

Abstract
Regulated or programmed cell death plays a critical role in the development and tissue organization and function. In addition, it is intrinsically connected with immunity and host defence. An increasing cellular and molecular findings cause a change in the concept of cell death, revealing an expanding network of regulated cell death modalities and their biochemical programmes. Likewise, recent evidences demonstrate the interconnection between cell death pathways and how they are involved in different immune mechanisms. This work provides an overview of the main cell death programmes and their implication in innate immunity not only as an immunogenic/inflammatory process, but also as an active defence strategy during immune response and at the same time as a regulatory mechanism.
Keywords: programmed cell death, molecular pathways, apoptosis, regulated, necrosis, innate immunity
This paper gives an integrated view of the involvement of programmed cell death in innate immunity not only as an active antimicrobial defence but also as a regulatory mechanism.

Abbreviations
- 3‐MA
3‐methyladenine
- ACD
Autophagic cell death
- AIM2
Protein absent in melanoma 2
- ASC
Apoptosis‐associated speck‐like protein
- ATG
Autophagy‐related genes
- Bcl‐2
B‐cell lymphoma 2
- Bid
BH3 interacting domain death agonist
- CALCOCO 2
calcium binding and coiled‐coil domain‐containing protein 2
- CALR
Calreticulin
- CGD
Chronic granulomatous disease
- CIAS1
Cold‐induced autoinflammatory syndrome protein 1
- CTL
Cytotoxic T lymphocytes
- CYPD
Cyclophilin D
- DAI
DNA‐dependent activator of IFN
- DAMP
Danger‐associated molecular pattern
- DAP3
Death‐associated protein 3
- DC
Dendritic cells
- ECM
Extracellular matrix
- ERK
Extracellular signal‐regulated kinase
- OSCC
Oesophageal squamous cell carcinoma
- FAS
FS‐7‐associated surface antigen
- GM‐CSF
Granulocyte–macrophage colony‐stimulating factor
- GPX4
Glutathione peroxidase 4
- GSDM
Gasdermin
- GSH
Reduced glutathione
- HNSCC
Head and neck squamous cell carcinoma
- HMGB1
High‐mobility group box 1
- HSP
Heat‐shock protein
- IFN
Interferon
- IGF1R
Insulin‑like growth factor 1
- IKK
IkB kinase
- IPS‐1
Interferon‐β promoter stimulator 1
- IRF3
Interferon‐regulatory factor 3
- LC3
Light chain 3
- LDH
Lactate dehydrogenase
- LPS
Lipopolysaccharide
- MAPK
Mitogen‐activated protein kinase
- MLKL
Mixed lineage kinase domain‐like protein
- MOMP
Mitochondrial outer membrane permeabilization
- MPO
Myeloperoxidase
- MPT
Mitochondrial permeability
- mtDNA
Mitochondrial DNA
- NAIP
NLR family, apoptosis inhibitory protein
- NE
Neutrophil elastase
- NEK7
NIMA‐related kinase 7
- NETosis
Regulated necrosis with realizing of neutrophil extracellular traps
- NF‐kB
Nuclear factor‐kB
- NINJ1
Nerve injury‐induced protein 1
- NK
Natural killer
- NLR
NOD‐like receptor
- NLRP
Nod‐like receptor pyrin‐containing protein
- NOD
Nucleotide‐binding oligomerization domain
- NOX
NADPH oxidase
- NSCLC
Non‐small‐cell lung cancer
- OSCC
Oral squamous cell carcinoma
- PAD4
Peptidylarginine deiminase 4
- PAF
Platelet‐activating factor
- PAMP
Pathogen‐associated molecular pattern
- PARP1
Poly(ADP‐ribose) polymerase 1
- PCD
Programmed cell death
- PKR
RNA‐dependent protein kinase
- PMA
Phorbol myristate acetate
- PR3
Proteinase 3
- PRR
Pathogen recognition receptor
- RHIM
RIP homotypic interaction motif
- RIPK
Receptor‐interacting protein kinase
- ROCK
Rho‐associated coiled‐coil containing protein kinase
- ssRNA
Single‐strand RNA
- TCR
T‐cell receptor
- TLR
Toll‐like receptors
- TNF
Tumour necrosis factor
- TNFR1
TNF receptor 1
- TRAIL
TNF‐related apoptosis‐inducing ligand
- TRAILR
TRAIL receptor
- TRIF
TIR domain‐containing adapter‐inducing interferon‐β
- ZBP1
Z‐DNA binding protein 1
INTRODUCTION
Cell death is a physiological event essential for homeostasis in multicellular organisms. However, it is also implicated in multiple pathological conditions such as neurodegenerative and ischaemic disorders, cancer, and autoinflammatory and autoimmune diseases [1, 2, 3]. Consequently, cell death is tightly regulated by a strict biochemical programme, which depends on regulatory pathways and protein interaction networks. This programmed cell death (PCD) exhibits various types of modalities, according to the stimulus that triggers it and the molecular pathways that result in activated cell death protein [4, 5]. The multiple forms of PCD are in constant update by recent findings in this field, which are revealing the connections and molecular crosstalk between cell death programmes and their implication in a wide variety of biological processes [6].
PCD is involved in several physiological functions such as organ development, tissue remodelling and epithelial cell renewal. It is also essential for cellular homeostasis and cell response to stress, acting as an intrinsic mechanism to prevent malignant transformation and cancer development [2]. In addition to its role in tissue maintenance, PCD is a crucial biological response for immunity and host defence. It is part of various innate and adaptive immune mechanisms/processes such as antiviral defence, killing of intracellular pathogens, inflammation, chemo‐attraction, lymphocyte selection and immune tolerance [7, 8, 9]. An increasing evidence reveals novel functions of different cell death types in innate immunity, including the regulation and/or amplification of the inflammatory response, the modulation of cytokine production and release and the diverse microbial killing strategies in specialized immune cells [10, 11, 12, 13]. This review aimed to focus on important findings that highlight the interconnection between various forms of PCD and their roles in innate immunity.
DIFFERENT TYPES OF CELL DEATH
Cell death is a multifactorial process that involves several molecular events and depends on multiple physiological conditions; thus, it can be presented in a variety of fashions/modalities [14, 15]. The different forms of cell death are driven by distinct molecular pathways, although they share common regulatory factors and can be triggered by the same stimuli depending on the cellular context. This section will present a general background of the main cell death programmes currently identified.
Apoptosis and necroptosis
Apoptosis is the best‐characterized form of cell death, which is under molecular control and differs histologically and biochemically from necrosis, an unregulated or accidental cell death (reviewed in [16]). Until recently, apoptosis was considered the only form of PCD; however, the discovery of specific inhibitors of necrotic processes together with a systematic biochemical and genetic analysis has redefined necrosis as a programmed and regulated cell death, also termed necroptosis (reviewed in [15, 16]).
Unlike accidental necrosis, which is mainly caused by trauma, necroptosis is a biochemically controlled process activated by receptor‐interacting protein kinases 1 and 3 (RIPK1 and RIPK3) and mediated by the mixed lineage kinase domain‐like protein (MLKL) [3, 17, 18]. The executioner mechanism of MLKL in necroptosis is not clear; nevertheless, two independent and non‐exclusive models have been proposed to explain it. One of them is based on the recruitment of Ca2+ and Na+ ion channels by MLKL once inserted in plasma membrane, causing a massive osmotic imbalance that leads to membrane permeabilization and cell lysis, a distinctive feature of necroptotic cell death [19]. The other model proposes a pore‐forming mechanism by MLKL oligomerization at plasma membrane, which directly induces leaking of intracellular contents and cell lysis [20, 21]. Interestingly, a very recent report demonstrates the involvement of nerve injury‐induced protein 1 (NINJ1) in the plasma membrane rupture during necroptosis and other forms of programmed necrosis. This study shows that murine Ninj1‐/‐ macrophages treated with different inducers of programmed necrosis fail to die by the typical necrotic process with cell lysis and associated release of high‐mobility group box 1 (HMGB1) and lactate dehydrogenase (LDH), two main markers of plasma membrane rupture [22]. Instead, they die with a persistent round balloon‐like morphology, compared with stimulated dying cells with normal NINJ1 expression, which show the usual necrotic cell lysis. These results indicate that cell permeabilization and swelling occurs (probably mediated by MLKL and other pore‐forming proteins), but the final membrane destabilization and rupture requires NINJ1. Further research is necessary to define the precise sequence of events during PCD‐driven cell lysis, but this is the first insight suggesting that not only pore‐forming proteins or osmotic imbalance but also specific membrane proteins are required for cell rupture during programmed necrosis.
Necroptosis could be induced by the pro‐inflammatory cytokine tumour necrosis factor (TNF) under certain cellular conditions, through the signalling cascade of TNF receptor 1 (TNFR1), which lead to the activation of RIP kinases and formation of necrosome complex (reviewed in [4]). TNFR1 also activates the apoptotic cascade by the recruitment of the cytosolic complex IIb, in a mechanism that is regulated by RIPK1 [23, 24]. Other membrane receptors that induce necroptosis include FAS (FS‐7‐associated surface antigen), TRAILR1 and TRAILR2 (TNF‐related apoptosis‐inducing ligand (TRAIL) receptors 1 and 2), which normally induce apoptosis through the activation of caspase 8, but could recruit RIPK1 to initiate the formation of the necrosome under caspase 8 blockade [11, 25, 26]. Interestingly, RIPK1 seems to act as a molecular switch between apoptosis and necroptosis depending on the cellular conditions, a transition that is dynamically regulated by post‐translational modifications and protein expression [25, 27]. Furthermore, some Toll‐like receptors (TLRs) such as TLR3 and TLR4 can also activate necroptosis in response to poly(I:C) and LPS, respectively, interacting with RIPK3 through the adaptor protein TIR domain‐containing adapter‐inducing interferon‐β (TRIF). This has been demonstrated in mice‐derived macrophages with TIRF or RIPK3 deficiency [28]. The activation of this pathway seems to occur during caspase inhibition, particularly caspase 8, and is RIPK1‐independent at least in fibroblasts and endothelial cells but requires RIPK1 when is induced by TLR agonists in macrophages [29].
Both apoptosis and necroptosis can be presented in specific variants, which are triggered by particular events and possess additional biological consequences. For example, anoikis is a special type of apoptosis, which is induced by the loss of cell–cell/cell–extracellular matrix (ECM) contacts [30, 31]. When cell is detached from neighbouring cells or from the surrounding ECM, the disruption of cadherin or integrin interactions, respectively, triggers a caspase‐dependent mechanism that ends in a PCD [31]. Selected cells can be also excluded from epithelial layers due to malignant transformation or overcrowding by a process called ‘extrusion’, resulting in the activation of a biochemical programme that eliminates extruded and detached cells by anoikis [15]. Likewise, regulated or programmed necrosis includes a variety of cell death programmes with a common cellular phenotype but different molecular pathways.
Other forms of programmed necrosis
In general, all modes of regulated necrosis exhibit typical hallmarks such as cellular rounding and swelling (known as oncosis), granulation at cytoplasmic level and plasma membrane rupture; however, physiological and biochemical differences lead to a variety of subclasses with particular mechanisms (reviewed in [16]). These non‐apoptotic cell death modalities include the mitochondrial permeability transition‐mediated regulated necrosis (MPT‐mediated necrosis), parthanatos and ferroptosis. The first one is induced by a mitochondrial pore composed of at least cyclophilin D (CYPD) (the only component of the pore that has been identified so far), whereas parthanatos is caused by an excessive PARylation of intracellular proteins by Poly(ADP‐ribose) polymerase 1 (PARP1) provoking a depletion of NAD+ and ATP that leads to necrotic cell death [4, 5, 16]. On the other hand, ferroptosis involves an iron‐dependent oxidative stress that is produced by a decrease in cysteine uptake (the oxidized form of cysteine), a deficit of GSH (reduced glutathione) and a depletion of the enzyme glutathione peroxidase 4 (GPX4) [4, 32]. In neurons, it has been reported a form of ferroptosis called oxytosis, which occurs as a result of glutamate toxicity by the blockade of the antiporter system Xc‐ producing the deficit of cystine and the iron‐dependent production of reactive oxygen species [33].
In addition, pyroptosis and regulated necrosis associated with the release of neutrophil extracellular traps (termed as NETosis) are pro‐inflammatory and microbial‐induced forms of programmed necrosis, which occur in specialized immune cells [5, 16]. Pyroptosis is triggered by canonical or non‐canonical inflammasome stimulation, which induces activation of caspase 1 or caspase 11 [7, 14, 34]. The basic mechanism causing cell death in pyroptosis is based on osmotic imbalance and cellular swelling similar to those occurred in necroptosis, but the pore‐forming protein involved is Gasdermin D (GSDMD) instead of MLKL. GSDMD is proteolytically activated by active caspase 1 upon inflammasome stimulation, though the interaction with apoptosis‐associated speck‐like protein (ASC) after upstream stimulation mediated by intracellular sensors such as AIM2 (protein absent in melanoma 2) and Nod‐like receptor pyridine containing protein 1 or 3 (NLRP1 or NLRP3) [35, 36]. However, different studies have revealed the involvement of other caspases such as caspases 4 and 5 in GSDMD activation and pyroptosis induction in different cell types by non‐canonical inflammasome stimulation [37]. Moreover, recent findings demonstrate that pyroptosis can be also induced by caspase 8‐dependent cleavage of GSDMD and even by caspase 3, through the cleavage of Gasdermin E (GSDME), in a process that has been mostly observed in cancer cells. Other members of the Gasdermin protein family such as GSDMA, GSDMB and GSDMC can also lead to cell permeabilization and pyroptotic cell death, but the mechanism of activation seems to be caspase‐independent and their role in infection or malignant transformation is still not clear (reviewed in [36]).
Some authors also highlight the existence of an alternative necroptotic cell death with some pyroptosis‐like features, but without the release of IL‐1β or the involvement of GSDM pore formation [38, 39]. This type of PCD was formerly called ‘pyronecrosis’, a term that was introduced by Willingham et al. in 2007, to distinguish it from caspase 1‐dependent pyroptosis; however, although this definition has been used by some authors in different contexts, the term was not approved by the Nomenclature Committee on Cell Death 2018 [5]. This PCD has been reported in monocytes and macrophages in response to certain inflammatory stimuli or certain pathogens such as Shigella flexneri, and seems to be dependent of NLRP3, previously known as cold‐induced autoinflammatory syndrome protein 1 (CIAS1) or Cryopyrin [40, 41]. It shows a necrosis‐like morphology, but the molecular mechanism that governs this particular necrotic cell death is still poorly understood. It involves NLRP3 and ASC signalling, but rather than caspase activation, this event leads to lysosomal permeabilization and cathepsin release, which are the ultimate executors of this cell death, but, unlike pyroptosis or lysosomal cell death, is caspase‐independent, does not involve IL‐1β secretion and causes HMGB1 release to extracellular space [41]. A similar necrotic‐like cell death was reported in murine macrophages in response to Legionella pneumophila infection, with a rapid activation mechanism dependent on cathepsin B and accompanied by HMGB1 release and PARP cleavage [42]. These necrotic cell deaths do not fall into any of the defined categories for PCD, and there is not consensus in their molecular mechanism [5]. They have particular features similar to pyroptosis, necroptosis or lysosomal‐dependent cell death, but with substantial differences between them. Further research is needed to elucidate whether these are just particular variants of necroptosis, caspase‐independent forms of pyroptosis or caspase and mitochondrial‐independent types of lysosomal cell death, or even a crosstalk between these forms of PCD in a particular context.
On the other hand, NETosis, which is also another particular and interesting PCD, occurs primarily not only in neutrophils, but also in other innate immune cells, and is characterized by the release of chromatin structures with associated histones (called extracellular traps) that represents an efficient antimicrobial mechanism [1]. The molecular events underlying this form of PCD have been characterized, and it has been demonstrated that NADPH oxidase (NOX), a common enzyme in neutrophils, is a key component for the activation of this pathway. The hyperactivation of this enzyme in response to pathogens is mediated by the extracellular signal‐regulated kinase (ERK) and the mitogen‐activated protein kinase (MAPK) signal transduction pathways, changing the ROS balance within the neutrophil and inducing myeloperoxidase (MPO) and neutrophil elastase (NE) activity, a downstream event that leads to chromatin condensation and massive permeabilization (including nucleus, granules and plasma membrane), interestingly through the pore‐forming protein GSDMD, a common feature with pyroptosis. It has been described that NE is implicated in GSDMD processing and activation, as well as histone cleavage, a modification that together with histone citrullination (mediated by peptidylarginine deiminase 4 or PAD4) facilitates DNA and chromatin rearrange (reviewed in [13]). The final consequence is the extrusion of the extracellular trap with histones, proteases and granular proteins, resulting in the death of the neutrophil; however, under certain conditions, the extrusion can occur with neutrophil survival, a process termed as ‘vital NETosis’. [43, 44]. In contrast to the classical lytic NETosis that can be also initiated by chemical inducers of inflammation such as the phorbol myristate acetate (PMA), vital NETosis is a rapid and oxidative stress‐independent process that is stimulated by bacteria and parasites, immunocomplexes and a number of physiological and immune‐activating agonists such as TNF, IL‐8, granulocyte–macrophage colony‐stimulating factor (GM‐CSF), platelet‐activating factor (PAF) and the complement protein fragment C5a, and does not involve the permeabilization and death of neutrophils [44, 45, 46]. Other works have demonstrated alternative NETosis pathways, which are NOX‐ and ROS‐independent and do not require PAD4 enzymatic activity, but they are affected by calcium chelation and depend on NE function and nuclear translocation [47, 48]. This interesting form of PCD is still under research to elucidate the precise molecular mechanism operating in different conditions, but it seems to have a rapid non‐oxidative variant that could be a PAD4‐dependent or PAD4‐independent and a slower NOX‐ and ROS‐dependent form that involves PAD4 activity. In all cases, NE activity seems to be necessary for NET induction and release; thus, this enzyme seems to have a key role in all forms of NETosis.
Autophagic cell death
Beyond apoptosis and regulated necrosis, autophagic cell death (ACD) is receiving attention as an interesting third form of PCD. Autophagy is, in fact, an adaptive mechanism essential for cell survival under unfavourable conditions such as starvation, extracellular or intracellular stress, high temperatures, overcrowding, hypoxia and antiproliferative stimuli. During the autophagic mechanism, the cell orchestrates a complex response that includes different membrane rearrangements to form autophagosomes and ultimately autolysosomes, allowing the cell to digest and catabolize its own constituents to obtain energy and recycle intracellular molecules (reviewed in [49]). However, this process can lead eventually to a form of cell death, in which the cell ‘eat itself’, causing irreversible damage such as loss of membrane and organelle integrity [15, 50]. Therefore, autophagy is tightly regulated and depends on a sequence of specific intracellular events. This multistep process is mediated by a gene family termed as ATG (autophagy‐related genes) and involves protein–protein interactions and post‐translational modifications, such as the lipidation of the microtubule‐binding protein LC3 (light chain 3) with phosphatidylethanolamine and the degradation of p62 (reviewed in [51]). Autophagy is a classic pathway that has been studied for years, but more recently, ACD is getting attention as a defensive and regulatory mechanism rather than a deregulation of the process [14, 52]. According to this, ACD can act in a similar way to apoptosis or necroptosis, representing another self‐induced type of cell death in response to extracellular stressors or pathological stimuli.
Unconventional cell death programmes
In addition to the classical forms of PCD and their emerging submodalities, other authors are reporting ‘particular’ or ‘unconventional’ ways in which cells die, involving distinct molecular events not fully understood yet. In line with this, phagoptosis and entosis have been described as specific forms of cell death, which implicate the assimilation of a cell by other cell, a phenomenon that could lead to different outcomes (reviewed in [15]). When a viable cell is phagocyted by other active live cell, it fails to undergo a process called phagoptosis, different from phagocytosis of apoptotic or dead cells by macrophages. It could be homotypic or heterotypic, if the fused cells are of the same or different type, respectively. The molecular pathway has not been characterized in deep, but it requires the exposure of phosphatidylserine in cell surface and the loss of CD47, as occurs with the typical phagocytosis. The physiological importance of this process includes the turnover of erythrocytes and neutrophils, and it has been described in some pathological conditions such as neuroinflammation [15]. On the other hand, entosis is the opposite process, when a viable cell invades another life cell, penetrating directly into the cytoplasm where it is vacuolized, turning into an internalized or entosed cell. An entosed cell could be released resulting in survival or killed by a lysosomal‐dependent degradation that involves LC3‐mediated vacuole targeting, recruitment and fusion. The entotic cell requires Rho‐associated coiled‐coil containing protein kinase (ROCK) activity and actin–myosin structures that facilitate the cell‐into‐cell penetration. This cell death modality is mainly implicated in epithelial tissue removal and embryo implantation [5].
Another particular form of PCD has been described in Kupffer cells (resident liver macrophages) in response to human adenovirus and Listeria monocytogenes, which does not depend on caspase, RIPK, ASC, cathepsins or NOX activity and does not involve the mitochondrial pore‐forming proteins BAX, BAK or CYPD [53]. This cell death variant shows a necrotic‐like morphology, with mitochondrial swelling, plasma membrane permeabilization and LDH release but with a very rapid kinetics independent of the main molecular mediators or apoptosis, lysosomal cell death, necroptosis, pyroptosis and other forms or programmed necrosis, in addition to specific morphological changes that do not correspond to an autophagic cell death. This specific PCD requires the interferon‐regulatory factor 3 (IRF3) but, interestingly, does not involve Ser396 IRF3 phosphorylation or the activation of IRF3 downstream genes, which only occurs when the pathogen escape from phagocytic or vacuolar compartments [53]. All these results suggest a novel role for IRF3 in viral and parasite infection beyond gene transcription, as an inductor of rapid necrotic cell death to prevent viral or parasite replication and spread of the infection.
Some recent studies also describe other vacuole‐dependent forms of PCD termed as methuosis and paraptosis. The first one involves Ras hyperactivation and a massive accumulation of large single‐membrane vacuoles full of extracellular fluid, which are derived from macropinosomes. The second variant is associated with cytoplasmic vacuolization as well, but these vacuoles are derived from expansion of endoplasmic reticulum and mitochondria. It could be driven by misfolded protein accumulation or Ca2 + overload and involves insulin‑like growth factor 1 receptor (IGF1R) activation (reviewed in [6]).
CELL DEATH AS PART OF INNATE IMMUNITY
As was described above, cell death is a dynamic phenomenon with specific purposes that is involved in a variety of biological functions. All forms of PCD represent biochemically controlled events with specific cellular functions, which are activated or downregulated depending on the physiological context. They can be also manipulated by pharmacological interventions, representing interesting targets to modulate different pathological conditions. The role of the main cell death modalities in particular biological functions, with special focus on innate immunity, is discussed in this section.
Apoptosis and innate immunity
Apoptosis, the most classical and the best‐characterized form of PCD, represents a biological strategy that selectively eliminates infected cells to restrict the reproduction and propagation of viruses and intracellular pathogens [12]. In this context, apoptosis seems to be stimulated by pathogen–cell interactions leading to caspase 8 activation and initiation of the extrinsic apoptotic pathway [54]. This evolutionary mechanism is in fact highly effective, and many intracellular pathogens have evolved to suppress apoptosis in a caspase 8‐dependent manner [55, 56, 57]. The intrinsic apoptotic pathway is also important in the elimination of infected cells; thus, there are different pathogen‐mediated strategies to interfere with mitochondrial activation of apoptosis, by the cleavage of pro‐apoptotic B‐cell lymphoma 2 (Bcl‐2) protein family members or the inhibition of mitochondrial outer membrane permeabilization (MOMP) (reviewed in [57]) (Figure 2). In addition, apoptosis is an essential part of the cytotoxic mechanism displayed by innate immune cells such as natural killer (NK) lymphocytes. It has been described that these effector cells induce apoptosis in microbe‐infected cells through specific serine proteases, called granzymes [58]. In the current model proposed to explain this process, perforins and granzymes released from cytoplasmic granules of NK cells are internalized by target cells during immune synapse, through endocytosis. Once in the endocytic compartment, membrane pore‐forming proteins perforins facilitate the release of granzymes into the cytosol, where they induce apoptosis proteolytically activating executioner caspases (such as caspase 3) or members of Bcl‐2 protein family, such as Bid (BH3 interacting domain death agonist) (reviewed in [59]). These lymphocytes also eliminate target cells by apoptosis, expressing Fas and TRAIL receptors, which induce the apoptotic process by the intrinsic pathway via caspase 8 activation [59].
FIGURE 2.
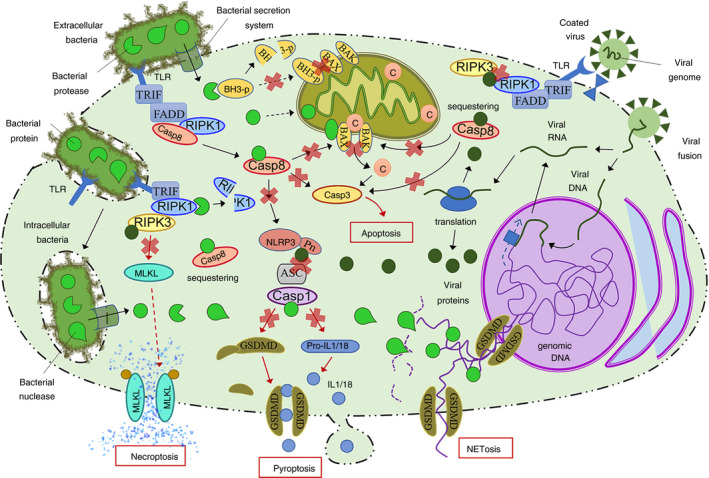
Overview of the main bacterial and viral mechanisms to suppress PCD such as apoptosis and major forms of programmed necrosis. Both extracellular and intracellular bacteria have developed pore‐forming secretion systems to deliver specific proteins inside the cell to inhibit the main targets of multiple processes including cell death. Bacterial suppression of PCD is based on proteases and inhibitory proteins that decrease the integrity/activity of host cell factors. Some bacteria can also produce nucleases and DNA binding proteins that impair NET formation. On the other hand, viruses (including DNA and RNA viruses) deliver their genetic material into the cell where it is processed by the host cell machinery to undergo transcription and translation or direct translation, leading to the expression of viral proteins. These virulence factors suppress PCD mainly interacting with target proteins to sequester them or occluding binding domains to impair protein–protein interaction. Black arrows represent sequential steps in a process/pathway, and red arrows represent the final steps in a given pathway. Red crosses represent inhibition or blockade of a protein/pathway. ASC, apoptosis‐associated speck‐like protein; BH3‐p, BH3 only domain‐containing proteins; C, cytochrome c; Casp, caspase; FADD, Fas‐associated protein with death domain; GSDMD, Gasdermin D; IL, interleukin; MLKL, mixed lineage kinase domain‐like protein; NET, neutrophil extracellular trap; NLRP3, Nod‐like receptor pyrin‐containing protein 3; Pn, pyrin domain; RIPK, receptor‐interacting protein kinases; TLRs, Toll‐like receptors; TRIF, TIR domain‐containing adapter‐inducing interferon‐β
The molecular crosstalk between apoptosis and innate immunity is not limited to the executioner mechanisms, and it also occurs during the regulation of cell response to infection. For example, the stimulator of the mitochondrial apoptotic pathway Bid is also involved in pathogen recognition receptor (PRR) signalling, inflammation and immunity. Bid seems to be recruited by nucleotide‐binding and oligomerization domain (NOD) proteins after microbial DNA recognition, to form a complex with IκB kinase (IKK) and promote the activation of nuclear factor‐κB (NF‐κB) and extracellular signal‐regulated kinase (ERK) pathways [54]. Through this dual role, Bid regulates how cells react to infection, either dying by an apoptotic process or surviving and displaying a pro‐inflammatory and antimicrobial response. In a similar way, TNF could stimulate pro‐inflammatory and antimicrobial defensive pathways or induce apoptotic cell death, depending on the cellular conditions [4]. Additional examples of this interplay between apoptosis induction and innate immune response are related to the multifunctional role of other proteins that interconnect both processes, such as the interferon promoter stimulator 1 (IPS‐1) and heat‐shock proteins (HSPs). IPS‐1 is involved in mitochondrial antiviral signalling and virus‐induced interferon‐ β (IFN‐β) stimulation; however, it is also crucial to apoptosis and anoikis induction after cell detachment [60]. It has been experimentally demonstrated that IPS‐1, once inserted in mitochondrial outer membrane, is able to recruit and activate caspase 8 to induce anoikis by a distinct pathway that is independent of death receptor signalling or death‐associated protein 3 (DAP3) function [60]. On the other hand, HSPs, which are apoptosis inhibitors and cytoprotective chaperones that promote cell survival to stress, can be translocated to plasma membrane or secreted to extracellular space to stimulate the immune system and enhance the immune response [61]. HSP70 and HSP90 inhibit apoptosis interfering with death receptor signalling, restricting the mitochondrial permeabilization and cytochrome c release and preventing caspase activation, but in their extracellular form, they display a variety of immune functions including antigen presentation, cell recruitment and activation and a cytokine‐like behaviour (reviewed in [61]).
Other studies have revealed that apoptosis also activates the NLRP3 inflammasome, which contributes to immune activation and immune cell recruitment, but is also a crosstalk between apoptosis and pyroptosis in response to infections and cellular stress. Shimada et al. in 2012 demonstrated that oxidized mitochondrial DNA (mtDNA) released during apoptosis activates NLPR3‐stimulated secretion of IL‐1β in macrophages by direct binding to NLRP3 protein, a process that can be inhibited by Bcl‐2 or the oxidized nucleotide 8‐OH‐dG [62]. Different subsequent researches have established that NLPR3 inflammasome localization and activation are actively regulated by compartmentalization and interaction between cytosol, endoplasmic reticulum and mitochondria and NLRP3 protein can be activated during apoptosis‐driven MOMP by various factors released from permeabilized mitochondria such as ROS, mtDNA and cardiolipin (reviewed in [63]). In addition, a recent work revealed the activation of NLRP3 inflammasome in extrinsic and intrinsic apoptosis by a mitochondrial‐independent mechanism, based on the pore‐forming protein Pannexin 1 [64]. Chen et al. experimentally demonstrated that this protein, which is proteolytically activated by executioner caspases during apoptosis, is involved not only in caspase 8‐dependent NLRP3 activation in response to TNF‐induced apoptosis but also in NLRP3 activation during intrinsic apoptosis, in a mechanism that does not depend on GSDM activity. They propose a model in which Pannexin 1 is activated by both apoptotic pathways through caspase 3. These additional evidences reveal the highly connected interplay between cell death programmes during immune response. In fact, this idea has been explored in deep by some authors, such as Zheng and Devi‐Kanneganti in their recent paper. They propose the term ‘PANoptosis’ to define a functional inter‐regulation and co‐ordination between pyroptosis, apoptosis and necroptosis in the context of viral infection, for example during influenza infection, through the NLRP3 inflammasome activation by the DNA‐dependent activator of IFN‐regulatory factors (DAI) (also known as Z‐DNA binding protein 1 or ZBP1) [65].
Apoptosis not only controls infected cells, but also transformed and malignant cells, an important function that is tightly related to immune surveillance and primary immune response to cancer [21]. The intrinsic apoptotic pathway is triggered by irreversible events such as irreparable DNA damage, disruption of cell division or cell cycle arrest, representing a crucial process to prevent genomic instability, increase in mutation rate and, consequently, oncogenesis [66]. Likewise, anoikis is a very effective mechanism to prevent metastasis, eliminating misplaced and detached cells [67]. During tumour development, cancer cells must survive in pro‐apoptotic conditions such as hypoxia, growth factor deprivation, oxidative stress and metabolic deregulation; therefore, apoptosis evasion is considered a hallmark of cancer [21, 66]. Malignant cells have to avoid apoptosis in several stages to successfully become a tumour and colonize a distant organ. In this sense, this form of PCD acts as an intrinsic defence to suppress oncogenesis and neoplastic growth. In line with this, different researches have illustrated the multiple strategies of cancer cells to avoid or delay apoptosis in order to survive and proliferate (Table 1). For example, the overexpression of anti‐apoptotic factors of Bcl‐2 protein family as an apoptosis evasion mechanism has been observed in multiple cancer types [68]. In the same way, the complex metabolic reprogramming that takes place in the mitochondria during malignant transformation not only allows cancer cells to grow under anaerobic and hypoxic conditions, but also desensitizes mitochondria to internal danger signals, impairs mitochondrial permeabilization and inactivates pro‐apoptotic factors such as cytochrome c (reviewed in [69]).
TABLE 1.
Different mechanisms of cancer cells to block or suppress PCD
| Cancer type | Targeted PCD | Mechanism of suppression | Reference |
|---|---|---|---|
| Colorectal, breast, lung | Apoptosis | MOMP impairment and cytochrome c inactivation | [69] |
| Lymphoma, colorectal, breast, ovarian, prostate, lung | Apoptosis | Overexpression of Bcl‐2 anti‐apoptotic members | [67] |
| Colorectal, gastric, breast | Apoptosis/pyroptosis | Low expression of NLRs | [77] |
| Myeloma, glioma, gastric, breast, ovarian, prostate | Apoptosis | Overexpression of G1P3 | [74, 75, 76] |
| Non‐small‐cell lung carcinoma | Necroptosis | Low expression of CALR | [105] |
| Breast carcinoma | Necroptosis | Low expression of HMGB1 | [106] |
| Leukaemia, colorectal, breast | Necroptosis | Low expression of RIPK3 | [107, 108, 109] |
| Gastric | Pyroptosis | Low expression of GSDMD | [118] |
| Melanoma | Pyroptosis | Low expression of GSDME | [120] |
| Breast, ovarian, prostate | Autophagic cell death | Low Beclin 1 expression | [135] |
| Head and neck squamous cell carcinoma, gastric | Autophagic cell death | Upregulation of G9a | [138, 139] |
Abbreviations: Bcl‐2, B‐cell lymphoma 2; Beclin 1, an autophagy‐related protein; CALR, calreticulin; G1P3, an IFN‐stimulated product; G9a, a histone methyltransferase;GSDMD/E, Gasdermin D/E; HMGB1, high‐mobility group box 1; MOMP, Mitochondrial outer membrane permeabilization; NLRs, Nod‐like receptors; RIPK3, receptor‐interacting protein kinase 3.
Apoptosis is definitely a key mechanism linked to immune response to tumours. As was described above, apoptotic cell death is crucial to NK cell‐mediated cytotoxicity. This primary immune mechanism is critical to antitumor defence, particularly during carcinogenesis and early tumorigenesis [70]. On the other hand, different molecules usually implicated in innate immune response to infection are also involved in apoptosis induction in the context of cancer. For example, ISG12a and G1P3, two IFN‐stimulated products, actively participate in the induction, maintenance or reversion of apoptosis in virus‐infected and transformed cells. Both proteins are expressed in response to IFNs and modulate mitochondrial integrity and function; however, ISG12a displays strong pro‐apoptotic effects mediated by mitochondrial membrane destabilization, whereas G1P3 acts as an anti‐apoptotic factor that recovers and maintains mitochondrial membrane potential [71]. The induction or overexpression of hISG12a decreases viability of cancer cells and sensitizes them to etoposide‐induced apoptosis [72]. Likewise, the transient expression of ISG12a arrest cell cycle in G1 phase can induce apoptosis in human cervical and breast cancer cell lines, in a caspase‐dependent and p53‐independent manner [73]. In contrast, G1P3 exerts anti‐apoptotic effects in gastric cancer cells and myeloma, preventing apoptosis induction by cytotoxic agents such as 5‐fluorouracil and cycloheximide or endogenous factors such as TRAIL [74, 75]. G1P3 is also overexpressed in ovarian cancer in response to paclitaxel treatment and in gliomas, and breast and prostate cancer after radiation [71]. The overexpression of this factor and its capacity to inhibit apoptosis, anoikis of detached cells, affect the normal growth morphology and induce tamoxifen resistance were demonstrated in different human breast cancer cell lines in comparison with human breast epithelial cells. In addition, this overexpression correlated with reduced relapse‐free and overall survival in oestrogen receptor positive breast cancer patients [76].
More recently, the role of the NOD‐like receptors (NLRs) in cancer has been also revised. These intracellular PRRs are crucial to innate immunity against invader pathogens but sense danger signals and cell abnormalities during tumorigenesis, to trigger apoptosis in transformed cells. NAIP (NLR family, apoptosis inhibitory protein), NOD1, NOD2 and NLRC4 have been demonstrated to be required for tumour apoptosis induction in colorectal and breast cancer mouse models, and lack of expression of these receptors conferred susceptibility to cancer progression and poor disease outcome [77]. In the same way, lower expression of these proteins and mutations/polymorphisms in its encoding sequences has been seen in tumour samples of colon, breast and gastric cancer patients, correlating with a bad prognosis and inefficient response to treatments [78, 79, 80]. In spite of that, overexpression of these receptors and other NLRs has been found in other tumour variants, such as prostate and certain types of breast cancer, suggesting that the precise implication of these molecules in disease progression should be explored in deep (reviewed in [77]).
Interestingly, as occurs with other physiological mechanism, tumour cells not only evade apoptosis but also manipulate it in its favour, as an immune evasion mechanism. In fact, it is known that cancer cells beyond suppressed intrinsic or cell‐induced apoptosis are also able to trigger apoptosis and impair antitumor capacity of immune cells such as cytotoxic CD8+ T lymphocytes and NK cells, through the overexpression of programmed cell death‐1 ligand 1 (PD‐L1) [81, 82, 83].
In addition to the above‐described functions, apoptosis also plays a key role in regulation and homeostatic balance after immune response. This is the biochemical programme by which immune cells die while the infection is resolved, ensuring the decrease in circling immune cells at the end of the response and avoiding an excessive damage to local tissues. Thus, the population of activated immune cells is regulated by the own infection, via dead receptors and caspase 8 [58, 84]. Apoptotic death of neutrophils during bacterial, fungal or protozoal infection is a clear example of this scenario. Engulfed microorganisms accelerate neutrophil apoptosis ensuring a secure disposal of the phagocyted materials and ultimately the termination of the response, limiting the release of reactive oxygen species and at the same time recruiting and activating resident macrophages [85] (Figure 1).
FIGURE 1.
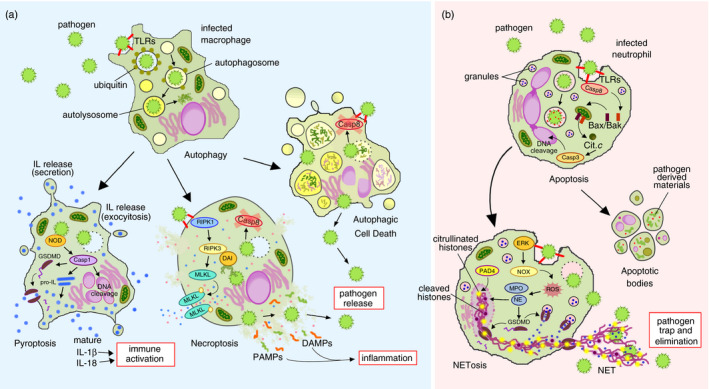
Schematic view of the interplay between different cell death modalities in the context of infection. During immune challenge, in infected macrophages (a) an autophagic process can be triggered by pathogen contact and the phagocyted microorganisms are targeted by an ubiquitin coat to autophagosomes and then to autolysosomes to be eliminated by acidic and enzymatic degradation. If microbes are persistent or escape from vacuolar compartments, under apoptosis blockade (e.g. by caspase 8 inactivation), alternative cell death programmes can be activated, releasing the pathogen to be neutralized by other immune cells, enhancing inflammation and activating the immune system. Thus, the cell can go undergo autophagic cell death, necroptosis induced by TLRs under caspase 8 inhibition, through the RIPK1‐RIPK3‐MLKL pathway or by microbial DNA through DAI‐RIPK3 interactions, or caspase 1‐dependent pyroptosis triggered by NOD receptors. On the other hand, in infected neutrophils (b) pathogen contact triggers an apoptotic process that ensures the safe disposal of the microbial components and toxins after its degradation and at the same time regulates neutrophil population during immune response. Alternatively, persistent pathogens can also activate NETosis, through the ERK‐NOX4 pathway, massive permeabilization and releasing of granule contents that cause neutrophil death, but the released extracellular traps enclose and destroy invader pathogens. Casp, caspase; DAI, DNA‐dependent activator of IFN‐regulatory factors; DAMPs, danger‐associated molecular patterns; ERK, extracellular signal‐regulated kinase; GSDMD, Gasdermin D; IL, interleukin; MLKL, mixed lineage kinase domain‐like protein; MPO, myeloperoxidase; NE, neutrophil elastase; NET, neutrophil extracellular trap; NOD, nucleotide‐binding and oligomerization domain; NOX, NAPH oxidase; PAD4, peptidylarginine deiminase 4; PAMPs, pathogen‐associated molecular patterns; RIPK, receptor‐interacting protein kinases; ROS, reactive oxygen species; TLRs, Toll‐like receptors
Regulated necrosis and innate immunity
Unlike apoptosis, which is considered a less immunogenic variant of PCD, all forms of regulated necrosis are highly inflammatory [4, 16]. This type of PCD involves the massive release of intracellular molecules, which act as danger‐associated molecular patterns (DAMPs), activating the immune system and amplifying the inflammatory response [3, 14]. The same process is triggered during innate immune response to microbes, inducing the release of pathogen‐associated molecular patterns (PAMPs) from infected cells, leading to immune cell recruitment and activation [86] (Figure 1). This concept was introduced by certain studies that demonstrated that unlike apoptotic cells, necrotic cells are able to release specific molecules that activate immune cells such as macrophages and antigen‐presenting cells. For example, the research of Basu and colleges in 2000 demonstrates that necrotic but no apoptotic E.G7 cells release HSPs such as HSP90, HSP70 and Gp96, which can activate macrophages and dendritic cells through the stimulation of the NF‐κB pathway.
Although in this work, they do not induce an specific necroptotic process (just a mimic of a regular necrosis), this was a first approach to the idea that during oncosis and non‐apoptotic cell death, the release of these intracellular factors is a crucial internal signalling to activate the immune response [87]. Necroptotic cell death has demonstrated to be one of the most immunogenic forms of PCD, inducing the release of important danger signals such as calreticulin (CALR) and HMGB1, which activate antigen uptake and processing, respectively, in immune cells [88]. This has been experimentally corroborated in the context of inflammatory and pathogen‐induced necroptosis. Mouse embryonic fibroblasts and myoblasts die through a necroptotic process in response to Shigella infection and Clostridium septicum‐derived α‐toxin, accompanied by the secretion of KC (a neutrophil‐attracting chemokine) and the immunostimulatory protein HMGB1, respectively [89, 90]. Likewise, Salmonella induces type I interferon‐dependent assembly of RIPK1 and/or RIPK3 in macrophages, leading to necroptosis and the release of pro‐inflammatory cytokines such as TNF, KC, IL‐1α, IL‐1β and IFN‐γ [91]. In the same way, murine cancer cell lines such as TC‐1 and CT26 undergo chemically induced necroptosis, but not accidental necrosis (e.g. freeze‐thawing‐driven necrosis) are able to activate immune cells in vitro inducing several markers for cell–cell recognition and antigen processing and ‘vaccinate’ mice in vivo, against challenge with the same cell type, inducing IFN‐γ and CD8+ T‐cell responses [92, 93].
According to this, necroptotic machinery is required for an effective response to pathogenic entities. For example, in spite of their pleiotropic role in apoptosis–necrosis transition, inflammasome activation and TNF signalling, the kinases RIPK1 and RIPK3 are actively involved in antiviral and antibacterial response in a necroptotic‐dependent way (reviewed in [11, 23, 24]). It has been demonstrated in vitro and in vivo that certain viral entities such as cytomegalovirus or herpes simplex virus regulate the necroptotic response in murine infected cells through the interaction between virus‐encoded proteins and the RIP homotypic interaction motif (RHIM) domains of RIPK1 and RIPK3 [94, 95, 96] (Figure 2). This has also been observed in human‐infected cells [97, 98]. A recent study has revealed a similar mechanism to inactivate RIPK3 as a viral necroptosis‐resistance strategy. The authors demonstrate that poxviruses encode MLKL homologues, which are able to interact with RIPK3 and block necroptosis in vitro when they are expressed in U937 (human myeloid cells), HT29 (human colon carcinoma) and mouse dermal fibroblasts [99]. Viral MLKL‐like proteins seem to act as competitive inhibitors that sequester RIPK3 to impair the binding with cellular MLKL and subsequent necroptosis activation. Some bacteria have also evolved to suppress necroptosis as an infective mechanism, that is the case of Porphyromonas gingivalis, which is able to cleave RIPK1 in human‐infected endothelial cells via a lysine‐specific (Kgp) protease [100] (Figure 2). This example demonstrates the ancestral role of necroptosis in the control of viral and bacterial infections and its efficiency as an antimicrobial mechanism. However, necroptosis can be also manipulated as a microbial strategy to impair immune function. In line with this, intracellular bacteria such as Salmonella enterica are also strong inducers of RIPK3‐dependent necroptosis in infected macrophages [91]. The same mechanism is used by other pathogenic entities, which induce necroptosis in immune cells to interfere with its defensive function and amplify the inflammatory loop [101].
In spite of that, necroptosis is still an efficient mechanism to eliminate and expose intracellular pathogens. The key role of RIP kinases and necroptosis in the maintenance of an immunocompetent status has been confirmed in vivo in RIPK3‐knockout mice, which are more susceptible to viral and bacterial infections [102, 103]. Moreover, the role of RIP kinases in innate immunity is beyond necroptosis induction. RIPK1 and RIPK3 are key regulators of cell final destination during an infection or immune challenge, but they can also participate in signal transduction for the downstream activation of NF‐κB acting as adaptor proteins in the signalling cascade of nucleic acid‐sensing proteins such as the DAI or TLRs such as TLR3 and TLR4 [11, 29].
Necroptosis is also important in the antitumor response. As this type of PCD can be induced in conditions of apoptosis blockade or inhibition, it represents an important therapeutic alternative in apoptosis‐resistant malignancies [104]. The relevance of necroptosis in tumour inhibition has been highlighted by the increasing capacity of different cancers to develop resistance mechanism to avoid necroptosis (Table 1), which decreases their response to treatments, increases their resilience and makes their prognosis worst. For example, the loss of CALR and HMGB1 expression in lung and breast cancers, respectively, is positively correlated with tumour size, aggressiveness and a bad disease outcome [105, 106]. Other types of cancer also suppress necroptosis via loss of RIPK3 expression. This has been observed in vitro in more than 40 cancer cell lines and in vivo in three different cohorts with patients of acute myeloid leukaemia, breast carcinoma and colorectal cancer [107, 108, 109].
Necroptosis has shown to be also important for adaptive antitumor immunity, particularly for dendritic cell (DC) antigen processing and presentation and CD8+ T‐cell activation; however, it is also a process that restraint T‐cell populations, causing cell death of tumour‐reactive T cells that recognize cognate antigens upon T‐cell receptor (TCR) restimulation [110]. According to this, blocking TCR restimulation‐driven necroptosis can improve the survival and reactivity of tumour‐infiltrating T cells achieving a better tumour growth inhibition, a fact that has been experimentally demonstrated in adoptively transferred T cells using RIPK1 inhibitors [111]. On the other hand, necroptotic cell death creates a pro‐inflammatory environment that can not only help to activate and recruit immune cells, but also contribute to tumour proliferation and metastasis (reviewed in [110]). It seems that the role of necroptosis in cancer is contradictory. As a process that can be pharmacologically manipulated, it offers important advantages to cancer immunotherapy, but at the same time, it can cause certain complications and variable response to treatments. As with other therapeutic approaches, the tissue specificity and targeting and the selectivity to induce the process under certain conditions and inhibit it in others seem to be the key for the therapeutic success.
Not only necroptosis but also other forms of regulated necrosis are also implicated in particular and interesting mechanisms of innate immunity. That is the case of the IRF3‐induced necrotic cell death, which is a rapid and efficient mechanism to impair viral and parasitic replication with Kupffer cells and prevent the subsequent spread of the infection from the liver to other parts of the organism. The efficacy of this particular form of PCD has been tested in vivo in mice infected with human adenovirus and L. monocytogenes [53]. Cell death is rapidly triggered in Kupffer cells in response to these pathogens when they escape from vacuolar or phagocytic compartments, as a ‘suicidal necrosis’, which depends on IRF3 expression but not transcriptional activity and allows the release of the pathogens to be processed by other immune cells.
Another clear example of the active role of regulated necrosis in innate immune response to pathogens is pyroptosis, which receives this denomination due to its final and distinctive molecular event: the release of pyrogenic cytokines. This special form of PCD occurs in different cell types but primarily in immune cells, mainly macrophages, as a direct response to pathogens leading to the caspase 1‐dependent processing and release of IL‐1β, IL‐α and IL‐18, which have important immune‐activating functions [34] (Figure 1). IL‐1 family are strong inflammation mediators that induce cyclooxygenase type 2, and increase expression of adhesion molecules and nitric oxide synthesis [112]. On the other hand, IL‐18 is a potent immune stimulator that induces interferon‐γ (IFN‐γ) production in TH1 cells, NK cells and cytotoxic T lymphocytes (CTLs), and also promotes TH2 cell development [113]. The induction of pyroptosis and the active secretion of ILs in response to bacterial and viral infections have been corroborated in vitro and in vivo. Intracellular bacteria such as Burkholderia pseudomallei induce NLRP3‐ and NLRC4‐dependent pyroptosis with IL‐β secretion in infected macrophages and DCs in vitro, and Nlrp3‐/‐ mice are more susceptible to B. pseudomallei due to IL‐β ‐ and IL‐18‐deficient secretion [114]. This study demonstrates that not particularly IL‐1β but IL‐18 has a crucial role in mice survival against B. pseudomallei infection due to its IFN‐γ stimulatory activity. Likewise, pyroptosis of macrophages and IL‐1β active secretion in response to viral haemorrhagic infection have been monitored in vivo, in zebrafish models [115].
However, besides its function in cytokine maturation and release, pyroptosis itself seems to be an efficient mechanism to eliminate and release intracellular bacteria from infected macrophages, exposing pathogens to neutrophil‐mediated clearance through a molecular mechanism that relies on caspase 1 activity. This has been demonstrated in vivo for Salmonella typhimurium, L. pneumophila and Burkholderia thailandensis in Casp1 knockout mice [7]. The effectiveness of pyroptosis in the control of bacterial and viral infections is evident by the different mechanisms that these pathogens develop to inhibit the inflammasome‐mediated induction of this PCD (Figure 2). Extracellular bacteria such as Yersinia spp., Vibrio parahaemolyticus and Pseudomonas aeruginosa or intracellular bacteria such as Chlamydia trachomatis, Francisella tularensis and L. pneumophila secrete specific proteins that interfere with the formation and activation of the ASC‐NLRC4/NLRP3 inflammasome or directly inhibit caspase 1 to prevent IL‐1/IL‐18 and GSDMD processing (reviewed in [116]). In the same way, H5N1 and H3N2 influenza A viruses produce a small accessory protein (PB1‐F2) that binds to NLRP3 to induce a conformational state that impairs its interaction with the NIMA‐related kinase 7 (NEK7) and to prevent exposure of its pyrin domain required for ASC interaction and inflammasome assembly [117].
It has been demonstrated that not only caspase 1 but also caspase 3 has a role in pyroptosis induction via GSDME, the newly identified pyroptosis executioner of the Gasdermin protein family, but this process seems not to be directly connected to pathogen clearance, but more to antitumor responses in certain cell types with high GSDME expression [36]. Pyroptosis has been recently recognized by its role in the immune response against bacterial infections of the gastrointestinal tract and even in the control of transformed cells in the context of gastrointestinal cancer [9]. In fact, the expression of GSDMD, the major executor of pyroptosis, is decreased in gastric cancer compared with normal gastric epithelium [118] and its downregulation in experimental models in vitro and in vivo is related to an enhanced tumour proliferation [119]. In spite of the increased levels of this protein in some types of tumours such as non‐small‐cell lung cancer (NSCLC), GSDMD‐driven pyroptosis is induced by several chemotherapeutic agents, small molecules and natural products in a wide variety of cancers including oesophageal squamous cell carcinoma (OSCC), lung epithelial adenocarcinomas, triple negative breast cancer, oral squamous cell carcinoma (OSCC), myeloid acute leukaemia and ovarian cancer, with important antitumor activities. Likewise, decreased GSDME expression has been found in melanoma cells as a factor associated with etoposide resistance [120]. This protein has shown anticancer effects and sensitization to chemotherapeutic agents in melanomas and ESCC through pyroptotic cell death induction (reviewed in [36]).
On the other hand, NETosis is another important example of how regulated necrosis actively mediates microbial killing and immune enhancement. This cell death modality is known to be induced in specialized immune cells such as neutrophils and other polymorphonuclear cells, leading to the release of the so‐called ‘extracellular traps’, complex structures composed of chromatin fragments and histones, which opsonize and enclose circling pathogens, limiting their spreading within the body and facilitating their clearance by phagocytosis [1, 10] (Figure 1). These extracellular traps also contain antimicrobial proteins such as MPO, NE, proteinase 3 (PR3), cathepsin G, lysozyme and α‐defensins representing a non‐phagocytic mode to neutralize and degrade invading pathogens [121, 122], and are able to stimulate the production of IFN‐α, a strong antiviral cytokine [123].
In the past 10 years, this form of PCD has been extensively studied to elucidate the sequence of its molecular events and its role in immune response, demonstrating that it is actually one of the most specialized immune mechanisms, which can be activated depending on microbe size and location, and is a strategy to control pathogens that are able to escape from neutrophil phagocytosis (reviewed in [13]). The effectiveness of this antimicrobial defence is evident in some pathogens such as Group A Streptococcus and Staphylococcus aureus, which produce nucleases and DNA binding proteins to impair NET formation and block NETosis as an active evasion mechanism [124] (Figure 2). Experimental findings have also demonstrated the efficiency of this antimicrobial mechanism against bacteria such as S. aureus and parasites such as Leishmania amazonensis and Entamoeba histolytica through a rapid NET formation and releasing mechanism independent of oxidative stress [44, 47, 48]. In the same way, it has been reported that patients with chronic granulomatous disease (CGD) are more susceptible to Aspergillus nidulans growth, due to their deficiency in NADPH oxidase function and consequently in NET formation, at least by the classical pathway [125]. Invasive aspergillosis is a leading cause of death in CGD patients, suggesting the importance of NETosis in pathogen control and immunocompetent status.
In contrast to its evident role in defence against sepsis and infection, the involvement of NETosis in cancer has not been completely elucidated. Some studies have revealed that NET formation could be a risk factor in cancer and other chronic inflammatory conditions mainly due to excessive coagulation and thrombotic vascular complications (reviewed in [126]). It is not clear how the use of NETosis inhibitors could improve the outcome of these diseases and whether this inhibition could immunocompromise the patient or lead to other complications; however, some authors propose the NETs in blood and organs as a biomarker for certain cancer stages and even as target to ameliorate vascular complications and metastatic potential [126, 127].
Autophagic cell death and innate immunity
Different experimental evidences have revealed the presence of autophagosomes and the rearrangement of the cytoskeleton in dying cells, but wherever this is a strategy of cell survival rather than an active death mechanism has been a controversy. Conclusive data, using apoptosis inhibitors and Bax/Bak‐deficient mouse embryonic fibroblasts, have shown that autophagic cell death or ACD is an alternative to apoptosis in which cell dies in response to chemical stressors or cytotoxic substances such as staurosporine or etoposide, through a process that involves the formation of multiple vesicular organelles and can be reversed using classical autophagy inhibitors such as 3‐methyladenine (3‐MA) or silencing ATG genes (reviewed in [50]). It seems that like regulated necrosis, autophagic death is activated under certain stimulus including infection when apoptosis is blocked or inhibited, playing a protective role in host defence. In line with this, different in vivo studies have demonstrated the importance of the ATG genes against bacterial and viral infections such as L. monocytogenes, Toxoplasma gondii, herpes simplex virus 1 and Sindbis virus in mice [8, 128, 129].
As occurs with apoptosis and regulated necrosis, ACD can be activated by PRRs as a result of direct contact with pathogens, inducing an alternative defence that helps cells to deal with viruses or intracellular microorganisms, a key element of innate immune response. The activation of the autophagic death by TLRs such as TLR1/2, TLR3, TLR4 and TLR7 has been corroborated in mice‐derived macrophages and dendritic cells, in response to agonists such as bacterial lipopolysaccharide (LPS) or under Mycobacterium tuberculosis infection. In the same way, peptidoglycan has been demonstrated to induce autophagy through NOD proteins, mainly Nod1 and Nod2 in murine myeloid and epithelial cells (reviewed in [130]). Interestingly, muramyldipeptide, a peptidoglycan‐derived product, also activates autophagy in human dendritic cells, in a process that requires NOD2, RIP2 and the ATG genes ATG5, ATG7 and ATG16L1, increasing MHC Class II antigen expression and CD4+ T lymphocyte proliferation [131]. Not only bacterial but viral infections also induce an autophagic defensive response in host cells. Viral single‐strand RNAs (ssRNAs) are recognized by TLR7‐ and NOD2‐stimulating antiviral defence in RAW 264·7 macrophages and type I IFN production in dendritic cells. The ssRNAs can additionally activate other intracellular sensors such as RNA‐dependent protein kinase (PKR), which activate autophagy via Beclin‐1 in mouse embryonic fibroblasts and primary neurons infected with herpes simplex virus 1 [132].
The process of autophagy by itself is intrinsically connected to innate immune clearance of intracellular pathogens. Thus, when a pathogen interacts with a host cell, it triggers an autophagic process that could lead to its own phagocytosis or eventually to the death of the infected cell, exposing this microbe to other effector cells of the immune system (Figure 1). Experimental results with intracellular Salmonella, enteropathogenic Escherichia coli, Streptococcus pyogenes and L. monocytogenes show that engulfed bacteria are targeted to autophagosomes and subsequently to autolysosomes by a coat of ubiquitin that interacts with the cytoskeleton protein LC3. This was corroborated by colocalization and confocal microscopy studies, together with the use of Atg5‐ or p62‐deficient cells and genetically modified microbial strains insensitive to ubiquitin recognition [130].
In the context of infection, different autophagic signalling proteins such as p62, CALCOCO 2 (calcium binding and coiled‐coil domain‐containing protein 2) and optineurin can act as adaptor proteins in the inflammatory pathway ultimately activating NF‐κB and cytokine secretion. Additionally, p62 is also involved in the recruitment of cytosolic proteins such as ubiquitin or ribosomal proteins to autolysosomes, to be converted by internal proteolysis in antimicrobial peptides, highly effective to intracellular pathogens such as M. tuberculosis (reviewed in [133]). Interestingly, ACD can also release and expose DAMPs to activate the immune system, a process usually associated with necrotic forms of PCD. Ayna et al. demonstrated that autophagic dying Ba/F3 cells (cell death induced by IL‐3 deprivation), but not living, apoptotic or necroptotic cells, activate mouse macrophages in coculture, inducing the NLRP3 inflammasome and IL‐1β secretion. The activation occurs during the phagocytosis of autophagic cells and is mediated by the release of ATP (acting as a danger signal) through Pannexin‐1 channels in Ba/F3 dying cells that activate P2X7 purinergic receptor in macrophages [134]. This is another evidence of the crosstalk between pyroptosis and other forms of PCD in macrophages under immunostimulatory conditions, induced by Pannexin‐1‐mediated activation of NLRP3. This was corroborated in vivo, injecting autophagic dying Ba/F3 cells intraperitoneally in mice, leading to acute inflammation and neutrophil recruitment [134].
The processes of autophagy and ACD have been also revised due to its controversial role in cancer. Research findings reveal that autophagy actually contribute to survival, metabolic reprogramming and tumour development. Interestingly, the same process can lead to the elimination of cancer cells by autophagic death [51]. Some studies have established a negative correlation between autophagy and malignant transformation by the fact that important components of the autophagy machinery such as Beclin 1 are deficient or have lower expression in tumours of different origin including breast, ovarian and prostate cancer [135]. In addition, ACD is involved in the cytotoxic and antitumor effects displayed by some chemotherapeutic drugs such as tamoxifen or arsenic trioxide in human breast carcinoma and glioma and can be induced by radiation in breast, prostate and colon cancers [136]. More recently, other anticancer agents have demonstrated to induce ACD as a potent antineoplastic mechanism. For example, the compound F0911‐7667 was discovered by a screening of small molecules targeting the protein deacetylase Sirtuin 1 with potential anticancer properties. This chemical agent acts as a Sirtuin 1 activator, inducing mitophagy and ACD and repressing glioblastoma proliferation in vitro and in vivo [137]. In the same way, the natural flavonoid kaempferol induces ACD in gastric cancer cells, modulating the endoplasmic reticulum stress response and downregulating the histone methyltransferase G9a, an important target in other human cancers [138]. In fact, G9a is upregulated in head and neck squamous cell carcinoma (HNSCC) and its expression correlates with cancer progression and poor prognosis [139]. This study also demonstrates that downregulation of G9a enhances the antitumor response inducing ACD.
Autophagy is also required for immune cell activation, antigen presentation, thymus selection and cytokine release in different cell types such as NK cells, DCs and T lymphocytes, but represents a ‘double‐edge sword’ that can also regulate these cell populations by ACD. Current research aimed to focus on the understanding of this dual role and its potential application in cancer immunotherapy [140]. Autophagy can be induced in cancer cells as a survival strategy in response to stressful conditions of their microenvironment such as hypoxia and nutrient deficiency; however, ACD seems to act as an alternative cell death pathway that can subvert tumour progression by elimination of transformed cells. In line with this and as was described above, multiple anticancer agents are showing important pharmacological effects on tumour regression and cancer proliferation inhibition, which are based on ACD induction. The capacity to target autophagic death in cancer cells over autophagy to disrupt the balance between the anti‐ and pro‐survival effects of autophagic process seems to be the key for a successful anticancer therapy based on this form of PCD.
CONCLUSIONS AND PERSPECTIVES
Programmed cell death (PCD) in its different modes of presentation and acting through different pathways has proved to be more than that of the final fate of a damaged or a senescent cell. This is a complex process, which can be induced, sustained, adjusted and even reversed according to the cell type, the intra‐ and extracellular conditions, the stimulus that triggers it and the interactions with other surrounding cells. Several experimental data have demonstrated that PCD is actively involved in a variety of biological functions but especially in innate immunity and host defence. As it has been discussed throughout this review, PCD is induced by pathogen presence and is part of the immune mechanisms involved in their detection and elimination. Different types of pathogens are able to induce different cell death modalities, indicating that is a specialized process rather than just a physiological consequence of microbe invasion to host cells. PCD is no longer seen as the result of an inflammatory process but as fine‐tuned mechanism that can induce, amplify or modulate this inflammation, during either physiological or pathological conditions, including infection and malignant transformation. The expanding network of cell death programmes is revealing novel and complex variants or PCD and their key role in innate immunity and immune cell homeostasis. The existence of multiple forms of a cell to die and their involvement in different immune mechanisms offer interesting ways to selectively modulate and enhance immune response to infections, chronic inflammation and cancer, through pharmacological interventions and immunotherapy.
CONFLICT OF INTEREST
There are not conflicts of interest associated with this work.
REFERENCES
- 1. Allam R, Kumar SV, Darisipudi MN, Anders H‐J. Extracellular histones in tissue injury and inflammation. J Mol Med. 2014;92:465–72. [DOI] [PubMed] [Google Scholar]
- 2. Godlewski M, Kobylińska A. Programmed cell death‐strategy for maintenance cellular organisms homeostasis. Postepy Hig Med Dosw(Online). 2016;70:1229. [PubMed] [Google Scholar]
- 3. Linkermann A, Stockwell BR, Krautwald S, Anders H‐J. Regulated cell death and inflammation: an auto‐amplification loop causes organ failure. Nat Rev Immunol. 2014;14:759–67. [DOI] [PubMed] [Google Scholar]
- 4. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–20. [DOI] [PubMed] [Google Scholar]
- 5. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yan G, Elbadawi M, Efferth T. Multiple cell death modalities and their key features. World Acad Sci J. 2020;2:39–48. [Google Scholar]
- 7. Miao EA, Rajan JV, Aderem A. Caspase‐1‐induced pyroptotic cell death. Immunol Rev. 2011;243:206–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Orvedahl A, MacPherson S, Sumpter R Jr, Tallóczy Z, Zou Z, Levine B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe. 2010;7:115–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou C‐B, Fang J‐Y. The role of pyroptosis in gastrointestinal cancer and immune responses to intestinal microbial infection. Biochimica et Biophysica Acta (BBA)‐Reviews Cancer. 2019;1872:1–10. [DOI] [PubMed] [Google Scholar]
- 10. Andrade F, Darrah E. NETs: the missing link between cell death and systemic autoimmune diseases? Front Immunol. 2013;3:428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Humphries F, Yang S, Wang B, Moynagh PN. RIP kinases: key decision makers in cell death and innate immunity. Cell Death Differ. 2015;22:225–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen H, Ning X, Jiang Z. Caspases control antiviral innate immunity. Cell Mol Immunol. 2017;14:736–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Burgener SS, Schroder K. Neutrophil extracellular traps in host defense. Cold Spring Harb Perspect Biol. 2020;12:a037028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. D’Arcy MS. Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int. 2019;43:582–92. [DOI] [PubMed] [Google Scholar]
- 15. Gudipaty SA, Conner CM, Rosenblatt J, Montell DJ. Unconventional ways to live and die: cell death and survival in development, homeostasis, and disease. Annu Rev Cell Dev Biol. 2018;34:311–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vanden Berghe T, Kaiser WJ, Bertrand MJ, Vandenabeele P. Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol Cell Oncol. 2015;2:e975093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Galluzzi L, Kepp O, Krautwald S, Kroemer G, Linkermann A. Molecular mechanisms of regulated necrosis. Semin Cell Dev Biol. 2014;35:24–32. [DOI] [PubMed] [Google Scholar]
- 18. Vandenabeele P, Galluzzi L, Berghe TV, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–14. [DOI] [PubMed] [Google Scholar]
- 19. Cai Z, Jitkaew S, Zhao J, Chiang H‐C, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF‐induced necroptosis. Nat Cell Biol. 2014;16:55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7:971–81. [DOI] [PubMed] [Google Scholar]
- 21. Su Z, Yang Z, Xu Y, Chen Y, Yu Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer. 2015;14:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kayagaki N, Kornfeld OS, Lee BL, Stowe IB, O’Rourke K, Li Q, et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature. 2021;591:131–136. 10.1038/s41586-021-03218-7 [DOI] [PubMed] [Google Scholar]
- 23. Silke J, Rickard JA, Gerlic M. The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol. 2015;16:689–97. [DOI] [PubMed] [Google Scholar]
- 24. Silke J, Rickard JA, Gerlic M. Erratum: the diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol. 2015;16:889. [DOI] [PubMed] [Google Scholar]
- 25. Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, et al. cIAPs block Ripoptosome formation, a RIP1/caspase‐8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell. 2011;43:449–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vanlangenakker N, Bertrand M, Bogaert P, Vandenabeele P, Berghe TV. TNF‐induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis. 2011;2:e230‐e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Geng J, Ito Y, Shi L, Amin P, Chu J, Ouchida AT, et al. Regulation of RIPK1 activation by TAK1‐mediated phosphorylation dictates apoptosis and necroptosis. Nat Commun. 2017;25:359. 10.1038/s41467-017-00406-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. He S, Liang Y, Shao F, Wang X. Toll‐like receptors activate programmed necrosis in macrophages through a receptor‐interacting kinase‐3–mediated pathway. Proc Natl Acad Sci USA. 2011;108:20054–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll‐like receptor 3‐mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288:31268–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gilmore A. Anoikis. Cell Death Differ. 2005;12:1473–7. [DOI] [PubMed] [Google Scholar]
- 31. Paoli P, Giannoni E, Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta (BBA)‐Molecular Cell Res. 2013;1833:3481–98. [DOI] [PubMed] [Google Scholar]
- 32. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron‐dependent form of nonapoptotic cell death. Cell 2012;149:1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Albrecht P, Lewerenz J, Dittmer S, Noack R, Maher P, Methner A. Mechanisms of oxidative glutamate toxicity: the glutamate/cystine antiporter system xc as a neuroprotective drug target. CNS Neurol Disord‐Drug Targets (Formerly Current Drug Targets‐CNS & Neurological Disorders) 2010;9:373–82. [DOI] [PubMed] [Google Scholar]
- 34. Kepp O, Galluzzi L, Zitvogel L, Kroemer G. Pyroptosis–a cell death modality of its kind? Eur J Immunol. 2010;40:627–30. [DOI] [PubMed] [Google Scholar]
- 35. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome‐activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ruan J, Wang S, Wang J. Mechanism and regulation of pyroptosis‐mediated in cancer cell death. Chem Biol Interact. 2020;323:109052. [DOI] [PubMed] [Google Scholar]
- 37. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5. [DOI] [PubMed] [Google Scholar]
- 38. Ting JP‐Y, Willingham SB, Bergstralh DTJNRI. NLRs at the intersection of cell death and immunity. Nat Rev Immunol. 2008;8:372–9. [DOI] [PubMed] [Google Scholar]
- 39. Nirmala JG. Lopus M. Cell death mechanisms in eukaryotes. Cell Biol Toxicol. 2020;36:145–64. [DOI] [PubMed] [Google Scholar]
- 40. Fujisawa A, Kambe N, Saito M, Nishikomori R, Tanizaki H, Kanazawa N, et al. Disease‐associated mutations in CIAS1 induce cathepsin B–dependent rapid cell death of human THP‐1 monocytic cells. Blood. 2007;109:2903–11. [DOI] [PubMed] [Google Scholar]
- 41. Willingham SB, Bergstralh DT, O'Connor W, Morrison AC, Taxman DJ, Duncan JA, et al. Microbial pathogen‐induced necrotic cell death mediated by the inflammasome components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe. 2007;2:147–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Morinaga Y, Yanagihara K, Nakamura S, Hasegawa H, Seki M, Izumikawa K, et al. Legionella pneumophila induces cathepsin B‐dependent necrotic cell death with releasing high mobility group box1 in macrophages. Respir Res. 2010;11:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13:463–9. [DOI] [PubMed] [Google Scholar]
- 44. Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus . J Immunol. 2010;185:7413–25. [DOI] [PubMed] [Google Scholar]
- 45. Kraaij T, Tengström FC, Kamerling SW, Pusey CD, Scherer HU, Toes RE, et al. A novel method for high‐throughput detection and quantification of neutrophil extracellular traps reveals ROS‐independent NET release with immune complexes. Autoimmun Rev. 2016;15:577–84. [DOI] [PubMed] [Google Scholar]
- 46. Tatsiy O, McDonald P. Physiological stimuli induce PAD4‐dependent, ROS‐independent NETosis, with early and late events controlled by discrete signaling pathways. Front Immunol. 2018;9:2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Díaz‐Godínez C, Fonseca Z, Néquiz M, Laclette JP, Rosales C, Carrero J, et al. Entamoeba histolytica trophozoites induce a rapid non‐classical NETosis mechanism independent of NOX2‐derived reactive oxygen species and PAD4 activity. Front Cell Infect Microbiol. 2018;8:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rochael NC, Guimarães‐Costa AB, Nascimento MT, DeSouza‐Vieira TS, Oliveira MP, Souza LFG, et al. Classical ROS‐dependent and early/rapid ROS‐independent release of neutrophil extracellular traps triggered by Leishmania parasites. Sci Rep. 2015;5:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tsujimoto Y, Shimizu S. Another way to die: autophagic programmed cell death. Cell Death Differ. 2005;12:1528–34. [DOI] [PubMed] [Google Scholar]
- 51. Yang X, Yu D‐D, Yan F, Jing Y‐Y, Han Z‐P, Sun K, et al. The role of autophagy induced by tumor microenvironment in different cells and stages of cancer. Cell Biosci. 2015;5:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yonekawa T, Thorburn A. Autophagy and cell death. Essays Biochem. 2013;55:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Di Paolo NC, Doronin K, Baldwin LK, Papayannopoulou T, Shayakhmetov D. The transcription factor IRF3 triggers “defensive suicide” necrosis in response to viral and bacterial pathogens. Cell Rep. 2013;3:1840–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yeretssian G, Correa RG, Doiron K, Fitzgerald P, Dillon CP, Green DR, et al. Non‐apoptotic role of BID in inflammation and innate immunity. Nature. 2011;474:96–9. [DOI] [PubMed] [Google Scholar]
- 55. Weng D, Marty‐Roix R, Ganesan S, Proulx MK, Vladimer GI, Kaiser WJ, et al. Caspase‐8 and RIP kinases regulate bacteria‐induced innate immune responses and cell death. Proc Natl Acad Sci. 2014;111:7391–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mocarski ES, Upton JW, Kaiser WJJNRI. Viral infection and the evolution of caspase 8‐regulated apoptotic and necrotic death pathways. Nat Rev Immunol. 2012;12:79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Robinson KS, Aw R. The commonalities in bacterial effector inhibition of apoptosis. Trends Microbiol. 2016;24:665–80. [DOI] [PubMed] [Google Scholar]
- 58. Abbas AK, Lichtman AH, Pillai S. Cellular and molecular immunology E‐book, 8th edn. Saunders: Philadelphia, 2014. [Google Scholar]
- 59. Prager I, Liesche C, van Ooijen H, Urlaub D, Verron Q, Sandström N, et al. NK cells switch from granzyme B to death receptor–mediated cytotoxicity during serial killing. J Exp Med. 2019;216:2113–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li HM, Fujikura D, Harada T, Uehara J, Kawai T, Akira S, et al. IPS‐1 is crucial for DAP3‐mediated anoikis induction by caspase‐8 activation. Cell Death Differ. 2009;16:1615–21. [DOI] [PubMed] [Google Scholar]
- 61. Joly A‐L, Wettstein G, Mignot G, Ghiringhelli F, Garrido C. Dual role of heat shock proteins as regulators of apoptosis and innate immunity. J Innate Immun. 2010;2:238–47. [DOI] [PubMed] [Google Scholar]
- 62. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liu Q, Zhang D, Hu D, Zhou X, Zhou Y. The role of mitochondria in NLRP3 inflammasome activation. Mol Immunol. 2018;103:115–24. [DOI] [PubMed] [Google Scholar]
- 64. Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, et al. Extrinsic and intrinsic apoptosis activate pannexin‐1 to drive NLRP 3 inflammasome assembly. EMBO J. 2019;38:e101638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zheng M, Kanneganti TDJIR. The regulation of the ZBP1‐NLRP3 inflammasome and its implications in pyroptosis, apoptosis and necroptosis (PANoptosis). Immunol Rev. 2020;297:26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 67. Zörnig M, Hueber A‐O, Baum W, Evan G. Apoptosis regulators and their role in tumorigenesis. Biochim Biophys Acta (BBA)‐Reviews Cancer. 2001;1551:F1–F37. [DOI] [PubMed] [Google Scholar]
- 68. Wilson T, Johnston P, Longley D. Anti‐apoptotic mechanisms of drug resistance in cancer. Curr Cancer Drug Targets. 2009;9:307–19. [DOI] [PubMed] [Google Scholar]
- 69. Porporato PE, Filigheddu N, Bravo‐San Pedro JM, Kroemer G, Galluzzi L. Mitochondrial metabolism and cancer. Cell Res. 2018;28:265–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pistritto G, Trisciuoglio D, Ceci C, Garufi A, D'Orazi G. Apoptosis as anticancer mechanism: function and dysfunction of its modulators and targeted therapeutic strategies. Aging (Albany NY). 2016;8:603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cheriyath V, Leaman DW, Borden E. Emerging roles of FAM14 family members (G1P3/ISG 6–16 and ISG12/IFI27) in innate immunity and cancer. J Interferon Cytokine Res. 2011;31:173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rosebeck S, Leaman DWJA. Mitochondrial localization and pro‐apoptotic effects of the interferon‐inducible protein ISG12a. Apoptosis. 2008;13:562–72. [DOI] [PubMed] [Google Scholar]
- 73. Makovitzki‐Avraham E, Daniel‐Carmi V, Alteber Z, Farago M, Tzehoval E, Eisenbach L. The human ISG12a gene is a novel caspase dependent and p53 independent pro‐apoptotic gene, that is overexpressed in breast cancer. Cell Biol Int Rep. 2013;20:37–46. [Google Scholar]
- 74. Tahara E, Tahara H, Kanno M, Naka K, Takeda Y, Matsuzaki T, et al. G1P3, an interferon inducible gene 6‐16, is expressed in gastric cancers and inhibits mitochondrial‐mediated apoptosis in gastric cancer cell line TMK‐1 cell. Cancer Immunol Immunother. 2005;54:729–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cheriyath V, Glaser KB, Waring JF, Baz R, Hussein MA, Borden E. G1P3, an IFN‐induced survival factor, antagonizes TRAIL‐induced apoptosis in human myeloma cells. J Clin Invest. 2007;117:3107–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Cheriyath V, Kuhns M, Jacobs B, Evangelista P, Elson P, Downs‐Kelly E, et al. G1P3, an interferon‐and estrogen‐induced survival protein contributes to hyperplasia, tamoxifen resistance and poor outcomes in breast cancer. Oncogene. 2012;31:2222–36. [DOI] [PubMed] [Google Scholar]
- 77. Velloso FJ, Trombetta‐Lima M, Anschau V, Sogayar MC, Correa R. NOD‐like receptors: major players (and targets) in the interface between innate immunity and cancer. Biosci Rep. 2019;39:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Endo T, Abe S, Seidlar H, Nagaoka S, Takemura T, Utsuyama M, et al. Expression of IAP family proteins in colon cancers from patients with different age groups. Cancer Immunol Immunother. 2004;53:770–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kutikhin A. Role of NOD1/CARD4 and NOD2/CARD15 gene polymorphisms in cancer etiology. Hum Immunol. 2011;72:955–68. [DOI] [PubMed] [Google Scholar]
- 80. Li Z‐X, Wang Y‐M, Tang F‐B, Zhang L, Zhang Y, Ma J‐L, et al. NOD1 and NOD2 genetic variants in association with risk of gastric cancer and its precursors in a Chinese population. PLoS One. 2015;10:e0124949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zitvogel L, Kroemer G. Targeting PD‐1/PD‐L1 interactions for cancer immunotherapy. Oncoimmunology. 2012;1:1223–1225. 10.4161/onci.21335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Niu C, Li M, Zhu S, Chen Y, Zhou L, Xu D, et al. PD‐1‐positive Natural Killer Cells have a weaker antitumor function than that of PD‐1‐negative Natural Killer Cells in. Lung Cancer. 2020;17:1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Liu Y, Cheng Y, Xu Y, Wang Z, Du X, Li C, et al. Increased expression of programmed cell death protein 1 on NK cells inhibits NK‐cell‐mediated anti‐tumor function and indicates poor prognosis in digestive cancers. Oncogene. 2017;36:6143–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Feig C, Peter ME. How apoptosis got the immune system in shape. Eur J Immunol. 2007;37(S1):S61–S70. [DOI] [PubMed] [Google Scholar]
- 85. Geering B, Simon H‐U. Peculiarities of cell death mechanisms in neutrophils. Cell Death Differ. 2011;18:1457–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage‐associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–23. [DOI] [PubMed] [Google Scholar]
- 87. Basu S, Binder RJ, Suto R, Anderson KM, Srivastava PK. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF‐kappa B pathway. Int Immunol. 2000;12:1539–46. [DOI] [PubMed] [Google Scholar]
- 88. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97. [DOI] [PubMed] [Google Scholar]
- 89. Carneiro LA, Travassos LH, Soares F, Tattoli I, Magalhaes JG, Bozza MT, et al. Shigella induces mitochondrial dysfunction and cell death in nonmyleoid cells. Cell Host Microbe. 2009;5:123–36. [DOI] [PubMed] [Google Scholar]
- 90. Kennedy CL, Smith DJ, Lyras D, Chakravorty A, Rood J. Programmed cellular necrosis mediated by the pore‐forming α‐toxin from Clostridium septicum. PLoS Pathog. 2009;5:e1000516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol. 2012;13:954–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Aaes TL, Kaczmarek A, Delvaeye T, De Craene B, De Koker S, Heyndrickx L, et al. Vaccination with necroptotic cancer cells induces efficient anti‐tumor immunity. Cell Rep. 2016;15:274–87. [DOI] [PubMed] [Google Scholar]
- 93. Yang H, Ma Y, Chen G, Zhou H, Yamazaki T, Klein C, et al. Contribution of RIP3 and MLKL to immunogenic cell death signaling in cancer chemotherapy. Oncoimmunology 2016;5:e1149673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Huang Z, Wu S‐Q, Liang Y, Zhou X, Chen W, Li L, et al. RIP1/RIP3 binding to HSV‐1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe. 2015;17:229–42. [DOI] [PubMed] [Google Scholar]
- 95. Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3‐dependent necrosis. Cell Host Microbe. 2010;7:302–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Kaiser WJ, Upton JW, Mocarski ES. Receptor‐interacting protein homotypic interaction motif‐dependent control of NF‐κB activation via the DNA‐dependent activator of IFN regulatory factors. J Immunol. 2008;181:6427–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Guo H, Omoto S, Harris PA, Finger JN, Bertin J, Gough PJ, et al. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe. 2015;17:243–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Omoto S, Guo H, Talekar GR, Roback L, Kaiser WJ, Mocarski ES. Suppression of RIP3‐dependent necroptosis by human cytomegalovirus. J Biol Chem. 2015;290:11635–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Petrie EJ, Sandow JJ, Lehmann WI, Liang L‐Y, Coursier D, Young SN, et al. Viral MLKL homologs subvert necroptotic cell death by sequestering cellular RIPK3. Cell Rep. 2019;28:3309–19.e5. [DOI] [PubMed] [Google Scholar]
- 100. Madrigal AG, Barth K, Papadopoulos G, Genco CA. Pathogen‐mediated proteolysis of the cell death regulator RIPK1 and the host defense modulator RIPK2 in human aortic endothelial cells. PLoS Pathog. 2012;8:e1002723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. 2017;18:127–36. [DOI] [PubMed] [Google Scholar]
- 102. Kaiser WJ, Upton JW, Mocarski ES. Viral modulation of programmed necrosis. Curr Opin Virol. 2013;3:296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol. 2017;17:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Fulda S. editor Therapeutic exploitation of necroptosis for cancer therapy. In: Seminars in cell & developmental biology. Elsevier; 2014. [DOI] [PubMed] [Google Scholar]
- 105. Fucikova J, Becht E, Iribarren K, Goc J, Remark R, Damotte D, et al. Calreticulin expression in human Non‐Small cell lung cancers correlates with increased accumulation of antitumor immune cells and favorable prognosis. Cancer Res. 2016;76:1746–56. [DOI] [PubMed] [Google Scholar]
- 106. Yamazaki T, Hannani D, Poirier‐Colame V, Ladoire S, Locher C, Sistigu A, et al. Defective immunogenic cell death of HMGB1‐deficient tumors: compensatory therapy with TLR4 agonists. Cell Death Differ. 2014;21:69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Feng X, Song Q, Yu A, Tang H, Peng Z, Wang XJN. Receptor‐interacting protein kinase 3 is a predictor of survival and plays a tumor suppressive role in colorectal cancer. Neoplasma. 2015;62:592–601. [DOI] [PubMed] [Google Scholar]
- 108. Nugues A‐L, El Bouazzati H, Hetuin D, Berthon C, Loyens A, Bertrand E, et al. RIP3 is downregulated in human myeloid leukemia cells and modulates apoptosis and caspase‐mediated p65/RelA cleavage. Cell Death Dis. 2014;5:e1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Koo G‐B, Morgan MJ, Lee D‐G, Kim W‐J, Yoon J‐H, Koo JS, et al. Methylation‐dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015;25:707–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Najafov A, Chen H, Yuan J. Necroptosis and cancer. Trends Cancer. 2017;3:294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kesarwani P, Chakraborty P, Gudi R, Chatterjee S, Scurti G, Toth K, et al. Blocking TCR restimulation induced necroptosis in adoptively transferred T cells improves tumor control. Oncotarget. 2016;7:69371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Dinarello CA. Immunological and inflammatory functions of the interleukin‐1 family. Annu Rev Immunol. 2009;27:519–50. [DOI] [PubMed] [Google Scholar]
- 113. Liu B, Mori I, Hossain MJ, Dong L, Takeda K, Kimura Y. Interleukin‐18 improves the early defence system against influenza virus infection by augmenting natural killer cell‐mediated cytotoxicity. J Gen Virol. 2004;85:423–8. [DOI] [PubMed] [Google Scholar]
- 114. Ceballos‐Olvera I, Sahoo M, Miller MA, Del Barrio L, Re FJPP. Inflammasome‐dependent pyroptosis and IL‐18 protect against Burkholderia pseudomallei lung infection while IL‐1β is deleterious. PLoS Pathog. 2011;7:e1002452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Varela M, Romero A, Dios S, van der Vaart M, Figueras A, Meijer AH, et al. Cellular visualization of macrophage pyroptosis and interleukin‐1β release in a viral hemorrhagic infection in zebrafish larvae. J Virol. 2014;88:12026–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Cunha LD, Zamboni D. Subversion of inflammasome activation and pyroptosis by pathogenic bacteria. Front Cell Infect Microbiol. 2013;3:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Boal‐Carvalho I, Mazel‐Sanchez B, Silva F, Garnier L, Yildiz S, Bonifacio JP, et al. Influenza A viruses limit NLRP3‐NEK7‐complex formation and pyroptosis in human macrophages. EMBO Rep. 2020;21:e50421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Saeki N, Usui T, Aoyagi K, Kim DH, Sato M, Mabuchi T, et al. Distinctive expression and function of four GSDM family genes (GSDMA‐D) in normal and malignant upper gastrointestinal epithelium. Genes Chromosomes Cancer. 2009;48:261–71. [DOI] [PubMed] [Google Scholar]
- 119. Wang WJ, Chen D, Jiang MZ, Xu B, Li XW, Chu Y, et al. Downregulation of gasdermin D promotes gastric cancer proliferation by regulating cell cycle‐related proteins. J Dig Dis. 2018;19:74–83. [DOI] [PubMed] [Google Scholar]
- 120. Lage H, Helmbach H, Grottke C, Dietel M, Schadendorf D. DFNA5 (ICERE‐1) contributes to acquired etoposide resistance in melanoma cells. FEBS Lett. 2001;494(1–2):54–9. [DOI] [PubMed] [Google Scholar]
- 121. Branzk N, Papayannopoulos V, editors. Molecular mechanisms regulating NETosis in infection and disease. In: Seminars in immunopathology. Springer; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Urban CF, Ermert D, Schmid M, Abu‐Abed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009;5:e1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Garcia‐Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med 2011;3:73ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Storisteanu DM, Pocock JM, Cowburn AS, Juss JK, Nadesalingam A, Nizet V, et al. Evasion of neutrophil extracellular traps by respiratory pathogens. Am J Respir Cell Mol Biol. 2017;56:423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Bianchi M, Hakkim A, Brinkmann V, Siler U, Seger RA, Zychlinsky A, et al. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood, J Am Soc Hematol. 2009;114:2619–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Cedervall J, Olsson A‐KJO. NETosis in cancer. Oncoscience. 2015;2:900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Decker AS, Pylaeva E, Brenzel A, Spyra I, Droege F, Hussain T, et al. Prognostic role of blood NETosis in the progression of head and neck cancer. Cells. 2019;8:946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Orvedahl A, Alexander D, Tallóczy Z, Sun Q, Wei Y, Zhang W, et al. HSV‐1 ICP34. 5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23–35. [DOI] [PubMed] [Google Scholar]
- 129. Zhao Z, Fux B, Goodwin M, Dunay IR, Strong D, Miller BC, et al. Autophagosome‐independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe. 2008;4:458–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Sumpter R Jr, Levine B. Autophagy and innate immunity: triggering, targeting and tuning. Semin Cell Dev Biol. 2010;21:699–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16:90–7. [DOI] [PubMed] [Google Scholar]
- 132. Gu H, Wang X, Yang P. Virus‐Induced Autophagy in Innate Immunity. Clinical Microbiology: Open Access. 2014.
- 133. Deretic V. Autophagy as an innate immunity paradigm: expanding the scope and repertoire of pattern recognition receptors. Curr Opin Immunol. 2012;24:21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Ayna G, Krysko DV, Kaczmarek A, Petrovski G, Vandenabeele P, Fésüs L. ATP release from dying autophagic cells and their phagocytosis are crucial for inflammasome activation in macrophages. PLoS One. 2012;7:e40069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6. [DOI] [PubMed] [Google Scholar]
- 136. Gozuacik D, Kimchi AJO. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–906. [DOI] [PubMed] [Google Scholar]
- 137. Yao Z‐q, Zhang X, Zhen Y, He X‐Y, Zhao S, Li X‐F, et al. A novel small‐molecule activator of Sirtuin‐1 induces autophagic cell death/mitophagy as a potential therapeutic strategy in glioblastoma. Cell Death Dis. 2018;9:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Kim TW, Lee SY, Kim M, Cheon C, Ko S. Kaempferol induces autophagic cell death via IRE1‐JNK‐CHOP pathway and inhibition of G9a in gastric cancer cells. Cell Death Dis. 2018;9:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Li K‐C, Hua K‐T, Lin Y‐S, Su C‐Y, Ko J‐Y, Hsiao M, et al. Inhibition of G9a induces DUSP4‐dependent autophagic cell death in head and neck squamous cell carcinoma. Mol Cancer. 2014;13:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Jiang G‐M, Tan Y, Wang H, Peng L, Chen H‐T, Meng X‐J, et al. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol Cancer. 2019;18:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]