

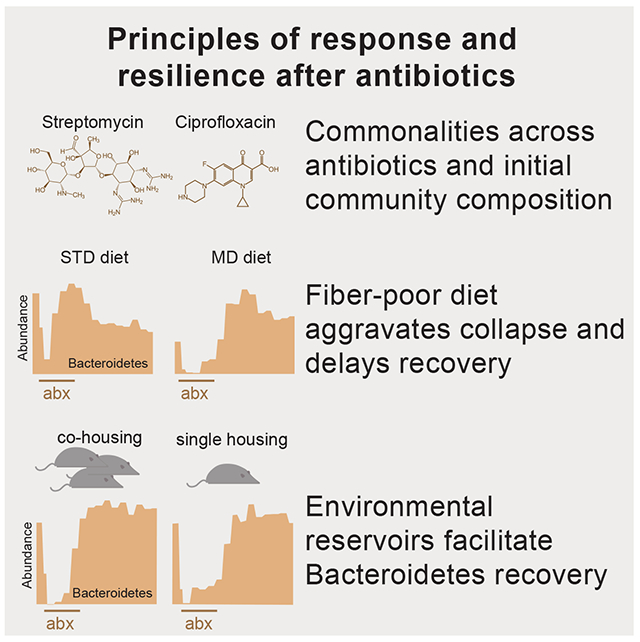
Summary
Antibiotics alter microbiota composition and increase infection susceptibility. However, the generalizable effects of antibiotics on gut commensals and the contribution of environmental variables remain unclear. To address this, we tracked microbiota dynamics with high temporal and taxonomic resolution during antibiotic treatment in a controlled murine system, isolating variables such as diet, treatment history, and housing co-inhabitants. Human microbiota were remarkably resilient, and recovered during antibiotic treatment with transient dominance of resistant Bacteroides and taxa-asymmetric diversity reduction. In certain cases, in vitro sensitivities were not predictive of in vivo responses, underscoring the significance of host and community context. A fiber-deficient diet exacerbated microbiota collapse and delayed recovery. Species replacement through cross-housing after ciprofloxacin treatment established resilience to a second treatment. Single-housing drastically disrupted recovery, highlighting the importance of environmental reservoirs. Our findings highlight deterministic microbiota adaptations to perturbations, and the translational potential for modulating diet, sanitation, and microbiota composition during antibiotics.
Keywords: gut microbiota, antibiotics, resilience, reseeding, co-housing, coprophagia, microbiota perturbations
Graphical Abstract
eToC Blurb:
Ng et al. systematically compare microbiota responses to antibiotics in mice. A fiber-deficient diet or single-housing aggravated otherwise rapid recovery, while microbiota reprogramming through repeated treatment or transmission between hosts enhanced resilience. This work highlights the translational potential for modulating diet, sanitation, and microbiota composition during antibiotic treatment.
Introduction
The gut microbiota performs functions crucial to the host, guiding development of the immune system and tuning inflammation (Ivanov et al., 2008; Mazmanian et al., 2008). Antibiotic treatment perturbs community structure and function, often resulting in prolonged pathogen susceptibility (Doorduyn et al., 2006; Pavia et al., 1990; Stecher et al., 2007). Moreover, antibiotic treatment has long-lasting effects on physiological processes such as adiposity, insulin resistance, and cognitive function (Cho et al., 2012; Cox et al., 2014; Frohlich et al., 2016; Hwang et al., 2015). The ability to accurately predict how a given antibiotic will affect a community may reveal control mechanisms for these wide-ranging effects.
The manner and extent to which complex communities respond to and recover from antibiotics is not consistent. For example, while the aminoglycoside streptomycin shifted the balance between the two dominant phyla in the mouse gut toward a Bacteroidetes-dominated microbiota in one study (Thompson et al., 2015), Firmicutes dominated in others (Ng et al., 2013; Stecher et al., 2007). The fluoroquinolone ciprofloxacin elicited highly individualized responses in humans (Dethlefsen et al., 2008; Dethlefsen and Relman, 2011). Many factors across studies and between hosts could account for variable outcomes, motivating a systematic analysis of treatment outcomes. Moreover, few studies have focused on recovery dynamics after treatment despite the relevance to pathogen susceptibility (Doorduyn et al., 2006).
Antibiotic response may depend on environmental perturbations that multifactorially impact the microbiota and the intestinal environment. Removal of microbiota-accessible carbohydrates (MACs) from the mouse diet thins the mucosal layer surrounding the gut epithelium (Desai et al., 2016; Earle et al., 2015), increasing sensitivity to certain pathogens (Desai et al., 2016), and reduces microbiota transmission between generations (Sonnenburg et al., 2016). Fecal microbiota transplants can resolve recurrent infections of C. difficile in humans (van Nood et al., 2013), and are effective at restoring species after diet- (Sonnenburg et al., 2016) or osmotic diarrhea-mediated loss (Tropini et al., 2018), suggesting the importance of environmental reservoirs for commensal recovery. In some humans, a second round of ciprofloxacin resulted in larger disruption of the gut community than the first treatment (Dethlefsen and Relman, 2011). The factors that influence community disruption with multiple treatments and the microbiota response to combinations of antibiotics with other gut perturbations are underexplored and likely critical for understanding how antibiotics reshape the gut.
Can in vitro data on the antibiotic susceptibility of model organisms be used to predict the response of communities within a host? A recent screen of 40 commensals across a range of antibiotics and human drugs revealed that sensitivity profiles can be highly species-specific (Maier et al., 2018), indicating that phylogeny is not generally useful for inferring responses. Moreover, growth rate can greatly affect minimum inhibitory concentrations (MICs) in vitro (Evans et al., 1991), suggesting that the spatially heterogeneous and dynamic nutrient environment of the gut further confounds predictions. In a few cases, mechanisms such as drug metabolism (Onderdonk et al., 1979) have been identified by which one species can alter the sensitivity of another species. Lastly, community interactions may perturb sensitivity in a widespread manner. For example, the cell wall-targeting vancomycin primarily affects only Gram-positive species in vitro due to the barrier function of the Gram-negative outer membrane, but nevertheless reduces the levels of Gram-negative species in the mouse gut (Ivanov et al., 2008). These factors motivate detailed analyses of species-level dynamics throughout antibiotic treatment and comparisons with in vitro sensitivities.
To address these knowledge gaps, we tracked microbiota dynamics with high temporal and taxonomic resolution during antibiotic treatment in a controlled model system, while isolating variables that influence collapse and recovery such as diet, treatment history, and housing co-inhabitants. Our results emphasize that the response of individual bacterial species can be highly affected by the host, the rest of the gut community, and available environmental reservoirs. Our systematic investigation of the resilience and recovery of particular bacterial taxa to antibiotic perturbation lays the foundation for strategies that could mitigate long-term effects of antibiotic treatment on the gut microbiota.
Results
Microbial load recovers rapidly after massive, transient disturbance from streptomycin
We first sought to determine the extent to which the gut microbiota is disrupted during antibiotic treatment and the timescale of recovery in total abundance and composition. Fifteen germ-free mice across 3 cages were equilibrated with human feces (‘humanized’) from a single donor (Methods) and gavaged with 20 mg/day streptomycin for 5 days (days 0-4). Feces were collected for 16S rRNA sequencing immediately before the first gavage, 12 h later (day 0.5), and every day for 14 days. Three mice were sacrificed on days 0, 1, 5, 8, and 14 for other analyses (Fig. 1A).
Figure 1: Rapid recovery during antibiotic administration in a human microbiota across antibiotics, with common trajectories of collapse and recovery but cage-specific end points.
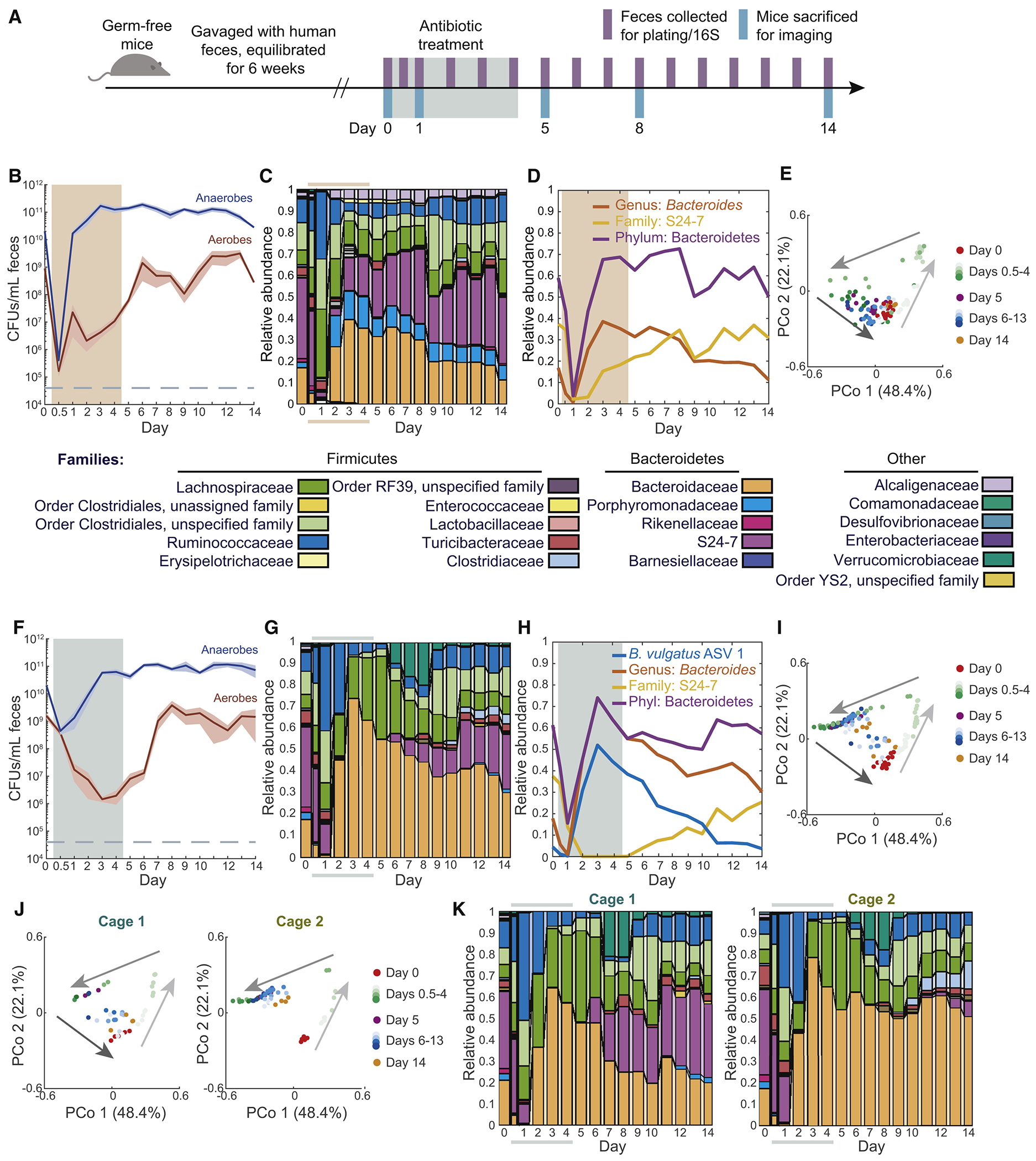
See also Figures S1 and S4.
(A) Schematic of humanization of germ-free mice, antibiotic treatment, and sampling.
(B,F) Culturable anaerobic and aerobic fecal densities in humanized mice treated for 5 days with (B) 20 mg streptomycin daily (n = 15), and (F) 3 mg ciprofloxacin twice daily (n = 14). Bacterial loads recovered during treatment (colored area). Error bars: S.E.M.
(C,G) Family-level composition in feces was similar in (C) streptomycin-treated and (G) ciprofloxacin-treated humanized mice (colored bars denote treatment period).
(D,H) Relative abundance of Bacteroides, S24-7, and Bacteroidetes in (D) streptomycin-treated and (H) ciprofloxacin-treated humanized mice.
(E,I) PCoA of community composition in humanized mice during (E) streptomycin or (I) ciprofloxacin treatment reveal a conserved trajectory. Analyses used weighted UniFrac distances.
(J) PCoA of humanized mice during ciprofloxacin treatment in two cages uncovered cage-specific differences.
(K) Family-level composition from the two cages in (H) revealed stochasticity of S24-7 recovery.
We measured the culturable colony forming units (CFU) per mL in each fecal sample and observed a drop from ~1010 to 105-106 by day 0.5 (Fig. 1B). Nonetheless, the anaerobic load recovered to >1010 CFU/mL by day 1 and remained thereafter at 1010-1011 despite continued drug administration on days 2-4. These rapid dynamics illustrate that even daily sampling could miss a rapid, antibiotic-induced density decrease. The density of aerobes was much lower than that of the anaerobes (hence made minimal contribution to total load) and did not recover until day 6 (Fig. 1B), indicating that treatment continually reconfigured microbiota composition despite rapid recovery in bacterial load.
Streptomycin elicits a common trajectory of compositional recovery despite hysteresis and kinetic variation
Using 16S sequencing, we identified and mapped amplicon sequence variants (ASVs) using DADA2, which uses error correction to overestimation of diversity (Callahan et al., 2016); these ASVs represent predicted species. The collapse and recovery of microbiota composition were similar across all mice. Before treatment, mice had similar abundances of Bacteroidetes and Firmicutes, with these two phyla accounting for >95% abundance (Fig. 1C). By day 1, Bacteroidetes dropped to <10% (Fig. 1D), while Firmicutes increased (Fig. 1C). By day 2, Bacteroidetes returned to pre-antibiotic relative abundances (Fig. 1D), as did total culturable load (Fig. 1B). Betaproteobacteria increased from ~2% to ~5% during treatment and then persisted at that level (Fig. 1C), suggesting that they opportunistically filled a niche opened by treatment.
These dynamics were evident in a principal coordinates analysis (PCoA), which measures similarity in composition: all mice reached a common point of maximal disturbance along coordinate 1 by day 1 (Fig. 1E). Thus, despite the initial, massive decrease in load (Fig. 1B), the path to maximum disturbance was deterministic. After day 1, the microbiotas re-equilibrated to the pre-treatment composition along a different path than the one traversed to the point of maximal disturbance (Fig. 1E). In addition to this hysteresis, recovery kinetics varied. Some mice had already returned to the day 0 state by day 5, while others returned more slowly (Fig. 1E), perhaps due to their reliance on re-seeding from other mice. Nonetheless, all mice followed a similar trajectory in the space of coordinates 1 and 2 (Fig. 1E), suggesting that intermediate recovery states were similar despite kinetic variation.
Ciprofloxacin treatment results in similar microbiota dynamics to streptomycin
To determine whether the rapid recovery of bacterial load and composition was antibiotic-specific, we treated 14 humanized mice (co-housed in 3 cages) twice daily (Fig. S1A) for 5 days with 3 mg ciprofloxacin, a broad-spectrum DNA gyrase inhibitor. The anaerobic compartment of culturable bacteria initially dropped by 100- to 1000-fold by day 1 but recovered by day 3 (Fig. 1F), before administration ended as in streptomycin-treated mice (Fig. 1B). The phylum-level effects of ciprofloxacin were similar to streptomycin (Fig. 1G), particularly the PCoA trajectory despite differences in kinetics (Fig. 1E,I). The similarity was driven at least in part by the initial replacement of Bacteroidetes with Firmicutes (Fig. 1C,G,S1B). Bacteroidetes relative abundance dropped to 10-25% by day 1, but recovered by day 3 and remained stable thereafter (Fig. 1G). This recovery was largely associated with a single ASV matching Bacteroides vulgatus (Bv) that was at 4% prior to antibiotics but bloomed to >50% in all mice during treatment (Fig. 1H). Once treatment ended, Bv was replaced by S24-7 family members (Fig. 1H), suggesting a competition between these two families for a common niche. We isolated 12 colonies from a mouse sample during Bv dominance on Bacteroides-selective plates; 16S Sanger sequencing revealed that all isolates were Bv. Whereas Betaproteobacteria increased in abundance during streptomycin, Verrucomicrobiae bloomed after ciprofloxacin was removed (Fig. 1G), again indicating opening of favorable niches. Thus, ciprofloxacin and streptomycin influence composition in surprisingly similar manners despite highly distinct molecular effects, although distinctions emerge during recovery.
Cage-specific effects during recovery from ciprofloxacin indicate importance of environmental microbial reservoirs
After maximal disturbance, one subset of ciprofloxacin-treated mice continued to follow the recovery trajectory of streptomycin-treated mice, while the rest remained in a disrupted state (Fig. 1I). Interestingly, these subsets were entirely cage-determined (Fig. 1J,K), suggesting reseeding plays a major role in recovery. This difference was not apparent in class-level composition (Fig. S1C). However, an obvious distinguishing feature within ciprofloxacin-treated mice was the dynamics of the family S24-7, a Bacteroidetes member abundant prior to treatment (~35%) and highly prevalent in homeothermic animals (Ormerod et al., 2016). In one cage that exhibited complete recovery in PCoA space (Fig. 1J), S24-7 disappeared in the latter stages of treatment, but re-emerged directly after ciprofloxacin was removed and re-established at pre-treatment levels (Fig. 1K,S1D). By contrast, in another cage S24-7 was undetectable until day 10, and only recovered to ~5% by day 14 (Fig. 1K,S1D). In streptomycin-treated mice, S24-7 recovered in all cages, even before cessation of antibiotics (Fig. S1E). A likely explanation for the heterogeneity is that each mouse has a low probability of retaining S24-7 or being recolonized from the cage reservoir, but afterward the rest of the cage shares in S24-7 recovery. These data suggest that gamma diversity of host microbiotas within a cage is an important driver of recovery after antibiotics.
Antibiotics cause sustained loss in alpha diversity, driven by loss of Bacteroidetes members
Although the culturable densities and phylum-level relative abundances of Bacteroidetes and Firmicutes returned to pre-antibiotic states regardless of antibiotic (Fig. 1B,C,F,G), we were interested in long-term effects suggested by the loss of S24-7 in some mice (Fig. 1K, S1D) and oligodominance of Bacteroides spp. (Fig. 1H). To identify one such effect—extinction—we quantified alpha diversity (number of observed species), a measure of community resilience (Lozupone et al., 2012). For streptomycin and ciprofloxacin, alpha diversity dropped during treatment (Fig. 2A,B), with a larger decrease in ciprofloxacin-treated mice despite the smaller decrease in bacterial load (Fig. 1B,F). Alpha diversity slowly increased after either antibiotic was removed, and re-equilibrated at a level significantly lower than pre-treatment (Fig. 2A,B).
Figure 2: Antibiotic administration alters the state of the microbiota via reductions in Bacteroidetes diversity.
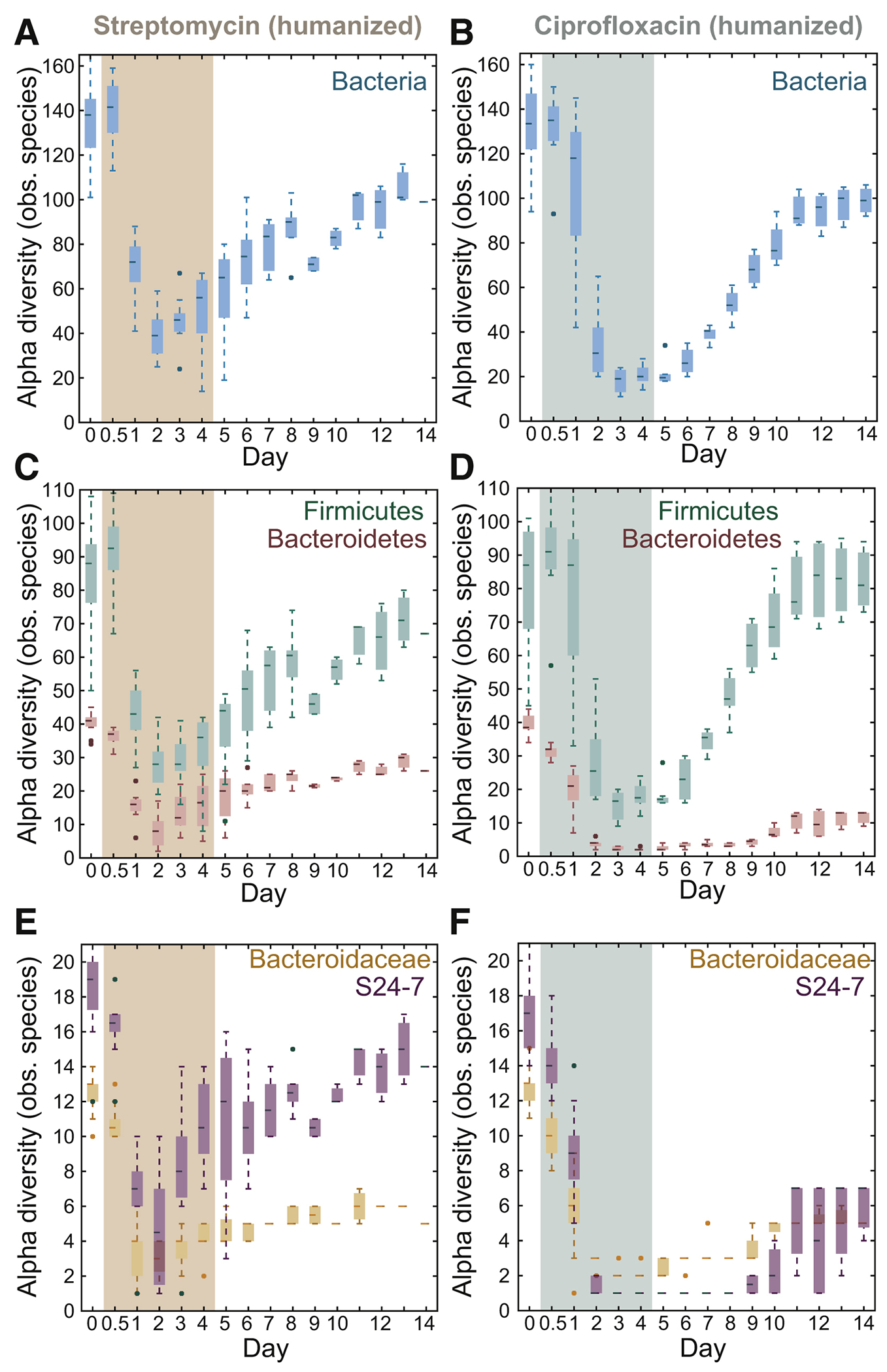
See also Figure S2.
(A,B) After treatment (colored area), the microbiota stabilized at lower alpha diversity.
(C,D) Alpha diversity of the Bacteroidetes phylum was impacted much more than the Firmicutes for both treatments.
(E,F) S24-7 and Bacteroidaceae alpha diversities were both impacted, particularly S24-7 by ciprofloxacin.
The sustained decrease was driven largely by Bacteroidetes taxa, which permanently decreased in diversity by 36% and 70% in streptomycin and ciprofloxacin-treated mice, respectively (Fig. 2C,D). By contrast, Firmicutes diversity was only slightly affected (Fig. 2C,D). In streptomycin-treated mice, the decrease in Bacteroidetes diversity was largely accounted for by the Bacteroidaceae family, which includes the Bacteroides genus (Fig. 2E); S24-7 diversity recovered to near pre-treatment levels (Fig. 2E). By contrast, in ciprofloxacin-treated mice, both Bacteroidaceae and S24-7 diversity experienced a large decrease (Fig. 2E,F) and remained low after ciprofloxacin was removed; S24-7 diversity decreased to as low as 1 observed ASV from ~17 at the start of the experiment, (Fig. 1F), suggesting complete extinction of some species. Faith’s phylogenetic diversity gave similar conclusions to observed ASVs (Fig. S2A–F). Together, our results reveal that the two dominant phyla exhibit asymmetric vulnerability to two different antibiotics, with Bacteroidetes loss and recovery likely influenced by cage mates.
Antibiotic resistance and intra-genus competition during and after ciprofloxacin treatment determines the relative abundances of Bacteroides species
The transient decrease in relative abundance and extinction of taxa within the Bacteroidaceae (Fig. 1C,1G,2E,2F) suggests that these species experience selection during antibiotics. To determine whether this behavior was largely driven by the selection for resistance, we performed a second antibiotic treatment (Fig. 3A). We employed ciprofloxacin due to the greater drop in alpha diversity compared with streptomycin (Fig. 2).
Figure 3: The microbiotas of conventional and humanized mice display distinct signatures under repeated ciprofloxacin treatment.
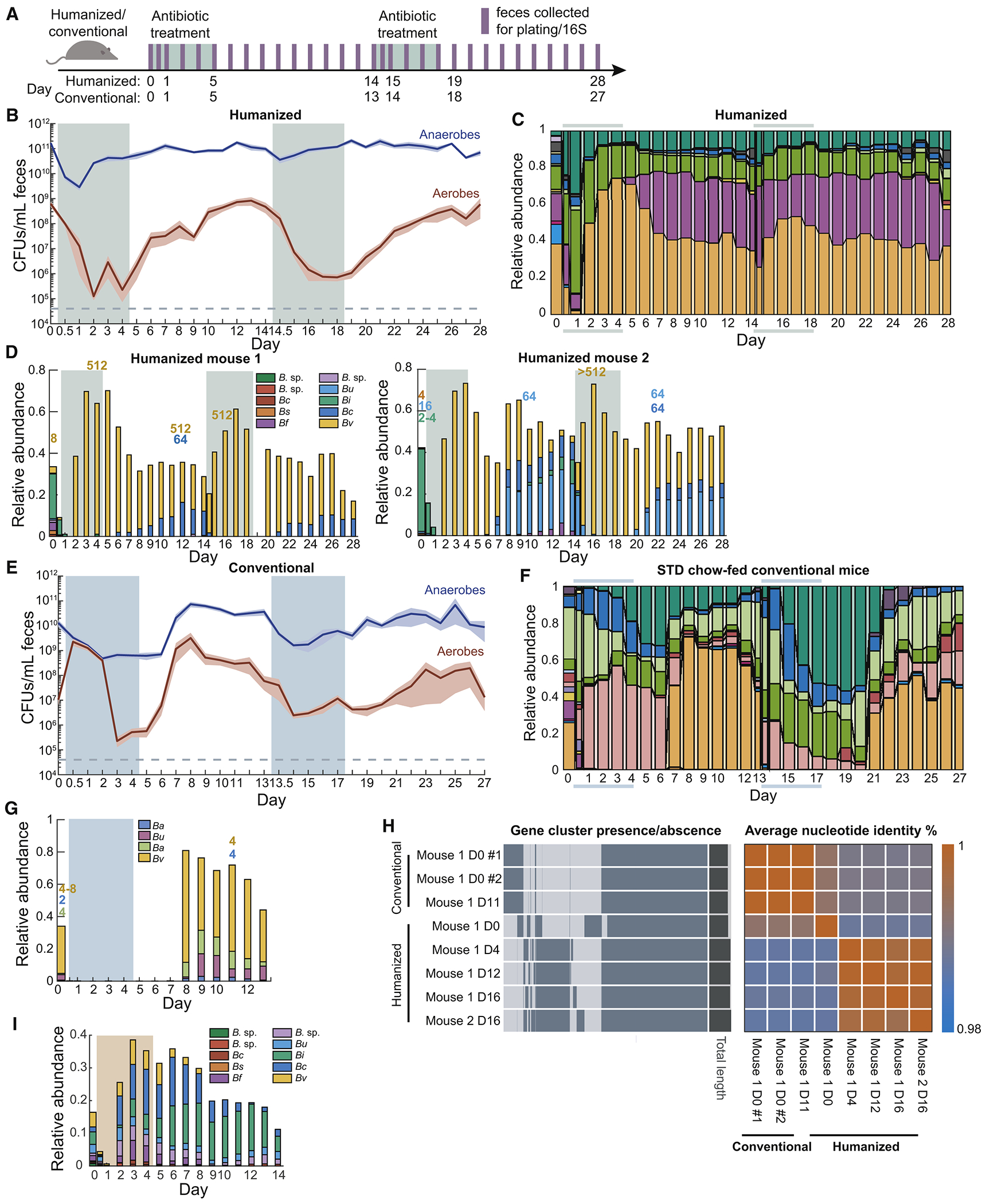
See also Figure S3.
(A) Schematic for treatment with two courses of ciprofloxacin.
(B,E) Culturable anaerobic and aerobic fecal densities in (B) humanized and (E) conventional mice treated with ciprofloxacin (treatment marked by colored areas) revealed that conventional mice responded similarly to both treatments, but humanized mice had a more robust second response. Error bars: S.E.M.
(C,F) Family-level composition in feces of (C) humanized and (F) conventional mice further demonstrated robustness of humanized mice to a second treatment.
(D,G,I) With differential resistance to antibiotics, resistant strains dominate during antibiotic treatment; other Bacteroides spp. expand after antibiotics. Relative abundance of the 10 most abundant Bacteroides ASVs in (D) ciprofloxacin-treated humanized mice, (G) ciprofloxacin-treated conventional mice and (I) streptomycin-treated humanized mice. B. sp: unknown Bacteroides spp.; Bc: B. caccae; Bs: B. salyersiae; Bf: B. fragilis; Bu: B. uniformis; Bi: B. intestinalis; Bv: B. vulgatus; Ba: B. acidifaciens. MICs are labeled above days of isolation, and colored by the appropriate ASV based on 16S Sanger sequencing. Treatment adenoted by shaded areas.
(H) Comparison of gene clusters and average nucleotide identity of Bv isolates revealed that day 0 (D0) isolate is different from isolates from other days, and that Bv isolates from conventional mice also cluster.
The anaerobic compartment was negligibly affected by the second treatment (Fig. 3B), consistent with a lack of change in family-level composition (Fig. 3C). By day 1 of the second treatment, the class-level composition was almost indistinguishable from the stabilized end of recovery after the first treatment, although different from the baseline community before treatment (Fig. S3A), demonstrating that the first treatment selected for a more resilient community.
Despite the lack of family-level changes during the second treatment, throughout both treatments Bacteroidaceae dropped to a single, dominant ASV (Bv) and then recovered to 2-5 ASVs after ciprofloxacin was removed (Fig. 3D). This domination during antibiotic treatment was coupled to decreased alpha diversity (Fig. S3B). To examine the extent to which the emergence of ciprofloxacin resistance explains the response of humanized mice to a second treatment, we isolated strains from fecal samples of two mice. We identified strains as Bacteroides spp. via 16S sequencing and measured their MICs to ciprofloxacin. The MICs of pre-antibiotic Bacteroides isolates (Table S1) were within the reported range of literature values (2-16 μg/mL) (Goldstein and Citron, 1992). Bv isolates from samples during or after antibiotic treatment displayed >100-fold increases relative to the isolates from day 0 (Fig. 3D). Whole-genome sequencing of Bv isolates from one mouse revealed that day 4, 12, or 16 isolates, but not the day 0 isolate, possessed a gyrA mutation that confers ciprofloxacin resistance. The genotypes of the day 4, 12, and 16 isolates were highly distinct from the day 0 isolate (Fig. 3H), suggesting that the initial microbiota included multiple Bv strains with varying ciprofloxacin sensitivity and that treatment reconfigured their relative abundances. The domination occurred only during antibiotic treatment (Fig. 3D). After treatment ceased, although total Bacteroidaceae relative abundance stabilized, the dominance of Bv son day 6 rapidly transitioned to multiple Bacteroides ASVs (Fig. 3D). Thus, other Bacteroides are able to reseed and/or recover and then compete effectively with Bv in the absence of antibiotics.
Conventional and humanized mice have similar recovery trajectories but different kinetics during ciprofloxacin
To determine whether the compositional response of the humanized microbiota was influenced by antibiotic history and/or the nature of humanization, we carried out a double-treatment experiment in conventional mice with a mouse-adapted microbiota that has likely not experienced as much antibiotics as the human donor for the humanized mice. Conventional samples mapped onto approximately the same PCoA trajectories as humanized mice (Fig. S3C), with slower dynamics: conventional mice on day 8 resembled humanized mice on day 3 (Fig. S3C). Mirroring this delay, the bacterial load (Fig. 3E) and Bacteroidetes abundance (Fig. 3F) did not recover during antibiotic treatment in any mice. Instead, Bacteroidetes recovered 1 day after treatment cessation, matching the time scale of ciprofloxacin clearance from feces (Fig. S3E), with recovery almost entirely due to increased Bacteroidaceae abundance.
In contrast to a humanized community that exhibited enrichment and domination of a resistant strain during treatment (Fig. 3D), all Bacteroides isolates from conventional mice before or after treatment had MICs of 4-8 μg/mL (Fig. 3G); the lack of resistance was consistent with the similar decreases in bacterial load during the two treatments (Fig. 3E). Whole-genome sequencing demonstrated that the day 11 isolate was virtually identical to the day 0 isolate (Fig. 3H), with no obvious resistance-conferring mutations. Consistent with the lack of selection, multiple Bacteroides spp. were maintained in the period after antibiotic treatment. Thus, despite identical dosing conditions between humanized and conventional mice, the selection for Bacteroides resistance in a complex community is not pre-destined; the large decrease in bacterial load and similar compositional kinetics during the second treatment in conventional mice suggests a lack of resistance in the community at large.
Concomitant with the sensitivity of Bacteroides, S24-7 was highly responsive, but never recovered to the high densities seen in some humanized mice (Fig. 3C,F). In sum, our data suggest that the conventional mouse microbiota is more sensitive than the humanized mouse microbiota. The effects of the second treatment were remarkably similar to those of the first treatment: ~10-100-fold drop in CFU/mL (Fig. 3E), and Bacteroidetes were replaced by Firmicutes and Verrucomicrobiae (Fig. 3F). Alpha diversity failed to recover to the same level as before the second treatment (Fig. S3D), suggesting further elimination of community members. Bacteroidetes were undetectable until 3 days after treatment stopped (Fig. 3F), and S24-7 was driven to undetectable levels in all mice immediately upon the second treatment (Fig. 3F). Thus, an inability to recover completely from the first treatment predisposes the conventional microbiota to a more profound disturbance upon the second treatment.
In vitro resistance of the Bacteroides genus contrasts with its in vivo response to streptomycin
Antibiotic resistance appears to explain the Bv response to ciprofloxacin, hence we wondered whether the recovery of Bacteroides after day 1 of streptomycin treatment (Fig. 1D) was also due to resistance. Previous studies suggested that anaerobes are resistant to aminoglycosides such as streptomycin in vitro (Bryan et al., 1979), making it surprising that the Bacteroidetes decreased dramatically in abundance during the first 12 h of streptomycin treatment (Fig. 1C). We measured in vitro streptomycin MICs of Bacteroides type strains and isolates from the pre-treatment donor sample (SI). The majority exhibited no adverse growth effects at 512 μg/mL, the highest dose tested (Table S1), supporting the resistance of the Bacteroides genus. Despite being present at 17% before treatment, surprisingly they experienced a similar ~105-fold initial decrease in total abundance as the rest of the community upon treatment, given that they are straightforwardly cultured. To test whether the drop arose indirectly from the killing of other species, affecting cross-feeding and nutrient availability, or from host-related effects, we monocolonized germ-free mice with B. thetaiotaomicron and treated with streptomycin. CFUs dropped during the first 24 hours to a similar extent as in humanized mice (Fig. S4A), indicating that the host somehow reversed the natural resistance of B. thetaiotaomicron to streptomycin. These data demonstrate that in vitro susceptibility measurements are not necessarily predictive of in vivo response.
Additionally, we did not observe dominance of Bv (or any other Bacteroides spp.) in streptomycin-treated mice, in contrast to ciprofloxacin-treated humanized mice (Fig. 3I), supporting the hypothesis that dominance arises out of selection of resistant strains during antibiotic treatment. In conventional mice, dominance was not observed (Fig. 3G) because there did not appear to be resistant strains that could outcompete sensitive strains during antibiotics. In the case of streptomycin, the lack of selection is likely due to the near-universal resistance across the Bacteroides.
Antibiotic treatment increases luminal mucus
Given that domination of ciprofloxacin-resistant Bacteroides spp. occurred only during treatment, after which sensitive strains expanded, we wondered whether factors involving the host environment such as nutrient availability affected taxonomic dynamics. Since many Bacteroides spp. are known mucus utilizers (Salyers et al., 1977; Sonnenburg et al., 2005), we hypothesized their recovery might be spurred by that increased mucus availability during antibiotic treatment. We harvested and fixed tissues of humanized mice sacrificed on days 0, 1, 5, 8, and 14 and performed DNA and mucus staining and imaging (Earle et al., 2015; Johansson and Hansson, 2012) (Fig. 4A,B).
Figure 4: Antibiotic treatment increases loose intestinal mucus.
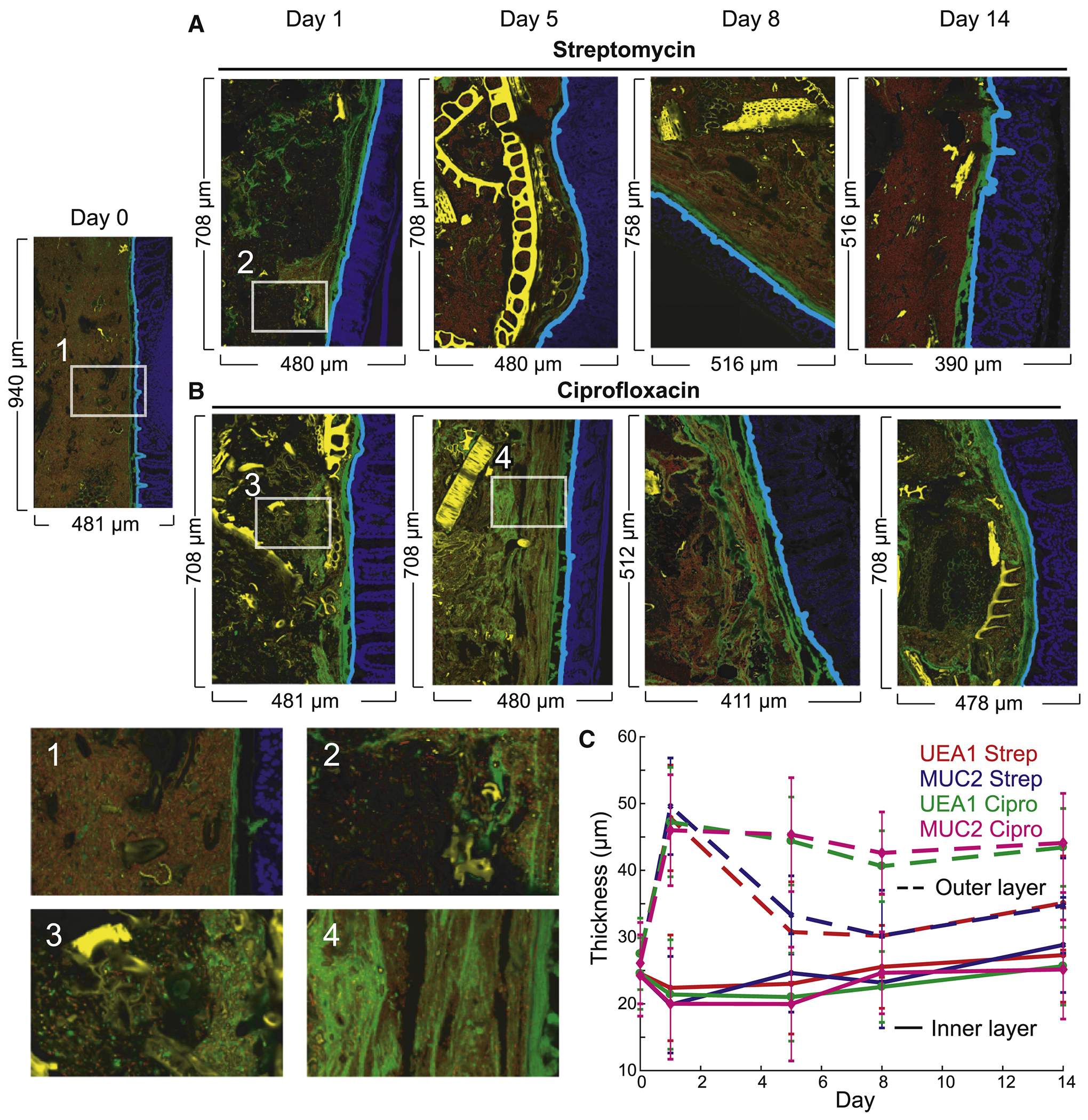
See also Figure S4.
(A,B) Imaging of the distal colon of humanized mice before, during, and after treatment with (A) streptomycin and (B) ciprofloxacin revealed sustained increase in loose luminal mucus. Sections were stained with DAPI (epithelial DAPI, blue; luminal DAPI, red) and MUC2 (green). Computationally segmented debris appears in yellow. Zooms: (1) Dense community before antibiotics with little luminal mucus and a tight mucus layer. (2,3) Massive decrease in luminal DAPI 1 day after treatment. (4) Recovery of luminal DAPI but persistence of loose luminal mucus.
(B) Thickness of the tight inner mucus layer remained approximately constant during treatment, while the looser outer luminal mucus proliferated during treatment with both antibiotics and persisted after ciprofloxacin. Error bars represent S.D.
We utilized the software BacSpace (Earle et al., 2015) to quantify the thickness of the mucus layer. For both antibiotics, the thickness of the tight mucus layer was relatively constant over time (Fig. 4C). However, there was an obvious expansion of Muc2 signal into the lumen upon treatment (Fig. 4C), similar to the increase in mucus in germ-free mice (Johansson et al., 2008). In ciprofloxacin-treated humanized mice, this loose-mucus increase persisted throughout treatment and recovery, suggesting that the increase was not due to direct effects of ciprofloxacin on mucus as ciprofloxacin is cleared by day 7 (Fig. S3E); in streptomycin-treated mice, the loose mucus returned to baseline by day 5 (Fig. 4C). Verrucomicrobiae, which includes the mucin-degrading Akkermansia muciniphila, did not noticeably change in abundance during or after streptomycin treatment (Fig. 1C), but bloomed after ciprofloxacin treatment was removed (Fig. 1G). Additionally, whole genome sequencing revealed that Bacteroides strains isolated from post-ciprofloxacin fecal samples possessed more mucin and host carbohydrate-degrading genetic modules than strains isolated before antibiotic treatment (Fig. S4B), suggesting that the capability to eat loosened mucus may drive the expansion of Bacteroides spp. and other mucus-responsive phyla (Fig. 1C,G) and counteract the domination of Bv (Fig. 1H,3D).
A polysaccharide-deficient diet disrupts recovery after ciprofloxacin treatment
Feeding mice a diet lacking MACs thins the tight mucus layer (Desai et al., 2016; Earle et al., 2015); bacteria deprived of dietary carbohydrates must utilize mucin-derived polysaccharides instead. A MAC-deficient diet also selects for mucin utilizers like A. muciniphila (Earle et al., 2015; Marcobal et al., 2013), which may compete with Bacteroides spp. for mucus carbohydrates. Thus, we hypothesized that the MAC-deficient diet-induced mucus reduction would antagonize the proliferation of loose mucus during antibiotic treatment (Fig. 4C) and thereby hamper the recovery of mucin utilizers such as Bacteroidaceae.
We transitioned humanized mice with the same microbiota as in Fig. 1–3 onto a MAC-deficient diet for two weeks, and then began ciprofloxacin treatment (Fig. 5A–D). As predicted, the Bacteroidaceae did not completely recover until after cessation of antibiotics in MAC-deficient diet-fed (MD) mice, unlike standard diet-fed (STD) mice (Fig. 5D,1G). This delayed recovery was accompanied by incomplete recovery of other families within the Bacteroidetes; S24-7 was completely eliminated (Fig. 5D). Moreover, the alpha diversity of MD mice started somewhat lower than STD mice (Fig. 5C, p = 0.02), and dropped even more with treatment (~6-fold in MD mice vs. ~4-fold in STD mice; Fig. 5C), indicating that ciprofloxacin acted additively with the diet shift. In MD mice, the anaerobic (and total) bacterial load (Fig. 5B) exhibited similar dynamics to STD mice (Fig. 1F), although the aerobic compartment decreased more and recovery was slower than in STD mice (Fig. 5B, 1F), mirroring recovery delays in alpha diversity (Fig. 5C) and composition (Fig. 5D).
Figure 5: A MAC-deficient diet sensitizes the microbiota to ciprofloxacin.
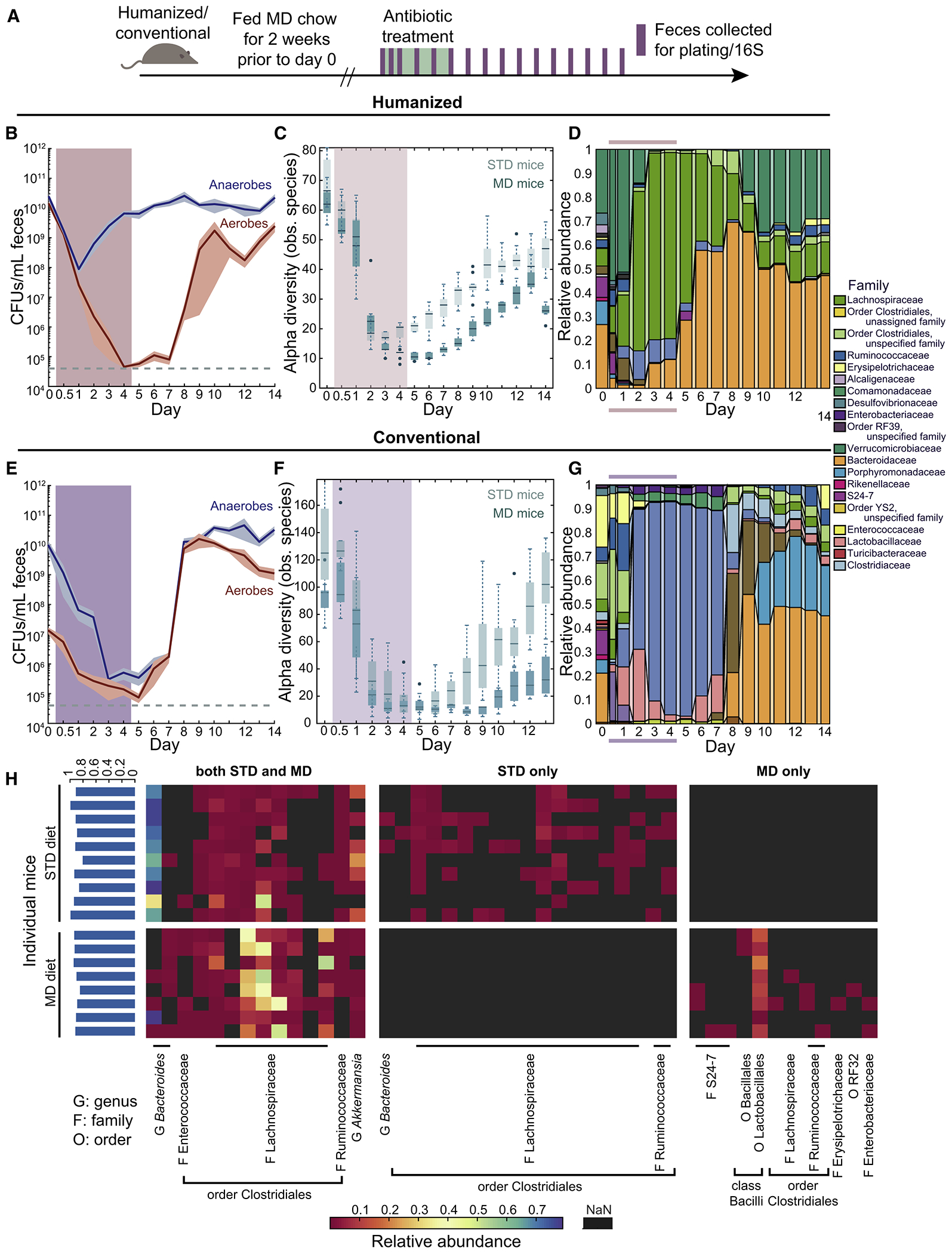
(A) Schematic for treatment of humanized/conventional STD or MD mice.
(B,E) Culturable anaerobic and aerobic fecal densities in (B) humanized or (E) conventional MD mice revealed that the aerobic compartment was sensitized in humanized mice, and both compartments were sensitized in conventional mice. Error bars: S.E.M.; treatment period marked by colored area.
(C,F) Alpha diversity decreased more in MD mice than in STD mice after treatment.
(D,G) Family-level composition in feces of MD mice substantially differed from STD mice (Fig. 1F,K,L).
(H) Heatmap of the core microbiome at the nadir of alpha diversity in STD and MD humanized mice. Shared ASVs are clustered on the left, and STD- and MD-specific ASVs appear on the right. The percentage of total relative abundance accounted for by the shared core is ~90% across most mice (blue bars, left).
To determine whether the effects of the diet shift were microbiota-dependent, we carried out a similar experiment with conventional mice (Fig. 5A). In these mice, the bacterial load was dramatically affected by ciprofloxacin, with a >10,000-fold decrease in anaerobic density (Fig. 5E), in contrast to the ~10- to 100-fold drop in conventional STD mice (Fig. 3E). Similar to humanized mice (Fig. 5B–D), recovery was delayed in MD versus STD conventional mice (Fig. 5E–G). The decrease in alpha diversity was even more prolonged in MD conventional mice than in MD humanized mice, despite a higher starting diversity (Fig. 5F). Taken together, these data indicate that the shift to a MAC-deficient diet exacerbates the effects of ciprofloxacin, particularly for the more sensitive conventional mouse microbiota.
Ciprofloxacin selects for a common core of species in humanized mice regardless of diet
Given the diversity decrease after ciprofloxacin treatment due to a diet switch, we wondered if species extinction exhibited increased stochasticity in MD compared with STD mice. Thus, we focused on the species present at the time of minimum alpha diversity in humanized mice (Fig. 5H). Some species showed diet-specific sensitivity: 6 ASVs in >50% of STD mice and 3 ASVs found in >50% of MD mice were not present in the other diet (Fig. 5H). Nonetheless, a core of 14 ASVs was present at the time of lowest diversity in at least one MD and one STD mouse, with 6-11 of the core ASVs in each STD mice and 8-12 in each MD mice (Fig. 5H). Four of the 14 were detected in all STD mice, and five were in all MD mice (Fig. 5H). Overall, this core represented ~92% of the total abundance in STD mice and ~90% in MD mice (Fig. 5H). Thus, ciprofloxacin selects for a specific cohort at the point of maximal disturbance regardless of diet, even with a larger decrease in load and diversity in MD compared with STD mice.
An initial antibiotic treatment can condition the Bacteroides response of a human microbiota to a second treatment
To determine whether the response of the Bacteroides and other taxa to ciprofloxacin depends on the overall microbiota composition, we humanized mice for 6 weeks with a sample from a different donor. The microbiota of mice from this second donor (H2) had a higher fraction of Bacteroidetes than the first donor (H1) (Fig. 6A,B). We treated H2 mice with ciprofloxacin as above. As previously, Bacteroidetes relative abundance dropped during the first day with a concurrent bloom of Verrucomicrobiae (Fig. 6B). However, the response thereafter was dramatically different than in H1 mice. Instead of recovery of the Bacteroidetes during days 2-5, the Lachnospiraceae bloomed to >70% on day 3 (Fig. 6B). On day 4, the Barnesiellaceae, a sister family of S24-7 and Bacteroidaceae, began to recover (Fig. 6B). S24-7 did not begin recovery until day 6, and the Bacteroidaeceae were undetectable until day 9 (Fig. 6B), similar to H1 mice after a MAC-deficient diet shift (Fig. 5D), suggesting that the H2 microbiota is sensitized to ciprofloxacin treatment.
Figure 6: Cross-housing-mediated invasion of Bacteroides improves microbiota resilience.
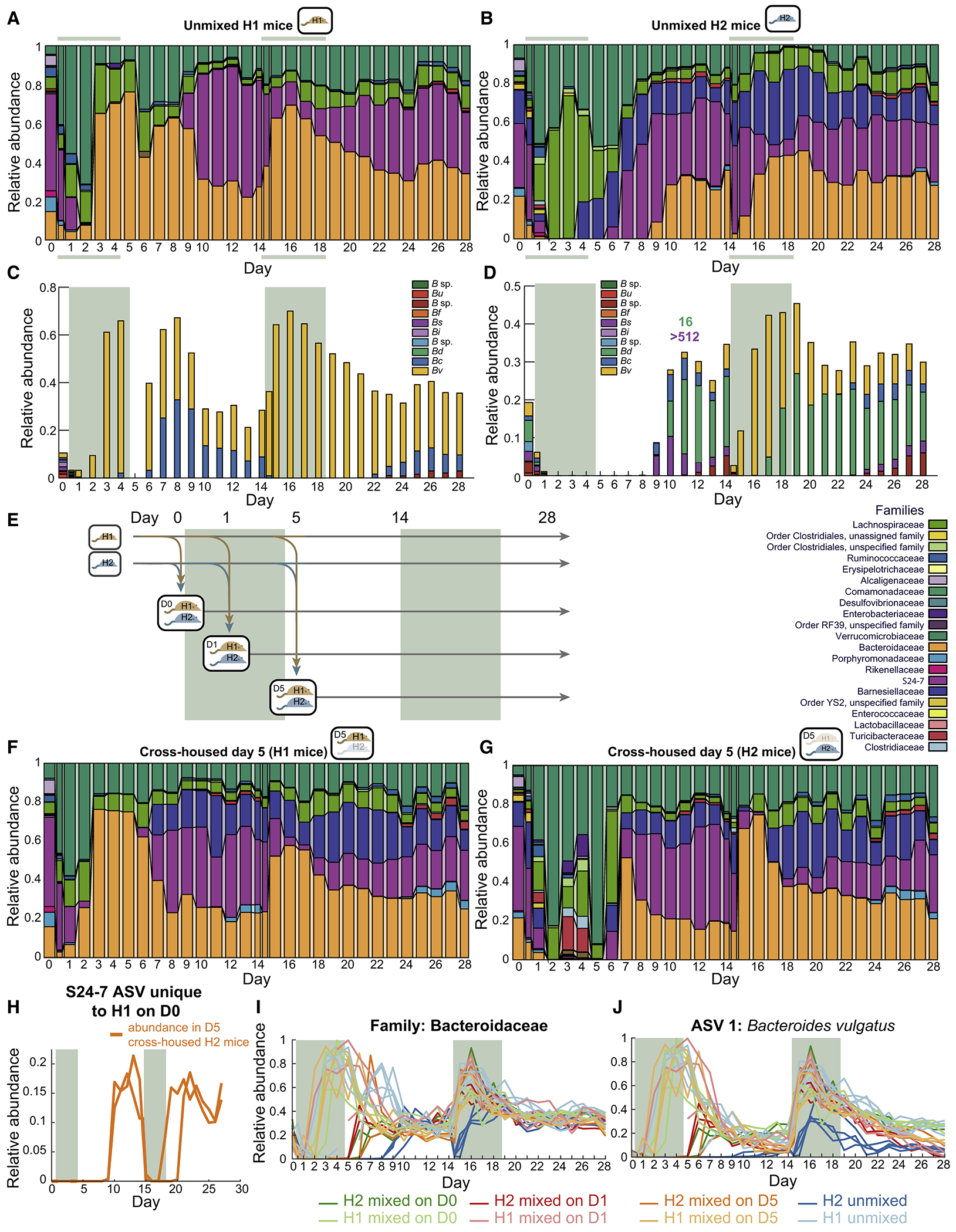
See also Figure S5.
(A,B) Family-level composition in feces of mice colonized with donor samples 1 (H1 (A), n=5) or 2 (H2, (B), n=4) during two ciprofloxacin treatments revealed delayed Bacteroidetes recovery in H2 mice during the first treatment but not the second.
(C,D) Patterns of recovery of the 10 most abundant Bacteroides ASVs (abbreviations same as in Fig. 5H,I) differed between (C) H1 and (D) H2 mice. MICs were calculated for two isolates from H2 mouse samples (labeled above the day of isolation). Treatment periods denoted by colored areas.
(E) Schematic for cross-housing of mice with different microbiotas before, during, and ciprofloxacin treatment.
(F,G) Family-level composition in feces in (F) H1 mice mixed with H2 mice on day 5 (n=3) and (H) H2 mice mixed with H1 mice on day 5 (n=2). Cross-housing stimulated Barnesiellaceae blooming after the first treatment in H1 mice, and resilience of the Bacteroides during the second treatment in H2 mice.
(H) Relative abundance of an S24-7 ASV that was only present in H1 mice on day 0 but then bloomed in H2 mice, highlighting potential transmission .
(I,J) Relative abundances of (I) Bacteroidaceae and (J) the dominant Bv ASV in all H1 and H2 mice, including cross-housed and unmixed mice, during a double course of ciprofloxacin treatment. The Bv ASV became dominant in all H1 mice during both treatments, but only during the second treatment in H2 mice. Bv abundance increased more rapidly and reached higher levels in cross-housed H2 mice compared with unmixed H2 mice.
The Bv ASV that was dominant in H1 mice throughout antibiotic treatments (Fig. 6C) was initially present at similar levels in H2 mice (~3.5% in H2 vs. 1.8% in H1), but did not recover until day 10, and then only to ~6.5% by day 14 (Fig. 6D). A second treatment resulted in a Bacteroidaceae response similar to that of H1 mice during a first treatment: relative abundance dropped rapidly but then recovered to levels comparable to H1 mice by the second day (day 16) (Fig. 6B), with dominance of the Bv ASV (Fig. 6D). Notably, we also isolated a B. salyersiae from day 11 samples of H2 mice with MIC >512 μg/mL that decreased to almost undetectable levels during the second treatment (Fig. 6D), indicating that in vitro resistance is not necessarily predictive of the response in vivo, much like the initial Bacteroides drop during streptomycin. These observations suggest an ecological landscape with distinct Bacteroidaeceae recovery behaviors, and that a microbiota can move between them through antibiotics.
Invasion during cross-housing accelerates recovery from ciprofloxacin
The large dynamics in load and taxonomic composition during ciprofloxacin suggested the potential for invasion from environmental reservoirs. Anticipating the microbiota-specific responses to ciprofloxacin that we identified between H1 and H2 mice (Fig. 6A,B), we also co-housed subsets of H1 and H2 mice (henceforth referred to as cross-housed) on days 0, 1, and 5 (Fig. 6E). Mice underwent two ciprofloxacin treatments as above. With the exception of the transfer and blooming of Barnesiellaceae, which was not initially present, the compositional dynamics of H1 mice were relatively unaffected by cross-housing with H2 mice (Fig. 6F). By contrast, Bacteroidaceae recovery in H2 mice was accelerated by cross-housing (Fig. 6G,I, S5A,B). Cross-housing on day 5 led to multiple waves of transient blooming involving families that were not major components of the stabilized community (Fig. 6G). Moreover, an S24-7 ASV that was initially absent in all H2 mice invaded to ~15% abundance from H1 mice (Fig. 6H), suggesting that S24-7 survival is dependent on reseeding.
By day 10, the family-level composition of all H1 and H2 mice was similar regardless of cross-housing (Fig. 6A,B,F,G). However, the decrease in Bacteroidaceae in H2 mice upon the second antibiotic treatment that occurred in non-cross-housed mice (Fig. 6B,I) was avoided by cross-housing (Fig. 6G,I, S5A,B). In all cross-housed H2 mice, the Bv ASV that dominanted H1 mice during treatment bloomed to 20-30% directly after the first treatment (Fig. 6J), similar to the levels in H1 mice and consistent with the transfer of the resistant strain from H1 mice. After the first treatment, the dynamics of this ASV in cross-housed H2 mice were the same as in H1 mice, with a quantitatively similar increase upon start of the second treatment (Fig. 6J); in non-cross-housed H2 mice, the relative abundance of this ASV initially decreased (Fig. 6J), suggesting that community context and antibiotic resistance may both play a role in Bv dynamics and dominance. Thus, cross-housing effectively shifts the H2 microbiota toward the response of the H1 microbiota, indicating that environmental reservoirs can reprogram the microbiota toward more robust antibiotic recovery.
Reduction of environmental reservoirs impairs microbiota recovery and increases stochasticity
The observation of invasion from cagemates (Fig. 6F,H) indicates that microbes from other hosts are an important resource for recovery after antibiotics. Thus, we hypothesized that singly housing mice to mimic the increased sanitation prevalent in Western society would negatively impact recovery. We compared the effects of streptomycin treatment on co-housed and singly housed mice; streptomycin was selected based on the large initial drop in bacterial load (Fig. 1B,F) that could result in stochastic extinction. We randomly selected 6 mice from a cohort of 11 co-housed ex-germ-free, conventionalized mice and individually housed them (Fig. 7A). In all mice, anaerobes decreased by 102-105 over the first 24 h and aerobes decreased by ~103 (Fig. 7B). The rapid recovery thereafter in co-housed mice was absent in singly housed mice (Fig. 7B), indicating the absence of species that could flourish in the presence of streptomycin.
Figure 7: Co-housing enables robust recovery after streptomycin treatment.
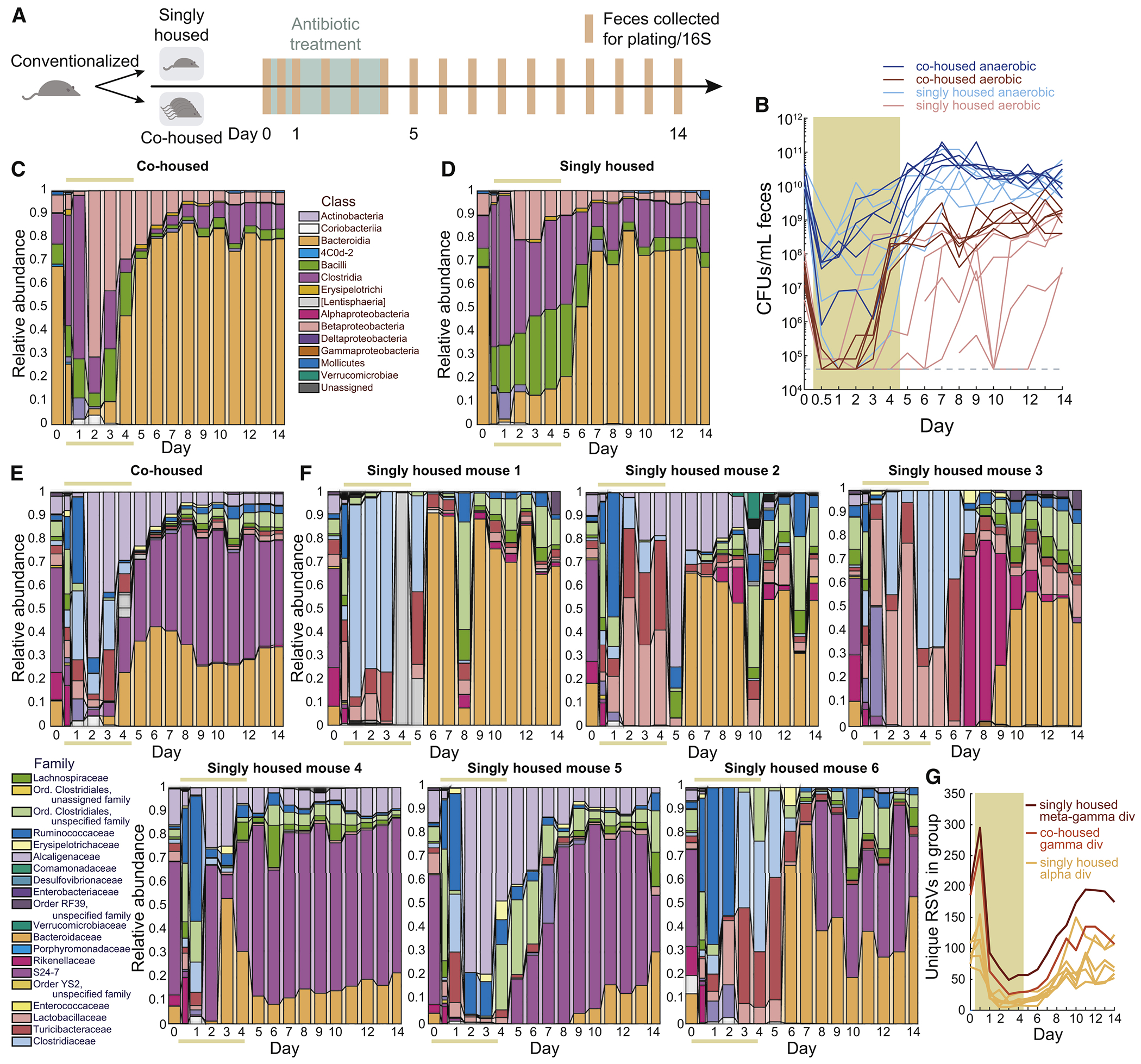
See also Figure S6.
(A) Schematic for housing experiment.
(B) Anaerobic and aerobic culturable fecal densities in co-housed and singly housed streptomycin-treated conventionalized mice (colored area denotes antibiotic treatment period) revealed delayed recovery.
(C,D) Class-level composition in feces in (C) co-housed (n=5) and (D) singly housed (n=6) mice revealed delayed Bacteroidetes recovery in singly housed mice.
(E) Family-level composition in co-housed mice showed robust Bacteroidaeceae recovery during streptomycin.
(F) Family-level composition demonstrated heterogeneous recovery patterns across singly housed mice.
(G) The noisiness of reseeding in singly housed mice was evident from the lower gamma diversity (unique ASVs) of co-housed mice compared with the meta gamma diversity of singly housed mice treated as if they were a group, despite the low alpha diversity of each singly housed mice.
The co-housed conventionalized mice showed similar compositional dynamics (Fig. 7C,E) to co-housed, streptomycin-treated humanized mice (Fig. 1C), indicating that humanized and conventional microbiotas have similar responses to streptomycin. By contrast, singly housed mice exhibited dramatically different compositions from co-housed mice (Fig. 7C–E) and from each other (Fig. 7F); no two mice were qualitatively similar. While the Bacteroidaceae recovered during treatment in co-housed mice (Fig. 7E, S6A), they did not recover in many singly housed mice until after treatment (Fig. 7F,S6A). S24-7 recovered in all co-housed mice (Fig. 7E,S6B) but in only half of the singly housed mice (Fig. 7F,S6B); in some cases singly housed mice experienced transient blooms of families that were at low or zero abundance in co-housed mice.
After streptomycin cessation, the diversity of 5 of 6 singly housed mice was significantly lower than the gamma diversity of the co-housed mice (Fig. 7G). We hypothesized that the slower and more variable recovery dynamics of singly housed mice was a result of stochastic extinctions in the absence of a distinct microbial reservoir, and predicted that we would observe different cohorts of species across individual mice after treatment. Indeed, the total number of unique ASVs found in singly housed mice, akin to gamma diversity had the mice been co-housed, was higher than the gamma diversity of the co-housed mice (Fig. 7G). Taken together, these findings reinforce the importance of environmental reservoirs for robust recovery and colonization resistance of the microbiota.
Discussion
Our study provides important insights into the antibiotic response of individual strains, taxa, and the entire community. The death of virtually all Bacteroides in streptomycin-treated mice (Fig. 1B,C) despite predicted resistance (Table S1) illustrates that in vitro sensitivity is not necessarily predictive of the response within the context of a complex ecosystem in a mammalian host. The death of Bacteroides spp. during streptomycin treatment in all colonization conditions tested (humanization (Fig. 1B,C), conventionalization (Fig. 7B–D), monocolonization with B. thetaiotaomicron (Fig. S4A) suggests that the drug may be disrupting the host (e.g. through mucosal damage), and Bacteroides dynamics may be the consequences of that disruption and the subsequent repair. Tissue imaging revealed increases in mucus thickness due to streptomycin and ciprofloxacin treatment (Fig. 4) that could affect community dynamics, motivating future studies of gut biogeography during perturbations. We uncovered several common features of the response to treatment across microbiotas and between streptomycin and ciprofloxacin. One of the most striking was the recovery of bacterial load during antibiotic treatment, an effect common to all six antibiotic treatments tested in humanized mice (Fig. 1G, S7A,B). Thus, despite the lack of predictability based on the behavior of isolated species, community features such as density may have predictable responses independent of mechanism of action.
The phylum Bacteroidetes, particularly the S24-7 family and Bacteroides genus, was of interest throughout our study due to notable dynamics. In all antibiotics, Bacteroidetes relative abundance was either maintained throughout, or in the case of the transcriptional inhibitor rifaximin showed dynamics similar to streptomycin and ciprofloxacin (Fig. 1C,F, S7B). Dominance of a single Bacteroides species was observed in ciprofloxacin (Bv, Fig. 1H) and in clindamycin (B. ovatus, Fig. S7C). Based on our ciprofloxacin analysis, we surmise that B. ovatus domination likely results from intrinsic resistance to clindamycin (Karlowsky et al., 2012). Nonetheless, the elimination of a resistant B. salyersiae during ciprofloxacin treatment (Fig. 6D), and perhaps other resistant taxa, motivates future studies of interactions among antibiotic resistant strains. The lack of domination of a single Bacteroides when there is pan-resistance (streptomycin) or likely pan-susceptibility (rifaximin) (Finegold et al., 2009) suggests that coexistence is maintained during a perturbation as long as the playing field is levels. Just as we identified a core set of species that survived ciprofloxacin regardless of host diet (Fig. 5H), there was a distinct core of 24 ASVs that appears in at least 5 of 10 mice in our pilot study that accounts for 70.4±13.1% of the microbiota on day 3 of treatment, with 88.6±6.0% accounted for by Bacteroides or S24-7 species (Fig. S7D). These data suggest that particular interspecies interactions generally establish resilience to perturbations, and that focus on S24-7 and Bacteroides is likely warranted for other antibiotics.
Our results indicate that caution must be taken in antibiotic experiments. In cases such as streptomycin, it is critical to sample at high frequency to capture the large changes in bacterial load and composition (Fig. 1B,C). Earlier sampling may capture potential effects of antibiotics such as metronidazole, which displayed remarkable resilience in our pilot (Fig. S7A,B). Our compositional results during streptomycin are qualitatively consistent with some previous studies (Stecher et al., 2007) but differ from others (Thompson et al., 2015), perhaps reflecting microbiota-specific or housing effects. By controlling for community composition as well as facility, our experimental design allowed us to robustly identify key roles of diet (Fig. 5) and co-housing (Fig. 7) in the microbiota response to antibiotics.
Our findings have a wide range of translational implications. The error-correction methodology of DADA2 for 16S analysis provides a stark view of the precarious loss in diversity experienced by certain taxa during antibiotics (Fig. 2G-I), in contrast to OTU-picking based methods (Caporaso et al., 2010) that suggested that hundreds of species remained (Fig. S2G) and did not reveal Bv dominance during ciprofloxacin (Fig. 5H). There is a seemingly permanent loss of diversity after antibiotics (Fig. 2, 5, S3), reflecting the extinction of sensitive Bacteroidetes species. Transplants of these species may hasten microbiota recovery after treatment; for example, Bacteroides are amenable to transfer to Bacteroides-lacking hosts (Fig. 6). Loss of taxa may open niches for opportunistic pathogens, highlighting the importance of autologous fecal transplants and/or targeted microbial therapies to restore colonization resistance. A MAC-deficient diet sensitizes the microbiota to ciprofloxacin (Fig. 5) and may increase the potential for pathogen invasion. In future studies, it will be intriguing to probe whether dietary interventions (e.g., high fiber) can improve resilience and recovery, similar to the reversal of a persistent Clostridium difficile infection (Hryckowian et al., 2018). Our experiments suggest that antibiotics can also recalibrate microbiotas for future treatments, such as the Bacteroides increase in H2 miec during the first treatment that potentiates rapid recovery during a second treatment (Fig. 6D,I).
Our study highlights similarities in the responses of conventional and humanized mice, such as the dominance during recovery of Bv strains in both colonization states. Nonetheless, several observations suggest that conventional mice are more sensitive than humanized mice to ciprofloxacin treatment. First, there was essentially no recovery of culturable densities in conventional mice during treatment (Fig. 3E), potentially due to the lack of Bacteroidetes throughout treatment (Fig. 3F). Second, the decrease in alpha diversity was larger and more prolonged in conventional mice (Fig. S3D) vs. humanized mice (Fig. 2B,S3B). Finally, the response to a second treatment mirrored the first treatment in conventional mice (Fig. 3E,F), whereas humanized mice displayed a more resilient response to a second treatment (Fig. 5B,C). While this resilience may be due to the emergence of resistant strains in the humanized microbiota, it is remarkable that family-level abundances remained virtually unchanged throughout the second treatment (Fig. 3F). The lack of complete immune function in humanized mice may partially account for these differences. Nonetheless, our data suggest that human microbiotas may be more resilient to antibiotics than mouse microbiotas, consistent with the dramatically increased disruption caused by ciprofloxacin in MD vs. STD conventional mice.
Antibiotic treatment has interesting commonalities with other common gut microbiota perturbations. Both antibiotics and osmotic diarrhea (Tropini et al., 2018) revealed sensitivity of S24-7 (Fig. 1G, 5D,G), its competition with Bacteroidaceae for niches, the key role of sanitation in recovery (Fig. 7), and transitions to substantially altered steady states (Fig. 3) (Tropini et al., 2018). There were surprising similarities among the responses to streptomycin, ciprofloxacin, and rifaximin (Fig. 1C,G, S7B), suggesting a stereotyped microbiota collapse mechanism (Fig. 1B). However, the increased heterogeneity among singly housed mice (Fig. 7) suggests that collapse and recovery may rely on interactions that cannot be maintained after substantial extinction. Our findings paint the picture of a microbial community that bends with global shifts in diet, antibiotics, and composition, yet exhibits resilience if the community can be reinforced through environment-mediated recolonization. Future studies investigating how microbiotas respond to perturbations on multiple time scales should enable mapping of the landscape of community states and trajectories, empowering the design of resilient communities and reprogramming of dysbiosis.
STAR Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
This study generated reagents in the form of isolated Bacteroides strains. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kerwyn Casey Huang (kchuang@stanford.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse strains
All mouse experiments were conducted in accordance with the Administrative Panel on Laboratory Animal Care, Stanford University’s IACUC. Experiments involved Swiss-Webster mice of 6-12 weeks of age, and both genders were used. For all experiments, mice were co-housed unless stated otherwise.
For humanized mouse experiments, mice were gavaged with a human fecal sample obtained from a healthy anonymous donor (American male living in the San Francisco Bay Area, CA, age 42, omnivorous diet) as previously described (Kashyap et al., 2013). The microbiota was allowed to equilibrate for 6-8 weeks before perturbation experiments commenced. For monocolonization experiments, mice were gavaged with an overnight culture of Bacteroides thetaiotaomicron and allowed to equilibrate for 2 weeks before streptomycin treatment commenced. Conventional Swiss-Webster mice (RFSW, Taconic) were bred from an in-house colony. Mice were orally gavaged with antibiotics (Sigma Aldrich) dissolved in 200 μL water for five days. For experiments involving a dietary switch, mice were first fed a standard diet (Purina LabDiet 5010) rich in MACs, and then a defined low-MAC diet (Harlan TD.86489) in which the sole carbohydrates are sucrose (31% w/w), cornstarch (31% w/w), and cellulose (5% w/w).
Mice were euthanized with CO2 and death was confirmed via cervical dislocation. After sacrifice, sections of the ileum, proximal colon, and distal colon were preserved, embedded, sectioned, stained and imaged as previously described (Earle et al., 2015).
Bacterial strains
For isolation of Bacteroides strains, frozen mouse fecal samples were resuspended in PBS and plated onto Bacteroides Bile Esculin agar/ Brucella Laked Blood agar with kanamycin and vancomycin (BBE/LKV) plates (Anaerobe Systems). This medium selects for Bacteroides spp. Plates were incubated at 37 °C in an anaerobi c chamber. After 2 days, colonies were grown in PYG (Anaerobe Systems) for 2 days and glycerol stocked.
For MIC measurements, all isolates and type strains were grown anaerobically in a plate reader with constant shaking in pre-reduced BHIS (BHI broth supplemented with 0.5 μg/mL porcine hemin and 0.5 μg/mL vitamin K1). After 24 h of growth, cultures were diluted 100-fold into fresh medium and grown for another 24 h. Cultures were then diluted 200-fold into fresh medium containing antibiotics (ciprofloxacin, streptomycin, and rifaximin) at various concentrations in 384-well plates and grown at 37 °C. After 48 h of growth, absorbance was measured in a plate reader. The minimum dose of antibiotic at which absorbance was indistinguishable from media controls was used to determine the MIC.
METHOD DETAILS
Quantification of bacterial densities
Anaerobic and aerobic bacterial densities were quantified by spot plating on duplicate Brain Heart Infusion agar plates supplemented with 10% defibrinated horse blood. Aerobic plates were incubated in a 37 °C warm room and anaerobic plates were placed in a 37 °C incubator inside an anaerobic chamber (C oy Laboratory Products).
Tissue collection and imaging
After sacrifice, sections of the colon and cecum were collected and immediately fixed in methacarn (Methanol-Carnoy) solution (60% anhydrous methanol, 30% chloroform, and 10% glacial acetic acid) and processed in paraffin as previously described (Earle et al., 2015; Johansson and Hansson, 2012). Briefly, after 3 h to 2 weeks of fixation, sections were washed twice in anhydrous methanol for 30 min, twice in anhydrous ethanol for 20 min, and twice in xylene for 15 min. The sections were then submerged in paraffin at 70 °C for two hours. Paraffin blocks were sliced into 4-μm sections and deparaffinized for immunofluorescence as described below (Earle et al., 2015; Johansson and Hansson, 2012).
For immunofluorescence, sections were dewaxed by incubation at 60 °C, and two additional incubations in xylene for 10 min each. The samples were then hydrated in solutions with decreasing concentration of ethanol (100, 95, 70, 50, and 30%) for 5 min each. Sections were washed in PBS and circled with a PAP (liquid blocker) pen. Blocking solution (5% w/v fetal bovine serum in PBS) was added and incubated in darkness in a humid chamber for 30 min at room temperature. The specific antibody for MUC2 (Santa Cruz Biotechnology, diluted 1:100 in block solution), the primary intestinal mucin, was added to the slide and incubated in darkness for 4–24 h at 4 °C. Slides were washed three times in PBS. The secondary antibody was diluted in block solution 1:100, combined with DAPI (Sigma, 1:500 dilution) and UEA-1 (rhodamine-labeled Ulex Europaeus Agglutinin I; Vector labs, 1:50 dilution), and added to the sample. After incubation in the dark in a humid environment at room temperature for 2 h, slides were washed three times in PBS for 10 min each. Slides were allowed to dry completely, and the sections were mounted using Prolong antifade mounting media and set at room temperature. The slides were thereafter stored at 4 °C.
Images were collected using a Zeiss LSM 880 inverted confocal microscope with ZEN Black software. Mucus thickness was quantified using the analysis platform BacSpace (Earle et al., 2015).
16S rRNA analyses
DNA was extracted from whole fecal pellets with the PowerSoil and PowerSoil-htp kits (MO BIO, San Diego, CA). 16S rRNA amplicons were generated using the Earth Microbiome Project-recommended 515F/806R primer pairs. PCR products were cleaned, quantified, and pooled using the UltraClean 96 PCR Cleanup kit (MO BIO) and Quant-It dsDNA High Sensitivity Assay kit (Invitrogen). Samples were sequenced with 250- or 300-bp reads on a MiSeq (Illumina).
Samples were de-multiplexed and analyzed using DADA2 and QIIME v. 1.8 as previously described (Callahan et al., 2016; Caporaso et al., 2010). Custom MATLAB (MathWorks) scripts were used to analyze OTU distributions during antibiotic timecourses. Glycoside hydrolase (GH) imputations were performed as previously described (Zhang et al., 2018) using the dbCAN2 meta server on 11/19/2018. Collections of CAZy types were visualized using Anvi’o v. 5.2 (Eren et al., 2015). Anv’io was also used in conjunction with DIAMOND, MCL, and PyANI to perform gene clustering and calculation and visualization of average nucleotide identity (Buchfink et al., 2015; Pritchard et al., 2016; van Dongen and Abreu-Goodger, 2012).
Preparation of mouse fecal pellets for mass spectrometry analysis
Fecal samples were taken from mice at indicated times and stored at −80 ° until the time of mass spectrometry analysis. Fecal pellets were placed in tared, 2-mL mass spectrometer-compatible tubes (USA Scientific, cat. # 1620-2700). 200 μL of methanol were added to each pellet. A set of 2.8-mm zirconium oxide ceramic beads (VWR, cat. # 10144-494) were added to each tube and samples were disrupted for 20 min at a frequency of 20 Hz on a TissueLyser II (Qiagen, cat. # 85300). After disruption, supernatants were obtained via centrifugation of samples for 15 min at 16,300g. Ten microliters of cleared supernatant were removed and added to 10 μL of water + 0.1 % formic acid, mixed well, and placed in glass vials for analysis via mass spectrometry. If analysis was not immediately performed, isolated samples were stored at −20 °C.
UPLC/electrospray ionization-MS analysis
LC-MS analyses were performed on an Agilent 6530 Accurate Mass QToF LC/MS with a dual electrospray ionization source (Agilent) connected to a Agilent 1290 Infinity II UPLC front-end equipped with an SB C18 column (1.8 μm, 3.0x100 mm, Agilent cat. #828975-302). The gas temperature was set to 350 °C . The VCap was set to 4000 V. The instrument was operated in positive-ion mode under MS conditions. The absorbance threshold was set at 5000 and the relative threshold was set at 1%. Data were collected and stored in centroid form. The mobile phase system was made up of solvent A (water + 0.1% formic acid) and solvent B (95:5 acetonitrile:water + 0.1% formic acid), with a flow rate of 0.2 mL/min. The method and gradient used to elute the fecal extracts was: 0 min to 2 min 0% B, 2 min to 12 min 0% B to 100%, 12 min to 13 min held at 100% B, 13 min to 14 min 100% B to 0% B, 14 min to 15 min 0% B to re-equilibrate the column. All gradients were linear. The first 2 min of the run were sent to waste to remove salt. MS data were collected from 1.2 min until 12.5 min. Two microliters of each sample were injected/run. Analysis was performed using the Agilent MassHunter Qualitative Analysis Software. Exact masses were calculated using ChemDraw and analyzing standards. TICs were extracted using these exact masses ±100 ppm, the preset default by MassHunter to generate EICs. EICs were manually integrated and the area under the peak was calculated by MassHunter. Area under the curve was then adjusted to be per gram of fecal matter input during fecal preparations.
QUANTIFICATION AND STATISTICAL ANALYSIS
For two-condition comparisons, two-sample t-tests were applied. Significant changes were defined as p<0.05. Data were plotted using custom MATLAB software. In figures with boxplots, the central line represents the median and the top and bottom edges indicate the 25th and 75th percentiles. Whiskers extend to the most extreme data points (excluding outliers, which are plotted using the ‘+’ symbol).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse Mucin 2 Antibody (H-300) | Santa Cruz Biotechnology | sc-15334 |
| Alexa Fluor® 488-AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch | 711-545-152 |
| Bacterial and Virus Strains | ||
| Bacteroides thetaiotaomicron VPI-5482 | ATCC | ATCC 29148 |
| Bacteroides fragilis NCTC 9343 | ATCC | ATCC 25285 |
| Bacteroides vulgatus ATCC 8482 | ATCC | ATCC 8482 |
| Bacteroides distasonis ATCC 8503 | ATCC | ATCC 8503 |
| Bacteroides caccae ATCC 43185 | ATCC | ATCC 43185 |
| Bacteroides ovatus ATCC 8483 | ATCC | ATCC 8483 |
| Bacteroides uniformis ATCC 8492 | ATCC | ATCC 8492 |
| Bacteroides intestinalis DSM 17393 | DSMZ | DSM 17393 |
| Bacteroides cellulosilyticus CL02T12C19 | BEI (HMP) | HM-726 |
| Bacteroides salyersiae CL02T12C01 | BEI (HMP) | HM-725 |
| Bacteroides stercoris CC31F | BEI (HMP) | HM-1036 |
| Bacteroides vulgatus CL09T03C04 | BEI (HMP) | HM-720 |
| Bacteroides vulgatus (humanized mouse 1, D0) | This study | |
| Bacteroides vulgatus (humanized mouse 1, D4) | This study | |
| Bacteroides vulgatus (humanized mouse 1, D12) | This study | |
| Bacteroides caccae (humanized mouse 1, D12) | This study | |
| Bacteroides vulgatus (humanized mouse 1, D16) | This study | |
| Bacteroides salyersiae (humanized mouse 2, D0) | This study | |
| Bacteroides stercoris (humanized mouse 2, D0) | This study | |
| Bacteroides xylanisolvens (humanized mouse 2, D0) | This study | |
| Bacteroides intestinalis (humanized mouse 2, D0) | This study | |
| Bacteroides intestinalis (humanized mouse 2, D0) | This study | |
| Bacteroides uniformis (humanized mouse 2, D0) | This study | |
| Bacteroides uniformis (humanized mouse 2, D10) | This study | |
| Bacteroides uniformis (humanized mouse 2, D10) | This study | |
| Bacteroides vulgatus (humanized mouse 2, D16) | This study | |
| Bacteroides caccae (humanized mouse 2, D22) | This study | |
| Bacteroides uniformis (humanized mouse 2, D22) | This study | |
| Bacteroides uniformis (humanized mouse 2, D22) | This study | |
| Bacteroides vulgatus (conventional mouse 1, day 0) | This study | |
| Bacteroides vulgatus (conventional mouse 1, day 0) | This study | |
| Bacteroides acidifaciens (conventional mouse 1, day 0) | This study | |
| Bacteroides uniformis (conventional mouse 1, day 0) | This study | |
| Bacteroides vulgatus (conventional mouse 1, day 11) | This study | |
| Bacteroides acidifaciens (conventional mouse 1, day 11) | This study | |
| Bacteroides uniformis (conventional mouse 1, day 11) | This study | |
| Bacteroides sp. D1 (H2 microbiota mouse 1, day 11) | This study | |
| Bacteroides sp. D1 (H2 microbiota mouse 1, day 11) | This study | |
| Bacteroides salyersiae (H2 microbiota mouse 1, day 11) | This study | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Rhodamine-labeled Ulex Europaeus Agglutinin I | Vector Labs | Cat# RL-1062 |
| Bacteroides Bile Esculin Agar/Laked Brucella Blood Agar with Kanamycin and Vancomycin (BBE/LKV) | Anaerobe Systems | AS-242 |
| BD Difco™ Brain Heart Infusion Agar | BD | 241830 |
| Standard MAC-rich diet | Purina | LabDiet 5010 |
| Defined low-MAC diet | Harlan | TD.86489 |
| Critical Commercial Assays | ||
| PowerSoil HT DNA Isolation Kit | Qiagen | 12955 |
| UltraClean 96 PCR Cleanup Kit | Qiagen | 12596-4 |
| Quant-iT™ dsDNA Assay Kit, high sensitivity | Invitrogen | Q33120 |
| Deposited Data | ||
| 16S and whole genome sequencing datasets | Stanford Digital Repository | https://purl.stanford.edu/rm696bw4911 |
| Experimental Models: Organisms/Strains | ||
| Swiss Webster Mice – Germ Free | Taconic | SW GF |
| Swiss Webster Mice – Restricted Flora | Taconic | SW RF |
| Software and Algorithms | ||
| QIIME v.1.8 | (Caporaso et al., 2010) | N/A |
| BacSpace | (Earle et al., 2015) | N/A |
| FastQC v. 0.11.4 | FastQC | N/A |
| R v. 3.2.1 | (Ihaka and Gentleman, 1996) | N/A |
| MATLAB 2017a | Mathworks | N/A |
| bowtie2 v. 2.3.09 | (Langmead and Salzberg, 2012) | N/A |
| dbCAN2 meta server | (Zhang et al., 2018) | N/A |
| ResFinder | (Zankari et al., 2012) | N/A |
| Diamond v. 0.8.18 | (Buchfink et al., 2015) | N/A |
| MCL | (van Dongen and Abreu-Goodger, 2012) | N/A |
| Trimmomatic 0.36 | (Bolger et al., 2014) | N/A |
| Anvi’o v. 3 | (Eren et al., 2015) | N/A |
Highlights.
Human microbiotas were resilient and recovered rapidly during antibiotic administration
A low fiber diet aggravated microbiota collapse and delayed recovery from ciprofloxacin
Microbiota reprogramming and transmission conferred resilience to repeated treatment
Single-housing disrupted recovery, highlighting roles of reservoirs and sanitation
Acknowledgments
The authors thank Michelle St. Onge and Marina Grunina for helpful discussions and advice and Nassos Typas for careful reading of the manuscript. A.A.-D. is a Howard Hughes Medical Institute International Student Research Fellow and a Stanford Bio-X Bowes Fellow. M.A.F., J.L.S, and K.C.H. are Chan Zuckerberg Biohub Investigators. K.B.X. and R.A.O. acknowledge Fundação para a Ciência e Tecnologia for individual grants IF/00831/2015 and PD/BD/106000/2014.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
DATA AND CODE AVAILABILITY
Whole genome sequencing of isolates and 16S sequencing data are available at the Stanford Digital Repository: https://purl.stanford.edu/rm696bw4911
References
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan LE, Kowand SK, and Van Den Elzen HM (1979). Mechanism of aminoglycoside antibiotic resistance in anaerobic bacteria: Clostridium perfringens and Bacteroides fragilis. Antimicrob Agents Chemother 15, 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B, Xie C, and Huson DH (2015). Fast and sensitive protein alignment using DIAMOND. Nat Methods 12, 59–60. [DOI] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, and Holmes SP (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13, 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7, 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho I, Yamanishi S, Cox L, Methe BA, Zavadil J, Li K, Gao Z, Mahana D, Raju K, Teitler I, et al. (2012). Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 488, 621–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox LM, Yamanishi S, Sohn J, Alekseyenko AV, Leung JM, Cho I, Kim SG, Li H, Gao Z, Mahana D, et al. (2014). Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 158, 705–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, Pudlo NA, Kitamoto S, Terrapon N, Muller A, et al. (2016). A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 167, 1339–1353 e1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, Huse S, Sogin ML, and Relman DA (2008). The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol 6, e280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, and Relman DA (2011). Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A 108 Suppl 1, 4554–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doorduyn Y, Van Den Brandhof WE, Van Duynhoven YT, Wannet WJ, and Van Pelt W (2006). Risk factors for Salmonella Enteritidis and Typhimurium (DT104 and non-DT104) infections in The Netherlands: predominant roles for raw eggs in Enteritidis and sandboxes in Typhimurium infections. Epidemiol Infect 134, 617–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earle KA, Billings G, Sigal M, Lichtman JS, Hansson GC, Elias JE, Amieva MR, Huang KC, and Sonnenburg JL (2015). Quantitative Imaging of Gut Microbiota Spatial Organization. Cell Host Microbe 18, 478–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren AM, Esen OC, Quince C, Vineis JH, Morrison HG, Sogin ML, and Delmont TO (2015). Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ 3, e1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DJ, Allison DG, Brown MR, and Gilbert P (1991). Susceptibility of Pseudomonas aeruginosa and Escherichia coli biofilms towards ciprofloxacin: effect of specific growth rate. J Antimicrob Chemother 27, 177–184. [DOI] [PubMed] [Google Scholar]
- Finegold SM, Molitoris D, and Vaisanen ML (2009). Study of the in vitro activities of rifaximin and comparator agents against 536 anaerobic intestinal bacteria from the perspective of potential utility in pathology involving bowel flora. Antimicrob Agents Chemother 53, 281–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohlich EE, Farzi A, Mayerhofer R, Reichmann F, Jacan A, Wagner B, Zinser E, Bordag N, Magnes C, Frohlich E, et al. (2016). Cognitive impairment by antibiotic-induced gut dysbiosis: Analysis of gut microbiota-brain communication. Brain Behav Immun 56, 140–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein EJ, and Citron DM (1992). Comparative activity of ciprofloxacin, ofloxacin, sparfloxacin, temafloxacin, CI-960, CI-990, and WIN 57273 against anaerobic bacteria. Antimicrob Agents Chemother 36, 1158–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hryckowian AJ, Van Treuren W, Smits SA, Davis NM, Gardner JO, Bouley DM, and Sonnenburg JL (2018). Microbiota-accessible carbohydrates suppress Clostridium difficile infection in a murine model. Nat Microbiol 3, 662–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang I, Park YJ, Kim YR, Kim YN, Ka S, Lee HY, Seong JK, Seok YJ, and Kim JB (2015). Alteration of gut microbiota by vancomycin and bacitracin improves insulin resistance via glucagon-like peptide 1 in diet-induced obesity. FASEB J 29, 2397–2411. [DOI] [PubMed] [Google Scholar]
- Ihaka R, and Gentleman R (1996). R: a language for data analysis and graphics. Journal of computational and graphical statistics 5, 299–314. [Google Scholar]
- Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, and Littman DR (2008). Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 4, 337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson ME, and Hansson GC (2012). Preservation of mucus in histological sections, immunostaining of mucins in fixed tissue, and localization of bacteria with FISH. Methods Mol Biol 842, 229–235. [DOI] [PubMed] [Google Scholar]
- Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, and Hansson GC (2008). The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A 105, 15064–15069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlowsky JA, Walkty AJ, Adam HJ, Baxter MR, Hoban DJ, and Zhanel GG (2012). Prevalence of antimicrobial resistance among clinical isolates of Bacteroides fragilis group in Canada in 2010-2011: CANWARD surveillance study. Antimicrob Agents Chemother 56, 1247–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashyap PC, Marcobal A, Ursell LK, Larauche M, Duboc H, Earle KA, Sonnenburg ED, Ferreyra JA, Higginbottom SK, Million M, et al. (2013). Complex interactions among diet, gastrointestinal transit, and gut microbiota in humanized mice. Gastroenterology 144, 967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, and Knight R (2012). Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier L, Pruteanu M, Kuhn M, Zeller G, Telzerow A, Anderson EE, Brochado AR, Fernandez KC, Dose H, Mori H, et al. (2018). Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 555, 623–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcobal A, Southwick AM, Earle KA, and Sonnenburg JL (2013). A refined palate: bacterial consumption of host glycans in the gut. Glycobiology 23, 1038–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazmanian SK, Round JL, and Kasper DL (2008). A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453, 620–625. [DOI] [PubMed] [Google Scholar]
- Ng KM, Ferreyra JA, Higginbottom SK, Lynch JB, Kashyap PC, Gopinath S, Naidu N, Choudhury B, Weimer BC, Monack DM, et al. (2013). Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 502, 96–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onderdonk AB, Louie TJ, Tally FP, and Bartlett JG (1979). Activity of metronidazole against Escherichia coli in experimental intra-abdominal sepsis. J Antimicrob Chemother 5, 201–210. [DOI] [PubMed] [Google Scholar]
- Ormerod KL, Wood DL, Lachner N, Gellatly SL, Daly JN, Parsons JD, Dal’Molin CG, Palfreyman RW, Nielsen LK, Cooper MA, et al. (2016). Genomic characterization of the uncultured Bacteroidales family S24-7 inhabiting the guts of homeothermic animals. Microbiome 4, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavia AT, Shipman LD, Wells JG, Puhr ND, Smith JD, McKinley TW, and Tauxe RV (1990). Epidemiologic evidence that prior antimicrobial exposure decreases resistance to infection by antimicrobial-sensitive Salmonella. J Infect Dis 161, 255–260. [DOI] [PubMed] [Google Scholar]
- Pritchard L, Glover RH, Humphris S, Elphinstone JG, and Toth IK (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Analytical Methods 8, 12–24. [Google Scholar]
- Salyers AA, West SE, Vercellotti JR, and Wilkins TD (1977). Fermentation of mucins and plant polysaccharides by anaerobic bacteria from the human colon. Appl Environ Microbiol 34, 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg ED, Smits SA, Tikhonov M, Higginbottom SK, Wingreen NS, and Sonnenburg JL (2016). Diet-induced extinctions in the gut microbiota compound over generations. Nature 529, 212–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg JL, Xu J, Leip DD, Chen CH, Westover BP, Weatherford J, Buhler JD, and Gordon JI (2005). Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science 307, 1955–1959. [DOI] [PubMed] [Google Scholar]
- Stecher B, Robbiani R, Walker AW, Westendorf AM, Barthel M, Kremer M, Chaffron S, Macpherson AJ, Buer J, Parkhill J, et al. (2007). Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol 5, 2177–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JA, Oliveira RA, Djukovic A, Ubeda C, and Xavier KB (2015). Manipulation of the quorum sensing signal AI-2 affects the antibiotic-treated gut microbiota. Cell Rep 10, 1861–1871. [DOI] [PubMed] [Google Scholar]
- Tropini C, Moss EL, Merrill BD, Ng KM, Higginbottom SK, Casavant EP, Gonzalez CG, Fremin B, Bouley DM, Elias JE, et al. (2018). Transient Osmotic Perturbation Causes Long-Term Alteration to the Gut Microbiota. Cell 173, 1742–1754 e1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dongen S, and Abreu-Goodger C (2012). Using MCL to extract clusters from networks. Methods Mol Biol 804, 281–295. [DOI] [PubMed] [Google Scholar]
- van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, Visser CE, Kuijper EJ, Bartelsman JF, Tijssen JG, et al. (2013). Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med 368, 407–415. [DOI] [PubMed] [Google Scholar]
- Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, Aarestrup FM, and Larsen MV (2012). Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother 67, 2640–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Yohe T, Huang L, Entwistle S, Wu P, Yang Z, Busk PK, Xu Y, and Yin Y (2018). dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res 46, W95–W101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Whole genome sequencing of isolates and 16S sequencing data are available at the Stanford Digital Repository: https://purl.stanford.edu/rm696bw4911