

Abstract

Pregnane X receptor (PXR) that orchestrates the intricate network of xeno- and endobiotic metabolism is considered as a promising therapeutic target for cholestasis. In this study, the human PXR (hPXR) agonistic bioassay-guided isolation of Euphorbia lathyris followed by the structural modification led to the construction of a lathyrane diterpenoid library (1–34). Subsequent assay of this library led to the identification of a series of potent hPXR agonists, showing better efficacy than that of typical hPXR agonist, rifampicin. The most active compound, 8, could dose-dependently activate hPXR at micromolar concentrations and significantly up-regulate the expressions of PXR downstream genes CYP3A4, CYP2B6, and MDR1. The structure–activity relationships (SARs) studied in combination with molecular modeling suggested that acyloxy at C-7 and the presence of 14-carbonyl were essential to the activity. These findings suggested that lathyrane diterpenoids could serve as a new type of hPXR agonist for future anticholestasis drug development.
Keywords: hPXR agonist, lathyrane diterpenoid, structural modification, structure−activity relationships
Cholestasis is a clinical condition where bile flow is defective, either due to impaired secretion by hepatocytes or by obstruction to the bile flow, resulting in intrahepatic accumulation of toxins such as bile acids (BAs).1 The progression of cholestasis would induce hepatocellular damage, causing a series of cholestatic liver diseases such as primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC).1,2 Thus, elimination of excess BAs or maintaining the homeostasis of bile flow is considered as the prevailing therapeutic strategy for the treatment of various cholestatic conditions. Until now, only two drugs, ursodeoxycholic acid (UDCA) and obeticholic acid (OCA), have been approved by the FDA for the treatment of cholestatic conditions. UDCA is the first choice of PBC drug that could stimulate secretion of hepatocytes and cholangiocytes via stabilization of the “biliary HCO3– umbrella”.3 OCA is a farnesoid X receptor (FXR) agonist that inhibits bile acid synthesis and uptake by activation of BAs detoxification pathways.3 However, only 50–60% PBC patients are able to respond to these drugs,1,4 leaving the nonresponders a high risk of progression to biliary fibrosis, cirrhosis, end-stage liver disease, and death.5 Therefore, there remains an urgent need for development of novel therapeutic agents with different mechanism to delay or relieve cholestasis-associated symptoms.
Pregnane X receptor (PXR) is a member of the nuclear receptors (NRs) superfamily that orchestrates the intricate network of endobiotic and xenobiotic metabolism.6,7 Compared with other NRs, PXR contains an N-terminal regulatory domain, a highly conserved DNA-binding domain (DBD), a hinge region, a less conserved ligand-binding domain (LBD), and a C-terminal domain.8 PXR is predominantly distributed in the liver and intestine, where it regulates the expression of phases I and II drug-metabolizing enzymes, such as cytochrome P450 (CYPs), alcohol and aldehyde dehydrogenases, sulfotransferases (SULTs) and glucuronyltransferases (UGTs),9 and numerous phase III efflux transporters (e.g., MDR1 and MRP) to control the metabolism of endobiotics (e.g., BAs, glucose, and lipid) and xenobiotics (e.g., therapeutic agents).10−13 Intensive evidences have proved the important role of PXR signaling in the maintenance of BA homeostasis.13 Activated-PXR could promote BA detoxification by activation of the hydroxylation and conjugation pathways. The former converts BAs into its hydroxyl derivatives with the assistance of enzymes such as CYP3A and CYP2B, whereas the latter further transforms these hydroxyl derivatives to more hydrophilic conjugates under the catalysis of enzymes UGTs, GSTs, and SULTs. Meanwhile, the activation of PXR could upregulate transporters such as P-glycoprotein (P-gp, encoded by MDR1), which finally transports the detoxified BA metabolites into the bile or urine.14 Strikingly, a recent study has shown that rifampicin, a typical human PXR (hPXR) agonist, could completely reverse severe persistent hepatocellular secretory failure induced by drugs or transient biliary obstruction in formerly healthy individuals, an enormous relief for otherwise desperate patients.15 Thus, PXR has emerged as a promising therapeutic target in cholestasis, and the discovery of novel PXR agonist is of great value in anticholestasis drug development.
Natural products have historically played an important and irreplaceable role in drug discovery. This role has been underscored in anticholestasis drug development, as the only two approved PBC drugs, UDCA and OCA, are all derived from natural products. In recent years, plenty of natural hPXR agonists, such as tanshinone IIA, schisandrol B, solomonsterols A and B, have been reported from various medicinal plants,16−18 suggesting that the search for hPXR agonists from natural source might be a shortcut in anticholestasis drug campaign. In the frame of our interest in the discovery of biologically significant macrocyclic diterpenoids from Euphorbiaceae species,19,20 we conducted the hPXR agonistic bioassay-guided isolation on a traditional Chinese medicine, Euphorbia lathyris, which led to the isolation of 16 lathyrane diterpenoids. The subsequent structural modification generated a lathyrane library containing 34 compounds for systematic investigation. Herein, we described the bioassay-guided isolation, structural elucidation, modification, hPXR agonistic activity, and SARs of these diterpenoids, as well as the molecular recognition mechanism between 8 and hPXR.
The dried seeds of E. lathyris (8 kg) were extracted with 95% ethanol at room temperature (rt) to give a crude extract, which was suspended in water and then successively partitioned with petroleum ether, EtOAc, and n-BuOH. Each fraction was tested for agonistic activity on the hPXR, and the EtOAc fraction which displayed a promising hPXR agonistic activity (2.1-fold increasing at 50 μg/mL) was selected for further chemical investigation. Subsequent systematic purification of this fraction using various chromatographic methods led to the identification of 16 structurally related lathyrane diterpenoids (1–16) (Figure 1), of which 1–5 were new compounds.
Figure 1.

Structures of compounds 1–34.
Compound 1 exhibited a molecular formula of C29H34O7 as determined by HRESIMS ion at m/z 517.2193 [M + Na]+ (calcd 517.2197). The 1D NMR spectra of 1 were similar to those of a known compound 15,17-di-O-acetyl-3-O-benzoyl-17-hydroxyjolkinol,21 with the major differences being the absence of the 17-acetyl group and the presence of the C-7 ketone group (δC 203.7) in 1. This was supported by the upfield-shifted carbon signal of C-17 and the downfield-shifted carbon signals of C-5 and C-8 in 1 with respect to those in known compound [δC 58.0 (C-17), 137.1 (C-5), 36.3 (C-8) in 1; δC 64.0 (C-17), 124.9 (C-5), 28.5 (C-8) in known compound]. The planar structure of 1 was further secured by detailed analyses of its 2D NMR data (Figure S2.1). The stereochemistry of 1 was assigned to be the same as that of 15,17-di-O-acetyl-3-O-benzoyl-17-hydroxyjolkinol by a NOESY experiment and comparison of their 1D NMR data (Table S1). Therefore, the structure of 1 was elucidated as 15-acetoxy-3-benzoyloxy-17-hydroxy-14-oxolathyra-5E,12E-diene and was given the trivial name euphlathyrinoid A.
Compound 2 had the molecular formula of C27H34O5 as determined by HRESIMS and 13C NMR data. The 1D NMR data of 2 bore a high resemblance to those of known compound 15,17-diacetoxy-3-hydroxy-14-oxolathyra-5E,12E-diene,22 except for the presence of a benzoyl group in 2 instead of two acetyl group in the known compound. The benzoyl group was located at C-17 by the HMBC correlation from H2-17 (δH 4.61 and 4.32) to the benzoyl carbonyl carbon (δC 166.5). The stereochemistry of 2 was assigned to be the same as that of known compound by comparison of their NOE correlations and 1D NMR data, except for the Z configuration of Δ5 was assigned by the NOESY correlations of H-4/H-17a (Figure S2.2). Thus, the structure of 2 was elucidated as 17-benzoyloxy-3,15-dihydroxy-14-oxolathyra-5Z,12E-diene and was given a trivial name euphlathyrinoid B.
Compound 3 had a molecular formula of C31H36O8 as established by HRESIMS data at m/z 559.2306 [M + Na]+ (calcd 559.2302). The NMR data of 3 showed high similarity to those of coisolated known compound 5,15-diacetoxy-3-benzoyloxy-7-hydroxy-14-oxolathyra-6(17),12E-diene (9),22 except for the presence of a ketone signal (δC 199.9) in 3 instead of an oxygenated-methine signal for C-7 (δH 4.23; δC 78.2) in 9, indicating that 3 was a C-7 oxidized derivative of 9. This was supported by the downfield-shifted carbon signal of C-17 in 3 as compared to those in 9 (δC 128,0 in 3; δC 118.7 in 9) and further confirmed by detailed 2D NMR analyses (HSQC, HMBC, and 1H–1H COSY). The structure of 3 was further secured by chemical correlation of 14 to 3 via sequential oxidation. In brief, oxidation of the coisolated known compound 14 with selenium dioxide (SeO2) afforded 9, which was oxidized under Dess-Martin periodinane reagent to obtain 3 (Figure S2.4). Thus, the structure of 3 was identified as 5,15-diacetoxy-3-benzoyloxy-7,14-dioxolathyra-6(17),12E-diene and was named euphlathyrinoid C.
Compound 4 exhibited a molecular formula of C33H40O7 as determined by HRESIMS ion at m/z 571.2659 [M + Na]+ (calcd 571.2666). The NMR data of 4 were very similar to those of coisolated known compound 15-acetoxy-3-cinnamoyloxy-5-hydroxy-14-oxolathyra-6(17),12E-diene (13),22 except for the presence of an additional acetyl group in 4 [δH 1.92 (s); δC 21.1 and 170.3], indicating 4 was an acetylated derivative of 13. The acetyl group was located at 5-OH by the HMBC correlation from H-5 (δH 6.17) to the acetyl carbonyl carbon as well as the downfield-shifted H-5 signal in 4 with respect to that in 13 [δH 6.17 (1H, d, J = 10.0 Hz) in 4; δH 4.51 (1H, d, J = 9.8 Hz) in 13]. The structure of 4 was further secured by the chemical correlation of 13 to 4 via acetylation (Figure S2.4). Thus, the structure of 4 was elucidated as 5,15-diacetoxy-3-cinnamoyloxy-14-oxolathyra-6(17),12E-diene and was given a trivial name euphlathyrinoid D.
Compound 5 possessed a molecular formula of C26H36O8 as determined by HRESIMS data at m/z 499.2295 [M + Na]+ (calcd 499.2302). The NMR data of 5 were very similar to those of known compound 5,17-diacetoxy-3-benzoyloxy-15-hydroxy-14-oxolathyra-6Z,12E-diene,22 with the only differences being the replacement of C-3 benzoyl group in known compound by an acetyl group in 5. This was confirmed by HMBC correlation from H-3 (δH 5.66) to acetyl carbonyl carbon (δC 169.4). The structure of 5 was further secured by chemical correlation with the coisolated known analogue 15,17-di-O-acetyl-3-O-benzoyl-5,17-dihydroxyisolathyol (10).21 The alkaline hydrolysis of 5 and 10 generated the same product, 35, which was verified by comparison of their 1H NMR spectra and optical rotation data. Thus, the structure of 5 was elucidated as 3,5,17-triacetoxy-15-hydroxy-14-oxolathyra-6Z,12E-diene and was given the trivial name euphlathyrinoid E.
The known compounds, Euphorbia Factor L8 (6),23Euphorbia Factor L11 (7),24Euphorbia Factor L9 (8),25 5,15-diacetoxy-3-benzoyloxy-7-hydroxy-14-oxolathyra-6(17),12E-diene (9),22 15,17-di-O-acetyl-3-O-benzoyl-5,17-dihydroxy-isolathyol (10),21 15-O-acety-3-O-nicotinoy-jolkinol-5β,6β-oxide (11),26Euphorbia Factor L31 (12),27Euphorbia Factor L30 (13),27Euphorbia Factor L3 (14),25Euphorbia Factor L2 (15),23 and Euphorbia Factor L1 (16),23 were identified by comparison of their spectroscopic data with those in the literatures.
To clarify the SARs of lathyrane diterpenoids related to the hPXR, we used the major components, 14 and 15, as the starting materials for the design of various derivatives (Scheme 1). Briefly, the structural modifications were mainly deployed on the Δ6(17) terminal double bond, α,β-unsaturated ketone, cyclopropane ring as well as substituents on C-3, C-5, C-7, and C-15. First, alkaline hydrolysis of 15 and 14 afforded 17 and 27, respectively, to increase their hydrophilicity. Then, the acylation of the free hydroxyls in 17 with acetic anhydride or 2-furoyl/2-thiophenecarbonyl/benzoyl chloride yielded corresponding esters 18–22. The partial palladium catalyzed hydrogenation of 14 at Δ6(17) or Δ12 afforded 30 or 29, respectively. The complete hydrogenation of 15 via an excess of palladium generated 26. The oxidation of 15 with meta-chorobenzoic acid (m-CPBA) gave the epoxide derivatives 24 and 25. Reduction of 14 and 15 with the sodium borohydride generated 31 [possessing a rare CH(12)–O–C(15) linkage] and 23, respectively. Finally, the treatment of 14 with sodium borohydride followed with dilute hydrochloric acid afforded the cyclopropane-opening products 32, 33, and 34.
Scheme 1. Synthesis of Lathyrane Diterpenoid Derivatives.

Reagents and conditions: (a) 1% NaOH in MeOH (m/v), rt, 1 h; (b) acetic anhydrides/2-furoyl/2-thiophenecarbonyl/benzoyl chlorides, Pyr, rt or 0 °C, 2 h; (c) sodium borohydride, MeOH, rt, 15 min; (d) 10% Pd/C, H2, EtOAc, 50 °C, 24 h; (e) m-CPBA, CH2Cl2, 60 °C, 1 h; (f) (1) sodium borohydride, MeOH, rt, 15 min; (2) 10% HCl in MeOH (m/v), 30 min.
The cytotoxicity of 1–34 was initially performed on HEK293T cells to exclude the cytotoxic compounds 3, 12, 13, and 28 (Figure S2.5). Then, the remaining compounds were subjected to the hPXR agonistic screening by using a dual-luciferase reporter gene system constructed in HEK293T cells via transient transfection with reporter plasmids.17 Rifampicin (RIF), a classical hPXR agonist, was used as the positive drug. The results showed most of these lathyrane diterpenoids exhibited potent hPXR agonistic activity at the concentration of 10 μM (Figure 2A). Among them, 8, 26, and 30 could significantly enhance the hPXR reporter gene activity by 6.9, 3.4, and 4.9 fold, respectively, being more active than that of RIF (activation fold = 2.9). Then, the dose–response assays of 8, 26, 30, and RIF were performed. As shown in Figure 2B, all of these compounds could dose-dependently enhance the hPXR reporter gene activity.
Figure 2.

Effects of 8, 26, and 30 on human PXR activation. (A) Dual-luciferase reporter gene assay was performed in HEK293T cells transiently transfected with expression vectors encoding human PXR (pSG5-hPXR), reporter plasmid (pGL3-CYP3A4-XREM-Luc), and the control plasmid (pRL-TK). Cells were treated with the positive agonist RIF (10 μM) or compounds 1–34 (10 μM) for 24 h. (B) Dose–response assay for 8, 26, 30, and RIF (0.1, 0.5, 1, 5, 10, and 50 μM). Data are presented as mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, versus the vehicle.
To confirm the hPXR activation effects of 8, 26, and 30, we further evaluated their regulation on hPXR downstream key genes that are responsible for BAs metabolism and transport. HepaRG cells were incubated with 8, 26, and 30, and the mRNA expressions of CYP3A4, CYP2B6, and MDR1 were measured by RT-qPCR. The results indicated that 8 remarkably increased the expressions of CYP3A4, CYP2B6, and MDR1, suggesting that 8 may activate hPXR to promote BA detoxification (Figure 3).
Figure 3.

RT-qPCR analysis was used to detect the expressions of the PXR downstream genes (A) CYP3A4, (B) CYP2B6, and (C) MDR1 in HepaRG cells after treatment of RIF (10 μM) and compounds 8, 26, and 30 (10 μM). Values were relative to house-keeping gene β-ACTIN. Data were presented as mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, versus the vehicle.
In general, the hydrolysis products showed a dramatic decrease in the activity, as shown by 14 vs 27 and 15 vs 17, indicating that proper lipophilicity is necessary for activity. On this basis, the different substituents on C-3, C-5, C-7, and C-15 endowed with different agonistic activity on hPXR. In the 3-O-acyls series, the activities were ranked as 3-O-cinnamoyl (4) ≈ 3-O-benzoyl (14) > 3-O-nicotinoyl (6). In the C-7 substitutes series, different moiety contributed to the activity was ranked as 7-acyloxy > 7-hydroxyl > 7-alkyl (8 and 15 > 9 > 14; 18, 20, and 21 > 17 > 27), indicating the presence of acyloxy moieties at C-7 were beneficial to activity. Remarkably, 7-O-nicotinoyl significantly increased the activity as compared to 7-O-benzoyl (6.93-fold in 8 vs 2.03-fold in 15). The acylation of OH-15 was detrimental to activity, as shown by 7 vs 15. In addition, the presence of 5-O-benzoyl led to a dramatic decrease of the activity, as shown by 18, 20, and 21 vs 22, suggesting the unfavorableness of large substituent at C-5. In α,β-unsaturated ketone group, the hydrogenation or epoxidation of Δ12 generally had little influence on the activity as shown by 29 vs 14 and 25 vs 15, whereas the reduction of 14-carbonyl decreased the activity (23 vs 7). The hydrogenation of Δ6(17) dramatically increased the activity as shown by 30 vs 14 and 26 vs 15, whereas epoxidation or migration had little influence (24 vs 25; 16 vs 14; 10 vs 14). In addition, the cyclopropane ring-opening seemed indifferent to the activity, as shown by 32, 33, and 34 vs 14. The above-mentioned SARs information is summarized in Figure 4.
Figure 4.

SARs of lathyrane diterpenoids on hPXR agonistic activity. ↑, increased activity; ↓, decreased activity; →, little influence on activity.
To further explore the potential molecular recognition mechanism between these lathyrane diterpenoids and hPXR, we simulated the binding modes of 8 and RIF with hPXR, respectively, by docking these agonists into the hPXR ligand-binding domain (LBD, PDB ID: 1SKX) using MOE2014.0901. As shown in Figure 5, 8 was docked well into the LBD of hPXR. It engaged one hydrogen bond with His407 via 5-OAc, one hydrogen bond with Gln285 via 7-ONic, and two favorable π–π stacking interactions with Phe420 and Phe251 via 3-OBz and 7-ONic, respectively. These interactions were different to those of RIF, which formed two hydrogen bonds with Gln 285 and one CH-π interaction with Phe288. Generally, the calculated binding energy of 8 with the hPXR [S-score, S(Max) = −13.1065] was better than that of RIF [S(Max) = −12.8002], explaining the higher potency of 8. Among the interactions between 8 and hPXR, the 7-ONic of 8 contributed a hydrogen bond and a π–π stacking interaction to the hPXR binding, suggesting its important role for activity. This was consistent with the above-mentioned SARs and also explained that the replacement of 7-ONic by a 7-OBz led to a dramatic decrease in the activity.
Figure 5.
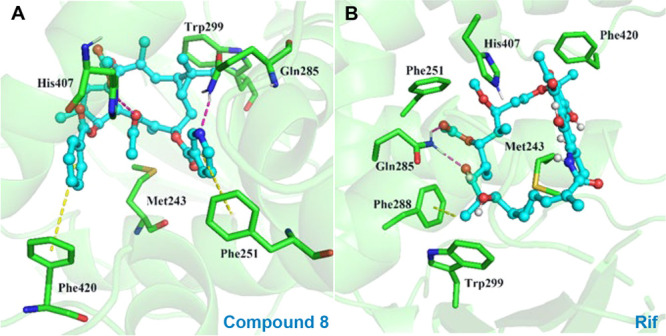
Binding modes of ligands with hPXR derived from docking simulations (red dashed lines for hydrogen bond and yellow dashed lines for π–π stacking interaction or CH−π interaction. (A) Binding mode of compound 8. (B) Binding mode of RIF.
In summary, a lathyrane diterpenoid library (1–34), containing 19 new compounds, was constructed by bioassay-guided phytochemical investigation of E. lathyris and subsequent structural modification. The hPXR agonistic assay of this library led to the identification of a promising hPXR agonist, 8. Compound 8 could significantly activate hPXR as evidenced by the hPXR reporter gene activity (6.9-fold), and up-regulate the expressions of hPXR downstream key genes CYP3A4, CYP2B6, and MDR1. The SARs indicated that acyloxy substituents on C-7, and the presence of 14-carbonyl were essential to activity. The molecular recognition mechanism between 8 and hPXR further underscored the importance of C-7-acyloxy and commendably explained that the replacement of 7-ONic by a 7-OBz led to a dramatic decrease of the activity. Lathyrane diterpenoids featuring a 5/11/3-tricyclic carbon framework are the major components of E. lathyris. Our current study revealed for the first time that the lathyrane diterpenoids could serve as a new type of hPXR agonist. The easily accessible natural source and the clear SARs may provide opportunity for us to rationally design more potent lathyrane-type hPXR agonists and verify its in vivo anticholestatic effect in the near future.
Acknowledgments
This work was supported by Natural Science Foundation of China (81973195 and 81973203), the Fundamental Research Funds for the Central Universities (20ykjc04), the Guangdong Provincial Key Laboratory of Construction Foundation (2017B030314030), the China Postdoctoral Science Foundation (2020M683138), the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01Y093), the Shenzhen Science and Technology Program (KQTD20190929174023858), and the 111 project (B16047).
Glossary
Abbreviations
- hPXR
human pregnane X receptor
- BAs
bile acids
- PBC
primary biliary cholangitis
- PSC
primary sclerosing cholangitis
- UDCA
ursodeoxycholic acid
- OCA
obeticholic acid
- FXR
farnesoid X receptor
- DBD
DNA-binding domain
- LBD
ligand-binding domain
- CYPs
cytochrome P450
- SULTs
sulfotransferases
- UGTs
glucuronyltransferases
- m-CPBA
meta-chorobenzoic acid
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00277.
Full experimental procedures; 1D and 2D NMR, IR, MS, and HPLC spectra of 1–34; X-ray crystallographic data of 26, 30, and 31 (PDF)
Author Contributions
† D.H. and R.-M.W. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Goldstein J.; Levy C. Novel and emerging therapies for cholestatic liver diseases. Liver Int. 2018, 38, 1520–1535. 10.1111/liv.13880. [DOI] [PubMed] [Google Scholar]
- Ghonem N. S. D.; Assis N.; Boyer J. L. On fibrates and cholestasis: a review. Hepatology 2015, 62, 635–643. 10.1002/hep.27744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuers U.; Trauner M.; Jansen P.; Poupon R. New paradigms in the treatment of hepatic cholestasis: from UDCA to FXR, PXR and beyond. J. Hepatol. 2015, 62, S25–S37. 10.1016/j.jhep.2015.02.023. [DOI] [PubMed] [Google Scholar]
- Nevens F.; Andreone P.; Mazzella G.; Strasser S. I.; Bowlus C.; Invernizzi P.; Drenth J. P. H.; Pockros P. J.; Regula J.; Beuers U.; Trauner M.; Jones D. E.; Floreani A.; Hohenester S.; Luketic V.; Shiffman M.; van Erpecum K. J.; Vargas V.; Vincent C.; Hirschfield G. M.; Shah H.; Hansen B.; Lindor K. D.; Marschall H.-U.; Kowdley K. V.; Hooshmand-Rad R.; Marmon T.; Sheeron S.; Pencek R.; MacConell L.; Pruzanski M.; Shapiro D. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N. Engl. J. Med. 2016, 375, 631–643. 10.1056/NEJMoa1509840. [DOI] [PubMed] [Google Scholar]
- Harms M. H.; Lammers W. J.; Thorburn D.; Corpechot C.; Invernizzi P.; Janssen H. L. A.; Battezzati P. M.; Nevens F.; Lindor K. D.; Floreani A.; et al. Major hepatic complications in ursodeoxycholic acid-treated patients with primary biliary cholangitis: risk factors and time trends in incidence and outcome. Am. J. Gastroenterol. 2018, 113, 254–264. 10.1038/ajg.2017.440. [DOI] [PubMed] [Google Scholar]
- Kandel B. A.; Thomas M.; Winter S.; Damm G.; Seehofer D.; Burk O.; Schwab M.; Zanger U. M. Genomewide comparison of the inducible transcriptomes of nuclear receptors CAR, PXR and PPARalpha in primary human hepatocytes. Biochim. Biophys. Acta, Gene Regul. Mech. 2016, 1859, 1218–1227. 10.1016/j.bbagrm.2016.03.007. [DOI] [PubMed] [Google Scholar]
- Kliewer S. A.; Goodwin B.; Willson T. M. The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr. Rev. 2002, 23, 687–702. 10.1210/er.2001-0038. [DOI] [PubMed] [Google Scholar]
- Watkins R. E.; Wisely G. B.; Moore L. B.; Collins J. L.; Lambert M. H.; Williams S. P.; Willson T. M.; Kliewer S. A.; Redinbo M. R. The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science 2001, 292, 2329–2333. 10.1126/science.1060762. [DOI] [PubMed] [Google Scholar]
- Rosenfeld J. M.; Vargas R. Jr; Xie W.; Evans R. M. Genetic profiling defines the xenobiotic gene network controlled by the nuclear receptor pregnane X receptor. Mol. Endocrinol. 2003, 17, 1268–1282. 10.1210/me.2002-0421. [DOI] [PubMed] [Google Scholar]
- Synold T. W.; Dussault I.; Forman B. M. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat. Med. 2001, 7, 584–590. 10.1038/87912. [DOI] [PubMed] [Google Scholar]
- Staudinger J. L.; Goodwin B.; Jones S. A.; Hawkins-Brown D.; MacKenzie K. I.; LaTour A.; Liu Y.; Klaassen C. D.; Brown K. K.; Reinhard J.; Willson T. M.; Koller B. H.; Kliewer S. A. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 3369–3374. 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson H. I.; Wada T.; Xie W.; Renga B.; Zampella A.; Distrutti E.; Fiorucci S.; Kong B.; Thomas A. M.; Guo G. L.; Narayanan R.; Yepuru M.; Dalton J. T.; Chiang J. Y. L. Role of nuclear receptors in lipid dysfunction and obesity-related diseases. Drug Metab. Dispos. 2013, 41, 1–11. 10.1124/dmd.112.048694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Tang Y.; Guo C.; Wang J.; Boral D.; Nie D. Nuclear receptors in the multidrug resistance through the regulation of drug-metabolizing enzymes and drug transporters. Biochem. Pharmacol. 2012, 83, 1112–1126. 10.1016/j.bcp.2012.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonker J. W.; Liddle C.; Downes M. FXR and PXR: Potential therapeutic targets in cholestasis. J. Steroid Biochem. Mol. Biol. 2012, 130, 147–158. 10.1016/j.jsbmb.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk R.; Kremer A. E.; Smit W.; van den Elzen B.; van Gulik T.; Gouma D.; Lameris J. S.; Bikker H.; Enemuo V.; Stokkers P. C. F.; Feist M.; Bosma P.; Jansen P. L. M.; Beuers U. Characterization and treatment of persistent hepatocellular secretory failure. Liver Int. 2015, 35, 1478–1488. 10.1111/liv.12603. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Ma Z.; Liang Q.; Tang X.; Hu D.; Liu C.; Tan H.; Xiao C.; Zhang B.; Wang Y.; Gao Y. Tanshinone IIA exerts protective effects in a LCA-induced cholestatic liver model associated with participation of pregnane X receptor. J. Ethnopharmacol. 2015, 164, 357–367. 10.1016/j.jep.2015.01.047. [DOI] [PubMed] [Google Scholar]
- Zeng H.; Jiang Y.; Chen P.; Fan X.; Li D.; Liu A.; Ma X.; Xie W.; Liu P.; Gonzalez F. J.; Huang M.; Bi H. Schisandrol B protects against cholestatic liver injury through pregnane X receptors. Br. J. Pharmacol. 2017, 174, 672–688. 10.1111/bph.13729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festa C.; De Marino S.; D’Auria M. V.; Bifulco G.; Renga B.; Fiorucci S.; Petek S.; Zampella A. Solomonsterols A and B from Theonella swinhoei. the first example of C-24 and C-23 sulfated sterols from a marine source endowed with a PXR agonistic activity. J. Med. Chem. 2011, 54, 401–405. 10.1021/jm100968b. [DOI] [PubMed] [Google Scholar]
- Zhu J.; Wang R.; Lou L.; Li W.; Tang G.; Bu X.; Yin S. Jatrophane diterpenoids as modulators of P-glycoprotein-dependent multidrug resistance (MDR): advances of structure-activity relationships and discovery of promising MDR reversal agents. J. Med. Chem. 2016, 59, 6353–6369. 10.1021/acs.jmedchem.6b00605. [DOI] [PubMed] [Google Scholar]
- Li W.; Tang Y.-Q.; Sang J.; Fan R.-Z.; Tang G.-H.; Yin S. Jatrofolianes A and B: two highly modified lathyrane diterpenoids from Jatropha gossypiifolia. Org. Lett. 2020, 22, 106–109. 10.1021/acs.orglett.9b04029. [DOI] [PubMed] [Google Scholar]
- Lu J.; Li G.; Huang J.; Zhang C.; Zhang L.; Zhang K.; Li P.; Lin R.; Wang J. Lathyrane-type diterpenoids from the seeds of Euphorbia lathyrism. Phytochemistry 2014, 104, 79–88. 10.1016/j.phytochem.2014.04.020. [DOI] [PubMed] [Google Scholar]
- Zhang C.-Y.; Wu Y.-L.; Zhang P.; Chen Z.-Z.; Li H.; Chen L.-X. Anti-inflammatory Lathyrane Diterpenoids from Euphorbia lathyrism. J. Nat. Prod. 2019, 82, 756–764. 10.1021/acs.jnatprod.8b00600. [DOI] [PubMed] [Google Scholar]
- Appendino G.; Tron G. C.; Cravotto G.; Palmisano G.; Jakupovic J. An Expeditious Procedure for the Isolation of Ingenol from the Seeds of Euphorbia lathyrism. J. Nat. Prod. 1999, 62, 76–79. 10.1021/np980218n. [DOI] [PubMed] [Google Scholar]
- Liao S.-G.; Zhan Z.-J.; Yang S.-P.; Yue J.-M. Lathyranoic Acid A: First Secolathyrane Diterpenoid in Nature from Euphorbia lathyrism. Org. Lett. 2005, 7, 1379–1382. 10.1021/ol050206a. [DOI] [PubMed] [Google Scholar]
- Itokawa H.; Ichihara Y.; Yahagi M.; Watanabe K.; Takeya K. Lathyrane diterpenes from Euphorbia lathyris. Phytochemistry 1990, 29, 2025–2026. 10.1016/0031-9422(90)85062-K. [DOI] [Google Scholar]
- Seip E. H.; Hecker E. Lathyrane type diterpenoid esters from Euphorbia characias. Phytochemistry 1983, 22, 1791–1795. 10.1016/S0031-9422(00)80273-3. [DOI] [Google Scholar]
- Wang Q.; Zhen Y.-Q.; Gao F.; Huang S.; Zhou X.-L. Five new diterpenoids from the seeds of Euphorbia lathyrism. Chem. Biodiversity 2018, 15, e1800386 10.1002/cbdv.201800386. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.