

Ageing is characterized by the development of persistent pro-inflammatory responses that contribute to atherosclerosis, metabolic syndrome, cancer and frailty1–3. The ageing brain is also vulnerable to inflammation, as demonstrated by the high prevalence of age-associated cognitive decline and Alzheimer’s disease4–6. Systemically, circulating pro-inflammatory factors can promote cognitive decline7,8, and in the brain, microglia lose the ability to clear misfolded proteins that are associated with neurodegeneration9,10. However, the underlying mechanisms that initiate and sustain maladaptive inflammation with ageing are not well defined. Here we show that in ageing mice myeloid cell bioenergetics are suppressed in response to increased signalling by the lipid messenger prostaglandin E2 (PGE2), a major modulator of inflammation11. In ageing macrophages and microglia, PGE2 signalling through its EP2 receptor promotes the sequestration of glucose into glycogen, reducing glucose flux and mitochondrial respiration. This energy-deficient state, which drives maladaptive pro-inflammatory responses, is further augmented by a dependence of aged myeloid cells on glucose as a principal fuel source. In aged mice, inhibition of myeloid EP2 signalling rejuvenates cellular bioenergetics, systemic and brain inflammatory states, hippocampal synaptic plasticity and spatial memory. Moreover, blockade of peripheral myeloid EP2 signalling is sufficient to restore cognition in aged mice. Our study suggests that cognitive ageing is not a static or irrevocable condition but can be reversed by reprogramming myeloid glucose metabolism to restore youthful immune functions.
The underlying mechanisms that are responsible for the development of maladaptive myeloid phenotypes in ageing are not well understood; however, previous work suggests that cellular energy metabolism has an important role in regulating the activation state and function of the immune system12–16. To maintain homeostasis, immune cells require robust glycolytic and mitochondrial metabolism to meet the demand for energy and biosynthetic precursors. In line with this, recent studies indicate that ageing macrophages show marked decreases in glycolysis and mitochondrial oxidative phosphorylation that cause immune dysfunction17.
PGE2–EP2 signalling drives brain ageing
The lipid messenger PGE2 is a downstream product of the cyclooxygenase 2 (COX-2) pathway (Extended Data Fig. 1a) and a major modulator of inflammation11. Levels of PGE2 increase in ageing and in neurodegenerative disease18–20. We hypothesized that increases in PGE2 might underlie the development of age-associated maladaptive inflammation and cognitive decline. We identified a significant increase in PGE2 synthesis in human monocyte-derived macrophages (MDMs) from individuals of over 65 years of age, compared to those from younger individuals (below 35 years of age) (Fig. 1a; Extended Data Fig. 1b). Given the link between cellular metabolism and myeloid cell function17, we tested whether PGE2 signalling affected macrophage bioenergetics. Dose-dependent stimulation with PGE2 decreased glycolysis (extra-cellular acidification rate (ECAR)) and suppressed the mitochondrial oxygen consumption rate (OCR) in human MDMs (Fig. 1b, c). Although PGE2 signals through four G-protein-coupled receptors—EP1, EP2, EP3 and EP421—this suppressive effect of PGE2 was mediated specifically by the EP2 receptor, the expression of which increased markedly in aged human MDMs (Extended Data Fig. 1c–f). In contrast to PGE2 and the EP2 agonist butaprost (Fig. 1d), treatment with the EP2 inhibitors PF-0441894822 and compound 52 (C52)23 led to an increase in OCR and ECAR in macrophages (Extended Data Fig. 1g, h). These data suggest that inhibition of PGE2–EP2 signalling might enhance energy production in ageing myeloid cells.
Fig. 1 |. PGE2 EP2 receptor regulates myeloid metabolism and inflammation in ageing.
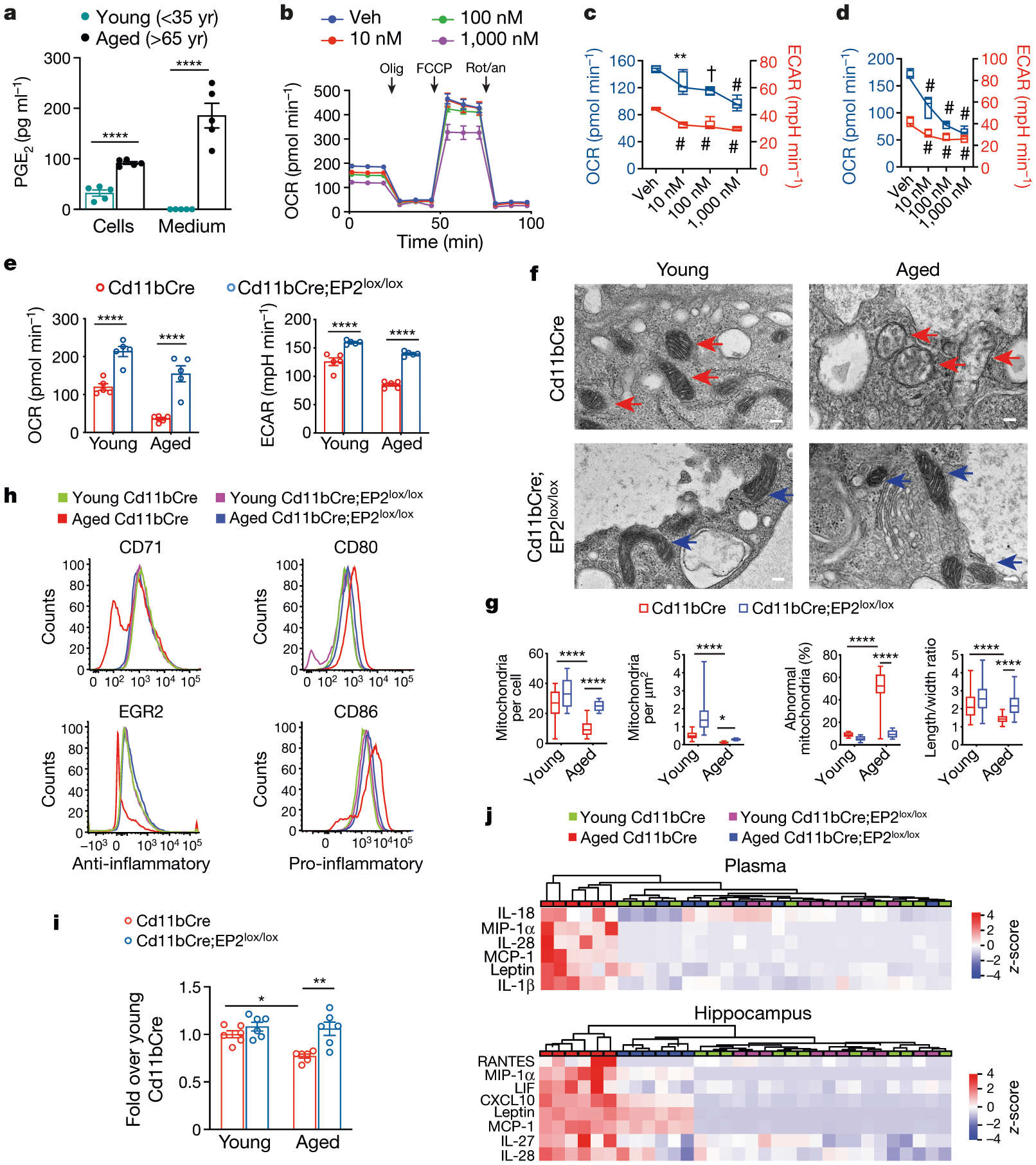
Data are mean ± s.e.m. unless otherwise specified. a, Levels of PGE2 from young and aged human MDMs cultured for 20 h; ****P < 0.0001 by two-tailed Student’s t-test. b, Real-time changes in the OCR of human MDMs in response to treatment with the indicated concentrations of PGE2 for 20 h. Cells were treated with 1 μM oligomycin (olig), 2 μM carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) and 0.5 μM rotenone and antimycin (rot/an), as indicated by the three black arrows. Veh, vehicle. c, Basal respiration (OCR) (blue) and ECAR (red) in human MDMs stimulated with the indicated concentrations of PGE2 for 20 h. Box plots (upper box limit, 5th percentile; lower box limit, 95th percentile; centre line, median; upper whisker, maximum value; lower whisker, minimum value); one-way analysis of variance (ANOVA), P < 0.0001; Tukey’s post hoc test, **P = 0.0085, †P = 0.0001, #P < 0.0001. d, Basal respiration and ECAR in human MDMs stimulated with the indicated concentrations of the EP2 agonist butaprost for 20 h. Box plots (5th–95th percentile); one-way ANOVA, P < 0.0001; Tukey’s post hoc test #P < 0.0001. In a–d, n = 5 donors per group; age (mean ± s.e.m.) 47.8 ± 2.105 years. e, Basal respiration and ECAR in peritoneal macrophages from young (3–4 months old) and aged (20–23 months old) Cd11bCre and Cd11bCre;EP2lox/lox mice. Two-way ANOVA, age and genotype P < 0.0001; Tukey’s post hoc test, ****P < 0.0001 (n = 5 mice per group). f, TEM of peritoneal macrophage mitochondria from young and aged Cd11bCre (red arrows) and Cd11bCre;EP2lox/lox (blue arrows) mice; two independent experiments (n = 6 mice per group). Scale bars, 100 nm. g, Quantification of TEM mitochondrial metrics from f. Box plots (5th–95th percentile); two-way ANOVA, age and genotype P < 0.0001; Tukey’s post hoc test, ****P < 0.0001 (n = 106 cells per group). h, Flow cytometry histograms of peritoneal macrophages for the anti-inflammatory markers CD71 and EGR2 and the pro-inflammatory markers CD80 and CD86 from young and aged Cd11bCre and Cd11bCre;EP2lox/lox mice; three independent experiments (n = 10,000–20,000 cells per sample; n = 3 mice per group). i, Phagocytosis of fluorescent Escherichia coli particles in peritoneal macrophages from young and aged Cd11bCre and Cd11bCre;EP2lox/lox mice. Two-way ANOVA, age P = 0.0147 and genotype P = 0.0008; Tukey’s post hoc test, *P = 0.0127, **P = 0.0017 (n = 6 mice per group). j, Hierarchical clustering of significantly regulated immune factors in the plasma and hippocampus from young and aged Cd11bCre and Cd11bCre;EP2lox/lox mice (n = 10 young, n = 6 aged mice per group).
Plasma and brain PGE2 levels, as well as macrophage EP2 levels, also increased in aged (20-month-old) mice relative to young mice (Extended Data Fig. 1i–k). Myeloid-cell-specific deletion of EP2 in ageing Cd11bCre;EP2lox/lox mice, in which levels of myeloid EP2 are reduced by 50%24, increased OCR and ECAR in peritoneal macrophages (Fig. 1e). Transmission electron microscopy (TEM) revealed age-associated abnormalities in the morphology, number and density of mitochondria in peritoneal macrophages from aged Cd11bCre mice; these mitochondrial abnormalities were not present in aged Cd11bCre;EP2lox/lox mice (Fig. 1f, g). Aged Cd11bCre macrophages exhibited a pro-inflammatory phenotype compared to young macrophages; however, the polarization state of aged Cd11bCre;EP2lox/lox macrophages was indistinguishable from that of young macrophages of either genotype (Fig. 1h, Extended Data Fig. 2a). Knockdown of myeloid EP2 in aged mice also restored phagocytic capability to a level comparable to that of young mice (Fig. 1i). Multiplex profiling of immune factors showed that there was a distinctive pattern of pro-inflammatory factor enrichment in the plasma and hippocampus of aged Cd11bCre mice, whereas aged Cd11bCre;EP2lox/lox mice had profiles more similar to those of young mice (Fig. 1j, Extended Data Fig. 2b, c). Thus, myeloid knockdown of EP2 prevents age-associated changes in cell bioenergetics, polarization state and phagocytic capability.
Given the association between inflammation and cognitive impairment, we reasoned that a reduction in myeloid EP2 signalling and the consequent rejuvenation of myeloid metabolism and function might improve cognitive function in ageing mice. Hippocampal-dependent spatial memory is particularly vulnerable to ageing, so we tested the performance of mice in the object location memory task and the Barnes maze task. In both tasks, the performance of aged Cd11bCre;EP2lox/lox mice was indistinguishable from that of young mice of either genotype—in sharp contrast to aged Cd11bCre control mice (Fig. 2a–d, Extended Data Figs. 2d, e, 9a). Hippocampal pre- and postsynaptic proteins, the levels of which decrease in ageing25,26, were increased in aged Cd11bCre;EP2lox/lox hippocampi (Extended Data Fig. 2f).We assessed hippocampal synaptic plasticity in aged Cd11bCre;EP2lox/lox mice using electrophysiological recordings of the CA3 to CA1 Schaffer collateral pathway, and found that these mice showed a robust improvement in long-term potentiation—a cellular correlate of learning and memory that deteriorates with ageing27 (Fig. 2e, Extended Data Fig. 2g). Thus, reduction of myeloid EP2 signalling in ageing mice restores hippocampal plasticity and memory function to youthful levels.
Fig. 2 |. Myeloid knockdown of the EP2 receptor prevents cognitive ageing.
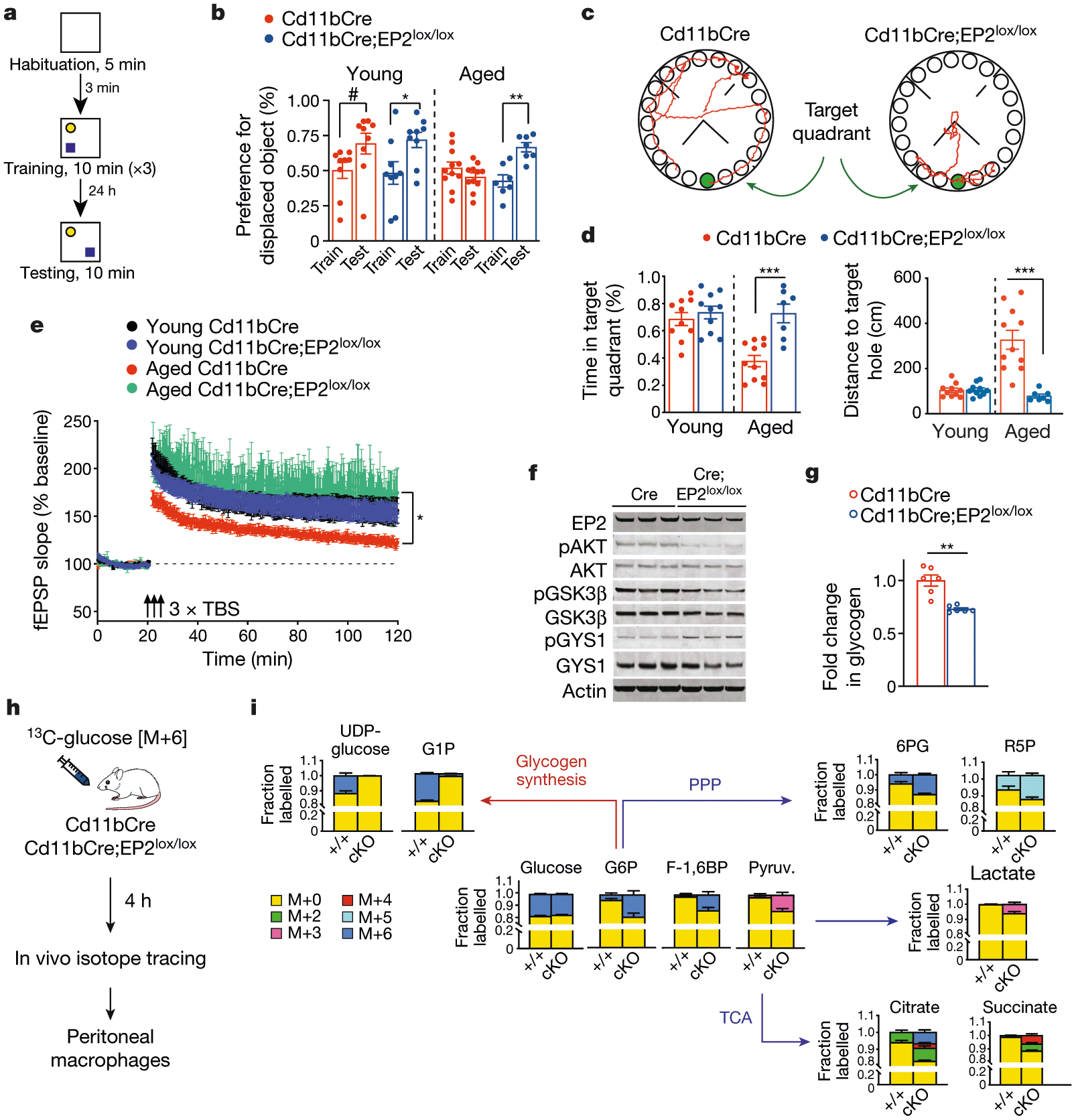
Experiments were performed in young (3–4-month-old) and aged (20–23-month-old) Cd11bCre and Cd11bCre;EP2lox/lox mice. Data are mean ± s.e.m. unless otherwise specified. a, Schematic depicting the object location memory task. b, Fraction of time spent exploring the displaced object is expressed as the per cent preference for the displaced object. Paired t-test, #P = 0.0280, *P = 0.0188, **P = 0.0034 (n = 9 young, n = 11 aged Cd11bCre, n = 7 aged Cd11bCre;EP2lox/lox mice). c, Representative traces of paths to target hole (green) by aged mice on the day of testing in the Barnes maze. d, Quantification of percentage of time in the target quadrant and distance travelled to the target hole in the Barnes maze. Two-tailed unpaired Student’s t-test, ***P = 0.0003, (n = 10 young, n = 11 aged Cd11bCre, n = 7 aged Cd11bCre;EP2lox/lox mice). e, Long-term potentiation, measured as the change in field excitatory postsynaptic potential (fEPSP), in the CA1 hippocampal region over 120 min. Three episodes of theta-burst stimulation (3 × TBS; black arrows) were applied. Two-way ANOVA, effects of time and genotype P < 0.0001; Sidak’s multiple comparisons test with Geisser–Greenhouse correction, *P = 0.0272 (n = 8 slices, n = 3 mice per group). f, Representative immunoblots of 2 independent experiments showing EP2 signalling via the AKT–GSK3β–GYS1 pathway in peritoneal macrophages from 6-month-old Cd11bCre and Cd11bCre;EP2lox/lox mice (n = 6 mice per group). g, Glycogen levels in peritoneal macrophages from aged Cd11bCre and Cd11bCre;EP2lox/lox mice. Two-tailed Student’s t-test, **P = 0.0031 (n = 6 mice per group). h, Mice were administered 13C-glucose (1 g per kg by oral gavage) and peritoneal macrophages were isolated 4 h later for isotope tracing. i, Quantification of 13C-glucose metabolism in mouse peritoneal macrophages demonstrates a shift away from glycogen synthesis and towards glycolysis, the pentose phosphate pathway (PPP) and the TCA cycle in aged Cd11bCre;EP2lox/lox (cKO) mice relative to Cd11bCre (+/+) mice (n = 6 mice per group). G1P, glucose 1-phosphate; G6P, glucose 6-phosphate; F-1,6BP, fructose 1,6-bisphosphate; pyruv., pyruvate; 6PG, 6-phosphogluconate; R5P, ribulose 5-phosphate.
PGE2–EP2 signalling suppresses glucose flux
To understand how inhibition of EP2 signalling elicited these beneficial effects in the context of ageing, we examined signalling cascades downstream of EP2 (Extended Data Fig. 2h). PGE2–EP2 signalling led to the activation of protein kinase B (also known as AKT), which then phosphorylated and inactivated GSK3β at Ser928,29, leading to the activation of glycogen synthase (GYS1) and glycogen synthesis. Reduction of EP2 signalling in Cd11bCre;EP2lox/lox macrophages led to GSK3β-mediated inactivation of GYS1 and decreased levels of intracellular glycogen (Fig. 2f, g, Extended Data Fig. 2i). Pharmacological inhibition of EP2 yielded similar results in mouse peritoneal macrophages and human MDMs (Extended Data Fig. 3a–c, Supplementary Fig. 2). Untargeted metabolomics in human MDMs treated with EP2 inhibitor revealed increased levels of glucose 6-phosphate and fructose 6-phosphate in these cells compared to vehicle-treated human MDMs (Extended Data Fig. 3d, e), suggesting that EP2 signalling has an effect on glucose flux down the glycolytic pathway.
To further explore the metabolic effects of EP2 signalling in macrophages, we administered [U-13C]glucose to aged Cd11bCre and Cd11bCre;EP2lox/lox mice, isolated macrophages four hours later and measured labelled metabolites from the glycolytic and tricarboxylic acid (TCA) cycle pathways (Fig. 2h, i, Extended Data Fig. 3f). Knockdown of EP2 in macrophages reduced the in vivo incorporation of 13C-glucose into uridine diphosphate glucose (UDP-glucose), the precursor of glycogen, and increased the incorporation of 13C-glucose into glycolytic and TCA cycle intermediates—consistent with increased glucose flux. We also performed 13C-glucose labelling in human MDMs treated with the EP2 inhibitor C52, and obtained similar results (Extended Data Fig. 3g, h). Previous studies have shown that cellular bioenergetics regulate the polarization state of macrophages through the accumulation of TCA cycle intermediates such as pro-inflammatory succinate12,16,30–33. Consistent with this, inhibition of EP2 shifted the polarization of human MDMs to a more anti-inflammatory activation state with increased phagocytic capacity (Extended Data Fig. 3i, j). These genetic and pharmacological findings link EP2-driven changes in glycogen synthesis and glucose flux to the activation state and phagocytic ability of macrophages.
We next directly tested the effect of reducing glycogen synthesis on the energy metabolism, cytokine production and polarization state of macrophages. Knockdown of rate-limiting GYS1 in human MDMs decreased glycogen levels and increased basal respiration and ECAR (Extended Fig. 4a–c). Consistent with this, GYS1-depleted human MDMs showed improved bioenergetics compared to control cells, with increases in glycolytic intermediates, NAD+, NADH and NADPH, and reduced glutathione levels (targeted metabolomics analysis) (Extended Data Fig. 4d). A shift away from glycogen synthesis and towards glycolysis was further confirmed using 13C-glucose isotope tracing in human MDMs, which was associated with a more anti-inflammatory activation state (Extended Data Fig. 4e, f). We then tested whether GYS1 deficiency in aged macrophages could restore youthful mitochondrial function and polarization state. Indeed, knockdown of GYS1 in human MDMs from aged individuals (over 65 years of age) restored glycolysis, mitochondrial respiration and polarization state to levels observed in human MDMs from young individuals (below 35 years of age) (Fig. 3a–c, Extended Data Fig. 4g, h). Notably, activation of EP2 signalling with butaprost did not alter the bioenergetic or immune phenotypes of aged human MDMs that lack GYS1 (Fig. 3d, e, Extended Data Fig. 4i), confirming that GYS1 is a critical effector of EP2-mediated effects on myeloid metabolism and immune state.
Fig. 3 |. EP2-directed glycogen synthesis regulates macrophage bioenergetics and immune responses.
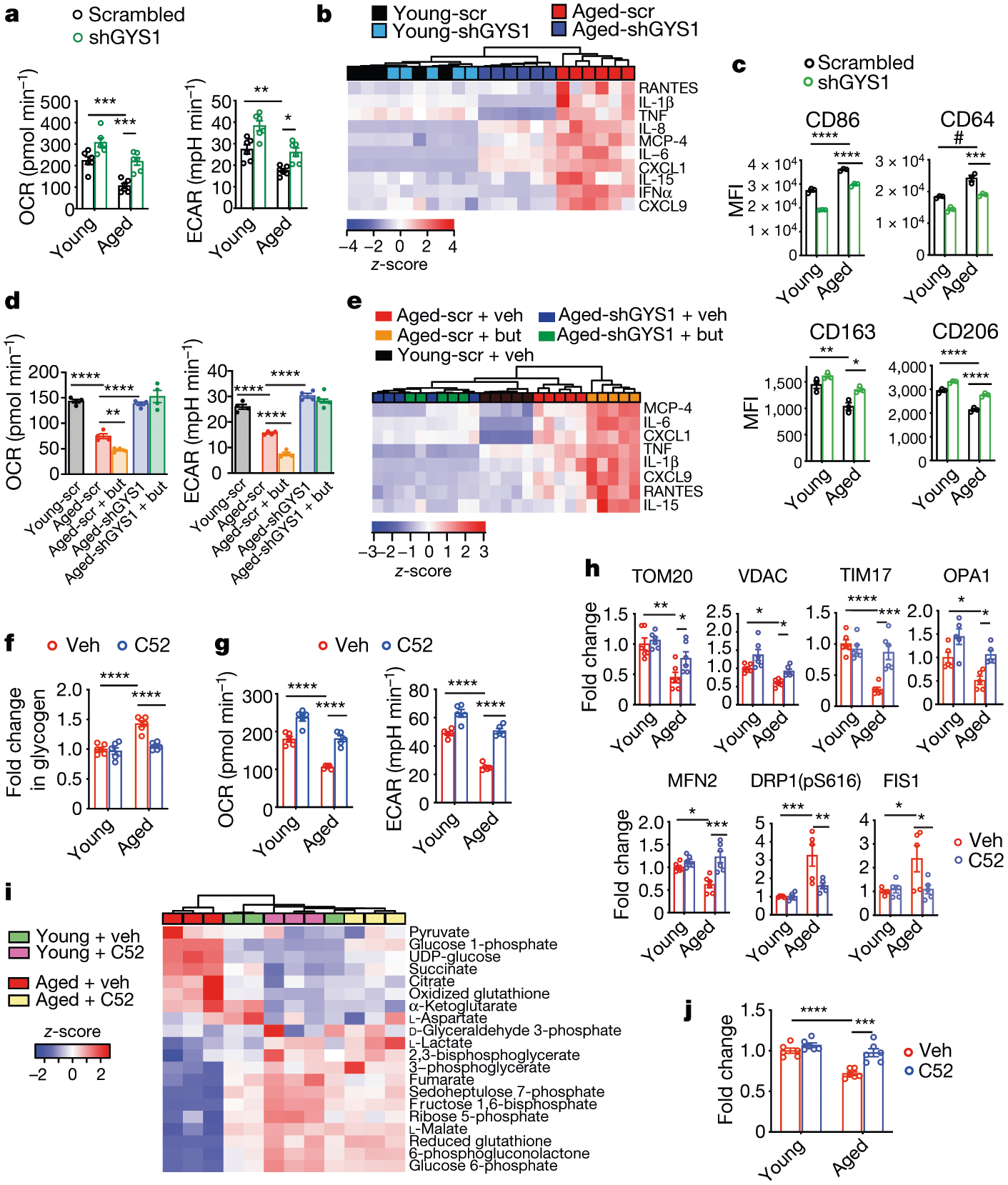
Human MDMs were derived from young (below 35 years of age) and aged (over 65 years of age) donors. Data are mean ± s.e.m. unless otherwise specified. a, Basal respiration and ECAR in human MDMs transfected with short hairpin RNA (shRNA) against GYS1 (shGYS1) or with control scrambled shRNA; three independent experiments. Two-way ANOVA, age and genotype P < 0.0001; Tukey’s post hoc test *P = 0.0225, **P = 0.0075, ***P < 0.001 (n = 6 donors per group). b, Hierarchical clustering of significantly regulated immune factors in human MDMs 20h after transfection of shGYS1 or scrambled shRNA (scr) (n = 5 young, n = 6 aged donors). c, Mean fluorescence intensity (MFI) of three independent flow cytometry experiments in young and aged human MDMs transfected with shGYS1 or with control scrambled shRNA. Two-way ANOVA, age and shRNA P < 0.0001; Tukey’s post hoc test, *P = 0.0178, **P = 0.0041, #P = 0.0003, ***P = 0.0006, ****P < 0.0001 (n = 20,000–40,000 cells per point; n = 3 donors per group). d, Basal respiration and ECAR of young and aged human MDMs transfected with shGYS1 or control scrambled shRNA and stimulated 8 h later with vehicle or butaprost (but; 100 nM, 20 h). Two-tailed Student’s t-test, **P = 0.0018, ****P < 0.0001 (n = 4 donors per group). e, Hierarchical clustering of significantly regulated immune factors in human MDMs from d (n = 5 donors per age group). f, Glycogen levels in young and aged human MDMs treated with vehicle or C52 (100 nM, 20 h). Two-way ANOVA, age and treatment P < 0.0001; Tukey’s post hoc test, ****P < 0.0001 (n = 6 donors per group). g, Basal respiration and ECAR in young and aged human MDMs treated with vehicle or C52 (100 nM, 20 h). Two-way ANOVA, age and treatment P < 0.0001; Tukey’s post hoc test, ****P < 0.0001 (n = 5 donors per group). h, Levels of mitochondrial proteins quantified by immunoblot in young and aged human MDMs treated with vehicle or C52 (100 nM, 20 h). Two-way ANOVA with Tukey’s post hoc test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (n = 6 donors per group). i, Liquid chromatography coupled to mass spectrometry (LC–MS) measurements of glycolytic and TCA cycle metabolites in young and aged human MDMs treated with vehicle or C52 (100 nM, 20 h; n = 3 donors per group). j, Phagocytosis of fluorescent E. coli particles in young and aged human MDMs treated with vehicle or C52 (100 nM, 20 h). Two-way ANOVA with Tukey’s post hoc test, ***P = 0.0006, ****P < 0.0001 (n = 6 donors per group).
We then tested the effect of pharmacological inhibition of inflammatory EP2 in aged human MDMs. PGE2 levels and GYS1 activation increased in human MDMs derived from aged compared to young individuals (Extended Data Fig. 5a–c). Consistent with our genetic data in aged Cd11bCre;EP2lox/lox mice, pharmacological inhibition of EP2 in aged human MDMs normalized glycogen levels and restored glycolysis and mitochondrial OCR to levels similar to those observed in young human MDMs (Fig. 3f, g, Extended Data Fig. 5d). EP2 blockade in aged human MDMs restored mitochondrial protein levels, membrane potential and reactive oxygen species to youthful levels34 (Fig. 3h, Extended Data Fig. 5e–g). Targeted metabolomics and 13C-glucose tracing confirmed the rescue of glucose flux in aged human MDMs treated with EP2 inhibitor, and phagocytosis also improved in these human MDMs (Fig. 3i, j, Extended Data Fig. 5h, i). Inhibition of inflammatory COX-2 also improved mitochondrial respiration in aged human MDMs (Extended Data Fig. 5j, k).
The activity of respiratory complex II of the electron transport chain (succinate dehydrogenase (SDH)) is suppressed in aged macrophages17. Low SDH activity leads to the accumulation of succinate—a TCA cycle metabolite that stabilizes the activity of hypoxia-inducible factor 1α (HIF-1α), which is an activator of pro-inflammatory cytokine expression16,31–33. Accumulation of succinate is also observed in macrophages stimulated with lipopolysaccharide (LPS); in this context, itaconate, an endogenous inhibitor of SDH, is generated from the TCA cycle intermediate cis-aconitate by cis-aconitate decarboxylase, which is highly induced in settings of inflammation31. Here, LPS-mediated changes in OCR and ECAR or succinate accumulation did not change with EP2 blockade, and nor did the levels of aconitate and itaconate (Extended Data Fig. 6a–c). These data indicate that the metabolic state of aged macrophages is distinct from that of LPS-activated macrophages (Extended Data Fig. 6d).
Glycogen is a fuel source that is used by many cell types, including immune cells35,36. In ageing macrophages, an increase in the activity of PGE2, EP2 and GYS1 enhanced the sequestration of glucose into glycogen, leading to lower glucose flux and bioenergetic insufficiency. As cells can normally use alternative fuel sources that bypass GYS1—for example, glutamine or lactate—we wondered whether aged macrophages were unable to metabolize other fuel substrates, thus rendering them more metabolically vulnerable than young macrophages (Extended Data Fig. 6e–i). Indeed, labelling of human MDMs with 13C-isotopes of glutamine, pyruvate, lactate and glucose revealed a fundamental dependence of aged human MDMs on glucose as a fuel source. Whereas young macrophages stably incorporated 13C derived from glucose, pyruvate, lactate and glutamine into glycolytic and TCA cycle intermediates, aged macrophages metabolized only glucose. Furthermore, assessment of fuel flexibility confirmed the dependence of aged macrophages on glucose when pyruvate transport was inhibited, indicating that glucose availability and flux are essential for oxidative phosphorylation in aged macrophages (Extended Data Fig. 6j, k).
EP2 blockade reverses cognitive ageing
We then tested whether in vivo pharmacological inhibition of EP2 signalling in aged mice might elicit similar effects on inflammation and cognitive function to those that were observed in aged Cd11bCre;EP2lox/lox mice. EP2 expression localized specifically to IBA1-positive microglia in the hippocampal CA1 area and the parietal cerebral cortex (Extended Data Fig. 7a–d). Administration of the brain-penetrant EP2 inhibitor C52 for one month restored pro- and anti-inflammatory factors in plasma and hippocampus to youthful levels and reduced the levels of CD68, a lysosomal protein associated with microglial activation (Fig. 4a–c, Extended Data Fig. 7e–h). Brain microglia and peritoneal macrophages assayed for in vivo incorporation of 13C-glucose showed reduced glycogen synthesis and enhanced glycolytic and TCA cycle labelling after in vivo EP2 inhibition (Fig. 4d, Extended Data Fig. 8a, Supplementary Fig. 3). TEM of hippocampi from aged mice treated with C52 showed a resolution of mitochondrial morphology to a more youthful state (Extended Data Fig. 8f–h). Functionally, EP2 blockade reversed age-associated spatial memory deficits in the novel object location and Barnes maze tasks and restored synaptic proteins to similar levels to those of young mice (Fig. 4e, f, Extended Data Figs. 7i, j, 8b, 9b). Electrophysiological recordings in aged mice also showed that treatment with C52 restored hippocampal CA1 long-term potentiation to youthful levels (Fig. 4g, Extended Data Fig. 8c). Together, these results with C52 recapitulate our findings in aged Cd11bCre;EP2lox/lox mice.
Fig. 4 |. EP2 blockade reverses age-associated inflammation and spatial memory loss.
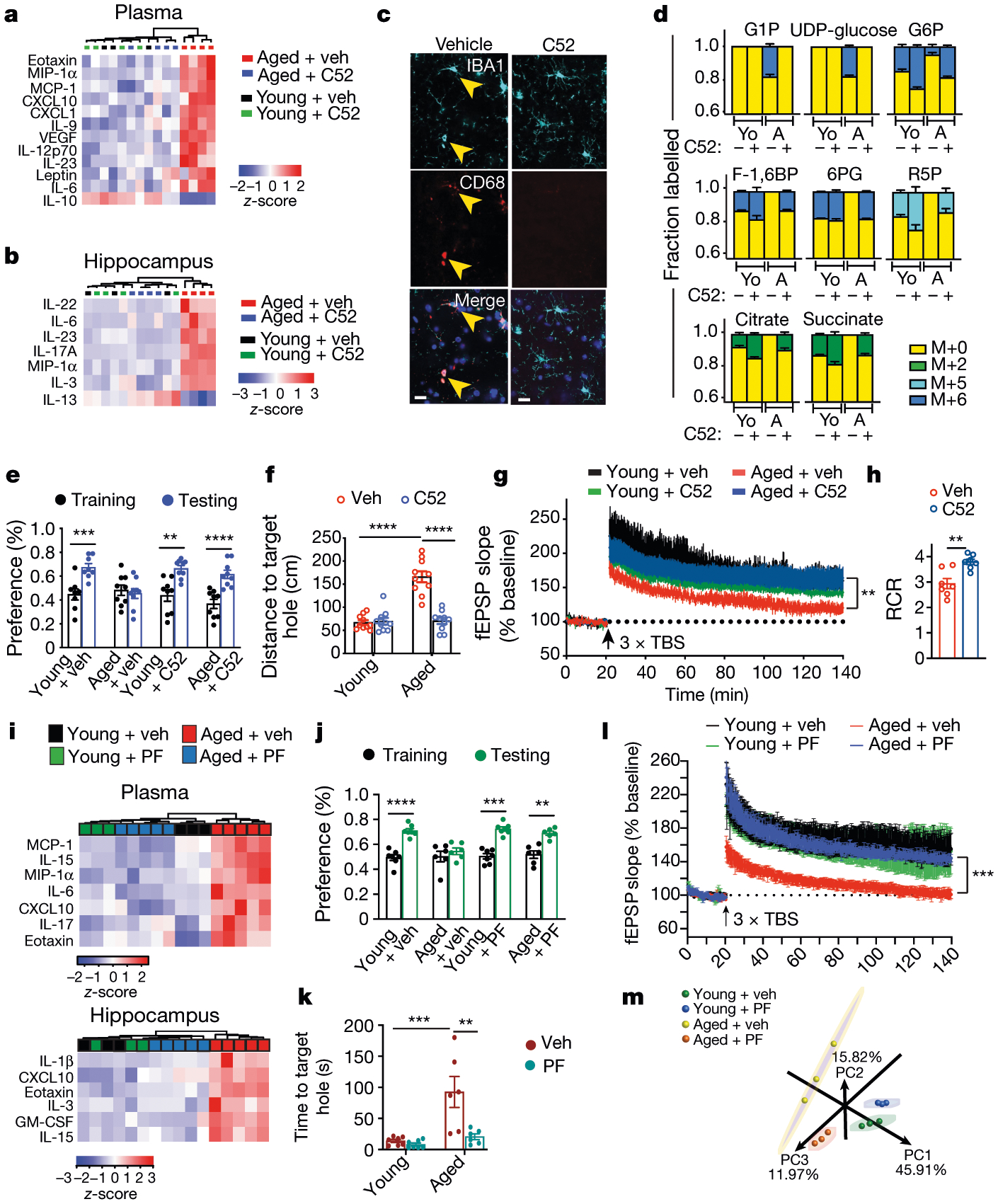
Data are mean ± s.e.m. a–g, Young (3–4-month-old) and aged (22–24-month-old) mice were administered vehicle or brain-penetrant C52 (10 mg per kg per day) for 1 month (a–c), 10 days (d) and 2 weeks (e–g). a, b, Hierarchical clustering of significantly regulated immune factors in the plasma (a) and hippocampus (b) (n = 3 young + veh, n = 4 mice per group for all other groups). c, Immunofluorescent staining of the hippocampal CA1 region for IBA1+ microglia. Scale bars, 10 μm. d, Mass labelling of brain microglia from mice treated with vehicle or C52 and administered U-13C-glucose 4 h before collection. Aged microglia increase incorporation of 13C-glucose into glycogen precursors and reduce incorporation into TCA cycle intermediates; this is reversed with EP2 blockade (n = 3 mice per group). Yo, young; A, aged. e, Per cent preference in the novel displacement object task. Paired two-tailed Student’s t-test, **P = 0.0014, ***P = 0.0003, ****P < 0.0001 (n = 8 young + veh, n = 9 mice per group for all other groups). f, Distance travelled to the target hole in the Barnes maze. Two-way ANOVA, age and treatment P < 0.0001; Tukey’s post hoc test, ****P < 0.0001 (n = 10 mice per group). g, Long-term potentiation in the CA1 hippocampal region. Two-way ANOVA, age and treatment P < 0.0001; Sidak’s multiple comparisons with Geisser–Greenhouse correction, **P = 0.0096 (n = 10–12 slices, n =5 mice per group). h, Synaptic mitochondria of 16-month-old mice treated with vehicle or C52 (10 mg per kg per day, 10 days), assayed for respiration control ratio (RCR).Two-tailed Student’s t-test, **P = 0.0025 (n = 7 mice per group). i–l, Young (3–4-month-old) and aged (22–24-month-old) mice were administered vehicle or brain-impermeant PF-04418948 (PF; 2.5 mg per kg per day, 6 weeks). i, Hierarchical clustering of significantly regulated immune factors in plasma and hippocampus (n = 3 young, n = 5 aged mice per group). j, Per cent preference in novel object location task. Paired two-tailed Student’s t-test, **P = 0.0074, ***P = 0.0004, ****P < 0.0001 (n = 7 young, n = 6 aged mice). k, Time to target hole in the Barnes maze. Two-way ANOVA, age (P = 0.0008) and treatment (P = 0.0034); Tukey’s post hoc test, ***P = 0.0005, **P = 0.0022 (n = 7 young, n = 6 aged mice). l, Hippocampal CA1 long-term potentiation; two-way ANOVA, age and treatment P < 0.0001; Sidak’s multiple comparisons with Geisser–Greenhouse correction, ***P = 0.002 (n = 6 slices, n = 5 mice per group). m, Principal component (PC) analysis of peritoneal macrophage transcriptomics. Ellipses represent 95% confidence intervals.
Healthy mitochondria are critical for synaptic neurotransmission and plasticity. We assessed the integrity of synaptic mitochondria by determining the extent of coupling between the electron transport chain and oxidative phosphorylation of ADP to ATP in synaptosomes37 (Fig. 4h, Extended Data Fig. 8d, e). The respiratory control ratio, represented as the ratio of state III/state IVo, was significantly increased after inhibition of EP2, indicating improved mitochondrial coupling of electron transport and ATP synthesis. These data demonstrate that inhibition of inflammatory EP2 improves the mitochondrial health of aged synapses.
As the EP2 receptor is highly expressed in aged peripheral myeloid cells as well as in aged brain microglia (Extended Data Figs. 1c, k, 9d, e, l), we examined whether peripheral EP2 blockade could reverse age-associated inflammation and hippocampal memory deficits. Administration of the brain-impermeant EP2 antagonist PF-0441894822 for six weeks reduced levels of pro-inflammatory factors not only in the blood but also in the hippocampus, and—notably—restored hippocampal memory function as well as long-term potentiation to youthful levels (Fig. 4i–l, Extended Data Figs. 1g, 9c, f–m). Transcriptomic analysis of peritoneal macrophages revealed a marked restoration of youth-associated immune and metabolic signatures in aged mice that were treated with PF-04418948 (Fig. 4m, Extended Data Fig. 10).
Discussion
Our study suggests that the development of maladaptive inflammation and cognitive decline in ageing may not be a static or permanent condition, but rather that it can be reversed by inhibiting inflammatory PGE2 signalling through the myeloid EP2 receptor. First, we find that ageing is associated with a significant increase in pro-inflammatory PGE2 signalling in myeloid cells, which drives the sequestration of glucose into glycogen through the AKT–GSK3β–GYS1 pathway and away from the generation of ATP. Second, we uncover a fundamental vulnerability of ageing myeloid cells, in which they become dependent on glucose and are unable to use alternative energy sources to support mitochondrial respiration. These two mechanisms converge, leading to a reduction of glucose flux and the development of an energy-depleted state that drives pro-inflammatory immune responses. Third, by directing glucose towards the production of ATP, as opposed to glycogen storage, inhibition of myeloid EP2 in ageing cells reverts the polarization state to a more homeostatic anti-inflammatory state that prevents age-associated cognitive decline. We also demonstrate that peripheral EP2 blockade is sufficient to re-establish youthful immune homeostasis not only in the blood, but also in the brain, and to restore hippocampal function and plasticity in aged mice. The mechanisms underlying this rescue are probably distinct from those that underlie the effect of a brain-penetrant EP2 inhibitor that can directly target microglial EP2 signalling. Peripheral EP2 blockade may lead to changes in the composition of blood, components of which could beneficially affect the ageing cerebrovascular endothelium or penetrate the brain parenchyma to improve neuronal function.
Finally, our findings are consistent with a feedforward loop involving the inflammatory COX-2–PGE2–EP2 cascade, in which increasing PGE2 signalling via the EP2 receptor induces additional COX-2 expression and activity, further amplifying downstream PGE2 generation and signalling24,38,39. Thus, inhibition of EP2-dependent changes in myeloid metabolism may represent a new approach to disorders of ageing, with greater specificity than the use of non-steroidal anti-inflammatory drugs that target COX-2 and COX-1 and suppress both beneficial and toxic prostaglandin signalling pathways40.
Methods
Mice
This study was conducted in accordance with National Institutes of Health (NIH) guidelines and the Institutional Animal Care and Use Committee at Stanford University approved protocols. All mice were housed in an environmentally controlled, pathogen-free barrier facility on a 12-h light–dark cycle, with food and water available ad libitum. The Stanford Veterinary Service Center monitors and maintains pathogen-free mouse housing. C57BL/6J mice were bred in our laboratory using mice purchased from The Jackson Laboratory or obtained from the NIH aged rodent colony. For genetic and pharmacological experiments, both control and experimental genotypes were housed together in the same cages. Young (2–4-month-old) and aged (20–24-month-old) C57BL/6J mice used in each experiment were age- and source-matched. Cd11bCre and Cd11bCre;EP2lox/lox mice have been previously described24. For confirmation of EP2 expression in microglia, C57BL/6J ROSA-CreERT2 mice (Jackson Laboratories) were crossed to C57BL/6J EP2lox/lox mice24 or wild-type mice for generation of ROSA-CreERT2;EP2lox/lox and ROSA-CreERT2;EP2+/+ mice. Tamoxifen (2 mg intraperitoneally for 5 days; Sigma) was administered to mice of all Rosa-CreERT2 genotypes at 8 weeks of age following established protocols41.
Materials
PGE2, Butaprost, PF-04418948 and Iloprost were purchased from Millipore-Sigma. ONO-AE248 and ONO-AE1–329 were gifts from Ono Pharmaceuticals. C52 (Charnwood Molecular) and PF-04418948 (Millipore-Sigma) were resuspended in 40% PEG (Sigma-Aldrich) and 60% of a 30% Kolliphor HS15 solution (Sigma-Aldrich) and administered orally at 10 mg/kg/d and 2.5 mg/kg/d, respectively. U-13C-glucose, U-13C-lactate, U-13C-glutamine and U-13C-pyruvate were purchased from Cambridge Isotopes. HUSH-29 plasmids containing shRNAs to human GYS1 were purchased from Origene Technologies. Human MDMs were incubated with 1640 medium with GlutaMAX + HEPES without sodium pyruvate (Thermo Fisher Scientific, 72400146). Mouse macrophages were incubated in DMEM without sodium pyruvate (Sigma-Aldrich, D5796).
OCR and ECAR
Cells were counted and plated at 1.8 × 105 (mouse peritoneal macrophages) or 1.0 × 105 (human MDMs) cells per well in a Seahorse XF24 Cell Culture Microplate for all experiments (Agilent). Cells were then treated with indicated inhibitors or agonists in each experiment for 20 h. Cells were washed twice with Agilent Seahorse XF medium (Agilent) supplemented with 1 mM pyruvate, 2 mM l-glutamine and 10 mM d-glucose; a final volume of 525 μl was placed in each well. Cells were then incubated in a 0% CO2 chamber at 37 °C for 1 h before being placed into a Seahorse XFe24 Analyzer (Agilent). For OCR and ECAR mitostress test experiments, cells were treated with 1 μM oligomycin, 2 μM FCCP and 0.5 μM rotenone or antimycin (indicated by three black arrows in representative OCR traces). For mitoFlex Fuel test experiments, cells were treated with UK5099 (200 μM) and BPTES (200 μM) or Etomoxir (80 μM). A total of three OCR and pH measurements were taken after each compound was administered. All Seahorse experiments were repeated at least three times unless otherwise indicated. All OCR and ECAR data were normalized to cell number per well using CyQUANT (Thermo Fisher Scientific).
Transmission electron microscopy
Primary peritoneal mouse macrophages from young (3–4-month-old) and aged (20–23-month-old) Cd11bCre;Ep2lox/lox and C11bCre mice were plated in 6-well plates at 2 × 106 cells per well; 2 × 106 cells ml−1 were isolated for electron microscopy processing. Hippocampi were isolated from young (3–4-months) and aged (22–24 months) mice treated with C52 (10 mg/kg/d, 1 month). Cells and hippocampi were fixed in 2% glutaraldehyde and 4% paraformaldehyde in 0.1 M sodium cacodylate (pH 7.4), treated with 10% gelatin solution in sodium cacodylate buffer and incubated with 2% osmium tetroxide. Cell pellets were stained with 1% uranyl acetate, dehydrated in ethanol and embedded in resin (EPON epoxy resin). Sections were generated using a Leica EM UC7 ultramicrotome (Leica Microsystems) at 50 nm and placed within grids stained with a 1:1 mix of 3% uranyl acetate, 50% acetone for 30–60 s. Grids were imaged with a JEM 1400 transmission electron microscope (JEOL) at × 1,200 for low magnification and × 12,000 for high magnification (unless otherwise noted) using Gatan Microscopy Suite software (Gatan). Images were quantified by an individual blinded to experimental conditions in ImageJ. Abnormal mitochondria were defined as containing at least one of the following features: paracrystalline inclusions, linearization of cristae and abnormal angular features, concentric layering of cristae membranes, matrix compartmentalization or nanotunnelling, in combination with doughnut-shaped mitochondria. Microglia from hippocampal sections were identified in TEM by presence of the following characteristics: relative size (3–10 μm), degenerating myelin vacuoles, lipid inclusions, prominent heterochromatin nets throughout the nucleus and electron-dense cytoplasm.
Flow cytometry
Human MDMs were plated in 10-cm plates at 10 × 106 cells per well and collected using 0.25% trypsin-EDTA at 37 °C. Cells were washed with flow cytometry buffer (PBS with 2% FCS, 2 mM EDTA and 25 mM HEPES, pH 7.4), and then incubated with blocking buffer (5% mouse serum in flow cytometry buffer) for 15 min at 4 °C. Cells were then stained with the desired antibodies for 30 min at 4 °C. Dead cells were identified and excluded using 0.5 μg/ml propidium iodide. The following controls were used: unstained cells; single-stained cells; and dead cells. The cells were gated using forward and side scatter, as well as live–dead staining using 4,6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific). Cells were analysed on a BD FACSAria II (BD Biosciences). Raw FCS files were analysed with FlowJo software (FlowJo). Antibodies to the following markers were obtained from BD Biosciences (listed as: catalogue number, channel, dilution, clone): Human CD14 (557831, APC-Cy7, 1:50, MφP9); human CD64 (560970, FITC, 1:50, 10.1); human CD206 (566281, FITC, 1:50, 19.2); human CD68 (565595, PE-Cy7, 1:200, Y1/82A); human CD163 (556018, PE, 1:50, GHI/61); human CD86 (555660, APC, 1:50, FUN-1); mouse CD80 (564160, BV421, 1:50, 16-10A1); mouse CD14 (553740, PE-Cy7, 1:50, rmC5-3); mouse CD71 (561936, FITC, 1:50, C2F2); and mouse CD86 (563055, BV605, 1:50, GL1). Anti-EGR2 was obtained from Invitrogen (1706691-82, APC, 1:50, ERONGR2).
Chemokine and cytokine multiplex assay
Hippocampal lysates or mouse plasma were stored at −80 °C and cytokine analysis was carried out at the Human Immune Monitoring Core (Stanford University) or Eve Technologies (Calgary) using Luminex mouse 39-plex kits and human 71-plex kits. Plates were read using a Luminex LabMap200 instrument or a MSD chemiluminescence instrument with a lower bound of 100 beads per sample per measured cytokine. Each sample was tested in triplicate. MFI was averaged over duplicate wells for each cytokine per sample on each plate. All were perfused with PBS before isolation of hippocampi and other brain tissue.
Novel object location
The novel object location (object location memory task) protocol was adopted from a previous study42 with minor modifications. Mice interacted with the chamber (a 16 × 16 × 15-inch white box made from PVC) over the course of 2 days involving 1 habituation period, 3 training sessions and 1 testing session. On the day of training (day 1) mice were placed in the middle of an empty chamber and given 5 min to explore the chamber. Mice were then placed in an independent holding cage for an inter-training interval (ITI) of 3 min. The objects used were a plastic bottle and seasoning shaker of similar size (3 inch H × 1 inch W × 1 inch L). After the habituation session, the mice then underwent three 10-min training sessions each with a 3-min ITI in between sessions. Twenty-four hours after the last training session, a testing session was conducted in which one of the objects was displaced to a new location. Mice were recorded using a JVC Everio HD camcorder GZE200 and analysed with Kinovea video tracking software. Exploration of the objects was defined as the amount of time mice were oriented towards an object with their nose within 1 cm of it, and was scored by an experimenter blind to experimental group.
Barnes maze
The Barnes maze protocol was adopted from a previous study43 with minor modifications. The maze was made from a circular, 8-mm thick, white PVC slab with a diameter of 36 inches. Twenty holes with a diameter of 3 inches were made on the perimeter at a distance of 1 inch from the edge. This circular platform was then mounted on top of a rotating stool, 30 inches above the ground.
The escape cage was made by using a mouse cage and assembling a platform and ramp 2 inches below the surface of the maze. The outside of the walls of the cage were covered with black tape so as to prevent light for entering the escape cage. The maze placed in the centre of a dedicated room and two 120-W lights were placed on the edges of the room facing towards the maze to provide an aversive stimulus for the mice. Eight simple coloured-paper shapes (squares, rectangles and circles) were mounted on the walls of the room as visual cues.
After testing each mouse, the maze was cleaned with 70% ethanol and rotated clockwise after every mouse to avoid intra-maze odour or visual cues. All sessions were recorded using a JVC Everio HD camcorder GZ-E200 and analysed with Kinovea video tracking software.
The mice interacted with the Barnes maze in three phases: habituation (day 1), training (days 2–3) and probe (day 4). Before starting each experiment, mice were acclimated to the testing room for 1 h. Then all mice from one cage (n = 4 or 5) were placed in individual holding cages, where they remained until the end of their testing sessions each day. On habituation day, the mice were placed in the centre of the maze within a vertically oriented black PVC pipe 4 inches in diameter and 7 inches in height for 15 s. The mice were then guided slowly to the hole that led to the escape cage over the course of 10–15 s. The mice were given 3 min to independently enter the target hole, and if they did not, they were nudged with the PVC pipe to enter. The 120-W lights were then shut off and mice were allowed to rest in the escape cage for 2 min.
The training phase occurred 24 h after the habituation phase and was split across 2 days (days 2 and 3), with 3 trials on the first day and 2 trials on the second day. During each trial, the mice were placed in the centre of the maze within the PVC pipe for 15 s and afterwards were allowed 3 min to explore the maze. If mice found and entered the target hole before 3 min passed, the lights were shut off and the training trial ended. Mice were allowed to rest in the escape cage for 2 min. If at the end of the three minutes the mice had not entered the target hole, they were nudged with the PVC pipe. A total of five trials were conducted. During each trial, latency (time) to enter the target hole as well as distance travelled were recorded.
The probe phase occurred 24 h after the training phase and was conducted on the last day (day 4). Mice were placed in the centre of the maze within the PVC pipe for 15 s and afterwards were allowed 3 min to explore the maze. The probe session ended whenever the mouse entered the target hole or if 3 min had passed. During the probe phase, measures of time spent per quadrant, latency to enter the target hole and distance travelled were recorded.
Electrophysiology
To measure the cellular mechanism of learning and memory, we used a modified protocol previously described44. In brief, mice were euthanized by cervical dislocation, and the hippocampus was rapidly dissected out into ice-cold (4 °C) artificial cerebrospinal fluid (ACSF), saturated with carbogen (95% O2/5% CO2). ACSF consisted of (in mM): 124 NaCl, 4.9 KCl, 24.6 NaHCO3, 1.20 KH2PO4, 2.0 CaCl2, 2.0 MgSO4 and 10.0 glucose, pH 7.4. Transverse hippocampal slices (350 μm thick) were prepared from the dorsal area of the hippocampus with the McIlwain tissue chopper and transferred to a recovery chamber for at least 1.5 h with oxygenated ACSF at room temperature before being placed into a submerged-type chamber in which they were kept at 32 °C and continuously perfused with ACSF at a flow-rate of 1.5 ml/min. Slices were carefully positioned on a R6501A multi-electrode array (Alpha MED Scientific) with electrodes arrayed in an 8 × 2 matrix with interpolar distance of 150 μm; each matrix measured 50 μm × 50 μm. After a 30-min incubation, the fEPSPs in CA1 were recorded by stimulating downstream electrodes in the CA1 and CA3 regions along the Schaffer collateral pathway. Signals were acquired using the MED64 System (AlphaMED Sciences, Panasonic). The time course of the fEPSP was calculated as the descending slope function for all experiments. Input/output curves were established by applying increasing stimulus currents to the pathway from 10 μA to 90 μA (in 5-μA increments) and recording evoked responses. After input/output curves had been established, the stimulation strength was adjusted to elicit a fEPSP slope at 35% maximal value, which was maintained throughout the experiment. During baseline recording, a single response was evoked at a 30-s interval for at least 20 min. To induce a strong form of long-term potentiation, three episodes of theta-burst stimulation (TBS) were used, each TBS consisting of 10 bursts of 4 stimuli at 100 Hz separated by 200 ms (double pulse-width), followed by recording evoked responses beginning 1 min after the induction of long-term potentiation and continuing every 30 s until the end of the experiments. Experiments using control and transgenic mice were interleaved with each other. The mean baseline fEPSP value was calculated and percentage change from baseline after the TBS was analysed for long-term potentiation.
Quantitative immunoblotting
Quantitative immunoblotting was carried out as previously described38. Mouse anti-β-actin (1:10,000; Sigma-Aldrich) or anti-β-tubulin was used as an internal loading control. Imaging of blots was carried out using a Licor CLX-1306 with fluorescent secondary antibodies. Analysis was carried out in Image Studio Lite and signal intensities were normalized to loading controls, when applicable. Antibodies and their concentrations are listed as follows (antibody, company, catalogue number, dilution): anti-synapsin (Millipore Sigma; AB1543P; 1:1,000); anti-SNAP25 and anti-SNAP25 (Abcam ab41455 and ab2723, respectively, 1 μg/ml); anti-CamKIIa (pan; Cell Signaling Technology; 3362S; 1:1,000); anti-EP1 and anti-EP3 (Cayman Chemical, 101740 and 101760, respectively, 1:200); anti-EP2 (Abcam, Ab167171, 1:1,000); anti-EP4 (Santa Cruz, Sc-55596, 1:100); anti-p(Ser473)AKT, anti-AKT (pan), anti-p(Ser9) GSK3β (Cell Signaling Technology, 4060S, 2920S and 9336, respectively, 1:1,000, 1:1,000 and 1:500, respectively); anti-GSK3β (Abcam, ab93926, 1:500); anti-p(Ser641,645,649)GYS1 (Millipore Sigma, 07-817, 1:500); anti-GYS1 (human) and anti-GYS1 (mouse) (Thermo Fisher Scientific, MA5-15802 and MA5-15022, respectively, 1:500 and 1:1,000, respectively); anti-TOM20 and anti-TIM17 (Santa Cruz, sc-17764 and sc-271152, respectively, 1:500); anti-VDAC, anti-PGES and anti-β-tubulin (rb host) (Abcam, ab14734, ab62050 and ab6406, respectively, 1:500); anti-OPA1 (BD Biosciences, 612607, 1:1,000); anti-MFN2 (Abnova, 11325-6A8, 1:1,000); anti-p(Ser616)DRP1 (Cell Signaling Technology, 4494S, 1:250); anti-FIS1 (Proteintech, 10956-1-AP, 1:1,000); anti-COX2 (Cayman Chemical, 160126, 1:1,000); anti-β-tubulin (ms host) (EMD Millipore, 05-661, 1:5,000); and anti-β-actin (Millipore Sigma, A5441, 1:10,000).
Glycogen quantification
Glycogen was quantified according to the manufacturer’s instructions using a commercially available colorimetric kit (ab65620, Abcam).
TMRE quantification
Mitochondrial membrane potential was measured according to the manufacturer’s instructions using the tetramethylrhodamine ethyl ester (TMRE) mitochondrial membrane potential assay kit (ab113852, Abcam). Fluorescence was analysed using a SpectraMax M2e microplate reader (Molecular Devices; excitation, 549 nm; emission, 575 nm).
Quantification of cellular reactive oxygen species
Cellular reactive oxygen species (ROS) were measured according to the manufacturer’s instructions using the cellular ROS assay kit (ab186027, Abcam). Fluorescence was analysed using a SpectraMax M2e microplate reader (Molecular Devices; excitation, 520 nm; emission, 605 nm).
Peritoneal macrophages
Peritoneal macrophages were collected from 2–4-, 6- and 20–24-month-old Cd11bCre;EP2lox/lox, Cd11bCre and wild-type mice. Mice were injected intraperitoneally with 1.5 ml 3% (w/v) thioglycolate medium (BD Biosciences), and primary macrophages were isolated 3–4 days later by flushing with ice-cold 1× PBS buffer (Corning). Cells were seeded at a density of 3 × 106 cells per well in DMEM supplemented with 10% heat-inactivated fetal bovine serum (FBS; Sigma-Aldrich) and 100 U ml−1 penicillin and streptomycin, and maintained at 5% CO2 at 37 °C. After overnight culture, cells were washed twice with medium to remove non-adherent cells.
Neuron and astrocyte culture
Hippocampi were dissected from embryonic day 17.5 mouse embryos, dissociated using trypsin (2 mg/ml) and DNase I (0.6 mg/ml), and plated at a density of 100,000 cells per well in Seahorse XF24 culture plate coated with poly-l-lysine. Neurons were maintained in neurobasal medium, B27 (Invitrogen) and penicillin–streptomycin (Invitrogen) at 37 °C in a humidified atmosphere containing 5% CO2. The medium was refreshed twice weekly by replacing half the medium with fresh medium. After 12–14 days in vitro, cells underwent real-time oxygen consumption analysis with the Seahorse XFe24 machine and the MitoStress test kit.
Primary astrocyte cultures were prepared from cerebral cortices of postnatal day 1–2 C57BL/6J mice. In brief, dissociated cortical cells were suspended in DMEM/F12 50/50 (Life Technologies, 11320-033) containing 25 mM glucose, 4 mM glutamine, 1 mM sodium pyruvate and 10% FBS, and plated on uncoated 75-cm2 flasks at a density of 1.5 × 105 cells per cm2. Monolayers of astrocytes were obtained 12–14 days after plating. Cultures were gently shaken, and floating cells (microglia) were collected, resulting in a more than 95% pure culture of astrocytes. Astrocytes were dissociated by trypsinization and then reseeded at 4 × 104 cells per well in a XF24-well cell culture microplate, and cells underwent real-time oxygen consumption analysis with the Seahorse XFe24 machine and the MitoStress test kit.
Human MDMs
Peripheral blood mononuclear cells from de-identified healthy donors (young, below 35 years old; aged, above 65 years old) were obtained from the Stanford Blood Center and transferred to 50-ml conical tubes. Samples were diluted with 20 ml PBS and layered onto 10 ml of Ficoll-Paque (GE Healthcare) using a Pasteur pipette. Tubes were centrifuged at 1,500 rpm for 25 min without brake at 20 °C. The mononuclear cell layer was transferred to a new 50-ml conical tube, resuspended in 50 ml 1× PBS and centrifuged at 1,500 rpm for 10 min, repeated twice. After centrifugation, monocytes were isolated using a Monocyte Isolation Kit, human (130-096-537, Miltenyi Biotech). Cells were then plated 10 × 106 per 10-cm Petri dish and differentiated for 7 days in Roswell Park Memorial Institute (RPMI) medium supplemented with 10% FBS, 1% penicillin–streptomycin and 50 ng ml−1 M-CSF (Peprotech).
In vivo mass labelling of macrophages and microglia
Young and aged mice were administered C52 for 10 days at 10 mg/kg/day by oral gavage. On day 10, U-13C-glucose (1g/kg by gavage) was administered for in vivo isotope tracing of brain microglia and peritoneal macrophages were collected 4 h later. Microglia were isolated by flow cytometry (see ‘Microglial isolation’) and peritoneal macrophages were isolated by lavage and processed by LC–MS for measurement of metabolites (see ‘LC–MS measurement of metabolites for in vitro and in vivo studies’).
LC–MS measurement of metabolites for in vitro and in vivo studies
Isotope labelling was performed as previously described45. Labelled compounds U-13C-glucose (Cambridge Isotope Laboratories) were added to customized RPMI medium lacking glucose (customized RPMI 1640 medium + Gibco GlutaMAX supplement + HEPES; Thermo Fisher Scientific) or prepared in 0.9% saline for oral gavage.
Water soluble metabolites: Human MDMs were grown on 10-cm plates (Corning). For steady-state labelling of metabolites, U-13C-glucose (11.1 mM)-labelled medium was replaced every day and then 20 h before metabolic analysis, at which point cellular metabolism was quenched by rapidly cooling cells on dry ice. Cells were washed with 1× PBS twice by aspirating the medium and immediately adding 1 ml −80 °C 80:20 methanol/water. After 20 min of incubation on dry ice, the resulting mixture was scraped, collected into a centrifuge tube and centrifuged at 10,000g for 5 min at 4 °C. Pellets were then extracted again with 500 μl −80 °C 80:20 methanol/water, incubated for 5 min and centrifuged at 10,000g for 5 min at 4 °C. Both extractions were combined into a 1.5-ml microcentrifuge tube. The supernatants were centrifuged at 16,000g for 20 min to remove any residual debris before analysis. Supernatants were analysed within 24 h by LC–MS. The LC–MS method involved hydrophilic interaction chromatography (HILIC) coupled to the Q Exactive PLUS mass spectrometer (Thermo Fisher Scientific). The LC separation was performed on a XBridge BEH Amide column (150 mm 3 2.1 mm, 2.5 mm particle size, Waters). Solvent A is 95%: 5% H2O: acetonitrile with 20 mM ammonium bicarbonate, and solvent B is acetonitrile. The gradient was 0 min, 85% B; 2 min, 85% B; 3 min, 80% B; 5 min, 80% B; 6 min, 75% B; 7 min, 75% B; 8 min, 70% B; 9 min, 70% B; 10 min, 50% B; 12 min, 50% B; 13 min, 25% B; 16 min, 25% B; 18 min, 0% B; 23 min, 0% B; 24 min, 85% B; 30 min, 85% B. Other LC parameters were: flow rate 150 ml/min, column temperature 25 °C, injection volume 10 ml and autosampler temperature 5 °C. The mass spectrometer was operated in both negative- and positive-ion mode for the detection of metabolites. Other MS parameters were: resolution of 140,000 at m/z 200, automatic gain control (AGC) target at 3 ×106, maximum injection time of 30 ms and scan range of m/z 75–1,000. Data were analysed using MAVEN software, and isotope labelling was corrected for natural 13C abundance in the tracer experiments.
For identification and isolation of hexose phosphates and glycolytic intermediates, capillary electrophoresis mass spectroscopy was used as previously described46. In brief, adherent cells on dishes were washed with 5% mannitol aqueous solution at room temperature. The cells were immersed in 400 μl methanol for 30 s, and 275 μl of the Internal Standard Solution (10 μM, Solution ID: H3304-1002, Human Metabolome Technologies) for 30 s. The extraction liquid was centrifuged at 2,300g for 5 min at 4 °C. The supernatant (400 μl) was centrifugally filtered at 9,100g for 4 h at 4 °C through a 5-kDa cut-off filter (Millipore) to remove proteins, and then the filtrate was lyophilized and suspended in 25 μl Milli-Q water. The metabolite suspension was analysed by capillary electrophoresis time of flight mass spectrometry (CE-TOF/MS) using an Agilent capillary electrophoresis (CE) system equipped with an Agilent 6210 TOFMS, an 1100 isocratic high-performance liquid chromatography pump, a G1603A CE-MS adaptor kit and a G1607A CE-electrospray ionization-mass spectrometry (ESI-MS) sprayer kit (Agilent Technologies). The system was controlled using G2201AA ChemStation software v.B.03.01 for CE (Agilent).
Nucleofection
Human GYS1 shRNA sequences are as follows: shRNA 1: 5′-UCAACAG CAGUGCCGACCGACCGACCGUGAAGGUG-3′; shRNA 2: 5′-GAUCGAAA GACAGCCUGGUCA-3′. Plasmids containing human GYS1 shRNA or scramble (2 μg) (HuSH shRNA shGYS1 Lenti Cloning Vector (pGFP-C-shLenti)) were incubated with a mixture of nucleofection solution and P4 primary cell supplement (82 μl:18 μl nucleofection solution: supplement) and placed in nucleofection cuvettes. A total of 1 × 106 human MDMs were added to each cuvette and subjected to program DP-148 for the Nucleofector 4D X-unit device (Lonza). Immediately afterwards, 500 μl of DMEM (preincubated at 37 °C under 5% CO2 and supplemented with 20% FBS and 1% penicillin–streptomycin) was added. Cells were then plated in 10-cm plates and incubated 37 °C under 5% CO2 for 8 h before GYS1 protein expression was analysed by quantitative immunoblotting.
Phagocytosis assay
Human MDMs were grown in 10-cm plates at 10 × 106 cells per well and then trypsinized using 0.25% trypsin-EDTA at 37 °C. Cells were then plated in 96-well plates at 80,000 cells per well and the Vybrant Phagocytosis Assay Kit (Thermo Fisher Scientific) with E. coli particles was carried out following the manufacturer’s protocol.
PGE2 LC–MS detection
Brain tissue was homogenized with 500 μl of MeOH:formic acid (100:0.2) containing internal standard consisting of a mixture of deuterium-labelled prostaglandins, using microtip sonication. The samples were submitted to solid phase extraction using an Oasis HLB cartridge (5 mg; Waters) as previously described47,48. In brief, samples were diluted with water:formic acid (100:0.03) to give a final MeOH concentration of 20% by volume, applied to preconditioned cartridges and washed serially with water:formic acid (100:0.03), water:ethanol:formic acid (90:10:0.03), and petroleum ether. Samples were then eluted with 200 μl of MeOH:formic acid (100:0.2). The filtrate was concentrated with a vacuum concentrator (SpeedVac, Thermo Fisher Scientific). The concentrated filtrate was dissolved in 20 μl methanol and used for LC–MS/MS. The amount of prostaglandins in brain tissue was quantified using a previously described method48. In brief, a triple-quadrupole mass spectrometer equipped with an electrospray ionization (ESI) ion source (LCMS-8060; Shimadzu Corporation) was used in the positive and negative-ESI and multiple reaction monitoring (MRM) modes.
PGE2 ELISA detection
Cell lysates and medium were prepared according to the manufacturer’s instructions for PGE2 detection by enzyme-linked immunosorbent assay (ELISA) (KGE004B, Research & Diagnostic Systems).
Immunostaining
Quantification of microglia in free-floating 40-μm coronal sections of young and aged mice was carried out using anti-IBA1 (1:500; NB100-1028; Novus Biologicals), anti-CD68 (1:1,000; MCA1957GA; BioRad), anti-NeuN (1:1,000, MAB377, Millipore-Sigma) and anti-EP2 (1:200, ab167171, Abcam). For quantification of IBA1 and CD68, every eighth section through the brain was stained and imaged. Images were captured using a Keyence BZ-X800 fluorescence microscope (Keyence) using the Z-stack function in the BZ-X analyser software (Keyence).
Microglial isolation
Mice were anaesthetized and transcardially perfused with ice-cold PBS containing EDTA. Brains were minced with a razor blade and a single-cell suspension obtained by Dounce homogenization in a solution of HBSS containing HEPES, glucose and DNase I. The suspension was filtered through a 70-μm strainer. Myelin was removed using myelin removal beads (Miltenyi Biotec). Cells were stained with CD45–FITC (BioLegend 30-F11) and CD11b–PE Dazzle 594 (BioLegend M1/70) for microglia identification through flow cytometry for mass-labelling experiments. Live cells were identified by 7-AAD exclusion. For quantitative western blot experiments, microglia were isolated from whole brains using a tissue dissociation and Dounce homogenization protocol coupled with myelin removal following a previously established protocol49.
Isolation of synaptic mitochondria
Synaptic mitochondria were isolated as previously described37. In brief, brain cortices were removed and added to cold freshly prepared 9 ml of mitochondrial isolation buffer (225 mM mannitol, 75 mM sucrose, 2 mM K2PO4, 0.1% BSA, 5 mM HEPES and 1 mM EGTA (pH 7.2). The tissues were homogenized using a Dounce homogenizer. The resultant homogenate was centrifuged at 13,000g at 4 °C and layered on top of 3 × 2-ml discontinuous gradient of 15%, 23% and 40% Percoll (GE) and centrifuged at 34,000g for 14 min. After centrifugation, the band between 15% and 23% containing synaptosomes and the band between 23% and 40% containing nonsynaptic mitochondria were removed and subjected to wash in isolation buffer with 0.02% digitonin. The isolates were then pelleted by centrifugation at 16,500g for 15 min. The pellets were resuspended in IB and layered over another discontinuous gradient similar to described above. The band between 23% and 40% containing synaptic mitochondria was obtained and washed in ice-cold IB. Protein estimation was performed using a BioRad Bradford assay (BioRad Laboratories). Isolated mitochondria were immediately used for analysis. Ten micrograms of freshly isolated synaptic mitochondria were plated in XFe24 cell culture microplates in a volume of 50 μl mitochondrial assay solution (MAS: 70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA and 0.2% BSA with 10 mM succinate and 2 μM rotenone) and attached to the wells by spinning down the plates at 2,000 rpm at 4 °C. After attaching mitochondria to the plate wells, the volume of wells was made up to 450 μl of MAS containing substrate. In the meantime, Seahorse XF24 Flux Analyzer was equilibrated to 37 °C overnight a day before the assay. The final concentrations of substrates and inhibitors added to the wells were 4 mM ADP, 2.5μg/ml oligomycin A, 4 μM FCCP and 4 μM antimycin A. The coupling assays were run in 2–3 replicate wells for each independent biological sample. XFe24 data were collected according to Seahorse software. The point-to-point run for each well was exported and statistical analysis was conducted in Prism (GraphPad software) using two-tailed unpaired t-tests.
Measurement of PF-04418948 in plasma and brain
The concentration of PF-04418948 was measured in aged (20–22 months) mice treated with PF-04418948 2.5 mg/kg/d for 6 weeks in plasma and perfused brain using LC–MS (Shimadzu LC-20AD and ABSciex 4000 QTRAP) at Quintara Discovery. In brief, brain samples were first homogenized in two volumes of ice-cold water, then further twofold diluted in blank plasma. An aliquot of 20 μl of each plasma sample or plasma-diluted tissue homogenate was treated with 100 μl acetonitrile containing internal standard (Terfenadine). The mixture was vortexed on a shaker for 15 min and subsequently centrifuged at 4,000 rpm for 15 min. An aliquot of 70 μl of the extract was transferred to an injection plate and reconstituted in 70 μl of water for LC–MS/MS injection. Calibration standards and quality control samples were prepared by spiking the testing compound into blank plasma followed by processing with the samples.
Nanostring transcriptomics
For gene expression analysis on the NanoString nCounter system, 100 ng of RNA was hybridized to a multiplexed nucleotide probe pool for 16 h at 65 °C. Enriched targets were purified and quantified using the nCounter MAX Analysis System. NanoString data analysis was performed by Canopy Biosciences. Raw counts were normalized using the geometric mean of both positive control probes (technical variability) and housekeeping gene probes (assay input variability). Normalized data were uploaded to the interactive analysis platform Rosalind (https://rosalind.onramp.bio/), with a HyperScale architecture developed by OnRamp BioInformatics. Read distribution percentages, violin plots, identity heat maps and sample multidimensional scaling (MDS) plots were generated as part of the quality controlstep. The limma R library 1 was used to calculate fold changes and P values and perform optional covariate correction. Clustering of genes for the final heat map of differentially expressed genes was done using the PAM (Partitioning Around Medoids) method using the fpc R library 2 that takes into consideration the direction and type of all signals on a pathway, the position, role and type of every gene, and so on. Functional enrichment analysis of pathways, gene ontology, domain structure and other ontologies was performed using HOMER3. Several database sources were referenced for enrichment analysis, including InterPro v.4, NCBI v.5, MSigDB v.6 and 7, REACTOME v.8 and WikiPathways v.9. Enrichment was calculated relative to a set of background genes of relevance to the experiment.
Statistics and reproducibility
Data are expressed as mean ± s.e.m. unless otherwise indicated. Statistical comparisons were made in the Prism software using a Student’s t-test (for two groups meeting the normal distribution criteria, according to the Shapiro–Wilk normality test), Mann–Whitney U-test (for two groups not meeting the normal distribution criteria) or ANOVA with Tukey’s multiple comparison test (for groups across variables, with multiple comparisons between groups). Data were subjected to Grubbs’ test to identify the presence or absence of outlier data points; no outliers were excluded in our analyses. For all tests, P < 0.05 was considered significant, except for targeted metabolomics in which Q < 0.05. All experiments were repeated in at least duplicate, unless otherwise indicated. No statistical methods were used to predetermine sample size. The experiments were not randomized.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Raw data for nanostring analysis are available from the Gene Expression Omnibus under accession code GSE155992. Untargeted metabolomics data are provided in the source data file for Extended Data Fig. 3d. Source data are provided with this paper.
Extended Data
Extended Data Fig. 1 |. PGE2 regulates macrophage energy metabolism via the EP2 receptor.
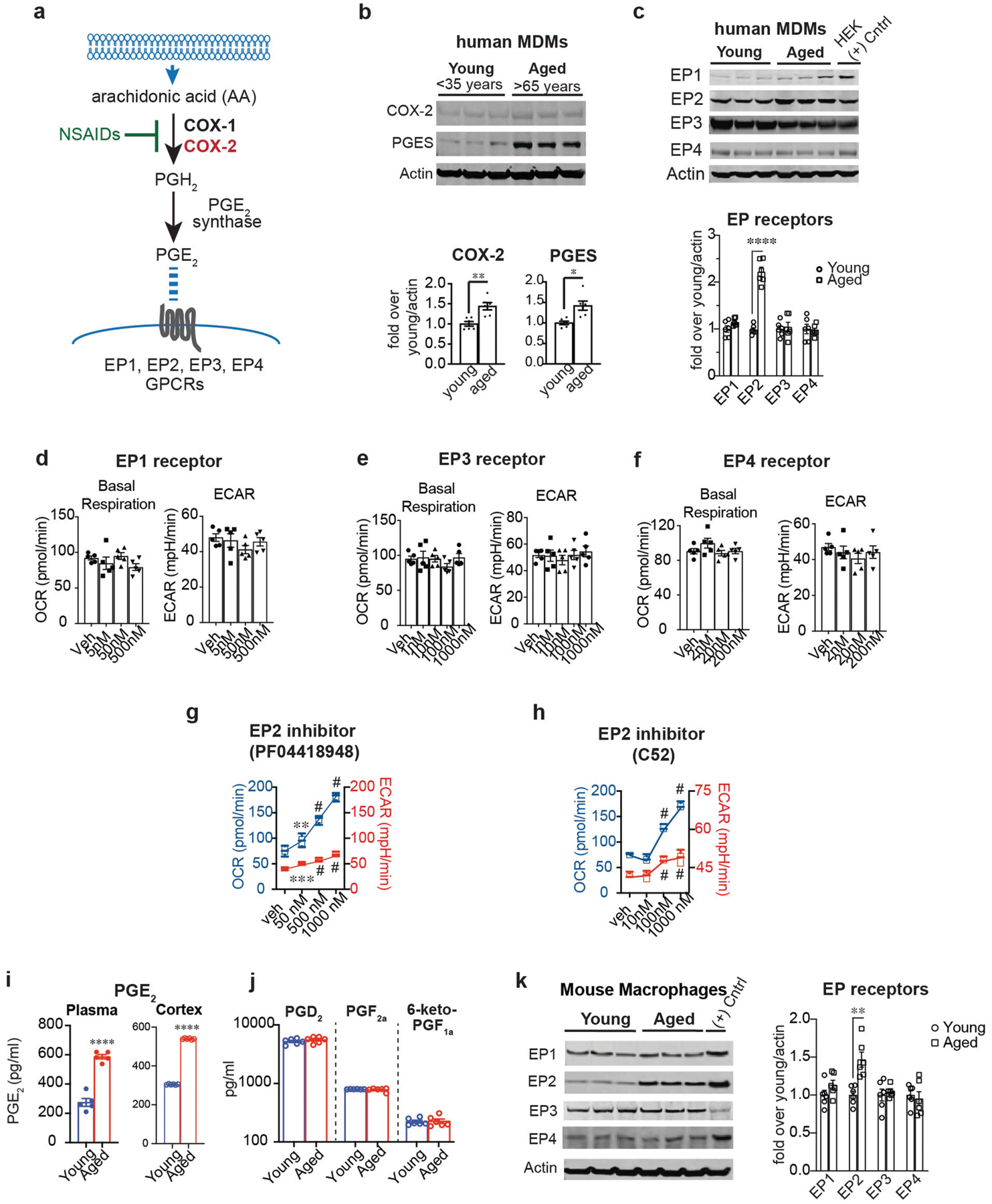
Data are mean ± s.e.m. unless otherwise specified. a, The PGE2 synthetic pathway. Arachidonic acid is converted to the prostaglandin precursor PGH2 by the action of constitutive COX-1 and/or inducible COX-2; this step is inhibited by non-steroidal anti-inflammatory drugs (NSAIDs). PGE2 synthase converts PGH2 to PGE2 which can bind to four distinct G-protein-coupled receptors (EP1–EP4 receptors). b, Top, representative immunoblot of two independent experiments quantifying COX-2 and PGE2 synthase in young and aged human MDMs. Bottom, quantification shows higher COX-2 and mPGE2 synthase levels in aged (above 65 years) compared to young (below 35 years) human MDMs (n = 6 donors per group; **P = 0.0030, *P = 0.0106, Student’s two tailed t-test). c, Top, representative immunoblot of two independent experiments quantifying EP1–EP4 in young and aged human MDMs; last lane, HEK cells transfected with EP receptor cDNA as a positive control. Bottom, quantification shows increased EP2 in aged (over 65 years) compared to young (below 35 years) human MDMs (n = 6 donors per group; ****P < 0.0001, Student’s two tailed t-test). d–f, Quantification of basal respiration and ECAR of human MDMs treated with ascending doses of the EP1 agonist iloprost (d), the EP3 agonist AE248 (e) and the EP4 agonist AE1329 (f) for 20 h (n = 5 donors per group). g, h, Dose response of the brain-impermeant EP2 antagonist PF-04418948 (g) and the brain-penetrant EP2 antagonist C52 (h) in human MDMs at 20 h. Data are represented as box plots (5–95 percentile); one-way ANOVA for OCR and ECAR, P < 0.0001; Tukey’s post hoc test for PF-04418948: **P = 0.0083, ***P = 0.0005, #P < 0.0001; Tukey’s post hoc test for C52: #P < 0.0001 (n = 5 donors per group). i, LC–MS quantification of PGE2 levels in the plasma and cerebral cortex of aged (18–20 months) and young (3–4 months) mice. ****P < 0.0001 by two-tailed Student’s t-test (n = 6 mice per group) j, LC–MS quantification of PGD2, PGF2a and prostacyclin (measured as 6-keto-PGF1a) in the cerebral cortex of young and aged mice (n = 5–6 mice per group). k, Left, Representative immunoblot of EP1–EP4 levels in mouse peritoneal macrophages isolated from young (3–4 months) and aged (18–20 months) mice; last lane, HEK cells transfected with EP receptor cDNAs as a positive control (n = 6 mice per group). Right, EP2 is selectively upregulated in aged mouse peritoneal macrophages, similar to aged human MDMs in c; **P = 0.0018 (n = 6 mice per group).
Extended Data Fig. 2 |. Myeloid EP2 receptor knockdown prevents age-associated inflammation.
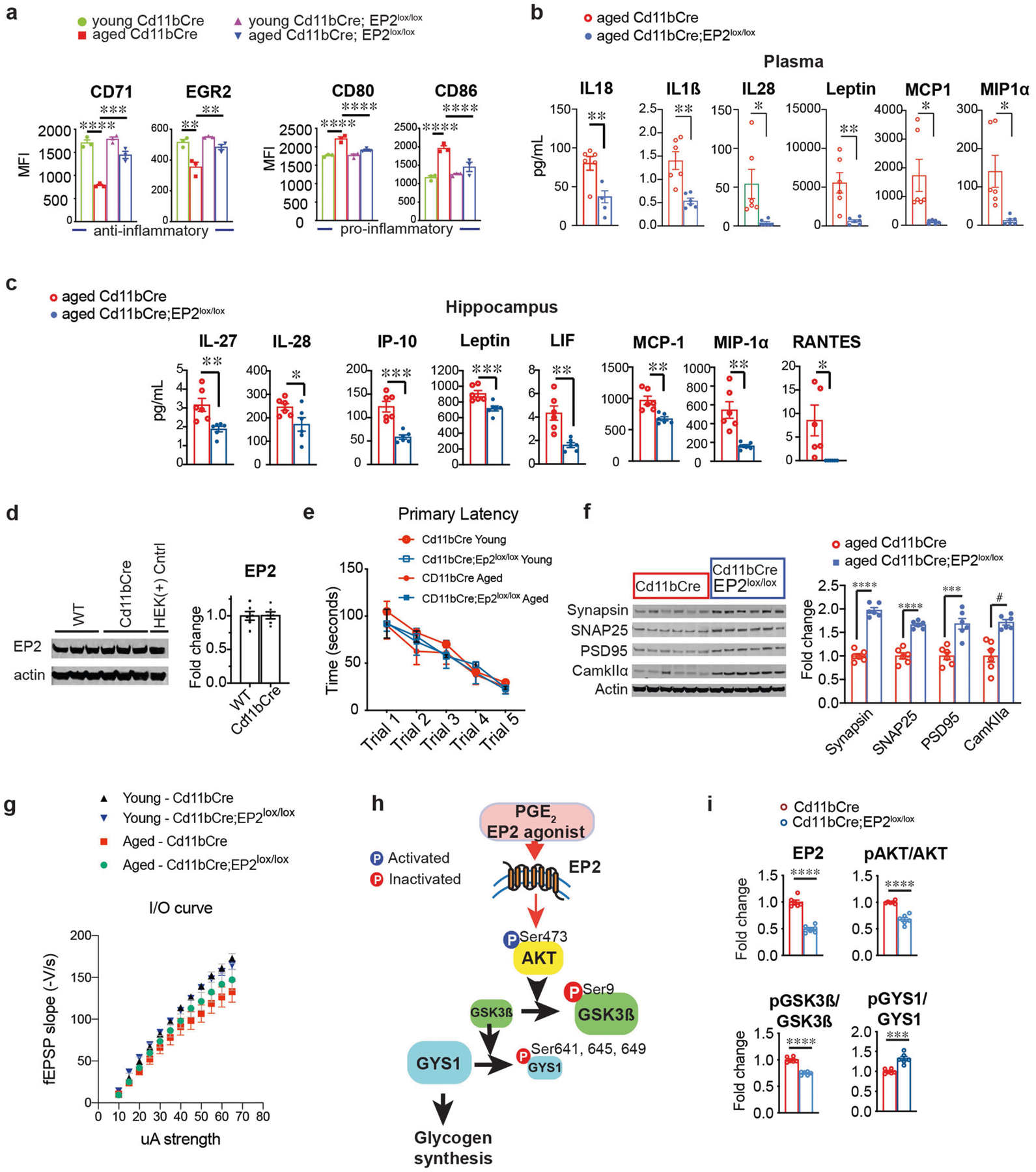
Data are mean ± s.e.m. unless otherwise specified. a, MFI quantification from three independent experiments for the anti-inflammatory markers CD71 and EGR2 and pro-inflammatory markers CD80 and CD86 in young (3–4 months) and aged (20–23 months) Cd11bCre and Cd11bCre;EP2lox/lox mice (n = 10,000–20,000 cells per point; n = 3 mice per group). Two-way ANOVA: effects of genotype and age P < 0.0001; Tukey’s post hoc test **P < 0.01, ***P < 0.001, ****P < 0.0001. b, c, Quantification of immune factors in plasma (b) and hippocampus (c) comparing aged Cd11bCre and aged Cd11bCre;EP2lox/lox mice; *P < 0.05, **P < 0.01 by two-tailed Student’s t-test (n = 6 mice per group). d, Representative immunoblot and quantification from two independent experiments comparing EP2 levels in 6-month-old Cd11bCre and wild-type mice (n = 6 per group). e, Primary latency in the Barnes maze for the five learning trials (n = 10 (young), n = 11 (aged Cd11bCre), n = 7 (aged Cd11bCre;EP2lox/lox). f, Representative immunoblot and quantification of hippocampal presynaptic proteins synapsin and SNAP-25 and postsynaptic proteins PSD95 and CamKIIα; #P = 0.0005, ***P = 0.0006, ****P < 0.0001 by two-tailed Student’s t-test (n = 6 mice per group; 20–23-month-old mice). g, Input/output curves as a measure of basal synaptic transmission in the CA1 region of the hippocampus (n = 8 slices, 3 mice per group). h, PGE2 activation of the EP2 receptor activates AKT signalling through phosphorylation of Ser473. Activated pAKT (Ser473) inactivates GSK3β through inhibitory phosphorylation of Ser9. Inactivation of GSK3β leads to constitutive activity of glycogen synthase 1 (GYS1) and glycogen synthesis. Conversely, deletion or antagonism of EP2 receptor signalling leads to downstream inhibitory phosphorylation of Ser641, Ser645 and Ser649 on GYS1 by activated, non-phosphorylated GSK3β. i, Quantification of EP2, pAKT (Ser 473)/total AKT, pGSK3β (Ser9)/total GSK3β and pGYS1 (Ser 641, 645, 649)/GYS1 levels in peritoneal macrophages from 6-month-old Cd11bCre and Cd11bCre;EP2lox/lox mice; ***P = 0.0002, ****P < 0.0001 by two-tailed Student’s t-test (n = 6 mice per group).
Extended Data Fig. 3 |. Macrophage EP2 signalling increases glycogen synthesis.
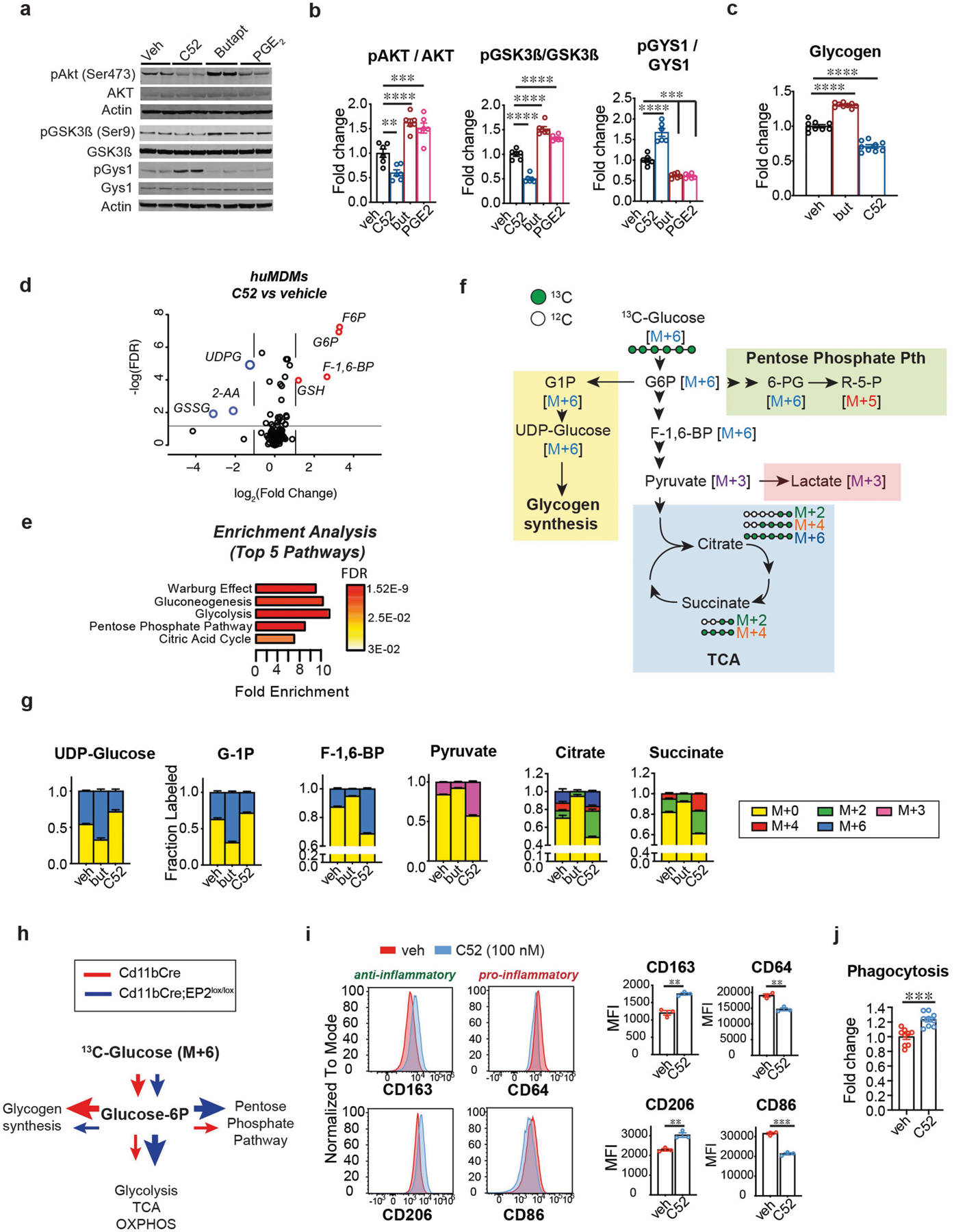
Data are mean ± s.e.m. unless otherwise specified. a, Representative immunoblots and quantification of the EP2–AKT–GSK3β–GYS1 signalling pathway in mouse peritoneal macrophages isolated from 6-month-old wild type C57B6/J mice treated with EP2 antagonist C52 (100 nM), EP2 agonist butaprost (100 nM) or PGE2 (100 nM) for 20 h. b, Quantification of a; P < 0.0001 by one-way ANOVA; Tukey’s post hoc test **P = 0.0072, ***P < 0.001, ****P < 0.0001 (n = 6 mice per group). c, Quantification of glycogen levels in human MDMs (age (mean ± s.e.m.) 43.9 ± 3.451 years) treated with EP2 agonist butaprost (100 nM) or EP2 antagonist C52 (100 nM) for 20 h. P < 0.001 by one-way ANOVA; Tukey’s post hoc test ****P < 0.0001 (n = 9 per group). d, LC–MS analysis of human MDMs (age (mean ± s.e.m.) 43.9 ± 3.451 years) treated with C52 (100 nM, 20 h) demonstrates upregulation of proximal glycolytic pathway (G6P, F6P, F-1,6-BP) and GSH and downregulation of UDPG and GSSG. Red circles represent metabolites with fold change >1.5, blue circles with fold change <1.5; q-value < 0.05 with false discovery rate (FDR) correction (n = 6 donors per group). e, Enrichment pathway analysis of d using MetaboAnalyst. f, Schematic depicting U-13C-glucose metabolism via glucose-1P (G1P) and UDP-glucose to glycogen synthesis (yellow shaded box) versus flux towards the pentose phosphate pathway (green shaded box), glycolysis or lactate, and the TCA cycle (blue shaded box) with associated mass-labelled molecules. Glucose is labelled at all 6 carbons (in green; M+6) and is converted via the glycolytic pathway to two molecules of pyruvate (or M+3). Pyruvate is transported into the mitochondria and undergoes oxidative decarboxylation to yield acetyl-CoA (M+2) which enters the TCA cycle. Successive additions of labelled acetyl-CoA through the TCA cycle yield M+2, M+4 and M+6. g, Isotope tracing of U-13C-glucose metabolism was performed in human MDMs (age (mean ± s.e.m.) 42.13 ± 3.674 years) treated with EP2 agonist butaprost (100 nM, 20 h) or EP2 inhibitor C52 (100 nM, 20 h; n = 6 donors per group). Activation of EP2 signalling with butaprost increases incorporation of heavy glucose in glycogen precursors G1P and UDP-glucose and reduces labelling of glycolytic intermediates (F-1,6-BP and pyruvate) as well as TCA cycle intermediates (citrate and succinate); inhibition of EP2 with C52 conversely reduces synthesis of glycogen precursors and increases glycolytic and TCA cycle intermediates. h, Schematic depicting changes in glucose metabolism in Fig. 2h, i. i, Representative flow cytometry histograms and corresponding MFI quantification of human MDMs with or without the EP2 inhibitor C52 (100 nM, 20 h) from three independent experiments. Surface levels of anti-inflammatory markers CD206 and CD163 increase with inhibition of EP2 signalling whereas levels of pro-inflammatory markers CD86 and CD64 decrease. **P < 0.01, ***P = 0.002 by two-tailed Student’s t-test (n = 30,000 – 40,000 cells per point; n = 3 donors per group). j, Quantification of phagocytosis of fluorescent E. coli particles in human MDMs treated with EP2 inhibitor C52 (100 nM, 20 h) from two independent experiments. ***P = 0.0002 by two-tailed Student’s t-test (n = 9 donors per group).
Extended Data Fig. 4 |. Knockdown of GYS1 promotes macrophage glucose metabolism and anti-inflammatory polarization.
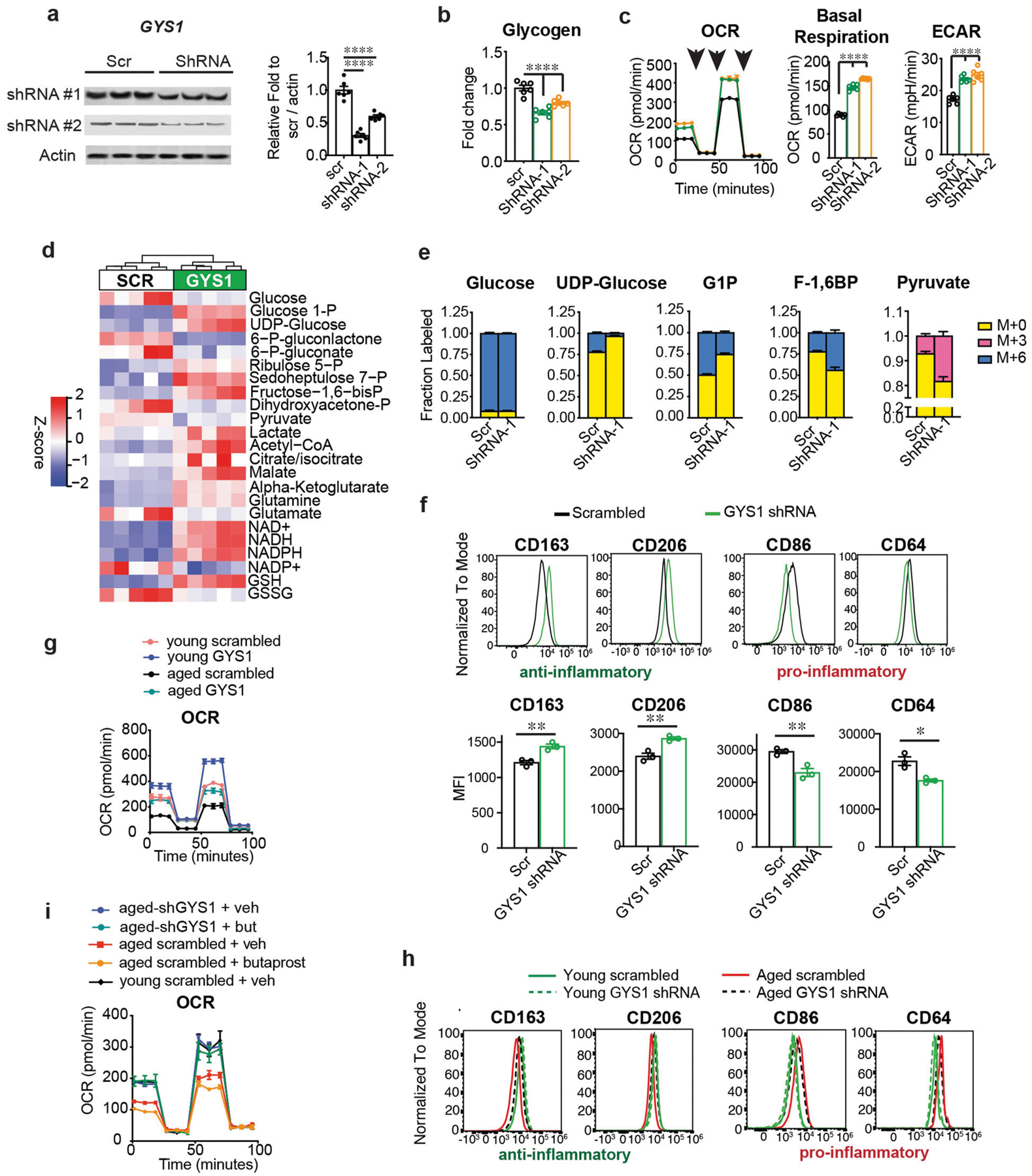
Data are mean ± s.e.m. unless otherwise specified. a–f, Human MDMs are from donors (age (mean ± s.e.m.) 47.2 ± 1.582 years); g–i, Human MDMs are from young (below 35 years) and aged (over 65 years) donors. a, Representative immunoblots and quantification of human MDMs transfected with two different shRNAs to human GYS1 at 8 h. P < 0.0001 by one-way ANOVA; Tukey’s post hoc test ****P < 0.0001 (n = 6 donors per group). b, Quantification of glycogen levels in human MDMs transfected with shRNAs to GYS1 at 8 h. P < 0.0001 by one-way ANOVA; Tukey’s post hoc test ****P < 0.0001 (n = 6 donors per group). c, Representative traces and quantification of OCR and ECAR for three independent experiments in human MDMs transfected with shRNAs for GYS1 at 8 h (n = 5 donors per group). P < 0.0001 by one-way ANOVA; Tukey’s post hoc test ****P < 0.0001 (n = 6 donors per group). Black arrows represent addition of oligomycin (1 μM), FCCP (2 μM), and rotenone/antimycin (500 nM), respectively, at time points indicated. d, Hierarchical clustering of targeted metabolomics for glycolysis, pentose phosphate shunt and TCA cycle metabolites in human MDMs transfected with shRNA to GYS1 at 8h (n = 5 donors per group). e, Isotope tracing of U-13C-glucose in human MDMs transfected with shRNA to GYS1 at 8 h reveals a decreased labelling in the glycogen precursor UDP-glucose and an increase in glycolytic intermediates F-1,6-BP and pyruvate (n = 6 donors per group). f, Representative flow cytometry histograms of three independent experiments for the pro-inflammatory markers CD86 and CD64 and anti-inflammatory markers CD206 and CD163 in human MDMs treated with or without shRNA to GYS1. Bottom, quantification of MFI, two-tailed Student’s t-test, *P < 0.05, **P < 0.01 (n = 20,000–40,000 cells per point, n = 3 donors per group). g, Representative trace of real-time changes in OCR from three independent experiments of young (below 35 years) and aged (over 65 years) human MDMs treated with or without shRNA to GYS1 (n = 5 young, n = 6 aged donors). h, Representative histograms of anti- and pro-inflammatory surface markers in young and aged human MDMs treated with or without shRNA to GYS1 (n = 3 donors per group). i, Representative traces of real-time changes in OCR from two independent experiments in young (below 35 years) and aged (over 65 years) human MDMs transfected with shRNA to GYS1 and treated 8 h later with butaprost (100 nm) for 20 h. OCR in GYS1-deficient aged human MDMs does not change with activation of the EP2 receptor by butaprost (n = 4 donors per group).
Extended Data Fig. 5 |. EP2 blockade restores AKT–GSK3β–GYS1 signalling, glycolysis and mitochondrial respiration to youthful levels.
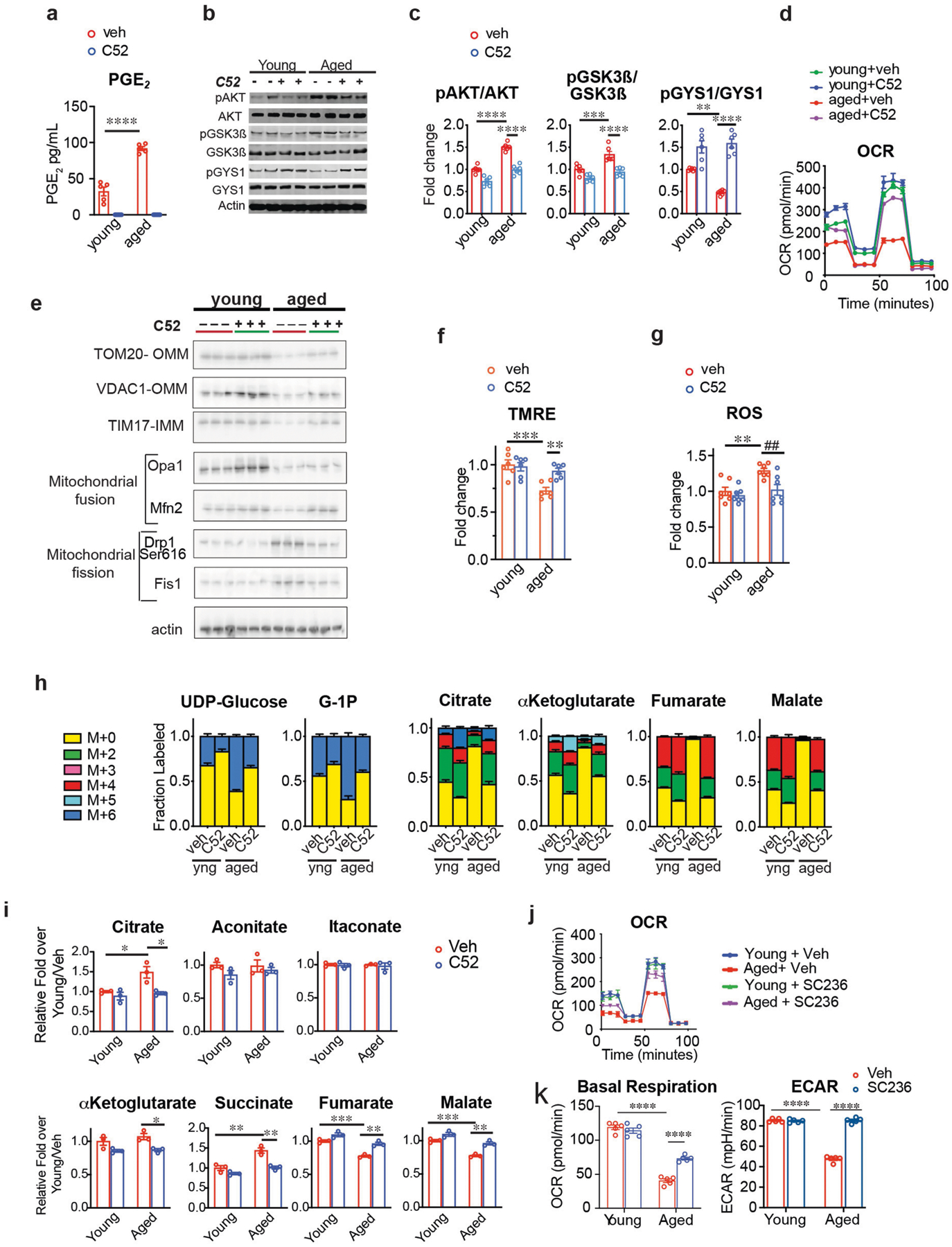
Data are mean ± s.e.m. unless otherwise specified. a, Quantification of PGE2 levels in young (below 35 years) and aged (over 65 years) human MDMs treated with or without C52 (100 nM, 20 h). Effects of age and treatment P < 0.0001 by two-way ANOVA; Tukey’s post hoc test ****P < 0.0001 (n = 5 donors per group). b, c, Representative immunoblots (b) and quantification (c) of effects of C52 treatment (100 nM, 20 h) in human MDMs from young (below 35 years) and aged (over 65 years) donors. Two-way ANOVA, age and treatment, P < 0.0001; Tukey’s post hoc test **P = 0.002, ***P = 0.0005, ****P < 0.0001 (n = 6 donors per group). d, Real-time changes in OCR from three independent experiments of young and aged human MDMs treated with or without C52 (100 nM, 20 h; n = 5 donors per group). e, Representative immunoblots of effects of EP2 inhibition on mitochondrial protein levels in young and aged human MDMs (n = 6 donors per group; OMM, outer mitochondrial membrane; IMM, inner mitochondrial membrane). See Fig. 3h for quantification. f, Membrane potential (TMRE) in young and aged human MDMs treated with or without C52 (100 nM, 20 h). Two-way ANOVA: age P = 0.0009 and treatment P = 0.0333; Tukey’s post hoc test, ***P = 0.0006, **P = 0.0088 (n = 6 donors per group). g, ROS in young and aged human MDMs treated with or without C52 (100 nM, 20 h). Two-way ANOVA: age P = 0.0022 and treatment P = 0.0055; Tukey’s post hoc test **P = 0.0045, ##P = 0.0084 (n = 7 donors per group). h, Young and aged human MDMs were incubated with U-13C-glucose for 20 h with or without C52. Aged human MDMs showed increased labelling in glycogen precursors (G1P and UDP-glucose) and decreased labelling in TCA cycle intermediates. This was prevented with EP2 inhibition; n = 3 donors per group. i, Quantification of TCA cycle metabolites from Fig. 3i. Note normalization of citrate, α-ketoglutarate (α-KG), succinate, fumarate and malate in aged human MDMs with EP2 inhibition. Also note that itaconate, which is increased in models of acute macrophage stimulation with LPS, is not changed with ageing (n = 3 donors per group). Two-way ANOVA with Tukey’s multiple comparisons test; *P < 0.05, **P < 0.01, ***P < 0.001. j, k, Representative traces of real-time changes in OCR (j) and quantification of basal respiration and ECAR (k) from two independent experiments in peritoneal macrophages isolated from young (3–4 months) and aged (18–20 months) mice treated with COX-2 inhibitor SC236 (100 nM, 20 h). Two-way ANOVA: age and treatment, P < 0.0001 and P = 0.004, respectively; Tukey’s post hoc test, ****P < 0.0001 (n = 5 mice per group).
Extended Data Fig. 6 |. EP2 blockade does not alter LPS-mediated glucose metabolism reprogramming, and aged macrophages lose the capacity to use alternative fuel sources.
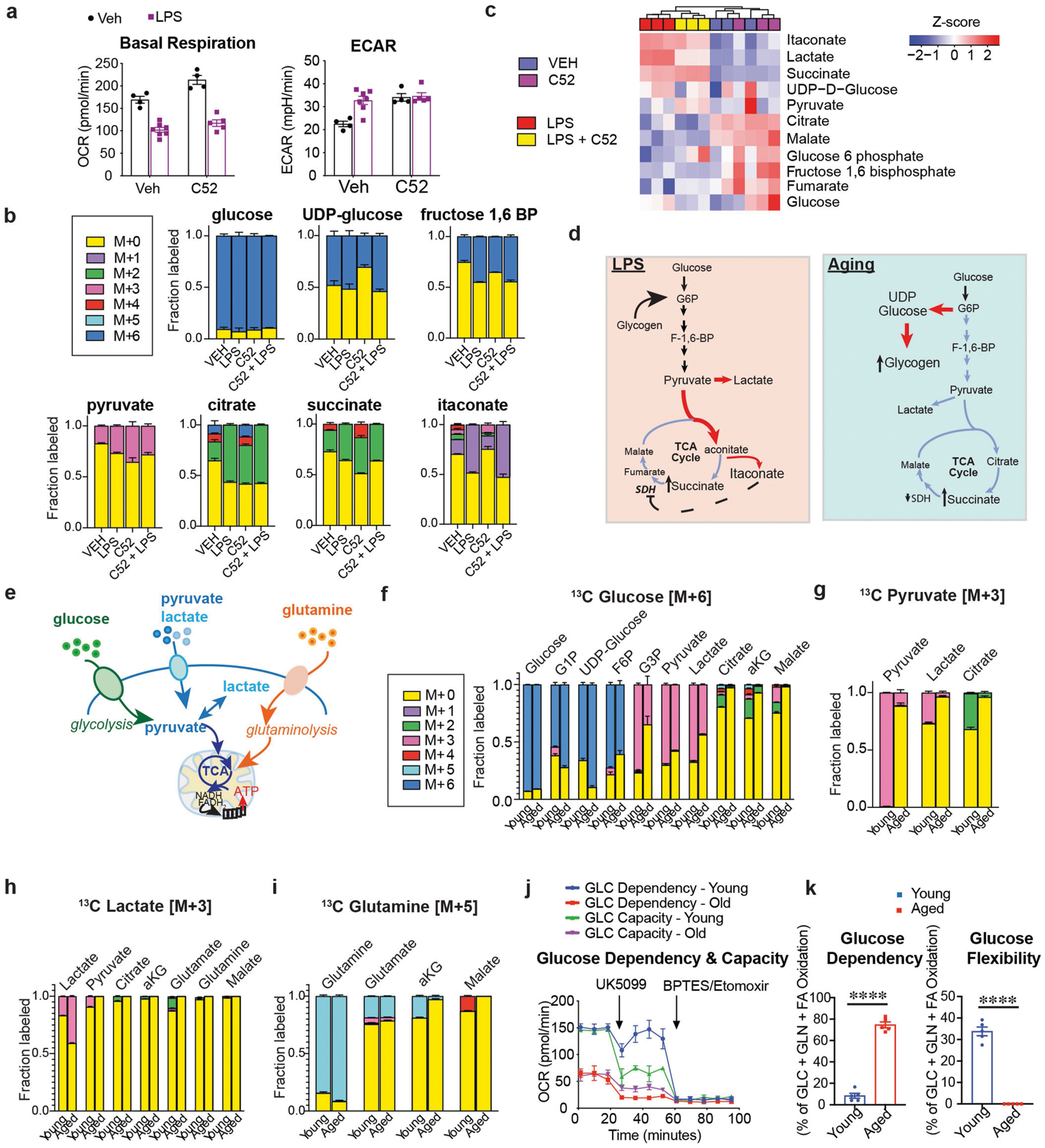
Data are mean ± s.e.m. unless otherwise specified. a, Human MDMs with or without LPS treatment (100 ng ml−1, 20 h; n = 4 (veh), n = 4 (veh + C52), n = 5 (LPS), n = 7 (veh + LPS) donors per group, age (mean ± s.e.m.) 42.3 ± 8.212 years) were stimulated with or without C52 (100 nM, 20 h). Blockade of EP2 does not rescue OCR and ECAR in LPS-treated human MDMs. b, 13C-glucose isotope tracing of human MDMs with or without LPS (100 ng ml−1, 20 h) and with or without C52 (100 nM, 20 h), demonstrates reprogramming of the TCA cycle towards increased production of itaconate with LPS stimulation that is not reversed with EP2 inhibition (n = 3 donors per group). c, Hierarchical clustering of glycolytic and TCA cycle metabolites in human MDMs with or without LPS (100 ng ml−1, 20 h) and with or without C52 (100 nM, 20 h) (n = 3 donors per group). d, Model highlighting differences in glucose metabolism and the TCA cycle in LPS-stimulated macrophages versus aged macrophages. LPS stimulation upregulates glycolysis and lactate production and increases itaconate production from aconitate by cis-aconitate decarboxylase, the protein product of Irg1, which is highly induced in settings of inflammation; increased itaconate suppresses SDH. In aged human MDMs, glucose is diverted from glycolysis to glycogen, reducing glucose flux into the TCA cycle; ageing is characterized by low SDH activity arising from deficient SIRT3 deacetylation of complex II subunits as a result of declining NAD+ levels17. In both cases, lower succinate dehydrogenase activity leads to accumulation of the pro-inflammatory TCA cycle intermediate succinate, which enhances pro-inflammatory gene expression by stabilizing the transcription factor HIF-1α. e, Diagram illustrating fuel substrates for the TCA cycle and mitochondrial OXPHOS. Glucose and pyruvate/lactate can feed into the TCA cycle via acetyl-CoA; glutamine is metabolized via anaplerosis to α-KG, a TCA cycle intermediate. f, Young (below 35 years) and aged (over 65 years) human MDMs received U-13C-glucose [M+6] for 20 h. Isotope tracing reveals young macrophages can incorporate significantly higher 13C from glucose into the TCA cycle compared to aged macrophages (P = 0.0026 by Student’s two-tailed t-test with Welch’s correction for 13C citrate[M+2] proportion in young versus aged cells; n = 3 donors per group). g, Young (below 35 years) and aged (over 65 years) human MDMs received U-13C-pyruvate [M+3] for 20 h. Isotope tracing reveals young macrophages are able to incorporate significantly higher 13C from pyruvate into the TCA cycle compared to aged macrophages (P < 0.0001 by Student’s two-tailed t-test with Welch’s correction for 13C -citrate[M+2] in young versus aged cells; n = 3 donors per group). h, Young (below 35 years) and aged (over 65 years) human MDMs received U-13C-lactate [M+3] for 20 h. Isotope tracing reveals young macrophages are able to incorporate significantly higher 13C from lactate into the TCA cycle than aged macrophages (P = 0.0083 by Student’s two-tailed t-test with Welch’s correction for 13C citrate[M+2] in young versus aged cells; n = 3 donors per group). i, Young (below 35 years) and aged (over 65 years) human MDMs received U-13C-glutamine [M+5] for 20 h. Isotope tracing reveals young macrophages are able to incorporate significantly higher 13C from glutamine into the TCA cycle than aged macrophages (P < 0.0001 by Student’s two-tailed t-test with Welch’s correction for13C-α-KG [M+5] in young versus aged cells; n = 3 donors per group). j, Fuel flexibility was assayed by inhibiting fatty acid oxidation, glutamine metabolism, and pyruvate transport into the mitochondria. Representative trace of real-time changes in OCR from two independent experiments demonstrating increased dependence on glucose and reduced capacity by aged mouse macrophages (20–23 months) for oxidative phosphorylation (n = 5 donors per group). UK5099: mitochondrial pyruvate carrier inhibitor; BPTES: glutaminase inhibitor; etomoxir: inhibitor of carnitine palmitoyltransferase 1 (CPT1) which transports fatty acids into mitochondrial matrix. For dependency traces cells received UK5099 in first injection and BPTES/Etomoxir in second injection as indicated on figure. For capacity traces cells received BPTES/Etomoxir in first injection and UK5099 in second injection. k, Quantification of j; n = 5 donors per group; ****P < 0.0001 by two-tailed Student’s t-test.
Extended Data Fig. 7 |. EP2 is expressed in hippocampal CA1 microglia.
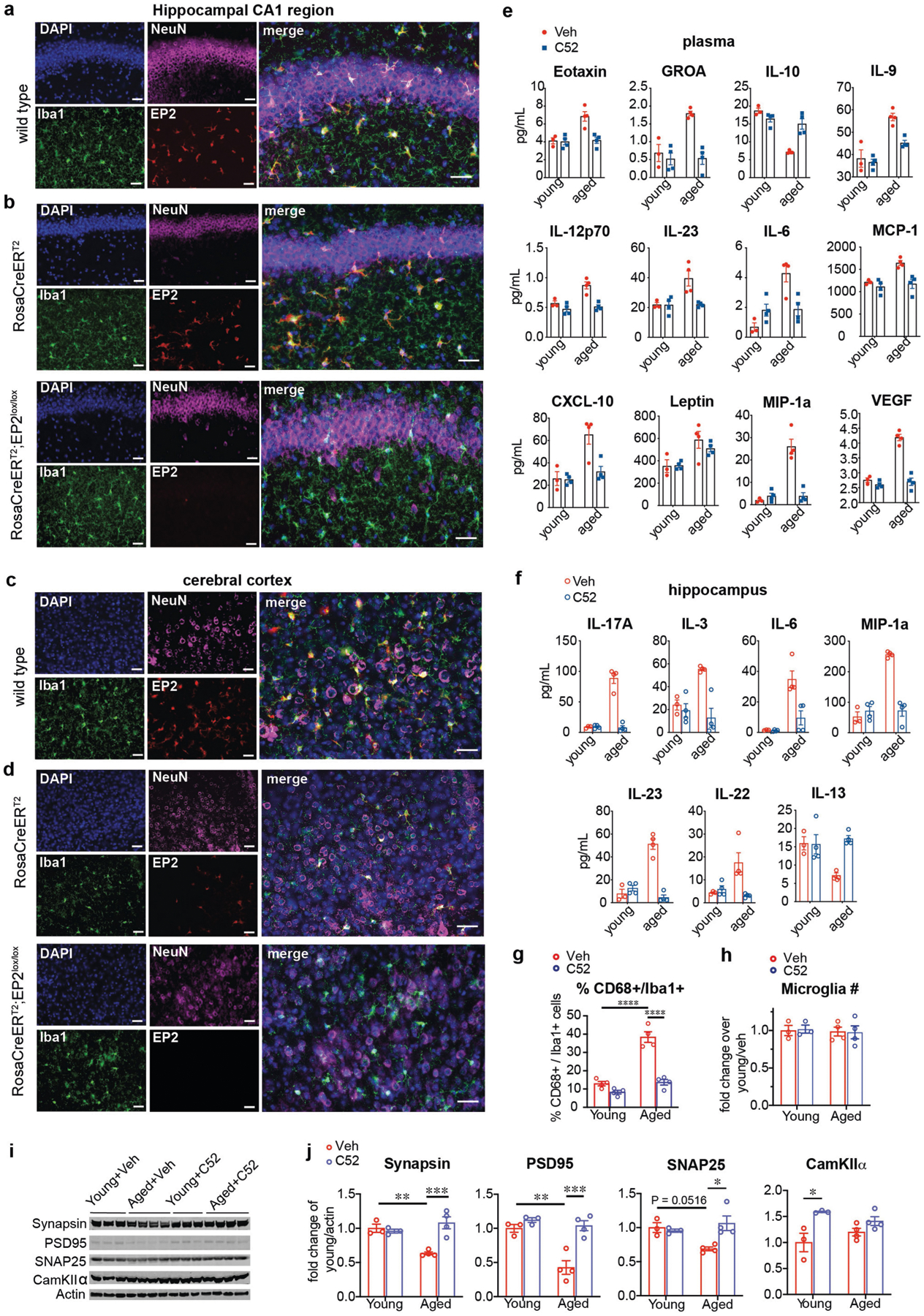
Data are mean ± s.e.m. unless otherwise specified. Scale bars, 40 μm. a, b, Images are from the CA1 region of the hippocampus. a, Colocalization of EP2 immunofluorescence with IBA1 in microglia (18-month-old mice). b, Microglial EP2 is not detected in RosaCreERT2;EP2lox/lox mice (15–16-month-old mice). c, d, Images are from the cerebral cortex. c, Colocalization of EP2 immunofluorescence with IBA1 in microglia (18-month-old mice). d, Microglial EP2 is not detected in RosaCreERT2;EP2lox/lox mice (15–16-month-old mice). e, f, Quantification of immune factors in the plasma (e) and hippocampus (f) from young (3–4 months) and aged (22–24 months) mice with or without C52 treatment (10 mg/kg/d for 1 month; n = 3 mice/young + veh group; n = 4 mice per group for all other groups). g, Quantification of percent CD68+/IBA1+ cells in CA3 hippocampus from young and aged mice with or without C52 treatment (10 mg/kg/day, 1 month); effect of age and treatment P < 0.0001 by two-way ANOVA; Tukey’s post hoc test ****P < 0.0001 (n = 8 slices, n =4 mice per group). h, Quantification of microglial numbers from young and aged mice with or without C52 treatment (10mg/kg/day, 1 mo; n = 8 slices, n =3 young and n =4 aged mice per group). i, Representative immunoblot of the pre- and postsynaptic proteins synapsin, PSD95, SNAP25 and CamKIIa in young and aged hippocampi in mice with or without C52 treatment (10 mg/kg/day, 1 month); n =3 young and n =4 aged mice per group. j, Quantification of i. Synapsin: effects of age (P = 0.0685) and treatment (P = 0.0072) by two-way ANOVA; Tukey’s post hoc test **P = 0.0069, ***P = 0.0010. PSD95: effects of age (P = 0.0019) and treatment (P = 0.0009) by two-way ANOVA; Tukey’s post hoc test **P = 0.0020, ***P = 0.0007. SNAP25: effects of age (P = 0.1930) and treatment (P = 0.0463) by two-way ANOVA; Tukey’s post hoc test *P = 0.0121. CamKIIα: effects of age (P = 0.9210) and treatment (P = 0.0025) by two-way ANOVA; Tukey’s post hoc test *P = 0.0132 (n = 3 young and 4 aged mice per group).
Extended Data Fig. 8 |. Effects of in vivo EP2 inhibition on macrophage metabolism, synaptic mitochondria and mitochondrial morphology in young and aged mice.
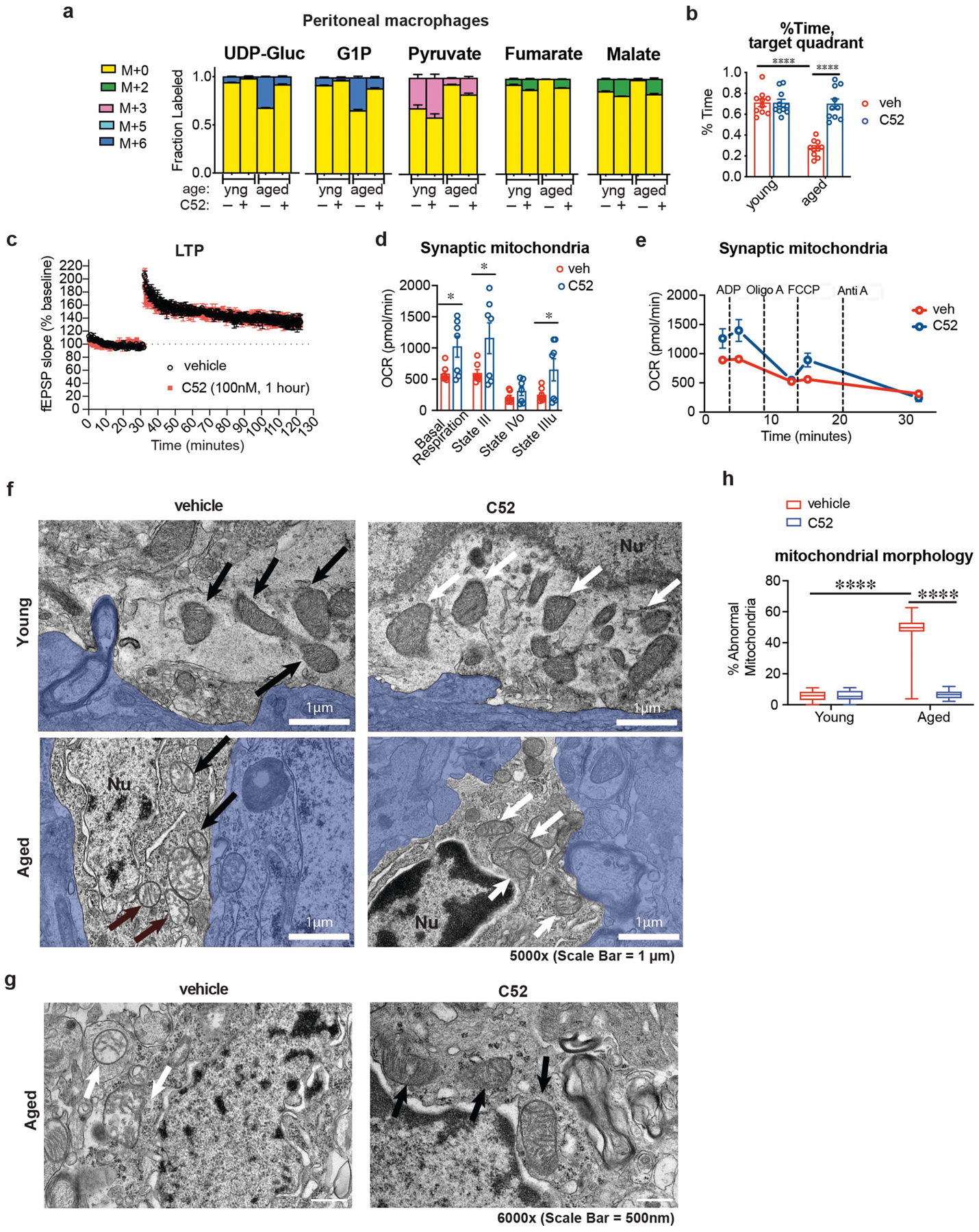
Data are mean ± s.e.m. unless otherwise specified. a, Peritoneal macrophage metabolite labelling following in vivo U-13C-Glucose isotope tracing (n = 6 mice per group). b, Quantification of per cent time in the target quadrant of the Barnes maze. Two-way ANOVA, effect of age or treatment ****P < 0.0001, Tukey’s post hoc test ****P < 0.0001 (n = 10 mice per group). c, Long-term potentiation (LTP) in the CA1 hippocampal region over a 120-min recording interval. Acute administration of C52 (100 nM, 1 h) before TBS does not alter LTP in aged mice (20–22 mo mice, n = 3 mice per group). d, Synaptic mitochondria were isolated from synaptosome fractions prepared from 16–month-old mice treated with or without C52 (10 mg/kg/d, 10 days). Coupling between the mitochondrial electron transport chain and oxidative phosphorylation (using succinate as the substrate) was determined using the Seahorse XFe24 analyser. Basal respiration (state II) and ADP-supplemented respiration (state III) reflect both electron transport and ATP generation and increased by twofold with EP2 blockade. Maximal respiration (state IIIu) after application of the H+ gradient uncoupler FCCP was also higher with EP2 inhibition, and state IVo, reflecting blockade of ATP synthase with oligomycin, was unchanged. *P < 0.05, two-tailed Student’s t-test (n = 7 mice per group). e, Coupling assay trace of synaptic mitochondria from aged mice treated with or without C52 for 10 days. Rates of basal complex II respiration as well as states III (ADP stimulated respiration), IV (oligomycin) and IIIu (FCCP) were consecutively measured (n = 7 mice per treatment group). f, TEM images at 5,000× magnification of microglia in the CA3 region of the hippocampus from young (3–4 months) and aged (22–24 months) mice treated with or without C52 (10 mg/kg/d, 1 month). Aged mice exhibit abnormal, non-electron-dense mitochondria; these features are rescued with C52 treatment. Arrows (black for vehicle, white for C52) point to mitochondria within microglia; blue shaded areas are non-microglial cells. Nu, nuclei; white scale bars, 1 μm. g, Higher-power representative TEM images of aged (22–24 months) mice treated with or without C52 (10 mg/kg/d, 1 month) showing differences in cristae and electron density. White scale bars, 500 nm. h, Quantification of per cent abnormal mitochondria in microglia of young (3–4 months) and aged (22–24 months) mice treated with or without C52 (10 mg/kg/d, 1 month); n = 66 mitochondria per group, 2-way ANOVA with Tukey’s post hoc test, ****P < 0.001.
Extended Data Fig. 9 |. Peripheral EP2 blockade restores youthful inflammatory profile and cognitive status.
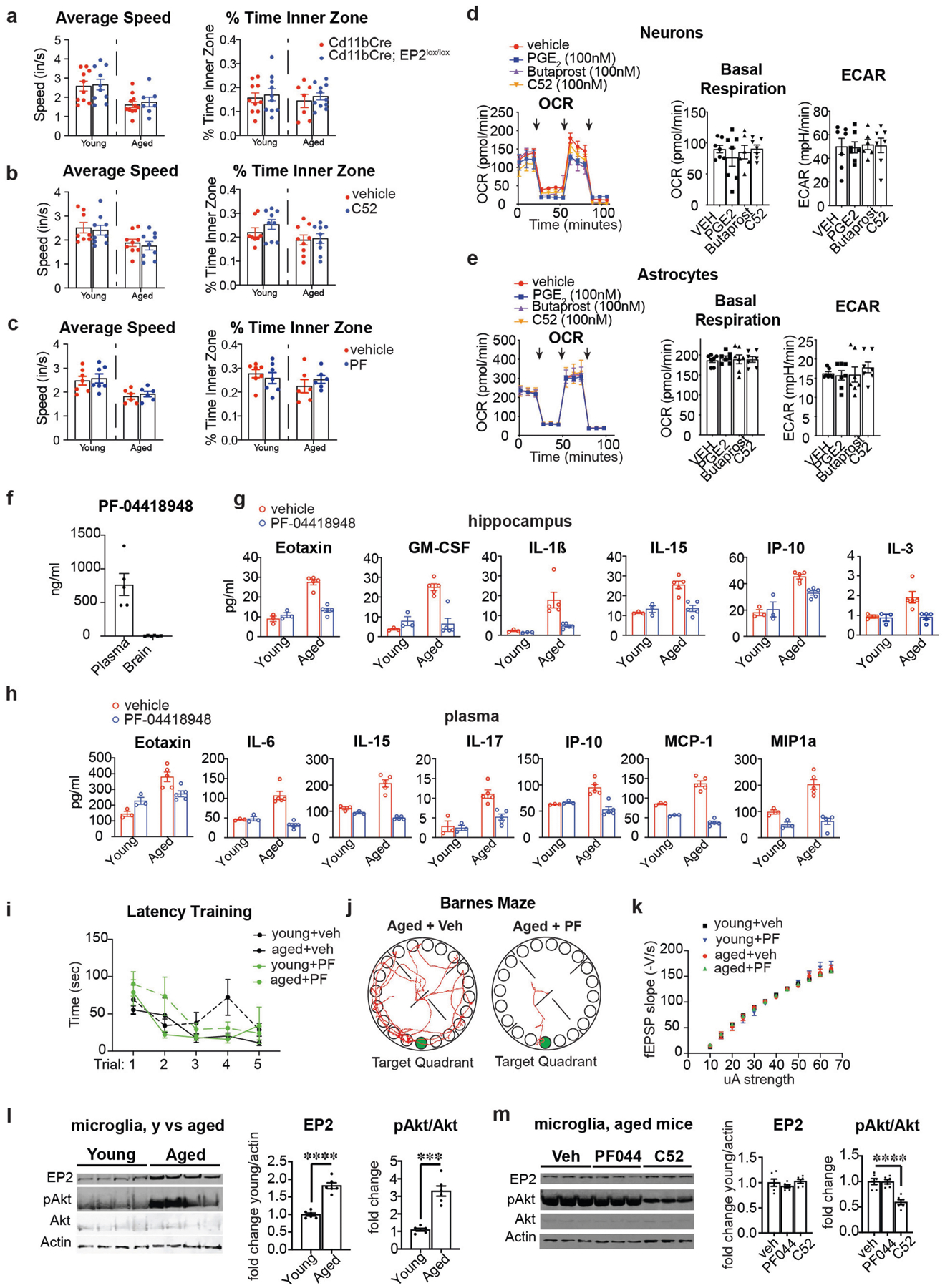
Data are mean ± s.e.m. unless otherwise specified. a, Average speed (inches per second) and percentage of time spent in the inner zone in young (3–4 months) and aged (20–23 months) Cd11bCre;EP2lox/lox mice. There are no significant differences between age-matched Cd11bCre and Cd11bCre;EP2lox/lox mice (n = 10 mice (young); n = 7 mice (aged Cd11bCre;EP2lox/lox); n = 11 mice (aged Cd11bCre)). b, Average speed (inches/second) and percentage of time spent in the inner zone in young (3–4 months) and aged (22–24 months) mice treated with C52 compound (10 mg/kg/d, 1 month). There are no significant differences between age-matched vehicle and C52-treated mice (n = 8 mice for young + veh; n = 9 mice per group for all other groups). c, Average speed (inches per second) and percentage of time spent in the inner zone in young (3–4 months) and aged (20–22 months) mice treated with PF compound (2.5 mg/kg/d, 6 weeks.) There are no significant differences between age-matched vehicle or PF-treated mice (n = 7 mice for young groups; n = 6 mice for aged groups). d, Real-time changes in OCR and quantification of basal respiration and ECAR of three independent experiments on mouse hippocampal neurons treated with PGE2 (100 nM, 20 h), butaprost (100 nM, 20 h) and C52 (100 nM, 20 h) (n = 6 (butaprost), n = 7 (all others) biologically independent samples per group). Black arrows represent addition of oligomycin (1 μM), FCCP (2 μM), and rotenone/antimycin (500 nM), respectively, at time points indicated. e, Real-time changes in OCR and quantification of basal respiration and ECAR of three independent experiments on mouse astrocytes treated with PGE2 (100 nM, 20 h), butaprost (100 nM, 20 h) and C52 (100 nM, 20 h) (n = 7 biologically independent samples per group). Black arrows represent addition of oligomycin (1 μM), FCCP (2 μM), and rotenone/antimycin (500 nM), respectively, at time points indicated. f, LC–MS analysis of plasma and brain levels of PF-04418948 (2.5 mg/kg/d, 6 weeks). PF-04418948 was not detected in whole-brain lysates of treated mice (n = 5–6 mice per group, 20–22 mo). g–k, Young (3–4 months) and aged (20–22 months) mice were treated with vehicle or PF-04418948 at 2.5 mg/kg/d for 6 weeks. g, Quantification of significantly regulated immune factors in hippocampi (n = 3 young mice; n = 5 aged mice per group). h, Quantification of significantly regulated immune factors in plasma (n = 3 young mice; n = 5 aged mice per group). i, Primary latency in the Barnes maze for the five learning trials (n = 7 young mice; n = 6 aged mice per group). j, Representative traces of paths taken to the target hole (green) on the day of testing in the Barnes maze comparing aged mice with or without PF-04418948. k, Input/output curves as a measure of basal synaptic transmission in the CA1 region of the hippocampus (n = 8 slices, 3 mice per group). l, Microglia were isolated from brains of young (2–3 months) and aged (20–22 months) mice and assayed for EP2 receptor, pAKT and total AKT levels. Left, representative western blot; right, quantification. Two-tailed Student’s t-test, ***P < 0.001, **** P < 0.0001 (n = 6 mice per group). m, Microglia were isolated from brains of aged (20–24 months) mice treated with vehicle, PF-04418948 (10 mg/kg/day for 10 days) or C52 (10 mg/kg/day for 10 days). Left, representative western blot; right, quantification with two-tailed Student’s t-test, **** P < 0.0001 (n = 6 mice per group).
Extended Data Fig. 10 |. Transcriptomics of primary peritoneal macrophages from young and aged mice treated with vehicle or with the non-brain-penetrant EP2 inhibitor PF-04418948.
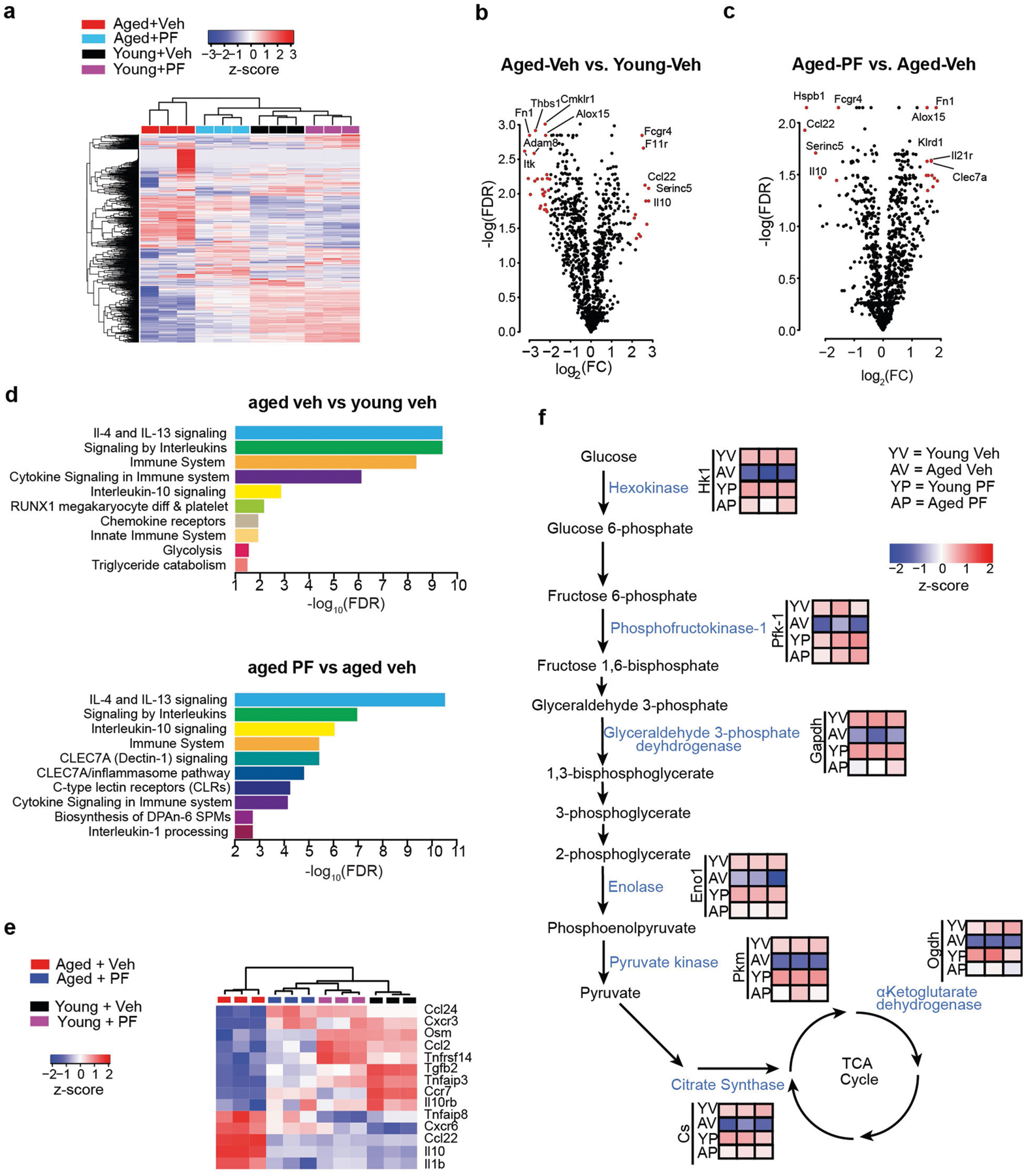
Data are mean ± s.e.m. unless otherwise specified. a, Heat map of 1448 genes from Nanostring assay reveals that aged mice treated with PF-04418948 cluster with young mice treated with vehicle or PF-04418948. b, Volcano plot of peritoneal macrophages collected from aged versus young vehicle-treated mice. Red dots indicate genes that are absolute-value[log2(FC)] ≥ 2 and FDR < 0.05 by t-test with Benjamini–Hochberg correction. c, Volcano plot of peritoneal macrophages collected from aged + PF-04418948-treated versus aged + vehicle-treated mice. Red dots indicate genes that are absolute-value[log2(FC)] ≥ 1.5 and FDR < 0.05 by t-test with Benjamini–Hochberg correction. d, Top 10 signalling pathways from the Reactome pathway database for FDR < 0.05 genes comparing aged +veh versus young + v eh mice (top) and aged + PF-04418948 versus aged + veh mice (bottom). e, Hierarchical clustering of top differentially regulated chemokine and cytokines transcripts (FDR < 0.05) demonstrates that PF-04418948 treatment shifts expression towards young macrophage levels. f, Nanostring analysis of significantly regulated genes (FDR < 0.05) demonstrates differential expression of bioenergetic transcripts in peritoneal macrophages isolated from young (3–4 months) and aged (20–22 months) mice with or without PF-04418948 (2.5 mg/kg/d for 6 weeks). Aged peritoneal macrophages exhibit suppressed expression of genes that encode critical glycolytic enzymes, including the rate-limiting enzyme phosphofructokinase-1 (PFK-1) as well as the rate-setting TCA cycle enzyme, citrate synthase. Peripheral myeloid EP2 inhibition with PF-04418948 corrects the age-associated suppression of myeloid glycolytic and TCA cycle gene expression.
Supplementary Material
Acknowledgements
This work was supported by RO1AG048232 (K.I.A.), RF1AG058047 (K.I.A.), R21NS087639 (K.I.A.), American Heart Association 19PABH134580007 (K.I.A.), 1P50 AG047366 (K.I.A.), 1P30 AG066515 (K.I.A.), Bright Focus (K.I.A.), the Paul and Daisy Soros Fellowship for New Americans (P.S.M.), the Gerald J. Lieberman Fellowship (P.S.M.), DP1DK113643 (M.R.M., L.L. and J.D.R.), the HHMI Hanna H. Gray Fellows Program (M.R.M.), the Burroughs Wellcome Fund PDEP (M.R.M.), the Stanford Innovation Fund (A.U.J. and D.M.-R.), the Takeda Pharmaceuticals Science Frontier Fund (D.M.-R.), the Ludwig Cancer Foundation (I.L.W.), NIH/NCI F30 CA228215 (J.Q.H), NIH/NCI R35CA220434 (I.L.W.), the Japan Science and Technology Agency’s Exploratory Research for Advanced Technology (JST-ERATO) Suematsu Gas Biology Project (Y.S. and M.S.), the Scully Family Initiative (F.M.L.), the Taube Family Foundation (F.M.L.) and the Jean Perkins Foundation (F.M.L.). The authors thank J. Perrino at the Stanford Cell Sciences Imaging Facility (supported by NIH 1S10RR02678001) and the Stanford Human Immune Monitoring Center.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-020-03160-0.
Competing interests The authors declare no competing interests.
Supplementary information The online version contains supplementary material available athttps://doi.org/10.1038/s41586-020-03160-0.
References
- 1.Franceschi C et al. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev 128, 92–105 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Nikolich-Žugich J The twilight of immunity: emerging concepts in aging of the immune system. Nat. Immunol 19, 10–19 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Ferrucci L & Fabbri E Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol 15, 505–522 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindenberger U Human cognitive aging: corriger la fortune? Science 346, 572–578 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Brayne C The elephant in the room—healthy brains in later life, epidemiology and public health. Nat. Rev. Neurosci 8, 233–239 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Cunningham C & Hennessy E Co-morbidity and systemic inflammation as drivers of cognitive decline: new experimental models adopting a broader paradigm in dementia research. Alzheimers Res. Ther 7, 33 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Villeda SA et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477, 90–94 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Villeda SA et al. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat. Med 20, 659–663 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarlus H & Heneka MT Microglia in Alzheimer’s disease. J. Clin. Invest 127, 3240–3249 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mhatre SD, Tsai CA, Rubin AJ, James ML & Andreasson KI Microglial malfunction: the third rail in the development of Alzheimer’s disease. Trends Neurosci 38, 621–636 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ricciotti E & FitzGerald GA Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol 31, 986–1000 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pearce EL & Pearce EJ Metabolic pathways in immune cell activation and quiescence. Immunity 38, 633–643 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biswas SK & Mantovani A Orchestration of metabolism by macrophages. Cell Metab 15, 432–437 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Ganeshan K & Chawla A Metabolic regulation of immune responses. Annu. Rev. Immunol 32, 609–634 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelly B & O’Neill LA Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res 25, 771–784 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mills EL, Kelly B & O’Neill LAJ Mitochondria are the powerhouses of immunity. Nat. Immunol 18, 488–498 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Minhas PS et al. Macrophage de novo NAD+ synthesis specifies immune function in aging and inflammation. Nat. Immunol 20, 50–63 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Casolini P, Catalani A, Zuena AR & Angelucci L Inhibition of COX-2 reduces the age-dependent increase of hippocampal inflammatory markers, corticosterone secretion, and behavioral impairments in the rat. J. Neurosci. Res 68, 337–343 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Montine TJ et al. Elevated CSF prostaglandin E2 levels in patients with probable AD. Neurology 53, 1495–1498 (1999). [DOI] [PubMed] [Google Scholar]
- 20.Wu D & Meydani SN Mechanism of age-associated up-regulation in macrophage PGE2 synthesis. Brain Behav. Immun 18, 487–494 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Breyer RM, Bagdassarian CK, Myers SA & Breyer MD Prostanoid receptors: subtypes and signaling. Annu. Rev. Pharmacol. Toxicol 41, 661–690 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Forselles KA et al. In vitro and in vivo characterisation of PF-04418948, a novel, potent and selective prostaglandin EP2 receptor antagonist. Br. J. Pharmacol 164, 1847–1856 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fox BM et al. A selective prostaglandin E2 receptor subtype 2 (EP2) antagonist increases the macrophage-mediated clearance of amyloid-beta plaques. J. Med. Chem 58, 5256–5273 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Johansson JU et al. Suppression of inflammation with conditional deletion of the prostaglandin E2 EP2 receptor in macrophages and brain microglia. J. Neurosci 33, 16016–16032 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sidhu VK, Huang BX, Desai A, Kevala K & Kim HY Role of DHA in aging-related changes in mouse brain synaptic plasma membrane proteome. Neurobiol. Aging 41, 73–85 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.VanGuilder HD, Yan H, Farley JA, Sonntag WE & Freeman WM Aging alters the expression of neurotransmission-regulating proteins in the hippocampal synaptoproteome. J. Neurochem 113, 1577–1588 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lynch MA Long-term potentiation and memory. Physiol. Rev 84, 87–136 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Castellone MD, Teramoto H, Williams BO, Druey KM & Gutkind JS Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-β-catenin signaling axis. Science 310, 1504–1510 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Jiang J & Dingledine R Prostaglandin receptor EP2 in the crosshairs of anti-inflammation, anti-cancer, and neuroprotection. Trends Pharmacol. Sci 34, 413–423 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jha AK et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42, 419–430 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Lampropoulou V et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab 24, 158–166 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mills EL et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167, 457–470 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tannahill GM et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanin DE et al. Mitochondrial membrane potential regulates nuclear gene expression in macrophages exposed to prostaglandin E2. Immunity 49, 1021–1033 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thwe PM et al. Cell-intrinsic glycogen metabolism supports early glycolytic reprogramming required for dendritic cell immune responses. Cell Metab 26, 558–567 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma J et al. Glycogen metabolism regulates macrophage-mediated acute inflammatory responses. Nat. Commun 11, 1769 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gauba E, Chen H, Guo L & Du H Cyclophilin D deficiency attenuates mitochondrial F1Fo ATP synthase dysfunction via OSCP in Alzheimer’s disease. Neurobiol. Dis 121, 138–147 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johansson JU et al. Prostaglandin signaling suppresses beneficial microglial function in Alzheimer’s disease models. J. Clin. Invest 125, 350–364 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aoki T et al. Prostaglandin E2–EP2–NF-κB signaling in macrophages as a potential therapeutic target for intracranial aneurysms. Sci. Signal 10, eaah6037 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Woodling NS & Andreasson KI Untangling the web: toxic and protective effects of neuroinflammation and PGE2 signaling in Alzheimer’s disease. ACS Chem. Neurosci 7, 454–463 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Monvoisin A et al. VE-cadherin-CreERT2 transgenic mouse: a model for inducible recombination in the endothelium. Dev. Dyn 235, 3413–3422 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Wimmer ME, Hernandez PJ, Blackwell J & Abel T Aging impairs hippocampus-dependent long-term memory for object location in mice. Neurobiol. Aging 33, 2220–2224 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Attar A et al. A shortened Barnes maze protocol reveals memory deficits at 4-months of age in the triple-transgenic mouse model of Alzheimer’s disease. PLoS ONE 8, e80355 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Latif-Hernandez A, Faldini E, Ahmed T & Balschun D Separate ionotropic and metabotropic glutamate receptor functions in depotentiation vs. LTP: a distinct role for group1 mGluR subtypes and NMDARs. Front. Cell. Neurosci 10, 252 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Su X, Lu W & Rabinowitz JD Metabolite spectral accuracy on Orbitraps. Anal. Chem 89, 5940–5948 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamashita A et al. Increased metabolite levels of glycolysis and pentose phosphate pathway in rabbit atherosclerotic arteries and hypoxic macrophage. PLoS ONE 9, e86426 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamada M et al. A comprehensive quantification method for eicosanoids and related compounds by using liquid chromatography/mass spectrometry with high speed continuous ionization polarity switching. J. Chromatogr. B 995–996, 74–84 (2015). [DOI] [PubMed] [Google Scholar]
- 48.Kita Y, Takahashi T, Uozumi N & Shimizu T A multiplex quantitation method for eicosanoids and platelet-activating factor using column-switching reversed-phase liquid chromatography-tandem mass spectrometry. Anal. Biochem 342, 134–143 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Zhou L & Li Q Isolation of region-specific microglia from one adult mouse brain hemisphere for deep single-cell RNA sequencing. J. Vis. Exp 154, e60347 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data for nanostring analysis are available from the Gene Expression Omnibus under accession code GSE155992. Untargeted metabolomics data are provided in the source data file for Extended Data Fig. 3d. Source data are provided with this paper.