

Abstract
Microanatomical organization of innate immune cells within lymph nodes (LNs) is critical for the generation of adaptive responses. In particular, steady-state LN-resident dendritic cells (Res cDCs) are strategically localized to intercept lymph-draining antigens. If myeloid cell organization changes during inflammation and how that might impact the generation of immune responses is unknown. Here, we report that during Type-I, but not Type-II, inflammation after adjuvant immunization or viral infection, antigen-presenting Res cDCs undergo CCR7-dependent intranodal repositioning from the LN periphery into the T cell zone (TZ) to elicit T cell priming. Concurrently, inflammatory monocytes infiltrate the LNs via local blood vessels, enter the TZ, and cooperate with Res cDCs by providing polarizing cytokines to optimize T cell effector differentiation. Monocyte infiltration is nonuniform across LNs, generating distinct microenvironments with varied local innate cell composition. These spatial microdomains are associated with divergent early T cell effector programming, indicating that innate microenvironments within LNs play a critical role in regulating the quality and heterogeneity of T cell responses. Together, our findings reveal that dynamic modulation of innate cell microenvironments during Type-I inflammation leads to optimized generation of adaptive immune responses to vaccines and infections.
Keywords: Lymph node microanatomy, innate immunity, adaptive immunity, dendritic cells, monocytes, T cells, antigen presentation, effector T cell differentiation, vaccines, infections, quantitative imaging
One Sentence Summary:
Distinct myeloid cell microenvironments in LNs promote divergent T cell programming during Type-I inflammation.
Introduction
Spatiotemporal coordination and crosstalk between various innate and adaptive immune cells is critical for timely generation of protective immunity (1). During infection or vaccination, conventional dendritic cells (cDCs) migrate from peripheral tissues to draining LNs (dLNs) in a C-C chemokine receptor type 7 (CCR7) dependent manner to present captured antigens on major histocompatibility complexes (MHC) to induce T cell responses (2–5). In addition, much faster transport of microbes, vaccine components and soluble antigens into dLNs occurs via lymphatic transport (6–9). Within LNs, steady-state LN-resident cDCs (Res cDC), and in particular Res cDC2s, localize near lymphatic sinuses (10). This promotes robust sampling of draining antigens, and in combination with the enhanced capacity of cDC2s for MHC-II presentation, allows efficient induction of CD4 T cell responses (6–8). Res cDC1s, cells specialized in MHC-I cross-presentation, are more homogeneously distributed throughout the TZ (10–12). This reduces efficiency of antigen capture after subunit vaccination (6, 7), and instead promotes the interception of infected migratory cDCs and dying cells, leading to enhanced CD8 T cell responses during viral infections or cancer (13–15).
In response to Toll-like receptor (TLR) stimulation, cDCs increase expression of costimulatory molecules and produce various inflammatory cytokines, such as interleukin 12 (IL-12), to elicit the differentiation of effector T cells (2). Current models propose that a single cDC delivers all three signals (peptide-MHC, costimulation and polarizing cytokines) to cognate T lymphocytes to induce their programming (2). However, T cells can also integrate signals from serial interactions with multiple cDCs or other innate cell populations (13, 14, 16). In particular, inflammatory monocytes (MOs) can migrate to dLNs after infection or vaccination to modulate T cell responses (17–19). Detailed mechanisms of whether MOs transport antigens from peripheral tissues or directly traffic into LNs via high endothelial venules (HEVs), and if they present antigens or promote cytokine-driven T cell responses are less clear (17–23).
Even at steady state, myeloid cells generate intricate spatial patterns within LNs (10, 24, 25). During inflammation, they can undergo further reorganization and together with influx of new myeloid cells are likely to generate additional microenvironments (11, 26, 27). T cells activated within different microenvironments may thus receive distinct stimuli and undergo divergent programs of activation. This is supported by substantial heterogeneity in clonal expansion and effector differentiation observed for individual T cells bearing identical T cell receptors (TCRs) (28–31). However, studying how local tissue microenvironments influence T cell priming has been challenging, as this requires use of advanced in situ imaging approaches capable of simultaneously mapping multiple myeloid and lymphoid cell populations, as well as their activation, differentiation, and functional states.
Here, we utilized advanced analytical imaging to characterize the spatial organization of innate and adaptive responses in dLNs during Type-I or Type-II inflammation elicited with distinct vaccine adjuvants or infections (32–35). We found that Type-I inflammation was associated with a signature of myeloid cell organization in dLNs, with rapid repositioning of Res cDCs from the LN periphery into the deep TZ, as well as robust infiltration of the TZ by inflammatory MOs. These changes were not observed during Type-II inflammation, suggesting specificity in myeloid cell patterning based on inflammatory conditions. Within the TZ, spatially juxtaposed Res cDCs and MOs functionally cooperated with one another, providing complementary stimuli to T cells to promote optimization of cellular responses. Moreover, MO infiltration was nonuniform across the LNs, leading to the formation of MO-rich vs cDC-dominated microenvironments. Priming of T cells within these different microenvironments led to generation of distinct early effector populations, allowing for formation of effector heterogeneity. Thus, our findings reveal a signature of myeloid cell organization during Type-I inflammation, as well as demonstrate that innate cell microenvironments influence the magnitude, quality and heterogeneity of adaptive responses.
Results
DC relocalization and Monocyte influx in LNs during Type-I inflammation
We previously reported that preferential localization of Res cDC2s in lymphatics-proximal regions promoted robust T cell responses to subunit vaccination (6, 7). However, naïve T cells primarily reside in the TZ, and how efficient T cell activation was achieved remained unclear. One possibility is that during inflammation, innate cells are spatially reorganized to promote physical interactions with T cells. To investigate how intranodal positioning of myeloid cells changes during inflammation, we injected mice with a broad array of vaccine adjuvants, including Type-I inflammatory TLR agonists: CpG (TLR9), R848 (TLR7/8), Flagellin (TLR5), LPS (TLR4), and pIC (TLR3); Type-II adjuvants: Alhydrogel (Alum) and Addavax (MF59 analog), as well as the protease allergen, papain. Adjuvants were initially used as they are clinically relevant and have well-defined drainage and inflammatory properties. One day post immunization (subcutaneous, footpad or intradermal), dLNs were isolated and the localization of myeloid cell subsets was visualized with multiparameter confocal imaging and analyzed with histocytometry. Multiplexed staining for CD11c, MHC-II, CD169, CD64, SIRPα, Clec9a and other myeloid markers allowed clear discrimination of migratory and Res cDC populations as well as other myeloid cell types (fig. S1A and table S1). We observed that all tested TLR agonists elicited marked repositioning of Res cDC2s from the peripheral LN regions into the deep TZ (Fig. 1A). This was quantified using a positional TZ gate generated with CD3 and B220 staining delineating the B cell follicles and TZ, respectively (Fig. 1B and fig. S1B). Although Res cDC1s were less abundant in the lymphatics-proximal regions as compared to cDC2, these cells re-localized to the deep TZ after TLR agonist immunization (Fig. 1, A and B). cDC repositioning was similarly detected by calculating the distance of cells from the deep TZ (fig. S2A). As expected, after repositioning, Res cDC1s and cDC2s did not occupy the same regions within the TZ (Fig. 1A and fig. S2A), supporting previous observations of Ebi2-mediated compartmentalization (36, 37). Notably, none of the tested Type-II stimuli elicited Res cDC repositioning, suggesting that this response is specific to Type-I inflammatory conditions (Fig. 1, A and B, and fig. S2, B and C). Kinetics studies revealed that Res cDC relocalization began by 9h post immunization (Fig. 1C and fig. S2D), indicating that this was a relatively rapid phenomenon likely associated with the drainage of soluble TLR agonists into the dLNs (38).
Figure 1. MO influx and Res cDC TZ relocalization during Type 1 inflammation.

A-B) B6 mice were injected in the ears with the indicated adjuvants and dLNs were analyzed 24h later by histocytometry. A) Representative images depicting Imaris 3D surfaces for indicated cell types, overlayed with B220 staining. White circles denote the TZ boundary. [See fig. S1A for gating]. B) Frequency of Res cDCs and MOs within the TZ, as well as MO density. Data analyzed using Brown-Forsythe and Welch ANOVA tests with Dunnett’s multiple comparison. C-D) B6 mice were immunized with CpG in both ears and dLNs were analyzed at indicated time points. C) Histocytometry for Res cDC localization and MO density. D) MOs in dLNs enumerated by flow cytometry. Data representative of 5 independent experiments. E-F) B6 mice were infected in the footpad with 100 pfu of West Nile virus-TX and dLNs were analyzed by histocytometry. E) Representative images from mock infected and 36h post infection. F) Localization of Res cDCs and MO density at the indicated time points. Scale bars denote 200 μm. Data represent at least 2 independent experiments.
In addition, TLR agonist immunization elicited the appearance of large numbers of CD64+SIRPa+CD169- cells, markers associated with inflammatory MOs (39, 40), within the deep TZ of dLNs (Fig. 1A and B, and fig. S2A). This was confirmed by flow cytometry, which demonstrated robust influx of Ly6C+CD64+CD11b+Ly6G-CD3-CD19-NK1.1- MOs into immunized dLNs (fig. S2E–G and table S1), beginning as early as 8h, and peaking at 12–16h post immunization (Fig. 1, C and D, and fig. S2D). Some differences in MO recruitment between the different TLR-agonists were noted, with LPS inducing comparably lower responses. Minor increases in MO number were also seen after papain administration, albeit these cells did not efficiently penetrate the deep TZ (Fig. 1, A and B). Both the centralization of Res cDCs and presence of MOs within the TZ were observed on day 2 post immunization (see below), suggesting that Type-I inflammation is associated with lasting changes in the composition and organization of innate cells within the dLNs. Immunization also led to increased number of migratory cDCs, and as expected these cells were localized within the TZ (fig. S2, A and H).
We next investigated whether these phenomena also applied to the Type-I inflammatory settings seen during infection. Indeed, robust Res cDC relocalization and MO recruitment were seen after administration of heat-killed Escherichia coli (fig. S2I) and after West Nile virus infection (Fig. 1, E and F). In contrast, infection with Nippostrongylus brasiliensis, which is associated with Type-II inflammation, did not elicit these effects in skin-draining LNs (fig. S2J). Together, these data demonstrated that Type-I inflammation induced by TLR-agonist administration, bacterial inoculation, and West Nile virus infection induced the redistribution of Res cDCs into the deep TZ and robust recruitment of MOs into the dLN.
CCR7-mediated Res cDC intranodal repositioning during inflammation
We hypothesized that intranodal Res cDC repositioning was driven by chemotaxis (41). Peripheral tissue cDCs up-regulate CCR7 expression for migration into the dLNs (42, 43). Splenic cDCs also utilize CCR7 to relocalize into the white pulp during systemic inflammation (36, 37, 44–46). We thus examined CCR7 expression on Res cDCs after immunization. We found that after Type-I, but not Type-II, adjuvant administration CCR7 expression was markedly increased on both Res cDC subsets (Fig. 2, A and B, and fig. S3A). This was also associated with higher surface expression of MHC-II, indicative of Res cDC maturation (Fig. 2A and fig. S3A). Both CCR7 and MHC-II expression increased as early as 4h post immunization, and rapidly approached levels found on migratory cDCs (Fig. 2A). In contrast, expression of Ebi2, which can also influence cDC localization, was not substantially changed with immunization (fig. S3B).
Figure 2. CCR7-mediated Res cDC intranodal relocalization following immunization.
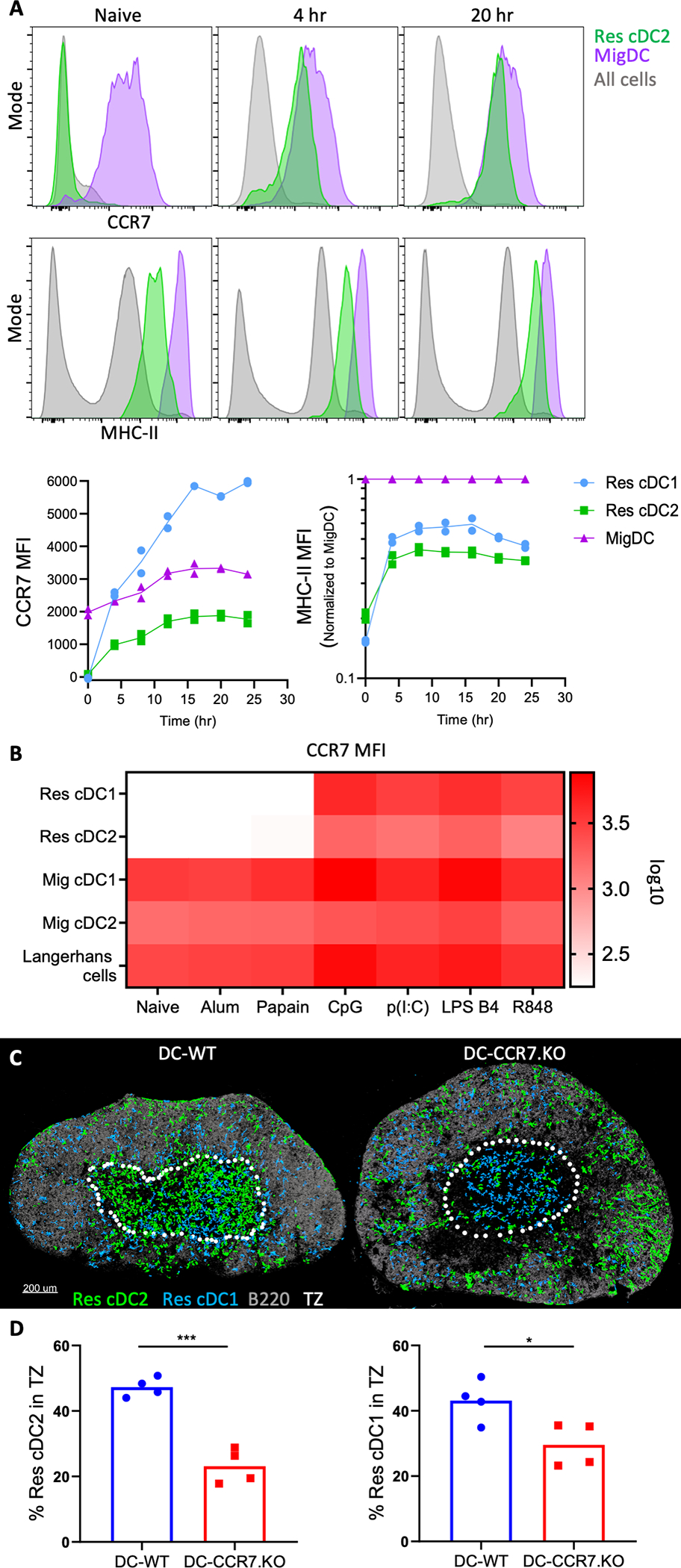
A) B6 mice were injected in the ears with CpG and dLNs were analyzed for cDC expression of MHC-II and CCR7 by flow cytometry [See fig. S3A for gating scheme]. MHC-II geometric mean fluorescence intensity (MFI) was normalized to MHC-II on migratory cDCs (Mig cDC). B) Heatmap depicting CCR7 geometric MFI on cDCs 24h after immunization with indicated adjuvants. Each square represents mean of n=4. C-D) DC-CCR7.KO and DC-WT mixed BMCs were administered DT and injected with CpG 1d later in the footpad. Histocytometry of popliteal dLNs 1.5d later. Data analyzed by unpaired t test with Welch’s correction. Data represent at least 2 independent experiments.
To test the role of CCR7 in intranodal relocalization of Res cDCs, we generated mixed bone marrow chimeras (BMCs) consisting of a 50:50 mix of CCR7.KO, or control WT, and Zbtb46-DTR BM (fig. S3C). This allowed specific depletion of Zbtb46-DTR donor cDCs using diphtheria toxin (DT), while leaving the CCR7.KO (DC-CCR7.KO) or control WT (DC-WT) cDCs intact (47, 48). This setup maintains normal LN architecture (fig. S3D), which is otherwise disrupted in CCR7 deficient animals (42). No notable changes in the total number of Res cDCs or the size of the LNs were detected between DC-CCR7.KO and DC-WT BMCs after DT administration, though as expected, migratory cDCs were absent in the DC-CCR7.KO LNs (fig. S3D). The DC-CCR7.KO and DC-WT mice were treated with DT and one day later immunized with CpG (fig. S3C). Disruption of CCR7 upregulation after immunization in DC-CCR7.KO was verified by flow cytometry (fig. S3E). As expected, Res cDCs in the control DC-WT BMCs efficiently relocalized into the deep TZ. In contrast, Res cDCs in DC-CCR7.KO BMCs were largely excluded from the TZ, and instead were frequently observed proximal to B cell follicles and medullary sinuses (Fig. 2, C and D, and fig. S3F). In contrast to cDCs, MOs still efficiently infiltrated the TZ in DC-CCR7.KO dLNs (fig. S3G), indicating that MO trafficking to and localization within dLNs was not interdependent with Res cDC repositioning. Mixed BMCs, consisting of a 50:50 mix of CCR7.KO and WT BM, also showed that CCR7.KO MOs did not display major defects in TZ infiltration (fig. S3H), confirming previous reports that MOs do not require CCR7 for migration into LNs (17, 20). Collectively, these data demonstrated that during Type-I inflammation, upregulation of CCR7 expression by Res cDCs leads to intranodal relocalization from the LN periphery into the deep TZ.
HEV-mediated recruitment of MOs into the dLNs
We next studied the mechanisms of MO recruitment during Type-I inflammation. MOs enter the circulation from the BM in a CCR2-dependent manner (23). Indeed, blockade of CCR2 using an anti-CCR2 antibody significantly reduced MO recruitment into dLNs after immunization (Fig. 3A), without affecting the total numbers and localization of Res cDCs (fig. S4A). Further trafficking of MOs into dLNs could have occured via two distinct routes: 1) initial MO recruitment into inflamed peripheral sites of immunization, followed by continued migration into dLNs via the afferent lymphatics, or 2) direct MO entry into dLNs from blood via HEVs (23). To investigate the former possibility, mice were administered CpG in the ear, and the site of immunization was surgically removed 1–2h later. This allows for drainage of administered TLR agonists to dLNs during the initial hour after immunization, while the site removal precludes the subsequent migration of cells from the immunization site. This is especially true for MOs, which require additional time to traffic to the inflamed skin (49, 50). We observed similar numbers of MOs in both site-intact and site-removed dLNs 24h post immunization, suggesting that peripheral tissue trafficking was not the primary mechanism of early MO recruitment (Fig. 3B). Nevertheless, surgical trauma associated with site removal could independently promote MO mobilization and confound the results. We thus injected mice with a blocking antibody against P-selectin glycoprotein ligand-1 (PSGL-1), an adhesion molecule predominantly involved in trafficking to inflamed peripheral tissues (51), and again immunized the animals in the ear, as well as performed site removal in some of the mice (Fig. 3B). PSGL-1 blockade, with or without site removal, led to a modest, but non-significant decrease in MO numbers in the dLNs, corroborating that early MO trafficking into dLNs was not predominantly occurring via the peripheral tissues. To test the possibility of direct migration of MOs via the HEVs, mice were treated with an anti-CD62L blocking antibody at the time of immunization, as CD62L is essential for HEV-mediated trafficking (52). CD62L blockade led to a dramatic loss in MO cellularity (Fig. 3B). Combined anti-PSGL-1 and anti-CD62L blockade caused more minor additional decreases (Fig. 3B). This indicated that although minor trafficking of MOs via the afferent lymphatics may occur (20), HEV-mediated entry was the dominant pathway of early MO recruitment into the dLNs during Type-I inflammation.
Figure 3: HEV-mediated recruitment of MOs into the dLNs.
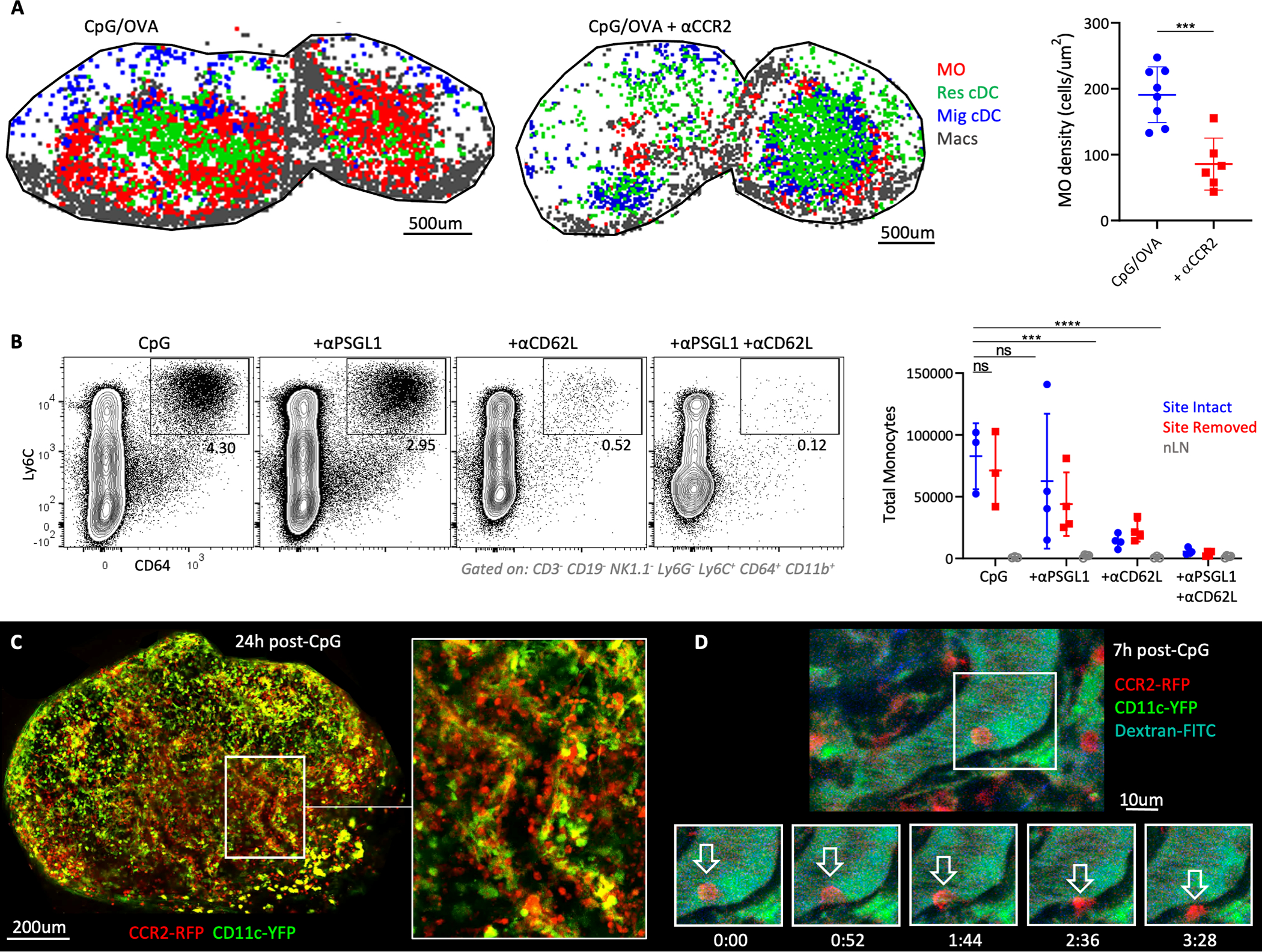
A) OT-II-transferred B6 mice were immunized with CpG plus OVA in ears and footpads. Some animals were treated with αCCR2. Myeloid cell localization (left), and MO density (right) in dLNs was assessed by histocytometry 1.75d later. Data analyzed by unpaired t test with Welch’s correction. Macs = macrophages. B) B6 mice were immunized with CpG in the ears. Some mice had the immunization site surgically removed 1–2h later, and/or were treated with αPSGL1 and/or αCD62L. Total MOs in dLNs and non-draining LNs (nLNs) were quantified 1d later by flow cytometry. Data analyzed via one-way ANOVA with Dunnett’s multiple comparisons test. C-D) CCR2-RFP x CD11c-YFP mice were administered CpG in the footpad and popliteal LNs were imaged by 2-photon intravital microscopy C) 24h or D) 7h after immunization. Representative time-lapse images demonstrate MO extravasation. Data represent at least 2 independent experiments.
To directly visualize the dynamics of MO trafficking, we performed 2-photon intravital microscopy of dual-reporter CCR2-RFP x CD11c-YFP mice, which allowed concurrent visualization of both MOs and cDCs, respectively. LN vasculature was also intravenously labeled with fluorescently-labeled anti-CD31 antibody or dextran, which do not alter cellular trafficking (53). As anticipated, unimmunized mice exhibited very few MOs (RFP+YFP-) in the vasculature, and the few detected cells rapidly moved through the vessels (movie S1). In contrast, markedly increased numbers of RFP+ MOs were observed in the dLN blood vessels starting 6h post immunization, and many cells exhibited slow rolling-type behavior and frequently extravasated into the parenchyma (Fig. 3, C and D, and movie S1). CCR2 can also be expressed by cDCs or effector T cells (54). However, the rolling and extravasating RFP+ cells were CD11c-YFP-negative. Additional intravenous labeling for CD90, a T cell specific molecule, also did not label most RFP+ cells (fig. S4B). Moreover, the timing of MO detection within the HEVs with intravital microscopy correlated with the kinetics data obtained by confocal imaging and flow cytometry (Fig. 1, C and D, and fig. S2D), collectively suggesting that Type-I inflammation elicits rapid MO recruitment into the dLNs via HEVs.
We next analyzed MO morphology and expression of CD11c and MHC-II, markers associated with MO differentiation. At early time points after immunization, MOs within the LN parenchyma exhibited a round morphology (fig. S4C), similar to cells within the HEV lumen (Fig. 3, C and D, and fig. S4B). Moreover, these recently arrived MOs were CD11c and MHC-II negative and were mainly localized in the TZ periphery (fig. S4C). Over time, MOs progressively displayed reduced sphericity and acquired a dendritic-like appearance, expressed CD11c, and migrated into the deeper LN paracortex (fig. S4, C and D). Flow cytometry confirmed that the early infiltrating MOs phenocopied intravenously-labeled blood MOs, displaying minimal surface levels of CD11c and MHC-II (fig. S4E), and that with time they gradually increased expression of both markers (fig. S4, F–H). Together, these data indicated that MOs were recruited into dLNs via local vasculature and underwent differentiation over time.
Antigen presentation and initiation of T cell priming by repositioned Res cDCs
We next studied the functional roles of Res cDCs and MOs in the generation of adaptive immunity. Considering that Res cDC2s specialize in MHC-II presentation, while MOs have delayed kinetics of MHC-II expression (fig. S4, F–H), we hypothesized that early antigen presentation was not equivalent between these populations. To test this, mice were immunized with CpG plus EαGFP, a model protein which allows detection of antigen uptake via GFP fluorescence and MHC-II presentation using a YAe antibody, specific for the Eα peptide-MHC-II complex (49). One day post immunization, Res cDC2s and migratory cDC2s were the dominant cell types that captured and presented antigen (Fig. 4A). While some MOs had detectable GFP fluorescence, they did not stain positive with the YAe antibody, indicating minimal MHC-II antigen processing (Fig. 4A). Similar results were seen 2 days post immunization (fig. S5A). Notably, a minor fraction of CD64+ GFP+YAe+ cells were observed (fig. S5A), albeit these cells were phenotypically distinct from inflammatory MOs, expressing lower levels of Ly6C and higher levels of MHC-II (fig. S5A). A similar population was found in steady state LNs, and their numbers demonstrated a much more modest increase following vaccination as compared to the Ly6CHIGH MOs (fig. S5B), indicating that they are distinct from inflammatory MOs and may represent tissue-derived cells or inflammatory cDCs (20, 23, 55, 56). These results indicated that cDCs, but not the early-recruited inflammatory MOs, were the dominant antigen presenting cells in dLNs after Type-I agonist immunization.
Figure 4. Induction of T cell priming and clonal expansion via Res cDC repositioning.
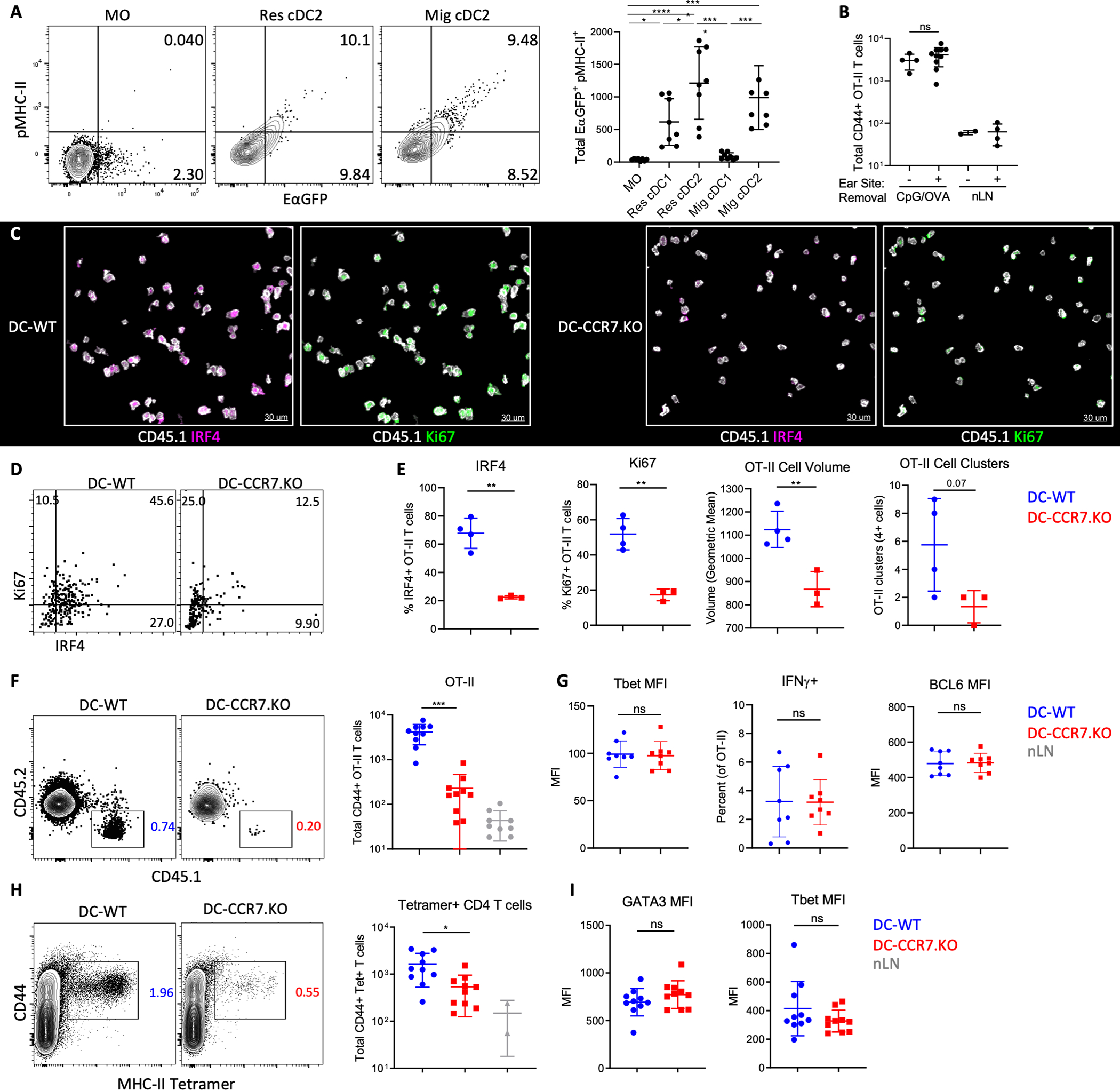
A) B6 mice were immunized with CpG plus EαGFP in both ears and analyzed by flow cytometry for GFP fluorescence and peptide-MHC-II staining (YAe) 1d later. Data analyzed by one-way ANOVA with Tukey’s multiple comparison test. B) DC-WT mixed BMCs were transferred with 105 OT-II T cells and 1d later ears were immunized with CpG plus OVA. Some mice also had site removal 1–2h later. 4d post-immunization, LNs were analyzed by flow cytometry for T cell cellularity. C-G) DC-WT and DC-CCR7.KO mixed BMCs were transferred with 106 (C-D) or 105 (F-G) OT-II T cells, treated with DT, immunized with CpG plus OVA in both ears, and had site removal. dLNs were assessed (C-E) 1.5d later by histocytometry or (F-G) 4d later by flow cytometry. C) Representative areas in the TZ, with the presented IRF4 and Ki67 signal masked outside of CD45.1+ OT-II T cells for visual clarity. D) Representative gating and E) quantification of OT-II T cell expression of IRF4 and Ki67, as well as average T cell volume and number of OT-II clusters (>4 cells in direct contact) per section. F) Total CD44+ OT-II T cells and (G) Tbet and Bcl6 geometric MFI, and percentage of IFNγ+ after restimulation. G-H) DT-treated DC-CCR7.KO and DC-WT mixed BMCs were immunized with CpG plus GPC in the ear, followed by site removal 2h later. (H) Cellularity and (I) differentiation of CD44+ GPC-tetramer binding T cells were quantified in dLNs 7d post-immunization. B, E-I) Data analyzed by unpaired t test with Welch’s correction. Data represent at least two independent experiments.
To test how the repositioning of antigen-presenting Res cDCs influences T cell activation, we adoptively transferred 1×106 naïve, OVA-specific CD4 OT-II T cells into DC-WT or DC-CCR7.KO mixed BMCs and immunized these mice with OVA plus CpG. dLNs were analyzed 1.5–2 days after, just prior to extensive T cell proliferation. Although migratory cDCs presented antigens on MHC (Fig. 4A and fig. S5A), removal of the immunization site 1–2h after vaccination recapitulated previous findings that migratory cDCs were largely dispensable for CD4 T cell responses (Fig. 4B). Nevertheless, we performed site removal in all animals, as this restricted effects of CCR7 deficiency to intranodal Res cDC repositioning and minimized additional changes in migratory cDC trafficking. As expected, OT-II T cells in DC-WT animals were robustly activated, as demonstrated by T cell clustering, enlarged cell volume, as well as interferon regulatory factor 4 (IRF4) and Ki67 expression, indicating TCR engagement and proliferation, respectively (Fig. 4, C–E) (57, 58). In contrast, early OT-II T cell activation was drastically impaired in the DC-CCR7.KO mice (Fig. 4, C–E). DC-CCR7.KO mice also elicited substantially reduced clonal expansion of OT-II T cells 4 days after immunization, when using a more physiological (105) cell transfer number (Fig. 4F). While the numbers of expanded T cells were markedly different between DC-WT and DC-CCR7.KO mice, there were no major differences in their expression of T-box transcription factor (Tbet) or B-cell lymphoma 6 (BCL6), or in their ability to produce interferon gamma (IFNγ) post restimulation (Fig. 4G and fig. S5C). This indicated that the few CD4 T cells that were activated in the DC-CCR7.KO settings underwent relatively normal effector differentiation, albeit additional differences cannot be ruled out. We also examined polyclonal CD4 T cell responses by immunizing mice with lymphocytic choriomeningitis virus (LCMV) -derived recombinant glycoprotein complex (GPC) and CpG. Seven days post immunization and site removal, large numbers of GPC MHC-II tetramer positive CD44+ CD4 T cells were detected in DC-WT but not DC-CCR7.KO mice (Fig. 4H). T cells in both expressed equivalent levels of Tbet and Gata binding protein 3 (Gata3) proteins (Fig. 4I).
Given that the Res cDC1 also upregulated CCR7 to move into the deep TZ (Fig. 1 and 2), and these cells specialize in cross-presentation, we also examined CD8 T cell priming using OVA-specific CD8 OT-I T cells. Site removal again did not impact CD8 T cell expansion, indicating minimal contribution of migratory cDCs (fig. S5D). Similar to CD4 T cells, we observed a drastic reduction in OT-I T cell clustering and expression of activation markers in DC-CCR7.KO animals 1.5d after immunization (fig. S5, E and F). A significant reduction in OT-I T cell expansion in DC-CCR7.KOs was also observed 4d after immunization (fig. S5G). The few responding OT-I T cells in DC-CCR7.KO mice again displayed no major differences in expression of Tbet and TCF1, nor in their ability to produce IFNγ post restimulation (fig. S5H). Together, these data demonstrated that CCR7-mediated relocalization of both Res cDC2s and cDC1s into the TZ during Type-I inflammation controls the activation and clonal expansion of both CD4 and CD8 T cells.
Production of IL-12 by TZ-localized monocytes during Type-I inflammation
Activated MOs can express inflammatory cytokines, such as IL-12, although where in LNs MOs generate this cytokine and whether this coincides with early T cell differentiation is not known (17, 18, 59). To this end, we first looked for expression of IL-12p40 by various myeloid cell subsets using flow cytometry, as verified using IL-12.KO BMC mice (Fig. S6, A and B). We found that 1 day post CpG immunization, some of the MOs produced copious quantities of IL-12p40 and this became more apparent on day 2 (Fig. 5, A and B, and fig. S6, A–C). As previously described, migratory and Res cDC1 cells also produced large amounts of IL-12p40 (60–62). While the frequency of IL-12-positive MOs was relatively modest compared to cDCs, due to substantial increases in cellularity, MOs represented the most numerically dominant IL-12-producing cell type within the dLNs.
Figure 5: IL-12 production by TZ-localized inflammatory MOs.

A-B) B6 mice were given CpG plus EαGFP in both ears and dLNs were analyzed A) 1d or B) 2d later by flow cytometry. Percent IL-12p40+ (left) and total IL-12p40+ cells (right) is shown. C-E) OT-I-transferred B6 mice were immunized with CpG plus OVA in ears and footpads. dLNs were analyzed by histocytometry 1.75d later. C) Representative images demonstrating IL-12p40 expressing CD64+ MOs (arrows), and D) positional analysis of all myeloid cells (left) and IL-12p40+ myeloid cells (right). Dashed line demarcates the TZ. E) Quantification of total IL-12p40+ cells within the TZ for the indicated subsets. F) OT-I transferred-B6 mice were immunized with indicated adjuvants plus OVA. dLNs were analyzed 1.5d later by histocytometry for total IL-12p40+ myeloid cells within the TZ. For each group, n=3–6. G) B6 mice were infected in the footpad with 100 pfu of West Nile virus-TX. dLNs were analyzed by histocytometry 24h later for total IL-12p40+ cells within the TZ. H) OT-I transferred B6 mice were immunized with CpG plus OVA in ears and footpads. Some mice were also treated with αCCR2. Histocytometry analysis of total IL-12p40+ cells within the TZ of dLNs 1.75d after immunization. I) B6 mice were immunized with CpG in ears and footpads and dLNs were harvested 2d later. Some mice were treated with αCCR2. ELISA quantification of total IL-12p70 signal in dLNs. Data analyzed via paired t test with Welch’s correction. Individual points represent means from four independent experiments. Lines connect experimental means, with the αCCR2 group normalized to the CpG group. Data in A,B,E,G) analyzed by one-way ANOVA and Tukey’s multiple comparisons test. Linked points in E and G indicate data from the same LNs. All data represent at least 2 independent experiments.
We next used histocytometry to visualize where IL-12 producing cells were located. IL-12p40 staining, as verified using IL-12.KO BMC mice, was detectable in small quantities in steady state LNs, primarily associated with migratory cDC1 and some Res cDC1 (fig. S6, D and E), as described (63, 64). A substantial increase in MO-associated IL-12p40 signal was observed in dLNs 1.75 days after CpG immunization and was localized within the TZ (Fig. 5C and D, and fig. S6E). Of note, the frequency of IL-12p40 positive cells appeared higher by imaging vs. flow cytometry. This may represent difficulties in cellular extraction, potential cell death, or degranulation during enzymatic tissue digestion for flow cytometry (10, 65, 66). Quantification of the imaging data revealed that MOs were a dominant IL-12p40-producing cell population in the TZ after immunization with multiple TLR agonists and following West Nile virus infection (Fig. 5. E–G). Blockade of MO trafficking with anti-CCR2 resulted in substantial loss of MO-derived, but not cDC-derived, IL-12p40 in the TZ (Fig. 5H). In contrast, little MO-associated IL-12p40 signal was seen within the TZ during Type-II inflammation (fig. S6F). Since IL-12p40 does not necessarily reflect the immunologically relevant IL-12p70 (IL-12p40 and IL-12p35 heterodimer), we examined IL-12p70 levels within dLNs in the presence or absence of MOs with ELISA. IL-12p70 was markedly increased 2 days after CpG immunization, reflective of adjuvant induced inflammation (fig. S6G). Blockade of MO trafficking led to a partial and highly significant decrease in IL-12p70 signal (Fig. 5I and fig. S6G). Collectively, these data indicated that in addition to the conventional contributions by cDCs, MOs provide a substantial source of IL-12p70 in the TZ of dLNs during Type-I inflammation.
Association of early effector T cells with MO-rich LN microenvironments
IL-12 plays a critical role in T cell differentiation (67, 68). Given the spatiotemporal coalescence of antigen-presenting Res cDCs and IL-12 expressing MOs in the TZ of dLNs, we hypothesized that both cell types may functionally cooperate to modulate effector T cell responses. Supporting this notion, 1.5–1.75d after OVA plus CpG immunization, we observed large numbers of activated OT-I and OT-II T cells embedded within a dense, interwoven network of both Res cDCs and CD11c-expressing CD64+ MOs (fig. S7, A and B). To quantify these observations, we calculated the spatial correlation of different immune cells with respect to one another using CytoMAP (25). Use of 30 μm radius spatial neighborhoods for this also accommodated for potential effects of cellular motility or cytokine diffusion which may occur at these time points (69). This analysis confirmed that the spatial distribution of activated OT-I and OT-II T cells was positively correlated with the location of both Res cDC and MOs, but not migratory cDCs or macrophages (fig. S7, C and D), indicating preferential cell-cell interactions. The positive spatial correlation of activated T cells with Res cDC was reduced in DC-CCR7.KO mixed BMCs as compared to the DC-WT counterparts, reflective of poor Res cDC repositioning in these settings (Fig. S7E). In contrast, OT-I T cells still maintained a positive correlation with MOs in DC-CCR7.KOs (Fig. S7E), corroborating the findings that MOs still efficiently entered the T zone irrespective of Res cDC relocalization (fig. S3G).
A closer examination of dLNs across multiple experiments also revealed a nonuniform, polarized distribution of MOs across the tissues, being particularly noticeable at later time points and in larger, bilobular auricular and brachial dLNs (Fig. 1A, 5C, 5D, and 6A, and fig. S2I and S7A). In contrast to MOs, both lobes in these polarized dLNs displayed a more homogeneous centralization of Res cDCs. The polarization of MOs likely corresponds to non-uniform TLR agonist biodistribution and inflammation across the dLNs after afferent drainage (7). We hypothesized that this myeloid heterogeneity could impact the local signals that T cells encounter during priming. To investigate this, we analyzed the expression of various markers associated with effector differentiation in responding OT-I T cells in relation to the different myeloid populations (Fig. 6A). Considerable heterogeneity in expression of Tbet, TCF1, and CD25 by the activated (Ki67+) OT-I T cells was observed, suggesting early bifurcation of responses (Fig. 6, A and B). Importantly, we observed distinct spatial distribution patterns for the early effector subpopulations, with the more differentiated Tbet+TCF1-CD25+ OT-I T cells preferentially localizing in regions heavily infiltrated by MOs, and with the less differentiated Tbet-TCF1+ OT-I T cells predominantly distributed in areas with more Res cDCs and fewer MOs (Fig. 6C). Correlation analysis of multiple tissues confirmed these observations, demonstrating that the more differentiated effector OT-I T cells (Tbet+TCF1-) were spatially correlated with (i.e. more proximal to) MOs, while the less differentiated T cells (Tbet+TCF1-) were correlated with both MOs and Res cDCs (fig. S7F). We examined the relative myeloid cell composition within regions that were dominantly populated by either the Tbet+TCF1- or Tbet-TCF1+ OT-I T cells (fig. S7G). Examination of larger regions also further accommodated for effects of T cell motility across the tissues. While MOs and Res cDCs, and in particular cDC1s, were found in relatively equal proportions in Tbet-TCF1+ OT-I regions, MOs greatly outnumbered other myeloid cells in regions populated by Tbet+TCF1- early effector T cells (Fig. 6D and fig. S7H). Notably, there was no difference between the regions in the frequency of OT-I T cells expressing IRF4 and phosphorylation of the S6 ribosomal protein (pS6), suggesting that early TCR engagement was more homogenous across the compartments (Fig. 6E, fig. S7I). Polarized distribution of Tbet+TCF1- and Tbet-TCF1+ OT-I T cells was also observed at later time points, when T cells begin to undergo extensive proliferation and start to leave the tissues (fig. S7J). In addition, we found a similar spatial heterogeneity of OT-I effector cells after administration of OVA plus p(I:C), with marked segregation of TbetHI and TbetLO T cells across the polarized dLNs (Fig. 6F). TbetHI regions were again dominantly populated by MOs, while the TbetLO regions were more equally populated by cDCs and MOs (Fig. 6, G and H). Collectively, these data indicate that distinct myeloid microenvironments in dLNs, as determined by the local density of MOs, are associated with distinct patterns of CD8 T cell effector differentiation.
Figure 6: Regulation of T cell response heterogeneity by myeloid cell microenvironments.
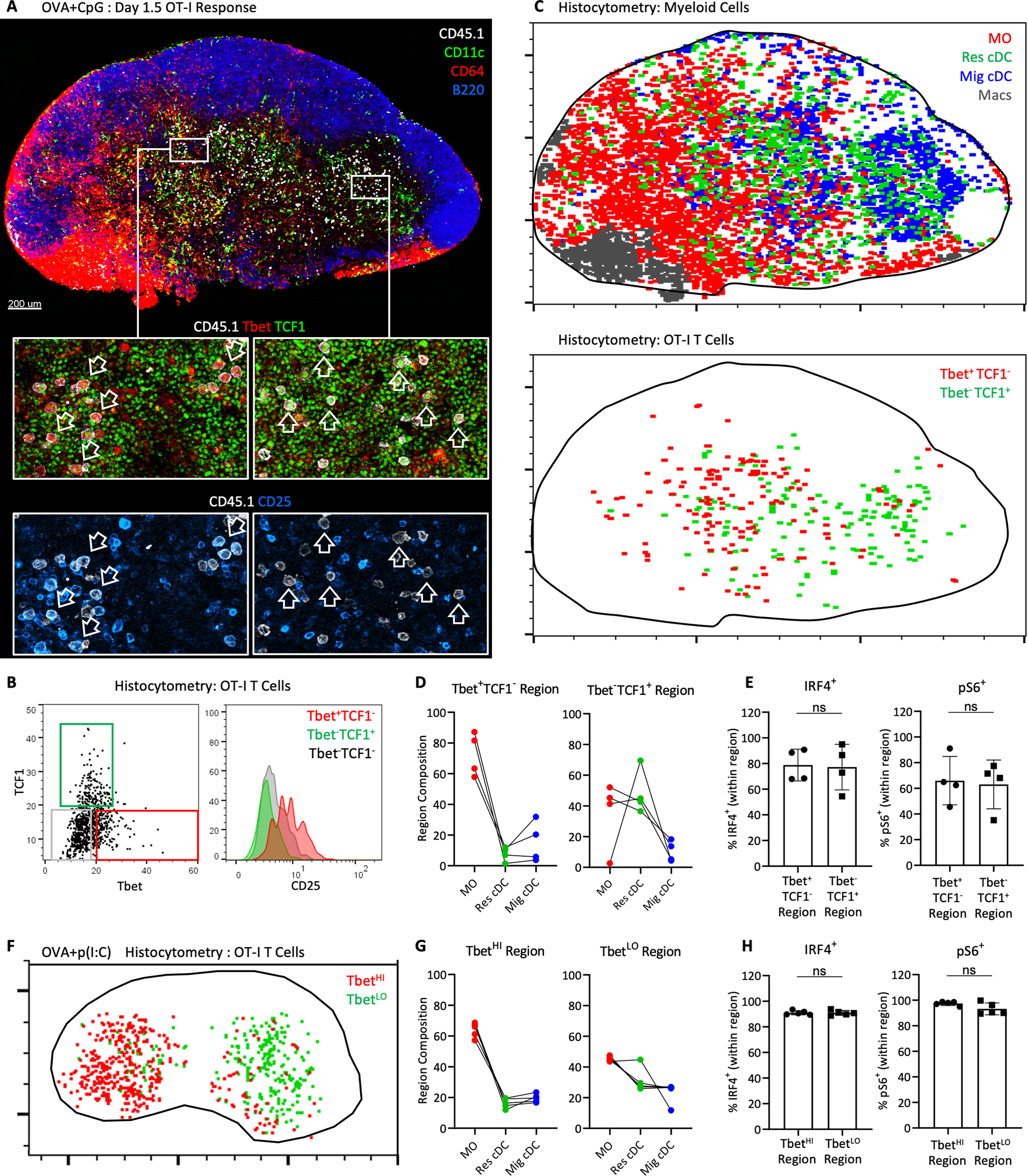
A-H) B6 mice were transferred with 106 OT-I T cells and immunized with (A-E) CpG plus OVA in ears and footpads or (F-H) p(I:C) plus OVA in ears. Auricular and brachial dLNs were analyzed 1.5–1.75d later by histocytometry. A) Representative dLN image demonstrating staining for various myeloid cells (top), as well as markers of T cell differentiation (bottom). Left vs. right zoom-in panels demonstrate differential staining of CD45.1+ OT-I cells for Tbet, TCF1 and CD25. B) Representative histocytometry gating of OT-I cells based on Tbet and TCF1 expression, and the corresponding CD25 expression. C) Histocytometry analysis of the image from (A), showing the distribution of myeloid cell populations (top) or OT-I cell subsets (bottom). D-E) dLNs were subdivided into regions dominantly associated with either Tbet+TCF1- or Tbet-TCF1+ Ki67+ OT-I T cells [representative spatial gating shown in fig. S7G]. D) Composition of myeloid cells and E) percent of IRF4+ and pS6+ OT-I cells within each region. F) Representative positional analysis of TbetHI and TbetLO Ki67+ OT-I cells in dLN after p(I:C) plus OVA immunization. G-H) dLNs were subdivided into TbetHI or TbetLO regions. G) Composition of myeloid cells, and H) percent of IRF4+ and pS6+ OT-I cells within each region. Data analyzed by unpaired t test with Welch’s correction. In D and G, linked points indicate data from the same LNs. All data represent at least 2 independent experiments.
MO-dependent differentiation of effector T cells
To test the contribution of MOs to CD4 and CD8 T cell responses, we transferred OT-I and OT-II T cells, immunized the mice with OVA plus CpG, and in some of the animals also blocked MO recruitment with anti-CCR2. In these experiments, we also injected a higher dose of OVA (10ug) to provide MOs with maximal opportunity for antigen presentation. Nevertheless, minimal impact of MO blockade was seen on T cell expansion and expression of Ki67, IRF4 or pS6, indicating that MOs play a negligible role in MHC-I and MHC-II antigen presentation (fig. S8, A and B). In contrast, we observed a drastic reduction in the spatial polarization and generation of TbetHITCF1LO OT-I cells in dLNs of mice that received CCR2-blocking antibody (Fig. 7A), suggesting a major role for MOs in driving localized effector differentiation of CD8 T cells. Similar decreases in the generation of Tbet+ effector OT-II CD4 T cells were seen following CCR2 blockade (fig. S8B). Reduced CD8 and CD4 T cell differentiation, but not clonal expansion, was also observed on day 4 (Fig. 7, B and C). Similarly, blockade of MO trafficking resulted in marked attenuation in Tbet and CXCR3 expression, as well as IFNg production upon restimulation by polyclonal GPC-specific CD4 T cells after GPC plus CpG immunization (fig. S8C).
Figure 7: Optimization of CD4 and CD8 T cell effector differentiation by MOs.
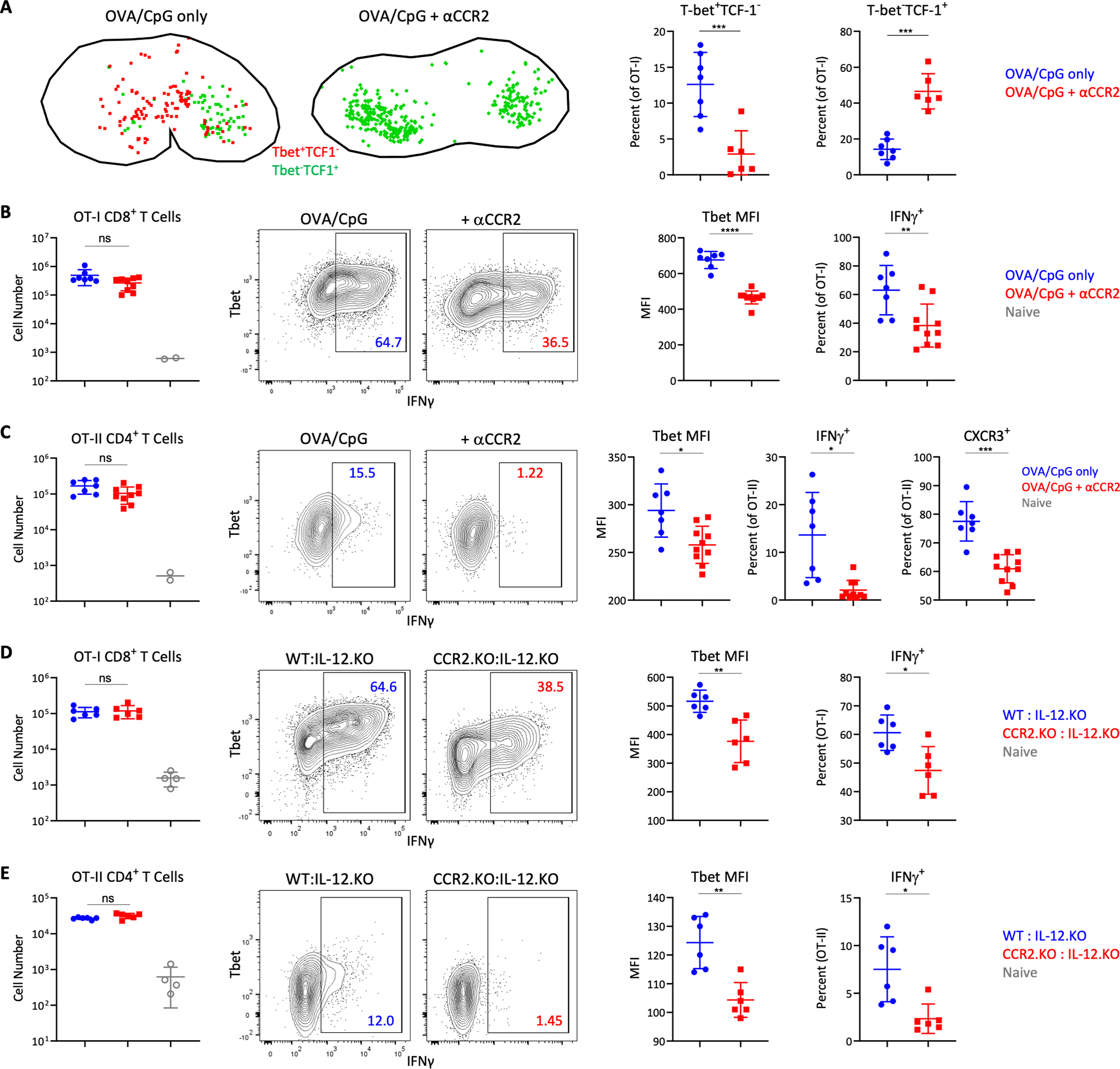
A) B6 mice were transferred with 106 OT-I T cells and immunized with CpG plus OVA in ears and footpads. Some animals were treated with αCCR2. dLNs were analyzed 1.75d later by histocytometry for location (left) and frequency (right) of Tbet+TCF1- and Tbet-TCF1+ OT-I cells. B-C) B6 mice were co-transferred with 105 OT-I and OT-II T cells and immunized with CpG plus OVA in the ears. Some mice were treated with αCCR2. dLNs were analyzed 4d later by flow cytometry. Total CD44+ cells, Tbet geometric MFI, percentage of IFNγ+ Τ cells following restimulation, and percent CXCR3+ for B) OT-I and C) OT-II cells. D-E) WT:IL-12p40.KO and CCR2.KO:IL-12p40.KO mixed BMCs were co-transferred with 105 OT-I and OT-II T cells and immunized with CpG plus OVA 1d later. D) OT-I and E) OT-II cellularity and differentiation in dLNs was assessed by flow cytometry 4d later. Data analyzed by unpaired t test with Welch’s correction. Data represent at least 2 independent experiments.
Finally, we tested the direct contribution of IL-12 production by MOs using two distinct mixed BMC models. In the former, we reconstituted irradiated B6 recipients with a 50:50 mix of CCR2-DTR BM and either WT or IL-12.KO BM (fig. S8, D and E). Ablation of MOs using DT administration allowed examination of T cell responses in the presence of MOs derived only from either the WT or the IL-12.KO donors. In the latter model, B6 recipients were reconstituted with IL-12.KO BM together with either WT or CCR2.KO BM, which allowed testing the role of MO-derived IL-12 in the absence of DT-mediated cell toxicity or possible off-target effects (Fig. 7, D and E). In both models, no major changes in T cell expansion were observed 4 days after immunization (Fig. 7, D and E, and fig. S8, D and E). In contrast, we observed significantly reduced OT-I and OT-II T cell effector differentiation and IFNγ production upon restimulation, indicating that MO-derived IL-12 directly contributes to effector T cell programming. Similar results were seen for polyclonal T cell responses (fig. S8F), as well as after p(I:C) plus OVA immunization (fig. S8G). Collectively, these findings demonstrated that MOs provided a localized source of IL-12 to T cells to drive optimized effector differentiation during Type-I inflammation.
Discussion
Here, we defined several critical features of myeloid cell organization that shape T cell responses during Type-I but not Type-II inflammation (fig. S9). We found rapid repositioning of Res cDCs from the LN periphery and concomitant influx of blood-derived inflammatory MOs into the deep TZ of dLNs. The repositioning of Res cDCs allowed for efficient MHC presentation of draining antigens in the appropriate anatomical compartment to elicit early T cell activation and differentiation. MO influx on the other hand provided an additional source of signal-3 cytokines to responding T cells, promoting generation of fully differentiated effector responses. Together, these distinct innate cell types functionally cooperated with one another to promote the generation of optimized adaptive immunity. We found extensive heterogeneity in MO infiltration across the TZ, which generates distinct microenvironments with varied MO and cDC abundance. Priming of T cells within these microenvironments influenced the exposure of T cells to different activating stimuli, such as MO-produced IL-12, and promoted localized generation of different effector subsets. Thus, inflammatory innate microenvironments within dLNs regulated the overall quality and heterogeneity of the resulting T cell responses.
The finding that LN Res cDCs respond to inflammatory stimuli and use CCR7 to reposition within the dLNs is consistent with the migration programs of peripheral tissue and splenic cDC subsets (5, 36, 37, 44). Our data connect these observations, demonstrating that cDCs across multiple different tissues utilize the CCR7 axis during inflammation for migration into the most proximal T cell compartments. These observations also highlight caveats in studies using CCR7.KO animals to dissect the role of migratory cDCs, as we showed that the same pathway was utilized by LN Res cDCs for intra-nodal repositioning. Similar considerations likely apply to other models in which the migratory capacity of multiple cDC populations may be affected (44, 70–72). Our findings do not argue against the role of migratory cDCs in T cell immunity, as these cells are critical during highly tropic infections (73, 74), and for promoting Th2 and TfH responses (71, 75, 76). Migratory cDCs and pDCs can also cargo antigens to Res cDCs (3, 4, 14, 77–79). Indeed, we did not find efficient Res cDC repositioning or MO recruitment in Type-II inflammatory conditions, supporting the importance of migratory cDCs in these settings. Instead, our observations indicated that in conditions of ample antigen drainage after vaccination or during certain microbial infections, Res cDCs are exceptionally potent at inducing T cell activation (6, 7, 27, 49, 80). A better understanding of the distinct contributions of peripheral vs. Res cDC populations in different immunologic settings will aid in vaccine design (81). Of note, the responses seen for LN-res cDC1s, cDC2s and MOs occurred in response to multiple Type-I agonists regardless of TLR expression on these populations (82–84). This suggests that secondary inflammatory mediators, such as IFN-I or TNFα, may be involved (50, 85–87).
In addition, we provided substantial insight into the organization and role of MOs during Type-I inflammation. As early as 6h after immunization, MOs entered the dLNs, primarily via the local HEVs instead of the afferent lymphatics, and rapidly occupied various LN regions, including the medulla, B cell follicles and parts of the TZ. CCR2 appears critical for this process and it is likely that CCR2 ligands in both the BM and inflamed dLNs are involved (17, 22, 50). In contrast, CCR7 was not essential for MO trafficking to the dLNs during inflammation, and if anything, previous studies have reported increased numbers in CCR7.KO mice (17, 20). There was substantial heterogeneity in MO infiltration across the TZ, likely driven by localized afferent drainage and polarized dispersal of inflammatory stimuli (7). This heterogeneity generated distinct microenvironments in dLNs which were directly associated with different effector CD8 T cell subtypes, with more differentiated effector T cells primarily observed within MO-rich regions, and less differentiated cells found in cDC1-dominated regions with lower MO abundance. Divergent T cell programming appeared to be at least in part MO-derived IL-12 mediated, as blockade of MO trafficking or IL-12 production resulted in loss of effector T cell spatial polarity and reduced differentiation. Thus, MOs generated highly inflammatory microenvironments in LNs to promote the generation of fully differentiated effector T cells in a localized fashion. Our findings do not contradict previous observations that cDC1s can produce IL-12 and promote effector T cell differentiation (60, 61, 63, 64, 88, 89). Indeed, we observed potent IL-12 production by Res and migratory cDC1s, and MO blockade suppressed but did not abolish effector T cell responses. Instead, our data suggested that cDCs provide an early source of IL-12 and this is further amplified by the IL-12 derived from MOs in a localized fashion. Therefore, the collaborative action of both cDCs and MOs was important for optimized generation of adaptive immunity.
It is important to note that our spatial analysis was largely limited to use of tissue sections from individual timepoints, and that generation of effector T cells in MO-rich regions could be in part influenced by cellular repositioning after initial priming. This may particularly be true for CD4 T cell responses, which have distinct kinetics of effector programming and additional chemotactic properties as compared to CD8 T cells (47, 90, 91). Statistical region analysis, as performed here, minimized this issue, but the development of next generation technologies capable of comprehensive spatiotemporal tracking of T cell fates across entire organs and over time is necessary. In addition, our study did not thoroughly investigate all possible immunologic settings. Whether similar processes occur in other conditions, such as autoimmunity or cancer, and how distinct myeloid cell microenvironments influence T cell priming remain to be investigated. Our findings do highlight the concept that rational vaccine strategies to simultaneously target multiple innate cell populations, including MOs, should be explored (92, 93). MOs are a highly plastic innate cell population capable of a wide spectrum of functions (94). Given their abundance and ability to infiltrate most tissues, strategies to modulate MO trafficking and function may prove of value in a variety of settings. In conclusion, our findings revealed fundamental features of innate-adaptive cell crosstalk during Type-I inflammation, demonstrating that the spatial coordination and functional cooperation of innate cells within dLNs was critical for the generation and shaping of adaptive responses to vaccines and infections.
Materials and Methods
Study Design
The aim of this study was to investigate the role of innate cell LN microenvironments in shaping T cell immunity in mouse models of vaccination and infection. We performed multi-modal analysis with quantitative confocal imaging, intravital 2-photon microscopy and flow cytometry to examine the dynamic changes in spatial organization for various myeloid cell populations during inflammation, as well as their association with activated T cells. In vivo adoptive transfer assays, mixed BMC systems, and antibody blocking studies were utilized to investigate the role of cDCs and MOs in the generation of T cell responses. Detailed methods are described below and in the Supplementary Materials.
Mice
C57BL/6J, B6.129(Cg)-Ccr2tm2.1Ifc/J (CCR2-RFP), B6.Cg-Tg(Itgax-Venus)1Mnz/J (CD11c-YFP), B6(Cg)-Tyrc−2J/J (B6 albino), B6.Cg-Zbtb46tm4.1(HBEGF)MnzTyrc−2J/J (Zbtb46-DTR), B6.129P2(C)-Ccr7tm1Rfor/J (CCR7.KO), B6.129S1-Il12btm1Jm/J (IL-12p40.KO), and B6.129S4-Ccr2tm1Ifc/J (CCR2.KO) mouse strains were obtained from The Jackson Laboratory. CD45.1+ B6.Cg-Tg(TcraTcrb)425Cbn/J (OT-II), CD45.1+ C57BL/6-Tg(TcraTcrb)1100Mjb/J (OT-I), and B6.SJL-PtprcaPepcb/BoyCrl (CD45.1+) were obtained either from donating investigators (Dr. Pamela J. Fink, University of Washington) or Charles River. CD11c-YFP animals were crossed with B6 albino mice to homozygosity, and next crossed to CCR2-RFP mice to generate a CD11c-YFP x CCR2-RFPHetrozygous dual reporter mice. CCR2-DTR mice were obtained from donating investigators (Dr. Steven F. Ziegler, Benaroya Research Institute) and with approval from the originating investigators (Drs. Tobias M. Hohl and Eric G. Pamer, Memorial Sloan-Kettering Cancer Center) (95). 6–10 week-old male and female mice were kept in specific pathogen–free conditions at an Association for Assessment and Accreditation of Laboratory Animal Care–accredited animal facility at the University of Washington, South Lake Union campus. All procedures were approved by the University of Washington Institutional Animal Care and Use Committee.
Bone Marrow Chimeras and Adoptive Transfers
For BMCs, C57BL/6 mice were exposed twice to 600 rads of gamma irradiation from a cesium source separated by a 3-hour rest period, and injected with 2×106 donor BM cells intravenously the same day. Mice were kept on neomycin for 3 weeks or Baytril for 2 weeks, and used experimentally at least 6 weeks after transplant.
For CCR7 BMCs, irradiated recipients were reconstituted with a 50:50 mix of CCR7.KO or C57BL/6 and Zbtb46-DTR BM. For IL-12p40.KOs, irradiated recipients were reconstituted with IL-12p40.KO BM. For IL-12p40.KO:CCR2-DTR mixed BMCs, a 50:50 mix of IL-12p40.KO or C57BL/6 and CCR2-DTR BM was used for reconstitution. For CCR2.KO:IL-12p40.KO mixed BMCs, a 50:50 mix of CCR2.KO or C57BL/6 and IL-12p40.KO BM was used for reconstitution. For CCR7.KO:WT mixed BMCs, irradiated recipients were reconstituted with a 50:50 mix of congenically marked CCR7.KO BM and C57BL/6 BM. In experiments using DTR BMCs, mice were administered with DT in phosphate-buffered saline (PBS) at 20 ng / g bodyweight intraperitoneally every 2d.
For adoptive transfers, naïve CD45.1+ OT-II or CD45.1+ OT-I T cells were isolated from LNs and spleens using either the Naïve CD4+ or CD8+ T cell isolation kits (Miltenyi Biotec), respectively. Average purity of OT-II and OT-I cells was about 75% and 90%, respectively. The indicated number of cells were transferred into CD45.2+ C57BL/6 hosts intravenously.
Immunizations and Infections
The following adjuvants and amounts per immunization site were used: 20 μg CpG ODN 1668 (AdipoGen), 10 μg R848 (Invivogen), 10–20 μg poly(I:C) or ‘p(I:C)’ (Amersham), 10 μg Lipopolysaccharides (LPS) from Escherichia coli O111:B4 (Sigma-Aldrich), 20 μg flagellin (FliC) from Salmonella enterica Typhimurium (kind gift from Kelly D. Smith, University of Washington) (96), Alhydrogel or ‘Alum’ (Invivogen) diluted 1:2 with PBS, 50 μg papain (Sigma-Aldrich), and Addavax (Invivogen) diluted 1:2 with PBS. Except for Alum and Addavax, the indicated adjuvants were mixed with PBS (20 μl total volume). All adjuvants were injected in either the front or hind footpads, or intradermally in the ear pinnae (brachial, popliteal, or auricular dLNs, respectively) as indicated. In indicated studies, the immunized ear pinnae were surgically removed 1–2 hours post immunization.
In some studies, ~1.2×107 heat-killed Escherichia coli BioParticles (ThermoFisher) in 20 μl of PBS were injected into the ear. For West Nile Virus infections, 100 plaque forming units (pfu) of West Nile virus (Texas strain) in 20 μl of PBS were injected into the footpad. For Nippostrongylus brasiliensis infections, infectious third-stage larvae (L3) were raised and maintained as described (97). Mice were infected subcutaneously at the base of the tail (dLN, inguinal) with 500 Nippostrongylus brasiliensis L3.
For in vivo antibody blocking studies, 100 μg anti-CD62L (clone Mel-14, BioXCell) and/or 100 μg anti-PSGL-1 (clone 4RA10, BioXCell) were injected intraperitoneally at the time of immunization. In some experiments, 20 μg of MC-21 CCR2-blocking antibody (kind gift from Matthias Mack, University Hospital Regensburg) was injected intraperitoneally at the time of immunization, and administered daily for up to 3 days after (98). Intravascular labeling was achieved by injecting 2.5 μg of CD45.2-PE antibody 10 min prior to animal euthanasia (99). OVA immunizations used endotoxin-free OVA (InvivoGen), either 1 or 10 μg, as indicated, along with 20 μg of CpG or p(I:C). In some studies, 1 ug recombinant LCMV pre-glycoprotein GP complex (GPC, 10–90aa) (MyBioSource) along with 20 μg of CpG was injected intradermally in the ear pinnae. For antigen presentation studies, 10 μg of LPS-free EαGFP (gift from Marc K. Jenkins, University of Minnesota) plus 20 μg of CpG were injected intradermally in the ear pinnae.
ELISA
Mouse IL-12 p70 Quantikine ELISA kit (R&D Systems) was used on concentrated LN supernatants using the manufacturer’s protocol. Detailed methods can be found in the Supplementary Materials.
Confocal and two-photon intravital microscopy
For confocal imaging, fixed LN tissue sections were imaged as previously described using a Leica SP8 microscope (10). For two-photon intravital imaging, immunized or control mice were anesthetized and popliteal LNs were surgically exposed and imaged as previously described (100). Detailed methods can be found in the Supplementary Materials.
Histocytometry
Histocytometry analysis was performed as previously described (7, 10), with some modifications. Myeloid and T cell isosurface 3D objects were generated in Imaris, and object statistics were then exported to FlowJo software for gating and phenotypic characterization. Additional positional and spatial correlation analysis was performed in CytoMAP (25). For visual clarity, Imaris 3D surface objects were used for some data display (Fig. 1 and 2, fig. S1A, S2 and S3G). Detailed methods can be found in the Supplementary Materials.
Cell Isolation and Flow Cytometry
For myeloid cells, disrupted tissues were treated with 400 U/ml collagenase D (Roche Applied Science). In some studies, enhanced digestion was used for increased cell yield, as described (101, 102). T cell flow cytometry studies did not use enzymatic digestion. Data was acquired on an LSR-II flow cytometer (BD Biosciences) and analyzed using FlowJo software (TreeStar). Detailed methods, as well as antibody and staining reagent data can be found in the Supplemental Materials.
Antibodies, Staining Reagents, Microscopy and Image analysis
Detailed descriptions are available in Supplementary Methods.
Statistics
Statistical analysis was performed using GraphPad Prism software. The statistical significance of differences in mean values between two groups was analyzed by a two-tailed unpaired Student’s t test with Welch’s correction. When analyzing data with multiple side-by-side groups, a one-way ANOVA with Tukey’s multiple comparisons test. In multi-adjuvant comparison studies where variance between groups could not be assumed to be equal, the Brown-Forsythe and Welch ANOVA tests with Dunnett’s multiple comparison tests were performed. Paired t test was performed only when comparing responses within the same experimental animal or tissue. In bar graphs for all figures, bars represent data mean. Error bars represent standard deviation. ****, P ≤ 0.0001; ***, P ≤ 0.001; **, P ≤ 0.01; *, P ≤ 0.05; NS, P> 0.05. In all figures, unless otherwise noted, data points represent independent LNs.
Supplementary Material
Table S2: Raw data file
Fig. S1. Histocytometry analysis of myeloid cell positioning.
Fig. S2. Analysis of myeloid cell recruitment and localization within LNs in response to various stimuli.
Fig. S3. Chemokine receptors expressed by cDC and validation of DC-CCR7.KO bone marrow chimera system.
Fig. S4: HEV-mediated recruitment of MOs in the dLNs.
Fig. S5. Res cDC relocalization and antigen presentation in the regulation of CD8 T cell responses.
Fig. S6: IL-12 production by TZ-localized inflammatory MOs after immunization.
Fig. S7: Myeloid cell microenvironments in the generation of T cell responses.
Fig. S8: Regulation of T cell differentiation by inflammatory MOs.
Fig. S9: Model of innate and adaptive immune cell crosstalk during Type-I inflammation.
Table S1: Description of cellular phenotypes for flow and imaging studies
Video 1: 2-photon intravital microscopy visualization of MO trafficking into dLNs via local blood vessels.
Acknowledgements:
We thank Drs. Sunil Thomas, Jennifer Lund and Jessica B Graham for help with West Nile virus infections, and Dr. Jakob von Moltke and John W McGinty for help with Nippostrongylus brasiliensis infections. We are grateful to Dr. Matthias Mack for generously providing the CCR2 blocking antibody. We also thank Dr. Marion Pepper, Dr. Emily Hemann, Dr. Martin Prlic, Nicholas J Maurice, and Alexis K Taber for additional reagents and resources.
Funding:
This study was supported by the NIH grants R01AI134713 (M.Y.G), R21AI142667 (M.Y.G.), K22AI108628 (M.Y.G.), T32AI106677 (J.M.L.), T32GM007270 (J.M.L., J.H.), R01AI104002 and R01AI145296 (MG Jr.), and a Washington Research Foundation postdoctoral fellowship (C.S.). This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. (NSF DGE-1762114), (M.R.L.C).
Footnotes
Competing interests:
The authors declare no competing interests.
Data and materials availability:
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes:
- 1.Qi H, Kastenmüller W, Germain RN, Spatiotemporal Basis of Innate and Adaptive Immunity in Secondary Lymphoid Tissue. Annu. Rev. Cell Dev. Biol 30, 141–167 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Merad M, Sathe P, Helft J, Miller J, Mortha A, The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol 31, 563–604 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heath WR, Carbone FR, Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat. Immunol 10, 1237–1244 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Villadangos JA, Schnorrer P, Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat. Rev. Immunol 7, 543–555 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Worbs T, Hammerschmidt SI, Förster R, Dendritic cell migration in health and disease. Nat. Rev. Immunol 17, 30–48 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Gerner MY, Torabi-Parizi P, Germain RN, Strategically Localized Dendritic Cells Promote Rapid T Cell Responses to Lymph-Borne Particulate Antigens. Immunity. 42, 172–185 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Gerner MY, Casey KA, Kastenmuller W, Germain RN, Dendritic cell and antigen dispersal landscapes regulate T cell immunity. J. Exp. Med 214(10):3105–3122 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gonzalez SF, Lukacs-Kornek V, Kuligowski MP, Pitcher LA, Degn SE, Kim Y-A, Cloninger MJ, Martinez-Pomares L, Gordon S, Turley SJ, Carroll MC, Capture of influenza by medullary dendritic cells via SIGN-R1 is essential for humoral immunity in draining lymph nodes. Nat. Immunol 11, 427–434 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roozendaal R, Mempel TR, Pitcher LA, Gonzalez SF, Verschoor A, Mebius RE, von Andrian UH, Carroll MC, Conduits Mediate Transport of Low-Molecular-Weight Antigen to Lymph Node Follicles. Immunity. 30, 264–276 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerner MY, Kastenmuller W, Ifrim I, Kabat J, Germain RN, Histo-Cytometry: A Method for Highly Multiplex Quantitative Tissue Imaging Analysis Applied to Dendritic Cell Subset Microanatomy in Lymph Nodes. Immunity. 37, 364–376 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brewitz A, Eickhoff S, Dähling S, Quast T, Bedoui S, Kroczek RA, Kurts C, Garbi N, Barchet W, Iannacone M, Klauschen F, Kolanus W, Kaisho T, Colonna M, Germain RN, Kastenmüller W, CD8+ T Cells Orchestrate pDC-XCR1+ Dendritic Cell Spatial and Functional Cooperativity to Optimize Priming. Immunity. 0 (2017), doi: 10.1016/j.immuni.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kitano M, Yamazaki C, Takumi A, Ikeno T, Hemmi H, Takahashi N, Shimizu K, Fraser SE, Hoshino K, Kaisho T, Okada T, Imaging of the cross-presenting dendritic cell subsets in the skin-draining lymph node. Proc. Natl. Acad. Sci 113, 1044–1049 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eickhoff S, Brewitz A, Gerner MY, Klauschen F, Komander K, Hemmi H, Garbi N, Kaisho T, Germain RN, Kastenmüller W, Robust Anti-viral Immunity Requires Multiple Distinct T Cell-Dendritic Cell Interactions. Cell. 162, 1322–1337 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hor JL, Whitney PG, Zaid A, Brooks AG, Heath WR, Mueller SN, Spatiotemporally Distinct Interactions with Dendritic Cell Subsets Facilitates CD4+ and CD8+ T Cell Activation to Localized Viral Infection. Immunity. 43, 554–565 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Ruhland MK, Roberts EW, Cai E, Mujal AM, Marchuk K, Beppler C, Nam D, Serwas NK, Binnewies M, Krummel MF, Visualizing Synaptic Transfer of Tumor Antigens among Dendritic Cells. Cancer Cell. 37, 786–799.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Celli S, Garcia Z, Bousso P, CD4 T cells integrate signals delivered during successive DC encounters in vivo. J. Exp. Med 202, 1271–1278 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakano H, Lin KL, Yanagita M, Charbonneau C, Cook DN, Kakiuchi T, Gunn MD, Blood-derived inflammatory dendritic cells in lymph nodes stimulate acute T helper type 1 immune responses. Nat. Immunol 10, 394–402 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koker SD, Hoecke LV, Beuckelaer AD, Roose K, Deswarte K, Willart MA, Bogaert P, Naessens T, Geest BGD, Saelens X, Lambrecht BN, Grooten J, Inflammatory monocytes regulate Th1 oriented immunity to CpG adjuvanted protein vaccines through production of IL-12. Sci. Rep 7, 5986 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jakubzick CV, Randolph GJ, Henson PM, Monocyte differentiation and antigen-presenting functions. Nat. Rev. Immunol 17, 349–362 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, Ivanov S, Duan Q, Bala S, Condon T, van Rooijen N, Grainger JR, Belkaid Y, Ma’ayan A, Riches DWH, Yokoyama WM, Ginhoux F, Henson PM, Randolph GJ, Minimal Differentiation of Classical Monocytes as They Survey Steady-State Tissues and Transport Antigen to Lymph Nodes. Immunity. 39, 599–610 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leirião P, del Fresno C, Ardavín C, Monocytes as effector cells: Activated Ly-6Chigh mouse monocytes migrate to the lymph nodes through the lymph and cross-present antigens to CD8+ T cells. Eur. J. Immunol 42, 2042–2051 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Palframan RT, Jung S, Cheng G, Weninger W, Luo Y, Dorf M, Littman DR, Rollins BJ, Zweerink H, Rot A, von Andrian UH, Inflammatory chemokine transport and presentation in HEV: a remote control mechanism for monocyte recruitment to lymph nodes in inflamed tissues. J. Exp. Med 194, 1361–1373 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi C, Pamer EG, Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol 11, 762–774 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eisenbarth SC, Dendritic cell subsets in T cell programming: location dictates function. Nat. Rev. Immunol 19, 89–103 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stoltzfus CR, Filipek J, Gern BH, Olin BE, Leal JM, Wu Y, Lyons-Cohen MR, Huang JY, Paz-Stoltzfus CL, Plumlee CR, Pöschinger T, Urdahl KB, Perro M, Gerner MY, CytoMAP: A Spatial Analysis Toolbox Reveals Features of Myeloid Cell Organization in Lymphoid Tissues. Cell Rep. 31, 107523 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Groom JR, Regulators of T-cell fate: Integration of cell migration, differentiation and function. Immunol. Rev 289, 101–114 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Woodruff MC, Heesters BA, Herndon CN, Groom JR, Thomas PG, Luster AD, Turley SJ, Carroll MC, Trans-nodal migration of resident dendritic cells into medullary interfollicular regions initiates immunity to influenza vaccine. J. Exp. Med 211, 1611–1621 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grassmann S, Mihatsch L, Mir J, Kazeroonian A, Rahimi R, Flommersfeld S, Schober K, Hensel I, Leube J, Pachmayr LO, Kretschmer L, Zhang Q, Jolly A, Chaudhry MZ, Schiemann M, Cicin-Sain L, Höfer T, Busch DH, Flossdorf M, Buchholz VR, Early emergence of T central memory precursors programs clonal dominance during chronic viral infection. Nat. Immunol 21, 1563–1573 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Buchholz VR, Flossdorf M, Hensel I, Kretschmer L, Weissbrich B, Gräf P, Verschoor A, Schiemann M, Höfer T, Busch DH, Disparate Individual Fates Compose Robust CD8+ T Cell Immunity. Science. 340, 630–635 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Gerlach C, Rohr JC, Perié L, van Rooij N, van Heijst JWJ, Velds A, Urbanus J, Naik SH, Jacobs H, Beltman JB, de Boer RJ, Schumacher TNM, Heterogeneous Differentiation Patterns of Individual CD8+ T Cells. Science. 340, 635–639 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Plumlee CR, Sheridan BS, Cicek BB, Lefrançois L, Environmental cues dictate the fate of individual CD8+ T cells responding to infection. Immunity. 39, 347–356 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Annunziato F, Romagnani C, Romagnani S, The 3 major types of innate and adaptive cell-mediated effector immunity. J. Allergy Clin. Immunol 135, 626–635 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Gause WC, Wynn TA, Allen JE, Type 2 immunity and wound healing: evolutionary refinement of adaptive immunity by helminths. Nat. Rev. Immunol 13, 607–614 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spellberg B, Edwards JE, Type 1/Type 2 Immunity in Infectious Diseases. Clin. Infect. Dis. 32, 76–102 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Yip HC, Karulin AY, Tary-Lehmann M, Hesse MD, Radeke H, Heeger PS, Trezza RP, Heinzel FP, Forsthuber T, Lehmann PV, Adjuvant-Guided Type-1 and Type-2 Immunity: Infectious/Noninfectious Dichotomy Defines the Class of Response. J. Immunol 162, 3942–3949 (1999). [PubMed] [Google Scholar]
- 36.Lu E, Dang EV, McDonald JG, Cyster JG, Distinct oxysterol requirements for positioning naïve and activated dendritic cells in the spleen. Sci. Immunol 2 (2017), doi: 10.1126/sciimmunol.aal5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yi T, Cyster JG, EBI2-mediated bridging channel positioning supports splenic dendritic cell homeostasis and particulate antigen capture. eLife. 2, e00757 (2013).23682316 [Google Scholar]
- 38.Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B, Van Egeren DS, Park C, Irvine DJ, Structure-based programming of lymph-node targeting in molecular vaccines. Nature. 507, 519–522 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Langlet C, Tamoutounour S, Henri S, Luche H, Ardouin L, Grégoire C, Malissen B, Guilliams M, CD64 Expression Distinguishes Monocyte-Derived and Conventional Dendritic Cells and Reveals Their Distinct Role during Intramuscular Immunization. J. Immunol, 1102744 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Plantinga M, Guilliams M, Vanheerswynghels M, Deswarte K, Branco-Madeira F, Toussaint W, Vanhoutte L, Neyt K, Killeen N, Malissen B, Hammad H, Lambrecht BN, Conventional and Monocyte-Derived CD11b+ Dendritic Cells Initiate and Maintain T Helper 2 Cell-Mediated Immunity to House Dust Mite Allergen. Immunity. 38, 322–335 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Lämmermann T, Kastenmüller W, Concepts of GPCR-controlled navigation in the immune system. Immunol. Rev 289, 205–231 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Förster R, Schubel A, Breitfeld D, Kremmer E, Renner-Müller I, Wolf E, Lipp M, CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 99, 23–33 (1999). [DOI] [PubMed] [Google Scholar]
- 43.Ohl L, Mohaupt M, Czeloth N, Hintzen G, Kiafard Z, Zwirner J, Blankenstein T, Henning G, Förster R, CCR7 Governs Skin Dendritic Cell Migration under Inflammatory and Steady-State Conditions. Immunity. 21, 279–288 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Calabro S, Liu D, Gallman A, Nascimento MSL, Yu Z, Zhang T, Chen P, Zhang B, Xu L, Gowthaman U, Krishnaswamy JK, Haberman AM, Williams A, Eisenbarth SC, Differential Intrasplenic Migration of Dendritic Cell Subsets Tailors Adaptive Immunity. Cell Rep. 16, 2472–2485 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Becker G, Moulin V, Pajak B, Bruck C, Francotte M, Thiriart C, Urbain J, Moser M, The adjuvant monophosphoryl lipid A increases the function of antigen-presenting cells. Int. Immunol 12, 807–815 (2000). [DOI] [PubMed] [Google Scholar]
- 46.Trez CD, Pajak B, Brait M, Glaichenhaus N, Urbain J, Moser M, Lauvau G, Muraille E, TLR4 and Toll-IL-1 Receptor Domain-Containing Adapter-Inducing IFN-β, but Not MyD88, Regulate Escherichia coli-Induced Dendritic Cell Maturation and Apoptosis In Vivo. J. Immunol 175, 839–846 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Li J, Lu E, Yi T, Cyster JG, EBI2 augments Tfh cell fate by promoting interaction with IL-2-quenching dendritic cells. Nature. 533, 110–114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meredith MM, Liu K, Darrasse-Jeze G, Kamphorst AO, Schreiber HA, Guermonprez P, Idoyaga J, Cheong C, Yao K-H, Niec RE, Nussenzweig MC, Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J. Exp. Med 209, 1153–1165 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Itano AA, McSorley SJ, Reinhardt RL, Ehst BD, Ingulli E, Rudensky AY, Jenkins MK, Distinct Dendritic Cell Populations Sequentially Present Antigen to CD4 T Cells and Stimulate Different Aspects of Cell-Mediated Immunity. Immunity. 19, 47–57 (2003). [DOI] [PubMed] [Google Scholar]
- 50.Shi C, Jia T, Mendez-Ferrer S, Hohl TM, Serbina NV, Lipuma L, Leiner I, Li MO, Frenette PS, Pamer EG, Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating Toll-like receptor ligands. Immunity. 34, 590–601 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nourshargh S, Alon R, Leukocyte migration into inflamed tissues. Immunity. 41, 694–707 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Girard J-P, Moussion C, Förster R, HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat. Rev. Immunol 12, 762–773 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Woodfin A, Voisin M-B, Beyrau M, Colom B, Caille D, Diapouli F-M, Nash GB, Chavakis T, Albelda SM, Rainger GE, Meda P, Imhof BA, Nourshargh S, The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat. Immunol 12, 761–769 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang HH, Song K, Rabin RL, Hill BJ, Perfetto SP, Roederer M, Douek DC, Siegel RM, Farber JM, CCR2 identifies a stable population of human effector memory CD4+ T cells equipped for rapid recall response. J. Immunol. Baltim. Md 1950. 185, 6646–6663 (2010). [DOI] [PubMed] [Google Scholar]
- 55.Zigmond E, Varol C, Farache J, Elmaliah E, Satpathy AT, Friedlander G, Mack M, Shpigel N, Boneca IG, Murphy KM, Shakhar G, Halpern Z, Jung S, Ly6C hi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity. 37, 1076–1090 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Bosteels C, Neyt K, Vanheerswynghels M, van Helden MJ, Sichien D, Debeuf N, Prijck SD, Bosteels V, Vandamme N, Martens L, Saeys Y, Louagie E, Lesage M, Williams DL, Tang S-C, Mayer JU, Ronchese F, Scott CL, Hammad H, Guilliams M, Lambrecht BN, Inflammatory Type 2 cDCs Acquire Features of cDC1s and Macrophages to Orchestrate Immunity to Respiratory Virus Infection. Immunity. 52, 1039–1056.e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krishnamoorthy V, Kannanganat S, Maienschein-Cline M, face Cook SP, Chen J, Bahroos N, Sievert E, Corse E, Chong A, Sciammas R, The IRF4 Gene Regulatory Module Functions as a Read-Write Integrator to Dynamically Coordinate T Helper Cell Fate. Immunity. 47, 481–497.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Man K, Miasari M, Shi W, Xin A, Henstridge DC, Preston S, Pellegrini M, Belz GT, Smyth GK, Febbraio MA, Nutt SL, Kallies A, The transcription factor IRF4 is essential for TCR affinity-mediated metabolic programming and clonal expansion of T cells. Nat. Immunol 14, 1155–1165 (2013). [DOI] [PubMed] [Google Scholar]
- 59.Schreiber HA, Loschko J, Karssemeijer RA, Escolano A, Meredith MM, Mucida D, Guermonprez P, Nussenzweig MC, Intestinal monocytes and macrophages are required for T cell polarization in response to Citrobacter rodentium. J. Exp. Med 210, 2025–2039 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martínez‐López M, Iborra S, Conde‐Garrosa R, Sancho D, Batf3-dependent CD103+ dendritic cells are major producers of IL-12 that drive local Th1 immunity against Leishmania major infection in mice. Eur. J. Immunol 45, 119–129 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mashayekhi M, Sandau MM, Dunay IR, Frickel EM, Khan A, Goldszmid RS, Sher A, Ploegh HL, Murphy TL, Sibley LD, Murphy KM, CD8α+ Dendritic Cells Are the Critical Source of Interleukin-12 that Controls Acute Infection by Toxoplasma gondii Tachyzoites. Immunity. 35, 249–259 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reinhardt RL, Hong S, Kang S-J, Wang Z, Locksley RM, Visualization of IL-12/23p40 In Vivo Reveals Immunostimulatory Dendritic Cell Migrants that Promote Th1 Differentiation. J. Immunol 177, 1618–1627 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Conejero L, Khouili SC, Martínez-Cano S, Izquierdo HM, Brandi P, Sancho D, Lung CD103+ dendritic cells restrain allergic airway inflammation through IL-12 production. JCI Insight. 2, doi: 10.1172/jci.insight.90420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Everts B, Tussiwand R, Dreesen L, Fairfax KC, Huang SC-C, Smith AM, O’Neill CM, Lam WY, Edelson BT, Urban JF, Murphy KM, Pearce EJ, Migratory CD103+ dendritic cells suppress helminth-driven type 2 immunity through constitutive expression of IL-12. J. Exp. Med 213, 35–51 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Borges da Silva H, Wang H, Qian LJ, Hogquist KA, Jameson SC, ARTC2.2/P2RX7 Signaling during Cell Isolation Distorts Function and Quantification of Tissue-Resident CD8+ T Cell and Invariant NKT Subsets. J. Immunol. Baltim. Md 1950 202, 2153–2163 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyártó BZ, Southern PJ, Masopust D, Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell. 161, 737–749 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Curtsinger JM, Mescher MF, Inflammatory cytokines as a third signal for T cell activation. Curr. Opin. Immunol 22, 333–340 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhu J, Yamane H, Paul WE, Differentiation of Effector CD4 T Cell Populations. Annu. Rev. Immunol 28, 445–489 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mempel TR, Henrickson SE, von Andrian UH, T-cell priming by dendriticcells in lymph nodes occurs in three distinct phases. Nature. 427, 154–159 (2004). [DOI] [PubMed] [Google Scholar]
- 70.Bajaña S, Roach K, Turner S, Paul J, Kovats S, IRF4 Promotes Cutaneous Dendritic Cell Migration to Lymph Nodes during Homeostasis and Inflammation. J. Immunol, 1102613 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Krishnaswamy JK, Gowthaman U, Zhang B, Mattsson J, Szeponik L, Liu D, Wu R, White T, Calabro S, Xu L, Collet MA, Yurieva M, Alsén S, Fogelstrand P, Walter A, Heath WR, Mueller SN, Yrlid U, Williams A, Eisenbarth SC, Migratory CD11b+ conventional dendritic cells induce T follicular helper cell–dependent antibody responses. Sci. Immunol 2, eaam9169 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vander Lugt B, Khan AA, Hackney JA, Agrawal S, Lesch J, Zhou M, Lee WP, Park S, Xu M, DeVoss J, Spooner CJ, Chalouni C, Delamarre L, Mellman I, Singh H, Transcriptional programming of dendritic cells for enhanced MHC class II antigen presentation. Nat. Immunol 15, 161–167 (2014). [DOI] [PubMed] [Google Scholar]
- 73.Igyártó BZ, Haley K, Ortner D, Bobr A, Gerami-Nejad M, Edelson BT, Zurawski SM, Malissen B, Zurawski G, Berman J, Kaplan DH, Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific T helper cell responses. Immunity. 35, 260–272 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee HK, Zamora M, Linehan MM, Iijima N, Gonzalez D, Haberman A, Iwasaki A, Differential roles of migratory and resident DCs in T cell priming after mucosal or skin HSV-1 infection. J. Exp. Med 206, 359–370 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kumamoto Y, Linehan M, Weinstein JS, Laidlaw BJ, Craft JE, Iwasaki A, CD301b+ dermal dendritic cells drive T helper-2 cell mediated immunity. Immunity. 39 (2013), doi: 10.1016/j.immuni.2013.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Levin C, Bonduelle O, Nuttens C, Primard C, Verrier B, Boissonnas A, Combadière B, Critical Role for Skin-Derived Migratory DCs and Langerhans Cells in TFH and GC Responses after Intradermal Immunization. J. Invest. Dermatol 137, 1905–1913 (2017). [DOI] [PubMed] [Google Scholar]
- 77.Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, Zhan Y, Lew AM, Shortman K, Heath WR, Carbone FR, Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity. 25, 153–162 (2006). [DOI] [PubMed] [Google Scholar]
- 78.Kohli K, Janssen A, Förster R, Plasmacytoid dendritic cells induce tolerance predominantly by cargoing antigen to lymph nodes. Eur. J. Immunol. 46, 2659–2668 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Srivastava S, Ernst JD, Cell-to-cell transfer of M. tuberculosis antigens optimizes CD4 T cell priming. Cell Host Microbe. 15, 741–752 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hutchison S, Benson RA, Gibson VB, Pollock AH, Garside P, Brewer JM, Antigen depot is not required for alum adjuvanticity. FASEB J. 26, 1272–1279 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schudel A, Francis DM, Thomas SN, Material design for lymph node drug delivery. Nat. Rev. Mater. 4, 415–428 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, Mazloom AR, Ma’ayan A, Chua W-J, Hansen TH, Turley SJ, Merad M, Randolph GJ, Immunological Genome Consortium, Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol 13, 1118–1128 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, Pandey G, Leboeuf M, Elpek KG, Helft J, Hashimoto D, Chow A, Price J, Greter M, Bogunovic M, Bellemare-Pelletier A, Frenette PS, Randolph GJ, Turley SJ, Merad M, Immunological Genome Consortium, Deciphering the transcriptional network of the dendritic cell lineage. Nat. Immunol 13, 888–899 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Villani A-C, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, Griesbeck M, Butler A, Zheng S, Lazo S, Jardine L, Dixon D, Stephenson E, Nilsson E, Grundberg I, McDonald D, Filby A, Li W, De Jager PL, Rozenblatt-Rosen O, Lane AA, Haniffa M, Regev A, Hacohen N, Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. 356 (2017), doi: 10.1126/science.aah4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Blecher-Gonen R, Bost P, Hilligan KL, David E, Salame TM, Roussel E, Connor LM, Mayer JU, Bahar Halpern K, Tóth B, Itzkovitz S, Schwikowski B, Ronchese F, Amit I, Single-Cell Analysis of Diverse Pathogen Responses Defines a Molecular Roadmap for Generating Antigen-Specific Immunity. Cell Syst. 8, 109–121.e6 (2019). [DOI] [PubMed] [Google Scholar]
- 86.Longhi MP, Trumpfheller C, Idoyaga J, Caskey M, Matos I, Kluger C, Salazar AM, Colonna M, Steinman RM, Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J. Exp. Med 206, 1589–1602 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Spörri R, Reis e Sousa C, Inflammatory mediators are insufficient for full dendritic cell activation and promote expansion of CD4+ T cell populations lacking helper function. Nat. Immunol 6, 163–170 (2005). [DOI] [PubMed] [Google Scholar]
- 88.Hochrein H, Shortman K, Vremec D, Scott B, Hertzog P, O’Keeffe M, Differential Production of IL-12, IFN-α, and IFN-γ by Mouse Dendritic Cell Subsets. J. Immunol 166, 5448–5455 (2001). [DOI] [PubMed] [Google Scholar]
- 89.e Sousa CR, Hieny S, Scharton-Kersten T, Jankovic D, Charest H, Germain RN, Sher A, In Vivo Microbial Stimulation Induces Rapid CD40 Ligand–independent Production of Interleukin 12 by Dendritic Cells and their Redistribution to T Cell Areas. J. Exp. Med 186, 1819–1829 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Crotty S, Follicular Helper CD4 T Cells (TFH). Annu. Rev. Immunol 29, 621–663 (2011). [DOI] [PubMed] [Google Scholar]
- 91.Groom JR, Richmond J, Murooka TT, Sorensen EW, Sung JH, Bankert K, von Andrian UH, Moon JJ, Mempel TR, Luster AD, CXCR3 Chemokine Receptor-Ligand Interactions in the Lymph Node Optimize CD4+ T Helper 1 Cell Differentiation. Immunity. 37, 1091–1103 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lehmann CHK, Heger L, Heidkamp GF, Baranska A, Lühr JJ, Hoffmann A, Dudziak D, Direct Delivery of Antigens to Dendritic Cells via Antibodies Specific for Endocytic Receptors as a Promising Strategy for Future Therapies. Vaccines. 4, 8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Moyer TJ, Zmolek AC, Irvine DJ, Beyond antigens and adjuvants: formulating future vaccines. J. Clin. Invest 126, 799–808 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Guilliams M, van de Laar L, A Hitchhiker’s Guide to Myeloid Cell Subsets: Practical Implementation of a Novel Mononuclear Phagocyte Classification System. Front. Immunol 6 (2015), doi: 10.3389/fimmu.2015.00406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hohl TM, Rivera A, Lipuma L, Gallegos A, Shi C, Mack M, Pamer EG, Inflammatory Monocytes Facilitate Adaptive CD4 T Cell Responses during Respiratory Fungal Infection. Cell Host Microbe. 6, 470–481 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Smith KD, Andersen-Nissen E, Hayashi F, Strobe K, Bergman MA, Barrett SLR, Cookson BT, Aderem A, Toll-like receptor 5 recognizes a conserved site on flagellin required for protofilament formation and bacterial motility. Nat. Immunol 4, 1247–1253 (2003). [DOI] [PubMed] [Google Scholar]
- 97.Nadjsombati MS, McGinty JW, Lyons-Cohen MR, Jaffe JB, DiPeso L, Schneider C, Miller CN, Pollack JL, Nagana Gowda GA, Fontana MF, Erle DJ, Anderson MS, Locksley RM, Raftery D, von Moltke J, Detection of Succinate by Intestinal Tuft Cells Triggers a Type 2 Innate Immune Circuit. Immunity. 49, 33–41.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mack M, Cihak J, Simonis C, Luckow B, Proudfoot AEI, Plachý J, Brühl H, Frink M, Anders H-J, Vielhauer V, Pfirstinger J, Stangassinger M, Schlöndorff D, Expression and Characterization of the Chemokine Receptors CCR2 and CCR5 in Mice. J. Immunol 166, 4697–4704 (2001). [DOI] [PubMed] [Google Scholar]
- 99.Anderson KG, Mayer-Barber K, Sung H, Beura L, James BR, Taylor JJ, Qunaj L, Griffith TS, Vezys V, Barber DL, Masopust D, Intravascular staining for discrimination of vascular and tissue leukocytes. Nat. Protoc. 9, 209–222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bajénoff M, Egen JG, Koo LY, Laugier JP, Brau F, Glaichenhaus N, Germain RN, Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity. 25, 989–1001 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fletcher AL, Malhotra D, Acton SE, Lukacs-Kornek V, Bellemare-Pelletier A, Curry M, Armant M, Turley SJ, Reproducible Isolation of Lymph Node Stromal Cells Reveals Site-Dependent Differences in Fibroblastic Reticular Cells. Front. Immunol 2 (2011), doi: 10.3389/fimmu.2011.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sokol CL, Camire RB, Jones MC, Luster AD, The Chemokine Receptor CCR8 Promotes the Migration of Dendritic Cells into the Lymph Node Parenchyma to Initiate the Allergic Immune Response. Immunity. 49, 449–463.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2: Raw data file
Fig. S1. Histocytometry analysis of myeloid cell positioning.
Fig. S2. Analysis of myeloid cell recruitment and localization within LNs in response to various stimuli.
Fig. S3. Chemokine receptors expressed by cDC and validation of DC-CCR7.KO bone marrow chimera system.
Fig. S4: HEV-mediated recruitment of MOs in the dLNs.
Fig. S5. Res cDC relocalization and antigen presentation in the regulation of CD8 T cell responses.
Fig. S6: IL-12 production by TZ-localized inflammatory MOs after immunization.
Fig. S7: Myeloid cell microenvironments in the generation of T cell responses.
Fig. S8: Regulation of T cell differentiation by inflammatory MOs.
Fig. S9: Model of innate and adaptive immune cell crosstalk during Type-I inflammation.
Table S1: Description of cellular phenotypes for flow and imaging studies
Video 1: 2-photon intravital microscopy visualization of MO trafficking into dLNs via local blood vessels.
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.