

Abstract
Although there are many synthetic methods to produce fluorinated and trifluoromethylated organic structures, the construction of difluoromethylated compounds remains a synthetic challenge. We have discovered that using magnesium salts and organic bases, unactivated imines will react with difluoroenolates, generated under exceedingly mild conditions. We have applied this approach to the iminoaldol reaction to produce difluoromethylene groups as α,α-difluoro-β-amino-carbonyl groups. This method provides synthetically useful quantities of difficult to access α,α-difluoro-β-aminoketones without the need of protecting groups or the use of activated imines. Moreover, we have applied this strategy to create analogues of the dual orexin receptor antagonist, almorexant, in only two synthetic steps.
Graphical Abstract
INTRODUCTION
Organic compounds displaying fluorine are particularly valuable in the pharmaceutical, chemical, and agrochemical industries, because the presence of fluorine on a molecule can drastically improve its chemical and biological properties.1–3 Specifically in drug development, nearly 25% of all marketed drugs contain at least one fluorine atom.4–6 Numerous efforts for the synthesis of fluorine-containing organic compounds have been reported, yet it is challenging to create some complex fluorinated structures.7,8 For example, there are vastly more methods for the synthesis of molecules displaying a trifluoromethyl group or a single fluorine than a difluoromethylene group. A primary reason is the difluoromethyl group is not a terminal group; and therefore, newer strategies such as late-stage fluorination cannot be readily applied. A uniquely appealing type of difluorinated compound is a difluoro-β-amino acid, and the typical synthetic target is an α,α-difluoro-β-amino-carbonyl group. These building blocks have found potential use as difluorinated drug leads9,10 and enzyme inhibitors (Figure 1).11–13
Figure 1.
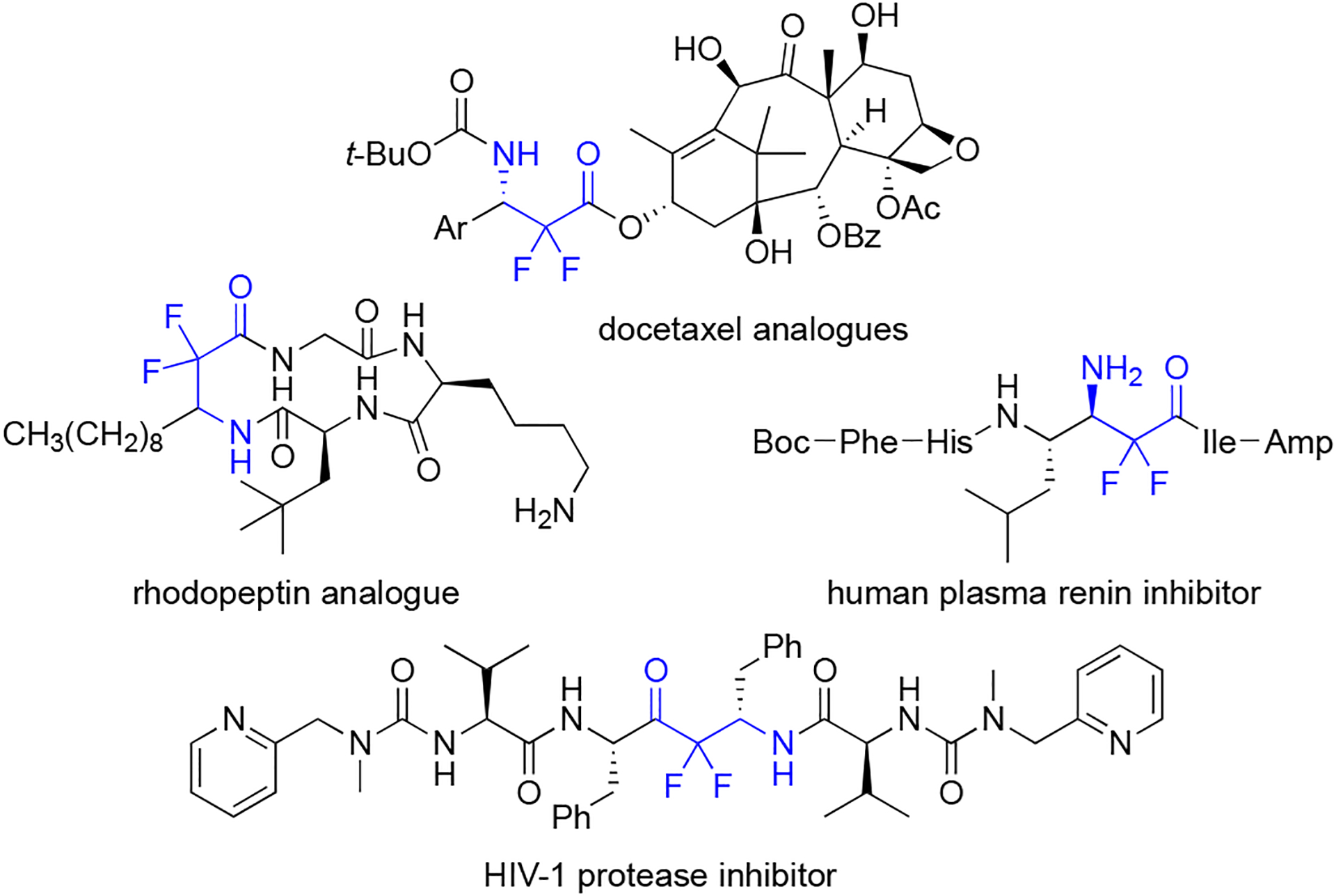
Examples of compounds displaying α,α-difluoro-β-amino-carbonyl groups.
The synthesis of α,α-difluoro-β-amino-carbonyl groups relies heavily on the addition of difluoroenolates to imines that are activated with protecting groups on nitrogen. Although the protecting groups assist in enhancing the reactivity (i.e., electrophilicity) of the imine, the utility of protecting groups continues to decline as two protection/deprotection steps must be added into the overall synthetic plan. The addition of difluoroenolates to unprotected, hence unactivated imines, is quite desirable but remains synthetically challenging. Currently, there are only two strategies to unite a difluoroenolate with an unactivated imine: 1) Reformatsky additions of bromodifluoroethyl acetate14–19 and bromodifluormethyl ketones20,21 2) Mukaiyama additions of difluoroenoxy silanes (Figure 2). Typically, Reformatsky reactions with bromodifluoroethyl acetate are vigorous and therefore provide the cyclized lactam.14,15 A diastereoselective variants have been reported,16–18 and an indium-promoted version of the Reformatsky process has been developed to favor the amino-adduct rather than the lactam.19 This chemistry has been extended to bromodifluoromethyl ketones in which the unactivated imines react in the presence of copper and zinc.20,21 Unfortunately, all of these reactions are restricted to a few esters or ketones that bear the requisite bromodifluoromethyl group. The traditional Mukaiyama addition of difluoroenoxysilanes to unactivated imines is limited to a single example in the literature,22 because these imines easily hydrolyze under typical reaction conditions.23 Although the addition of the Ruppert-Prakash reagent (i.e., CF3SiMe3) to acyl silanes followed by treatment of nucleophilic fluorine can generate reactive difluoroenoxysilanes in situ that add to unactivated imines,24 more effective methods are clearly needed. In order to address these deficiencies, we have devised a method that unleashes difluoroenolates under mild conditions yet tunes the reactivity to the unactivated imine. Moreover, we have applied this strategy to produce an α,α-difluoro-β-amino-carbonyl analogue of a drug candidate in only two synthetic steps.
Figure 2.
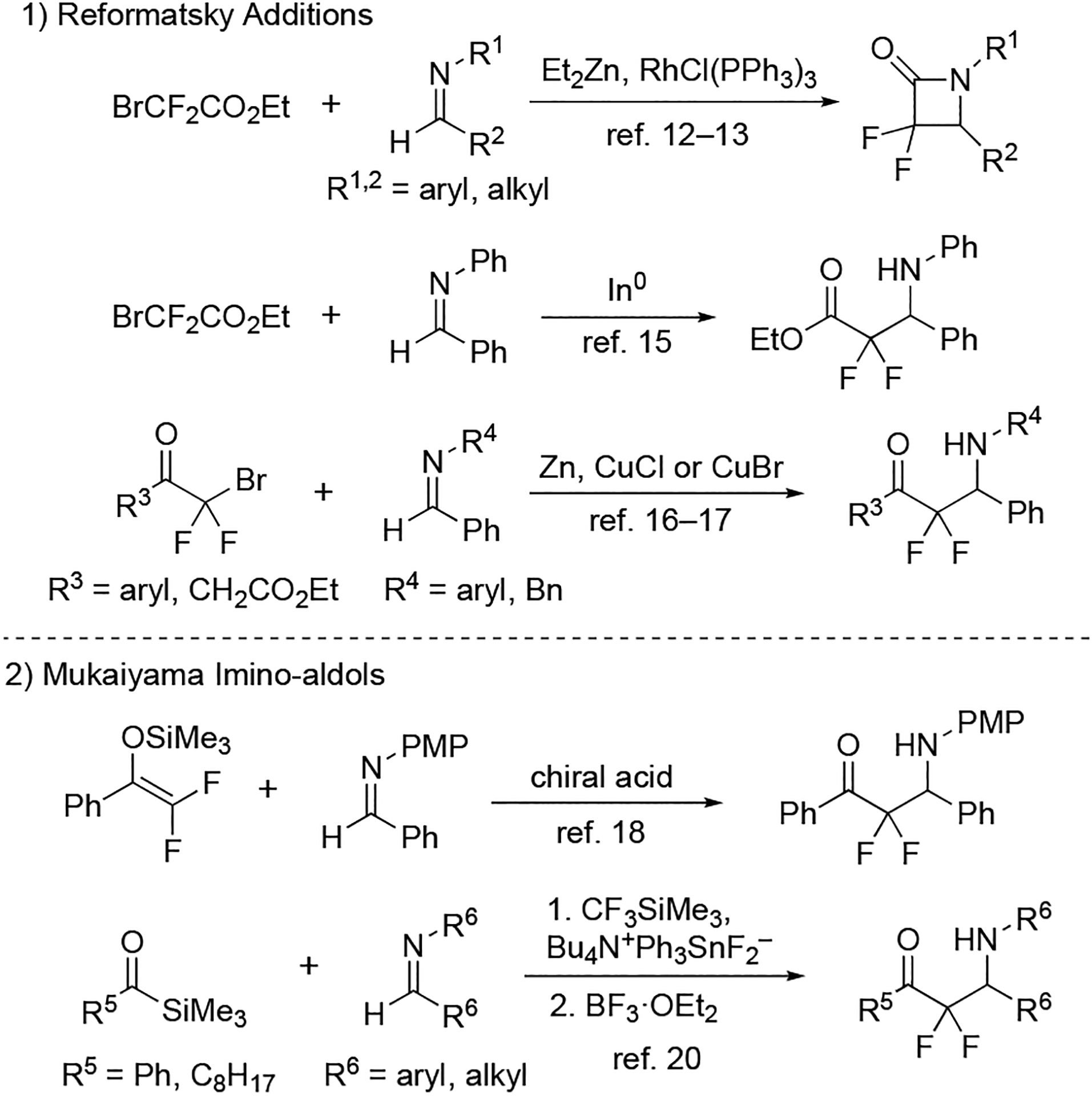
Two existing methods to prepare α,α-difluoro-β-amino-carbonyl groups from unactivated imines.
In 2011, we reported a mild approach for the generation of difluoroenolates from highly α-fluorinated gem-diols following the release of trifluoroacetate (Figure 3).25 These difluoroenolates subsequently react with aldehydes,25,26 halogenation reagents,27,28 trifluoromethyl ketones,29 and disulfides30 and can be quenched with water31 or D2O.32 This approach has been expanded to monofluoroenolates.33,34 The difluoroenolates also add to activated imines, such as N-sulfonyl,35,36 N-tosyl,36 N-Boc,35 and N-sulfinyl imines,37 using the originally reported conditions of LiBr/Et3N.25 These reactions provide the β-amino-α,α-difluoroketones, bearing the corresponding N-substituent, in high yields. Additional studies on the fragmentation of highly α-fluorinated gem-diols have described a novel photoredox-mediated addition to iminiums derived in situ from tetrahydroquinolines;38 however, a general method to add difluoroenolates produced from highly α-fluorinated gem-diols to imines without the need of protecting/activating groups is not currently available.
Figure 3.
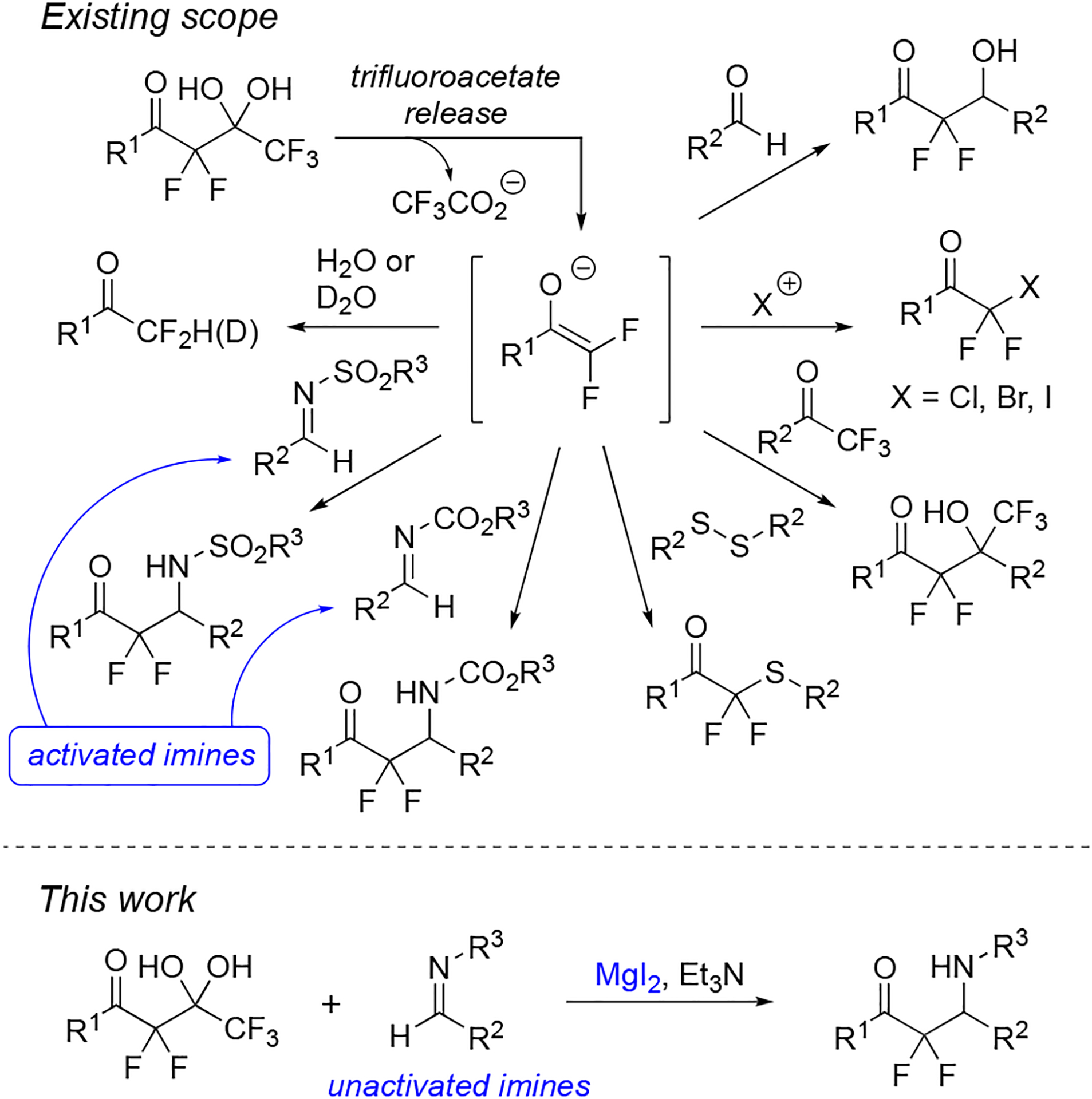
Preparation of α,α-difluoro-β-aminocarbonyl groups from highly α-fluorinated gem-diols.
Herein, we describe a simple and mild method to add difluoroenolates, produced from the release of trifluoroacetate from highly α-fluorinated gem-diols, to unactivated imines. This task is accomplished by using magnesium salts and base. Also, we have applied this methodology to synthesizing derivatives of tetrahydroisoquinolines, and particularly, we have produced an α,α-difluoro-β-amino-carbonyl derivative of the drug candidate, almorexant, using these iminoaldol reactions.
RESULTS AND DISCUSSION
Our initial studies began by examining the scope of reactivity of the fluorinated gem-diol 125 with activated imines using the reported conditions of LiBr/Et3N (Table 1).35–37 Imines 2–4 displaying an N-Boc,35 N-tosyl,36 or N-sulfonyl35,36 substituent participated in yields similar to the reported values. Moreover, the oxaziridine 5 also is a suitable electrophile for this process and provides the same product 9 as direct addition to the N-sulfonylimine 4. This type of addition to an oxaziridine has not been reported from a fluorinated gem-diol, to our knowledge. Also, the diazodicarboxylate 6 reacts with the difluoroenolate in the presence of LiBr and Et3N to produce hydrazine 10. This reactivity with azodicarboxylates has been recently reported using difluoroenoxysilanes as the difluoroenolate source,39 but it is not known in the literature from highly α-fluorinated gem-diols. Although these iminoaldol-type reactions demonstrate and expand the existing scope for the case of LiBr/Et3N using activated imines, additional reactivity can be easily tuned by selecting different reagents.
Table 1.
LiBr/Et3N Promoted Additions of Difluoroenolates Derived from Highly α-Fluorinated gem-Diols.
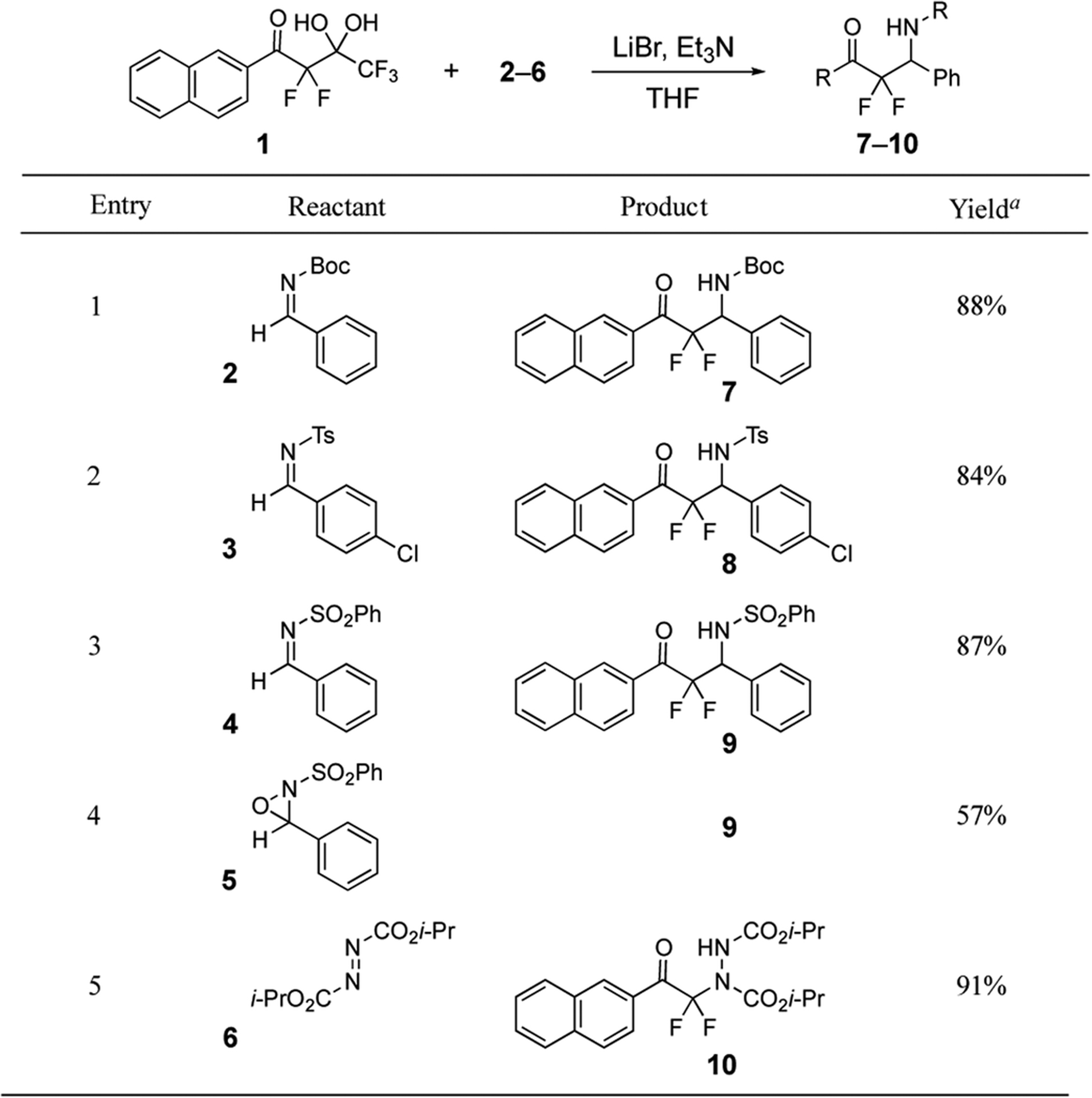
|
Yields determined by 19F NMR using trifluorotoluene as the internal standard.
The addition of the fluorinated gem-diol 125 and N-benzylidenebenzylamine 11 in the presence of Et3N and LiBr or LiCl failed to provide any of the desired product 12 (Table 2, entries 1–2). This lack of reactivity using lithium salts and unactivated imines has been previously reported.35 We hypothesized that these entries (i.e., 1–2, Table 2) and the results in Table 1 were due to the fact that lithium salts activate the lone pairs of electrons associated with oxygen atoms whereas magnesium salts activate lone pairs of electrons on nitrogen. Indeed, the advantages of magnesium salts have been demonstrated in similar carbon-carbon bond forming process.40,41 Accordingly, exchanging lithium salts with MgBr2, MgCl2, and MgI2 (i.e., entries 3–5, respectively) enabled addition to the unactivated imine 11 and production of the desired adduct 12 in 43–74% yields. Further optimizations revealed that 60 °C was the optimal temperature in the presence of MgI2 and product 12 was produced in 95% yield as determined by19F NMR. Isopropyl magnesium chloride also provided the product 12 (entry 10), as Grignard reagents are a functional class of bases for generating fluorinated nucleophiles.42
Table 2.
Optimization of Imino-aldol Process
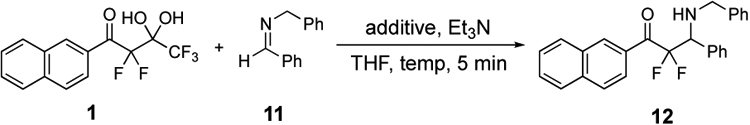 | ||||
|---|---|---|---|---|
| Entry | Additive | Imine (equiv.) | Temp | Yieldb |
| 1 | LiBr | 1.2 | rt | 0% |
| 2 | LiCl | 1.2 | rt | 0% |
| 3 | MgBr2 | 1.2 | 60 °C | 50%a |
| 4 | MgCl2 | 1.2 | 60 °C | 74%a |
| 5 | MgI2 | 1.2 | rt | 43% |
| 6 | MgI2 | 2.0 | rt | 62% |
| 7 | MgI2 | 2.0 | 40 °C | 66% |
| 8 | MgI2 | 2.0 | 60 °C | 73% (95%)a |
| 9 | i-PrNMgCl·LiClc | 2.0 | 60 °C | 4% |
| 10 | i-PrMgCl·LiBrc | 2.0 | rt | 54% |
Yields determined by 19F NMR using trifluorotoluene as the internal standard.
Isolated yields.
Et3N was not added.
We have previously reported the preparation of many highly α-fluorinated gem-diols 1, 13–17.25,27,28 The scope of the magnesium-promoted additions of unactivated imine 11 and difluoroenolates derived from these highly α-fluorinated gem-diols was characterized and is displayed in Table 3. A substantial range of isolated yields (15–85%) of the β-amino-α,α-difluoro ketones 12, 18–22 was observed. Typically, aromatic groups (1, 13, 15, and 16) were fully compatible with the imino-aldol process mediated by the release of trifluoroacetate and isolated yields were 49–85%; however, alkyl substrates 14 and 17 provided the lowest yields of 15% and 25%, respectively. This disparity demonstrates the scope of process when varying only the α-fluorinated gem-diol.
Table 3.
Magnesium-promoted Addition of Highly α-Fluorinated gem-Diols to the Unactivated Imine 11a
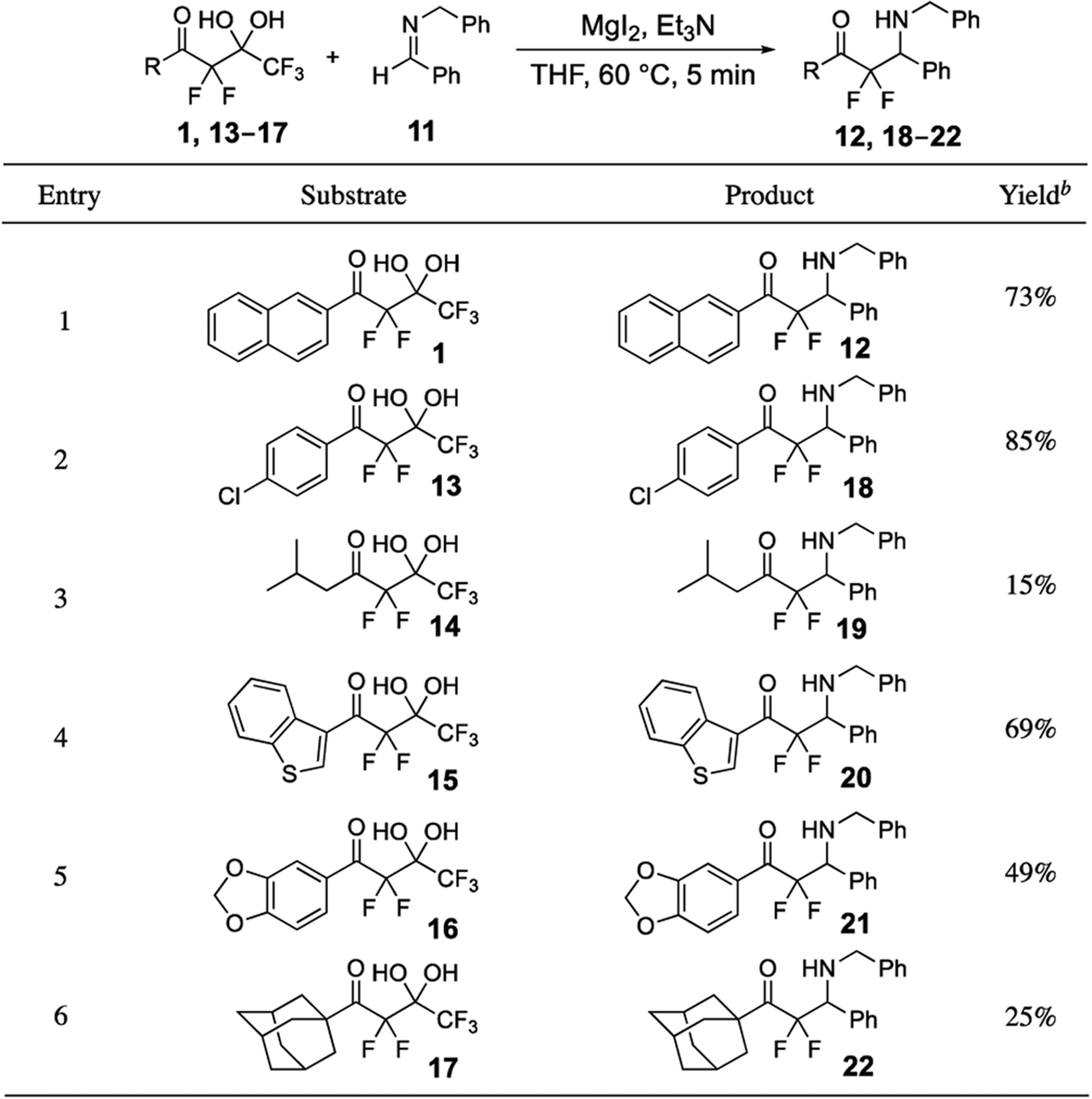
|
Reactions were typically performed with MgI2 (4.2 equiv.) and Et3N (2.1 equiv.), see Supporting Information for details.
Isolated yields.
Next, the scope of reactivity of unactivated imines 23–29 with the gem-diol 1 was investigated using the magnesium-promoted aldol reaction (Table 4). The effects of both of the substituents on the imine were examined by varying alkyl groups as well as aryl groups with electron-donating and electron-withdrawing substituents. Flame-dried magnesium iodide was needed to obtain good yields in some examples.40 In the case of imine 23, with a phenyl groups at each substituent, the product 30 was obtained in 59% isolated yield. The presence of an electron-donating methoxy group on phenyl imine 24 enabled a higher yield of 79% whereas the presence of an electron-withdrawing fluorine on the phenyl imine 32 resulted in a similar yield of 64%. On the hand, the methoxylphenyl imine 26 bearing a N-fluorophenyl substituent gave a high yield of 78%, yet the bis-methoxylphenyl imine 27 resulted in a lower 38% yield. Imines bearing a N-t-butyl group (28, entry 6) or N-benzyl group (29, entry 7) produced yields of 30 and 61%, respectively. This method provides quick access to useful quantities of compounds bearing α,α-difluoro-β-amino-carbonyl groups, and thus, X-ray structures of 30, 31, and 32 were obtained.
Table 4.
Magnesium-promoted Addition of Highly α-Fluorinated gem-Diol 1 to the Unactivated Imines 23–29a
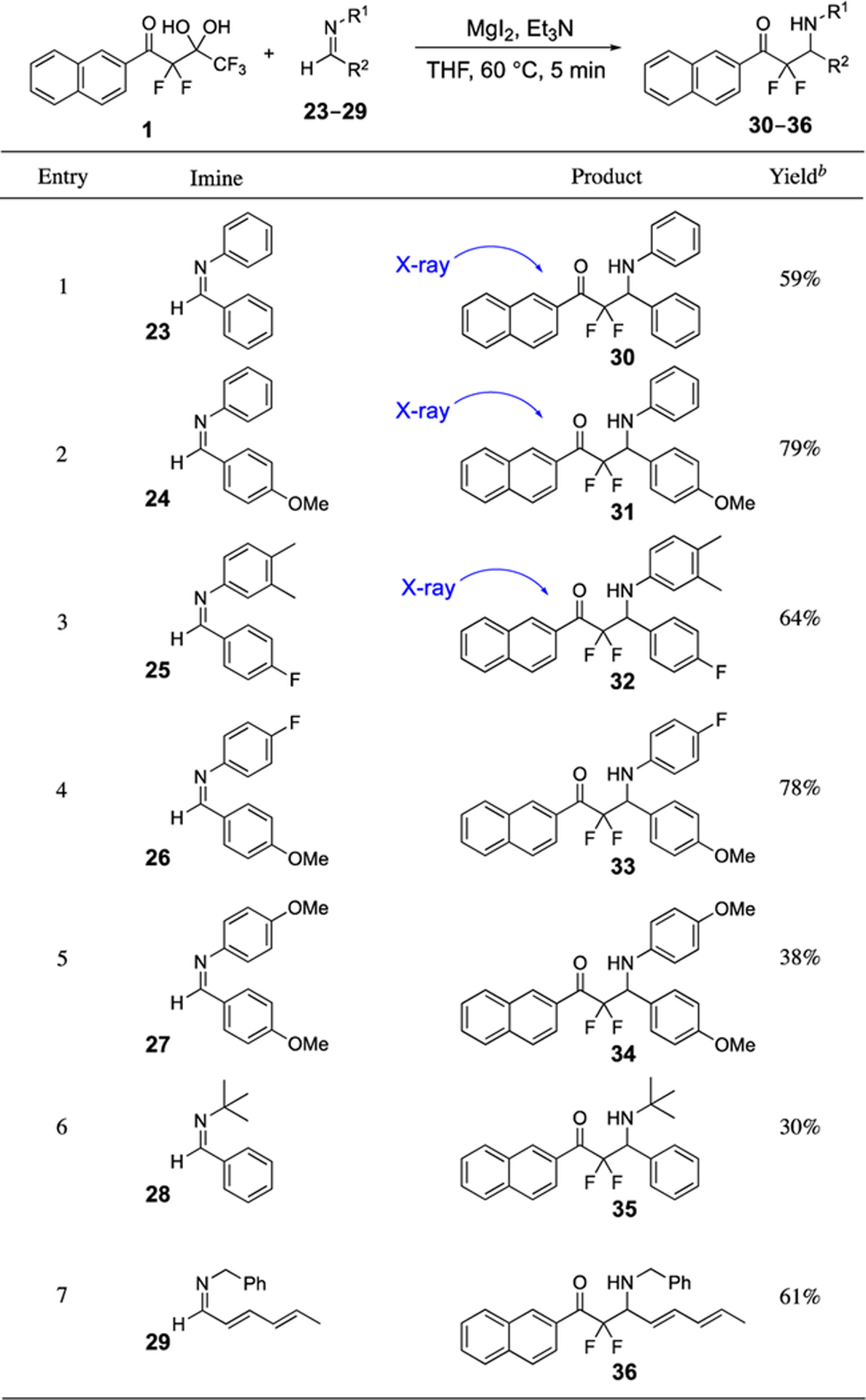
|
Reactions were typically performed with MgI2 (4.2 equiv.) and Et3N (2.1 equiv.), see Supporting Information for details.
Isolated yields.
The trifluoroacetate-release mediated imino aldol reaction using unactivated imines has substantial potential in the creation of α,α-difluoro-β-amino-carbonyl groups on organic molecules. A previous report demonstrated the utility of the trifluoroacetate-release aldol reaction in adding difluoroenolates to iminiums derived in situ from N-substituted tetrahydroisoquinoline using visible light-induced photoredox catalysis.38 We envisioned that the magnesium-initiated process could be analogously applied to dihydroisoquinolines to provide tetrahydroisoquinolines. Using this approach, the gem-diol 1 was added to dihydroisoquinoline 37 in the presence of MgI2 and Et3N to provide the corresponding tetrahydroisoquinoline α,α-difluoro ketones 38 in good 84% yield (Scheme 1). Suzuki–Miyaura cross-coupling of 38 with vinyltrifluoroborate43 provided the tetrahydroisoquinoline 39 in 40% yield and demonstrates structural variants of tetrahydroquinolines can be quickly created.
Scheme 1.
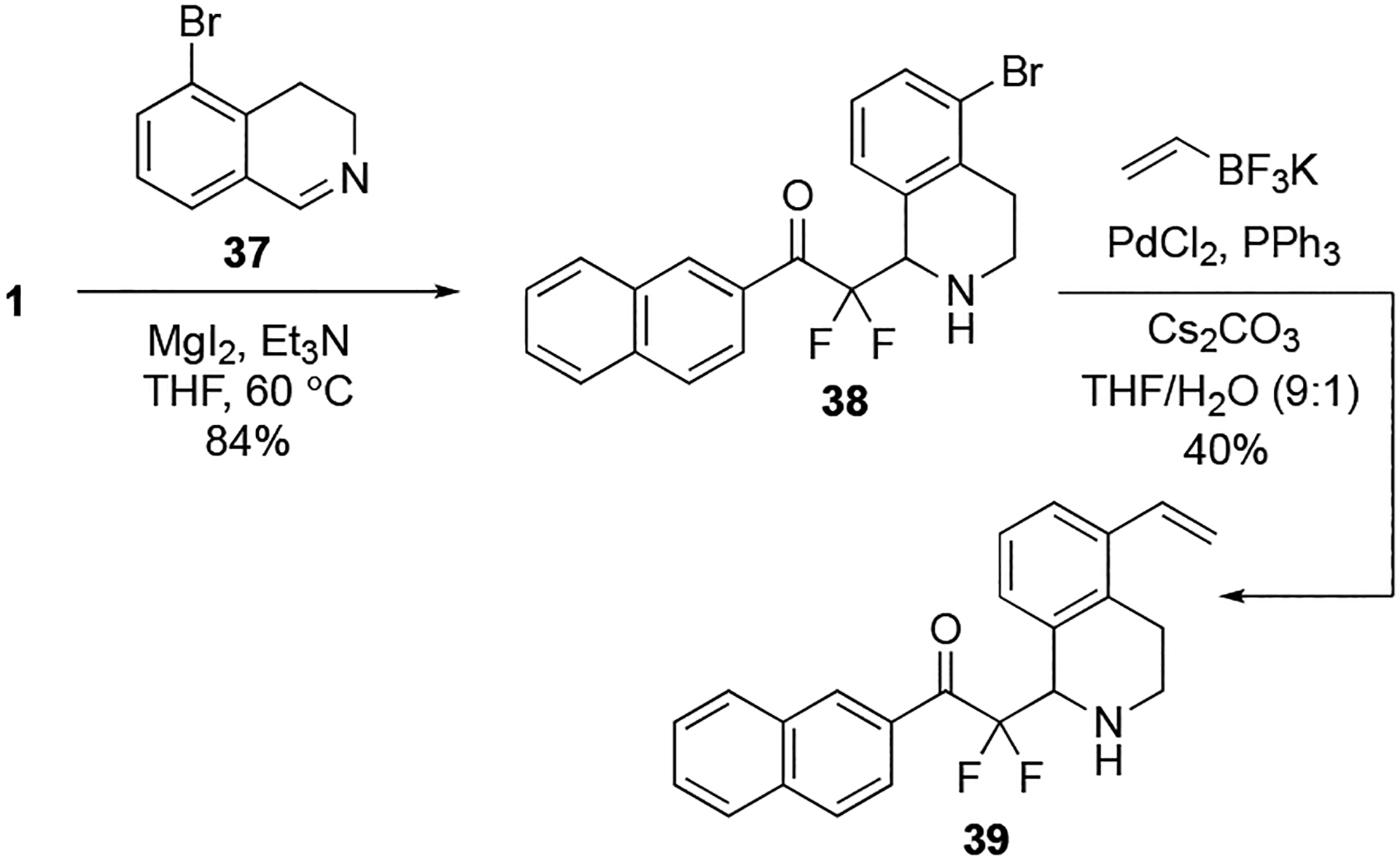
Imino-aldol addition of gem-diol 1 with dihydroisoquinoline 37
Tetrahydroisoquinolines are a valuable class of heterocycle that appears in bioactive natural products, such as the ecteinascidins,44 as well as drug discovery (Figure 4).45 Notably, almorexant belongs to a class of drugs that act as dual orexin receptor antagonist for treatment of sleep disorders and displays a key tetrahydroisoquinoline.46 There are few reported enantioselective syntheses of almorexant that involved key steps such as asymmetric transfer hydrogenation,47 asymmetric allylic amidation,48 and asymmetric induction with N-tert-butanesulfinamide.49 In addition, several almorexant analogues have been synthesized and reported.50
Figure 4.
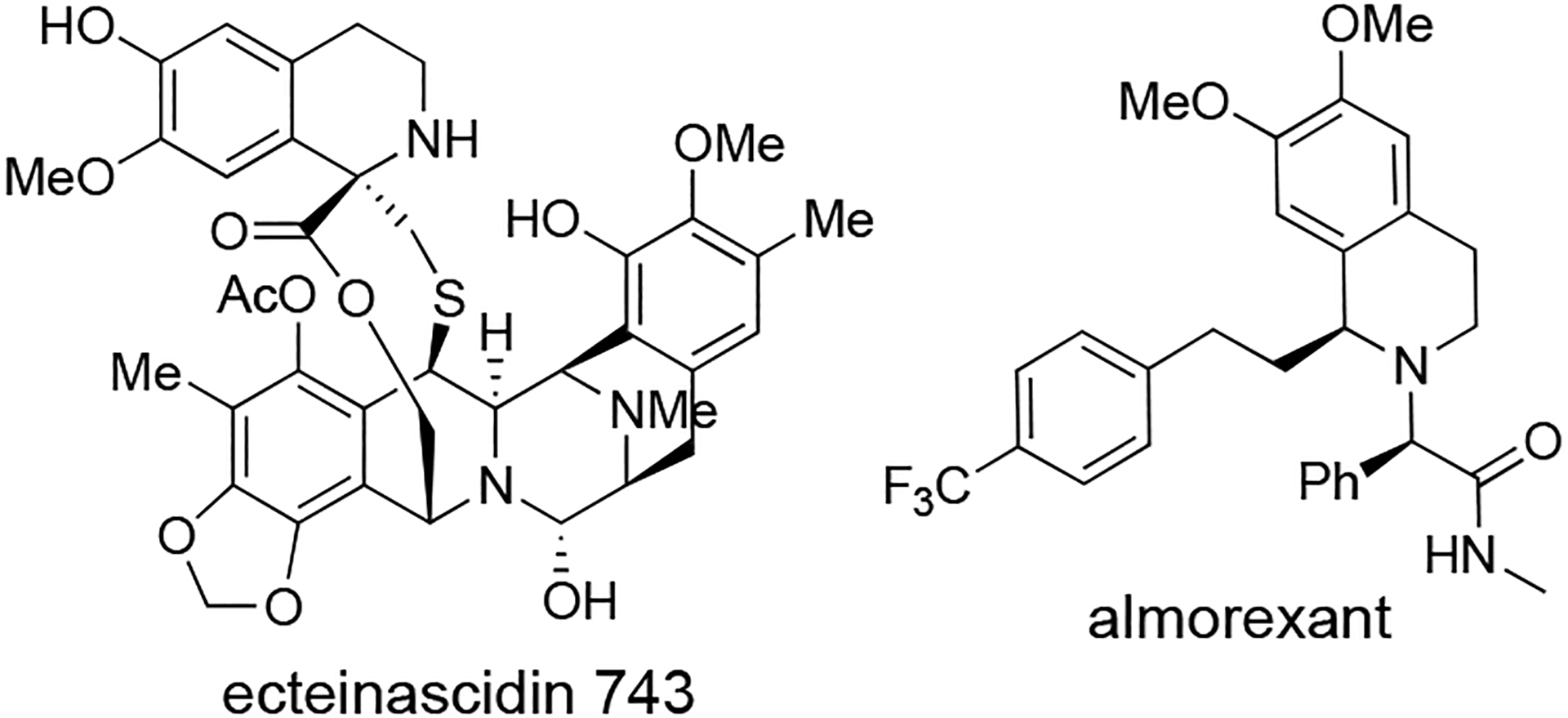
Structure of biologically active molecules displaying a tetrahydroisoquinoline.
We envisioned a strategy to produce derivatives of almorexant using the magnesium-promoted reaction between a fluorinated gem-diol and a dihydroquinoline (Scheme 2). Furthermore, this approach would be short and not require any protecting groups. Accordingly, the known fluorinated gem-diol 4038 and 6,7-dimethoxy-3,4-dihydroisoquinoline 41 were treated with MgI2 and Et3N and gave the α,α-difluoro-β-aminoketone 42. Next, 42 was coupled using the literature method48 to amide 4350 to give a 2.2:1 mixture of diastereomers and the major isomer was purified in 28% isolated yield. Accessing the almorexant derivative 44 in two synthetic steps is a functional demonstration of this method and displays how the unique α,α-difluoro-β-amino-carbonyl group integrates into known bioactive structures.
Scheme 2.
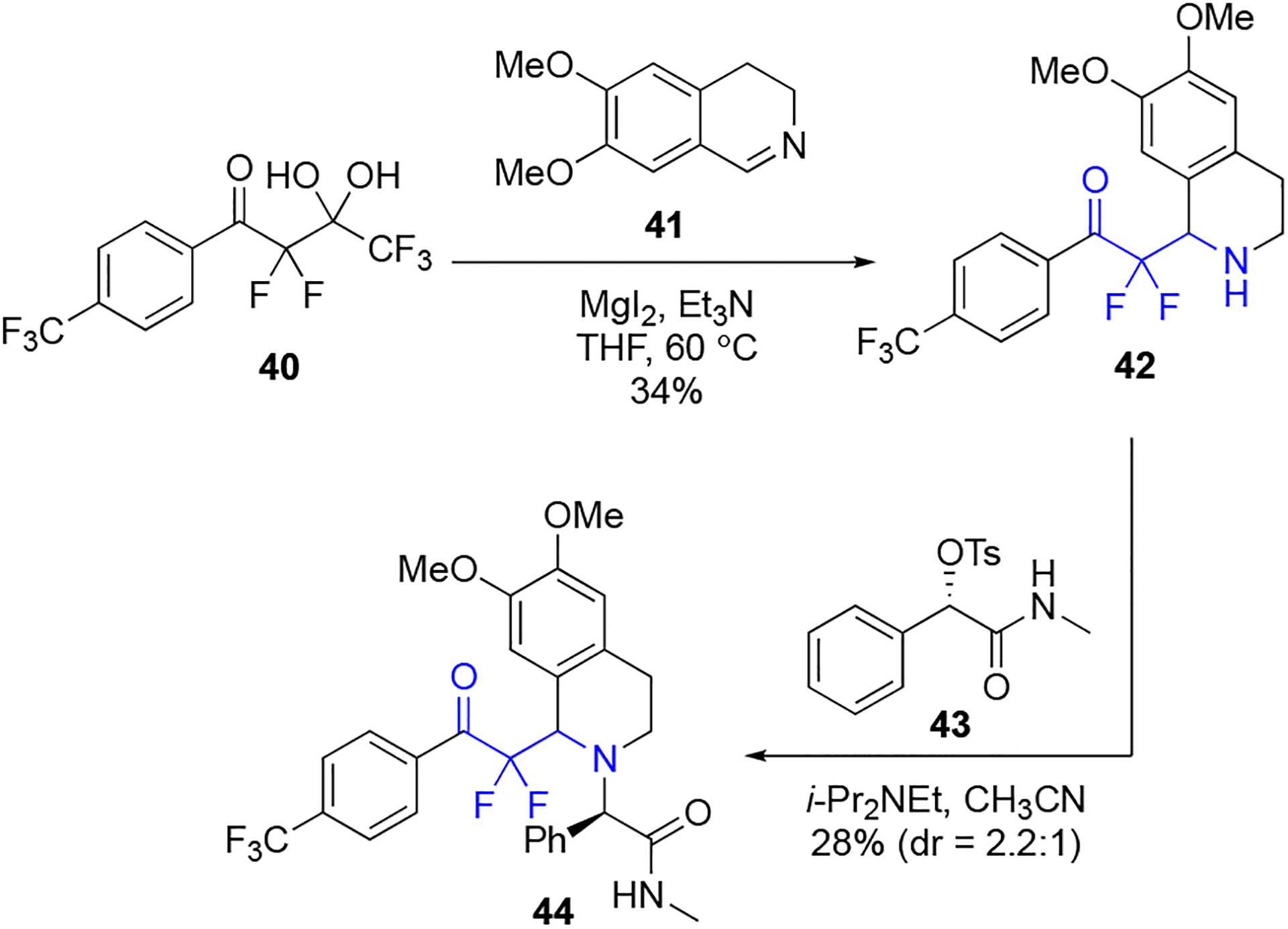
Two-step, protecting group-free synthesis of fluorinated almorexant analogue 44.
CONCLUSIONS
In summary, we have reported a new protocol to synthesize β-amino-α,α-difluoro ketones using magnesium-promoted additions of difluoroenolates with unactivated imines. The difluoroenolates were unleashed from highly α-fluorinated gem-diols using only MgI2 and Et3N following the release of trifluoroacetate. This process eliminates the need of an imine with an activating N-substituent or a protecting group. This approach is compatible with many alkyl and aryl substituted imines and gem-diols. It can be easily integrated into drug discovery, and we have demonstrated its potential use in the synthesis of the analogues of almorexant in only two steps. These reactions further establish the role of releasing trifluoroacetate in the presence of mild reagents, such as a salt and an organic base, to generate valuable and reactive difluoroenolates.
EXPERIMENTIAL SECTION
N-(1-(4-Chlorophenyl)-2,2-difluoro-3-(naphthalen-2-yl)-3-oxopropyl)-4-methylbenzene-sulfonamide 8.51
To a solution of 2,2,4,4,4-pentafluoro-3,3-dihydroxy-1-(naphthalen-2-yl)butan-1-one 125,27 (65.7 mg, 0.205 mmol), N-(4-chlorobenzylidene)-4-methylbenzenesulfonamide 3 (133 mg, 0.452 mmol) and LiBr (101 mg, 1.16 mmol) in THF (3 mL) was added was Et3N (60 μL, 0.43 mmol) dropwise. After 5 min, the reaction was quenched with saturated aqueous NH4Cl (5 mL), and the resultant mixture was extracted with EtOAc (5 mL × 5). The organics were dried over Na2SO4 and concentrated under reduced pressure. SiO2 flash column chromatography (9:1–8:2 hexanes/EtOAc) afforded the title compound 8 as a yellow solid in 84% yield (86.1 mg): mp 143–145 °C; 1H NMR (300 MHz, CDCl3) 8.47 (s, 1H), 8.00–7.82 (m, 4H), 7.72–7.50 (m, 4H), 7.19 (s, 4H), 7.09 (d, J = 8.1 Hz, 2H), 5.66 (d, J = 9.5 Hz, 1H), 5.26 (td, J = 12.2, 9.5 Hz, 1H), 2.32 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 188.3 (t, JCF = 29.1 Hz, 1C), 143.7, 136.9, 136.1, 134.9, 132.8, 132.1, 130.1, 130.0 (3C), 129.7, 129.4 (2C), 129.0, 128.6 (3C), 127.8, 127.0 (3C), 124.4, 116.1 (t, JCF = 262.1 Hz, 1C), 59.6 (t, JCF = 25.4 Hz, 1C), 21.4; 19F NMR (282 MHz, CDCl3) −103.87 (dd, JFF = 284.0, JHF = 11.8 Hz, 1F), −105.92 (dd, JFF = 284.2, JHF = 12.6 Hz, 1F); IR (film) νmax 3584, 3234, 3055, 2974, 2920, 1702 cm–1; HRMS (ESI) m/z calcd for C26H20O3NSClF2Na (M+Na)+ 522.0718, found 522.0721.
N-(2,2-Difluoro-3-(naphthalen-2-yl)-3-oxo-1-phenylpropyl)benzenesulfonamide 9.51
To a solution of 2,2,4,4,4-pentafluoro-3,3-dihydroxy-1-(naphthalene-2-yl)butan-1-one 125,27 (30 mg, 0.09 mmol) and N-benzylidenebenzenesulfonamide 4 (28 mg, 0.11 mmol) in THF (620 μL) was added LiBr (24 mg, 0.28 mmol), and the mixture was stirred for 10 min at rt. Next, Et3N (26 μl, 0.19 mmol) was added dropwise, and after 5 min, the reaction mixture was quenched with saturated aqueous NH4Cl (3 mL). The resultant mixture was extracted with EtOAc (2 mL × 3). The organics were dried over Na2SO4 and concentrated under reduced pressure. SiO2 flash chromatography (2:1:1 hexane/Et2O/CHCl3) afforded the title compound 9 as a colorless solid in 87 % yield (37 mg): mp 132–135 °C; 1H NMR (300 MHz, CDCl3) δ 8.46 (s, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.88–7.82 (m, 3H), 7.71–7.61 (m, 3H), 7.57 (t, J = 7.4 Hz, 1H), 7.41 (t, J = 7.1 Hz, 1H), 7.31–7.18 (m, 7H), 5.73 (m, 1H), 5.32 (td, J = 12.2, 9.9 Hz, 1H); 13C NMR (125 MHz CDCl3) 188.6 (t, JCF = 29.0 Hz, 1C), 140.1, 136.0, 132.7 (t, JCF = 4.3 Hz, 1C), 132.5, 132.4, 132.1, 130.1, 129.6, 129.3, 128.8, 128.7 (2C), 128.6, 128.5 (2C), 128.4 (2C), 127.7, 127.1, 126.9 (2C), 124.4, 116.3 (t, JCF = 261.9 Hz, 1C), 60.2 (t, JCF = 25.0 Hz, 1C); 19F NMR (282 MHz, CDCl3) δ −103.77 (dd, JFF = 280, JHF = 12.0 Hz, 1F), −105.48 (dd, JFF = 280, JHF = 12.8 Hz, 1F); IR (film) νmax 3272, 1696, 1331, 1284, 1166 cm–1; HRMS (ESI-TOF) m/z calcd for C25H19F2NO3SNa [M+Na]+ 474.0951, found 474.0962.
N-(2,2-Difluoro-3-(naphthalen-2-yl)-3-oxo-1-phenylpropyl)benzenesulfonamide 9.
2,2,4,4,4-Pentafluoro-3,3-dihydroxy-1-(naphthalen-2-yl)butan-1-one 125,27 (8 mg, 0.02 mmol) was azeotroped with toluene (3 × 1 mL) and dissolved in THF (0.3 mL). Next, the solution was added to a flask containing LiBr (8.0 mg, 0.92 mmol) and cooled to −78 °C under an argon atmosphere. A solution of Davis oxaziridine D1 (14 mg, 0.054 mmol), that was azeotroped with toluene (3 × 1 mL) and dissolved in THF (0.20 mL), was added dropwise to the reaction mixture and stirred for 15 min at −78 °C. Next, Et3N (5 μL, 0.04 mmol) was added, and the resultant mixture was allowed to warm slowly to rt and stirred for 24 h at rt. Then, the reaction mixture was quenched with saturated NH4Cl solution (5 mL), extracted with EtOAc (3 × 10 mL), washed with brine (3 × 5 mL), and dried over Na2SO4. The organics were concentrated under reduced pressure and purified by preparative TLC using 30% EtOAc in hexanes to give the title compound 9 as a colorless solid (6 mg, 57%).
Diisopropyl 1-(1,1-difluoro-2-(naphthalen-2-yl)-2-oxoethyl)hydrazine-1,2-dicarboxylate 10.
To a solution of 2,2,4,4,4-pentafluoro-3,3-dihydroxy-1-(naphthalen-2-yl)butan-1-one 125,27 (30 mg, 0.094 mmol) and LiBr (35 mg, 0.40 mmol) in THF (0.8 mL) at rt was added diisopropyl azodicarboxylate (40 μL, 0.20 mmol) followed by Et3N (28 μL, 0.20 mmol). The reaction mixture was warmed to 60 °C for 30 min. Then, the reaction mixture was cooled to rt, quenched with 1 mL of saturated NH4Cl solution (5 mL), extracted with EtOAc (3 × 10 mL), washed with brine (3 × 5 mL), dried over Na2SO4, and concentrated under reduced pressure. Purification by SiO2 flash chromatography using 5–10% EtOAc in hexanes provided the title compound 10 as a colorless oil (35 mg, 91 %): 1H NMR (500 MHz, CDCl3) δ 9.17 (s, 1H), 8.27 (d, J = 8.0 Hz, 1H), 8.09 (d, J = 7.2 Hz, 1H), 7.92 (d, J = 8.6 Hz, 1H), 7.86 (d, J = 8.1 Hz, 1H), 7.62 (t, J = 7.4 Hz, 1H), 7.56 (t, J = 7.5 Hz, 1H), 6.88 (s, 1H), 5.13 (qu, J = 6.2 Hz, 1H), 4.82 (t, J = 5.6 Hz, 1H), 1.34 (d, J = 4.6 Hz, 6H), 1.11 (d, J = 6.0 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 181.8 (t, JCF = 29.4 Hz, 1C), 155.5, 152.1, 135.8, 132.7, 132.4, 130.3, 129.1, 129.0, 128.4, 127.6, 126.8, 124.5, 112.7 (t, JCF = 259.6 Hz, 1C), 73.4, 70.8, 21.9, 21.8, 21.3, 21.2; 19F NMR (470 MHz, CDCl3) δ −86.96 (d, JFF= 190.5 Hz, 2F); IR νmax 3319, 2984, 2934, 1736, 1717, 1628, 1428, 1376, 1249, 1104, 1045 cm–1; HRMS (ESI-TOF) m/z calcd for C20H21F2N2O5 [M–H]– 407.1419, found 407.1418.
Representative Reaction Procedure for Trifluoroacetate Release/Imino-Aldol Reaction.
To a solution of 2,2,4,4,4-pentafluoro-3,3-dihydroxy-1-(naphthalene-2-yl)butan-1-one 125,27 (50 mg, 0.16 mmol) and N-benzylidinebenzylamine 11 (62 μL, 0.33 mmol) in THF (1.0 mL) was added MgI2 (182 mg, 0.656 mmol). The resultant mixture was stirred for 2 min at rt. Then, the mixture was warmed to 60 °C for 1 min and then Et3N (46 μL, 0.33 mmol) was added. After 5 min of stirring at 60 °C, the reaction was quenched with saturated aqueous NH4Cl (1.5 mL) and the resultant mixture was extracted with EtOAc (10 mL × 3). The organics were dried over Na2SO4 and concentrated under reduced pressure. The mixture was then treated with saturated NaHSO3 solution (15 mL) and stirred vigorously for 18 h. Next, the mixture was extracted with EtOAc (10 mL × 3). The organics were dried over Na2SO4 and concentrated under reduced pressure. SiO2 flash chromatography (9:1:0.01 hexanes/EtOAc/Et3N) afforded the product 12 as a colorless solid in 73% yield (45.8 mg).
3-(Benzylamino)-2,2-difluoro-1-(naphthalen-2-yl)-3-phenylpropan-1-one 12.51
See representative reaction procedure: mp 96–98 °C; 1H NMR (300 MHz, CDCl3) δ 8.49 (s, 1H), 8.00 (d, J = 8.7 Hz, 1H), 7.91–7.86 (m, 3H), 7.64 (m, 1H), 7.56 (m, 1H), 7.46‒7.36 (m, 5H), 7.09‒7.03 (m, 5H), 4.48 (dd, J = 20.1, 7.7 Hz, 1H), 3.76 (d, J = 13.1 Hz, 1H), 3.49 (d, J = 13.1 Hz, 1H), 1.82 (br s, 1H); 13C NMR (125 MHz, CDCl3) δ 190.3 (t, JCF = 28.4 Hz, 1C), 138.6, 135.7, 134.4, 132.2, 132.1, 130.4, 130.0, 129.1 (2C), 129.0, 128.7, 128.6 (2C), 128.4, 128.2 (4C), 127.7, 127.0, 126.8, 124.8, 118.1 (t, JCF= 258.5 Hz, 1C), 63.8 (t, JCF = 24.3 Hz, 1C), 50.9; 19F NMR (282 MHz, CDCl3) δ −102.7 (dd, JFF = 268.4, JHF = 7.6 Hz, 1F), −115.3 (dd, JFF = 268.4, JHF = 20.1 Hz, 1F); IR (film) νmax 1701, 1626, 1455, 1114, 698 cm–1; HRMS (EI-BE) m/z calcd for C26H22F2NO (M+H)+ 402.1669, found 402.1664.
3-(Benzylamino)-1-(4-chlorophenyl)-2,2-difluoro-3-phenylpropan-1-one 18.51
See representative reaction procedure. To a solution of 1-(4-chlorophenyl)-2,2,4,4,4-pentafluoro-3,3-dihydroxybutan-1-one 1325,27 (30.9 mg, 0.101 mmol) and N-benzylidinebenzylamine 11 (38 μL, 0.20 mmol) in THF (680 μL) was added MgI2 (112 mg, 0.404 mmol). The mixture was warmed to 60 °C for 1 min and then Et3N (30 μL, 0.20 mmol) was added. SiO2 flash chromatography (9:1:0.01 hexanes/EtOAc/Et3N) afforded the title compound 18 as a colorless oil (33.1 mg) in 85% yield: 1H NMR (300 MHz, CDCl3) δ 7.91 (d, J = 8.4 Hz, 2H), 7.47‒7.36 (m, 7H), 7.22‒7.17 (m, 3H), 7.10‒7.04 (m, 2H), 4.38 (dd, J = 20.7, 7.2 Hz, 1H), 3.77 (d, J = 13.1 Hz, 1H), 3.49 (d, J = 13.1 Hz, 1H), 2.11 (br s, 1H); 13C NMR (125 MHz, CDCl3) δ 189.3 (t, JCF = 27.5 Hz, 1C), 140.4, 138.5, 134.2, 131.6 (2C), 131.1, 129.0 (2C), 128.9 (2C), 128.8, 128.6 (2C), 128.3 (2C), 128.2 (2C), 127.2, 117.8 (t, JCF = 255.0 Hz, 1C), 63.4 (t, JCF = 22.5 Hz, 1C), 50.8; 19F NMR (282 MHz, CDCl3) δ −102.8 (dd, JFF = 268.7, JHF = 6.5 Hz, 1F), ‒116.4 (dd, JFF = 268.5, JHF = 18.6 Hz, 1F); IR (film) νmax 1693, 1444, 1266, 1039, 699 cm–1; HRMS (ESI-TOF) m/z calcd for C22H19ClF2NO (M+H)+ 386.1123, found 386.1131.
1-(Benzylamino)-2,2-difluoro-5-methyl-1-phenylhexan-3-one 19.
See representative reaction procedure. To a solution of 1,1,1,3,3-pentafluoro-2,2-dihydroxy-6-methylheptan-4-one 1425 (32.6 mg, 0.130 mmol) and N-benzylidinebenzylamine 11 (51 μL, 0.27 mmol) in THF (1.0 mL) was added MgI2 (152 mg, 0.547 mmol) and the mixture was stirred for 2 min at rt. The resultant mixture was warmed to 60 °C for 1 min and then Et3N (38 μL, 0.27 mmol) was added. SiO2 flash chromatography (100% hexanes–5:5:0.01 hexanes/EtOAc/Et3N) afforded the title compound 19 as a colorless oil (6.3 mg) in 15% yield: 1H NMR (400 MHz, CDCl3) δ 7.43–7.38 (m, 3H), 7.35 (d, J = 7.6 Hz, 2H), 7.32–7.25 (m, 3H), 7.19 (d, J = 6.9 Hz, 2H), 4.22 (dd, J = 20.6, 7.6 Hz, 1H), 3.73 (d, J = 13.0 Hz, 1H), 3.52 (d, J = 13.0 Hz, 1H), 2.52 (dd, J = 18.1, 6.9 Hz, 1H), 2.39 (dd, J = 18.1, 6.4 Hz, 1H), 2.16 (septet, J = 6.6 Hz, 1H), 1.68 (br s, 1H), 0.93 (d, J = 6.7 Hz, 3H), 0.89 (d, J = 6.7 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 201.5 (dd, JCF = 31.1, 26.5 Hz, 1C), 139.0, 134.4, 128.9 (2C), 128.7, 128.6 (2C), 128.4 (2C), 128.3 (2C), 127.3, 116.4 (dd, JCF = 260.0, 256.6 Hz, 1C), 62.8 (dd, JCF = 26.4, 21.6 Hz, 1C), 51.0, 46.7, 23.4, 22.4 (2C); 19F NMR (376 MHz, CDCl3) δ ‒109.9 (dd, JFF = 258.6, JHF = 7.5 Hz, 1F), ‒123.6 (dd, JFF = 258.8, JHF = 20.5 Hz, 1F); IR (film) νmax 2959, 1740, 1455, 1124 cm–1; HRMS (ESI-TOF) m/z calcd for C20H24F2NO (M+H)+ 332.1826, found 332.1828.
1-(Benzo[b]thiophen-3-yl)-3-(benzylamino)-2,2-difluoro-3-phenylpropan-1-one 20.
See representative reaction procedure. To a solution of 1-(benzo[b]thiophen-3-yl)-2,2,4,4,4-pentafluoro-3,3-dihydroxybutan-1-one 1527 (31 mg, 0.10 mmol) and N-benzylidinebenzylamine 11 (38 μL, 0.20 mmol) in THF (570 μL) was added MgI2 (112 mg, 0.402 mmol) and the mixture was stirred for 2 min at rt. The resultant mixture was warmed to 60 °C for 1 min and then Et3N (28 μL, 0.20 mmol) was added. SiO2 flash chromatography (9:1:0.01 hexanes/EtOAc/Et3N) afforded the title compound 20 as a pale yellow solid (26.7 mg) in 69% yield: mp 106–108 °C; 1H NMR (400 MHz, CDCl3) δ 8.73 (d, J = 8.2 Hz, 1H), 8.40 (s, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.56 (t, J = 7.6 Hz, 1H), 7.48 (t, J = 7.6 Hz, 1H), 7.44–7.35 (m, 5H), 7.16–6.99 (m, 5H), 4.43 (dd, J = 19.8, 8.0 Hz, 1H), 3.78 (d, J = 13.2 Hz, 1H), 3.51 (d, J = 13.2 Hz, 1H), 2.02 (br s, 1H); 13C NMR (125 MHz, CDCl3) δ 184.7 (dd, JCF = 29.0, 27.9 Hz, 1C), 140.0 (t, JCF = 9.0 Hz, 1C), 138.9, 138.7, 137.2, 134.5, 129.8, 129.1 (2C), 128.7, 128.6 (2C), 128.1 (4C), 127.1, 126.1, 125.7, 125.3, 122.1, 117.7 (dd, JCF = 260.4, 256.8 Hz, 1C), 63.7 (dd, JCF = 26.2, 22.0 Hz, 1C), 50.7; 19F NMR (376 MHz, CDCl3) δ −103.8 (dd, JFF = 263.0, JHF = 7.5 Hz, 1F), −116.1 (dd, JFF = 263.0, JHF = 18.8 Hz, 1F); IR (film) νmax 1675, 1458, 1106, 698 cm–1; HRMS (ESI-TOF) m/z calcd for C24H20F2NOS (M+H)+ 408.1234, found 408.1235.
1-(Benzo[d][1,3]dioxol-5-yl)-3-(benzylamino)-2,2-difluoro-3-phenylpropan-1-one 21.51
See representative reaction procedure. To a solution of 1-(benzo[d][1,3]dioxol-5-yl)-2,2,4,4,4-pentafluoro-3,3-dihydroxybutan-1-one 1625 (16 mg, 0.05 mmol) and N-benzylidinebenzylamine 11 (20 μL, 0.105 mmol) in THF (800 μL) was added MgI2 (57 mg, 0.205 mmol) and the mixture was stirred for 2 min at rt. The resultant mixture was warmed to 60 °C for 5 min and then Et3N (14 μL, 0.10 mmol) was added. Preparative TLC (8:2 hexanes/EtOAc) afforded the desired product 21 as a colorless solid (12 mg) in 61% yield: mp 89–91 °C; 1H NMR (500 MHz, CDCl3) δ 7.63 (d, J = 8.3 Hz, 1H), 7.45 (s, 1H), 7.40 (m, 5H), 7.24–7.19 (m, 3H), 7.14–7.09 (m, 2H), 6.82 (d, J = 8.3 Hz, 1H), 6.07 (dd, J = 5.7, 1.3 Hz, 2H), 4.39 (dd, J = 20.0, 7.6 Hz, 1H), 3.77 (d, J = 13.2 Hz, 1H), 3.50 (d, J = 13.2 Hz, 1H), 2.18 (br s, 1H); 13C NMR (125 MHz, CDCl3) δ 188.1 (t, JCF = 28.8 Hz, 1C), 152.5, 148.0, 138.8, 134.5, 129.1 (2C), 128.6, 128.5 (2C), 128.2 (4C), 127.5, 127.1, 126.7, 117.8 (t, JCF = 258.4 Hz, 1C), 109.5, 108.0, 102.0, 63.7 (t, JCF = 23.7 Hz, 1C), 50.8; 19F NMR (470 MHz, CDCl3) δ ‒102.5 (dd, JFF = 269.1, JHF = 7.3 Hz, 1F), ‒114.53 (dd, JFF = 269.1, 20.0 Hz, 1F); IR (film) νmax 1693, 1444, 1265, 1039, 699 cm–1; HRMS (ESI-TOF) m/z calcd for C23H20F2NO3 (M+H)+ 396.1411, found 396.1405.
1-(Adamantan-1-yl)-3-(benzylamino)-2,2-difluoro-3-phenylpropan-1-one 22.
See representative reaction procedure. To a solution of 1-(adamantan-1-yl)-2,2,4,4,4-pentafluoro-3,3-dihydroxybutan-1-one 1725,27 (35 mg, 0.11 mmol) and N-benzylidinebenzylamine 11 (42 μL, 0.23 mmol) in THF (570 μL) was added MgI2 (126 mg, 0.452 mmol) and the mixture was stirred for 2 min at rt. The resultant mixture was warmed to 60 °C for 1 min and then Et3N (31 μL, 0.23 mmol) was added. SiO2 flash chromatography (100% hexanes–5:5:0.01 hexanes/CH2Cl2/Et3N) afforded the title compound 22 as a colorless solid (10.8 mg) in 25% yield: mp 104–106 °C; 1H NMR (400 MHz, CDCl3) δ 7.43–7.16 (m, 10H), 4.39 (dd, J = 20.2, 8.4 Hz, 1H), 3.72 (d, J = 13.0 Hz, 1H), 3.54 (d, J = 13.0 Hz, 1H), 2.02 (s, 3H), 1.88 (q, J = 12.4 Hz, 6H), 1.79 (br s, 1H), 1.71 (q, J = 12.0 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 204.6 (dd, JCF = 28.4, 25.3 Hz, 1C), 139.3, 134.8, 129.1, 128.4 (4C), 128.3 (2C), 128.2 (2C), 127.1, 118.7 (dd, JCF = 264.1, 259.9 Hz, 1C), 62.9 (dd, JCF = 25.4, 21.1 Hz, 1C), 51.1, 46.5, 36.9 (3C), 36.4 (3C), 27.7 (3C); 19F NMR (376 MHz, CDCl3) δ −103.8 (dd, JFF = 270.7, JHF = 7.5 Hz, 1F), −116.9 (dd, JFF = 268.8, JHF = 20.7 Hz, 1F); IR (film) νmax 2907, 2852, 1716, 1454, 1114 cm–1; HRMS (ESI-TOF) m/z calcd for C26H30F2NO (M+H)+ 410.2295, found 410.2279.
2,2-Difluoro-1-(naphthalen-2-yl)-3-phenyl-3-(phenylamino)propan-1-one 30.
See representative reaction procedure. To a solution of 2,2,4,4,4-pentafluoro-3,3-dihydroxy-1-(naphthalene-2-yl)butan-1-one 125,27 (24 mg, 0.075 mmol), 4Å molecular sieves (100 mg), and N-benzylideneaniline 23 (27 mg, 0.15 mmol) in THF (1.5 mL) was added flame-dried MgI2 (84 mg, 0.302 mmol), and the mixture was stirred for 2 min at rt. The resultant mixture was warmed to 60 °C for 1 min and then Et3N (21 μL, 0.15 mmol) was added. The crude mixture was recrystallized from chloroform and hexanes to afford the title compound 30 as a yellow solid (17 mg) in 59% yield. Recrystallization from a solution of hexanes and CH2Cl2 (by slow evaporation) provided a crystalline solid suitable for X-ray structure analysis: mp 149–151 °C; 1H NMR (400 MHz, CDCl3) δ 8.50 (s, 1H), 7.98–7.83 (m, 4H), 7.64 (t, J = 7.5 Hz, 1H), 7.57 (t, J = 7.5 Hz, 1H), 7.48 (d, J = 7.3 Hz, 2H), 7.38–7.28 (m, 3H), 7.10 (t, J = 7.9 Hz, 2H), 6.71 (t, J = 7.3 Hz, 1H), 6.62 (d, J = 7.8 Hz, 2H), 5.38 (dt, J = 17.4, 8.9 Hz, 1H), 4.60 (d, J = 8.9 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 189.9 (dd, JCF = 29.5, 27.9 Hz, 1C), 145.5, 135.8, 134.5, 132.4 (dd, JCF = 5.0, 4.4 Hz, 1C), 132.2, 130.1, 130.0, 129.3, 129.2 (2C), 128.7, 128.7 (2C), 128.6 (2C), 128.5, 127.7, 127.0, 124.6, 118.9, 117.5 (dd, JCF = 260.8, 259.3 Hz, 1C), 114.2 (2C), 60.6 (dd, JCF = 25.7, 22.6 Hz); 19F NMR (376 MHz, CDCl3) δ ‒103.6 (dd, JFF = 272.0, JHF = 8.8 Hz, 1F), ‒112.2 (dd, JFF = 272.1, JHF = 17.1 Hz, 1F); IR (film) νmax 1694, 1455, 1285, 1105 cm–1; HRMS (ESI-TOF) m/z calcd for C25H20F2NO (M+H)+ 388.1513, found 388.1506.
2,2-Difluoro-3-(4-methoxyphenyl)-1-(naphthalen-2-yl)-3-(phenylamino)propan-1-one 31.
See representative reaction procedure. To a solution of 2,2,4,4,4-pentafluoro-3,3-dihydroxy-1-(naphthalene-2-yl)butan-1-one 125,27 (32 mg, 0.10 mmol) and N-(4-methoxybenzylidene)aniline 24 (44 mg, 0.21 mmol) in THF (1.0 mL) was added MgI2 (115 mg, 0.414 mmol) and the mixture was stirred for 2 min at rt. The resultant mixture was warmed to 60 °C for 1 min and then Et3N (29 μL, 0.21 mmol) was added. SiO2 flash chromatography (9:1:0.01 hexanes/EtOAc/Et3N) afforded the title compound 31 as a dark orange solid (32.5 mg) in 79% yield. Recrystallization from a solution of hexanes and CH2Cl2 (by slow evaporation) provided a crystalline solid suitable for X-ray structure analysis: mp 126–128 °C; 1H NMR (500 MHz, CDCl3) δ 8.52 (s, 1H), 7.98 (d, J = 8.7 Hz, 1H), 7.93 (d, J = 8.1 Hz, 1H), 7.89 (d, J = 5.7 Hz, 1H), 7.87 (d, J = 5.1 Hz, 1H), 7.64 (t, J = 7.4 Hz, 1H), 7.57 (t, J = 7.2 Hz, 1H), 7.41 (d, J = 8.5 Hz, 2H), 7.11 (t, J = 7.9 Hz, 2H), 6.88 (d, J = 8.7 Hz, 2H), 6.72 (t, J = 7.3 Hz, 1H), 6.63 (d, J = 7.9 Hz, 2H), 5.34 (dt, J = 16.3, 8.0 Hz, 1H), 4.58 (d, J = 7.3 Hz, 1H), 3.76 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 189.9 (dd, JCF = 29.2, 28.1 Hz, 1C), 159.8, 145.6, 135.8, 132.4 (t, JCF = 4.5 Hz, 1C), 132.1, 130.1, 130.0, 129.7 (2C), 129.3, 129.2 (2C), 128.5, 127.7, 127.0, 126.3, 124.6, 118.8, 117.5 (t, JCF = 259.8 Hz, 1C), 114.2 (2C), 114.1 (2C), 60.0 (dd, JCF = 25.7, 22.9 Hz, 1C), 55.2; 19F NMR (470 MHz, CDCl3) δ ‒104.2 (dd, JFF = 270.4, JHF = 9.1 Hz, 1F), ‒112.1 (dd, JFF = 270.4, JHF = 16.6 Hz, 1F); IR (film) νmax 1694, 1626, 1465, 1106, 692 cm–1; HRMS (ESI-TOF) m/z calcd for C26H22F2NO2 (M+H)+ 418.1619, found 418.1613.
3-((3,4-Dimethylphenyl)amino)-2,2-difluoro-3-(4-fluorophenyl)-1-(naphthalen-2-yl)propan-1-one 32.
See representative reaction procedure. To a solution of 2,2,4,4,4-pentafluoro-3,3-dihydroxy-1-(naphthalene-2-yl)butan-1-one 125,27 (29 mg, 0.09 mmol), 4Å molecular sieves (100 mg), and 3,4-dimethyl-N-(4-fluorobenzylidene)aniline 25 (44 mg, 0.19 mmol) in THF (1.7 mL) was added flame-dried MgI2 (107 mg, 0.385 mmol) and the mixture was stirred for 2 min at rt. The resultant mixture was warmed to 60 °C for 1 min and then Et3N (25 μL, 0.19 mmol) was added. The crude mixture was recrystallized from chloroform and hexanes to afford the title compound 32 as a pale yellow solid (25 mg) in 64% yield. Recrystallization from a solution of hexanes and CH2Cl2 (by slow evaporation) provided a crystalline solid suitable for X-ray structure analysis: mp 141–143 °C; 1H NMR (500 MHz, CDCl3) δ 8.54 (s, 1H), 7.96 (dd, J = 15.4, 8.6 Hz, 2H), 7.88 (t, J = 7.3 Hz, 2H), 7.65 (t, J = 7.4 Hz, 1H), 7.58 (t, J = 7.4 Hz, 1H), 7.45 (t, J = 6.1 Hz, 2H), 7.04 (t, J = 8.6 Hz, 2H), 6.84 (d, J = 8.1 Hz, 1H), 6.41 (s, 1H), 6.32 (d, J = 8.1 Hz, 1H), 5.32 (dt, J = 17.7, 7.3 Hz, 1H), 4.36 (d, J = 7.7 Hz, 1H), 2.10 (s, 3H), 2.08 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 189.6 (dd, JCF = 29.3, 27.7 Hz, 1C), 162.8 (d, JCF = 247.5 Hz, 1C), 143.3, 137.4, 135.9, 132.3 (t, JCF = 4.5 Hz, 1C), 132.2, 130.5 (d, JCF = 2.5 Hz, 1C), 130.2, 130.2 (d, JCF = 8.0 Hz, 2C), 130.1, 130.0, 129.4, 128.6, 127.7, 127.2, 127.0, 124.6, 117.4 (t, JCF = 248.5 Hz, 1C), 116.1, 115.6 (d, JCF = 21.6 Hz, 2C), 111.5, 60.1 (dd, JCF = 26.0, 22.7 Hz, 1C), 19.9, 18.6; 19F NMR (470 MHz, CDCl3) δ ‒103.0 (dd, JFF = 272.9, 1F), ‒113.3 (dd, JFF = 272.2, JHF = 15.7 Hz, 1F), ‒114.4 (s, 1F); IR (film) νmax 1692, 1507, 1212, 1063 cm–1; HRMS (ESI-TOF) m/z calcd for C27H23F3NO (M+H)+ 434.1732, found 434.1725.
2,2-Difluoro-3-((4-fluorophenyl)amino)-3-(4-methoxyphenyl)-1-(naphthalen-2-yl)propan-1-one 33.
See representative reaction procedure. To a solution of 2,2,4,4,4-pentafluoro-3,3-dihydroxy-1-(naphthalene-2-yl)butan-1-one 125,27 (32 mg, 0.10 mmol) and N-(4-methoxybenzylidene)-4-fluoroaniline 26 (48 mg, 0.21 mmol) in THF (570 μL) was added MgI2 (115 mg, 0.414 mmol) and the mixture was stirred for 2 min at rt. The resultant mixture was warmed to 60 °C for 1 min and then Et3N (29 μL, 0.21 mmol) was added. SiO2 flash chromatography (9:1:0.01 hexanes/EtOAc/Et3N) afforded the title compound 33 as a dark orange oil (33.5 mg) in 78% yield: 1H NMR (500 MHz, CDCl3) δ 8.50 (s, 1H), 7.97 (d, J = 8.7 Hz, 1H), 7.92 (d, J = 8.1 Hz, 1H), 7.89–7.81 (m, 2H), 7.64 (t, J = 7.5 Hz, 1H), 7.57 (t, J = 7.5 Hz, 1H), 7.37 (d, J = 7.8 Hz, 2H), 6.87 (d, J = 7.7 Hz, 2H), 6.80 (t, J = 8.7 Hz, 2H), 6.55 (m, 2H), 5.23 (dt, J = 17.0, 8.5 Hz, 1H), 4.44 (d, J = 8.6 Hz, 1H), 3.76 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 189.9 (t, JCF = 28.9 Hz, 1C), 159.9, 156.5 (d, JCF = 236.9 Hz, 1C), 141.9 (d, JCF = 1.9 Hz, 1C), 135.8, 132.4 (dd, JCF = 5.2, 4.3 Hz, 1C), 132.2, 132.0, 130.1, 130.0, 129.7 (2C), 128.5, 127.7, 127.0, 126.1, 124.6, 117.5 (t, JCF = 259.9 Hz, 1C), 115.7 (d, JCF = 22.5 Hz, 2C), 115.3 (d, JCF = 7.5 Hz, 2C), 114.1 (2C), 61.0 (dd, JCF = 25.9, 22.8 Hz, 1C), 55.2; 19F NMR (376 MHz, CDCl3) δ ‒103.9 (dd, JFF = 271.8, JHF = 8.6 Hz, 1F), ‒112.4 (dd, JFF = 271.8, JHF = 16.9 Hz, 1F), ‒127.1 (sept, J = 3.8 Hz, 1F); IR (film) νmax 1692, 1626, 1114 cm–1; HRMS (ESI-TOF) m/z calcd for C26H21F3NO2 (M+H)+ 436.1524, found 436.1515.
2,2-Difluoro-3-(4-methoxyphenyl)-3-((4-methoxyphenyl)amino)-1-(naphthalen-2-yl)propan-1-one 34.
See representative reaction procedure. To a solution of 2,2,4,4,4-pentafluoro-3,3-dihydroxy-1-(naphthalene-2-yl)butan-1-one 125,27 (35 mg, 0.11 mmol) and N-(4-methoxybenzylidene)-4-methoxyaniline 27 (55 mg, 0.23 mmol) in THF (570 μL) was added magnesium iodide (127 mg, 0.455 mmol) and the mixture was stirred for 2 min at rt. The resultant mixture was warmed to 60 °C for 1 min and then Et3N (32 μL, 0.23 mmol) was added. SiO2 flash chromatography (8:2:0.01 hexanes/EtOAc/Et3N) afforded the title compound 34 as a dark orange oil (12.3 mg) in 25% yield: 1H NMR (500 MHz, CDCl3) δ 8.51 (s, 1H), 7.98 (d, J = 8.7 Hz, 1H), 7.93 (d, J = 8.2 Hz, 1H), 7.88 (dd, J = 8.2, 5.5 Hz, 2H), 7.64 (t, J = 7.5 Hz, 1H), 7.57 (t, J = 7.5 Hz, 1H), 7.37 (d, J = 8.6 Hz, 2H), 6.87 (d, J = 8.7 Hz, 2H), 6.67 (d, J = 9.0 Hz, 2H), 6.55 (d, J = 8.9 Hz, 2H), 5.21 (dd, J = 17.6, 8.6 Hz, 1H), 4.25 (d, J = 9.0 Hz, 1H), 3.76 (s, 3H), 3.67 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 190.0 (dd, JCF = 29.5, 27.7 Hz, 1C), 159.8, 153.0, 139.5, 135.8, 132.3 (dd, JCF = 5.3, 3.9 Hz, 1C), 132.2, 130.3, 130.0, 129.7 (2C), 129.3, 128.5, 127.7, 127.0, 126.5, 124.7, 117.7 (dd, JCF = 260.8, 258.2 Hz, 1C), 115.9 (2C), 114.7 (2C), 114.1 (2C), 61.2 (dd, JCF = 26.0, 22.4 Hz, 1C), 55.6, 55.2; 19F NMR (470 MHz, CDCl3) δ ‒103.6 (dd, JFF = 270.1, JHF = 8.5 Hz, 1F), ‒113.3 (dd, JFF = 270.0, JHF = 17.6 Hz, 1F); IR (film) νmax 1693, 1509, 1114 cm–1; HRMS (ESI-TOF) m/z calcd for C27H24F2NO3 (M+H)+ 448.1724, found 448.1721.
3-(tert-Butylamino)-2,2-difluoro-1-(naphthalen-2-yl)-3-phenylpropan-1-one 35.
See representative reaction procedure. To a solution of 2,2,4,4,4-pentafluoro-3,3-dihydroxy-1-(naphthalene-2-yl)butan-1-one 125,27 (32 mg, 0.10 mmol) and N-benzylidene-tert-butylamine 28 (37 μL, 0.21 mmol) in THF (570 μL) was added MgI2 (115 mg, 0.414 mmol) and the mixture was stirred for 2 min at rt. The resultant mixture was warmed to 60 °C for1 min and then Et3N (29 μL, 0.21 mmol) was added. SiO2 flash chromatography (9:1:0.01 hexanes/EtOAc/Et3N) afforded the title compound 35 as a colorless solid (10.7 mg) in 30% yield: mp 106–108 °C; 1H NMR (400 MHz, CDCl3) δ 8.59 (s, 1H), 8.03 (d, J = 8.6 Hz, 1H), 7.97 (d, J = 8.1 Hz, 1H), 7.94–7.84 (m, 2H), 7.63 (t, J = 7.5 Hz, 1H), 7.57 (t, J = 7.5 Hz, 1H), 7.45–7.43 (m, 2H), 7.41–7.29 (m, 3H), 4.63 (dd, J = 22.0, 6.5 Hz, 1H), 1.83 (br s, 1H), 0.84 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 192.0 (dd, JCF = 30.5, 26.5 Hz, 1C), 138.3, 135.6, 132.2, 132.0 (t, JCF = 2.9 Hz, 1C), 131.9, 131.4, 129.9, 128.9, 128.7 (2C), 128.4 (2C), 128.2, 127.7, 126.8, 125.1, 118.3 (dd, JCF = 260.9, 257.3 Hz, 1C), 59.5 (dd, JCF = 26.9, 21.2 Hz, 1C), 51.5, 30.0 (3C); 19F NMR (376 MHz, CDCl3) δ ‒100.9 (dd, JFF = 258.8, JHF = 6.4 Hz, 1F), ‒117.5 (dd, JFF = 258.8, JHF = 22.2 Hz, 1F); IR (film) νmax 1698, 1456, 1108, 1077 cm–1; HRMS (ESI-TOF) m/z calcd for C23H24F2NO (M+H)+ 368.1826, found 368.1825.
(4E,6E)-3-(Benzylamino)-2,2-difluoro-1-(naphthalen-2-yl)octa-4,6-dien-1-one 36.
See representative reaction procedure. To a solution of 2,2,4,4,4-pentafluoro-3,3-dihydroxy-1-(naphthalene-2-yl)butan-1-one 125,27 (20 mg, 0.06 mmol) and N-(hexa-2,4-dien-1-ylidene)-1-phenylmethanamine 29 (30 mg, 0.16 mmol, 5:1 (2E,4E)-isomer/(2E,4Z)-isomer mixture*) in THF (1 mL) was added flame-dried MgI2 (56 mg, 0.20 mmol) and the mixture was stirred for 2 min at rt. The resultant mixture was warmed to 60 °C for 1 min and then Et3N (14 μL, 0.10 mmol) was added. SiO2 flash chromatography (9:1:0.01 hexanes/Et2O/Et3N) afforded the title compound 36 as a yellow oil in 61% yield (12 mg, 5:1 mixture of isomers*): 1H NMR (500 MHz, CDCl3) δ 8.57* (s, 1H), 8.55 (s, 1H), 8.03 (dd, J = 8.7, 1.4 Hz, 1H), 7.93 (d, J = 8.0 Hz, 1H), 7.89 (d, J = 8.8 Hz, 2H), 7.65 (td, J = 7.5, 1.1 Hz, 1H), 7.57 (td, J = 7.5, 1.2 Hz, 1H), 7.13–7.03 (m, 5H), 6.56* (dd, J = 15.2, 11.0 Hz, 1H), 6.24 (dd, J = 15.1, 10.4 Hz, 1H), 6.15 (qd, J = 12.5, 1.3 Hz, 1H), 5.79 (dq, J = 13.2, 6.6 Hz, 1H), 5.54 (dd, J = 14.7, 8.8 Hz, 1H), 3.86 (d, J = 13.4 Hz, 1H), 3.83 (m, 1H), 3.60 (d, J = 13.2 Hz, 1H), 1.80 (d, J = 6.6 Hz, 3H), 1.61 (br s, 1H); 13C NMR (125 MHz, CDCl3) δ 190.1 (t, JCF = 28.2 Hz, 1C), 138.9, 136.8, 135.7, 132.2, 132.0, 131.5, 130.5, 130.0, 129.1, 128.4, 128.1 (2C), 128.1 (2C), 127.7, 126.9, 126.8, 124.8, 123.0, 118.1 (t, JCF = 257.4 Hz, 1C), 62.2 (dd, JCF = 26.3, 22.4 Hz, 1C), 50.5, 18.2, 13.6; 19F NMR (470 MHz, CDCl3) δ ‒103.2 (dd, JFF = 268.9, JHF = 7.5 Hz, 1F), ‒115.5 (dd, JFF = 268.9, JHF = 18.8 Hz, 1F); IR (film) νmax 1700, 1454, 1116, 990, 699 cm–1; HRMS (ESI-TOF) m/z calcd for C25H24F2NO (M+H)+ 392.1826, found 392.1841. * denotes minor isomer.
2-(5-Bromo-1,2,3,4-tetrahydroisoquinolin-1-yl)-2,2-difluoro-1-(naphthalen-2-yl)ethenone 38.
See representative reaction procedure. To a solution of 2,2,4,4,4-pentafluoro-3,3-dihydroxy-1-(naphthalene-2-yl)butan-1-one 125,27 (92.6 mg, 0.289 mmol) and 5-bromo-3,4-dihydroisoquinoline 37 (127 mg, 0.605 mmol) in THF (1.7 mL) was added MgI2 (340 mg, 1.22 mmol) and the mixture was stirred for 5 min at rt. The resultant mixture was warmed to 60 °C for 5 min and then Et3N (85 μL, 0.61 mmol) was added. SiO2 flash chromatography (5:5:0.01 hexanes/CH2Cl2/Et3N) afforded the title compound 38 as a pale yellow solid (101.7 mg) in 84% yield: mp 112–114 °C; 1H NMR (500 MHz, CDCl3) δ 8.63 (s, 1H), 8.07 (d, J = 8.7 Hz, 1H), 7.96 (d, J = 8.2 Hz, 1H), 7.92 (d, J = 8.7 Hz, 1H), 7.89 (d, J = 8.2 Hz, 1H), 7.64 (t, J = 7.5 Hz, 1H), 7.58 (d, J = 7.9 Hz, 1H), 7.55 (d, J = 7.7 Hz, 1H), 7.41 (dd, J = 7.7, 2.7 Hz, 1H), 7.11 (t, J = 7.9 Hz, 1H), 4.89 (dd, J = 21.0, 8.2 Hz, 1H), 3.26 (m, 1H), 3.02 (m, 1H), 2.85–2.66 (m, 2H), 1.89 (br s, 1H); 13C NMR (100 MHz, CDCl3) δ 190.6 (dd, JCF = 29.8, 27.8 Hz, 1C), 136.8, 135.8, 132.2 (t, JCF = 8.3 Hz, 1C), 132.1, 131.9, 130.6, 130.0, 129.2, 128.5, 127.7 (2C), 127.6, 126.9 (2C), 125.7, 124.8, 118.9 (dd, JCF = 265.1, 258.5 Hz, 1C), 57.1 (t, JCF = 23.4 Hz, 1C), 39.5, 29.6; 19F NMR (376 MHz, CDCl3) δ ‒96.0 (dd, JFF = 270.5, JHF = 7.2 Hz, 1F), ‒110.4 (dd, JFF = 270.5, JHF = 21.0 Hz, 1F); IR (film) νmax 1698, 1440, 1173, 754 cm−1; HRMS (ESI-TOF) m/z calcd for C21H17BrF2NO (M+H)+ 416.0462, found 416.0448.
2,2-Difluoro-1-(naphthalen-2-yl)-2-(5-vinyl-1,2,3,4-tetrahydroisoquinolin-1-yl)ethan-1-one 39.
To a solution of 2-(5-bromo-1,2,3,4-tetrahydroisoquinolin-1-yl)-2,2-difluoro-1-(naphthalen-2-yl)ethenone 38 (23.5 mg, 0.056 mmol) in THF (0.3 mL) and water (40 μL), PdCl2 (4.1 mg, 0.023 mmol), PPh3 (5.8 mg, 0.02 mmol), Cs2CO3 (67.6 mg, 0.207 mmol), and potassium vinyltrifluoroborate (9.7 mg, 0.07 mmol) were added, and the resultant mixture was allowed to stir for 23 h at 85 °C in a sealed tube. The reaction mixture was warmed to rt, quenched with water (2 mL), and extracted with CH2Cl2 (2 mL). The aqueous layer was extracted with CH2Cl2 (2 mL × 3). The organics were dried over Na2SO4 and concentrated under reduced pressure. SiO2 flash chromatography (100:0.01 CH2Cl2/Et3N) afforded the title compound 39 as a pale yellow oil (8.3 mg) in 40% yield: 1H NMR (500 MHz, CDCl3) δ 8.61 (s, 1H), 8.08 (d, J = 8.6 Hz, 1H), 7.94 (d, J = 8.2 Hz, 1H), 7.91 (d, J = 8.8 Hz, 1H), 7.88 (d, J = 8.2 Hz, 1H), 7.63 (t, J = 7.4 Hz, 1H), 7.56 (t, J = 7.5 Hz, 1H), 7.45 (d, J = 7.6 Hz, 1H), 7.38 (d, J = 7.2 Hz, 1H), 7.23 (t, J = 7.6 Hz, 1H), 6.90 (dd, J = 17.3, 11.0 Hz, 1H), 5.62 (d, J = 17.3 Hz, 1H), 5.32 (d, J = 10.9 Hz, 1H), 4.93 (dd, J = 20.6, 8.3 Hz, 1H), 3.26 (m, 1H), 3.00 (m, 1H), 2.79–2.70 (m, 2H), 1.89 (br s, 1H); 13C NMR (100 MHz, CDCl3) δ 190.9 (dd, JCF = 29.7, 27.7 Hz, 1C), 137.2, 135.7, 134.7, 134.1, 132.2, 132.1 (dd, JCF = 5.1, 4.0 Hz, 1C), 130.8, 130.0, 129.7, 129.1, 128.4, 128.2, 128.1, 127.7, 126.9, 125.7, 125.3, 124.9, 116.3 (dd, JCF = 264.6, 258.5 Hz, 1C), 57.4 (t, JCF = 23.6 Hz, 1C), 39.6, 26.1; 19F NMR (376 MHz, CDCl3) δ ‒96.6 (dd, JFF = 268.8, JHF = 5.6 Hz, 1F), ‒110.6 (dd, JFF = 268.8, JHF = 20.5 Hz, 1F); IR (film) νmax 2923, 1698, 1627, 1466, 1074, 736 cm−1; HRMS (ESI-TOF) m/z calcd for C23H20F2NO (M+H)+ 364.1513, found 364.1510.
2-(6,7-Dimethoxy-1,2,3,4-tetrahydroisoquinolin-1-yl)-2,2-difluoro-1-(4-(trifluoromethyl)-phenyl)ethan-1-one 42.
See representative reaction procedure. To a solution of 2,2,4,4,4-pentafluoro-3,3-dihydroxy-1-(4-(trifluoromethyl)phenyl)butan-1-one 4038 (117 mg, 0.345 mmol) and 6,7-dimethoxy-3,4-dihydroisoquinoline 41 (139 mg, 0.728 mmol) in THF (2.5 mL) was added MgI2 (469.5 mg, 1.688 mmol) and the mixture was stirred for 5 min at rt. The resultant mixture was warmed to 60 °C for 5 min and then Et3N (101 μL, 0.723 mmol) was added. SiO2 flash chromatography (5:5:0.01 hexanes/EtOAc/Et3N) afforded the title compound 42 as a pale yellow solid (49.2 mg) in 34% yield: mp 128–130 °C; 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 8.1 Hz, 2H), 7.73 (d, J = 8.3 Hz, 2H), 6.88 (d, J = 2.4 Hz, 1H), 6.62 (s, 1H), 4.74 (dd, J = 21.2, 7.6 Hz, 1H), 3.87 (s, 3H), 3.86 (s, 3H), 3.12 (m, 1H), 2.95 (dt, J = 3.2, 1.3 Hz, 1H), 2.63 (dt, J = 1.1, 1.0 Hz, 2H), 1.62 (br s, 1H); 13C NMR (100 MHz, CDCl3) δ 191.2 (dd, JCF = 31.6, 27.8 Hz, 1C), 148.7, 147.2, 136.8, 134.6 (d, J = 32.9 Hz, 1C), 130.0 (td, JCF = 2.5, 1.2 Hz, 2C), 129.6, 125.4 (td, JCF = 3.7, 3.6 Hz, 2C), 124.8 (t, JCF = 287.7 Hz, 1C), 120.7, 118.9 (dd, JCF = 264.7, 256.9 Hz, 1C), 111.7, 111.1 (d, J = 5.6 Hz, 1C), 56.5 (t, JCF = 23.8 Hz, 1C), 56.0, 55.8, 39.8, 28.6; 19F NMR (376 MHz, CDCl3) δ −64.4 (s, 3F), ‒97.2 (dd, JFF = 265.9, JHF = 6.2 Hz, 1F), ‒113.3 (dd, JFF = 265.9, JHF = 21.1 Hz, 1F); IR (film) νmax 2927, 1714, 1518, 1465, 1129, 732 cm–1; HRMS (ESI-TOF) m/z calcd for C20H19F5NO3 (M+H)+ 416.1285, found 416.1281.
(2R)-2-(1-(1,1-Difluoro-2-oxo-2-(4-(trifluoromethyl)phenyl)ethyl)-6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)-N-methyl-2-phenylacetamide 44.
To a solution of 2-(6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-1-yl)-2,2-difluoro-1-(4-(trifluoromethyl)phenyl)ethan-1-one 42 (12.1 mg, 0.029 mmol) and (S)-2-(methylamino)-2-oxo-1-phenylethyl 4-methylbenzenesulfonate 4348 (10.2 mg, 0.032 mmol) in CH3CN (1.0 mL) was added i-Pr2NEt (10 μL, 0.06 mmol). The resultant mixture was stirred for 26 h at 85 °C. Then, reaction mixture was cooled to rt and concentrated under reduced pressure. The mixture was washed with saturated aqueous Na2CO3 (5 mL) and extracted with EtOAc (5 mL). The organics were dried over Na2SO4 and concentrated under reduced pressure to give a 2.2:1 mixture of diastereomers. SiO2 flash chromatography (5:5:0.01 hexanes/EtOAc/Et3N) afforded the title compound 44 as a colorless oil (4.6 mg, major isomer) in 28% yield: 1H NMR (500 MHz, CDCl3) δ 8.11 (d, J = 8.2 Hz, 2H), 7.80 (d, J = 8.4 Hz, 2H), 7.02 (t, J = 7.7 Hz, 2H), 6.69 (s, 1H), 6.52 (m, 1H), 6.45 (d, J = 7.5 Hz, 2H), 6.37 (d, J = 2.4 Hz, 1H), 4.19 (s, 1H), 3.91 (s, 3H), 3.89 (m, 1H), 3.79 (s, 3H), 3.52 (m, 1H), 3.12 (m, 1H), 2.92 (dd, J = 14.8, 7.0 Hz, 1H), 2.79 (d, J = 4.9 Hz, 3H), 2.58 (dd, J = 17.4, 5.6 Hz, 1H), 2.18 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 189.3 (t, JCF = 27.5 Hz, 1C), 171.2, 149.5, 147.4, 135.6, 135.5 (d, JCF = 32.9 Hz, 1C), 134.8, 129.4 (dt, JCF = 2.6, 2.3 Hz, 2C), 129.1 (2C), 128.9, 128.6 (2C), 128.5, 127.8, 126.2 (dt, JCF = 3.7, 3.6 Hz, 2C), 124.7 (t, JCF = 283.3 Hz, 1C), 118.3 (t, JCF = 263.3 Hz, 1C), 111.5, 111.1 (d, JCF = 3.3 Hz, 1C), 69.7, 60.5 (dd, JCF = 28.0, 23.1 Hz, 1C), 56.1, 55.8, 42.3, 29.7, 25.9; 19F NMR (376 MHz, CDCl3) δ ‒64.3 (s, 3F), ‒95.4 (dd, JFF = 259.9, JHF = 10.2 Hz, 1F), ‒106.6 (dd, JFF = 259.5, JHF = 20.9 Hz, 1F); IR (film) νmax 2922, 1712, 1664, 1466, 1116, 701 cm–1; HRMS (ESI-TOF) m/z calcd for C29H28F5N2O4 (M+H)+ 563.1969, found 563.1968.
Supplementary Material
ACKNOWLEDGMENT
The authors acknowledge funding from the University of Mississippi and the National Institute of General Medical Sciences (Grant P20GM104932). Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the National Institutes of Health (NIH). The Mass Spectrometry and Proteomic Facility of the University of Notre Dame and Mass Spectrometry Facility of the Louisiana State University are acknowledged for acquisition of high-resolution mass spectrometry data.
Footnotes
Supporting Information.
Spectroscopic data from 1H, 13C and 19F NMR, and X-ray experimental and data. The Supporting Information is available free of charge on the ACS Publications website.
The authors declare no competing financial interests.
REFERENCES
- (1).Begue J-P; Bonnet-Delpon D Bioorganic and Medicinal Chemistry of Fluorine; John Wiley and Sons: Hoboken, NJ, 2008. [Google Scholar]
- (2).Gillis EP; Eastman KJ; Hill MD; Donnelly DJ; Meanwell NA J. Med. Chem 2015, 58, 8315–8359. [DOI] [PubMed] [Google Scholar]
- (3).Zhu W; Wang J; Wang S; Gu Z; Acena JL; Izawa K; Liu H; Soloshonok VA J. Fluor. Chem 2014, 167, 37–54. [Google Scholar]
- (4).Müller K; Faeh C; Diederich F Science 2007, 317, 1881–1886. [DOI] [PubMed] [Google Scholar]
- (5).Zhou Y; Wang J; Gu Z; Wang S; Zhu W; Acena JL; Soloshonok VA; Izawa K; Liu H Chem. Rev 2016, 116, 422–518. [DOI] [PubMed] [Google Scholar]
- (6).Wang J; Sanchez-Rosello M; Acena JL; del Pozo C; Sorochinsky AE; Fustero S; Soloshonok VA; Liu H Chem. Rev 2014, 114, 2432–2506. [DOI] [PubMed] [Google Scholar]
- (7).Furuya T; Kamlet AS; Ritter T Nature 2011, 473, 470–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Tomashenko OA; Grushin VV Chem. Rev 2011, 111, 4475–4521. [DOI] [PubMed] [Google Scholar]
- (9).Uoto K; Ohsuki S; Takenoshita H; Ishiyama T; Iimura S; Hirota Y; Mitsui I; Terasawa H; Soga T Chem. Pharm. Bull 1997, 45, 1793–1804. [DOI] [PubMed] [Google Scholar]
- (10).Nakayama K; Kawato HC; Inagaki H; Nakajima R; Kitamura A; Someya K; Ohta T Org. Lett 2000, 2, 977–980. [DOI] [PubMed] [Google Scholar]
- (11).Silva AM; Cachau RE; Sham HL; Erickson JW J. Mol. Biol 1996, 255, 321–346. [DOI] [PubMed] [Google Scholar]
- (12).Thaisrivongs S; Schostarez HJ; Pals DT; Turner SR J. Med. Chem 1987, 30, 1837–1842. [DOI] [PubMed] [Google Scholar]
- (13).Vergely I; Boggetoo N; Okochi V; Golpayegani S; Reboud-Ravaux M; Kobaiter R; Joyeau R; Wakselman M Eur. J. Med. Chem 1995, 30, 199–208. [Google Scholar]
- (14).Sato K; Tarui A; Matsuda S; Omote M; Ando A; Kumadaki I Tetrahedron Lett. 2005, 46, 7679–7681. [Google Scholar]
- (15).Tarui A; Kondo K; Taira H; Sato K; Omote M; Kumadaki I; Ando A Heterocycles 2007, 73, 203–208. [Google Scholar]
- (16).Tarui A; Ozaki D; Nakajima N; Yokota Y; Sokeirik YS; Omote M; Kumadaki I; Ando A Tetrahedron Lett. 2008, 49, 3839–3843. [Google Scholar]
- (17).Sorochinsky A; Voloshin N; Markovsky A; Belik M; Yasuda N; Uekusa H; Ono T; Berbasov DO; Soloshonok VA J. Org. Chem 2003, 68, 7448–7454. [DOI] [PubMed] [Google Scholar]
- (18).Soloshonok VA; Ohkura H; Sorochinsky A; Voloshin N; Markovsky A; Belik M; Yamazaki Y Tetrahedron Lett. 2002, 43, 5445–5448. [Google Scholar]
- (19).Poisson T; Belhomme M-C; Pannecoucke XJ Org. Chem 2012, 77, 9277–9285. [DOI] [PubMed] [Google Scholar]
- (20).Wang Y; Zhu S Org. Lett 2003, 5, 745–748. [DOI] [PubMed] [Google Scholar]
- (21).Cao C; Jiang M; Liu J Eur. J. Org. Chem 2015, 1144–1151. [Google Scholar]
- (22).Kashikura W; Mori K; Akiyama T Org. Lett 2011, 13, 1860–1863. [DOI] [PubMed] [Google Scholar]
- (23).Yuan Z; Wei Y; Shi M Tetrahedron 2010, 66, 7361–7366. [Google Scholar]
- (24).Jonet S; Cherouvrier F; Brigaud T; Portella C Eur. J. Org. Chem 2005, 4304–4312. [Google Scholar]
- (25).Han C; Kim EH; Colby DA J. Am. Chem. Soc 2011, 133, 5802–5805. [DOI] [PubMed] [Google Scholar]
- (26).Zhang P; Wolf C Angew. Chem. Int. Ed 2013, 52, 7869–7873. [DOI] [PubMed] [Google Scholar]
- (27).John JP; Colby DA J. Org. Chem 2011, 76, 9163–9168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Hazlitt RA; John JP; Tran Q-L; Colby DA Tetrahedron Lett. 2016, 57, 1906–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zhang P; Wolf CJ Org. Chem 2012, 77, 8840–8844. [DOI] [PubMed] [Google Scholar]
- (30).Lin Y-M; Yi W-B; Shen W-Z; Lu G-P Org. Lett 2016, 18, 592–595. [DOI] [PubMed] [Google Scholar]
- (31).Leng DJ; Black CM; Pattison G Org. Biomol. Chem 2016, 14, 1531–1535. [DOI] [PubMed] [Google Scholar]
- (32).Sowaileh MF; Han C; Hazlitt RA; Kim EH; John JP; Colby DA Tetrahedron Lett. 2017, 58, 396–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Mei H; Xie C; Acena JL; Soloshonok VA; Roschenthaler G-V; Han J Eur. J. Org. Chem 2015, 6401–6412. [Google Scholar]
- (34).Zhu Y; Zhang W; Mei H; Han J; Soloshonok VA; Pan YJ Fluor. Chem 2017, 203, 99–103. [Google Scholar]
- (35).Xie C; Wu L; Zhou J; Mei H; Soloshonok VA; Han J; Pan YJ Fluorine Chem. 2015, 172, 13–21. [Google Scholar]
- (36).Xie C; Wu L; Mei H; Soloshonok VA; Han J; Pan Y Tetrahedron Lett. 2014, 55, 5908–5910. [Google Scholar]
- (37).Xie C; Wu L; Mei H; Soloshonok VA; Han J; Pan Y Org. Biomol. Chem 2014, 12, 7836–7843. [DOI] [PubMed] [Google Scholar]
- (38).Li W; Zhu X; Mao H; Tang Z; Cheng Y; Zhu C Chem. Commun 2014, 50, 7521–7523. [DOI] [PubMed] [Google Scholar]
- (39).Mamone M; Morvan E; Milcent T; Ongeri S; Crousse BJ Org. Chem 2015, 80, 1964–1971. [DOI] [PubMed] [Google Scholar]
- (40).Vazquez J; Prieto A; Fernandez R; Enders D; Lassaletta JM Chem. Commun 2002, 498–499. [DOI] [PubMed] [Google Scholar]
- (41).Truong N; Sauer SJ; Seraphin-Hatcher C; Coltart DM Org. Biomol. Chem 2016, 14, 7864–7868. [DOI] [PubMed] [Google Scholar]
- (42).Hazlitt RA; Tran Q-L; Sowaileh MF; Colby DA J. Org. Chem 2017, 82, 2231–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Molander GA; Brown AR J. Org. Chem 2006, 71, 9681–9686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Le VH; Inai M; Williams RM; Kan T Nat. Prod. Rep 2015, 32, 328–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Perrey DA; Decker AM; Li J-X; Gilmour BP; Thomas BF; Harris DL; Runyon SP; Zhang Y Bioorg. Med. Chem. Lett 2015, 23, 5709–5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Owen RT; Castañer R; Bolós J; Estivill C Drugs Future 2009, 34, 5–10. [Google Scholar]
- (47).Weller T; Koberstein R; Aissaoui H; Clozel M; Fischli W WO 2005/118548, 2005.
- (48).Fañanás-Mastral M; Teichert JF; Fernández-Salas JA; Heijnen D; Feringa BL Org. Biomol. Chem 2013, 11, 4521–4525. [DOI] [PubMed] [Google Scholar]
- (49).Reddy NSS; Reddy BVS Tetrahedron Lett. 2014, 55, 3157–3159. [Google Scholar]
- (50).Steiner MA; Gatfield J; Brisbare-Roch C; Dietrich H; Treiber A; Jenck F; Boss C ChemMedChem. 2013, 8, 898–903. [DOI] [PubMed] [Google Scholar]
- (51).Woods JR Ph.D. Dissertation, Purdue University. West Lafayette, Indiana, 2014. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.