H; Liu Z; Zhou R; Holthauzen LMF; Zhou J; Laezza F Design, synthesis, and pharmacological evaluation of analogues derived from the plev tetrapeptide as protein-protein interaction modulators of voltage-gated sodium channel 1.6. J. Med. Chem, 2020, 63(20), 11522–11547. 10.1021/acs.jmedchem.0c00531 [DOI] [PMC free article] [PubMed] [Google Scholar]