

Abstract
Osimertinib (OSI) is the first FDA-approved third-generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI). It can be used for treating non-small cell lung cancer (NSCLC) patients with activating EGFR mutation and for patients who are resistant to first-generation EGFR TKIs due to T790M resistance mutation. However, patients treated with OSI ultimately develop acquired resistance, which prevents its long-term benefit for patients. Therefore, the development of effective strategies to overcome OSI resistance will address a significant clinical challenge and benefit patients by prolonging their survival time. Our previous studies indicated that combination therapy was a promising strategy for overcoming OSI resistance. In this study, we developed nanoparticle (NP) formulations for co-delivery of osimertinib (OSI) and selumetinib (SEL) to treat OSI-resistant NSCLC effectively. We conjugated SEL with PEG through a reactive oxygen species (ROS)-responsive linker to generate polyethylene glycol (PEG)-SEL conjugate prodrug (PEG-S-SEL). Due to the amphiphilic nature of PEG-S-SEL, it can self-assemble in an aqueous solution to form micelle NP and serve as a delivery carrier for OSI. The ROS-responsive linker can facilitate the release of drugs in the tumor microenvironment with elevated ROS levels. OSI and SEL combination NP can overcome OSI resistance by simultaneously inhibiting both EGFR and mitogen-activated protein kinase (MEK), thus effectively inducing apoptosis in OSI-resistant NSCLC cells and inhibiting OSI-resistant tumors in vivo. In conclusion, the OSI+SEL NP combination therapy showed promising anticancer efficacy and demonstrated potential for treating NSCLC patients with OSI acquired resistance.
Keywords: Osimertinib, Selumetinib, Reactive Oxygen Species, Prodrug, Non-small Cell Lung Cancer, Acquired resistance
1. INTRODUCTION
Lung cancer is one of the leading causes of cancer death in both men and women in the USA, accounting for around 25% of all cancer-related deaths. The lung cancer-associated death exceeds that of breast, prostate, and colon cancers combined. The five-year survival rate for lung cancer patients has gone up to close 20% due to the advances in targeted therapies and immunotherapy. However, it is still much lower than patients with other cancers such as prostate cancer and breast cancer. More than 50% of patients with lung cancer die within one year after diagnosis [1]. Non-small cell lung cancer (NSCLC) is the most common lung cancer, which accounts for over 80% of lung cancers. For the past decades, small-molecule drugs targeting specific mutations have been developed to treat lung cancer patients effectively. Epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) belong to this group of drugs, which have excellent efficacy in lung cancer patients with activating EGFR mutations, including exon 19 deletion (19del) and exon 21 point mutation (L858R) in the receptor tyrosine kinase domain [2]. Currently, there are three generations of TKI, including first-generation TKIs (e.g., erlotinib), second-generation TKIs (e.g., afatinib), and third-generation TKIs (e.g., osimertinib), respectively.[2] Osimertinib (OSI; AZD9291 or tagrisso) is the first FDA-approved third-generation EGFR-TKI for treating NSCLC patients with activating EGFR mutations and for patients who are resistant to first-generation EGFR TKIs due to T790M drug resistance mutation. OSI has potent activities in cancer cells with EFGR-TKI sensitizing mutation and EGFR T790M resistance mutation. It has only limited activity against wide-type EGFR and thus has less toxic side effects [3, 4]. Patients treated with OSI showed significantly longer progression-free survival than those treated with other EFGR-TKIs. OSI treatment improved the median overall survival of patients [5]. Despite the success in clinical application, patients often become resistant to OSI, resulting in the relapse of cancer and limited prognosis [6, 7]. The acquired resistance to OSI is a significant cause, which prevents its long-term benefit for patients. Therefore, the development of effective strategies to overcome OSI resistance will address a significant clinical challenge and benefit patients through prolonging their survival time.
The abnormal EGFR can activate multiple downstream signaling pathways, including Raf/MEK/ERK and PI3K/Akt pathways. Among these pathways, the MEK/ERK signaling pathway plays a critical role in regulating tumor cell proliferation and survival [8–10]. We previously demonstrated that suppression of the MEK/ERK signaling and subsequent modulation of Bim and Mcl-1 levels were critical mechanisms for OSI to trigger apoptosis of EGFR-mutated NSCLC cells; these effects were lost once cells have become resistant to OSI. The inhibition of the MEK/ERK signaling with either a MEK inhibitor or an ERK inhibitor restored the sensitivity of OSI-resistant cells with different resistance mechanisms, including C797S mutation or MET amplification, as demonstrated in our in vitro and in vivo preclinical models [11, 12]. Impressively, this strategy also worked well to delay or abrogate the emergence of acquired resistance if we intervened at the early stage [13]. A recent clinical study reported that OSI-resistant NSCLC patients with BRAF fusion responded well to OSI and MEK inhibitor combination treatment [14]. Despite the promising activity of combination therapy, there is still a concern that this strategy may increase systemic toxicities or side effects since both drugs inhibit the same vertical pathway. Dose-limiting toxicities were observed in a phase Ib trial [15]. Targeting the MEK/ERK signaling pathway is an effective strategy for delaying and overcoming acquired resistance but with concern on potentially increased systemic toxicities.
In this study, we developed a novel combination nanomedicine to treat EGFR mutant NSCLC with acquired OSI resistance (Figure 1). (1) OSI and SEL combination therapy can overcome OSI resistance by simultaneously inhibiting both EGFR and MEK, and thus effectively inducing apoptosis in OSI-resistant NSCLC cells. (2) PEG-S-SEL conjugate will not only serve as a prodrug of SEL but also work as a delivery carrier for OSI. We will synthesize PEG-S-SEL conjugate as a prodrug of SEL. This conjugate prodrug is an amphiphilic macromolecule composed of a hydrophilic part (i.e., PEG) and a hydrophobic part (i.e., SEL). It forms micelle NPs through self-assembly. The PEG-S-SEL prodrug conjugate micelle can serve as a nanoscale delivery system and load the OSI drug through non-covalent interactions. (3) PEG-S-SEL conjugate will be synthesized with a tumor microenvironment ROS responsive linker to realize tumor-specific drug release. The high level of ROS in the tumor microenvironment can induce the cleavage of ROS responsive linker, promote the disassembly of NPs, and trigger the release of drugs. In contrast, the conjugate will be stable and have minimal drug leakage at normal tissues with a low ROS level. This feature will enhance tumor targeting specificity and reduce drug exposure in normal tissues. (4) Two drugs (i.e., OSI and SEL) simultaneously delivered in a single NP formulation will synchronize their exposure in tumor cells and achieve better synergistic anticancer efficacy. Also, the use of optimized drug delivery systems will reduce the dose needed to effectively inhibit the tumor growth, thus prevent potential dose-limiting toxicities in the clinical application of SEL and OSI combination therapy [15].
Figure 1. Synthesis and characterization of PEG-S-SEL conjugate.
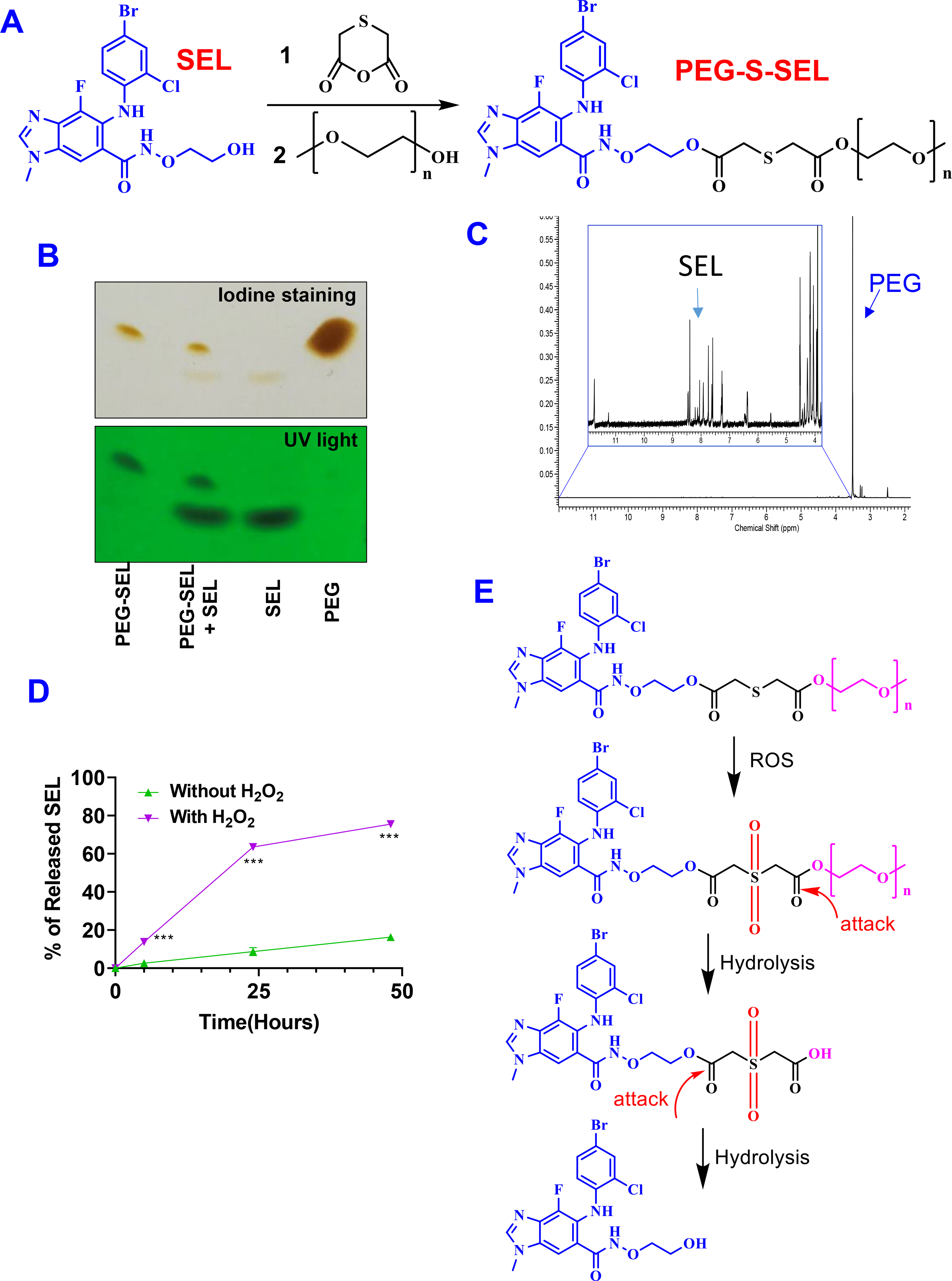
(A) Synthesis procedure. 1. EDCI, HOBT, DMF; 2. DCC, DMAP, DMF (B) TLC and (C) 1H-NMR characterization of PEG-S-SEL conjugate. (D) ROS triggered release of SEL from conjugate. Results are mean ± SD (n=3). ***, P < 0.001. (E) ROS-responsive drug release mechanism.
2. MATERIAL AND METHODS
2.1. Materials
PEG2k-OH and 4-dimethylaminopyridine (DMAP) was from Tokyo Chemical Industry (TCI) America. PEG2K −1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE) was from Laysan Bio, Inc. SEL and OSI were purchased from LC laboratories. 1,4-oxathiane-2,6-dione was purchased from Enamine LLC. Glutaric anhydride and N,N′-Dicyclohexylcarbodiimide (DCC) were purchased from Alfa Aesar. 1- Hydroxybenzotriazole hydrate (HoBt) and D-α-Tocopherol polyethylene glycol 1000 succinatewas (TPGS) were purchased from Sigma-Aldrich. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCl) was purchased from AK Scientific. 3-(4,5-dimethyl-thiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT) was purchased from Biosynth International Inc. Dialysis bag was purchased from Spectrum Labs. Calcein-AM was ordered from BD Biosciences, USA. Propidium iodide (PI) was ordered from Biotium. Annexin V-FITC/PI apoptosis detection kit was from Biolegend. Crystal violet was from Fisher Chemical. Dimethylformamide (DMF), methanol, and other reagents were ordered from VWR International.
2.2. Synthesis and characterization of PEG-SEL conjugates.
(1). Synthesis procedure
Synthesis of PEG-S-SEL:
SEL (50 mg), 1,4-oxathiane-2,6-dione (16 mg), EDCl (42 mg), and HoBt (25 mg) were dissolved in anhydrous DMF (5 ml). The mixture solution was stirred under N2 protection at room temperature for overnight. Then, PEG2k-OH (103 mg), DCC (45 mg), and DMAP (26 mg) dissolved in anhydrous DMF were added into the reaction mixture and stirred under N2 protection at room temperature for additional 48 hours. Then, the reaction mixture was transferred into a dialysis bag (MW 1,000 Da) and dialyzed against methanol and water. Finally, the sample was freeze-dried to obtain PEG-S-SEL (yield 47%). PEG-C-SEL was synthesized with a similar method by replacing 1,4-oxathiane-2,6-dione with glutaric anhydride in the reaction (yield 46%).
(2). Characterization
The PEG-S-SEL was characterized with the analytical thin-layer chromatography (TLC; chloroform/methanol, 9:1, vol/vol) and visualized by UV light at 254 nm and iodine staining, respectively. In addition, PEG-S-SEL and other controls were also characterized with H1-NMR (Bruker 600 MHz) using deuterated dimethyl sulfoxide as the solvent. The concentration of SEL conjugate was determined based on the UV absorption at 260 nm. To determine the ROS-responsive cleavage of PEG-S-SEL conjugate, it was dissolved in water containing 20% acetonitrile and 20% methanol or the same medium with H2O2 (3mM). Samples were kept in a shaker incubator (37 °C, 200 rpm). At pre-determined time points, the concentration of released SEL was determined with reverse-phase high-performance liquid chromatography (RP-HPLC; Shimadzu) with a diode array detector based on the UV absorption at 260 nm. A C18 column (3 μm, 50 × 4.6 mm, Shimadzu) was used. The mobile phase was composed of 20% acetonitrile, 20% methanol, 60% water at a flow rate of 0.5 mL/min.
2.3. Preparation and characterization of OSI+SEL NPs
The amphiphilic nature of PEG-S-SEL will self-assemble in an aqueous solution to form micelle NPs. We used a film-dispersion method to prepare OSI+SEL NPs [16]. Briefly, OSI (2.2 mg), PEG-S-SEL (5.2 mg), and DSPE-PEG (12 mg) were dissolved in dichloromethane (1 mL). Then, dichloromethane was removed with a rotavapor, and the formed film was dispersed in phosphate-buffered saline (PBS) with sonication. A trace amount of aggregations was removed by centrifugation for 10 minutes at 6,700 g and followed by filtration with a 0.45 μm filter. OSI NPs and PEG-S-SEL NPs with a single drug were prepared with a similar method. The particle sizes of different NPs were determined with dynamic light scattering (DLS; Zetasizer, Malvern, UK). The morphology of NPs was determined with atomic force microscopy (AFM, Anton Paar Tosca 400), operated in a constant tapping mode with silicon AFM probe (ARROW-NCR). The concentration of OSI in the NP formulations was determined with RP-HPLC with a diode array detector based on the UV absorption at 260 nm. A C18 column (3 μm, 50 × 4.6 mm, Shimadzu) was used. The mobile phase was composed of 20% acetonitrile, 20% methanol, 60% water with 0.5% trifluoroacetic acid (TFA) at the flow rate of 0.5 mL/min. The concentration of SEL in NP formulations was determined with the UV spectrometer based on the UV absorption at 260 nm. For SEL+OSI NP, the concentration of OSI was determined with the RP-HPLC method. The concentration of SEL was calculated based on its UV absorption at 260 nm. The drug loading efficiency was calculated with the following equation: Loading efficiency (%) = theoretical drug concentration/actual drug concentration x 100%. The drug loading (%) was calculated with the following equation: Drug loading (%) = amount of drug / amount of carrier x 100%. For in vitro drug release study, OSI+SEL NP in a dialysis bag (MW 2,000 Da) was incubated in phosphate-buffered saline (PBS, pH 7.4) containing 1% Tween 80 or same release medium with H2O2 (5 mM) and kept in a shaker incubator (37 °C, 200 rpm). At pre-determined time points, the concentration of SEL and OSI in the release medium was determined with RP-HPLC as described above. The percentage of cumulative drug release was calculated with the following equation: Drug release (%) = Amount of released drug/ Total amount of drug × 100%.
2.4. In vitro anticancer efficacy
(1). Cell lines and cell culture
OSI-resistant PC-9/AR and PC-9/GR/AR cells were established by exposing PC-9 cells or PC-9/GR cells to OSI with increasing concentration. OSI-resistant HCC827/AR cell line was established in our laboratory as described previously [17]. The cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 containing 5% fetal bovine serum (FBS) at 37°C in a humidified atmosphere of 95% air and 5% CO2.
(2). MTT assay
Cells were seeded into a 96-well plate (2000 cells/well) and incubated overnight. Then, cells were treated with different testing articles diluted in cell culture medium (100 μL/well). After 48 hours, the anticancer efficacy was determined with the MTT assay as described in previous reports [18]. The cell viability was determined based on the absorbance at 570 nm and a reference wavelength of 670 nm. Cell viability was calculated using the following equation: Cell Viability (%) = (ATest ⁄ Acontrol) × 100%. The combination index (CI) for combination therapy was calculated with CompuSyn software (ComboSyn, Inc). CI < 1, synergistic effects, CI =1, additive effects, and CI > 1, antagonistic effects. We also performed the MTT assay to compare the effects of PEG-S-SEL (with a ROS responsive linker) and PEG-C-SEL (with a non-ROS-responsive ester linker). In this case, cells were treated with PEG-S-SEL or PEG-C-SEL for 24 hours and followed by the treatment with OSI for additional 48 hours. Then, the cell viability was determined with the same approach.
(3). Sulforhodamine B (SRB) assay
Cells were seeded into a 96-well plate and incubated overnight. Then, cells were treated with different concentrations of OSI NP, SEL NP, and OSI+SEL NP for three days. The DSPE-PEG was substituted by TPGS in the preparation of NPs in this study. At the end of treatment, the cell number and growth inhibition was determined and calculated as previously described [11, 19].
(4). Calcein-AM/PI staining
Cells were seeded into a 12-well plate with a density of 50,000 cells in each well and incubated overnight. Then cells were treated with different formulations for 48 hours. At the end of the treatment, cells were stained with Calcium-AM/PI and observed with a fluorescence microscope (CYTATION 5 Imaging Reader).
(5). Colony-forming assay
Cells were seeded into a 12-well plate at a density of 200 cells per well and incubated overnight. Then, cells were treated with different formulations diluted in cell culture medium for 24 hours and cultured for additional seven days in cell culture medium without drug. At the end of treatment, cell colonies were fixed with methanol and stained with crystal violet. After the cell colony photos were taken, the crystal violet in each well was dissolved in 10% acetate acid, and the absorbance at 560 nm was determined [20].
(6). Determination of cell apoptosis with Annexin V-FITC/PI apoptosis detection kit
Cells were seeded into a 12-well plate at a density of 50,000 cells per well and incubated overnight. Then, cells were treated with different formulations for 48 hours. Cells were collected at the end of treatment, stained using Annexin V-FITC/PI apoptosis detection kit, and analyzed by flow cytometry (BD Accuri C6 Plus).
(7). Determination of cleavage of PARP and caspase-3 with Western blot.
PC-9/AR cells were exposed to free drugs (0.25 μM OSI or SEL) or NPs (0.25 μM SEL, 0.25 μM OSI, or SEL+OSI) for 48 hours. The DSPE-PEG was substituted by TPGS in the preparation of NPs in this study. At the end of treatment, whole-cell protein lysate was prepared. The cleavage of PARP and caspase-3 was determined with Western blot analysis as previously described [11, 21].
(8). Cellular uptake studies
Flow cytometry:
cells were seeded into a 12-well plate at a density of 100,000 cells per well and incubated overnight. Then, cells were treated with coumarin-6 loaded NPs for 3 hours. At the end of treatment, cells were washed, collected, and analyzed by flow cytometry (BD Accuri C6 Plus).
Fluorescence image:
Cells were seeded at a density of 20,000 cells per well into a 96-well plate and incubated overnight. Then, cells were treated with coumarin-6 loaded NPs for 3 hours. At the end of treatment, cells were washed with PBS, stained with DAPI, and fixed with methanol. The fluorescence images were acquired by CYTATION 5 Imaging Reader.
2.5. In vivo anticancer efficacy studies
Animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Emory University. Five to 6 week old female athymic (nu/nu) mice obtained from Charles River Labs were used in our studies. One million PC-9/AR cells suspended in serum-free medium were injected subcutanesouly into the flank of nude mice to establish OSI-resistant xenograft tumor model [11]. When tumor sizes were close to 100 mm3, tumor-bearing mice were randomly divided into four groups and treated with different formulations via intravenous (IV) injection once every three days for six injections. Tumor size (mm3) = [width (mm)2 × length (mm)] x 1/2. Bodyweight and tumor volume was monitored and recorded. At the end of the experiment, the mice were euthanized, and the weight of collected tumors was determined.
2.6. Statistical Analysis
Experiments were performed in triplicate or quadruplicate unless otherwise noted. Results were reported as mean ± standard deviation (SD). The difference between groups was analyzed with the unpaired t-test using GraphPad Prism 8.0 (GraphPad Software, Inc., San Diego, CA).
3. RESULTS
Synthesis and Characterization of PEG-S-SEL conjugate.
The PEG-S-SEL with a ROS-responsive linker was synthesized as shown in Figure 1A. Briefly, SEL, 2,2’-thiobisacetic acid anhydride, EDCI and HOBt were dissolved in DMF. The mixture was stirred at room temperature for overnight. Then, PEG2k-OH, DCC, and DMAP were added, and the mixture was stirred for an additional 48 hours. Finally, the PEG-S-SEL was purified through dialysis against purified methanol and water. The conjugation efficiency was around 80%. The formation of PEG-S-SEL conjugate was confirmed with TLC Figure 1B. The conjugation of PEG increased the hydrophilicity of SEL. Due to the significantly different properties of PEG-S-SEL and SEL, they could be separated in the TLC showing different retention factor (Rf). In addition, the PEG-S-SEL can be visualized with both UV light and iodine staining. In contrast, the PEG can only be visualized with iodine staining due to the lack of UV absorption. We also characterized the PEG-S-SEL with 1H-NMR (Figure 1C & Figure S1), which showed characteristic peaks from both SEL and PEG. The drug release study indicated that the PGE-S-SEL conjugate showed fast cleavage of the conjugate prodrug and release of SEL in the presence of H2O2. In contrast, the same conjugate was stable and showed significantly slower cleavage of conjugate and release of SEL in the absence of H2O2 (Figure 1D). We also synthesized PEG-C-SEL with an ester linker, which is not responsive to ROS (Figure S2). The PEG-C-SEL showed similar drug release profiles in the presence and absence of H2O2. (Figure S3). The conjugation efficiency was around 97%. Although the PEG-C-SEL and PEG-S-SEL showed a similar release profile in medium without H2O2, the PEG-S-SEL had significantly fast drug release than PEG-C-SEL in medium with H2O2. The ROS responsive mechanism of PEG-S-SEL was indicted in Figure 1E. In the presence of ROS, the thioether was converted into hydrophilic sulfone through oxidation reaction to facilitate the hydrolysis of the ester bond. The hydrolysis of the ester bond will remove PEG and release free SEL molecules from the conjugate.
Preparation and characterization of NPs.
A film dispersion method was used to prepare OSI+SEL NPs as well as NPs with a single drug (i.e., OSI NP, SEL NP). We selected ROS responsive PEG-S-SEL to prepare NPs in our study. Due to the amphiphilic nature of PEG-S-SEL, it will self-assemble in the aqueous solution to form micelle NPs. DSPE-PEG was used to prepare OSI NP; therefore it was also included in the SEL NP and OSI+SEL NP formulations for consistency. The optimized NP formulations have SEL concentration of around 1 mg/mL and OSI concentration of 2.3 mg/mL in both OSI+SEL NP or single drug-loaded NPs. The drug loading efficiencies were about 100% for the optimized NP formulations (Figure 2A). Our studies also demonstrated that the physical encapsulation of SEL free drug in DSPE-PEG micelle NP only achieved a relatively lower SEL drug concentration (0.6 mg/ml) and loading efficiency (about 60%). However, a significantly higher SEL drug concentration (1 mg/mL) and loading efficiency (around 100%) were achieved with the PEG-S-SEL conjugate NP formulation (Figure S4). The drug loading (%) of OSI and SEL in the optimized NP formulation was 13% and 6%, respectively. The particle sizes of different NPs were determined with DLS: SEL+SEL NP was 43 nm with PDI of 0.581; SEL NP was 77 nm with PDI of 0.413; and OSI NP was 17 nm with PDI of 0.595. These NPs were desirable for intravenous injection to enhance drug delivery into tumor tissues (Figure 2B) [22, 23]. The zeta potential for these NPs were −30 mV (OSI+SEL NP), −55 mV (SEL NP), and −41 (OSI NP), respectively (Figure 2C). The morphology of NPs was analyzed with AFM. The results confirmed the spherical shape of these three NPs (Figure S5). We also determined the cellular uptake of NPs labeled with a green fluorescence dye, coumarin-6. Their particle size and zeta potential were included in Figure S6. The fluorescence image showed that all three NPs could be efficiently uptake by PC-9/AR NSCLC cells after three hours’ incubation, and the green fluorescence signals were mainly located in the cytoplasm (Figure 2D). The cellular uptake efficiency was also determined quantitatively by flow cytometry, which demonstrated that these different NPs showed similar cellular uptake efficiency (Figure 2E). The in vitro drug release studies showed that the release of SEL was faster in the presence of H2O2, showing ROS-responsive release properties. However, the release of OSI was similar in medium with or without H2O2 (Figure S7).
Figure 2. (A).
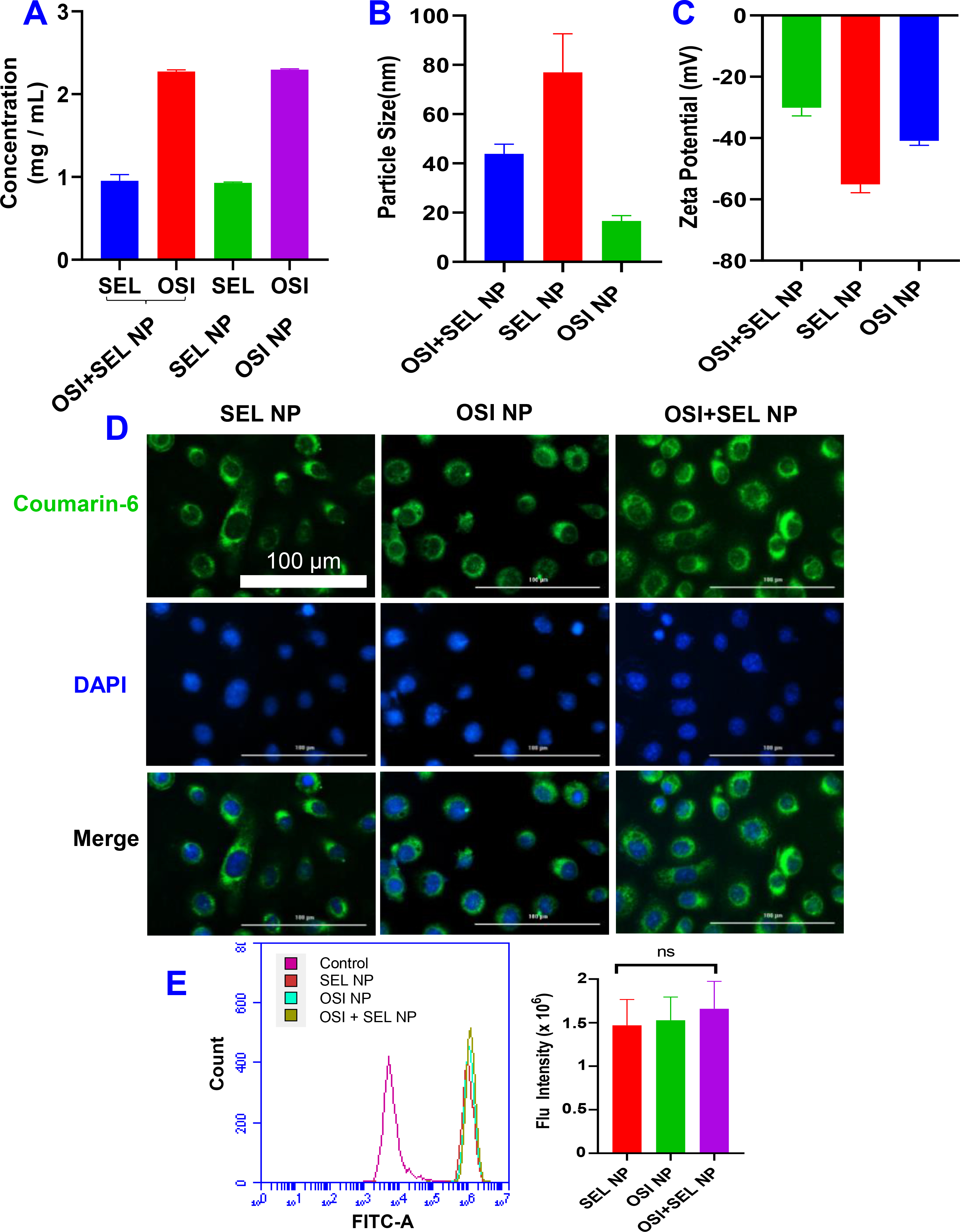
(A) Drug concentration, (B) Particle size, and (C) Zeta potential of different NPs. (D) Fluorescence image of PC-9/AR cells incubated with coumarin-6-labeled NPs for 3 hours. (E) Cellular uptake of NPs by PC-9/AR cells were determined by flow cytometry. Data are presented as the mean ± SD, n=3.
In vitro anticancer activities.
Once the OSI+SEL NP formulation was developed, we tested the anticancer activities of NP formulation with an OSI-resistant NSCLC cell line (i.e., PC-9/AR) with several in vitro assays (Figure 3). In addition, the free drug control groups were also included in selected representative experiments to test whether there was any significant difference between NPs and free drugs. As demonstrated in the MTT assay results (Figure 3A), PC-9/AR cells were resistant to the treatment of OSI or SEL monotherapy, while the OSI+SEL combination therapy showed significantly enhanced cytotoxicity. The IC50 for OSI and OSI NP was 2.95 ± 0.11and 2.94 ± 0.56 μM, respectively. IC50 for SEL free drug and SEL NP was not calculated due to their low toxicity at the tested range. The IC50 for free drug and NP combination therapy was 0.2 ± 0.02 / 0.1 ± 0.01 and 0.4 ± 0.03 / 0.2 ± 0.015 μM (OSI/SEL). The CI values for both free drug and combination therapy groups were smaller than 1 (ranges from 0.1 to 0.17 in at different concentrations), indicating the synergistic effects of combination therapy. We also compared the anticancer effects of PEG-S-SEL (with a ROS-responsive linker) and PEG-C-SEL (with a non-ROS-responsive ester linker) in combination with OSI. The PEG-S-SEL showed significantly better activities than PEG-C-SEL, due to the more efficient release of SEL from PGE-S-SEL (Figure S3 &S8). We also determined the anticancer effects of OSI NP, SEL NP, and OSI+SEL NPs with SRB assay, which measures the cell numbers to determine the cell growth inhibition effects of different NP formulations. Three types of OSI-resistant cell lines were tested. The results indicated that the OSI+SEL NP combination therapy showed significantly improved anticancer effects than OSI NP or SEL NP monotherapy in all of these three cell lines, demonstrating the capability of OSI+SEL NP in overcoming OSI resistance (Figure S9). We also tested the free drug and NP either as monotherapy or combination therapy in OSI-sensitive PC9 cells. Since this cell is sensitive to OSI treatment, the OSI monotherapy effectively killed PC9 cancer cells and the OSI+SEL combination therapy did not enhance its efficacy. Further, there was no significant difference between free drug and NP treatment groups (Figure S10).
Figure 3. In vitro anticancer efficacy against OSI-resistant PC-9/AR NSCLC cells.
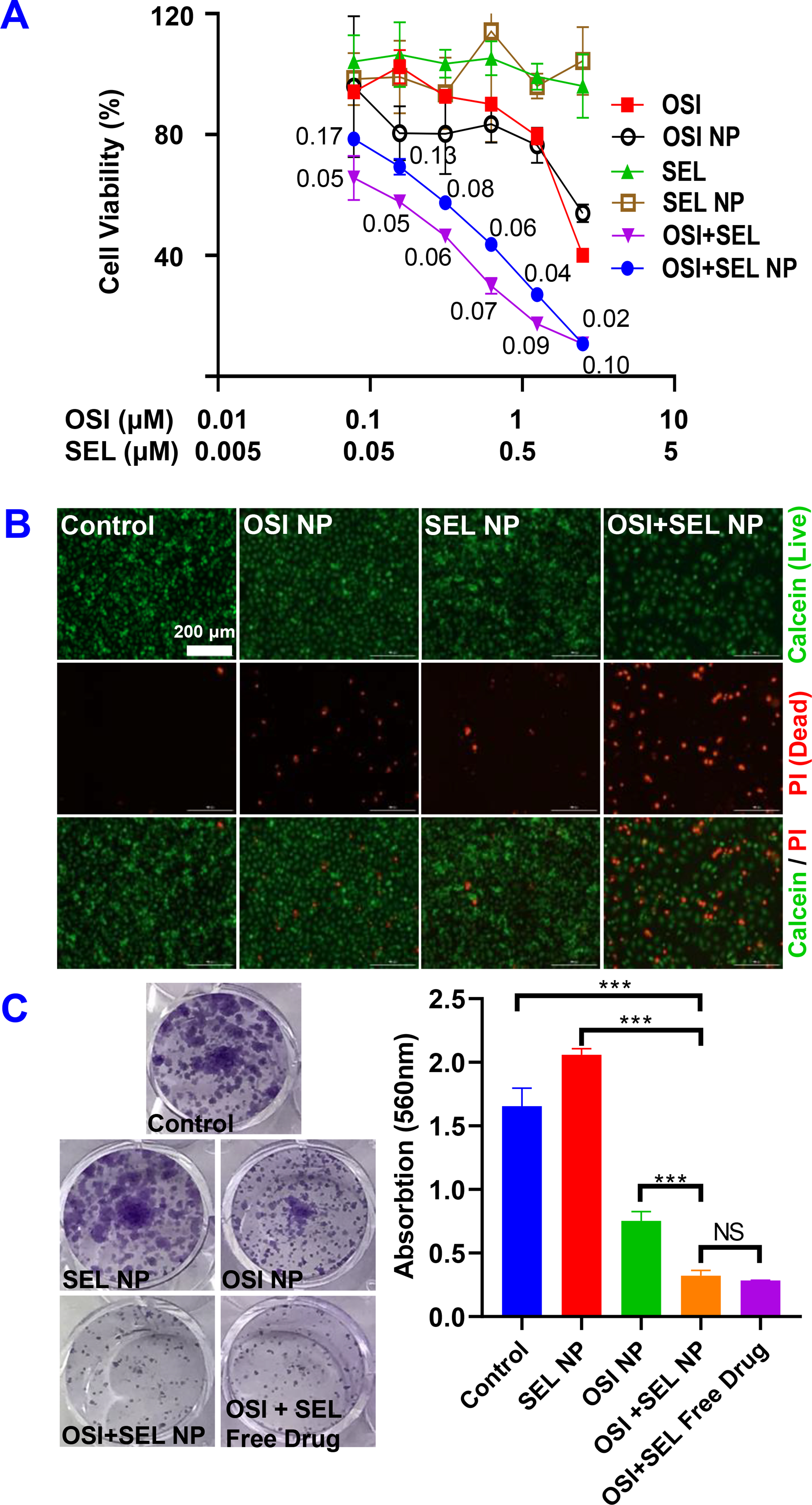
(A) Cell viability of PC-9/AR cells were determined with the MTT assay after treatment with different formulations for 48 hours. The numbers in the graphs are combination indices. (B) Calcein-AM/PI staining. Cells treated with different formulations for 48 hours and stained with Calcein-AM/PI to detect living (green) and dead (red) cells. (C) Inhibition of colony formation by OSI+SEL combination therapy. Cells were treated with different NP formulations and free drugs for a total of 7 days. Cell colonies were stained with crystal violet and photographed. Crystal violet in each groups were also dissolved by methanol and the absorption at 560 nm were determined. Results are mean ± SD (n=3). ***, P < 0.001
The anticancer effects were further confirmed with Calcein-AM/PI staining (Figure 3B). The treatment with OSI NP (1.25 μM) or SEL NP (0.625 μM) did not induce a significant change of living cells (green color) and dead cells (red color) when compared with the control group. In contrast, the OSI+SEL NP combination therapy group (OSI 1.25 μM & SEL 0.625 μM) showed significantly reduced living cells and increased dead cells. We also determined the anticancer efficacy with the colony formation assay of NPs and free drugs (Figure 3C). After treating cells with different formulations for seven days, cell colonies were stained with crystal violet and photographed. SEL-NP (0.625 μM) showed no significant effects on colony formation, and treatment with OSI-NPs (1.25 μM) slightly reduced the colony number and size. In contrast, OSI+SEL NPs combination therapy at the same concentration showed significant inhibition of colony formation. The combination treatment with OSI+SEL free drugs also showed a similar effect as the NP combination therapy. We also dissolved the crystal violet with methanol and determined the absorption at 560nm, which further confirmed the inhibition of colony formation by the combination therapy.
We also determined the cell apoptosis with an Annexin V-FITC/PI apoptosis detection kit (Figure 4A) and western blot analysis of apoptotic proteins (Figure 4B). The monotherapy with OSI NP (1.25 μM) or SEL NP (0.625 μM) treatment did not induce significant cell apoptosis. OSI NP treatment group showed 5.7% early-stage apoptosis cells (Annexin V-FITC+/PI−) and 10 % late-stage apoptosis (Annexin V-FITC+/PI+), respectively. Similarly, SEL NP treatment group showed 4.3% early-stage apoptosis and 8.7% late-stage apoptosis, respectively. In contrast, the OSI+SEL NP combination therapy resulted in a substantial increase of early-stage apoptotic cells (23.4%) and late-stage apoptotic cells (41.4%), respectively. The total apoptotic cells in the combination group were around 65%. These results indicated that the OSI+SEL NP could effectively kill OSI-resistant PC-9/AR NSCLC cells by inducing cell apoptosis. The western blot results also confirmed the induction of cell apoptosis by OSI+SEl NP. The treatment of PC-9/AR cells with OSI+SEL NP (0.25 μM OSI and 0.25 μM SEL) resulted in a significant increase of cleaved form of PARP and caspase 3, indicating cell apoptosis. However, the treatment of OSI NP (0.25 μM) or SEL NP (0.25 μM) did not increase the level of cleaved form PARP or caspase 3. We also determined the effects of OSI, SEL, and SEL+OSI free drug treatment on the cleavage of PARP and caspase 3. It showed similar results as observed in cells treated with corresponding NP formulations (Figure 4B).
Figure 4.
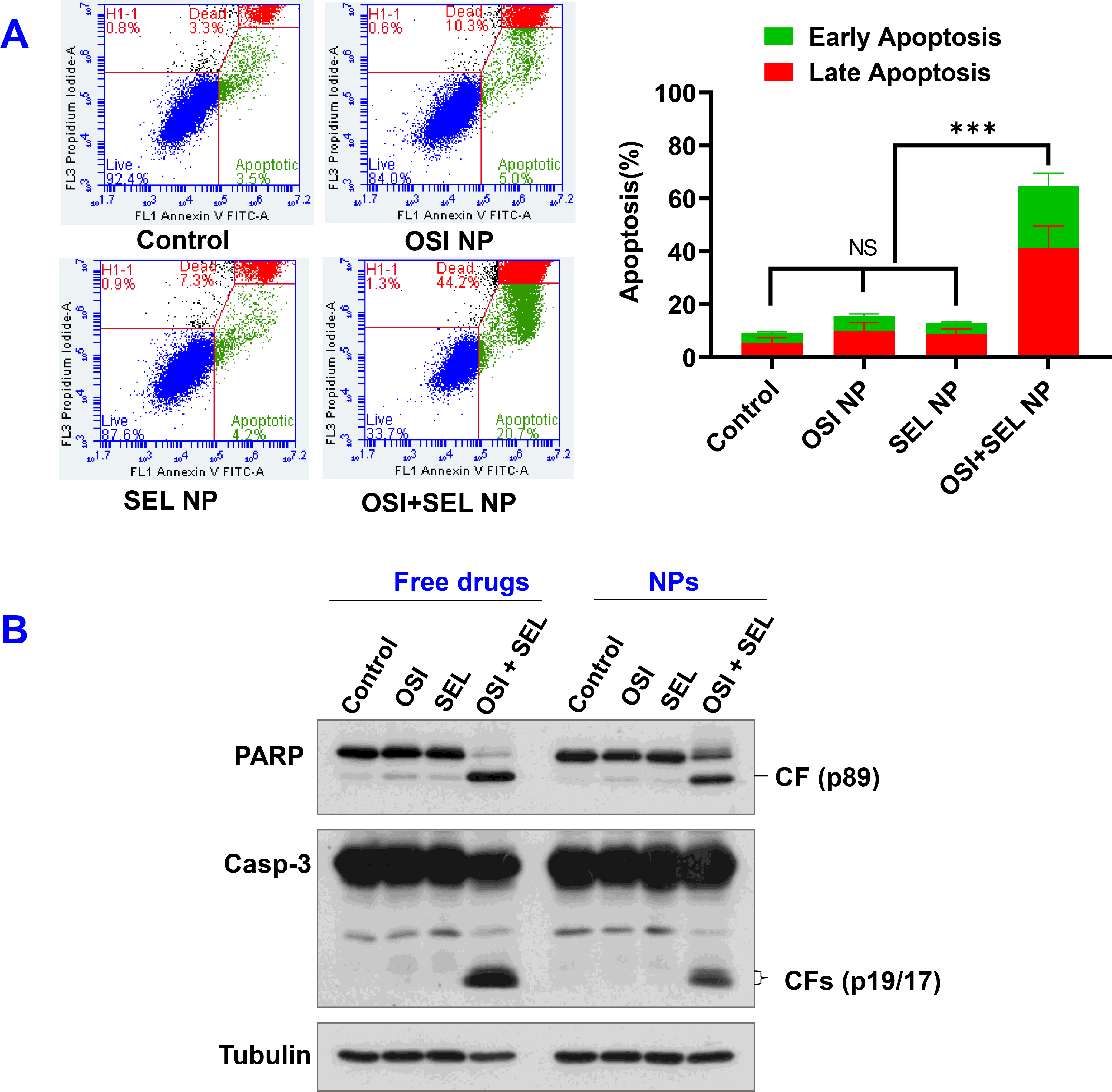
(A) Cell apoptosis was determined with flow cytometry. PC-9/AR were treated with OSI+SEL NP and other controls for 48 hours. The OSI +SEL NP induced significant increase of percentage of early apoptotic cells (Annexin V-FITC+/PI−) and late apoptotic cells (Annexin V-FITC+/PI+). Results are mean ± SD (n=3). ***, P < 0.001. (B) Western Blotting. PC-9/AR cells were exposed to the tested free drugs or NPs for 48 h and then harvested for Western blotting to detect cleavage of PARP and caspase-3. CF, cleaved form.
OSI+SEL NPs combination therapy effectively inhibited the growth of OSI-resistant NSCLC xenograft in nude mice.
Encouraged by the promising anticancer effects of OSI+SEL NPs combination therapy observed with in vitro cell-based studies, we decided to further investigate the in vivo anticancer efficacy of NPs with an OSI-resistant xenograft tumor model established with PC-9/AR cells on nude mice. When tumor sizes were close to 100 mm3, tumor-bearing mice were treated with different formulations via intravenous (IV) injection once every three days for six injections. As shown in Figure 5A, the monotherapy with OSI NP (10 mg/kg) or SEL NP (4.3 mg/kg)) showed almost no effects on tumor growth inhibition, while the OSI+SEL NP combination therapy (OSI 10 mg/kg & SEL 4.3 mg/kg) significantly suppressed the tumor growth. At the end of the study, tumors were collected and weighed (Figure 5B&C). The results further confirmed the potent anticancer efficacy of the OSI+SEL NP combination therapy. Although the tumor weight in OSI NP or SEL NP showed no significant difference compared with the control group, the tumor weight was significantly smaller in the OSI+SEL NP treatment group. The OSI+SEL NP and other NPs also demonstrated excellent safety profiles as measured by the change of body weight (Figure 5D). The body weight of mice in each group increased gradually during the course of in vivo studies, and no dramatic decrease in body weight was observed. Also, there was no significant difference in body weight between control groups and other treatment groups.
Figure 5. In vivo anticancer studies with PC-9/AR xenograft tumor model.
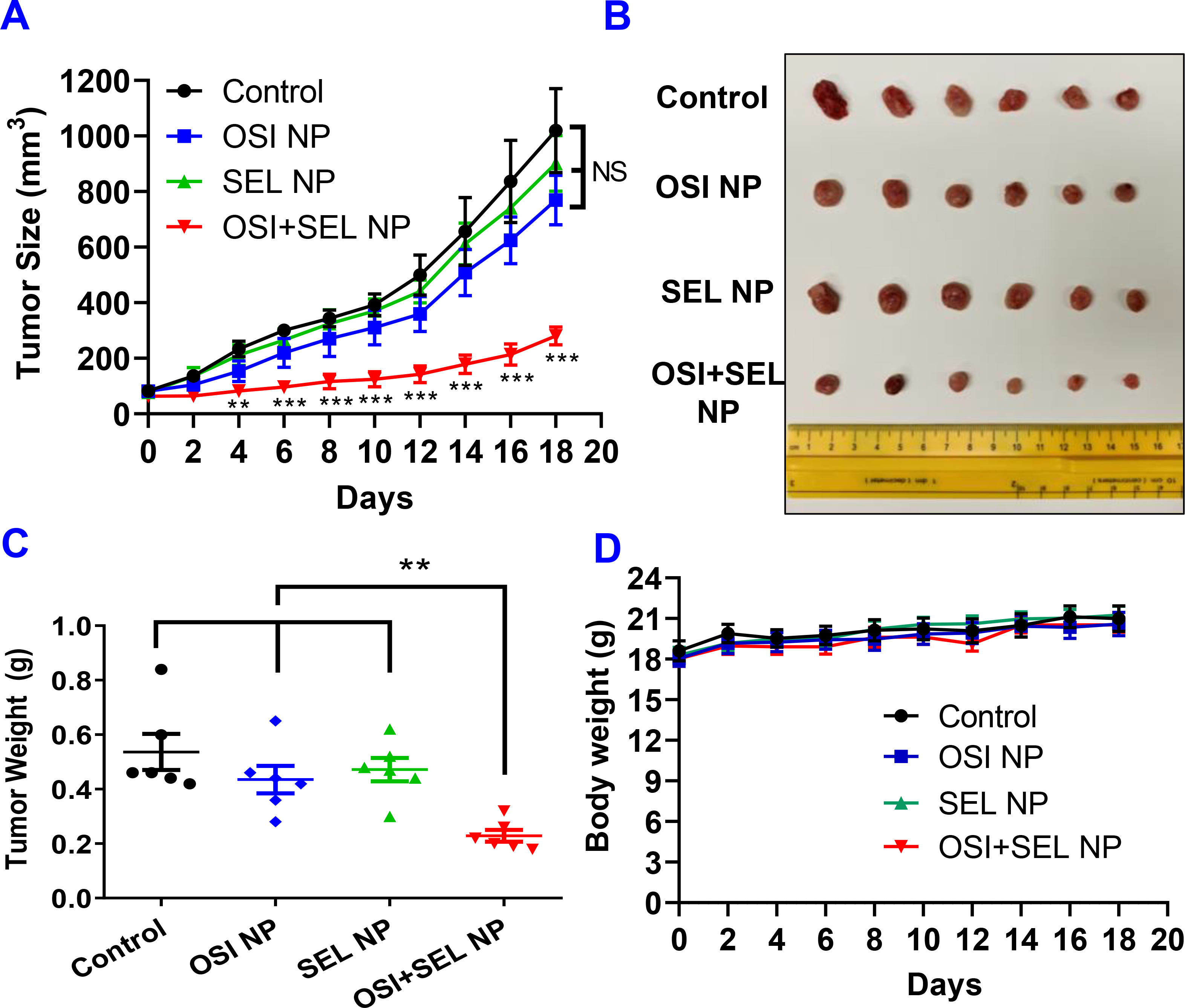
(A) Change of tumor size in tumor-bearing nude mice received intravenous injection of different formulations. At the end of the study, isolated tumors were photographed (B) and weighted (C). Body weight of PC-9/AR tumor-bearing mice during the course of study (D). Results are mean ± SD (n=6). **, P < 0.01; ***, P < 0.001, compared with control and monotherapy groups.
4. DISCUSSION
The development of drug resistance is a significant issue in cancer therapy. The prolonged treatment with a single drug often resulted in the activation of alternative survival pathways [24], occurrence of second mutations in target proteins, or impaired cell apoptosis [25]. Also, the upregulation of drug-resistant transporters could contribute to drug resistance by actively eliminating drugs from cancer cells [26]. The re-programming of tumor micro-environment and metabolism is another factor leading to the development of resistance to treatment [27, 28]. Combination therapy is a commonly used strategy to overcome drug resistance. The combinational use of two or more drugs can significantly improve the therapeutic effects and effectively treat resistant cancer through either additive or synergistic effects. Previous studies have explored various combination therapy strategies, including chemotherapy drugs plus drug-resistant transporter inhibitors [29, 30], drugs targeting different signaling pathways or different proteins in the same signaling pathway [31], drugs targeting different cell subpopulations in heterogeneous cancers [32], and drugs with varying mechanisms of action.
In the current study, we developed NP formulations for co-delivery of OSI and SEL to effectively treat OSI-resistant NSCLC as a combination therapy. Combination therapies have been frequently used to overcome TKI resistance and improve the anticancer efficacy in NSCLC by synergistically inhibiting multiple receptor tyrosine kinases in the ErbB family or inhibiting downstream pathways [33]. The abnormal EGFR can activate multiple downstream signaling pathways, including MAPK (mitogen-activated protein kinase) and AKT/PI3K/mTOR pathways. Among these pathways, MAPK signaling pathway plays a critical role in the regulation of tumor cell proliferation and survival [8–10]. Previous studies demonstrated that the combination therapies of OSI plus MEK inhibitors, ERK inhibitors, or other drugs could re-sensitize resistant NSCLC to OSI treatment in different preclinical models[11, 12, 34–37]. A clinical study reported that OSI-resistant NSCLC patients with BRAF fusion responded well to OSI and MEK inhibitor combination treatment [14]. These combinational therapies showed satisfactory performance in treating OSI-resistant NSCLC in preclinical studies, indicating their potential clinical use.
NPs are promising delivery systems for anticancer drugs [38]. NPs can deliver drugs into tumors through multiple mechanisms, including enhanced permeability and retention (EPR) effects [39] and transcytosis [40, 41]. NPs have also been used for co-delivering multiple drugs in combination cancer therapy and showed several benefits [42]. (1) NPs can alter the in vivo biodistribution and synchronize the pharmacokinetics profiles of co-delivered drugs; (2) NPs can be used to prepare formulations for drugs, which are otherwise unable to be used in patients due to low solubility, poor stability, or other formulation issues. Micelle-based NP is one of the most promising delivery systems. Micelle NPs are usually prepared with amphiphilic macromolecules having hydrophilic moieties (e.g., polyethylene glycol) and hydrophobic moieties. Amphiphilic macromolecules form NPs in an aqueous environment through self-assembly. Drugs are physically loaded into the hydrophobic core of micelle NPs. The hydrophilic shell of micelle NPs can improve the colloid stability and prevent NP aggregation during storage. The hydrophilic shell can also enhance the biocompatibility of NPs, reduce opsonization, and prolong NP circulation in the blood. Micelle NPs can be prepared with a simple process with high efficiency [16]. The simple formulation and straightforward preparation process make it possible to produce micelle NPs on a large scale and facilitate clinical translation and commercialization. In many studies, micelle NPs were often prepared with amphiphilic macromolecules as the delivery carriers. These macromolecule carriers usually were inert materials without any pharmacological activities. Recently, polymer-drug conjugate micelle NPs have been explored as a promising alternative [43–45]. In these systems, hydrophobic drug molecules were conjugated with hydrophilic polymers through covalent bonds to produce amphiphilic polymer-drug conjugates, which form NPs through self-assembly. The conjugated hydrophobic drugs functioned as the hydrophobic moiety to drive the self-assembly process and stabilize NPs through hydrophobic interactions. Since drug molecules are conjugated with polymers through covalent bonds, it can achieve high drug loading efficiency, have good drug loading stability, and minimize drug leakage or burst drug release. In the current study, we synthesized a PEG-SEL conjugate to prepare the NP formulation. This SEL conjugate NP showed significant higher drug concentration and loading efficiency than NPs prepared by physically encapsulation of SEL free drugs (Figure S4). In addition, the NP formulation prepared with PEG-SEL conjugate showed excellent colloid stability and could be stored as NP suspension for at least weeks with good stability. Furthermore, the PEG-SEL conjugate is an amphiphilic macromolecule and can also be used as the delivery carriers for co-delivering OSI and prepare “excipient-free or excipient-minimal” NP formulations [46, 47].
Conjugate prodrug is a promising drug delivery strategy to improve drug physicochemical properties, pharmacokinetics, biodistribution, metabolism, cellular uptake, and other properties [48–51]. Polymer-drug conjugate or other conjugate prodrugs can achieve extended drug release. This property is beneficial for applications that require prolonged exposure of drugs at a low concentration [52]. However, insufficient drug release is a notorious challenge for their use in cancer therapy. Also, premature drug leakage outside of the tumor tissues should be minimized to avoid toxic side effects. The selection of suitable linkers is critical for tumor-specific drug release to achieve efficient anticancer activities and reduce toxic side effects [48]. In previous studies, different types of linkers were explored to synthesize tumor microenvironment stimuli-responsive conjugate prodrugs [53]. PEG-SEL conjugate synthesized in this study could be efficiently cleaved in response to elevated ROS level and release SEL drug. The increased production of ROS is one of the hallmarks of cancer. ROS has a critical role in cancer progression and resistance [53]. Compared to normal tissues, tumor tissues often have much higher levels of ROS, such as hydrogen peroxides (H2O2), hydroxyl radicals (•OH), peroxynitrites (ONOO−), and superoxides (O2−) [54, 55]. The use of ROS-responsive prodrug conjugate is a promising strategy in cancer therapy, which could trigger the drug release in tumor microenvironment with elevated ROS levels and minimize drug leakage in normal tissues with lower levels of ROS [56, 57]. We also found that the OSI-resistant PC9/AR cells showed significantly elevated ROS than parenteral OSI-sensitive PC9 cells (Figure S11), indicating the potential use of ROS-responsive prodrug in this cells. The cleavage of ROS-responsive prodrug in cancer cells with low ROS levels will be insufficient and limit its application in this type of cancer. However, we could combine ROS-responsive prodrug with a ROS-inducing agent (e.g., lapachone) [58] which can bolster the ROS levels in the tumor microenvironment and enhance the efficacy of ROS prodrug. It will be helpful to develop a diagnostic tool to measure the ROS levels in tumors as a biomarker to assist in the selection of ROS-responsive drug formulations.
In the current study, the PEG-S-SEL conjugate was synthesized with a ROS-responsive thiothether linker, with PEG 2000 as the polymer, and at the SEL/PEG ratio of 1:1. In future studies, it will be worthwhile to explore PEG-SEL conjugates with different linkers, drug/PEG ratios, and PEG molecular weights. These design parameters will have a significant impact on the properties of PEG-SEL NPs. The knowledge about the structure-performance relationship of PEG-S-SEL conjugate will assist us in optimizing the formulation for OSI and SEL combination therapy.
In conclusion, we developed a prototype NP formulation for OSI+SEL combination therapy and performed proof-of-concept studies to evaluate its anticancer efficacies with in vitro studies and an in vivo tumor model. Although the use of OSI+SEL free drug combination therapy also showed promising activities, the combination therapy with free drugs might increase systemic toxicities or side effects as dose-limiting toxicities were reported in a recent clinical trial [15]. The OSI+SEL NP developed in this study will have the promise to targeted deliver OSI and SEL into tumor tissues and thus reduce the toxic side effects. The NP formulations will enhance the drug delivery into the tumor and minimize drug exposure in normal tissues. The use of ROS-responsive prodrug will further reduce the off-target drug exposure in the normal tissue with low ROS levels. Two drugs (i.e., OSI and SEL) simultaneously delivered in a single NP formulation will also synchronize their exposure in tumor cells and achieve better synergistic anticancer efficacy. Furthermore, the OSI+SEL NP developed in our study can be further modified with targeting ligand (e.g., transferrin receptor-binding peptide) to facilitate the transport cross blood-brain barrier and effectively treat brain metastatic NSCLC cancer [59]. The current proof-of-concept study demonstrated the feasibility of using the OSI+SEL NP to treat OSI-resistant NSCLC. In the future, additional studies should be performed to further develop this formulation and facilitate the clinical translation. Additional preclinical animal studies should be performed to evaluate the in vivo pharmacokinetics and biodistribution. Also, the safety of this NP formulation should be assessed with comprehensive pathological histology analysis and other proper assays. In the current study, we evaluated the efficacy of NP formulation with a xenograft tumor model established with PC9-AR OSI-resistant NSCLC cells. It is critical to use additional tumor models established with other OSI-resistant cells (e.g., HCC827/AR, PC-9/GR/AR) as well as NSCLC patient-derived xenografts (PDXs) to evaluate the anticancer efficacies of OSI+SEL NP formulation.
Supplementary Material
Scheme 1.
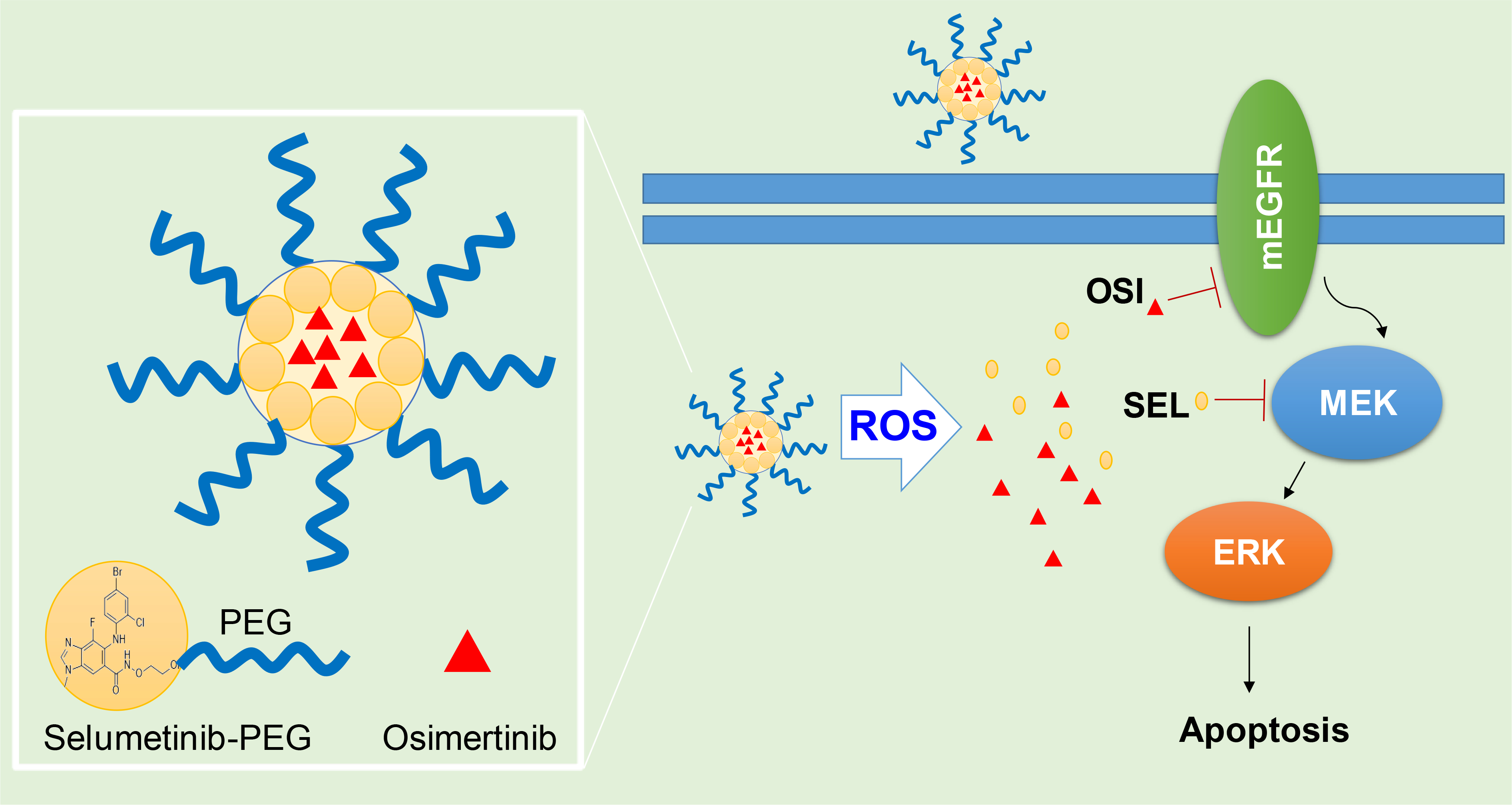
Nanoparticle for co-delivery of Selumetinib (SEL) and Osimertinib (OSI) to overcome drug resistance in NSCLC though synergistic effects on mEGFR and MEK.
Statement of Significance.
Osimertinib (OSI) is the first FDA-approved third-generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor. It has been successfully used for treating non-small cell lung cancer (NSCLC) patients with activating EGFR mutation. However, patients treated with OSI ultimately develop acquired resistance. This study developed OSI and selumetinib (SEL) co-delivering nanoparticles to overcome OSI-acquired resistance in NSCLC. PEG-SEL conjugate functions as reactive oxygen species (ROS)-responsive prodrug and forms micelle nanoparticles through self-assembly to deliver OSI. The combination NP can simultaneously inhibit EGFR and mitogen-activated protein kinase (MEK), thus effectively inducing apoptosis in OSI-resistant NSCLC cells. In summary, the OSI and SEL nanoparticle combination therapy showed promising anticancer efficacy and demonstrated potential for treating NSCLC patients with OSI acquired resistance.
ACKNOWLEDGEMENT
This work was financially supported by the following resources: Auburn University start-up fund (F. Li). SYS is a Georgia Research Alliance Distinguished Cancer Scientist and supported by NIH/NCI R01 CA223220, R01 CA245386, UG1 CA233259 and Lung Cancer SPORE P50 CA217691 DRP award and Emory University Winship Cancer Institute lung cancer pilot fund.
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
SUPPORTING INFORMATION
Additional Supporting material is available.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Siegel RL, Miller KD, Jemal A, Cancer statistics, 2018, CA: A Cancer Journal for Clinicians 68 (2018) 7–30. [DOI] [PubMed] [Google Scholar]
- [2].Andrews Wright NM, Goss GD, Third-generation epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of non-small cell lung cancer, Transl Lung Cancer Res 8 (2019) S247–s264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lim SM, Syn NL, Cho BC, Soo RA, Acquired resistance to EGFR targeted therapy in non-small cell lung cancer: Mechanisms and therapeutic strategies, Cancer Treat Rev 65 (2018) 1–10. [DOI] [PubMed] [Google Scholar]
- [4].Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ, Hughes G, Rahi A, Jacobs VN, Red Brewer M, Ichihara E, Sun J, Jin H, Ballard P, Al-Kadhimi K, Rowlinson R, Klinowska T, Richmond GH, Cantarini M, Kim DW, Ranson MR, Pao W, AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer, Cancer Discov 4 (2014) 1046–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, Zhou C, Reungwetwattana T, Cheng Y, Chewaskulyong B, Shah R, Cobo M, Lee KH, Cheema P, Tiseo M, John T, Lin MC, Imamura F, Kurata T, Todd A, Hodge R, Saggese M, Rukazenkov Y, Soria JC, Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC, N Engl J Med 382 (2020) 41–50. [DOI] [PubMed] [Google Scholar]
- [6].Ricordel C, Friboulet L, Facchinetti F, Soria J-C, Molecular mechanisms of acquired resistance to third-generation EGFR-TKIs in EGFR T790M-mutant lung cancer, Annals of Oncology 29 (2018) i28–i37. [DOI] [PubMed] [Google Scholar]
- [7].Ramalingam SS, Yang J, Lee CK, Kurata T, Kim D-W, John T, Nogami N, Ohe Y, Mann H, Rukazenkov Y, Osimertinib as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer, J Clin Oncol 36 (2018) 841–849. [DOI] [PubMed] [Google Scholar]
- [8].Rolfo C, Giovannetti E, Hong DS, Bivona T, Raez LE, Bronte G, Buffoni L, Reguart N, Santos ES, Germonpre P, Taron M, Passiglia F, Van Meerbeeck JP, Russo A, Peeters M, Gil-Bazo I, Pauwels P, Rosell R, Novel therapeutic strategies for patients with NSCLC that do not respond to treatment with EGFR inhibitors, Cancer Treatment Reviews 40 (2014) 990–1004. [DOI] [PubMed] [Google Scholar]
- [9].Wee P, Wang Z, Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways, Cancers 9 (2017) 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Eberlein CA, Stetson D, Markovets AA, Al-Kadhimi KJ, Lai Z, Fisher PR, Meador CB, Spitzler P, Ichihara E, Ross SJ, Ahdesmaki MJ, Ahmed A, Ratcliffe LE, O’Brien EL, Barnes CH, Brown H, Smith PD, Dry JR, Beran G, Thress KS, Dougherty B, Pao W, Cross DA, Acquired Resistance to the Mutant-Selective EGFR Inhibitor AZD9291 Is Associated with Increased Dependence on RAS Signaling in Preclinical Models, Cancer Res 75 (2015) 2489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Shi P, Oh YT, Deng L, Zhang G, Qian G, Zhang S, Ren H, Wu G, Legendre B Jr., Anderson E, Ramalingam SS, Owonikoko TK, Chen M, Sun SY, Overcoming Acquired Resistance to AZD9291, A Third-Generation EGFR Inhibitor, through Modulation of MEK/ERK-Dependent Bim and Mcl-1 Degradation, Clin Cancer Res 23 (2017) 6567–6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Li Y, Zang H, Qian G, Owonikoko TK, Ramalingam SR, Sun SY, ERK inhibition effectively overcomes acquired resistance of epidermal growth factor receptor-mutant non-small cell lung cancer cells to osimertinib, Cancer 126 (2020) 1339–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gu J, Yao W, Shi P, Zhang G, Owonikoko TK, Ramalingam SS, Sun S-Y, MEK or ERK inhibition effectively abrogates emergence of acquired osimertinib resistance in the treatment of epidermal growth factor receptor–mutant lung cancers, Cancer 126 (2020) 3788–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dagogo-Jack I, Piotrowska Z, Cobb R, Banwait M, Lennerz JK, Hata AN, Digumarthy SR, Sequist LV, Response to the Combination of Osimertinib and Trametinib in a Patient With EGFR-Mutant NSCLC Harboring an Acquired BRAF Fusion, J Thorac Oncol 14 (2019) e226–e228. [DOI] [PubMed] [Google Scholar]
- [15].Oxnard GR, Yang JC, Yu H, Kim SW, Saka H, Horn L, Goto K, Ohe Y, Mann H, Thress KS, Frigault MM, Vishwanathan K, Ghiorghiu D, Ramalingam SS, Ahn MJ, TATTON: a multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer, Ann Oncol 31 (2020) 507–516. [DOI] [PubMed] [Google Scholar]
- [16].Li F, Danquah M, Mahato RI, Synthesis and Characterization of Amphiphilic Lipopolymers for Micellar Drug Delivery, Biomacromolecules 11 (2010) 2610–2620. [DOI] [PubMed] [Google Scholar]
- [17].Shi P, Oh YT, Zhang G, Yao W, Yue P, Li Y, Kanteti R, Riehm J, Salgia R, Owonikoko TK, Ramalingam SS, Chen M, Sun SY, Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment, Cancer Lett 380 (2016) 494–504. [DOI] [PubMed] [Google Scholar]
- [18].Chang Y, Jiang J, Chen W, Yang W, Chen L, Chen P, Shen J, Qian S, Zhou T, Wu L, Hong L, Huang Y, Li F, Biomimetic metal-organic nanoparticles prepared with a 3D-printed microfluidic device as a novel formulation for disulfiram-based therapy against breast cancer, Applied Materials Today 18 (2020) 100492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sun SY, Yue P, Dawson MI, Shroot B, Michel S, Lamph WW, Heyman RA, Teng M, Chandraratna RA, Shudo K, Hong WK, Lotan R, Differential effects of synthetic nuclear retinoid receptor-selective retinoids on the growth of human non-small cell lung carcinoma cells, Cancer Res 57 (1997) 4931–9. [PubMed] [Google Scholar]
- [20].Chen W, Yang W, Chen P, Huang Y, Li F, Disulfiram Copper Nanoparticles Prepared with a Stabilized Metal Ion Ligand Complex Method for Treating Drug-Resistant Prostate Cancers, ACS Applied Materials & Interfaces 10 (2018) 41118–41128. [DOI] [PubMed] [Google Scholar]
- [21].Liu X, Yue P, Zhou Z, Khuri FR, Sun SY, Death receptor regulation and celecoxib-induced apoptosis in human lung cancer cells, J Natl Cancer Inst 96 (2004) 1769–80. [DOI] [PubMed] [Google Scholar]
- [22].Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, Terada Y, Kano M, Miyazono K, Uesaka M, Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size, Nature nanotechnology 6 (2011) 815–823. [DOI] [PubMed] [Google Scholar]
- [23].Yang X, Xue X, Luo Y, Lin T-Y, Zhang H, Lac D, Xiao K, He Y, Jia B, Lam KS, Li Y, Sub-100nm, long tumor retention SN-38-loaded photonic micelles for tri-modal cancer therapy, Journal of controlled release : official journal of the Controlled Release Society 261 (2017) 297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Nussinov R, Tsai C-J, Jang H, A New View of Pathway-Driven Drug Resistance in Tumor Proliferation, Trends in Pharmacological Sciences 38 (2017) 427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Morgillo F, Della Corte CM, Fasano M, Ciardiello F, Mechanisms of resistance to EGFR-targeted drugs: lung cancer, ESMO open 1 (2016) e000060–e000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gottesman MM, Fojo T, Bates SE, Multidrug resistance in cancer: role of ATP–dependent transporters, Nature Reviews Cancer 2 (2002) 48–58. [DOI] [PubMed] [Google Scholar]
- [27].Son B, Lee S, Youn H, Kim E, Kim W, Youn B, The role of tumor microenvironment in therapeutic resistance, Oncotarget 8 (2017) 3933–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Faubert B, Solmonson A, DeBerardinis RJ, Metabolic reprogramming and cancer progression, Science 368 (2020) eaaw5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang H, Li F, Du C, Wang H, Mahato RI, Huang Y, Doxorubicin and Lapatinib Combination Nanomedicine for Treating Resistant Breast Cancer, Molecular Pharmaceutics 11 (2014) 2600–2611. [DOI] [PubMed] [Google Scholar]
- [30].Li F, Danquah M, Singh S, Wu H, Mahato RI, Paclitaxel- and lapatinib-loaded lipopolymer micelles overcome multidrug resistance in prostate cancer, Drug Deliv Transl Res 1 (2011) 420–8. [DOI] [PubMed] [Google Scholar]
- [31].Danquah M, Li F, Duke CB, Miller DD, Mahato RI, Micellar Delivery of Bicalutamide and Embelin for Treating Prostate Cancer, Pharmaceutical Research 26 (2009) 2081. [DOI] [PubMed] [Google Scholar]
- [32].Batlle E, Clevers H, Cancer stem cells revisited, Nature Medicine 23 (2017) 1124–1134. [DOI] [PubMed] [Google Scholar]
- [33].Rolfo C, Giovannetti E, Hong DS, Bivona T, Raez LE, Bronte G, Buffoni L, Reguart N, Santos ES, Germonpre P, Taron M, Passiglia F, Van Meerbeeck JP, Russo A, Peeters M, Gil-Bazo I, Pauwels P, Rosell R, Novel therapeutic strategies for patients with NSCLC that do not respond to treatment with EGFR inhibitors, Cancer Treat Rev 40 (2014) 990–1004. [DOI] [PubMed] [Google Scholar]
- [34].Gu J, Yao W, Shi P, Zhang G, Owonikoko TK, Ramalingam SS, Sun SY, MEK or ERK inhibition effectively abrogates emergence of acquired osimertinib resistance in the treatment of epidermal growth factor receptor-mutant lung cancers, Cancer 126 (2020) 3788–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zang H, Qian G, Arbiser J, Owonikoko TK, Ramalingam SS, Fan S, Sun SY, Overcoming acquired resistance of EGFR-mutant NSCLC cells to the third generation EGFR inhibitor, osimertinib, with the natural product honokiol, Mol Oncol 14 (2020) 882–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zang H, Qian G, Zong D, Fan S, Owonikoko TK, Ramalingam SS, Sun SY, Overcoming acquired resistance of epidermal growth factor receptor-mutant non-small cell lung cancer cells to osimertinib by combining osimertinib with the histone deacetylase inhibitor panobinostat (LBH589), Cancer 126 (2020) 2024–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Liu Z, Gao W, Synergistic effects of Bcl-2 inhibitors with AZD9291 on overcoming the acquired resistance of AZD9291 in H1975 cells, Archives of Toxicology 94 (2020) 3125–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hartshorn CM, Bradbury MS, Lanza GM, Nel AE, Rao J, Wang AZ, Wiesner UB, Yang L, Grodzinski P, Nanotechnology Strategies To Advance Outcomes in Clinical Cancer Care, ACS Nano 12 (2018) 24–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Fang J, Islam W, Maeda H, Exploiting the dynamics of the EPR effect and strategies to improve the therapeutic effects of nanomedicines by using EPR effect enhancers, Adv Drug Deliv Rev 157 (2020) 142–160. [DOI] [PubMed] [Google Scholar]
- [40].Sindhwani S, Syed AM, Ngai J, Kingston BR, Maiorino L, Rothschild J, MacMillan P, Zhang Y, Rajesh NU, Hoang T, Wu JLY, Wilhelm S, Zilman A, Gadde S, Sulaiman A, Ouyang B, Lin Z, Wang L, Egeblad M, Chan WCW, The entry of nanoparticles into solid tumours, Nature Materials 19 (2020) 566–575. [DOI] [PubMed] [Google Scholar]
- [41].Liu X, Jiang J, Meng H, Transcytosis - An effective targeting strategy that is complementary to “EPR effect” for pancreatic cancer nano drug delivery, Theranostics 9 (2019) 8018–8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Rawal S, Patel MM, Threatening cancer with nanoparticle aided combination oncotherapy, Journal of Controlled Release 301 (2019) 76–109. [DOI] [PubMed] [Google Scholar]
- [43].Mutlu-Agardan NB, Sarisozen C, Torchilin VP, Cytotoxicity of Novel Redox Sensitive PEG2000-S-S-PTX Micelles against Drug-Resistant Ovarian and Breast Cancer Cells, Pharmaceutical Research 37 (2020) 65. [DOI] [PubMed] [Google Scholar]
- [44].Larson N, Ghandehari H, Polymeric conjugates for drug delivery, Chem Mater 24 (2012) 840–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Senevirathne SA, Washington KE, Miller JB, Biewer MC, Oupicky D, Siegwart DJ, Stefan MC, HDAC inhibitor conjugated polymeric prodrug micelles for doxorubicin delivery, Journal of Materials Chemistry B 5 (2017) 2106–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Yuan Y, Bo R, Jing D, Ma Z, Wang Z, Lin T.-y., Dong L, Xue X, Li Y, Excipient-free porphyrin/SN-38 based nanotheranostics for drug delivery and cell imaging, Nano Research 13 (2020) 503–510. [Google Scholar]
- [47].Zhang L, Zhou J, Yan Y, Zhou X, Zhou Q, Du R, Hu S, Ge W, Huang Y, Xu H, Kong Y, Zheng H, Ding Y, Shen Y, Wang W, Excipient-free nanodispersion of 7-ethyl-10-hydroxycamptothecin exerts potent therapeutic effects against pancreatic cancer cell lines and patient-derived xenografts, Cancer Letters 465 (2019) 36–44. [DOI] [PubMed] [Google Scholar]
- [48].Irby D, Du C, Li F, Lipid–Drug Conjugate for Enhancing Drug Delivery, Molecular Pharmaceutics 14 (2017) 1325–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Au - Li F, Au - Snow-Davis C, Au - Du C, Au - Bondarev ML, Au - Saulsbury MD, Au - Heyliger SO, Preparation and Characterization of Lipophilic Doxorubicin Pro-drug Micelles, JoVE (2016) e54338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Chen Y, Zhang M, Jin H, Tang Y, Wang H, Xu Q, Li Y, Li F, Huang Y, Intein-mediated site-specific synthesis of tumor-targeting protein delivery system: Turning PEG dilemma into prodrug-like feature, Biomaterials 116 (2017) 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bioconjugate Therapeutics: Current Progress and Future Perspective, Molecular Pharmaceutics 14 (2017) 1321–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ekladious I, Colson YL, Grinstaff MW, Polymer–drug conjugate therapeutics: advances, insights and prospects, Nature Reviews Drug Discovery 18 (2019) 273–294. [DOI] [PubMed] [Google Scholar]
- [53].Xie A, Hanif S, Ouyang J, Tang Z, Kong N, Kim NY, Qi B, Patel D, Shi B, Tao W, Stimuli-responsive prodrug-based cancer nanomedicine, EBioMedicine 56 (2020) 102821–102821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Trachootham D, Alexandre J, Huang P, Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach?, Nature Reviews Drug Discovery 8 (2009) 579–591. [DOI] [PubMed] [Google Scholar]
- [55].Yin W, Ke W, Chen W, Xi L, Zhou Q, Mukerabigwi JF, Ge Z, Integrated block copolymer prodrug nanoparticles for combination of tumor oxidative stress amplification and ROS-responsive drug release, Biomaterials 195 (2019) 63–74. [DOI] [PubMed] [Google Scholar]
- [56].Luo C, Sun J, Liu D, Sun B, Miao L, Musetti S, Li J, Han X, Du Y, Li L, Huang L, He Z, Self-Assembled Redox Dual-Responsive Prodrug-Nanosystem Formed by Single Thioether-Bridged Paclitaxel-Fatty Acid Conjugate for Cancer Chemotherapy, Nano Letters 16 (2016) 5401–5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Peiró Cadahía J, Previtali V, Troelsen NS, Clausen MH, Prodrug strategies for targeted therapy triggered by reactive oxygen species, MedChemComm 10 (2019) 1531–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Li Q, Hou W, Li M, Ye H, Li H, Wang Z, Ultrasound Combined with Core Cross-Linked Nanosystem for Enhancing Penetration of Doxorubicin Prodrug/Beta-Lapachone into Tumors, International journal of nanomedicine 15 (2020) 4825–4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhao P, Zhang J, Wu A, Zhang M, Zhao Y, Tang Y, Wang B, Chen T, Li F, Zhao Q, Huang Y, Biomimetic codelivery overcomes osimertinib-resistant NSCLC and brain metastasis via macrophage-mediated innate immunity, Journal of Controlled Release 329 (2021) 1249–1261. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.