

Summary
The hippocampus supports many facets of cognition, including learning, memory, and emotional processing. Anatomically, the hippocampus runs along a longitudinal axis, posterior-to-anterior in primates. The structure, function, and connectivity of the hippocampus vary along this axis. In human hippocampus, longitudinal functional heterogeneity remains an active area of investigation, and structural heterogeneity has not been described. To understand the cellular and molecular diversity along the hippocampal long axis in human brain and define molecular signatures corresponding to functional domains, we performed single-nuclei RNA-sequencing on surgically resected human anterior and posterior hippocampus from epilepsy patients, identifying differentially expressed genes at cellular resolution. We further identify axis- and cell-type specific gene expression signatures that differentially intersect with human genetic signals, identifying cell type-specific genes in the posterior hippocampus for cognitive function and in the anterior hippocampus for mood and affect. This data is accessible as a public resource through an interactive website.
eTOC Blurb
Ayhan et al. examined single-nuclei gene expression profiles from distinct regions of human hippocampus. Integrating these results with human genetic data reveals differential involvement of these regions in brain disorders. Comparisons between human and rodent data show that the results are partially conserved.
Introduction
The hippocampus plays a critical role in multiple cognitive functions including episodic memory (Scoville and Milner, 1957; Squire, 1992), spatial navigation (Buzsaki and Moser, 2013; Maguire et al., 1998; Morris et al., 1982), and the regulation of emotional responses (Jimenez et al., 2018; Kjelstrup et al., 2002; Xu et al., 2016). The involvement of the hippocampus in such diverse functions is partially explained by functional differences along its longitudinal axis that runs posterior-to-anterior in primates and dorsal-to-ventral in rodents. Connectivity differences reflect possible longitudinal functional specialization. The efferent (Dolorfo and Amaral, 1998; Witter et al., 1989) and afferent connections (Kishi et al., 2006; Risold and Swanson, 1996) to and from the hippocampus are topologically organized along the longitudinal axis suggesting dissociable functions for the dorsal and ventral hippocampus (Moser and Moser, 1998). Lesion studies support the anatomical segregation at the functional level. Experimental lesions in the dorsal hippocampus in mice affect spatial memory (Moser et al., 1993) while lesions in the ventral hippocampus result in emotional deficits (Henke, 1990; Kjelstrup et al., 2002). Such connectivity differences have also been observed in humans, although the functional significance of these has yet to be clarified (Bonner and Price, 2013; Bubb et al., 2017; Choi, 2020; Zeidman and Maguire, 2016).
The cellular and molecular underpinnings and consequences of this structural and functional segregation within the hippocampus have been studied in animal models, particularly in rodents. Gene expression studies using large-scale in situ hybridization (ISH) identified discrete molecular signatures in pyramidal neurons along the mouse dorsal-to-ventral axis (Dong et al., 2009; Thompson et al., 2008). More recently, bulk RNA sequencing has identified gene expression differences in the principal excitatory cell types of the hippocampus along the long axis (Cembrowski et al., 2016b). Despite this extensive research on the rodent hippocampus, many aspects of the hippocampal transcriptome in humans remain relatively unexplored, especially anterior versus posterior differences in gene expression patterns. Single-nuclei RNA-sequencing (snRNA-seq) profiling of the human hippocampus has identified major cell-class transcriptomes; however, these studies did not retain spatial information about the cells profiled (Franjic et al., 2020; Habib et al., 2017; Tran et al., 2020). Gene expression changes along the human hippocampal long axis have been examined using microarray datasets but these investigations lacked cell-type specificity (Vogel et al., 2020). Cellular and molecular heterogeneity along the hippocampal long axis in a cell-type specific manner in the human hippocampus has never been systematically analyzed.
Here, we investigated cell-type-specific variability across the human anterior (aHC) and posterior (pHC) hippocampus using snRNA-seq (Zheng et al., 2017). While all subjects were suffering from temporal lobe epilepsy, we excluded any subjects in whom there was any radiographic evidence of temporal sclerosis, as well as any subjects with microscopic evidence of loss of pyramidal neurons (characteristic of microscopic evidence of mesial temporal sclerosis (MTS)), cortical dysplasia, or any other lesion. Using this approach, we define unique transcriptional signatures present in pyramidal neurons, interneurons, and glial cells of the anterior and posterior hippocampus. First, we show that marked transcriptional differences exist in excitatory neurons. This axis-specific differential gene expression includes conserved axis-specific genes previously identified across the mouse dorsal-ventral axis (Cembrowski et al., 2016b) as well as axis markers unique to humans. Notably, in contrast to granule and pyramidal neurons, inhibitory interneuron populations do not show variability across the hippocampal long axis. Second, we present evidence for previously unrecognized overall heterogeneity in dentate gyrus granule cells. Last, we identify transcriptional states of astrocytes and microglia that are distributed equally along the hippocampal axis. Together, these data provide an important cell-type-specific publicly accessible transcriptomic resource (https://human-hippo-axis.cells.ucsc.edu/) of the human hippocampus that highlights human-relevant patterns of cellular identity that may underlie the distinct topological properties of the hippocampus.
Results
Cellular diversity of the anterior and posterior hippocampus
To characterize the cellular and transcriptional variability within and across the human anterior and posterior hippocampus, we processed single-nuclei isolated from 10 surgically resected hippocampal samples using the 10X Genomics platform (Zheng et al., 2017) (Table S1) (Figure 1A). Five aHC and five pHC samples were obtained from the same individuals. These samples were surgically removed from patients undergoing surgical treatment for epilepsy using an en bloc resection technique by which the hippocampus was dissected from its vascular pedicle immediately prior to tissue processing. The location of anterior and posterior specimens obtained along the hippocampal axis is highlighted on T1 magnetic resonance images for each subject (Figure S1). Thus, an important caveat of this dataset is that all tissue was obtained from patients affected by epilepsy. However, we implemented several steps in our analysis to minimize the impact of the underlying clinical condition on our findings. First, we excluded any subject in whom there was radiographic evidence of temporal sclerosis (or any other hippocampal lesion). All patients suffered from cryptogenic temporal lobe epilepsy; there were no cases with tumors, cortical dysplasia, or other underlying conditions. Further, we excluded any samples that exhibited microscopic evidence of temporal sclerosis, namely selective loss of pyramidal neurons identified on expert neuropathological review of tissue specimens (Table S1). Epilepsy duration and seizure frequency were also included as two of the covariates in the linear mixed model (see STAR Methods). All patients were on medication and this information is included in Table S1. Finally, only surgical specimens 3.5 cm in minimum length (from anterior to posterior as measured at the time of resection) were included, insuring adequate spatial separation for anterior versus posterior samples. After quality control including removal of the nuclei with <300 genes, >10000 UMIs, and >5% mitochondrial transcripts (see STAR Methods), we obtained 129,908 nuclei across ten samples (Fig S2A), with a median of 1,066 (2,101 for neuronal, 1000 for non-neuronal) genes per nucleus, and a median of 1,577 unique molecular identifiers (UMIs) per nucleus (4,216 for neuronal, 1,451 for non-neuronal), a depth sufficient to resolve cell-types and their transcriptional landscape in the human brain (Lake et al., 2018) (Fig S2B, C).
Figure 1. Unbiased snRNA-seq analysis identified 24 distinct cell types in human anterior and posterior hippocampus samples.
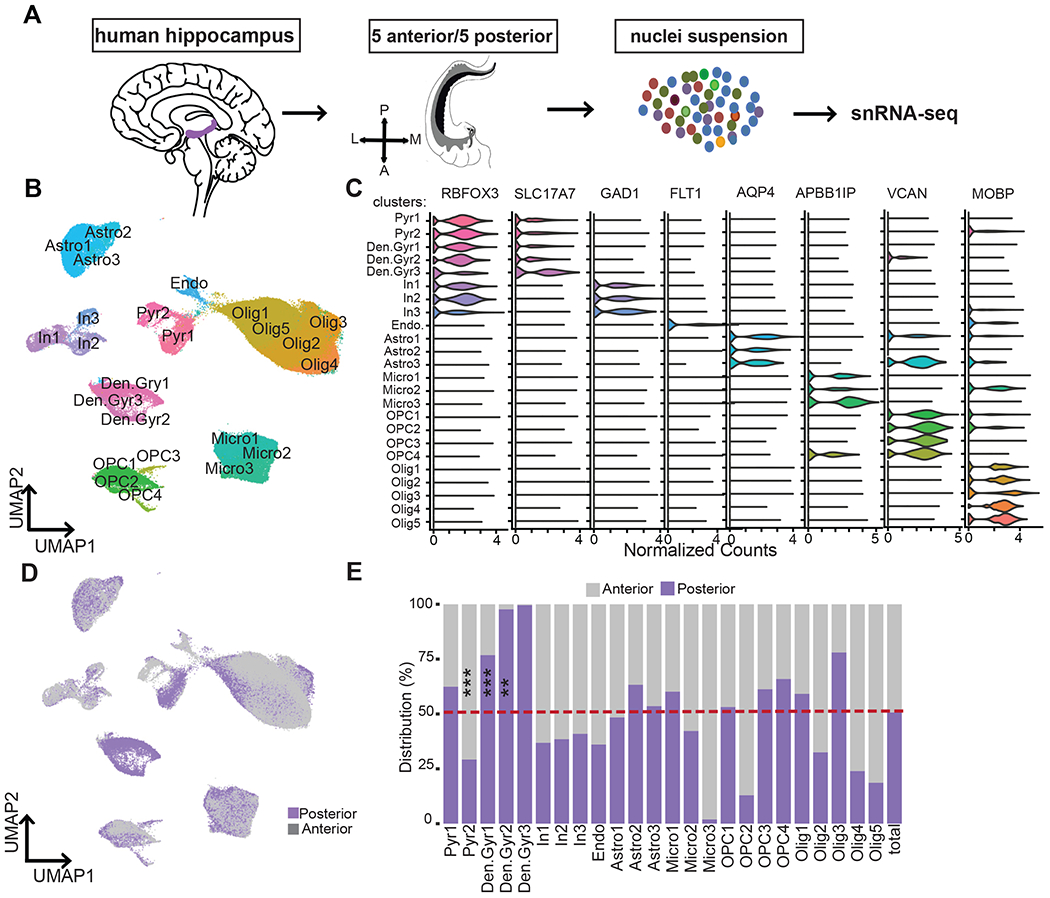
(A) Schematic overview of the experimental procedures used to extract nuclei from 5 anterior and 5 posterior samples, single-nuclei capture and barcoding using 10X Genomics Chromium, and Illumina next-generation sequencing. (B) UMAP plot of all cells analyzed from anterior (64,076) and posterior (65,832) hippocampus, colored by cluster identities and cell-type annotations. Pyr=Pyramidal neurons, Den.Gyr=dentate gyrus neurons, In=Interneurons, Endo=endothelial cells, Micro=microglia, Astro=astrocytes, OPCs=oligodendrocyte progenitor cells, and Olig=oligodendrocytes. (C) Violin plots of expression values for cell-type-specific marker genes. (D) UMAP plot of all cells analyzed, colored by the axis the cells were recovered from (posterior, purple; anterior, gray). (E) Bar chart showing the frequency distribution of all clusters between posterior (purple) and anterior (gray). *P < 0.01, ***P < 0.001, Robust generalized mixed model. See also Figure S2, S3, S4, and S5 and Table S2, S3.
To classify the major cell types, we clustered all the nuclei using an unsupervised graph-based approach with Seurat (Stuart et al., 2019) (see STAR Methods). This initially resulted in 24 major cell types in total (Figure 1B, Table S2), which were then annotated using a combination of known cell marker genes: broad markers for neurons (RBFOX3), excitatory neurons (SCL17A7), inhibitory neurons (GAD1), astrocytes (AQP4), oligodendrocytes (MOBP), oligodendrocyte precursor cells (OPCs) (VCAN), macrophages/microglia (APBB1IP) and endothelial cells (FLT1) (Figure 1C). To confirm our findings, we combined a subset of our dataset with previously published adult human hippocampus snRNA-seq (Habib et al., 2017) data using Integrated Anchors analysis followed by clustering with Seurat (Stuart et al., 2019). The cells from both datasets clustered together into cell-type-specific clusters indicating that our results are robust in spite of using epileptic brains (Figure S3A and B).
To understand the cellular diversity in anterior and posterior hippocampus, we next examined the distribution of nuclei isolated from anterior and posterior hippocampus within individual clusters (Figure 1D, E, S2D). Among the total nuclei sampled, 49.32% are anterior and 50.68% are obtained from posterior samples, allowing for approximately equivalent comparisons. All major cell types were found in both the anterior and posterior hippocampus (Figure S2D) across all five donors (Figure S2E). In general, the aHC and pHC distribution patterns within individual clusters (Figure 1E) are more variable than within cell-type distribution (Figure S2D). Statistical analysis using a robust linear mixed model (see STAR Methods) identified two clusters consisting of significantly greater number of nuclei from posterior hippocampus (Den.Gyr1 and Den.Gyr2 and one cluster with significantly greater number of nuclei from anterior hippocampus (Pyr2) (Figure 1E, S4, S5, Table S3). Three clusters (OPC2, Olig5, and Den.Gyr3) that were primarily driven by a single donor were not considered for this analysis (Table S3). Detection of greater numbers of posterior nuclei in dentate gyrus neuronal populations reflects the anatomical organization of hippocampal subfields along the longitudinal axis. Previous imaging studies of the human hippocampus have reported that the volumetric ratios of subfields vary along the axis with a decreasing ratio of the dentate gyrus subfield from posterior to anterior (Malykhin et al., 2010; Palomero-Gallagher et al., 2020).
Neuronal subpopulations in anterior and posterior hippocampus
To further understand the differences in neuronal subtypes along the axis, we subsetted and reclustered only the neuronal cells (clusters Pyr1-2, Den.Gyr1-3, In1-3) identified in our initial clustering (Figure 1B). Among these neurons, we identified 18 transcriptionally distinct clusters in the aHC and pHC (Figure 2A) across five donors (Figure 2B and S6) and determined cell type-specific marker genes with distinct levels of expression among these clusters (Figure S7A, B, Table S4). Annotation of these neuronal clusters included multiple strategies: 1) interrogating canonical mouse subfield markers identified via in situ hybridization (Lein et al., 2004) and next-generation sequencing (Cembrowski et al., 2018; Cembrowski et al., 2016b): granule cells of dentate gyrus (MAML2), broad markers for pyramidal neurons of CA (SV2B), CA1 neurons (SATB2), subiculum (FN1), CA1 & CA3 neurons (TYRO3), CA2 &3 neurons (PFKP), inhibitory neurons (CCK and SST) (Figure 2C, D, E) and 2) enrichment analysis using a hypergeometric test of unique cluster markers with mouse subfield markers identified via microdissection and bulk RNA-seq (Hipposeq dataset) (Cembrowski et al., 2016b) (Figure S8A, B). We reanalyzed the Hipposeq dataset to identify sets of genes enriched in DG, CA1, CA2, CA3, and CA4 subfields relative to all subfields (Figure S8A). Because this dataset shows a hierarchical structure reflecting the levels of transcriptional similarity and distance across hippocampal subfields (i.e. DG granule cells are most distant to pyramidal CA neurons) (Figure S8A), we used a step-wise strategy to identify subfield markers. We first curated the gene list distinguishing DG granule neuron from all CA neurons and annotated our excitatory clusters as DG or CA based on the subsequent hypergeometric test. Then, we further annotated the CA clusters by repeating this strategy with the differentially expressed genes CA4vsCA1/2/3, CA1vsCA2/3 and, CA2vsCA3 (Figure S8B). Among the clusters with CA identity, CA3.1 expresses HS3ST4 at relatively higher levels (Figure 2D). Because this gene is not one of the canonical markers of mouse hippocampus, we independently validated its enriched expression in CA3 pyramidal neurons via immunohistochemistry using a completely separate dataset consisting of postmortem hippocampal tissue (Figure S7C). Using this convergent strategy, we detected multiple clusters of dentate gyrus granule cells (clusters Gra.Neu1-7), pyramidal cells from CA1 (CA1.2, CA1.2), CA2, CA3 (CA3.1, CA3.2) regions, inhibitory interneurons (In.Neu1-4) and a cluster of cells from subiculum (Sub.).
Figure 2. snRNA-seq reveals neuronal cell heterogeneity and DEGs across the hippocampal poles.
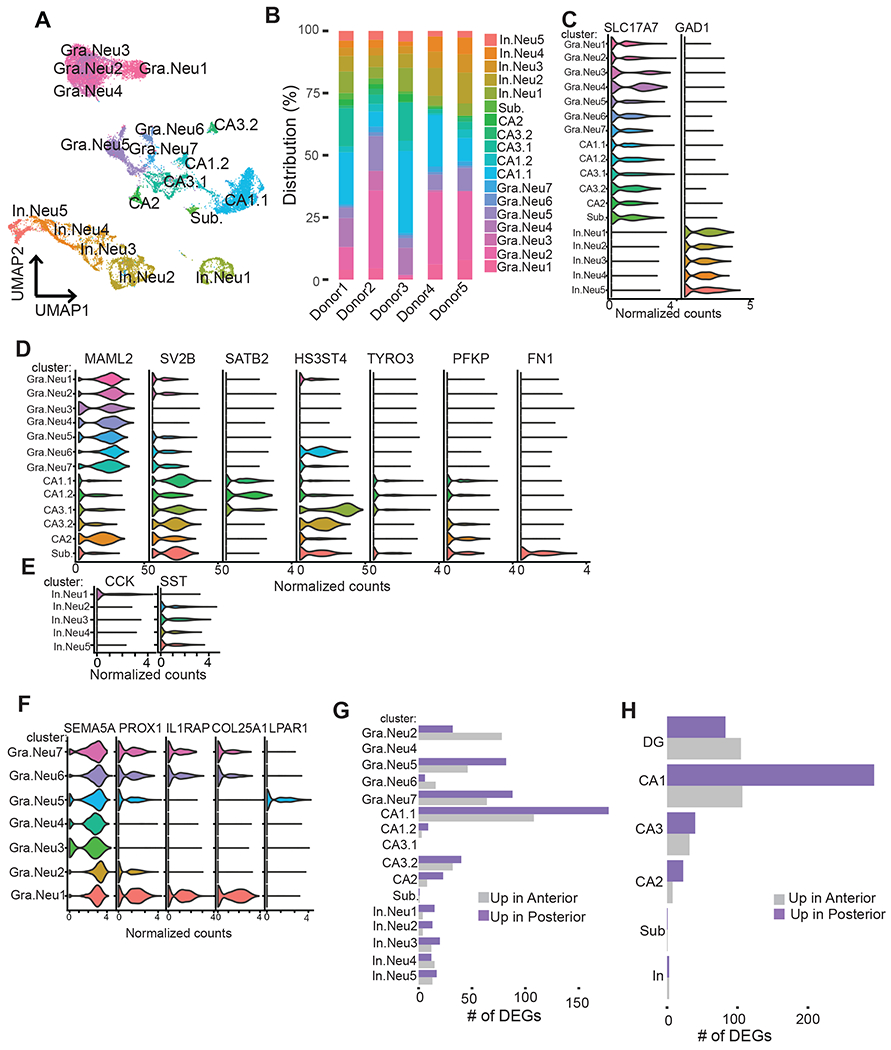
(A) UMAP plot of neuronal cells colored by cluster identities and cell-type annotations. (B) Frequency distribution of neuronal clusters in five donors. (C) Violin plots of expression values for markers for excitatory and inhibitory neurons, (D) for subfields markers, and (E) for inhibitory neuronal cell types across clusters. (F) Violin plots showing the normalized counts of shared and distinct markers (x-axis) of DG clusters (y-axis). (G) Within cluster differential gene expression analysis between anterior vs. posterior (adj. p-value<0.05, log2FC>0.3, percentage>25). (H) Within cell-type differential gene expression analysis between anterior vs. posterior (adj. p-value<0.05, log2FC>0.3, percentage>25). Gra.Neu=Granule Neurons, In.Neu=Inhibitory neurons, and Sub=subiculum. See also Figure S6, S7, S8, S9, S10 and Table S4, S5, S6, S7.
We identified seven clusters (Gra.Neu1-7) that were enriched for dentate gyrus granule (DG) cell identity (Figure 2A, S7A). Four of these clusters (Gran.Neu1-4) are distant from pyramidal neurons and inhibitory neurons in the UMAP space, while others (Gra.Neu5-7) are found closer to pyramidal neurons. To understand the distinguishing features of these granule cell subclusters, we identified enriched genes for these clusters. All of these clusters express a set of shared markers indicating DG cells such as SEMA5A (Duan et al., 2014) and MAML2 (Cembrowski et al., 2016b) (Figure 2D and 2F) uniformly confirming their granule cell identity. Additionally, we identified genes that were differentially expressed among these clusters, suggesting a transcriptionally diverse pool of DG neurons. For example, one of the canonical markers of mature granule cells, PROX1 show sparse expression in clusters Gra.Neu3 and Gran.Neu4. Clusters Gra.Neu1, Gra.Neu6, and Gra.Neu7 express IL1RAP and COL25A1 in addition to shared granule cell markers. Gra.Neu5 distinctly expresses a recently described neuronal stem cell marker LPAR1 (Figure 2F). (Walker et al., 2016). Another recent single-cell transcriptomics study of mouse dentate gyrus identified Lpar1+ radial glia-like cells in the adult hippocampus (Hochgerner et al., 2018). Given reports of neurogenesis in the adult dentate gyrus for both unaffected (Boldrini et al., 2018) and epileptic brains (Parent et al., 2006), the population of cells in Gra.Neu5 can potentially represent precursor granule cells with neurogenic capacity. Subtypes of granule neurons of the adult mouse dentate gyrus with distinct morphology and cellular activity were also recently described (Erwin et al., 2020). In this study, reanalysis of a previously published mouse hippocampal dataset (div-seq) (Habib et al., 2016) revealed Cck+ and Penk+ granule neuron populations. To understand the extent of similarity between the DG granule neurons we identified and mouse DG subtypes (Erwin et al., 2020), we also analyzed the div-seq (Habib et al., 2016) dataset in a similar way and identified Cck+ and Penk+ populations (Figure S9A and B). We then performed geneset enrichment between marker genes for the DG clusters we identified in human hippocampus with markers genes for the div-seq DG clusters (Figure S9C). While Gra.Neu1, Gra.Neu4, Gra.Neu5 and Gra.Neu7 show homology to div-seq DG clusters, three other human DG clusters did not show significant overlap with mouse data. Interestingly, none of our DG clusters map to the mouse Penk+ cluster (cluster 4). Taken together, our finding and the report from mouse hippocampus highlight a heterogeneous pool of granule neurons.
We additionally sought to determine the diversity of GABAergic interneurons further and performed reclustering on the GAD1+ interneurons in our neuronal clustering (Figure 2A). This analysis resulted in a more refined separation of inhibitory neuronal types including neuronal nitric oxide (NOS1)-positive, cholecystokinin (CCK)-positive (CCK/CXCL14 and CCK/CNR1), vasoactive intestinal peptide (VIP)-positive interneurons, somatostatin (SST)-positive, and parvalbumin (PVALB)-positive (Figure S10A). These clusters were identified by the respective marker genes: NOS1, CXCL14/CNR1, CALB2, SST, and SOX6 (Figure S10B). The PVALB-positive interneurons were identified by robust SOX6 expression without co-expression of SST as SOX6 labels both SST and PVALB-positive interneurons (Pelkey et al., 2017). We detected few PVALB transcripts in our dataset similar to a recent snRNA-seq study describing detection of PVALB-positive interneurons but not PVALB transcript in human striatum (Tran et al., 2020).To confirm robustness of the transcriptional signatures associated with these clusters, we compared our data (Table S5) with inhibitory neurons identified using human postmortem autopsy tissue (Figure S10C) (Habib et al., 2017). Overall, our surgical tissue derived interneurons agreed well with the interneurons captured from autopsy tissue. We detected specific enrichment between PVALB, SST, CCK/CNR1 and VIP/CALB2 interneurons. CCK/CXCL14 and NOS1 clusters showed enrichment for both CCK/CXCL14 and NOS1 positive cells, indicating shared markers between these cell-types. To understand the similarity between mouse and human interneurons, we performed gene set enrichment between the inhibitory neuronal clusters we identified and mouse GABAergic neurons (Habib et al., 2017) (Figure S10D). While PVALB, SST, and CCK/CNR1 clusters showed enrichment to corresponding mouse clusters, the enrichment for the other inhibitory neuronal clusters were less specific showing some degree of significant overlap to different mouse clusters.
Differential gene expression within neuronal clusters across the hippocampal axis
To identify specific genes that show enrichment in aHC versus pHC, we compared gene expression levels in neurons isolated from these regions using a linear mixed model (LMM) excluding any genes expressed in less than 25% of the cells in the cluster or cell-type tested (see STAR Methods). Using this approach, we identified cluster (Table S6) and cell-type-specific (Table S7) differentially expressed genes (DEGs) (log2FC>0.3, FDR <0.05) (Figure 2G and 2H). The number of DEGs found in excitatory neurons (granule cells and CA1 pyramidal neurons), in particular, was substantially higher compared to inhibitory neurons. Surprisingly, we did not detect many genes differentially expressed in CA3 neurons unlike a previous report using in situ hybridization across mouse CA3 longitudinal axis (Thompson et al., 2008), possibly because of the relatively low numbers of neurons sampled from the CA3 subfield in our study. It is also possible that human CA3 neurons exhibit a less degree of differential expression in comparison to mouse.
We next performed gene ontology (GO) analysis to elucidate the functional categories of all of the genes enriched in the human CA1 anterior and CA1 posterior hippocampus (Table S8). The functional categories for aHC and pHC-enriched genes exhibited a high degree of similarity (simRel = 0.6) (Schlicker et al., 2006) involving many common GO terms such as synaptic function, glutamate signaling, and transporter activity. The top GO categories for posterior and anterior-enriched genes are visualized in Figure S11. To examine the extent to which CA1 neuron transcriptomic heterogeneity might be similar between the human and mouse hippocampus, we analyzed the intersection between our CA1-specific differentially expressed genes with mouse CA1 axis markers identified by bulk RNA-sequencing (Cembrowski et al., 2016a). We found a significant overlap between the list of genes upregulated in dorsal CA1 in mouse and genes upregulated in the homologous human region, posterior CA1 (p=4.67e−05, hypergeometric test, Benjamini–Hochberg corrected, 9 overlapping genes (Figure 3A and Table S9). Similarly, genes upregulated in ventral CA1 of mice showed significant overlap with genes enriched in the corresponding anterior CA1 of humans (p=1.16e−06, hypergeometric test, Benjamini–Hochberg corrected, 9 overlapping genes) (Figure 3A and Table S9). Hence, this analysis identified conserved axis markers between human and mouse (Figure 3B). The conserved markers of anterior/ventral hippocampus are associated with a wide range of functional ontologies (Table S8) including synaptic function (GRIA4, SHISA9, NPTXR, LINGO2, RIT2), calcium signaling (NECAB1, SPOCK1), and cell-cell adhesion (PTPRK). Conserved markers of the posterior/dorsal hippocampus represent somewhat different functional ontologies: ion channels (CACNG8), synaptic function (CHRM3), protein tyrosine kinase (EPHA7), cytoskeletal organization (WIPF3), and transcriptional regulation (NR4A1, TFDP2). We also identified several genes differentially expressed in an axis-specific manner in humans but not in mice. CADPS2 encoding Ca2+-dependent activator protein for secretion 2 (CADPS2) is expressed at higher levels in the posterior hippocampus. Intriguingly, CADPS2 was shown to affect neurotransmitter release by stabilizing docked vesicles at hippocampal synapses in mice (Shinoda et al., 2016). We independently validated posteriorly enriched expression of CADPS2 using human post-mortem tissue without epilepsy or any other pathological conditions using immunohistochemistry (Figure 3C). We detected 1.57 fold higher levels (unpaired t test, p<0.05) of CADPS2 protein on the sections obtained from posterior hippocampus compared to the sections from anterior hippocampus on par with the differential expression detected by the LMM analysis (log2FC= 1.052, FDR=3.26e−149). Finally, we also observed some unexpected overlaps when we compared expression enrichment between species. This showed significant overlap between mouse ventral and human posterior CA1 (p=4.1e−04, hypergeometric test, Benjamini–Hochberg corrected, 10 overlapping genes) (Figure 3A and Table S9), suggesting distinct axis expression programs between species for some hippocampal genes.
Figure 3. Differential gene expression in CA1 neurons in anterior and posterior hippocampus.
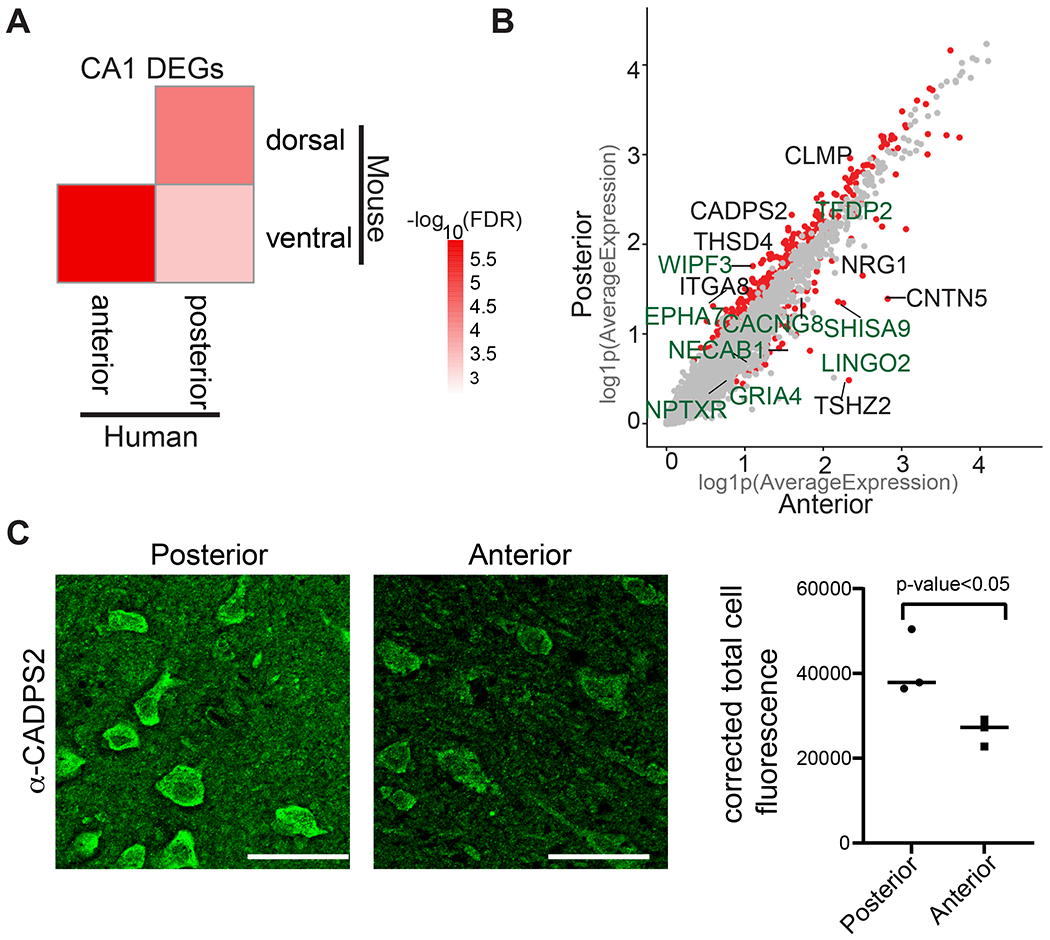
(A) Heatmap showing −log10(FDR) from a hypergeometric enrichment test for the overlaps between mouse CA1 dorsal-ventral enriched genes described in Cembrowski et al. (Cembrowski et al., 2016a) with human CA1 genes enriched in the anterior and posterior hippocampus. (B) Scatter plot showing log(1+x) (log1p) of the average expression for each gene in CA1 neurons in anterior (x-axis) and posterior (y-axis). Red dots represent the genes that are significantly differentially expressed across anterior CA1 vs posterior CA1 (adj. p-value<0.05, log2FC>0.3, percentage>25); gray dots represent the genes that are not differentially expressed. The names of genes that are differentially expressed both in mouse and human across the long axis are labeled green. The names of genes that are differentially expressed across the axis only in human are labeled black. (C) Immunohistochemistry demonstrates greater protein levels of CADPS2 in posterior compared to anterior hippocampus. Left panel: representative image of immunohistochemistry using α-CADPS2 in posterior or anterior hippocampal tissue. Right panel: quantification of corrected total cell fluorescence from immunohistochemistry using α-CADPS2 in posterior compared to anterior hippocampus. Individual points represent the average intensity values derived from each specimen (n=3, P< 0.05; unpaired t test). See also Figure S11 and Table S8, S9.
Transcriptomic heterogeneity of dentate gyrus granule cells in the hippocampus
Using the statistical approach described above, we identified differentially expressed genes in dentate gyrus granule cells in the anterior versus posterior hippocampus (log2FC>0.3, FDR <0.05) (Figure 2G and 2H). To examine the extent of DG granule neuron transcriptomic heterogeneity across the longitudinal axis between human and mouse hippocampus, we overlapped our DG-specific DEGs with mouse DG axis markers identified by two separate bulk RNA-sequencing studies (Cembrowski et al., 2016b; Zhang et al., 2018). One of these studies included comparison of dorsal and ventral DG in mice housed in a standard or enriched environment (Zhang et al., 2018). We found significant overlaps between the list of genes upregulated in dorsal DG in mouse and genes upregulated in human posterior DG by comparing our data to all three datasets: (p=2.86e−06 hypergeometric test, Benjamini–Hochberg corrected, 16 overlapping genes) (Figure 4A), (4.02e−22 hypergeometric test, Benjamini–Hochberg corrected, 42 overlapping genes) (Figure 4C) (p= 5.74e−23 hypergeometric test, Benjamini–Hochberg corrected, 43 overlapping genes) (Figure 4D). Similarly, genes upregulated in mouse ventral DG showed significant overlap with genes enriched in the corresponding human aHC (p=1.71e−07, hypergeometric test, Benjamini–Hochberg corrected, 20 overlapping genes) (Figure 4A), (0.02842, hypergeometric test, Benjamini–Hochberg corrected, 21 overlapping genes) (Figure 4C), (p= 0.0141, hypergeometric test, Benjamini–Hochberg corrected, 22 overlapping genes) (Figure 4D). The conserved markers of anterior/ventral hippocampus are associated with a wide range of functional ontologies including RNA binding proteins (RALYL, KHDRBS3), and cell-cell adhesion (PTPRD, CNTN4, CDH13, CADM1, PTPRG). Similarly, conserved markers of posterior/dorsal represent several functional ontologies: ion channels (KCNJ6), synapse organization (TANC1, NPTXR, SLIT1), protein tyrosine kinase (EPHA5), and cytoskeletal organization (CAP2, STXBP5L) (Table S8). We also identified DEGs that are not found as axis-specific in mouse including genes associated with cell adhesion (CNTN5, PCDH9), and transcription factors (RFX3) (Figure 4B).
Figure 4. Transcriptomic and cellular heterogeneity of dentate gyrus granule cells in the hippocampus.
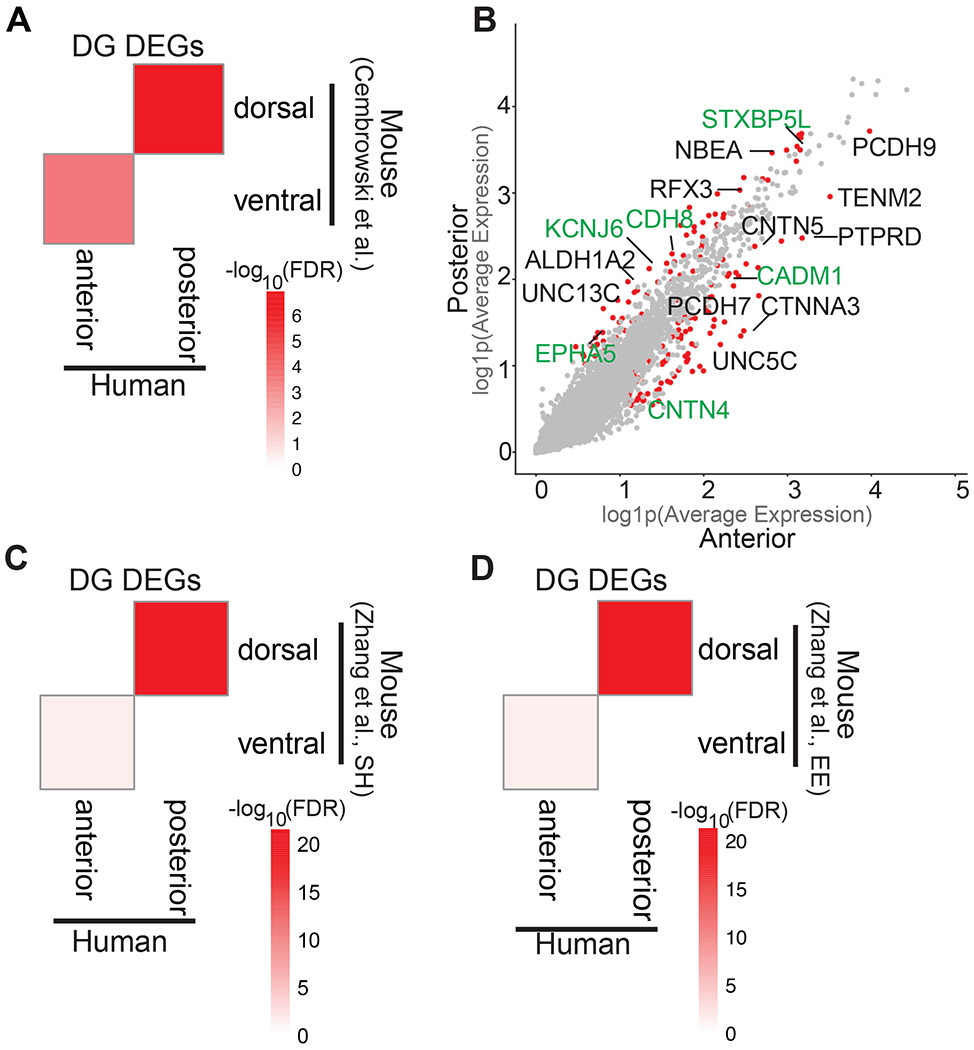
(A) Heatmap showing −log10(FDR) from a hypergeometric enrichment test for the overlaps between mouse DG dorsal-ventral enriched genes described in Cembrowski et al. (Cembrowski et al., 2016b) with human DG genes enriched in the anterior and posterior hippocampus. (B) Scatter plot showing log(1+x) (log1p) of the average expression for each gene in dentate gyrus (DG) granule neurons in anterior (x-axis) and posterior (y-axis). Red dots represent the genes that are significantly differentially expressed across anterior DG vs posterior DG (adj. p-value<0.05, log2FC>0.3, percentage>25); gray dots represent the genes that are not differentially expressed. The names of genes that are differentially expressed both in mouse and human across the long axis are labeled green. The names of genes that are differentially expressed across the axis only in human are labeled black. (C) Heatmap showing −log10(FDR) from a hypergeometric enrichment test for the overlaps between mouse DG dorsal-ventral enriched genes under standard housing environment described in Zhang et. al. (D) Heatmap showing −log10 (FDR) from a hypergeometric enrichment test for the overlaps between mouse DG dorsal-ventral enriched genes under enriched environment described in Zhang et. al (Zhang et al., 2018). SH= standard housing, EE=enriched environment. See also Table S8, S9.
Enrichment of cell-type and axis-specific genes for variants associated with cognitive traits and neuropsychiatric disorders
Impairments in hippocampal function have been observed and implicated in neurological and neuropsychiatric disorders including Alzheimer’s disease (AD), schizophrenia (SCZ) and major depressive disorder (MDD); however, cell-types and circuits underlying these distinct pathologies are not entirely understood (Small et al., 2011). Genome-wide association studies have identified hundreds of loci associated with these neurological disorders and complex traits. To test whether variants identified by GWAS for a specific disorder or trait converge on the hippocampal cell-subtypes axis, we performed Multi-marker Analysis of GenoMic Annotation (MAGMA) (de Leeuw et al., 2015) (see STAR Methods). MAGMA first identifies gene level localization of GWAS risk variants, and then enables integration of this gene level information with gene lists such as cell-type markers and differentially expressed genes we identified here. In this manner, we can examine the cell type-specific expression of genes that have variants associated with specific diseases or traits. We selected GWAS summary statistics for several neuropsychiatric disorders and complex traits and performed MAGMA gene set analysis (de Leeuw et al., 2015) (see STAR Methods). First, we sought to identify cell-types critically involved in disease pathogenesis and cognitive traits. Notably, neuronal cell-subtypes were enriched for risk variants (FDR < 0.05) linked with AD, autism spectrum disorder (ASD), bipolar disorder (BD), and MDD (Figure 5A). However, when a stricter Bonferroni correction is applied (Bonferroni < 0.05), we found a more specific enrichment for BD (Gra.Neu3), attention-deficit/hyperactivity disorder (ADHD) (CA1.1), ASD (CA1.2), and MDD (In.Neu5). Conversely, variants associated with cognitive traits including educational attainment, cognitive function, and intelligence as well as risk variants for SCZ showed a more widespread overlap in neuronal cell-types of the hippocampus with both relaxed (FDR) and stricter (Bonferroni) thresholds. These distinctive patterns suggest that while the majority of the neuronal cell-types in the hippocampus contribute to cognitive function, intelligence and SCZ risk, a more cell-type-specific pathophysiology is evident for specific disorders such as BD, ASD and MDD. Despite the widespread cellular enrichment of variants associated with cognitive traits in several clusters, we did not observe any enrichment for cognitive traits in LPAR+ neurogenic-like dentate gyrus cells, Gra.Neu5. Interestingly, both excitatory and inhibitory neurons are enriched for SCZ risk variants. This result underscores hippocampal vulnerability in SCZ, which is in line with previous bulk RNA-seq analysis of SCZ-affected hippocampal tissue (Collado-Torres et al., 2019; Perez et al., 2020). Both ASD- and ADHD-linked risk loci are enriched in pyramidal neurons of CA1 (CA1.1, CA1.2) and subiculum (Sub.) and inhibitory neuronal cluster In.Neu2, suggesting potential mechanistic overlap between these disorders (Salazar et al., 2015).
Figure 5. Enrichment of neuropsychiatric and cognitive trait variants to genes expressed in aHP and pHP.
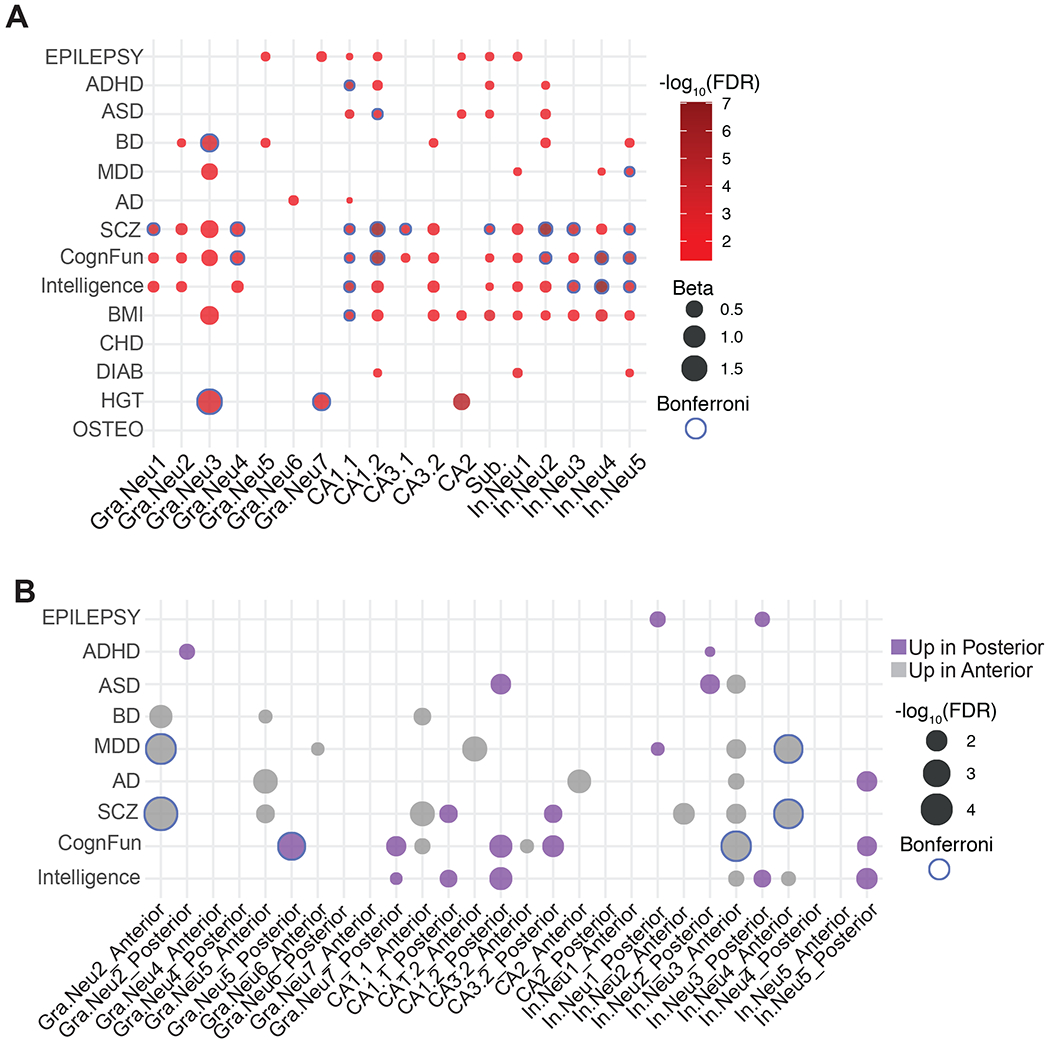
(A) GWAS enrichment in cluster markers. Bubble-chart highlighting the enrichment of human GWAS signals in the neuronal cluster marker genes identified in this study. Association analysis was performed using MAGMA. Gradient corresponds to the −log10(FDR) for each association test. Size corresponds to the effect size (Beta). Blue border corresponds to the Bonferroni correction threshold of p < 0.05. Y-axis lists the acronyms for the traits and diseases utilized for this analysis. X-axis lists the neuronal clusters with the corresponding cell-type annotation. (B) GWAS enrichment in cluster-specific DEGs. Bubble-chart highlighting the −log10(FDR) for the enrichment of human GWAS signal in the cluster-specific DEGs across aHP and pHP. Blue border corresponds to the Bonferroni correction threshold of p < 0.05. The y-axis shows the acronyms for the GWAS data utilized for this analysis. The x-axis shows aHP vs pHP DEGs in each cluster (Cluster#_Anterior=the list of genes expressed at higher levels in aHP in the given cluster, Cluster#_Posterior=the list of genes expressed at higher levels in pHP in the given cluster). ADHD=attention deficit hyperactivity disorder, ASD=autism spectrum disorders, AD=Alzheimer’s disease, BD=bipolar disorder, MDD=major depressive disorder, SCZ=schizophrenia, CognFunc=cognitive functions, BMI=body mass index, CHD=coronary artery disease, DIAB=diabetes, HGT=height, and OSTEO=osteoporosis. See also Table S10.
In addition, functional segregation along the hippocampal axis raises the possibility of differential involvement of aHP and pHP in these pathogenic states. To evaluate the anterior vs. posterior-specific involvement relative to each other in pathogenic risk and cognitive traits, we used MAGMA to identify whether differentially expressed genes across aHP and pHP are enriched for genetic risk linked to neuropsychiatric disorders and cognitive traits (Figure 5B). We found enrichment for cognitive function and intelligence loci specifically in pHP cell-subtypes. Furthermore, ASD- and ADHD-linked variants are enriched in pHP cell-subtypes possibly due to the relationship of cognitive dysregulation in these disorders. Conversely, MDD, BD and cognitive functions are enriched in aHP cell-subtypes (Figure 5B). This finding complements previous structural and cellular reports on the relative sensitivity of anterior hippocampus to chronic stress and depressed states (Hawley and Leasure, 2012; Szeszko et al., 2006; Tanti and Belzung, 2013; Willard et al., 2009) at the level of genomics as well as models of hippocampal longitudinal specialization incorporating affective processing distinctions (Strange et al., 2014). Moreover, because our data is epileptic patient-derived, we also analyzed the enrichment for epilepsy risk-loci. Even though we found enrichment for two sets of pHP inhibitory neuron DEGs, none of these cell-subtypes survived the stricter Bonferroni correction (Figure 5B). Comorbidities between ASD/ADHD and epilepsy have been reported (Bertelsen et al., 2016; Lo-Castro and Curatolo, 2014; Novarino et al., 2013). However, our data showed that there is no co-occurrence between epilepsy and ASD/ADHD risk-loci at the cell-subtype level, underscoring the limited amount of common variant genetic correlation between these disorders (Brainstorm et al., 2018). In conclusion, we show that gene expression at the single cell level allowed us to identify relevant hippocampal cell-types for complex traits and neuropsychiatric disorders. This is further refined by cell-subtypes differentially associated between anterior and posterior hippocampus.
Differential gene expression within glial clusters across the hippocampal axis
We also compared levels of gene expression in glial cells isolated from aHC versus pHC using a LMM (see STAR Methods). In contrast to excitatory neurons, glial cells did not exhibit transcriptomic diversity across the anterior-to-posterior axis, with only a few differentially expressed genes distinguishing these cells (Figure S12). However, we did identify multiple clusters of glial cells, suggesting human hippocampal glial cells do display transcriptomic diversity and these clusters could represent distinct cellular states for glial cells (Figure 1B). To gain insights into the transcriptomic diversity of astrocytes specifically, we assessed differential gene expression between astrocyte clusters. Our initial analysis identified 3 major astrocyte clusters (Figure 1B) with distinct gene expression profiles (Figure 6A). These clusters of astrocytes were detected at comparable levels in all five donors indicating these differences are not due to technical or biological variability (Figure 6B). Astro1 was marked by a relatively higher expression of classical astrocyte markers such as GFAP. Astro2 expressed astrocyte markers including AQP4 (Figure 1C) and was characterized by higher expression of ATP1B3 and NR4A3 (Figure 6A). Similar to Astro1 and Astro2 we identified, two distinct astrocyte populations with different levels of GFAP expression were found in the human cortex (Hodge et al., 2019). In this cortical dataset, the astrocytes with higher GFAP expression are located in layers I and II of the human middle temporal gyrus. In our hippocampal dataset, we have identified Astro1 (GFAP+) and Astro2 in both aHP and pHP (Figure 1E) suggesting similar distributions of these astrocyte cell types throughout the long axis. However, it is possible that astrocyte cell-types are differentially distributed in subfields of the hippocampus. In addition to these two subtypes, we detected a smaller astrocyte cluster, Astro3 (Figure 1B, 6B). This rare population differentially expresses TNR (Figure 6A), which was previously reported to be expressed in 10% of cultured mouse astrocytes and implicated in glutamate uptake (Okuda et al., 2014). To compare our astrocyte subclasses with the recently published astrocyte subclasses in mouse cortex and hippocampus (Batiuk et al., 2020), we performed gene-set enrichment using a hypergeometric test. The marker genes for our astrocyte Astro1 and Astro2 showed varying degrees of overlap with the markers of astrocyte clusters obtained from adult mouse cortex and hippocampus (Batiuk et al., 2020) (Figure 6C). Cluster Astro2 showed strong overlaps with mouse astrocyte subclasses AST1 and AST2, which are characterized as mature astrocytes in the mouse brain. Astro1 showed the highest overlap with AST4. Interestingly, AST4 was linked to neurogenesis based on marker gene expression and mainly localized to the hippocampus in the mouse dataset. Taken together, by carrying out comparative analyses of our data with recently published data, we identified transcriptionally distinct cellular states of astrocyte in the human hippocampus.
Figure 6. Transcriptomic and cellular heterogeneity of glial cells in the hippocampus.
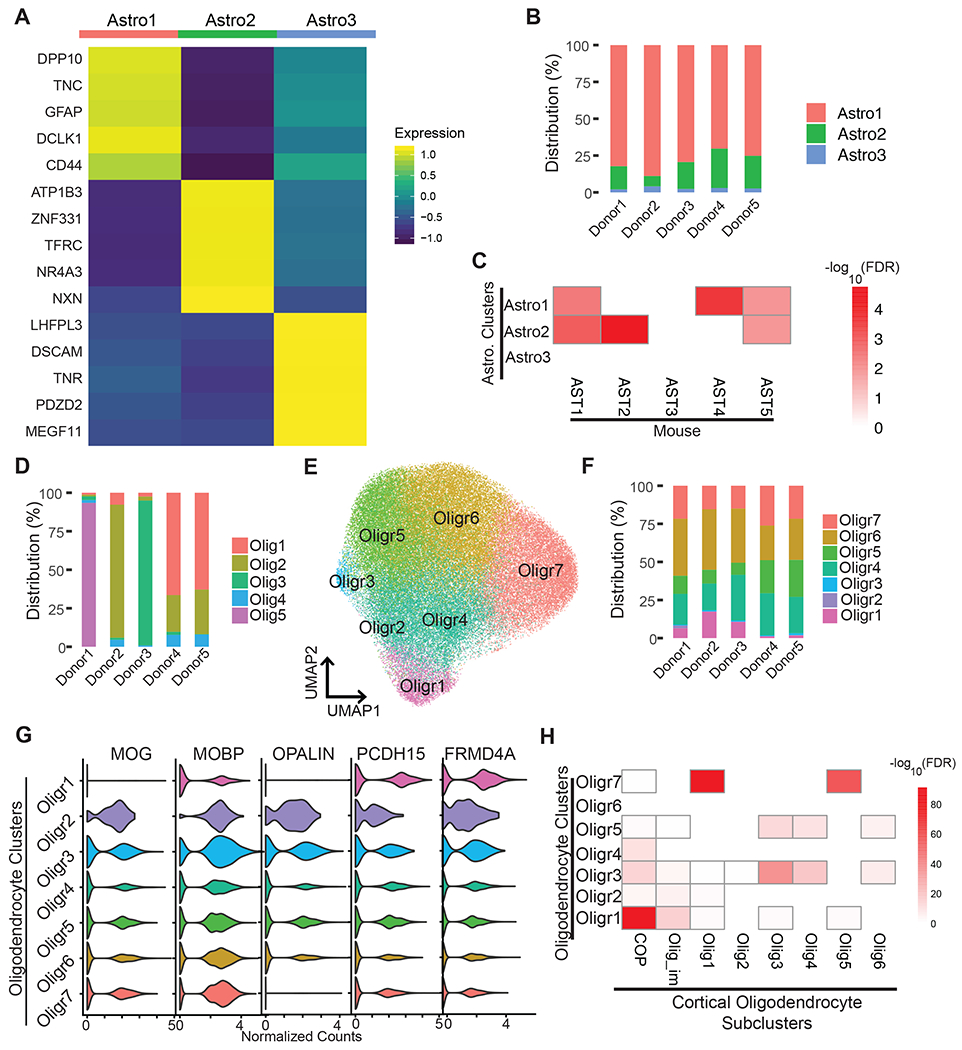
(A) Heatmap illustrating the average expression of the top 5 DEGs between astrocyte clusters. Color scheme corresponds to log-normalized and scaled values for average gene expression for each cluster. (B) Frequency distribution of astrocyte clusters in five donors. (C) Heat map illustrates −log10(FDR) of gene set enrichment (hypergeometric test) between the mouse astrocyte cluster marker genes with human astrocyte cluster marker genes. The x-axis lists the astrocyte populations identified in mouse (Batiuk et al., 2020). The y-axis lists the astrocyte clusters identified in this study. (D) Frequency distribution of oligodendrocyte clusters in five donors. (E) UMAP showing reclustering of oligodendrocytes independent of the rest of the dataset. (F) Frequency distribution of separately analyzed oligodendrocytes clusters in five donors. (G) Violin plots of normalized counts for mature and precursor oligodendrocyte marker genes. (H) Heat map illustrates −log10(FDR) of gene set enrichment (hypergeometric test) between the markers for oligodendrocyte types identified in human cortex (Jakel et al., 2019) and the markers for the oligodendrocyte clusters identified in this study. See also Figure S12, S13.
We identified several clusters of oligodendrocytes (Figure 1B) with varying distribution patterns across aHP and pHP (Figure 1E). In contrast to astrocytes, some of the oligodendrocyte clusters appeared to show a donor-specific distribution (Figure 6D), indicating some of these clusters might be driven by gene expression variability across donors. We hypothesized that our initial clustering of the entire dataset was not optimal for oligodendrocytes because of the lower number of genes expressed in oligodendrocytes resulting in overclustering of this cell-type. Thus, we subsetted and reclustered oligodendrocytes. This reclustering yielded seven oligodendrocyte clusters (Oligr1-7) with balanced distribution across the five donors (Figure 6E, F). To understand any differences among the oligodendrocyte clusters, we examined the expression of markers for genes known to be enriched in oligodendrocytes (MOG, MOBP, OPALIN) and committed oligodendrocyte precursors (COPs) (PCDH15, FRMD4A) (Marques et al., 2016) (Figure 6G). Clusters Oligr1 expresses higher levels of COP-enriched genes with less expression of mature oligodendrocyte markers. Oligr7 shows a MOG+/MOBP+/OPALIN-profile indicating a distinct subclass of mature oligodendrocytes. Additionally, we performed a hypergeometric test between our cluster markers and a recently published dataset of human oligodendrocyte subtypes (Jakel et al., 2019) (Figure 6H). Based on this analysis, Oligr1 show strong overlap with COPs. MOG+/MOBP+/OPALIN-Oligr7 showed variable degrees of similarity to the Olig1 and Olig5 subtypes from the cortex, which were characterized as the most mature oligodendrocytes. Thus, we are able to differentiate COPs (Oligr1) from mature oligodendrocytes (Oligr7), and other oligodendrocyte clusters may represent different states of oligodendrocyte maturation.
Similarly, multiple microglia transcriptional states are also evident among three distinct clusters: Micro1, Micro2, and Micro3 (Figure 1B, S13A). Among these, clusters Micro1 and Micro2 were detected in all five donors whereas Micro3 was primarily represented by Donor 1 (Figure S13B). Micro3 is similar to Micro2 based on marker gene expression (Figure S13C), and therefore could represent a similar cell-type with slightly different expression profiles among individuals based on some unknown biological covariate. However, because we do not know what biological factors might have impacted the distinguishing genomic profiles of Micro3, we caution interpreting whether this cluster truly represents a donor-specific biological state or some other gradient of gene expression among microglia. Although the impact of sex-specific differences on gene expression in microglia has been reported (Guneykaya et al., 2018), we detected three microglia subtypes at comparable levels in our male and female donors suggesting that sex-dependent transcriptomic diversity is subtle. Recent reports have transcriptomically grouped microglia in healthy brain into two categories: a resting homeostatic state and a pre-active state with elevated levels of chemokine and cytokine genes (Masuda et al., 2020). All of these three clusters express markers of homeostatic microglia, including P2RY12, and Micro1 expresses higher levels of EGR3, a gene enriched in pre-active state microglia (Masuda et al., 2019) (Figure S13C). Although there are an increasing number of snRNA-seq studies implicating distinct states of microglia (Mathys et al., 2017; Olah et al., 2020), a recent study reported limitations of snRNA-seq in detecting the whole repertoire of activated microglia genes (Thrupp et al., 2020). These two subclusters (Micro2, Micro3) of homeostatic microglia and one subtype of pre-active microglia (Micro1) represent previously unidentified cell-types in the human hippocampus for further investigation into the role of microglia in hippocampal function.
Discussion
Here, we describe the transcriptional and cellular landscape of the human anterior and posterior hippocampus. First, we demonstrate the transcriptional diversity of excitatory neurons in the anterior and posterior hippocampus, highlighting axis-enriched genes. While some of these genes coincide with known mouse axis markers, we also identified and validated axis genes not previously reported in rodents or humans. These genes are linked to various functional categories including ion channels, RNA-binding proteins, calcium signaling and cell-to-cell adhesion. Second, we describe the heterogeneous pool of dentate gyrus granule neurons distributed differentially along the axis and their shared and unique molecular signatures. Third, we identify subtypes of astrocytes and oligodendrocytes likely representing cellular states in the human hippocampus. This dataset provides entry points for understanding the differential brain circuits that the anterior and posterior hippocampus mediate. Moreover, the subtypes and axis-defined subtypes in the hippocampus can be integrated with ongoing efforts in the neocortex to understand shared and distinct developmental programs across the different regions of the human brain.
A necessary caveat to obtaining well-defined regions of the human hippocampus is the use of surgical specimens from patients with temporal lobe epilepsy. However, we show that our results have significant overlap with non-epileptic human datasets for which axis information was unknown (Habib et al., 2017) as well as with rodent datasets, including overlap of genes differentially expressed along the hippocampal axis (although cell type resolution was not generated in such data) (Cembrowski et al., 2016a; Cembrowski et al., 2016b). Further, we specifically excluded any specimens with radiographic or microscopic evidence of changes related to epilepsy (MTS), and the contribution of both anterior and posterior specimens from all individuals allowed us to use a paired design in our analysis. Stated another way, differential gene expression along the hippocampal axis is not a result of different subjects contributing anterior versus posterior specimens. Additionally, we were able to confirm enriched expression of CADPS2 and CA3-specific expression of HS3ST4 detected in our dataset on postmortem (non-epileptic) hippocampal tissue, highlighting that our findings are not solely driven via epileptic status and can give insights into hippocampal organization. While it is not possible to completely eliminate the possible influence of epilepsy-related factors in our dataset, these features of our analysis add confidence that our findings reflect more generalizable patterns. Theoretically, data comparing anterior versus posterior gene expression differences can be obtained from cadaveric specimens with careful dissection and separation of anterior and posterior structures at the time of harvest. However, the characteristic sensitivity of the hippocampus to hypoxic insults, and differential composition of cell type along the axis, raises the concern that the relatively uncontrolled period of perfusion changes occurring around the time of death and before tissue harvesting would introduce unacceptable confounds to longitudinal differences observed (Busl and Greer, 2010; Horstmann et al., 2010). In this regard, our surgical dataset potentially circumvents the technical noise derived from the peri- and postmortem interval associated with autopsy tissue.
Here, we identified the transcriptional diversity of neurons in aHC and pHC. Although several differentially expressed genes are present in DG and CA1 neurons across aHC and pHC, clustering analysis did not segregate neurons in an axis-dependent manner. Individual clusters are determined by cell-type-specific gene expression and contain the given cell-types from both aHC and pHC (Figure 1B, 2A). The fact that the axis-dependent differences are present in excitatory neurons suggests that segregation of functional properties across the axis is governed by the molecular composition of excitatory neurons. Finding genes involved in synaptic function suggests underlying differences in synaptic strength and connectivity.
Our integrative analysis of human GWAS with cell-type and axis-specific gene expression of human hippocampus shows that neurons of the aHP and pHP underlie complex hippocampal functions with variable vulnerability to cognitive and mood disorders. Because temporal lobe epilepsy is frequently associated with autism and/or cognitive function, at a first approximation, it might not be surprising to observe enrichment for disease variants within our hippocampal dataset that is derived from surgically resected tissues. However, none of the individuals who contributed tissue were comorbid for any other disease or cognitive impairment. In addition, a recent dataset derived from postmortem tissues of healthy individuals similarly found enrichment for disease variants within hippocampal cell types (Tran et al., 2020). Thus, our overall finding is independently confirmed and we further refine these enrichments by distinguishing which ones are more prominently associated with the anterior or posterior hippocampus.
Comparison of axis-enriched genes in DG and CA1 neurons with previously published mouse axis-genes shows significant overlap with some axis genes that have been reported in the mouse studies (Figure 3A, 4A, C, D). Similar to our findings in the adult hippocampus regarding DEGs along the long axis, a recent study reported gene expression differences in hippocampal subfields in developing mouse and human hippocampus (Zhong et al., 2020). Collectively, these results suggest that although the hippocampal anatomy and cytoarchitecture are conserved between rodents and primates (Manns and Eichenbaum, 2006), the hippocampus still shows organizational features distinguishing human hippocampus from rodents.
With the increasing number of single-cell RNA-sequencing based atlas efforts, it is critical to compare the results from these studies to come to a consensus regarding the novelty or similarity of cell-classes rather than having such cell types defined by only one study or atlas. Here, we attempted to integrate our data with other mouse and human brain single-cell datasets to assess the reproducibility of glial cell states across studies. Although these data came from different brain regions and species, we observed overlapping trends in subclusters of microglia, oligodendrocytes, and astrocytes substantiating increasing evidence for transcriptomically distinct states of glial cells in the mammalian brain. Future research is needed to determine whether these populations represent distinct glial cell-types or transitional transcriptional states.
While our work sheds light on the cellular and transcriptional diversity of the human hippocampus, there are aspects of the hippocampal transcriptome that are not examined here. First, this study focused on the anterior to posterior poles. Therefore, it lacks the differences at the intermediate locations along the axis as well as the differences along the other organizational dimensions such as the deep and superficial layers of the hippocampus. Second, snRNA-seq technology used here allowed us to perform a gene-level analysis, which misses differentially expressed isoforms present in hippocampal subfields (Farris et al., 2019). Third, our comparison between our excitatory neuronal markers with mouse subfield markers (Cembrowski et al., 2016b) did not show any enrichment for the CA4 subfield (Figure S8B). It is possible that CA4 transcriptional identity is similar to other CA neurons in human. Microdissection of CA subfields prior to snRNA-seq could be informative for more refined CA subfield annotation and identification of CA4 neurons. Last, while our dataset shows agreement with additional human and mouse datasets, highlighting the robustness of our findings, certain aspects of the data such as pre-active microglia signatures could be a result of epileptic states. While pre-active microglia have been defined for unaffected human brain (Masuda et al., 2020), it is hard to disentangle the cause of microglial activation by evaluating steady-state gene expression. These findings could also relate to the fact that we sampled from five individuals and we cannot completely rule out donor-specific contributions from such a limited number of donors. For example, we found that a few clusters such as Den.Gyr3, Olig5, OPC2, and Micro3 were primarily driven be a single donor. We believe that these particular examples could be caused by sampling only a proportion of nuclei from each region as the surgical method for resection was consistent for each subject and we had robust validation from both postmortem human and mouse studies. Similar to microglia, we cannot completely rule out a role for the epileptic status of our tissue driving the observation that cell types such as interneurons are not differentially distributed between the anterior/posterior axis of the human hippocampus. Future studies addressing these limitations will further elucidate additional aspects of hippocampal genomics.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Genevieve Konopka.
Materials availability
This study did not generate new unique reagents.
Data and code availability
The single-nuclei RNA-sequencing data reported in this paper can be accessed at NCBI GEO (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE160189) with the accession number GSE160189. Code that was used to perform data pre-processing, clustering and differential gene expression analysis is available at GitHub repository (https://github.com/konopkalab/10x_scRNAseqHippoAxisSeq). The data can also be accessed through an interactive R shiny application at the Github repository (https://github.com/konopkalab/10x_scRNAseq_HippoAxisSeq/tree/main/Shiny_App) and the interactive website (https://human-hippo-axis.cells.ucsc.edu/).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human surgical tissue
All hippocampal tissues in this study were obtained from patients with temporal lobe epilepsy with the informed consent of patients and approval of the UTSW Institutional Review Board (IRB). The surgical technique involved initial resection of the lateral temporal cortex to expose the hippocampus after opening the temporal horn of the ventricle. Following division of the amygdalo-hippocampal sulcus, the hippocampus was reflected inferiorly on its vascular pedicle until the fibria and adjacent choroidal fissure were visualized. After opening the alveus just lateral to the fimbria, an anterior-posterior line of dissection was carried along the hippocampal axis until the arteries feeding the hippocampus were identified and sequentially divided, freeing the hippocampus from its pedicle in an en bloc fashion. The tail of the hippocampus was severed posterior to the lateral geniculate nucleus in standard fashion. Only specimens greater than 3.5 cm in length in the AP dimension were included in the analysis. An approximately 0.5 cm segment was removed from the anterior and posterior poles of this specimen for inclusion in the study; the mid portion was sent for pathological analysis. Anterior specimens were taken from a location anterior to the uncal notch, following previously described demarcation (Poppenk and Moscovitch, 2011). Each specimen was dropped into ice-cold Neurobasal A Medium (Invitrogen, #10888-022) after removal from the patient. The tissue was transported from the operation room to the lab space within 20 mins, washed once in 1X PBS and flash-frozen in liquid nitrogen. All samples were evaluated for signs of pathology including mesial temporal sclerosis (MTS) by expert neuropathology review. Only samples lacking such pathology were included in this study. Demographic information and pathology reports are listed in Table S1.
Human autopsy tissue
10-μm cryosections of human postmortem anterior and posterior hippocampus samples were provided by the University of Texas Neuropsychiatry Research Program (Dallas Brain Collection). Surgical coordinates described above were used to dissect postmortem anterior and posterior hippocampus. Demographic information is listed in Table S1.
Sample size estimation
No statistical methods were used to pre-determine samples sizes because of the limited availability of human brain surgical tissues.
Allocation of samples to groups
Samples were not randomized or assigned to groups in any pre-determined manner. All samples that fit our quality criteria were included. Assignment of “anterior” or “posterior” notation was based on the neuroanatomical region from which the sample derived.
Effects of sex
We removed sex chromosomes from our analysis and included sex as a covariate in the linear mixed model for determining differential gene expression. We therefore did not observe significant effects of sex on the data.
METHOD DETAILS
Nuclei Isolation
Nuclei were isolated as previously described (Habib et al., 2017) https://www.protocols.io/view/rapid-nuclei-isolation-from-human-brain-scpeavn. Surgically resected cortical tissue was homogenized using a glass Dounce homogenizer in 2 ml of ice-cold Nuclei EZ lysis buffer (#EZ PREP NUC-101, Sigma) and was incubated on ice for 5 min. Nuclei were centrifuged at 500 × g for 5 min at 4 °C, washed with 4 ml ice-cold Nuclei EZ lysis buffer and, incubated on ice for 5 min. Nuclei were centrifuged at 500 × g for 5 min at 4 °C. After centrifugation, the nuclei were resuspended in 500 μl of nuclei suspension buffer (NSB) consisting of 1XPBS, 1%BSA (#AM2618, Thermo Fisher Scientific) and 0.2U/ul RNAse inhibitor (#AM2694, Thermo Fisher Scientific) and were filtered through a 40-μm Flowmi Cell Strainer (#H13680-0040, Bel-Art). Debris was removed with a density gradient centrifugation using the Nuclei PURE 2M Sucrose Cushion Solution and Nuclei PURE Sucrose Cushion Buffer from Nuclei PURE Prep Isolation Kit (#NUC201-1KT, Sigma Aldrich). Nuclei PURE 2M Sucrose Cushion Solution and Nuclei PURE Sucrose Cushion Buffer were first mixed in a 9:1 ratio. 500 μl of the resulting sucrose buffer was added to a 2 ml Eppendorf tube. 900 μl of the sucrose buffer was added to 500 μl of isolated nuclei in NSB. 1400 μl nuclei suspension was layered to the top of the sucrose buffer. This gradient was centrifuged at 13, 000 x g for 45 min at 4 °C. Nuclei pellet was resuspended, washed once in NSB and, filtered through a 40-μm Flowmi Cell Strainer (#H13680-0040, Bel-Art). Nuclei concentration was determined using 0.4% Trypan Blue (#15250061, Thermo Fisher Scientific). A final concentration of 1000 nuclei/μl was adjusted with NSB.
Droplet-based snRNA-seq
Droplet-based single-nuclei RNA-seq libraries were prepared using the Chromium Single Cell 3’ v2 or v3 (#120237, #1000153, 10x Genomics) according to the manufacturer’s protocol (Zheng et al., 2017). Libraries were sequenced using an Illumina NovaSeq 6000.
Sequence Alignment and Counting
Reads were aligned to the hg19-1.2.0 using CellRanger software (v.3.0.2) (10x Genomics). Because the nuclear transcriptome contains unspliced transcripts, reads mapping to a pre-mRNA reference file were counted.
Clustering Analysis
We used the R package Seurat (v3.0.1) and custom scripts to identify individual clusters. Cells with >300 genes, <10000 UMIs, and <5% mitochondrial transcripts were retained for downstream analysis. Genes located on the sex chromosomes and mitochondrial genes were removed. Potential doublets were removed using the DoubletFinder program (McGinnis et al., 2019). We merged anterior and posterior libraries obtained from a single donor. Each donor-specific merged dataset was log normalized with a scale factor of 10,000 using NormalizeData, and the top 2000 variable genes were identified with FindVariableGenes. Individual datasets were then combined using IntegrateData. We scaled the integrated datasets by regressing for UMI number, percent mitochondrial transcripts, batch, age, sex, epilepsy duration, and version of 10X chemistry. Based on JackStraw analysis, we selected principal components (PCs) 1:25 based on Elbow Plot shows the ranking of principle components based on the percentage of variance explained by each one (Figure S14), a resolution of 0.6 for Louvain clustering and UMAP. Two clusters from this clustering were removed from final analysis due to unknown identity and small number of cells (171 cells) (Figure S15). Cluster marker genes were identified using FindAllMarkers using Wilcoxon Rank Sum test with the following parameters: genes expressed at least minimum percentage of cells = 0.25, log fold change threshold = 0.25. For more refined neuronal clustering, cells belonging to RBFOX3+ clusters were separated and re-clustered using a resolution of 0.4 and PCs 1:64 based on significance of PCs shown in JackStraw Plot (Figure S16A). Three clusters from this clustering were removed from final analysis due to mixed identity with small number of cells (Figure S17). Detailed information, methods, and analysis are available on the GitHub repository (https://github.com/konopkalab/10x_scRNAseq_HippoAxisSeq). Processed data is also available as an interactive website (https://human-hippo-axis.cells.ucsc.edu/). For more refined inhibitory neuronal clusters, cells belonging to GAD1-positive interneurons in Figure 2 were separated and re-clustered using a resolution of 0.4 and PCs 1:10. Cells belonging to oligodendrocyte clusters in Figure 1 were separated and re-clustered using a resolution of 0.4 and PCs 1:10. Normalized counts calculated by the NormalizeData function were used for plotting marker gene expression in violin plots. Although we make reference to the relative distance between clusters within the UMAP plots, we note that such assessments are with regards to global distances with low-dimensional embeddings and such assessments should be treated with appropriate caution. Excitatory and inhibitory neuronal clusters were annotated based on marker gene expression (Figure 2D and Figure S10B) as well as hypergeometic enrichment of cluster markers we identified (Table S4 and S5) with previous mouse and human datasets (Figure S8B, S10C, and S10D).
Differential Gene Expression Analysis
To identify differentially expressed genes in anterior and posterior hippocampal samples in each cell type and cluster, MAST (v1.8.2) (Finak et al., 2015) was used to perform zero-inflated regression analysis by fitting a linear mixed model (LMM) as described previously (Velmeshev et al., 2019). LMM included age, sex, 10X chemistry version, epilepsy duration, batch, seizure frequency, mitochondrial gene percentage, and gene detection rate (cngeneson). Gene expression variance explained by each of these covariates in the entire dataset and per cluster is reported in Figure S18A and B.
zlm(“~axis + (1|ind) + cngeneson + age + sex + dur + mito_perc +version +batch +freq, method=”glmer”, ebayes=F, silent=T)
A likelihood ratio test (LRT) was performed by comparing the model with and without axis information. Genes expressed in at least 25% of tested cells with a log2 fold change of expression of at least ± 0.3 and FDR<0.05 were selected as differentially expressed (Anderson et al., 2020).
Because Gra.Neu1 cluster had very low number of cells from anterior hippocampus, we did not calculate and report DEGs for this cluster. LMM did not perform with Gra.Neu3 due to the low number of cells with certain covariates.
Functional Annotation of Differentially Expressed Genes
The functional annotation of differentially expressed genes was performed using ToppGene Suite (Chen et al., 2009). GO categories with Benjamini-Hochberg FDR<0.05 were summarized using REVIGO (Supek et al., 2011). Semantic similarity analysis for GO terms was performed using GOSemSim (Schlicker et al., 2006) based on Relevance method (Rel).
GWAS data and enrichment
We downloaded summary statistics for GWAS on risk and cognitive traits and non-brain disorders from Psychiatric Genomics Consortium and GIANT Consortium (Grove et al., 2019), (Jansen et al., 2019), (Bipolar et al., 2018; Davies et al., 2018; Estrada et al., 2012; Hoffmann et al., 2018; International League Against Epilepsy Consortium on Complex, 2018; Lee et al., 2018; Martin et al., 2018; Morris et al., 2012; Savage et al., 2018; Schunkert et al., 2011; Sohail et al., 2019; Wray et al., 2018). We used MAGMA (v1.07) (de Leeuw et al., 2015) for genome-wide gene-based association analysis. MAGMA statistics and −log10(FDR) are reported in Table S10 for each of the GWAS data analyzed. We used the 19,346 protein-coding genes from human gencode v19 as background for the gene-based association analysis. SNPs were selected within exonic, intronic, and UTR regions as well as SNPs within 10kb up/down-stream the protein-coding gene. SNP association revealed 18,988 protein-coding genes with at least one SNP. Gene based association tests were performed using linkage disequilibrium between SNPs. Beta value (effect size) from a linkage disequilibrium model was calculated. Benjamini-Hochberg correction was applied and significant enrichment is reported with a cut-off of FDR < 0.05. GWAS acronyms were used for the figures (ADHD = attention deficit hyperactivity disorder, ASD = autism spectrum disorders, AD = Alzheimer’s disease, BD = bipolar disorder, MDD = major depressive disorder, SZ = schizophrenia, Intelligence = Intelligence, CognFunc = cognitive functions, BMI = body mass index, CHD = coronary artery disease, DIAB = diabetes, HGT = height, OSTEO = osteoporosis).
Bulk RNA-seq differential gene expression analysis
We downloaded the count matrix for the mouse Hipposeq (Cembrowski et al., 2016b) dataset from (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE74985). DESeq2 (v1.20.0) (Love et al., 2014) was used for differential gene expression across subfields in this dataset. Genes were considered for differential expression analysis if the total FPKM was ≥ 10 across all samples. A stepwise comparison was performed by comparing DG versus CA, CA4 vs CA1/2/3, CA1 vs CA2/3, CA2 vs CA3. Genes with a log2 fold change of expression of at least ± 0.3 and FDR<0.05 were selected as differentially expressed. The mouse gene symbols were converted to the corresponding human gene symbols using the biomaRt (v2.38.0) (Durinck et al., 2009) package for comparison to our human dataset.
Hypergeometric overlap tests
The following hypergeometric overlap test function was used to assess the significance of the overlaps between human and mouse axis markers and subfield annotation.
library(gmp)
enrich_pvalue < - function(N, A, B, k)
{
m < - A + k
n < - B + k
i < - k:min(m,n)
as.numeric(sum(chooseZ(m,i)*chooseZ(N-m,n-i))/chooseZ(N,n))
}
enrich_pvalue(N,A,B,k)
The number of expressed genes in our neuronal dataset (16,932) was used for the background gene number for all hypergeometric overlap tests.
Immunohistochemistry
10-μm cryosections of human postmortem hippocampus were used for confirmatory immunohistochemistry. Heat induced antigen retrieval was performed by incubating the sections in citrate buffer (pH6.0) for 10 min at 95 C. Sections were blocked with 2% fetal bovine serum (FBS) in 0.1M Tris (pH 7.6) for 1 hour at room temperature. The sections were incubated with primary antibodies in 0.1M Tris pH 7.6/2% FBS overnight at 4C and subsequently incubated with secondary antibodies in 0.1M Tris pH pH 7.6/2% FBS for 1 hour at room temperature. Sections were immersed in 0.25% Sudan Black solution to quench lipofuscin auto-fluoresce and counterstained with 4′-6-diamidino-2-phenylindole (DAPI). The tissue was mounted and cover slipped using ProLong Diamond Antifade Mountant (#P36970, Thermo Fisher Scientific). The antibodies and working dilutions were as follows: rabbit α-CADPS2 (ab69794, Abcam, 1:100), mouse α-HS3ST4 (MA524332, Thermo Fisher Scientific, 1:80), species-specific secondary antibodies produced in donkey and conjugated to Alexa Fluor 488 and Alexa Fluor 555 (Thermo Fisher Scientific, 1:800). Images were captured using a Zeiss LSM 880 confocal microscope at the UT Southwestern Neuroscience Microscopy Facility. Hippocampal subfields were defined based on the DAPI staining highlighting the distinct organization of the dentate gyrus. For α-CADPS2 staining quantification, Z-stack images were acquired using ×20 magnification from n=3 individuals with the following settings: laser intensity: 1.1%, z-step size=0.2μm, pinhole=0.71 Airy unit, gain=707.0, detector offset=0, detector digital gain=1.0. Maximum intensity projection images were generated using 50 slices and used for measuring fluorescence intensity of the individual cells (n=39 for posterior, n=53 for posterior) using Image J (Schneider et al., 2012). Corrected total cell fluorescence was calculated by subtracting the background signal neighboring individual cells. Statistical significance was assessed by an unpaired t test on the average corrected total cell fluorescence for each individual using the software package Prism 5 (GraphPad Software Inc).
Statistical analysis of anterior:posterior cell abundance
Total number of cells were defined by cell identifier and donor. To calculate the differential abundance we applied a robust linear mixed model using the R library robustlmm. This method takes into account the inflation in the model estimates by outliers.
The model is as follows:
formula = log2(total_n_cell) ~ Axis*Cell + (1\donor)
rlmer(formula = formula, method=“DASvar”, data = Data)
R library emmeans was used to calculate the marginal means after contrast. Resultant p-values were corrected with the Benjamini-Hochberg false discovery rate. Code for this analysis is available in GitHub.
QUANTIFICATION AND STATISTICAL ANALYSIS
For snRNA-seq transcriptomic data the distribution was assumed to be normal but this was not formally tested. Non-parametric tests have been used to avoid uncertainty when possible. The methods for differential gene expression using linear mixed modeling in MAST software package are detailed in the Differential Gene Expression section. The results of differential gene expression analyses are listed in Table S6 and S7 and subsets of these comparisons are included in Figures 2G, 2H, 3B, 4B, and S12.
For statistical analysis of IHC quantification, three autopsy specimens for both anterior and posterior hippocampus were analyzed. The details regarding autopsy specimens used are listed in Table S1 and the number of cells analyzed for each group across these specimen is listed in the immunochemistry methods section. We first tested the normality of average intensity values derived from each specimen (n=3) using a Sharpio-Wilk test (data not shown) and software package Prism 5 (GraphPad Software Inc). Because this test shows that the average intensity values were normally distributed for both anterior and posterior samples, a t-test was used. The corresponding results section and the legend for Figure 3C include the results of this analysis.
Statistical analysis of anterior:posterior cell abundance was performed on n=5 anterior and posterior samples listed in Table S1. R packages robustlmm and emmeans were used and the results are shown in Figures 1E, S4, S5 and Table S3.
To calculate enrichment of overlapping datasets, hypergeometric tests were used. A Benjamini-Hochberg adjusted p-value was applied as a multiple comparisons adjustment. The results of these tests are shown in Figures 3A, 4A, 4C, 4D, 6C, 6H, S8, S9, S10, S11 and Table S9.
For gene ontology enrichments, a one-sided hypergeometric test was used to test overrepresentation of functional categories. A Benjamini-Hochberg adjusted p-value was applied as a multiple comparisons adjustment, and the results are shown in Figure S11 and Table S8.
ADDITIONAL RESOURCES
The data can also be accessed through an interactive R shiny application at the Github repository: https://github.com/konopkalab/10x_scRNAseq_HippoAxisSeq/tree/main/Shiny_App
The data can also be browsed through an interactive website: https://human-hippo-axis.cells.ucsc.edu/
Supplementary Material
Figure S1 (Related to STAR Methods). Magnetic resonance imaging (MRI).
Outer panels show sagittal plane T1 magnetic resonance images for the subjects contributing hippocampal tissue to the analysis. The location of anterior (orange) and posterior (blue) specimens obtained along the hippocampal axis are highlighted. Line segments represent approximately 3 cm of distance on the included images. The cartoon image shows a coronal section of the human brain indicating the position of the uncal apex, the posterior extent of which was used as a demarcation point for the anterior hippocampus. This structure can be identified relative to the oculomotor nerve and posterior cerebral artery in the ambient cistern, as shown in the cartoon.
Figure S2 (Related to Figure 1). Data quality.
(A) Bar chart showing the number of nuclei sampled from each donor.
(B) Violin plots showing the distribution of the number of genes in glial and neuronal cells separated by 10X Genomics chemistry versions used (v2 or v3).
(C) Violin plot showing the distribution of the number of transcripts in glial and neuronal cells separated by 10X Genomics chemistry version used.
(D) Bar charts showing the distribution of anterior and posterior nuclei in cell-type identified.
(E) Distribution of nuclei obtained from individual donors.
Figure S3 (Related to Figure 1). Integration of published human hippocampus dataset with our data.
(A) UMAP projections showing the clustering of a subset of our data combined with a published human hippocampus dataset (Habib et al., 2017) labeled by cluster annotations (left) and dataset (right).
(B) Bar plots showing the distribution of cells from our study and Habib for each cluster.
Figure S4 (Related to Figure 1). Anterior vs. posterior distribution in clusters.
Boxplots depicting the anterior:posterior cell proportions for each cluster. Each data point is a donor. Colors correspond to the axis. **P < 0.01, ***P < 0.001, Robust generalized mixed model. Clusters showing anterior or posterior enrichment driven by single donor (i.e Olig5, OPC2, Den.Gyr3) were not considered significant. Pyr=Pyramidal neurons, Den.Gyr=dentate gyrus neurons, In=Interneurons, Endo=endothelial cells, Micro=microglia, Astro=astrocytes, OPCs=oligodendrocyte progenitor cells, and Olig=oligodendrocytes.
Figure S5 (Related to Figure 1). Statistics for anterior vs. posterior distribution in clusters.
Volcano plot depicting the cell type differential abundance between axis. X-axis corresponds to the robust mixed model estimates. Y-axis corresponds to the −log10(p-value). Pyr=Pyramidal neurons, Den.Gyr=dentate gyrus neurons. Clustered labeled driven by a single (OPC2, Olig5, and Den.Gyr3) are labeled gray.
Figure S6 (Related to Figure 2). Neuronal clusters by donors.
UMAP plots showing neuronal clusters separated by corresponding donor tissue with cell type annotations. Gra.Neu=Granule Neurons, In.Neu=Inhibitory neurons, and Sub=subiculum.
Figure S7 (Related to Figure 2). Identification and validation of neuronal cluster markers.
(A) Dot plots showing the top 3 cluster markers in excitatory neuronal clusters.
(B) Dot plots showing the top 5 cluster markers in inhibitory neuronal clusters.
(C) Immunohistochemistry showing HS3ST4 expression in CA3 subfield of human postmortem hippocampus. Blue color indicates the nuclear stain DAPI and red color shows α-HS3ST4 staining detected with a fluorophore conjugated antibody.
Figure S8 (Related to Figure 2). Annotation of excitatory clusters.
(A) Dendrogram depicting the hierarchical structure of gene expression of the hippocampal subfields in the Hipposeq dataset (Cembrowski et al., 2016b).
(B) Hypergeometric overlaps of excitatory neuronal clusters with mouse subfield markers identified via bulk RNA-seq (Cembrowski et al., 2016b). Heat map illustrates the −log10(FDR) of gene set enrichment between the markers of the human excitatory neuronal clusters and the gene set lists distinguishing CA vs DG, CA4 vs CA1/2/3, CA1 vs CA2/3, and CA2 vs CA3 in mouse. The x-axis lists the excitatory neuronal clusters and y-axis lists the mouse subfields.
Figure S9 (Related to Figure 2). Human and mouse DG populations.
(A) UMAP plot of div-seq (Habib et al., 2016) dentate gyrus neuronal cells colored by cluster identities.
(B) Violin plots showing normalized counts for Cck and Penk showing DG populations identified by Erwin et al. (Erwin et al., 2020). (C) Heatmap showing −log10(FDR) from a hypergeometric enrichment test for the overlaps between the marker genes for DG granule neuron clusters identified in our human dataset study and the marker genes div-seq DG granule neuron clusters.
Figure S10 (Related to Figure 2). snRNA-seq reveals hippocampal GABAergic interneurons.
(A) UMAP plot of inhibitory neuronal cells colored by cluster identities and cell-type annotations. (B) Violin plots showing normalized counts for interneuron markers. (C) Heatmap showing −log10(FDR) from a hypergeometric enrichment test for the overlaps between the marker genes for inhibitory neuronal clusters identified in this study and the marker genes for inhibitory neuronal clusters identified in human autopsy brains by Habib et al. (Habib et al., 2017). (D) Heatmap showing −log10(FDR) from a hypergeometric enrichment test for the overlaps between the marker genes for inhibitory neuronal clusters identified in this study and the marker genes for inhibitory neuronal clusters identified in mouse hippocampus by Habib et al. (Habib et al., 2017).
Figure S11 (Related to Figure 3). Gene ontology analysis of CA1-specific DEGs.
Bubble chart showing the top gene ontology categories for genes with higher expression in posterior (left) and anterior (right) hippocampus. On the x-axis is the −log10(FDR) and on the y-axis is the number of genes per category. BP (blue)=biological function; MF (gray)=molecular function.
Figure S12 (Related to Figure 6). Glial specific DEGs across anterior and posterior hippocampus.
Upset plot showing the overlap of upregulated or downregulated DEGs in glial cell-types across aHP and pHP (adj. p-value<0.05, log2FC>0.3, percentage>25). Horizontal bars show the number of genes enriched in posterior (purple) and anterior (gray) in each cell type. Vertical black bars show the number of overlapping DEGs across the glial cell types. Oligo.=Oligodendrocytes
Figure S13 (Related to Figure 6). Microglia gene expression in human hippocampus.
(A) Heatmap illustrating the average expression of the top 5 DEGs between microglia clusters. Color scheme corresponds to log-normalized and scaled values for average gene expression for each cluster
(B) Frequency distribution of microglia clusters in five donors. (C) Violin plots showing the expression of homeostatic and pre-active microglia.
Figure S14 (Related to STAR Methods). JackStraw and Elbow Plots of the entire dataset.
(A) Scatter plot showing JackStraw statistics for each of the principal component (1-100) for PCA significance for dataset shown in Figure 1.
(B) Scree plot (Elbow Plot) showing the ranking of principle components based on the percentage of variance explained by each one for dataset shown in Figure 1. Arrow shows flattening around PC 25.
Figure S15 (Related to STAR Methods). Filtered clusters from entire dataset.
UMAP plot showing clustering the entire dataset with removed clusters labeled (left). UMAP plot final clustering after removal of two clusters (right). Py.Neu=Pyramidal neurons, Den.Gyr=dentate gyrus neurons, In=Interneurons, Astro=Astrocytes, Endo=endothelial cells, Micro=microglia, OPCs=oligodendrocyte progenitor cells, and Olig=oligodendrocytes.
Figure S16 (Related to STAR Methods). JackStraw and Elbow Plots of the neuronal cells.
(A) Scatter plot showing JackStraw statistics for each of the principal component (1-100) for PCA significance for dataset shown in Figure2.
(B) Scree plot (Elbow Plot) showing the ranking of principle components based on the percentage of variance explained by each one for dataset shown in Figure 2.
Figure S17 (Related to STAR Methods). Filtered neuronal clusters.
UMAP plot showing neuronal clustering with removed clusters labeled (left). UMAP plot final clustering after removal of three clusters (right). Gra.Neu=Granule Neurons, In=Inhibitory neurons, and Sub=subiculum.
Figure S18 (Related to STAR Methods). Variance explained by covariates.
(A) Barplot depicting the variance explained by each covariate. Y-axis represent the variance explained weighted across the first 10 PCs.
(B) Barplot depicting the variance explained by each covariate for each cluster. Clusters defined by one subject (Olig5, Den.Gyr3, OPC2, Micro3) were removed from the analysis. Y-axis represent the variance explained weighted across the first 10 PCs.
Table S1. Demographics of human tissue with the number of cells sampled and reads, PMI (Postmortem interval), and RIN (RNA integrity value), Related to STAR Methods
Table S3. Robust linear mixed model statistics for anterior:posterior, Related to Figure 1
Table S5. Cluster markers of inhibitory neuronal cells (Figure S9A), Related to Figure S9
Table S6. Cluster-specific differential gene expression, Related to Figure 2
Table S7. Cell-type specific differential gene expression, Related to Figure 2
Table S10. MAGMA statistics, Related to Figure 5
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse α-HS3ST4 antibody | Thermo Fisher Scientific | Cat# MA524332 |
| Rabbit α-CADPS2 | Abcam | Cat# ab69794 |
| Alexa Fluor 555 Donkey Anti-Mouse IgG | Thermo Fisher Scientific | Cat# A-31570 |
| Alexa Fluor 488 Donkey Anti-Rabbit IgG | Thermo Fisher Scientific | Cat# A-21206 |
| Biological Samples | ||
| Human hippocampal surgical specimens | Table S1 | N/A |
| Human hippocampal autopsy specimens | Table S1 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Nuclei EZ lysis buffer | Sigma | Cat# NUC-101 |
| UltraPure™ BSA (50 mg/mL) | Thermo Fisher Scientific | Cat #AM2618 |
| SUPERase•In™ RNase Inhibitor (20 U/μL) | Thermo Fisher Scientific | Cat #AM2694 |
| Nuclei PURE Prep Isolation Kit | Sigma | Cat# NUC201-1KT |
| Trypan Blue Solution, 0.4% | Thermo Fisher Scientific | Cat#15250061 |
| ProLong Diamond Antifade Reagent with DAPI | Life Technologies | Cat#P36971 |
| Critical Commercial Assays | ||
| Chromium Single Cell 3’ v2 | 10x Genomics | Cat#120237 |
| Chromium Single Cell 3’ v3 | 10x Genomics | Cat#1000153 |
| Deposited Data | ||
| Axis-specific hippocampus snRNA-seq data | This paper | GEO: GSE160189 |
| Human hippocampus snRNA-seq data | Habib et al., 2017 | https://www.gtexportal.org/home/datasets |
| Mouse Hipposeq RNA-seq data | Cembrowski et al., 2016b | GEO: GSE74985 |
| DEGs across mouse dorsal-ventral axis | Cembrowski et al., 2016a, Table S2 | N/A |
| Human cortical oligodendrocytes | Jakel et al., 2019, Supplementary Table 4 | N/A |
| Mouse cortex and hippocampus astrocytes |
Batiuk et al., 2020 Supplementary Data 2 |
N/A |
| Mouse DG axis DEGs in standard housing and enriched environment |
Zhang et, al. 2018 Supplementary Data 2 |
N/A |
| Mouse hippocampus div-seq data | Habib et al., 2016 | https://singlecell.broadinstitute.org/single_cell |
| Mouse interneuron markers |
Habib et al., 2017 Supplementary Table 5 |
N/A |
| Human autopsy interneuron markers |
Habib et al., 2017 Supplementary Table 9 |
N/A |
| Software and Algorithms | ||
| Adobe Illustrator 18.0.0 | Adobe | https://helpx.adobe.com/illustrator/user-guide.html/illustrator/system-requirements.ug.html |
| CellRanger v.3.0.2 | 10x Genomics | https://www.10xgenomics.com/solutions/single-cell/ |
| R version 3.5.2 | The R Project | https://www.r-project.org/ |
| Seurat_3.0.1 | Stuart et al., 2019 | https://github.com/satijalab/seurat |
| Toppgene | Chen et al., 2009 | https://toppgene.cchmc.org/ |
| MAST_1.8.2 | Finak et al., 2015 | https://github.com/RGLab/MAST/ |
| DESeq2_1.20.0 | Love et al., 2014 | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| biomaRt_2.38.0 | Durinck et al., 2009 | https://www.bioconductor.org/packages/release/bioc/html/biomaRt.html |
| REVIGO | Supek et al., 2011 | http://revigo.irb.hr/ |
| MAGMA v1.0735 | de Leeuw et al., 2015 | https://ctg.cncr.nl/software/magma |
| DoubletFinder_2.0.2 | McGinnis et al., 2019 | https://github.com/chris-mcginnis-ucsf/DoubletFinder |
| Image J | Schneider et al., 2012 | https://imagej.nih.gov/ij/download.html |
| GitHub repository of all code for data pre-processing, clustering and differential gene expression analysis | This manuscript | https://github.com/konopkalab/10x_scRNAseq_HippoAxisSeq |
| Other | ||
| Shiny app that permits exploration of the dataset | This manuscript | https://github.com/konopkalab/10x_scRNAseq_HippoAxisSeq/tree/main/Shiny_App |
| Interactive web site linked to UCSC single cell browser | This manuscript | https://human-hippo-axis.cells.ucsc.edu/ |
Highlights.
Analysis of single-nuclei transcriptomes along human hippocampal longitudinal axis
Excitatory neurons show transcriptional heterogeneity across the axis
Cell-type and axis-specific enrichment of disease associated genes
Acknowledgments
The authors thank the patients for consenting to participate in this research and Dr. Carol Tamminga and Kelly Gleason for providing the autopsy tissue. We also thank the UT Southwestern Neuroscience Microscopy Facility for providing imaging resources and Melissa Logies for generating the cartoon in Figure S1. G.K. is a Jon Heighten Scholar in Autism Research at UT Southwestern. This work was supported by the NIH (NS106447), a UT BRAIN Initiative Seed Grant (366582), the Chilton Foundation, and the National Center for Advancing Translational Sciences of the NIH under the Center for Translational Medicine’s award number UL1TR001105 to G.K. and B.L., the NIH (T32DA007290 and T32HL139438) to F.A., the NIH (NS107357) to B.L., and the Chan Zuckerberg Initiative, an advised fund of Silicon Valley Community Foundation (HCA-A-1704-01747) and the James S. McDonnell Foundation 21st Century Science Initiative in Understanding Human Cognition – Scholar Award (220020467) to G.K.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest
The authors declare no competing interests.
References
- Anderson AG, Kulkarni A, Harper M, and Konopka G (2020). Single-Cell Analysis of Foxp1-Driven Mechanisms Essential for Striatal Development. Cell Rep 30, 3051–3066 e3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batiuk MY, Martirosyan A, Wahis J, de Vin F, Marneffe C, Kusserow C, Koeppen J, Viana JF, Oliveira JF, Voet T, et al. (2020). Identification of region-specific astrocyte subtypes at single cell resolution. Nat Commun 11, 1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertelsen EN, Larsen JT, Petersen L, Christensen J, and Dalsgaard S (2016). Childhood Epilepsy, Febrile Seizures, and Subsequent Risk of ADHD. Pediatrics 138. [DOI] [PubMed] [Google Scholar]
- Bipolar D, Schizophrenia Working Group of the Psychiatric Genomics Consortium. Electronic address, d.r.v.e., Bipolar, D., and Schizophrenia Working Group of the Psychiatric Genomics, C. (2018). [Google Scholar]
- Genomic Dissection of Bipolar Disorder and Schizophrenia, Including 28 Subphenotypes. Cell 173, 1705–1715 e1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldrini M, Fulmore CA, Tartt AN, Simeon LR, Pavlova I, Poposka V, Rosoklija GB, Stankov A, Arango V, Dwork AJ, et al. (2018). Human Hippocampal Neurogenesis Persists throughout Aging. Cell Stem Cell 22, 589–599 e585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner MF, and Price AR (2013). Where is the anterior temporal lobe and what does it do? J Neurosci 33, 4213–4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brainstorm C, Anttila V, Bulik-Sullivan B, Finucane HK, Walters RK, Bras J, Duncan L, Escott-Price V, Falcone GJ, Gormley P, et al. (2018). Analysis of shared heritability in common disorders of the brain. Science 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubb EJ, Kinnavane L, and Aggleton JP (2017). Hippocampal - diencephalic - cingulate networks for memory and emotion: An anatomical guide. Brain Neurosci Adv 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busl KM, and Greer DM (2010). Hypoxic-ischemic brain injury: pathophysiology, neuropathology and mechanisms. NeuroRehabilitation 26, 5–13. [DOI] [PubMed] [Google Scholar]
- Buzsaki G, and Moser EI (2013). Memory, navigation and theta rhythm in the hippocampal-entorhinal system. Nat Neurosci 16, 130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cembrowski MS, Bachman JL, Wang L, Sugino K, Shields BC, and Spruston N (2016a). Spatial Gene-Expression Gradients Underlie Prominent Heterogeneity of CA1 Pyramidal Neurons. Neuron 89, 351–368. [DOI] [PubMed] [Google Scholar]
- Cembrowski MS, Wang L, Lemire AL, Copeland M, DiLisio SF, Clements J, and Spruston N (2018). The subiculum is a patchwork of discrete subregions. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cembrowski MS, Wang L, Sugino K, Shields BC, and Spruston N (2016b). Hipposeq: a comprehensive RNA-seq database of gene expression in hippocampal principal neurons. Elife 5, e14997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Bardes EE, Aronow BJ, and Jegga AG (2009). ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 37, W305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K, Lisa Bagen, Linley Robinson, Gray Umbach, Michael Rugg, Bradley Lega (2020). Longitudinal differences in human hippocampal connectivity during episodic memory processing. Cerebral Cortex. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado-Torres L, Burke EE, Peterson A, Shin J, Straub RE, Rajpurohit A, Semick SA, Ulrich WS, BrainSeq C, Price AJ, et al. (2019). Regional Heterogeneity in Gene Expression, Regulation, and Coherence in the Frontal Cortex and Hippocampus across Development and Schizophrenia. Neuron 103, 203–216 e208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies G, Lam M, Harris SE, Trampush JW, Luciano M, Hill WD, Hagenaars SP, Ritchie SJ, Marioni RE, Fawns-Ritchie C, et al. (2018). Study of 300,486 individuals identifies 148 independent genetic loci influencing general cognitive function. Nat Commun 9, 2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leeuw CA, Mooij JM, Heskes T, and Posthuma D (2015). MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol 11, e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolorfo CL, and Amaral DG (1998). Entorhinal cortex of the rat: organization of intrinsic connections. J Comp Neurol 398, 49–82. [DOI] [PubMed] [Google Scholar]
- Dong HW, Swanson LW, Chen L, Fanselow MS, and Toga AW (2009). Genomic-anatomic evidence for distinct functional domains in hippocampal field CA1. Proc Natl Acad Sci U S A 106, 11794–11799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y, Wang SH, Song J, Mironova Y, Ming GL, Kolodkin AL, and Giger RJ (2014). Semaphorin 5A inhibits synaptogenesis in early postnatal- and adult-born hippocampal dentate granule cells. Elife 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, and Huber W (2009). Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 4, 1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erwin SR, Sun W, Copeland M, Lindo S, Spruston N, and Cembrowski MS (2020). A Sparse, Spatially Biased Subtype of Mature Granule Cell Dominates Recruitment in Hippocampal-Associated Behaviors. Cell Rep 31, 107551. [DOI] [PubMed] [Google Scholar]
- Estrada K, Styrkarsdottir U, Evangelou E, Hsu YH, Duncan EL, Ntzani EE, Oei L, Albagha OM, Amin N, Kemp JP, et al. (2012). Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat Genet 44, 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farris S, Ward JM, Carstens KE, Samadi M, Wang Y, and Dudek SM (2019). Hippocampal Subregions Express Distinct Dendritic Transcriptomes that Reveal Differences in Mitochondrial Function in CA2. Cell Rep 29, 522–539 e526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, et al. (2015). MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol 16, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franjic D, Choi J, Skarica M, Xu C, Li Q, Ma S, Tebbenkamp ATN, Santpere G, Arellano JI, Gudelj I, et al. (2020). Molecular Diversity Among Adult Human Hippocampal and Entorhinal Cells. bioRxiv. [Google Scholar]
- Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, Pallesen J, Agerbo E, Andreassen OA, Anney R, et al. (2019). Identification of common genetic risk variants for autism spectrum disorder. Nat Genet 51, 431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guneykaya D, Ivanov A, Hernandez DP, Haage V, Wojtas B, Meyer N, Maricos M, Jordan P, Buonfiglioli A, Gielniewski B, et al. (2018). Transcriptional and Translational Differences of Microglia from Male and Female Brains. Cell Rep 24, 2773–2783 e2776. [DOI] [PubMed] [Google Scholar]
- Habib N, Avraham-Davidi I, Basu A, Burks T, Shekhar K, Hofree M, Choudhury SR, Aguet F, Gelfand E, Ardlie K, et al. (2017). Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat Methods 14, 955–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib N, Li Y, Heidenreich M, Swiech L, Avraham-Davidi I, Trombetta JJ, Hession C, Zhang F, and Regev A (2016). Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science 353, 925–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley DF, and Leasure JL (2012). Region-specific response of the hippocampus to chronic unpredictable stress. Hippocampus 22, 1338–1349. [DOI] [PubMed] [Google Scholar]
- Henke PG (1990). Hippocampal pathway to the amygdala and stress ulcer development. Brain Res Bull 25, 691–695. [DOI] [PubMed] [Google Scholar]
- Hochgerner H, Zeisel A, Lonnerberg P, and Linnarsson S (2018). Conserved properties of dentate gyrus neurogenesis across postnatal development revealed by single-cell RNA sequencing. Nat Neurosci 21, 290–299. [DOI] [PubMed] [Google Scholar]
- Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, Close JL, Long B, Johansen N, Penn O, et al. (2019). Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann TJ, Choquet H, Yin J, Banda Y, Kvale MN, Glymour M, Schaefer C, Risch N, and Jorgenson E (2018). A Large Multiethnic Genome-Wide Association Study of Adult Body Mass Index Identifies Novel Loci. Genetics 210, 499–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horstmann A, Frisch S, Jentzsch RT, Muller K, Villringer A, and Schroeter ML (2010). Resuscitating the heart but losing the brain: brain atrophy in the aftermath of cardiac arrest. Neurology 74, 306–312. [DOI] [PubMed] [Google Scholar]
- International League Against Epilepsy Consortium on Complex, E. (2018). Genome-wide mega-analysis identifies 16 loci and highlights diverse biological mechanisms in the common epilepsies. Nat Commun 9, 5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakel S, Agirre E, Mendanha Falcao A, van Bruggen D, Lee KW, Knuesel I, Malhotra D, Ffrench-Constant C, Williams A, and Castelo-Branco G (2019). Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 566, 543–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, Sealock J, Karlsson IK, Hagg S, Athanasiu L, et al. (2019). Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet 51, 404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez JC, Su K, Goldberg AR, Luna VM, Biane JS, Ordek G, Zhou P, Ong SK, Wright MA, Zweifel L, et al. (2018). Anxiety Cells in a Hippocampal-Hypothalamic Circuit. Neuron 97, 670–683 e676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi T, Tsumori T, Yokota S, and Yasui Y (2006). Topographical projection from the hippocampal formation to the amygdala: a combined anterograde and retrograde tracing study in the rat. J Comp Neurol 496, 349–368. [DOI] [PubMed] [Google Scholar]
- Kjelstrup KG, Tuvnes FA, Steffenach HA, Murison R, Moser EI, and Moser MB (2002). Reduced fear expression after lesions of the ventral hippocampus. Proc Natl Acad Sci U S A 99, 10825–10830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake BB, Chen S, Sos BC, Fan J, Kaeser GE, Yung YC, Duong TE, Gao D, Chun J, Kharchenko PV, et al. (2018). Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol 36, 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JJ, Wedow R, Okbay A, Kong E, Maghzian O, Zacher M, Nguyen-Viet TA, Bowers P, Sidorenko J, Karlsson Linner R, et al. (2018). Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat Genet 50, 1112–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Zhao X, and Gage FH (2004). Defining a molecular atlas of the hippocampus using DNA microarrays and high-throughput in situ hybridization. J Neurosci 24, 3879–3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo-Castro A, and Curatolo P (2014). Epilepsy associated with autism and attention deficit hyperactivity disorder: is there a genetic link? Brain Dev 36, 185–193. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire EA, Burgess N, Donnett JG, Frackowiak RS, Frith CD, and O’Keefe J (1998). Knowing where and getting there: a human navigation network. Science 280, 921–924. [DOI] [PubMed] [Google Scholar]
- Malykhin NV, Lebel RM, Coupland NJ, Wilman AH, and Carter R (2010). In vivo quantification of hippocampal subfields using 4.7 T fast spin echo imaging. Neuroimage 49, 1224–1230. [DOI] [PubMed] [Google Scholar]
- Manns JR, and Eichenbaum H (2006). Evolution of declarative memory. Hippocampus 16, 795–808. [DOI] [PubMed] [Google Scholar]
- Marques S, Zeisel A, Codeluppi S, van Bruggen D, Mendanha Falcao A, Xiao L, Li H, Haring M, Hochgerner H, Romanov RA, et al. (2016). Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science 352, 1326–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin J, Walters RK, Demontis D, Mattheisen M, Lee SH, Robinson E, Brikell I, Ghirardi L, Larsson H, Lichtenstein P, et al. (2018). A Genetic Investigation of Sex Bias in the Prevalence of Attention-Deficit/Hyperactivity Disorder. Biol Psychiatry 83, 1044–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda T, Sankowski R, Staszewski O, Bottcher C, Amann L, Sagar, Scheiwe C, Nessler S, Kunz P, van Loo G, et al. (2019). Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 566, 388–392. [DOI] [PubMed] [Google Scholar]
- Masuda T, Sankowski R, Staszewski O, and Prinz M (2020). Microglia Heterogeneity in the Single-Cell Era. Cell Rep 30, 1271–1281. [DOI] [PubMed] [Google Scholar]
- Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M, De Jager PL, Ransohoff RM, Regev A, and Tsai LH (2017). Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep 21, 366–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis CS, Murrow LM, and Gartner ZJ (2019). DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst 8, 329–337 e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris AP, Voight BF, Teslovich TM, Ferreira T, Segre AV, Steinthorsdottir V, Strawbridge RJ, Khan H, Grallert H, Mahajan A, et al. (2012). Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 44, 981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Garrud P, Rawlins JN, and O’Keefe J (1982). Place navigation impaired in rats with hippocampal lesions. Nature 297, 681–683. [DOI] [PubMed] [Google Scholar]
- Moser E, Moser MB, and Andersen P (1993). Spatial learning impairment parallels the magnitude of dorsal hippocampal lesions, but is hardly present following ventral lesions. J Neurosci 13, 3916–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser MB, and Moser EI (1998). Functional differentiation in the hippocampus. Hippocampus 8, 608–619. [DOI] [PubMed] [Google Scholar]
- Novarino G, Baek ST, and Gleeson JG (2013). The sacred disease: the puzzling genetics of epileptic disorders. Neuron 80, 9–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda H, Tatsumi K, Morita S, Shibukawa Y, Korekane H, Horii-Hayashi N, Wada Y, Taniguchi N, and Wanaka A (2014). Chondroitin sulfate proteoglycan tenascin-R regulates glutamate uptake by adult brain astrocytes. J Biol Chem 289, 2620–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ, Cimpean M, Khairallah A, Coronas-Samano G, Sankowski R, et al. (2020). Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat Commun 11, 6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomero-Gallagher N, Kedo O, Mohlberg H, Zilles K, and Amunts K (2020). Multimodal mapping and analysis of the cyto-and receptorarchitecture of the human hippocampus. Brain Struct Funct 225, 881–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Elliott RC, Pleasure SJ, Barbaro NM, and Lowenstein DH (2006). Aberrant seizure-induced neurogenesis in experimental temporal lobe epilepsy. Ann Neurol 59, 81–91. [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Chittajallu R, Craig MT, Tricoire L, Wester JC, and McBain CJ (2017). Hippocampal GABAergic Inhibitory Interneurons. Physiol Rev 97, 1619–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez JM, Berto S, Gleason K, Ghose S, Tan C, Kim TK, Konopka G, and Tamminga CA (2020). Hippocampal subfield transcriptome analysis in schizophrenia psychosis. Mol Psychiatry. [DOI] [PubMed] [Google Scholar]
- Poppenk J, and Moscovitch M (2011). A hippocampal marker of recollection memory ability among healthy young adults: contributions of posterior and anterior segments. Neuron 72, 931–937. [DOI] [PubMed] [Google Scholar]
- Risold PY, and Swanson LW (1996). Structural evidence for functional domains in the rat hippocampus. Science 272, 1484–1486. [DOI] [PubMed] [Google Scholar]
- Salazar F, Baird G, Chandler S, Tseng E, O’Sullivan T, Howlin P, Pickles A, and Simonoff E (2015). Co-occurring Psychiatric Disorders in Preschool and Elementary School-Aged Children with Autism Spectrum Disorder. J Autism Dev Disord 45, 2283–2294. [DOI] [PubMed] [Google Scholar]
- Savage JE, Jansen PR, Stringer S, Watanabe K, Bryois J, de Leeuw CA, Nagel M, Awasthi S, Barr PB, Coleman JRI, et al. (2018). Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat Genet 50, 912–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlicker A, Domingues FS, Rahnenfuhrer J, and Lengauer T (2006). A new measure for functional similarity of gene products based on Gene Ontology. BMC Bioinformatics 7, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schunkert H, Konig IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, Preuss M, Stewart AF, Barbalic M, Gieger C, et al. (2011). Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet 43, 333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoville WB, and Milner B (1957). Loss of recent memory after bilateral hippocampal lesions. J Neurol Neurosurg Psychiatry 20, 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda Y, Ishii C, Fukazawa Y, Sadakata T, Ishii Y, Sano Y, Iwasato T, Itohara S, and Furuichi T (2016). CAPS1 stabilizes the state of readily releasable synaptic vesicles to fusion competence at CA3-CA1 synapses in adult hippocampus. Sci Rep 6, 31540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small SA, Schobel SA, Buxton RB, Witter MP, and Barnes CA (2011). A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat Rev Neurosci 12, 585–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohail M, Maier RM, Ganna A, Bloemendal A, Martin AR, Turchin MC, Chiang CW, Hirschhorn J, Daly MJ, Patterson N, et al. (2019). Polygenic adaptation on height is overestimated due to uncorrected stratification in genome-wide association studies. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squire LR (1992). Memory and the hippocampus: a synthesis from findings with rats, monkeys, and humans. Psychol Rev 99, 195–231. [DOI] [PubMed] [Google Scholar]
- Strange BA, Witter MP, Lein ES, and Moser EI (2014). Functional organization of the hippocampal longitudinal axis. Nat Rev Neurosci 15, 655–669. [DOI] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bosnjak M, Skunca N, and Smuc T (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6, e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeszko PR, Betensky JD, Mentschel C, Gunduz-Bruce H, Lencz T, Ashtari M, Malhotra AK, and Bilder RM (2006). Increased stress and smaller anterior hippocampal volume. Neuroreport 17, 1825–1828. [DOI] [PubMed] [Google Scholar]
- Tanti A, and Belzung C (2013). Neurogenesis along the septo-temporal axis of the hippocampus: are depression and the action of antidepressants region-specific? Neuroscience 252, 234–252. [DOI] [PubMed] [Google Scholar]
- Thompson CL, Pathak SD, Jeromin A, Ng LL, MacPherson CR, Mortrud MT, Cusick A, Riley ZL, Sunkin SM, Bernard A, et al. (2008). Genomic anatomy of the hippocampus. Neuron 60, 1010–1021. [DOI] [PubMed] [Google Scholar]
- Thrupp N, Sala Frigerio C, Wolfs L, Skene NG, Fattorelli N, Poovathingal S, Fourne Y, Matthews PM, Theys T, Mancuso R, et al. (2020). Single-Nucleus RNA-Seq Is Not Suitable for Detection of Microglial Activation Genes in Humans. Cell Rep 32, 108189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran MN, Maynard KR, Spangler A, Collado-Torres L, Sadashivaiah V, Tippani M, Barry BK, Hancock DB, Hicks SC, Kleinman JE, et al. (2020). Single-nucleus transcriptome analysis reveals cell type-specific molecular signatures across reward circuitry in the human brain. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velmeshev D, Schirmer L, Jung D, Haeussler M, Perez Y, Mayer S, Bhaduri A, Goyal N, Rowitch DH, and Kriegstein AR (2019). Single-cell genomics identifies cell type-specific molecular changes in autism. Science 364, 685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel JW, La Joie R, Grothe MJ, Diaz-Papkovich A, Doyle A, Vachon-Presseau E, Lepage C, Vos de Wael R, Thomas RA, Iturria-Medina Y, et al. (2020). A molecular gradient along the longitudinal axis of the human hippocampus informs large-scale behavioral systems. Nat Commun 11, 960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker TL, Overall RW, Vogler S, Sykes AM, Ruhwald S, Lasse D, Ichwan M, Fabel K, and Kempermann G (2016). Lysophosphatidic Acid Receptor Is a Functional Marker of Adult Hippocampal Precursor Cells. Stem Cell Reports 6, 552–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willard SL, Friedman DP, Henkel CK, and Shively CA (2009). Anterior hippocampal volume is reduced in behaviorally depressed female cynomolgus macaques. Psychoneuroendocrinology 34, 1469–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witter MP, Van Hoesen GW, and Amaral DG (1989). Topographical organization of the entorhinal projection to the dentate gyrus of the monkey. J Neurosci 9, 216–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, Adams MJ, Agerbo E, Air TM, Andlauer TMF, et al. (2018). Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet 50, 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Krabbe S, Grundemann J, Botta P, Fadok JP, Osakada F, Saur D, Grewe BF, Schnitzer MJ, Callaway EM, et al. (2016). Distinct Hippocampal Pathways Mediate Dissociable Roles of Context in Memory Retrieval. Cell 167, 961–972 e916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidman P, and Maguire EA (2016). Anterior hippocampus: the anatomy of perception, imagination and episodic memory. Nat Rev Neurosci 17, 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang TY, Keown CL, Wen X, Li J, Vousden DA, Anacker C, Bhattacharyya U, Ryan R, Diorio J, O’Toole N, et al. (2018). Environmental enrichment increases transcriptional and epigenetic differentiation between mouse dorsal and ventral dentate gyrus. Nat Commun 9, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat Commun 8, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, Ding W, Sun L, Lu Y, Dong H, Fan X, Liu Z, Chen R, Zhang S, Ma Q, et al. (2020). Decoding the development of the human hippocampus. Nature. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (Related to STAR Methods). Magnetic resonance imaging (MRI).
Outer panels show sagittal plane T1 magnetic resonance images for the subjects contributing hippocampal tissue to the analysis. The location of anterior (orange) and posterior (blue) specimens obtained along the hippocampal axis are highlighted. Line segments represent approximately 3 cm of distance on the included images. The cartoon image shows a coronal section of the human brain indicating the position of the uncal apex, the posterior extent of which was used as a demarcation point for the anterior hippocampus. This structure can be identified relative to the oculomotor nerve and posterior cerebral artery in the ambient cistern, as shown in the cartoon.
Figure S2 (Related to Figure 1). Data quality.
(A) Bar chart showing the number of nuclei sampled from each donor.
(B) Violin plots showing the distribution of the number of genes in glial and neuronal cells separated by 10X Genomics chemistry versions used (v2 or v3).
(C) Violin plot showing the distribution of the number of transcripts in glial and neuronal cells separated by 10X Genomics chemistry version used.
(D) Bar charts showing the distribution of anterior and posterior nuclei in cell-type identified.
(E) Distribution of nuclei obtained from individual donors.
Figure S3 (Related to Figure 1). Integration of published human hippocampus dataset with our data.
(A) UMAP projections showing the clustering of a subset of our data combined with a published human hippocampus dataset (Habib et al., 2017) labeled by cluster annotations (left) and dataset (right).
(B) Bar plots showing the distribution of cells from our study and Habib for each cluster.
Figure S4 (Related to Figure 1). Anterior vs. posterior distribution in clusters.
Boxplots depicting the anterior:posterior cell proportions for each cluster. Each data point is a donor. Colors correspond to the axis. **P < 0.01, ***P < 0.001, Robust generalized mixed model. Clusters showing anterior or posterior enrichment driven by single donor (i.e Olig5, OPC2, Den.Gyr3) were not considered significant. Pyr=Pyramidal neurons, Den.Gyr=dentate gyrus neurons, In=Interneurons, Endo=endothelial cells, Micro=microglia, Astro=astrocytes, OPCs=oligodendrocyte progenitor cells, and Olig=oligodendrocytes.
Figure S5 (Related to Figure 1). Statistics for anterior vs. posterior distribution in clusters.
Volcano plot depicting the cell type differential abundance between axis. X-axis corresponds to the robust mixed model estimates. Y-axis corresponds to the −log10(p-value). Pyr=Pyramidal neurons, Den.Gyr=dentate gyrus neurons. Clustered labeled driven by a single (OPC2, Olig5, and Den.Gyr3) are labeled gray.
Figure S6 (Related to Figure 2). Neuronal clusters by donors.
UMAP plots showing neuronal clusters separated by corresponding donor tissue with cell type annotations. Gra.Neu=Granule Neurons, In.Neu=Inhibitory neurons, and Sub=subiculum.
Figure S7 (Related to Figure 2). Identification and validation of neuronal cluster markers.
(A) Dot plots showing the top 3 cluster markers in excitatory neuronal clusters.
(B) Dot plots showing the top 5 cluster markers in inhibitory neuronal clusters.
(C) Immunohistochemistry showing HS3ST4 expression in CA3 subfield of human postmortem hippocampus. Blue color indicates the nuclear stain DAPI and red color shows α-HS3ST4 staining detected with a fluorophore conjugated antibody.
Figure S8 (Related to Figure 2). Annotation of excitatory clusters.
(A) Dendrogram depicting the hierarchical structure of gene expression of the hippocampal subfields in the Hipposeq dataset (Cembrowski et al., 2016b).
(B) Hypergeometric overlaps of excitatory neuronal clusters with mouse subfield markers identified via bulk RNA-seq (Cembrowski et al., 2016b). Heat map illustrates the −log10(FDR) of gene set enrichment between the markers of the human excitatory neuronal clusters and the gene set lists distinguishing CA vs DG, CA4 vs CA1/2/3, CA1 vs CA2/3, and CA2 vs CA3 in mouse. The x-axis lists the excitatory neuronal clusters and y-axis lists the mouse subfields.
Figure S9 (Related to Figure 2). Human and mouse DG populations.
(A) UMAP plot of div-seq (Habib et al., 2016) dentate gyrus neuronal cells colored by cluster identities.
(B) Violin plots showing normalized counts for Cck and Penk showing DG populations identified by Erwin et al. (Erwin et al., 2020). (C) Heatmap showing −log10(FDR) from a hypergeometric enrichment test for the overlaps between the marker genes for DG granule neuron clusters identified in our human dataset study and the marker genes div-seq DG granule neuron clusters.
Figure S10 (Related to Figure 2). snRNA-seq reveals hippocampal GABAergic interneurons.
(A) UMAP plot of inhibitory neuronal cells colored by cluster identities and cell-type annotations. (B) Violin plots showing normalized counts for interneuron markers. (C) Heatmap showing −log10(FDR) from a hypergeometric enrichment test for the overlaps between the marker genes for inhibitory neuronal clusters identified in this study and the marker genes for inhibitory neuronal clusters identified in human autopsy brains by Habib et al. (Habib et al., 2017). (D) Heatmap showing −log10(FDR) from a hypergeometric enrichment test for the overlaps between the marker genes for inhibitory neuronal clusters identified in this study and the marker genes for inhibitory neuronal clusters identified in mouse hippocampus by Habib et al. (Habib et al., 2017).
Figure S11 (Related to Figure 3). Gene ontology analysis of CA1-specific DEGs.
Bubble chart showing the top gene ontology categories for genes with higher expression in posterior (left) and anterior (right) hippocampus. On the x-axis is the −log10(FDR) and on the y-axis is the number of genes per category. BP (blue)=biological function; MF (gray)=molecular function.
Figure S12 (Related to Figure 6). Glial specific DEGs across anterior and posterior hippocampus.
Upset plot showing the overlap of upregulated or downregulated DEGs in glial cell-types across aHP and pHP (adj. p-value<0.05, log2FC>0.3, percentage>25). Horizontal bars show the number of genes enriched in posterior (purple) and anterior (gray) in each cell type. Vertical black bars show the number of overlapping DEGs across the glial cell types. Oligo.=Oligodendrocytes
Figure S13 (Related to Figure 6). Microglia gene expression in human hippocampus.
(A) Heatmap illustrating the average expression of the top 5 DEGs between microglia clusters. Color scheme corresponds to log-normalized and scaled values for average gene expression for each cluster
(B) Frequency distribution of microglia clusters in five donors. (C) Violin plots showing the expression of homeostatic and pre-active microglia.
Figure S14 (Related to STAR Methods). JackStraw and Elbow Plots of the entire dataset.
(A) Scatter plot showing JackStraw statistics for each of the principal component (1-100) for PCA significance for dataset shown in Figure 1.
(B) Scree plot (Elbow Plot) showing the ranking of principle components based on the percentage of variance explained by each one for dataset shown in Figure 1. Arrow shows flattening around PC 25.
Figure S15 (Related to STAR Methods). Filtered clusters from entire dataset.
UMAP plot showing clustering the entire dataset with removed clusters labeled (left). UMAP plot final clustering after removal of two clusters (right). Py.Neu=Pyramidal neurons, Den.Gyr=dentate gyrus neurons, In=Interneurons, Astro=Astrocytes, Endo=endothelial cells, Micro=microglia, OPCs=oligodendrocyte progenitor cells, and Olig=oligodendrocytes.
Figure S16 (Related to STAR Methods). JackStraw and Elbow Plots of the neuronal cells.
(A) Scatter plot showing JackStraw statistics for each of the principal component (1-100) for PCA significance for dataset shown in Figure2.
(B) Scree plot (Elbow Plot) showing the ranking of principle components based on the percentage of variance explained by each one for dataset shown in Figure 2.
Figure S17 (Related to STAR Methods). Filtered neuronal clusters.
UMAP plot showing neuronal clustering with removed clusters labeled (left). UMAP plot final clustering after removal of three clusters (right). Gra.Neu=Granule Neurons, In=Inhibitory neurons, and Sub=subiculum.
Figure S18 (Related to STAR Methods). Variance explained by covariates.
(A) Barplot depicting the variance explained by each covariate. Y-axis represent the variance explained weighted across the first 10 PCs.
(B) Barplot depicting the variance explained by each covariate for each cluster. Clusters defined by one subject (Olig5, Den.Gyr3, OPC2, Micro3) were removed from the analysis. Y-axis represent the variance explained weighted across the first 10 PCs.
Table S1. Demographics of human tissue with the number of cells sampled and reads, PMI (Postmortem interval), and RIN (RNA integrity value), Related to STAR Methods
Table S3. Robust linear mixed model statistics for anterior:posterior, Related to Figure 1
Table S5. Cluster markers of inhibitory neuronal cells (Figure S9A), Related to Figure S9
Table S6. Cluster-specific differential gene expression, Related to Figure 2
Table S7. Cell-type specific differential gene expression, Related to Figure 2
Table S10. MAGMA statistics, Related to Figure 5
Data Availability Statement
The single-nuclei RNA-sequencing data reported in this paper can be accessed at NCBI GEO (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE160189) with the accession number GSE160189. Code that was used to perform data pre-processing, clustering and differential gene expression analysis is available at GitHub repository (https://github.com/konopkalab/10x_scRNAseqHippoAxisSeq). The data can also be accessed through an interactive R shiny application at the Github repository (https://github.com/konopkalab/10x_scRNAseq_HippoAxisSeq/tree/main/Shiny_App) and the interactive website (https://human-hippo-axis.cells.ucsc.edu/).