

Abstract
Insulin-producing pancreatic β-cells are central to glucose homeostasis, and their failure is a principal driver of diabetes development. To preserve optimal health β-cells must withstand both intrinsic and extrinsic stressors, ranging from inflammation to increased peripheral insulin demand, in addition to maintaining insulin biosynthesis and secretory machinery. Autophagy is increasingly being appreciated as a critical β-cell quality control system vital for glycemic control. Here we focus on the underappreciated, yet crucial, roles for selective and organelle-specific forms of autophagy as mediators of β-cell health. We examine the unique molecular players underlying each distinct form of autophagy in β-cells, including selective autophagy of mitochondria, insulin granules, lipid, intracellular amyloid aggregates, endoplasmic reticulum, and peroxisomes. We also describe how defects in selective autophagy pathways contribute to the development of diabetes. As all forms of autophagy are not the same, a refined view of β-cell selective autophagy may inform new approaches to defend against the various insults leading to β-cell failure in diabetes.
Introduction
Pancreatic β-cell loss or dysfunction is central to diabetes development, as β-cells are responsible for producing, storing, and secreting insulin to maintain normoglycemia. Numerous stressors lead to β-cell failure in diabetes, including reactive oxygen species (ROS), mitochondrial damage, endoplasmic reticulum (ER) stress, inflammation, and toxic amyloid aggregates. Therefore, stress responses and quality control pathways that mitigate these stressors, including autophagy, are vital for maintaining β-cell homeostasis.
Autophagy is an evolutionarily conserved process that degrades and recycles cytoplasmic components via the lysosome, allowing for nutrient reallocation. Disruption of autophagy is associated with numerous human diseases, including diabetes (Table 1), and plays critical roles in metabolism, inflammation/inflammasome action (reviewed in 1,2), neurodegeneration, and tumor suppression (3,4). Autophagy is often viewed as a nonselective bulk degradation response activated during starvation to compensate for nutrient deficiency. However, we now have a broader view of autophagy. Autophagy is critical for basal cellular function in the presence of nutrients and is important for cellular metabolism, proliferation, and survival (5,6). While autophagy was initially characterized as a nonselective pathway, emerging evidence demonstrates that selective organellar autophagy governs the balance between normal cellular function and the development of disease (7,8). We therefore posit that defects in selective β-cell autophagy could potentiate β-cell failure in diabetes.
Table 1.
Human autophagy gene and diabetes associations
| Nearby/mapped gene identified* | Disease association | Autophagy association | SNPs | Study accession no. |
|---|---|---|---|---|
| CDKN1B | T2D | Macroautophagy | rs2066827 | GCST009379 |
| BCL2 | Τ2D | Macroautophagy | rs12454712 | GCST004894, GCST009379, GCST010118, GCST007847 |
| CAMKK2 | T2D | Macroautophagy | rs3794205 | GCST005414 |
| MAP2K7 | T2D | Macroautophagy | rs4804833 | GCST009379 |
| CALCOCO2 | T2D | Selective autophagy (xenophagy) | rs10278 | GCST007517 |
| VEGFA | T2D | Macroautophagy | rs11967262 | GCST009379 |
| PTEN | T2D | Macroautophagy | rs1236816 | GCST010118 |
| CLN3 | T1D | Macroautophagy | rs151234 | GCST009875 |
| CLEC16A | T1D | Macroautophagy, selective autophagy (mitophagy) | rs741172 | GCST009875 |
| CTSH | T1D | Macroautophagy | rs34593439 | GCST009875 |
This table was generated by cross-referencing mapped/nearby genes from published T1D and T2D GWAS polymorphisms with the human autophagy database. This table reflects the identified gene, the autophagy pathway associated with that gene, and the nearby SNP used to identify the gene.
Recent studies suggest that several distinct forms of selective autophagy occur within β-cells (Fig. 1): mitochondria (mitophagy), secretory vesicle (crinophagy), lipid (lipophagy), aggregate (aggrephagy), ER (ER-phagy), and peroxisome (pexophagy). Each form of selective autophagy is tightly regulated and possesses its own unique autophagy receptors and machinery, thus distinguishing between forms of selective autophagy. In effect, autophagy is not a singular monolithic process, and all forms of autophagy are not the same. Our goal is to review the latest evidence supporting the underappreciated importance of selective autophagy to β-cell function, while underscoring the need for future work clarifying the significance of selective versus nonselective autophagy within β-cells.
Figure 1.

Overview of selective autophagy in β-cells. Selective autophagy relies on the formation of autophagosomes (blue circle) surrounding specific cellular cargo and subsequent fusion with lysosomes (red circle) for cargo degradation. In pancreatic β-cells, selective forms of autophagy that have been described to date include 1) mitophagy, 2) crinophagy, 3) lipophagy, 4) aggrephagy, and 5) ER-phagy and pexophagy.
Selective Versus Nonselective Autophagy
Nonselective/bulk macroautophagy (commonly referred to as autophagy) initiates sequestration of cellular contents following starvation by an expanding double-membraned phagophore, which engulfs cargo within an enclosed autophagosome. The autophagosome then fuses with the lysosome forming an autolysosome, which leads to degradation of autophagosome contents. It is clear, then, that lysosomal function is crucial for completion of the autophagic process, and any perturbations of lysosomal pH, function, or activity will impact autophagic flux. Decades of work have led to the elucidation of basic mechanisms and identification of the core autophagy machinery, also known as autophagy-related (ATG) genes. Autophagosome formation and cargo engulfment are regulated by ATG proteins, which are highly conserved from yeast to mammals (9). Interestingly, observations of selective autophagy actually predate the coining of the word autophagy. In the 1960s, autophagosomes were first described as double-membraned sequestration compartments delivering cytoplasmic cargo to the lysosome, whose cargo depended on both the cell-type and environmental conditions (10–12). Others types of autophagy in eukaryotic cells include microautophagy and chaperone-mediated autophagy, which bypass the need for a phagophore and instead directly deliver targets to the lysosome, and have previously been reviewed (13).
Selective macroautophagy requires the same core ATG proteins as nonselective/bulk autophagy and additionally uses a growing number of selective autophagy receptors (14). Selective autophagy receptors, including p62/SQSTM1, Nbr1, Optineurin, and Ndp52, bind proteins on specific target cargo and act as a scaffold between the cargo and the lipidated protein LC3 (LC3-II) on the expanding phagophore membrane. It is also important to note that specific individual proteins can be selectively targeted for degradation via these receptors, independently of entire organelles. While nonselective autophagy is principally a response to starvation, selective autophagy is important for both stress responses and cellular homeostasis, playing roles in the removal of damaged organelles, protein aggregates, target proteins, and pathogens. As such, it should not be surprising that disruption of selective autophagy is associated with numerous human diseases, including diabetes (15,16).
Initial β-cell autophagy studies showed that loss of a key ATG protein, Atg7, in mouse β-cells led to impaired glucose tolerance and insulin secretion, decreased β-cell mass due to apoptosis, and an accumulation of dysfunctional mitochondria (17,18). Furthermore, β-cells from patients with type 2 diabetes (T2D) have increased autophagosomes, though it is unclear whether this represents a protective response to stress or a pathogenic dysregulation of autophagy (19). Since these initial observations, numerous studies focusing on loss of Atg7 (20–25) and other upstream autophagy regulators (26–28) have been performed in β-cells. Overall, autophagy is pivotal for β-cell health and function, yet variations in the duration and context of autophagy gene deletion can exert dramatically different effects on β-cell function (29). However, these models disrupt both bulk and selective autophagy, making it difficult to discern the relative importance of either in β-cells.
Mitophagy
Observations of mitochondrial cargo within autophagosomes date back to the 1960s (12), followed shortly thereafter by further reports of specific mitochondrial autophagy (30,31). In the late 1990s, certain nutrient deficiencies in hepatocytes and neurons were found to induce mitochondrial depolarization, followed by mitochondrial autophagy (32,33). In 2005, the term “mitophagy” was coined and proposed as a mechanism for eliminating aged and damaged mitochondria that would otherwise cause oxidative stress (34).
Selective mammalian mitophagy was first observed in the context of the PTEN-induced kinase-1 (PINK1)-Parkin pathway. PINK1 acts as a sensor for mitochondrial health and accumulates on the surface of damaged mitochondria to recruit and phosphorylate the cytoplasmic E3 ligase Parkin (Fig. 2). Following its mitochondrial recruitment and activation, Parkin ubiquitinates several outer mitochondrial membrane (OMM) proteins, which leads to the binding of selective autophagic adaptor proteins, including p62/SQSTM1, Nbr1, Optineurin, and Ndp52, via ubiquitin-interacting motifs (35,36). These adaptors directly conjugate ubiquitinated OMM proteins to LC3-II on the autophagophore membrane via LC3-interacting regions, allowing for autophagosome engulfment and subsequent mitochondrial degradation in the lysosome. While PINK1/Parkin-mediated mitophagy is the most well-studied form of mitophagy, Parkin-independent mitophagy has also been observed through recruitment of alternative E3 ligases to mitochondria (37), transmembrane-receptor mediated mitophagy (reviewed in 38), cardiolipin-mediated mitophagy (39), and activation of the novel E3 ubiquitin ligase Clec16a (40,41).
Figure 2.
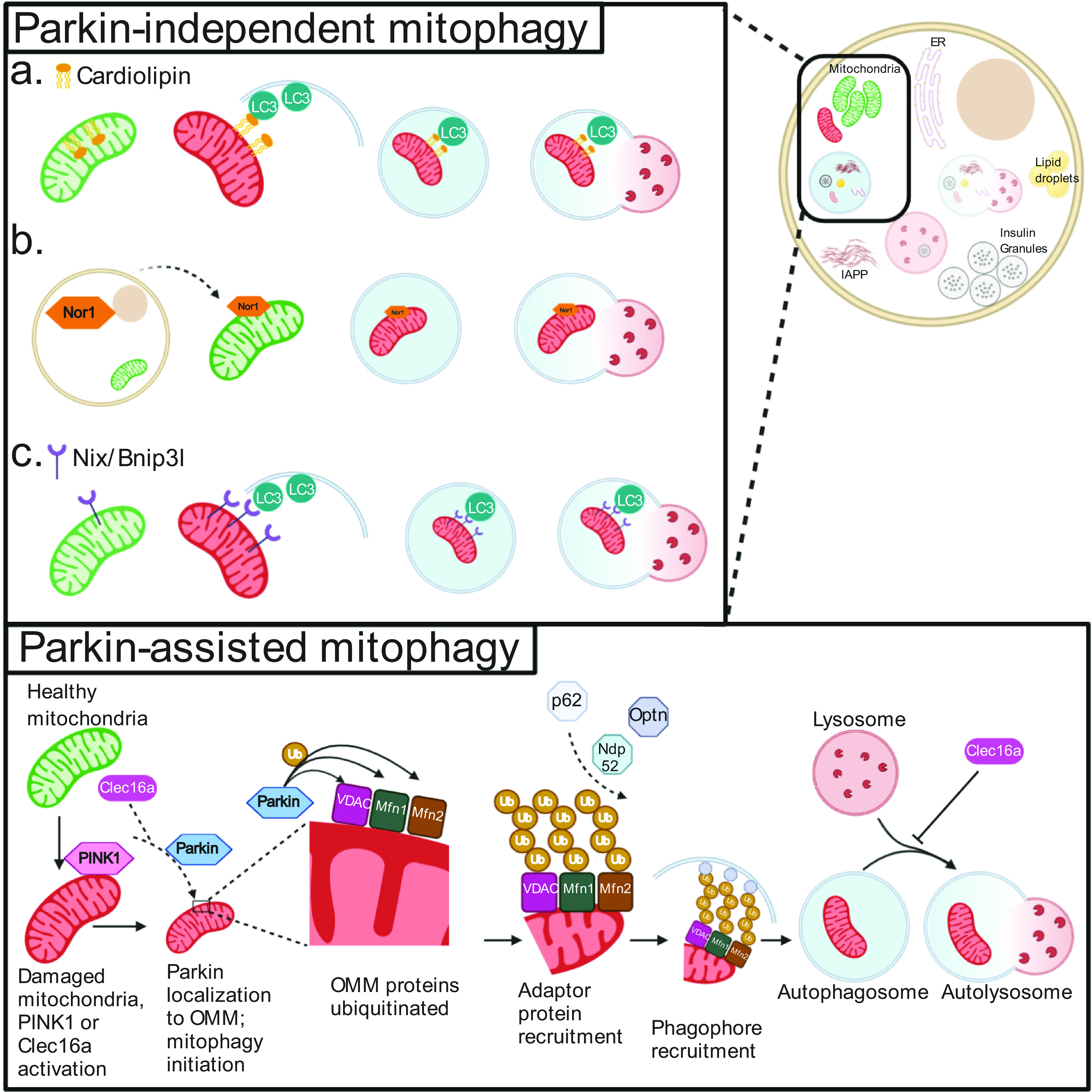
Mitophagy pathways in β-cells. Mitophagy can be initiated by a number of mechanisms, including those dependent (bottom) or independent (top) of the E3 ubiquitin ligase Parkin. During Parkin-mediated mitophagy, damaged mitochondria are recognized by activation of the Clec16a-Nrdp1-Usp8 complex, which leads Parkin to translocate to the OMM. Here, Parkin ubiquitinates a number of membrane proteins (including but not limited to VDAC, Mfn1, and Mfn2). The presence of ubiquitin (Ub) complexes signals to adaptor proteins capable of binding Ub and phagophore/autophagosome membrane resident proteins to traffic mitochondria to autophagosomes. Once the mitochondria are targeted to the phagophore, subsequent closure of the autophagosome and fusion with lysosomes result in selective mitochondrial clearance. These final steps are common to both Parkin-dependent and -independent mitophagy, as can be seen in a–c (top panel). Parkin-independent pathways of mitochondrial cargo recognition and clearance include the following (top panel): flipping of the phospholipid cardiolipin to the OMM, which can bind directly to LC3 on the expanding phagophore and is potentially targeted by the mitochondrial targeted tetrapeptide SS-31 (a); nuclear orphan receptor Nor1 translocation to the mitochondria (b); and increased expression of transmembrane receptor Nix/BNIP3L, which can again directly bind LC3 (c).
The ability of β-cells to secrete insulin in response to rising glucose levels depends on mitochondrial metabolism. Glucose metabolism increases mitochondrial ATP generation, altering the cytosolic ATP-to-ADP ratio and closing KATP channels, which is followed by cellular depolarization and insulin release. Thus, quality control mechanisms maintaining mitochondrial health and eliminating aged/damaged mitochondria are critical for β-cell function. Indeed, β-cells of human islet donors with T2D accumulate damaged and dysfunctional mitochondria (42). In the following sections, we will detail selective mitophagy pathways in β-cells, describing their role in cellular function and pathophysiology.
PINK1/Parkin-Mediated Mitophagy
The importance of the PINK1/Parkin pathway within β-cells to glucose homeostasis has been controversial. Hoshino et al. (43) observed that whole-body Parkin knockout mice have impairments in glucose tolerance and β-cell mitochondrial function and turnover and reduced glucose-stimulated insulin secretion (GSIS) following damage with the β-cell toxin streptozotocin. In contrast, using both pancreatic and β-cell–specific Parkin-deficient mouse models, we observed that Parkin was dispensable for glucose homeostasis, insulin secretion, β-cell mass, and islet architecture, both at baseline and in the context of diet-induced obesity (44) (summarized in Table 2). Of note, a similar inconsistency was observed when comparing the role of Parkin in adipocyte function in whole-body versus adipocyte-specific Parkin knockouts (44–46). These findings indicate that β-cell function may be increasingly dependent on Parkin when peripheral tissues concurrently lose their mitophagy machinery.
Table 2.
Mouse models of selective autophagy
| Gene | Model | Phenotype | Reference no. |
|---|---|---|---|
| Mitophagy | |||
| Bnipl3 (Nix) | Whole-body knockout on Pdx1+/− background | Restored glucose homeostasis and insulin secretion compared with Pdx1+/− alone | 65 |
| Clec16a | Pancreatic and β-cell–specific knockouts | Impaired GTT, loss of GSIS and mitochondrial respiration in β-cells | 40,41,53 |
| Pink1 | Whole-body knockout | Improved whole-body glucose tolerance and improved GSIS in isolated islets, reduced insulin islet content and mitochondrial respiration. Isolated islets have increased β-cell TFs | 49 |
| Prkn (Parkin) | Whole-body knockout | Impaired GTT and β-cell mitochondrial function, STZ treatment caused worse reduction in GSIS | 43 |
| Prkn (Parkin) | Pancreatic and β-cell–specific knockout | Normal GTT, insulin secretion, and β-cell mass on normal chow and high-fat diet | 44 |
| Rhot (Miro1) | β-cell–specific knockout | Impaired GTT after high-fat diet, reduced GSIS and mitochondrial function in β-cells | 48 |
| Tfb2m | β-cell–specific knockout | Progressive hyperglycemia and eventual diabetes due to impaired GSIS, reduced mitochondrial respiration, and loss of β-cell mass | 60 |
| Crinophagy | |||
| Atp6ap2 | β-cell–specific knockout | Impaired glucose tolerance, reduced GSIS and insulin content | 87 |
| Rab3a | Whole-body knockout | Glucose intolerance, reduced GSIS due to enhanced intracellular insulin degradation | 83 |
| Lipophagy | |||
| Lipa (Lal) | Whole-body knockout | Lower fasting glucose, increased glucose-stimulated plasma insulin levels, improved glucose tolerance, increased GSIS in isolated islets | 107 and unpublished observations* |
| Plin2 | Whole-body knockout on AkitaC96Y background | Protected from diabetes compared with Akita alone mice, lowered ER stress and β-cell death | 110 |
| Aggrephagy | |||
| hIAPP | β-cell–specific overexpression with Atg7 knockout | Diabetes development with decreased β-cell mass due to apoptosis and oxidative damage | 20,21,23 |
| Uchl1 | Whole-body Uchl1+/− nm3419 mutant with hIAPP overexpression | Accelerated diabetes onset compared with hIAPP overexpression alone, due to decreased β-cell mass via apoptosis | 123 |
GTT, glucose tolerance test; TFs, transcription factors; STZ, streptozotocin.
G.L.P. and T.J.B., unpublished observations.
Another player in the PINK1/Parkin pathway is Miro1, an OMM GTPase that serves as a mitochondrial docking site for Parkin (47). Miro1 loss reduces Parkin translocation and impairs mitophagy (47). Miro1 expression is reduced in human T2D islets and db/db mouse islets, and β-cell–specific loss in mice impairs glucose homeostasis following diet-induced obesity (48). Miro1 deficiency disrupts β-cell mitophagy, causing impaired insulin secretion and impaired mitochondrial function (48).
As for the upstream mitophagy effector PINK1, global PINK1 loss in mice surprisingly improves glucose tolerance (49). PINK1-deficient islets showed increased basal and glucose-stimulated insulin release, as well as reduced residual insulin stores in the context of high glucose and decreased mitochondrial glucose responsiveness (49). Therefore, it appears PINK1 loss impairs the ability of β-cells to couple glucose uptake and metabolism with appropriate insulin release, causing dysregulated basal insulin secretion and hypersecretion at high glucose (49). This suggests a role for PINK1 in β-cell metabolism independent of mitophagy. Further, McWilliams et al. (50) determined that PINK1 was dispensable for basal β-cell mitophagy in vivo. Interestingly, PINK1-deficient mice had increased expression of key β-cell transcription factors FoxA2, NeuroD, and Nkx6.1, which may explain their enhanced insulin secretion (49). Overall, there is no strong evidence to date that PINK1 controls β-cell function through mitophagy.
Clec16a-Mediated Mitophagy
Clec16a is an E3 ubiquitin ligase critical for regulating β-cell mitophagy (40,41,51). β-Cell–specific Clec16a loss impairs glucose homeostasis and insulin secretion and causes defects in mitochondrial turnover. Clec16a- deficiency results in increased Parkin levels and unrestrained Parkin activation, causing an accumulation of damaged mitochondria through overloaded mitophagy machinery (40). Clec16a forms a tripartite mitophagy complex with Nrdp1 and Usp8 to regulate mitophagic flux based on cellular demand. Specifically, Clec16a stabilizes the E3 ligase Nrdp1, which targets Parkin for proteasomal degradation to restrain Parkin activity during physiologically healthy states (41). Additionally, Usp8 contributes to the activation of Parkin following the induction of mitochondrial damage (41). Clec16a may also play a separate downstream role in mitophagy by promoting autophagosome-lysosome fusion (40) (Fig. 2).
Clec16a is implicated in the pathogenesis of type 1 diabetes (T1D), as it is a known T1D risk gene (52), and Clec16a deficiency sensitizes β-cells to inflammatory damage in both rodent and human islets (53). Further, Clec16a is transcriptionally regulated by the critical β-cell transcription factor and T2D susceptibility gene, PDX1, which may suggest a role for mitophagy in T2D pathogenesis (54). Indeed, the destabilization of the Clec16a-Nrdp1-Usp8 mitophagy complex in human islets by glucolipotoxicity further suggests that Clec16a-mediated mitophagy may be contributing to mitochondrial dysfunction in T2D (41).
Our recent studies of β-cell Clec16a deficiency suggest that excessive Parkin levels or activity could negatively impact β-cell function. In support of this concept, overexpression of Parkin reduced GSIS in Min6 insulinoma cells (55). Additionally, Kusminski et al. (56) determined that excess Parkin levels, caused by overexpression of the OMM protein mito-NEET, were detrimental to β-cell function. Mito-NEET overexpression in β-cells impaired mitochondrial oxidative capacity, leading to glucose intolerance and reduced GSIS. Mito-NEET induction increased β-cell Parkin expression and aberrantly activated Parkin-mediated mitophagy (56). Deletion of Parkin in the context of mito-NEET overexpression rescued glucose homeostasis and insulin secretion, indicating that excess Parkin activity impaired β-cell function (56). Collectively, these studies point to the importance of fine-tuning and restraining aberrant Parkin activity to maintain β-cell health.
Cardiolipin-Mediated Mitophagy
The phospholipid cardiolipin is found in the mitochondrial inner membrane, and its translocation to the OMM via phospholipid scramblase-3 and mitochondrial nucleoside diphosphate kinase-D promotes mitophagy in neuronal cells (39,57). Externalized cardiolipin initiates mitophagy by binding cardiolipin-interacting sites on LC3 to mediate autophagosome engulfment of mitochondria (39) (Fig. 2A). There is limited knowledge on the role of cardiolipin-mediated mitophagy in β-cells. The mitochondrial-localized tetrapeptide, elamipretide (SS-31), which is thought to bind cardiolipin on the inner mitochondrial membrane to prevent fragmentation (58), promotes mitophagy following glucolipotoxic stress in the IΝS1 β-cell line (59). However, elamipretide did not restore insulin secretion defects in INS1 cells following glucolipotoxicity (59). Thus, the impact of cardiolipin-mediated mitophagy on β-cell function remains unclear.
Transcriptional Regulators of Mitophagy
Beyond the regulation of Clec16a-mediated mitophagy by Pdx1 (see Clec16a-Mediated Mitophagy above [54]), other transcriptional regulators also influence β-cell mitophagy. Transcription factor B2 (TFB2M) is an RNA methyltransferase and a component of the mitochondrial transcription initiation complex. TFB2M is necessary to maintain β-cell mitochondrial DNA content, mitochondrial respiration, and GSIS (60). TFB2M deficiency causes an accumulation of mitophagy intermediates, suggesting impaired mitophagy (60). Nor1/NR4A3, an orphan nuclear receptor that functions as a transcriptional activator and also has proapoptotic extranuclear roles, is upregulated in T2D islets. Nor1/NR4A3 translocates to the mitochondria following exposure of INS1 832/13 cells to inflammatory cytokines, which aberrantly enhances mitophagy, and reduces mitochondrial function and GSIS (61,62) (Fig. 2B).
Transmembrane Receptor–Mediated Mitophagy
Selective mitophagy can also be mediated through outer mitochondrial transmembrane proteins directly binding LC3 on the phagophore (Fig. 2C), such as the BCL2/adenovirus E1B 19 kDa protein–interacting protein 3-like (BNIP3L) (also known as Nix) (63). Nix and the closely related protein BNIP3 are upregulated during hypoxia and mediate mitophagy as an adaptive response to metabolic stress (reviewed in 38). Interestingly, BNIP3 expression is upregulated in human islets following hypoxia, which may suggest the initiation of protective mitophagy (64). However, loss of Nix in mouse β-cells did not impair glucose homeostasis, insulin secretion, or autophagy, suggesting that Nix does not contribute to β-cell function through control of mitophagy (65). To our knowledge, the impact of BNIP3 in β-cell mitophagy is unexplored.
Mitochondrial Calcium and Mitophagy
The regulation of mitochondrial calcium has been connected to control of mitophagy in other metabolic tissues (66). In β-cells, mitochondrial calcium promotes respiration and ATP production by acting on calcium-sensitive enzymes (67,68). Calcium dysregulation disrupts mitochondrial membrane potential, increases ROS, and disrupts fission-fusion dynamics, all of which may potentiate mitophagy (68). Reducing mitochondrial calcium levels in primary mouse β-cells by silencing or genetic loss of the mitochondrial calcium uniporter (MCU) blunts glucose-stimulated ATP production and impairs ex vivo GSIS, indicating the importance of mitochondrial calcium dynamics to β-cell function (69,70). The direct impact of mitochondrial calcium dynamics on β-cell mitophagic flux, however, remains to be examined.
Mitochondrial-ER Contacts and Mitophagy
Mitochondrial-ER contact sites, which occur at mitochondria-associated membranes (MAMs), facilitate lipid and calcium exchange and are also proposed sites for autophagosome membrane formation (71). Following mitochondrial damage, PINK1/Parkin localize to MAMs and promote MAM formation, which is proceeded by autophagosome formation and mitophagy (72–74). The impact of MAMs on β-cell mitophagy is unclear. However, disruption of MAMs in primary mouse β-cells by selective EI24 loss impairs bulk autophagy (75). In INS1E β-cells, glucose stimulates MAM formation, and MAM disruption inhibits GSIS (76). Interestingly, chronic glucotoxicity also induces MAM formation in human islets (76). While a direct connection between MAMs and β-cell mitophagy is unclear, there is a significant role for MAMs in β-cell function, possibly through regulating β-cell mitophagy.
Selective mitochondrial turnover in β-cells is crucial to maintain β-cell health and function. Disruption of critical mitophagy regulators can precipitate apoptosis or hamper β-cell compensation for environmental stressors including insulin resistance or inflammation. It is also apparent that there are redundant β-cell mitophagy pathways (Fig. 2) that may compensate for each other to maintain β-cell mitochondrial health. While it is clear that mitochondrial structural and functional defects occur in human islets in T1D and T2D (42,53,77), future studies must resolve the importance of mitophagy in protection against the development of diabetes.
Crinophagy and Selective Insulin Granule Autophagy
Crinophagy refers to the selective degradation of secretory granules via direct fusion with lysosomes. It is therefore a specific form of microautophagy and thought to occur in the majority of secretory organ cells, including β-cells (78). Insulin granule crinophagy was first observed in 1980 in rat islets (79). Due to the high demand for insulin production, it is not surprising that β-cells use insulin turnover mechanisms to ensure that a functional insulin pool is always ready for release. Indeed, the number of insulin granules inside lysosomes was found to be increased as extracellular glucose concentrations declined (80,81), indicating a potential mechanism for eliminating older insulin stores when they are not readily needed. Defects in crinophagy have also been observed recently in the islets of T1D donors (82). Additionally, insulin granule turnover by macroautophagy has been proposed. Thus, both forms of selective insulin granule turnover (crinophagy and granule autophagy) will be discussed below (83–87).
To investigate the mechanisms by which insulin granules reached the lysosome, Marsh et al. (83) impaired insulin granule exocytosis through deletion of the insulin granule trafficking protein Rab3A. In response to impaired insulin exocytosis, insulin degradation via both crinophagy and granule autophagy was upregulated to ensure optimal insulin storage levels (83). Similarly, loss of BAIAP3, a Munc13 protein involved in dense core vesicle priming and docking for exocytosis, enhanced lysosomal degradation of insulin granules (86).
Selective insulin degradation is also observed in vitro during nutrient depletion, when insulin release/requirements should be at a nadir (84). This form of crinophagy appears to be dependent on protein kinase D and proximity to Golgi apparatus (88) and was dubbed starvation-induced nascent granule degradation (SINGD) (84). SINGD may share commonalities with another insulin granule degradation pathway, Golgi membrane–associated degradation (GOMED), which is induced upon glucose deprivation in autophagy-deficient β-cells (89). Intriguingly, mTOR-mediated macroautophagy was inhibited by the lysosomal products released from SINGD (84), suggesting that selective insulin degradation can act independently and upstream of bulk macroautophagy. It will be interesting to determine whether SINGD contributes to the dampened secretion of insulin that is associated with fasting or starvation in vivo.
The method of insulin granule degradation depends not only on upstream stimuli but also on whether proinsulin, insulin, or misfolded insulin is the target cargo (85,90). In INS-1E cells, insulin is degraded primarily by crinophagy, whereas proinsulin is cleared via an autophagosome-mediated route (85). In a mouse β-cell line established by Nozaki et al. (91), inhibition of autophagosome formation after cycloheximide treatment protected proinsulin levels, providing additional evidence that proinsulin is preferentially degraded by macroautophagy (90). However, in Min6 insulinoma cells, knockdown of Atg7 decreased autophagosome formation and increased total cellular insulin levels (25), suggesting that autophagosome-mediated pathways also regulate mature insulin degradation. Conversely, inhibition of autophagosome formation with the selective PI3K inhibitor 3-methyladenine in mouse islets reduced islet insulin content, yet increased GSIS (25). The opposing results of autophagy inhibition on insulin content could be due to differences in model systems and methodologies (3-methyladenine vs. Atg7 deficiency) or may be due to differences in crinophagy, which were not assessed. Further work is necessary to reconcile these results.
The proinsulin mutant Akita C(A7)Y was resistant to clearance by macroautophagy due to sequestration within the ER in INS-1E cells (85). However, Akita mice were protected from diabetes development when autophagy was induced by the mTOR inhibitor rapamycin, reducing ER stress and preventing β-cell apoptosis (27). This may be due to protective activation of other selective forms of autophagy, such as ER-phagy, which has recently been shown to selectively clear mutant proinsulin from the ER (92).
The lysosomal V-ATPase accessory protein Atp6ap2 restricts lysosomal fusion events upstream of macroautophagy activation. Atp6ap2 was found to be pivotal for control of insulin granule degradation (87). While the modification of lysosome function can control insulin degradation, the specific effects of lysosome function on crinophagy or selective granule autophagy were not addressed.
Recent studies demonstrate that younger insulin granules are preferentially released by β-cells over aged granules, consistent with the need for clearance of older, less dynamic insulin granules (93,94). The exact method of insulin granule degradation, whether through macroautophagy or crinophagy, depends on the specific cargo and stimulus inducing degradation. β-Cells appear to prioritize lysosomal degradation of insulin granules and even preferentially select for insulin granule degradation over other proteins including toxic IAPP aggregates (90). However, the specific effectors of this pathway remain a mystery. Interestingly, the small GTPases Rab2 and Rab7, a SNAP complex (consisting of Vamp7, SNAP29, and Syntaxin13), and the homotypic fusion and protein sorting (HOPS) complex have been implicated in selective crinophagy of salivary gland glue granules in Drosophila (95). Investigating roles for these and other targets in crinophagy and selective granule autophagy will be critical to understand how β-cells maintain an optimal insulin pool for glucose homeostasis.
Lipophagy
Lipid oversupply is a commonly observed destructive force in metabolic syndrome and T2D. Lipotoxicity is detrimental to the function and survival of β-cells (96). Exogenous treatment of β-cells with fatty acids causes a number of cellular responses, including alteration in lipid composition and induction of ER stress (96), which in and of themselves can impact bulk autophagy. However, specific lipid autophagy, or lipophagy, was first observed in 2009 when Singh et al. (97) found that lipid was a target of autophagic degradation in liver. Lipophagy has been extensively studied in the liver, hypothalamus, and fibroblasts (97–100), yet there has only been one article explicitly referencing β-cell lipophagy (25). However, electron microscopy images from murine and human islets after treatment with fatty acids frequently display lipid-containing autophagosomes (101), and many studies have explored the role of macroautophagy during lipid oversupply from high-fat feeding or ex vivo lipid exposure in β-cells (18,22,26,102,103). A recent proteomic screen identified a number of putative lipophagy-specific regulatory proteins (104), yet their role in β-cell function remains elusive. However, there are many inferences to the role of lipophagy in β-cell health and function to be found in the literature.
Some of the first insights into β-cell lipophagy arose from a study of high-fat diet feeding in β-cell–specific Atg7 knockout mice (18). High-fat diet increased autophagy in β-cells, and Atg7 deficiency led to apoptosis and impaired β-cell mass compensation to diet-induced obesity, suggesting that autophagy plays a critical protective role to lipid excess. The concept of lipid-induced β-cell lipophagy is further supported by the observations of lipid specifically within the autophagosomes of mouse and human islets after exogenous lipid exposure in isolated islets (101,105) and animal models (22,102). Indeed, in leptin-deficient ob/ob mice that develop extreme obesity, β-cell–specific Atg7 deficiency precipitated diabetes development due to β-cell apoptosis and loss of functional β-cell mass (22). β-cells appear to respond to lipid oversupply and obesity by increasing autophagy, while simultaneously decreasing lysosomal degradation of other cargo, including long-lived proteins (22), p62-conjugated aggregates (22,102), and mitochondria (26). These studies suggest that β-cells prioritize enhanced lipophagy at the expense of other degradative pathways, such as nonselective/bulk autophagy.
Lysosomal acid lipase (Lal), which is responsible for lipid turnover within the lysosome, has been observed to regulate β-cell function through lipophagy (25). Lipids can reach the lysosome in the form of cholesterol esters trafficked by direct endocytosis of LDL particles or cytosolic lipid droplets delivered by autophagosomes (106). Whole body loss of Lal caused reduced fasting glucose, and increased plasma insulin levels after a glucose bolus, along with a variety of lipid handling defects (107). In β-cells, inhibition of Lal promoted accumulation of cytosolic lipid droplets and enhanced GSIS, likely due to increased lipid intermediates amplifying insulin release (25). This study showed for the first time that impaired lipophagy caused lipid accumulation in pancreatic β-cells and positioned the lysosomal degradation of lipids as a key player in β-cell secretory function.
The maintenance and turnover of β-cell lipid droplets may also provide insights into lipophagy. Perilipin-2 (Plin2) is a membrane protein component of lipid droplets and recently has been implicated in regulating lipophagy in the liver and heart (108,109). Plin2 expression is increased following exogenous lipid exposure in β-cells (110), and Plin2 deficiency in Min6 insulinoma cells caused reciprocal upregulation of LC3-II with p62 downregulation following lysosomal inhibition with chloroquine, indicating increased autophagic flux (110). This study highlights that in the absence of a safe place to store exogenous lipid, i.e., lipid droplets, β-cells may upregulate autophagy/lipophagy to remove harmful lipid species.
Little is known regarding the function of lipophagy in human β-cells. Due to a higher frequency of lipid accumulation in human versus rodent islets, lipophagy could play a central role in human β-cell function. Indeed, initial observations of lipid droplets were made in human pancreatic tissue, demonstrating that human β-cells increase lipid storage with age (111). Later assessment of lipid storage in human β-cells using electron microscopy showed the presence of lipid/lysosomal structures, known as lipofuscin bodies (112). Lipofuscin bodies are waste storage organelles comprised of lipids with excessive peroxidation and cross-linking rendering them resistant to proteases and lipases within the lysosome. Lipofuscin bodies accumulated with age in human β-cells but were unchanged in T2D (112). Tong et al. (113) corroborated the increase in β-cell lipid droplets with age and also found lipid droplets increased substantially in T2D islets. While accumulation of indigestible lipofuscin bodies is a hallmark of aging β-cells, accumulation of potentially degradable lipid droplets in T2D islets may indicate changes in lipid metabolism and/or lipophagy. Indeed, a recent study demonstrated that glucose-stimulated lipolysis is impaired in human T2D islets (114), but the role of lipophagy in lipid handling in human β-cells and in T2D remains unexplored.
Aggrephagy of Toxic Islet Amyloid
Accumulation of protein aggregates is highly toxic and can disrupt cellular homeostasis. Many human diseases are associated with accumulation of intracellular aggregates, including neurodegenerative disorders and proteinopathies (reviewed in 115). While misfolded or damaged proteins can be degraded via the ubiquitin-proteasome system or by chaperone-mediated autophagy, large insoluble protein aggregates undergo selective aggregate autophagy, termed aggrephagy. Large protein aggregates are ubiquitinated and then associate with selective autophagy receptors (including p62/SQSTM1 or Nbr1), which recruit aggregates to the forming autophagosome (116). Both p62 and Nbr1 contain a C-terminal ubiquitin-associating domain that binds ubiquitinated aggregates, an LC3-interacting region that binds lipidated LC3 on the phagophore, and a Phox and Bem1 (PB1) domain, which assists in both phagophore recruitment and p62/Nbr1 self-interaction (116). While Nbr1 and p62 are implicated in aggrephagy, they also have roles in other types of selective autophagy including xenophagy and pexophagy (117,118).
Aggrephagy may play a critical role in eliminating toxic human islet amyloid polypeptide (hIAPP) aggregates found in T2D (119). hIAPP is a 37-residue protein that is cosecreted with insulin in β-cells. Increased levels of hIAPP, present as both intracellular β-cell oligomers and extracellular deposits, are found in islets in T2D (119). As rodent IAPP does not oligomerize, several transgenic hIAPP overexpression models have been developed to study the effects of hIAPP in β-cells (120). Transgenic overexpression of hIAPP in rat islets increased autophagosomes, seen by accumulation of LC3-II and p62 (121). hIAPP may stimulate aggrephagy, thereby increasing p62, or alternatively could inhibit aggrephagy, leading to accumulation of p62 due to decreased clearance. Following hIAPP overexpression, genetic loss of p62 and lysosomal inhibition each sensitized β-cells to hIAPP-induced toxicity (121). Interestingly, p62 is dispensable for normal β-cell function (122), suggesting a specific importance of p62 in the handling of hIAPP aggregates. These results suggest that hIAPP induction of p62 is protective and likely indicates increased autophagy/aggrephagy.
Further addressing the importance of autophagy/aggrephagy in compensation for hIAPP toxicity, three independent groups concurrently reported mouse models with β-cell–specific hIAPP overexpression together with Atg7 deficiency to impair autophagy/aggrephagy. All groups determined that aggrephagy protected β-cells from hIAPP toxicity (20,21,23). Mice with hIAPP overexpression and Atg7 loss developed overt diabetes, decreased β-cell mass and proliferation, increased oxidative damage, and increased apoptosis compared with hIAPP-alone controls (20,21,23). In all models, β-cells responded to hIAPP toxicity by enhancing autophagy and sequestering hIAPP in inert p62-positive inclusions, which were absent in autophagy-deficient mice. Together, these studies suggest that activation of aggrephagy is a protective response to combat toxic intracellular hIAPP aggregates.
Aggrephagy of hIAPP in β-cells is also modulated by a novel protein, ubiquitin carboxyl-terminal hydrolase isozyme L1 (Uchl1), without formerly known roles in autophagy. Uchl1 is a de-ubiquitinase that promotes hIAPP degradation and is deficient in T2D β-cells (123). β-Cell Uchl1 deficiency exacerbated hIAPP toxicity and impaired autophagy in vivo (123). Following hIAPP overexpression, Uchl1 deficiency decreased β-cell mass, increased β-cell apoptosis, and accelerated diabetes onset (123). Uchl1 loss also enhanced the accumulation of cytoplasmic p62-positive hIAPP inclusions, which could represent overloading of the aggrephagy pathway (123). While Uchl1 is classically implicated in the ubiquitin/proteasome system, this observation suggests that Uchl1 also plays a critical role in β-cell aggrephagy (123).
Together, these studies suggest that hIAPP aggregates are removed by aggrephagy, which may be critical to maintain β-cell health and function in T2D. However, many of these studies use genetic loss of Atg7 to study aggrephagy responses to hIAPP aggregates in β-cells, which disrupt both nonselective and selective autophagy. Thus, a focus on known regulators of aggrephagy, including the scaffolding protein ALFY (124) and the sorting protein Ubiquilin-1 (125), is critical to refine the understanding of aggrephagy of hIAPP in β-cells. Moreover, new technologies to study aggrephagy, including an inducible multimerization module to force protein aggregation, combined with fluorescent reporters to quantify aggregate delivery to the lysosome (126), may provide further insights into β-cell aggrephagy.
Other Selective Autophagy
Other forms of selective autophagy, including ER-phagy and pexophagy, have both very recently been reported in β-cells. Selective ER-phagy in mammalian cells has been linked to a number of ER-resident receptors, including Fam134b, Sec62, Ccpg1, and Reticulon 3 (Rtn3) (127), among others identified in a recent genome-wide CRISPR screen (128). In β-cells, autophagic ER degradation is critical for clearance of insoluble ER-resident aggregates of misfolded Akita proinsulin and wild-type proinsulin, along with canonical ER-associated degradation pathways (92). The degradation of these ER-resident aggregates required Rtn3. Overexpression of Rtn3 promoted aggregate clearance in INS1 832/13 cells, likely due to enhanced selective ER-phagy (92). The role of other ER-phagy receptor mediators (128) in normal β-cell function, or in diabetogenic conditions, remains unknown.
Peroxisomes are genuinely mysterious organelles, whose functions are speculated to be in fatty acid or ROS metabolism (129). β-cell–specific loss of the key peroxisomal protein, Pex5, led to β-cell dysfunction and obvious vacuolation by electron microscopy, potentially indicative of impaired autophagy (129). Acute β-cell–specific autophagy deficiency impacted peroxisomal function, with increases in specific akyl- or alkenyl-phospholipids (plasmalogens) following loss of Atg7 (29). Intriguingly, these plasmalogen species also appear to regulate GSIS (130). Together, these data indicate that peroxisomal function, and specifically pexophagy, may be more important in β-cells than previously appreciated.
Pharmacologic Manipulation of Selective Autophagy
Given the diverse roles of selective autophagy within β-cells, the development of pharmacologic agents targeting selective autophagy may improve β-cell health and function in diabetes. Urolithin A, a metabolite produced by gut bacteria, induces mitophagy and improves mitochondrial health in humans (59). Two similar benzothiophenes also induce basal mitophagy and improve insulin secretion in INS1 cells and mouse islets (131). Elamipretide (SS-31) also stimulates basal mitophagy and protects against mitochondrial fragmentation during nutrient excess in INS1 cells (59). Targeting mitochondrial calcium or mitochondrial ER contacts may be an additional mechanism to regulate mitophagy. Expanding the repertoire of drugs targeting other forms of selective autophagy, and understanding how these drugs impact human β-cell function, could provide a new avenue to treat diabetes.
Conclusions
Autophagy is often positioned as a bulk-degradative process activated in response to starvation. However, we are now are beginning to elucidate the nuanced roles autophagy plays in the calibrated and selective clearance of organelles/aggregates. The work described above lays the framework for recognizing the importance of selective autophagy in β-cells and moves beyond the cursory impression that all autophagy is the same. However, several studies on selective autophagy in β-cells still rely on manipulating machinery that also affects bulk autophagy. Thus, a coordinated focus on mechanisms underlying the selective turnover of cargo is needed to discern the roles of selective autophagy in β-cell health. More fully comprehending how, when, and why β-cells use selective autophagy pathways in response to distinct internal and external stressors will provide crucial insights to our understanding of β-cell failure in diabetes.
Article Information
Acknowledgments. Figures were created with BioRender (biorender.com).
Funding. S.A.S. was supported by the JDRF (CDA-2016-189, SRA-2018-539, COE-2019-861); the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institutes of Health (R01 DK108921, U01 DK127747, and P30 DK020572); and the U.S. Department of Veterans Affairs (I01 BX004444). G.L.P. was supported by the American Diabetes Association (19-PDF-063). M.A.G. was supported by the National Institute of General Medical Sciences, NIH (T32 GM008322), and the NIDDK, NIH (F31 DK122761). The JDRF Career Development Award to S.A.S. is partly supported by the Danish Diabetes Academy and the Novo Nordisk Foundation.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Footnotes
G.L.P. and M.A.G. contributed equally to this work.
References
- 1. Harris J, Lang T, Thomas JPW, Sukkar MB, Nabar NR, Kehrl JH. Autophagy and inflammasomes. Mol Immunol 2017;86:10–15 [DOI] [PubMed] [Google Scholar]
- 2. Rocha M, Apostolova N, Diaz-Rua R, Muntane J, Victor VM. Mitochondria and T2D: role of autophagy, ER stress, and inflammasome. Trends Endocrinol Metab 2020;31:725–741 [DOI] [PubMed] [Google Scholar]
- 3. Kim J, Lim Y-M, Lee M-S. The role of autophagy in systemic metabolism and human-type diabetes. Mol Cells 2018;41:11–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell 2019;176:11–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011;147:728–741 [DOI] [PubMed] [Google Scholar]
- 6. Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010;40:280–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol 2018;20:233–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011;7:279–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yin Z, Pascual C, Klionsky DJ. Autophagy: machinery and regulation. Microb Cell 2016;3:588–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hruban Z, Spargo B, Swift H, Wissler RW, Kleinfeld RG. Focal cytoplasmic degradation. Am J Pathol 1963;42:657–683 [PMC free article] [PubMed] [Google Scholar]
- 11. Ashford TP, Porter KR. Cytoplasmic components in hepatic cell lysosomes. J Cell Biol 1962;12:198–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deter RL, Baudhuin P, De Duve C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J Cell Biol 1967;35:C11–C16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yim WW, Mizushima N. Lysosome biology in autophagy. Cell Discov 2020;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shintani T, Huang WP, Stromhaug PE, Klionsky DJ. Mechanism of cargo selection in the cytoplasm to vacuole targeting pathway. Dev Cell 2002;3:825–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mizumura K, Choi AM, Ryter SW. Emerging role of selective autophagy in human diseases. Front Pharmacol 2014;5:244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van der Vaart A, Mari M, Reggiori F. A picky eater: exploring the mechanisms of selective autophagy in human pathologies. Traffic 2008;9:281–289 [DOI] [PubMed] [Google Scholar]
- 17. Jung HS, Chung KW, Won Kim J, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab 2008;8:318–324 [DOI] [PubMed] [Google Scholar]
- 18. Ebato C, Uchida T, Arakawa M, et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab 2008;8:325–332 [DOI] [PubMed] [Google Scholar]
- 19. Masini M, Bugliani M, Lupi R, et al. Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 2009;52:1083–1086 [DOI] [PubMed] [Google Scholar]
- 20. Shigihara N, Fukunaka A, Hara A, et al. Human IAPP-induced pancreatic β cell toxicity and its regulation by autophagy. J Clin Invest 2014;124:3634–3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rivera JF, Costes S, Gurlo T, Glabe CG, Butler PC. Autophagy defends pancreatic β cells from human islet amyloid polypeptide-induced toxicity. J Clin Invest 2014;124:3489–3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Quan W, Hur KY, Lim Y, et al. Autophagy deficiency in beta cells leads to compromised unfolded protein response and progression from obesity to diabetes in mice. Diabetologia 2012;55:392–403 [DOI] [PubMed] [Google Scholar]
- 23. Kim J, Cheon H, Jeong YT, et al. Amyloidogenic peptide oligomer accumulation in autophagy-deficient β cells induces diabetes. J Clin Invest 2014;124:3311–3324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abe H, Uchida T, Hara A, et al. Exendin-4 improves β-cell function in autophagy-deficient β-cells. Endocrinology 2013;154:4512–4524 [DOI] [PubMed] [Google Scholar]
- 25. Pearson GL, Mellett N, Chu KY, et al. Lysosomal acid lipase and lipophagy are constitutive negative regulators of glucose-stimulated insulin secretion from pancreatic beta cells. Diabetologia 2014;57:129–139 [DOI] [PubMed] [Google Scholar]
- 26. Las G, Serada SB, Wikstrom JD, Twig G, Shirihai OS. Fatty acids suppress autophagic turnover in β-cells. J Biol Chem 2011;286:42534–42544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bachar-Wikstrom E, Wikstrom JD, Ariav Y, et al. Stimulation of autophagy improves endoplasmic reticulum stress–induced diabetes. Diabetes 2013;62:1227–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blandino-Rosano M, Barbaresso R, Jimenez-Palomares M, et al. Loss of mTORC1 signalling impairs β-cell homeostasis and insulin processing. Nat Commun 2017;8:16014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chu KY, Mellet N, Thai LM, Meikle PJ, Biden TJ. Short-term inhibition of autophagy benefits pancreatic β-cells by augmenting ether lipids and peroxisomal function, and by countering depletion of n-3 polyunsaturated fatty acids after fat-feeding. Mol Metab 2020;40:101023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Beaulaton J, Lockshin RA. Ultrastructural study of the normal degeneration of the intersegmental muscles of Anthereae polyphemus and Manduca sexta (Insecta, Lepidoptera) with particular reference of cellular autophagy. J Morphol 1977;154:39–57 [DOI] [PubMed] [Google Scholar]
- 31. Remé CE, Young RW. The effects of hibernation on cone visual cells in the ground squirrel. Invest Ophthalmol Vis Sci 1977;16:815–840 [PubMed] [Google Scholar]
- 32. Lemasters JJ, Nieminen AL, Qian T, et al. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta 1998;1366:177–196 [DOI] [PubMed] [Google Scholar]
- 33. Xue L, Fletcher GC, Tolkovsky AM. Autophagy is activated by apoptotic signalling in sympathetic neurons: an alternative mechanism of death execution. Mol Cell Neurosci 1999;14:180–198 [DOI] [PubMed] [Google Scholar]
- 34. Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 2005;8:3–5 [DOI] [PubMed] [Google Scholar]
- 35. Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015;524:309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moore AS, Holzbaur EL. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci U S A 2016;113:E3349–E3358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Villa E, Marchetti S, Ricci JE. No Parkin zone: mitophagy without Parkin. Trends Cell Biol 2018;28:882–895 [DOI] [PubMed] [Google Scholar]
- 38. Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ 2009;16:939–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chu CT, Ji J, Dagda RK, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol 2013;15:1197–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Soleimanpour SA, Gupta A, Bakay M, et al. The diabetes susceptibility gene Clec16a regulates mitophagy. Cell 2014;157:1577–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pearson G, Soleimanpour SA. A ubiquitin-dependent mitophagy complex maintains mitochondrial function and insulin secretion in beta cells. Autophagy 2018;14:1160–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Anello M, Lupi R, Spampinato D, et al. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia 2005;48:282–289 [DOI] [PubMed] [Google Scholar]
- 43. Hoshino A, Ariyoshi M, Okawa Y, et al. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic β-cell function in diabetes. Proc Natl Acad Sci U S A 2014;111:3116–3121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Corsa CAS, Pearson GL, Renberg A, et al. The E3 ubiquitin ligase parkin is dispensable for metabolic homeostasis in murine pancreatic β cells and adipocytes. J Biol Chem 2019;294:7296–7307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim KY, Stevens MV, Akter MH, et al. Parkin is a lipid-responsive regulator of fat uptake in mice and mutant human cells. J Clin Invest 2011;121:3701–3712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu X, Altshuler-Keylin S, Wang Q, et al. Mitophagy controls beige adipocyte maintenance through a Parkin-dependent and UCP1-independent mechanism. Sci Signal 2018;11:eaap8526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Safiulina D, Kuum M, Choubey V, et al. Miro proteins prime mitochondria for Parkin translocation and mitophagy. EMBO J 2019;38:e99384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen L, Liu C, Gao J, et al. Inhibition of Miro1 disturbs mitophagy and pancreatic β-cell function interfering insulin release via IRS-Akt-Foxo1 in diabetes. Oncotarget 2017;8:90693–90705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Deas E, Piipari K, Machhada A, et al. PINK1 deficiency in β-cells increases basal insulin secretion and improves glucose tolerance in mice. Open Biol 2014;4:140051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McWilliams TG, Prescott AR, Montava-Garriga L, et al. Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab 2018;27:439–449.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gingerich MA, Sidarala V, Soleimanpour SA. Clarifying the function of genes at the chromosome 16p13 locus in type 1 diabetes: CLEC16A and DEXI. Genes Immun 2020;21:79–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hakonarson H, Grant SFA, Bradfield JP, et al. A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature 2007;448:591–594 [DOI] [PubMed] [Google Scholar]
- 53. Sidarala V, Pearson GL, Parekh VS, et al. Mitophagy protects β cells from inflammatory damage in diabetes. JCI Insight 2020;5:e141138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Soleimanpour SA, Ferrari AM, Raum JC, et al. Diabetes susceptibility genes Pdx1 and Clec16a function in a pathway regulating mitophagy in β-cells. Diabetes 2015;64:3475–3484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hofmeister-Brix A, Kollmann K, Langer S, Schultz J, Lenzen S, Baltrusch S. Identification of the ubiquitin-like domain of midnolin as a new glucokinase interaction partner. J Biol Chem 2013;288:35824–35839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kusminski CM, Chen S, Ye R, et al. MitoNEET-Parkin effects in pancreatic α- and β-cells, cellular survival, and intrainsular cross talk. Diabetes 2016;65:1534–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kagan VE, Jiang J, Huang Z, et al. NDPK-D (NM23-H4)-mediated externalization of cardiolipin enables elimination of depolarized mitochondria by mitophagy. Cell Death Differ 2016;23:1140–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Birk AV, Liu S, Soong Y, et al. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol 2013;24:1250–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Petcherski A, Trudeau KM, Wolf DM, et al. Elamipretide promotes mitophagosome formation and prevents its reduction induced by nutrient excess in INS1 β-cells. J Mol Biol 2018;430:4823–4833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nicholas LM, Valtat B, Medina A, et al. Mitochondrial transcription factor B2 is essential for mitochondrial and cellular function in pancreatic β-cells. Mol Metab 2017;6:651–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Close AF, Dadheech N, Villela BS, Rouillard C, Buteau J. The orphan nuclear receptor Nor1/Nr4a3 is a negative regulator of β-cell mass. J Biol Chem 2019;294:4889–4897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Close AF, Dadheech N, Lemieux H, Wang Q, Buteau J. Disruption of beta-cell mitochondrial networks by the orphan nuclear receptor Nor1/Nr4a3. Cells 2020;9:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Novak I, Kirkin V, McEwan DG, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 2010;11:45–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang M, Crager M, Pugazhenthi S. Modulation of apoptosis pathways by oxidative stress and autophagy in β cells. Exp Diabetes Res 2012;2012:647914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fujimoto K, Ford EL, Tran H, et al. Loss of Nix in Pdx1-deficient mice prevents apoptotic and necrotic β cell death and diabetes. J Clin Invest 2010;120:4031–4039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Favaro G, Romanello V, Varanita T, et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat Commun 2019;10:2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Georgiadou E, Rutter GA. Control by Ca2+ of mitochondrial structure and function in pancreatic β-cells. Cell Calcium 2020;91:102282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rimessi A, Bonora M, Marchi S, et al. Perturbed mitochondrial Ca2+ signals as causes or consequences of mitophagy induction. Autophagy 2013;9:1677–1686 [DOI] [PubMed] [Google Scholar]
- 69. Tarasov AI, Semplici F, Ravier MA, et al. The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced ATP increases in pancreatic β-cells. PLoS One 2012;7:e39722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Georgiadou E, Haythorne E, Dickerson MT, et al. The pore-forming subunit MCU of the mitochondrial Ca2+ uniporter is required for normal glucose-stimulated insulin secretion in vitro and in vivo in mice. Diabetologia 2020;63:1368–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hamasaki M, Furuta N, Matsuda A, et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013;495:389–393 [DOI] [PubMed] [Google Scholar]
- 72. Yang JY, Yang WY. Bit-by-bit autophagic removal of parkin-labelled mitochondria. Nat Commun 2013;4:2428. [DOI] [PubMed] [Google Scholar]
- 73. Gelmetti V, De Rosa P, Torosantucci L, et al. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 2017;13:654–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yang M, Li C, Yang S, et al. Mitochondria-associated ER membranes - the origin site of autophagy. Front Cell Dev Biol 2020;8:595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yuan L, Liu Q, Wang Z, Hou J, Xu P. EI24 tethers endoplasmic reticulum and mitochondria to regulate autophagy flux. Cell Mol Life Sci 2020;77:1591–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dingreville F, Panthu B, Thivolet C, et al. Differential effect of glucose on ER-mitochondria Ca2+ exchange participates in insulin secretion and glucotoxicity-mediated dysfunction of β-cells. Diabetes 2019;68:1778–1794 [DOI] [PubMed] [Google Scholar]
- 77. Lightfoot YL, Chen J, Lenchik N, et al. Decreased bioenergetic potential of β-cells in type 1 diabetes (T1D) (Abstract). Diabetes 2015;64(Suppl. 1):A89 [Google Scholar]
- 78. Weckman A, Di Ieva A, Rotondo F, et al. Autophagy in the endocrine glands. J Mol Endocrinol 2014;52:R151–R163 [DOI] [PubMed] [Google Scholar]
- 79. Halban PA, Wollheim CB. Intracellular degradation of insulin stores by rat pancreatic islets in vitro. An alternative pathway for homeostasis of pancreatic insulin content. J Biol Chem 1980;255:6003–6006 [PubMed] [Google Scholar]
- 80. Landström AHS, Westman J, Borg LAH. Lysosomes and pancreatic islet function. Time course of insulin biosynthesis, insulin secretion, and lysosomal transformation after rapid changes in glucose concentration. Diabetes 1988;37:309–316 [DOI] [PubMed] [Google Scholar]
- 81. Sandberg M, Borg LAH. Intracellular degradation of insulin and crinophagy are maintained by nitric oxide and cyclo-oxygenase 2 activity in isolated pancreatic islets. Biol Cell 2006;98:307–315 [DOI] [PubMed] [Google Scholar]
- 82. Muralidharan C, Conteh AM, Marasco MR, et al. Pancreatic beta cell autophagy is impaired in type 1 diabetes. Diabetologia 2021;64:865–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Marsh BJ, Soden C, Alarcón C, et al. Regulated autophagy controls hormone content in secretory-deficient pancreatic endocrine β-cells. Mol Endocrinol 2007;21:2255–2269 [DOI] [PubMed] [Google Scholar]
- 84. Goginashvili A, Zhang Z, Erbs E, et al. Insulin granules. Insulin secretory granules control autophagy in pancreatic β cells. Science 2015;347:878–882 [DOI] [PubMed] [Google Scholar]
- 85. Riahi Y, Wikstrom JD, Bachar-Wikstrom E, et al. Autophagy is a major regulator of beta cell insulin homeostasis. Diabetologia 2016;59:1480–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zhang X, Jiang S, Mitok KA, Li L, Attie AD, Martin TFJ. BAIAP3, a C2 domain-containing Munc13 protein, controls the fate of dense-core vesicles in neuroendocrine cells. J Cell Biol 2017;216:2151–2166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Binger KJ, Neukam M, Tattikota SG, et al. Atp6ap2 deletion causes extensive vacuolation that consumes the insulin content of pancreatic β cells. Proc Natl Acad Sci U S A 2019;1:19983–19988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Pasquier A, Vivot K, Erbs E, et al. Lysosomal degradation of newly formed insulin granules contributes to β cell failure in diabetes. Nat Commun 2019;10:3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yamaguchi H, Arakawa S, Kanaseki T, et al. Golgi membrane-associated degradation pathway in yeast and mammals. EMBO J 2016;35:1991–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Zhang X, Yuan Q, Tang W, Gu J, Osei K, Wang J. Substrate-favored lysosomal and proteasomal pathways participate in the normal balance control of insulin precursor maturation and disposal in β-cells. PLoS One 2011;6:e27647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Nozaki Ji, Kubota H, Yoshida H, et al. The endoplasmic reticulum stress response is stimulated through the continuous activation of transcription factors ATF6 and XBP1 in Ins2+/Akita pancreatic beta cells. Genes Cells 2004;9:261–270 [DOI] [PubMed] [Google Scholar]
- 92. Cunningham CN, Williams JM, Knupp J, Arunagiri A, Arvan P, Tsai B. Cells deploy a two-pronged strategy to rectify misfolded proinsulin aggregates. Mol Cell 2019;75:442–456.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Hoboth P, Müller A, Ivanova A, et al. Aged insulin granules display reduced microtubule-dependent mobility and are disposed within actin-positive multigranular bodies. Proc Natl Acad Sci U S A 2015;112:E667–E676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Yau B, Hays L, Liang C, et al. A fluorescent timer reporter enables sorting of insulin secretory granules by age. J Biol Chem 2020;295:8901–8911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Csizmadia T, Lőrincz P, Hegedűs K, Széplaki S, Lőw P, Juhász G. Molecular mechanisms of developmentally programmed crinophagy in Drosophila. J Cell Biol 2018;217:361–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Biden TJ, Boslem E, Chu KY, Sue N. Lipotoxic endoplasmic reticulum stress, β cell failure, and type 2 diabetes mellitus. Trends Endocrinol Metab 2014;25:389–398 [DOI] [PubMed] [Google Scholar]
- 97. Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature 2009;458:1131–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hernández-Gea V, Ghiassi-Nejad Z, Rozenfeld R, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012;142:938–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kaushik S, Rodriguez-Navarro JA, Arias E, et al. Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab 2011;14:173–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Rambold AS, Cohen S, Lippincott-Schwartz J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell 2015;32:678–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Martino L, Masini M, Novelli M, et al. Palmitate activates autophagy in INS-1E β-cells and in isolated rat and human pancreatic islets. PLoS One 2012;7:e36188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Chu KY, O’Reilly L, Ramm G, Biden TJ. High-fat diet increases autophagic flux in pancreatic beta cells in vivo and ex vivo in mice. Diabetologia 2015;58:2074–2078 [DOI] [PubMed] [Google Scholar]
- 103. Chu KY, O’Reilly L, Mellet N, Meikle PJ, Bartley C, Biden TJ. Oleate disrupts cAMP signaling, contributing to potent stimulation of pancreatic β-cell autophagy. J Biol Chem 2019;294:1218–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Robichaud S, Fairman G, Vijithakumar V, et al. Identification of novel lipid droplet factors that regulate lipophagy and cholesterol efflux in macrophage foam cells. Autophagy, 26 September 2008. Accessed 27 October 2020. Available from 10.1080/15548627.2021.1886839 [DOI] [PMC free article] [PubMed]
- 105. Chen YY, Sun LQ, Wang BA, Zou XM, Mu YM, Lu JM. Palmitate induces autophagy in pancreatic β-cells via endoplasmic reticulum stress and its downstream JNK pathway. Int J Mol Med 2013;32:1401–1406 [DOI] [PubMed] [Google Scholar]
- 106. Zhang H. Lysosomal acid lipase and lipid metabolism: new mechanisms, new questions, and new therapies. Curr Opin Lipidol 2018;29:218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Du H, Heur M, Duanmu M, et al. Lysosomal acid lipase-deficient mice: depletion of white and brown fat, severe hepatosplenomegaly, and shortened life span. J Lipid Res 2001;42:489–500 [PubMed] [Google Scholar]
- 108. Mardani I, Tomas Dalen K, Drevinge C, et al. Plin2-deficiency reduces lipophagy and results in increased lipid accumulation in the heart. Sci Rep 2019;9:6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Tsai TH, Chen E, Li L, et al. The constitutive lipid droplet protein PLIN2 regulates autophagy in liver. Autophagy 2017;13:1130–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Chen E, Tsai TH, Li L, Saha P, Chan L, Chang BH-J. PLIN2 is a key regulator of the unfolded protein response and endoplasmic reticulum stress resolution in pancreatic β cells. Sci Rep 2017;7:40855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Cnop M, Grupping A, Hoorens A, Bouwens L, Pipeleers-Marichal M, Pipeleers D. Endocytosis of low-density lipoprotein by human pancreatic β cells and uptake in lipid-storing vesicles, which increase with age. Am J Pathol 2000;156:237–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Cnop M, Hughes SJ, Igoillo-Esteve M, et al. The long lifespan and low turnover of human islet beta cells estimated by mathematical modelling of lipofuscin accumulation. Diabetologia 2010;53:321–330 [DOI] [PubMed] [Google Scholar]
- 113. Tong X, Dai C, Walker JT, et al. Lipid droplet accumulation in human pancreatic islets is dependent on both donor age and health. Diabetes 2020;69:342–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Liu S, Promes JA, Harata M, et al. Adipose triglyceride lipase is a key lipase for the mobilization of lipid droplets in human β-cells and critical for the maintenance of syntaxin 1a levels in β-cells. Diabetes 2020;69:1178–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Lamark T, Johansen T. Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol 2012;2012:736905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Lamark T, Kirkin V, Dikic I, Johansen T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 2009;8:1986–1990 [DOI] [PubMed] [Google Scholar]
- 117. Deosaran E, Larsen KB, Hua R, et al. NBR1 acts as an autophagy receptor for peroxisomes. J Cell Sci 2013;126:939–952 [DOI] [PubMed] [Google Scholar]
- 118. Hafrén A, Hofius D. NBR1-mediated antiviral xenophagy in plant immunity. Autophagy 2017;13:2000–2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Cooper GJ, Willis AC, Clark A, Turner RC, Sim RB, Reid KB. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc Natl Acad Sci U S A 1987;84:8628–8632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Westermark P, Engström U, Johnson KH, Westermark GT, Betsholtz C. Islet amyloid polypeptide: pinpointing amino acid residues linked to amyloid fibril formation. Proc Natl Acad Sci U S A 1990;87:5036–5040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Rivera JF, Gurlo T, Daval M, et al. Human-IAPP disrupts the autophagy/lysosomal pathway in pancreatic β-cells: protective role of p62-positive cytoplasmic inclusions. Cell Death Differ 2011;18:415–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Honda A, Komiya K, Hara A, et al. Normal pancreatic β-cell function in mice with RIP-Cre-mediated inactivation of p62/SQSTM1. Endocr J 2018;65:83–89 [DOI] [PubMed] [Google Scholar]
- 123. Costes S, Gurlo T, Rivera JF, Butler PC. UCHL1 deficiency exacerbates human islet amyloid polypeptide toxicity in β-cells: evidence of interplay between the ubiquitin/proteasome system and autophagy. Autophagy 2014;10:1004–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Filimonenko M, Isakson P, Finley KD, et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol Cell 2010;38:265–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. N’Diaye EN, Kajihara KK, Hsieh I, Morisaki H, Debnath J, Brown EJ. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep 2009;10:173–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Janssen AFJ, Katrukha EA, van Straaten W, Verlhac P, Reggiori F, Kapitein LC. Probing aggrephagy using chemically-induced protein aggregates. Nat Commun 2018;9:4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Grumati P, Dikic I, Stolz A. ER-phagy at a glance. J Cell Sci 2018;131:jcs217364. [DOI] [PubMed] [Google Scholar]
- 128. Liang JR, Lingeman E, Luong T, et al. A genome-wide ER-phagy screen highlights key roles of mitochondrial metabolism and ER-resident UFMylation. Cell 2020;180:1160–1177.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Baboota RK, Shinde AB, Lemaire K, et al. Functional peroxisomes are required for β-cell integrity in mice. Mol Metab 2019;22:71–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Pearson GL, Mellett N, Chu KY, Boslem E, Meikle PJ, Biden TJ. A comprehensive lipidomic screen of pancreatic β-cells using mass spectroscopy defines novel features of glucose-stimulated turnover of neutral lipids, sphingolipids and plasmalogens. Mol Metab 2016;5:404–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Cerqueira FM, Kozer N, Petcherski A, et al. MitoTimer-based high-content screen identifies two chemically-related benzothiophene derivatives that enhance basal mitophagy. Biochem J 2020;477:461–475 [DOI] [PubMed] [Google Scholar]