

Abstract
Summary
Mutational signatures are recurring DNA alteration patterns caused by distinct mutational events during the evolution of cancer. In recent years, several bioinformatics tools are available for mutational signature analysis. However, most of them focus on specific type of mutation or have limited scope of application. A pipeline tool for comprehensive mutational signature analysis is still lacking. Here we present Sigflow pipeline, which provides an one-stop solution for de novo signature extraction, reference signature fitting, signature stability analysis, sample clustering based on signature exposure in different types of genome DNA alterations including single base substitution, doublet base substitution, small insertion and deletion and copy number alteration. A Docker image is constructed to solve the complex and time-consuming installation issues, and this enables reproducible research by version control of all dependent tools along with their environments. Sigflow pipeline can be applied to both human and mouse genomes.
Availability and implementation
Sigflow is an open source software under academic free license v3.0 and it is freely available at https://github.com/ShixiangWang/sigflow or https://hub.docker.com/r/shixiangwang/sigflow.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Mutational signatures reflect the accumulated effects of both exogenous and endogenous mutational processes acting on cancer cells. These specific patterns of mutational processes have been initially identified by Alexandrov and colleagues with non-negative matrix factorization (NMF)-based matrix decomposition algorithm in 2013 (Alexandrov et al., 2013). Different other types of algorithms such as Bayesian NMF, expectation–maximization have also been built for do novo mutational signature extraction (Baez-Ortega and Gori, 2019). The application of mutational signature analysis to ever-growing amount of sequencing data leads to the formation of COSMIC signature database (Alexandrov et al., 2020).
Mutational signature analysis has been becoming a routine procedure after somatic variant calling in cancer genome study. This signature analysis can not only reveal the underlying mutational processes information but also provides biomarkers for cancer precision stratification and clinical response prediction (Davies et al., 2017; Ma et al., 2018; Wang et al., 2018). However, currently available mutational signature analysis tools either provide limited analysis features, or only focus on specific type of genome alterations, such as single base substitution (SBS; Baez-Ortega and Gori, 2019; Fischer et al., 2013; Gehring et al., 2015; Kim et al., 2016; Mayakonda et al., 2018; Rosenthal et al., 2016). In addition, the installation and application processes of currently available tools are complex and time-consuming.
Here, we present an open source pipeline tool Sigflow to provide a one-stop solution for efficient and reliable de novo signature extraction, reference signature fitting, signature exposure stability analysis, sample clustering based on signature exposure (Supplementary Table S1 and Fig. S1). Sample level and signature level results are properly visualized. The SBS, doublet base substitution (DBS), insertion and deletion (INDEL) signatures and the recent copy number signature analysis developed by our group are supported (Supplementary Fig. S2; Alexandrov et al., 2020; Wang et al., 2020). To solve the complex and time-consuming installation issues accompanying with current bioinformatics tools, a Docker image of Sigflow is constructed, and this enables good scalability for addition of other analysis features or other types of signatures in the future.
2 Tool description
Sigflow uses a command line-based interface and allows the user to efficiently and automatically perform the four workflows described below. Sigflow begins with importing somatic variant data in MAF (recommended), VCF or CSV/EXCEL format, and then parses the user input to select the workflow to run (Fig. 1 and Supplementary Fig. S1). Subsequently, a sample by mutation catalogue matrix is generated. Finally, a user specified workflow is performed to extract and analyze mutational signatures. Important immediate and final results are saved to the disk for general use. Comparisons between Sigflow and other mutational signature analysis tools are shown in Supplementary Table S1.
Fig. 1.
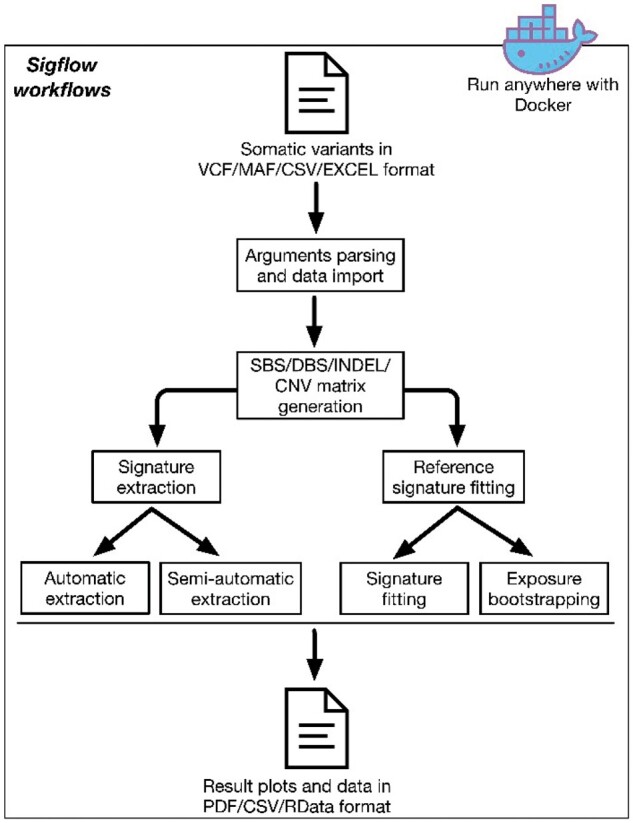
Overview of Sigflow pipeline
2.1 Automatic de novo signature extraction
Two approaches are available in Sigflow for automatic de novo signature extraction. In the first approach, a Bayesian variant of NMF algorithm is applied to enable optimal inferences for the number of signatures through the automatic relevance determination technique (Kim et al., 2016; Tan and Fevotte, 2013). This procedure starts from 30 signatures and reduces to an appropriate signature number which delivers highly interpretable and sparse representations for both signature profiles and exposures at a balance between data fitting and model complexity (Supplementary Figs S3 and S4). The whole procedure can be run at a specified number of times and the optimal solution is selected as the final output. In the second approach, Sigflow directly calls SigProfiler, which is the widely used software for de novo mutational signature extraction (Bergstrom, et al., 2019). The SigProfiler results are collected and transformed into the same format as in the first approach. After extracting signatures, the data of signature profile and absolute/relative exposure are generated, samples are clustered by relative signature exposures, and cosine similarity analysis is performed to match the extracted signatures to the COSMIC reference signatures (Supplementary Fig. S4).
2.2 Semi-automatic de novo signature extraction
Sigflow uses two-step strategy in semi-automatic signature extraction. In the first step, it runs NMF at a specified number of times for signature number range from 2 to a reasonable maximum value (30 for SBS and copy number signature, 15 for DBS signature and 20 for INDEL signature; this value can be modified by the user), then outputs some common measures (e.g. cophenetic correlation coefficient, silhouette and residual sum of squares) for each signature number to help the user to determine the number of signature to extract (Gaujoux and Seoighe, 2010). The key point is to select a signature number which results in high reproducible mutational signatures and low overall reconstruction error (Alexandrov et al., 2013). In the second step, Sigflow runs NMF at a specified number of times for the signature number from user input. Typically, 30–50 NMF runs can obtain a robust result (Gaujoux and Seoighe, 2010). This workflow has similar output files as the automatic signature extraction workflow.
2.3 Reference signature fitting
When the sample size is small (typically n < 50), the de novo workflows described above cannot properly decompose mutational signatures and their exposures. To extract signatures from single sample, an algorithm was designed to find a linear combination of the predefined signatures (such as COSMIC signatures) that best reconstructs the sample’s mutational profile. Here, Sigflow uses quadratic programming algorithm for reference signature fitting, and this algorithm is originally implemented in SignatureEstimation package and is fast and reliable (Huang et al., 2018). This workflow is computationally efficient (typically finished in several minutes for 100 samples) and is recommended for input data with small sample size (Supplementary Fig. S3).
2.4 Signature exposure stability analysis
The results from different signature analysis methods are not always consistent. Hence, one needs to be able to not only decompose a patient’s mutational profile into signatures but also establish the accuracy of such signature decomposition (Huang et al., 2018). Bootstrapping analysis is performed to quantify the confidences in the estimated exposure of each mutational signature. By repeatedly re-sampling original mutational catalogs for each tumor sample, this workflow generates estimated bootstrapping confidence intervals for each signature exposure and computes an empirical probability (P value) that a relative signature exposure is above a specific threshold. Signature instability is also measured as the root mean squared error of the exposure differences between bootstrapping estimates and the optimal solutions in the original data to test how much the bootstrapping exposures vary from original exposures (Supplementary Fig. S5). The outputs of this analysis include bootstrapping exposures, reconstruction errors and P values under different relative exposure cutoffs.
3 Implementation
Sigflow pipeline tool has been developed with R 4.0 following a clean, modular and robust design in concordance with best practice coding standards. Instructions on how to install and run Sigflow are presented in the public GitHub repository (https://github.com/ShixiangWang/sigflow). A detailed manual, which describes the workflows and operating parameters, is also provided in the GitHub README page. Sigflow is highly customizable with numerous parameter settings and is well supported for different input file formats, and all options are explained in the integrated help section or use cases. It has been designed to run as a command line-based program with a user-friendly interface, which allows non-expert users to become quickly familiarized. Sigflow allows keeping R-related data files, which can be easily loaded into R for flexible and interactive analysis and visualization.
To enable quick and reproducible research, we built a version-controlled Docker image for Sigflow to avoid the complex and time-consuming dependency issues in the installation of bioinformatics tools. Due to the flexibility of container technology, Sigflow can be easily deployed, managed and deleted on any operating system, thus it is convenient to be integrated with other cancer genome analysis platforms.
4 Conclusion
In the recent years, we have witnessed an increased number of tools and studies that explore and utilize mutational signatures in different aspects, including mutational etiologies exploration, biomarker discovery and cancer evolution. For better data integration and explanation, and higher computational efficiency, it is important to build robust, efficient and user-friendly tool that eventually allow a wide range of users to perform mutational signature analysis. Sigflow is a novel pipeline tool that provides comprehensive mutational signature analysis workflows, supports easy and quick tool deployment, and reproducible research.
Supplementary Material
Acknowledgement
We thank ShanghaiTech University High Performance Computing Public Service Platform for computing services.
Funding
This work was supported in part by The National Natural Science Foundation of China [31771373] and startup funding from ShanghaiTech University.
Conflict of Interest: none declared.
Contributor Information
Shixiang Wang, School of Life Science and Technology, ShanghaiTech University, Shanghai 201203, China; Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, Shanghai 200031, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Ziyu Tao, School of Life Science and Technology, ShanghaiTech University, Shanghai 201203, China; Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, Shanghai 200031, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Tao Wu, School of Life Science and Technology, ShanghaiTech University, Shanghai 201203, China; Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, Shanghai 200031, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Xue-Song Liu, School of Life Science and Technology, ShanghaiTech University, Shanghai 201203, China.
References
- Alexandrov L.B. et al. (2013) Deciphering signatures of mutational processes operative in human cancer. Cell Rep., 3, 246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov L.B. et al. ; PCAWG Mutational Signatures Working Group. (2020) The repertoire of mutational signatures in human cancer. Nature, 578, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baez-Ortega A., Gori K. (2019) Computational approaches for discovery of mutational signatures in cancer. Brief Bioinform., 20, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrom E.N. et al. (2019) SigProfilerMatrixGenerator: a tool for visualizing and exploring patterns of small mutational events. BMC Genomics, 20, 685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H. et al. (2017) HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med., 23, 517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A. et al. (2013) EMu: probabilistic inference of mutational processes and their localization in the cancer genome. Genome Biol., 14, R39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaujoux R., Seoighe C. (2010) A flexible R package for nonnegative matrix factorization. BMC Bioinformatics, 11, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring J.S. et al. (2015) SomaticSignatures: inferring mutational signatures from single-nucleotide variants. Bioinformatics, 31, 3673–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X. et al. (2018) Detecting presence of mutational signatures in cancer with confidence. Bioinformatics, 34, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. et al. (2016) Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat. Genet., 48, 600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J. et al. (2018) The therapeutic significance of mutational signatures from DNA repair deficiency in cancer. Nat. Commun., 9, 3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayakonda A. et al. (2018) Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res., 28, 1747–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal R. et al. (2016) DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol., 17, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan V.Y., Fevotte C. (2013) Automatic relevance determination in nonnegative matrix factorization with the beta-divergence. IEEE Trans. Pattern Anal. Mach. Intell., 35, 1592–1605. [DOI] [PubMed] [Google Scholar]
- Wang S. et al. (2018) APOBEC3B and APOBEC mutational signature as potential predictive markers for immunotherapy response in non-small cell lung cancer. Oncogene, 37, 3924–3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S. et al. (2020) Copy number signature analyses in prostate cancer reveal distinct etiologies and clinical outcomes. medRxiv.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.