

Abstract
ABCG2 is a member of the ATP-binding cassette transporter superfamily, and its overexpression causes multidrug resistance (MDR) in cancer chemotherapy. ABCG2 may also protect cancer stem cells by extruding cytotoxic materials. ABCG2 has previously been shown to exist as a high-order homo-oligomer consisting of possibly 8–12 subunits, and the oligomerization domain was mapped to the C-terminal domain, including TM5, ECL3, and TM6. In this study, we further investigate this domain in detail for the role of each segment in the oligomerization and drug transport function of ABCG2 using domain swapping and site-directed mutagenesis. We found that none of the three segments (TM5, TM6, and ECL3) is essential for the oligomerization activity of ABCG2 and that any one of these three segments in the full-length context is sufficient to support ABCG2 oligomerization. While TM5 plays an important role in the drug transport function of ABCG2, TM6 and ECL3 are replaceable. Thus, each segment in the TM5—ECL3—TM6 domain plays a distinctive role in the oligomerization and function of ABCG2.
Graphical Abstract
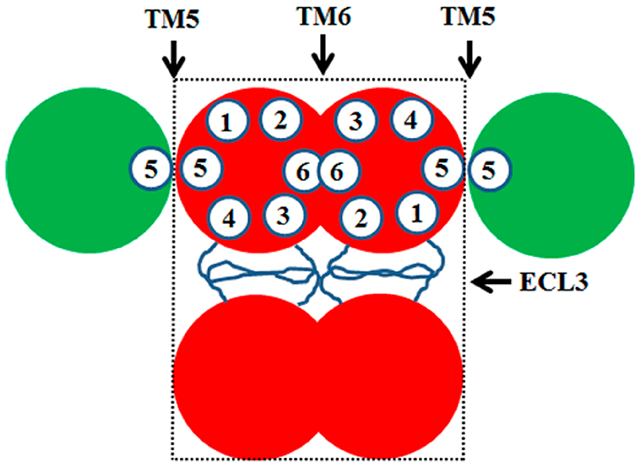
Multidrug resistance (MDR) is a major problem in the successful chemotherapy of human cancers. Studies with drug-resistant model cell lines have shown that overexpression of some members of the ATP-binding cassette (ABC) transporter superfamily such as ABCB1 (P-glycoprotein or Pgp), ABCC1 (multidrug resistance-associated protein or MRP1), and ABCG2 (breast cancer resistance protein or BCRP) represents one of the major mechanisms contributing to MDR.1–4 The increased levels of expression of these ABC transporters cause increased efflux and, thus, reduced levels of intracellular accumulation of anticancer drugs, conferring cellular resistance.
Of the 49 members of the human ABC transporter family, ABCG2 is particularly interesting because it is clinically significant in the prognosis prediction of both liquid and solid malignancies, in the development of both innate and acquired MDR, in the regulation of drug bioavailability, and possibly in protecting cancer stem cells.5–7 While the primary sequence of ABCG2 and its domain structure are approximately half as large as those of a traditional ABC transporter such as ABCB1,8 recent evidence suggests that ABCG2 may exist as a homo-oligomer consisting of 129 or 8 subunits,10 with a minimal stable complex of a tetramer.9 These findings suggest that ABCG2 may function in a more complicated manner than previously anticipated.
The oligomerization activity of ABCG2 has been mapped to a domain that includes TM5, ECL3, and TM6 (Figure 1A), and truncated ABCG2 with only this domain could function as a dominant negative molecule inhibiting the activity of full-length ABCG2.11 In this study, we further investigate this domain and map in detail the oligomerization activity and the role of each segment of this domain in the oligomerization and function of ABCG2. We found that all three segments (TM5, TM6, and ECL3) are sufficient to support ABCG2 oligomerization, although TM5 and TM6 each alone has reduced activity. However, none of them is essential for the oligomerization activity of ABCG2. While TM5 plays an important role in ABCG2 function, TM6 and ECL3 are replaceable. Taken together, we conclude that each segment in the TM5—ECL3—TM6 domain plays a distinctive role in the oligomerization and function of ABCG2.
Figure 1.
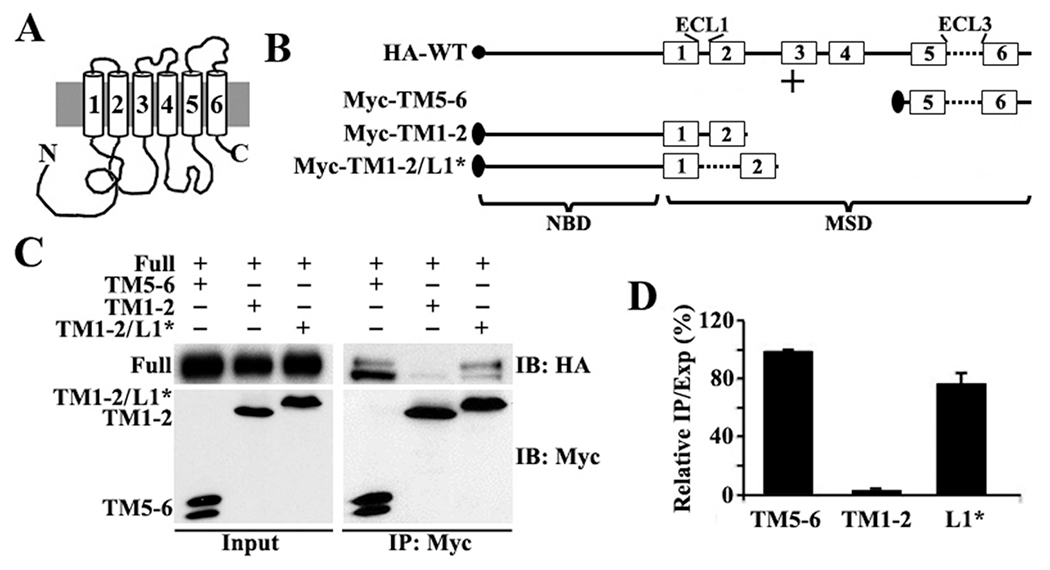
(A) Schematic presentation of ABCG2 topology. (B) Schematic presentation of HA-tagged full-length and Myc-tagged ABCG2 constructs. The solid circle and ovals represent HA and Myc tags, respectively. The numbered boxes and lines represent transmembrane (TM) segments and loops (L), respectively. Abbreviations: ECL, extracellular loop; NBD, nucleotide-binding domain; MSD, membrane-spanning domain. (C) Co-IP. HEK293 cells with stable expression of ABCG2HA-WT were transiently transfected with ABCG2Myc-TM5–6, ABCG2Myc-TM1–2, or ABCG2Myc-TM1–2/L1* followed by immunoprecipitation with the Myc antibody and Western blot analysis of the precipitates with HA and Myc antibodies. (D) Quantitative analysis of Co-IP. The expression and Co-IP level of constructs from three independent experiments as shown in panel C were quantified using ScnImage and calculation of the relative ratio of Co-IP to the expression level followed by normalization to that of the positive control construct ABCG2Myc-TM5–6.
MATERIALS AND METHODS
Materials.
All primers for engineering ABCG2 constructs via PCR (polymerase chain reaction) were obtained from Invitrogen, and pfu DNA polymerases were purchased from Stratagene. Cell culture medium DMEM, antibiotics, and trypsin were from Mediatech or Lonza. Anti-Myc antibody, anti-HA antibody, and protein G-PLUS agarose were purchased from Cell Signaling Technology, Covance, and Santa Cruz Biotechnology, respectively. DNA and protein molecular weight markers were obtained from Fermentas Life Sciences. Chemical cross-linking reagent disuccinimidyl suberate (DSS) and restriction endonucleases were from Pierce and New England Biolabs, respectively. ECL (enhanced chemiluminescence) Western Blot detection reagents were obtained from GE Healthcare. All other reagents of analytic grade or higher were purchased from Sigma-Aldrich or Fisher Scientific.
Engineering of Constructs.
Domain swapping and point mutation constructs of human ABCG2 were engineered using PCR as previously described,12–14 and the primers for each construct and template are listed in Table S1 of the Supporting Information. ABCG2Myc-TM1–2/L1* was engineered using ABCG2Myc-TM5–6 and ABCG2Myc-TM1–2 as templates from a previous study11 and P1/P2 and P3/P4 primer pairs, respectively. The two PCR products were then mixed with the P1/P5 primer pair for an overlap PCR. The second-round PCR product was digested with BamHI and EcoRI and cloned into pCDNA3.1 to generate ABCG2Myc-TM1–2/L1*.
To engineer ABCG2Myc-F-TM5*6*, ABCG2Myc-ECL3*, ABCG2Myc-L3*TM6*, and ABCG2Myc-TM5*L3*, we took advantage of a unique MluNI restriction site in TM4 of ABCG2. PCR was first performed as described above using ABCG2Myc-TM5–6 and ABCG2Myc-TM1–2 11 as templates and a primer containing an MluNI or EcoRI site and other primers (see Table S1 of the Supporting Information). The two PCR products were then used as templates for overlap PCR. The final products were digested with MluNI and EcoRI to replace the MluNI–EcoRI fragment in ABCG2Myc-WT, resulting in ABCG2Myc-F-TM5*6*, ABCG2Myc-ECL3*, ABCG2Myc-L3*TM6*, and ABCG2Myc-TM5*L3*.
ABCG2Myc-TM5*6*CL was engineered as described previously14 using the QuikChange multisite-directed mutagenesis kit. ABCG2Myc-TM5*6*CLM was engineered using the transformer site-directed mutagenesis kit (Clontech) with a mutagenic primer (5′-TGGCTGTCATGGCTTGCCTACTTCGCCATTCCACGATATGGA-3′) and a screening primer (5′-GCTTTTCTGTGACTGGTGAGGCCTCAACCAAGTCATTCTGAG-3′). All the constructs mentioned above were verified by double-stranded DNA sequencing.
Transient and Stable Cell Transfection.
HEK293 cells with stable expression of HA-tagged ABCG2 were established in previous studies9,11 and were cultured at 37 °C with 5% CO2 in DMEM, supplemented with 10% fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin, and 0.3 mg/mL G418. For transient transfection, the pcDNA 3.1(+) plasmid containing different Myc-tagged ABCG2 sequences or the vector control was transfected into HEK293 cells or HEK293 cells with stable expression of HA-tagged ABCG2 at 90% confluency using LipofectAMINE according to the manufacturer’s instructions. The cells were harvested for experiments 24 h after transient transfection.
To establish stable clones with expression of various Myc-tagged ABCG2 constructs, we first transfected the pcDNA 3.1(+) plasmid containing different Myc-tagged ABCG2 constructs or the vector control into HEK293 cells using LipofectAMINE according to the manufacturer’s instructions. Forty-eight hours following transfection, the transfected cells were diluted and selected with 0.8 mg/mL G418 for 3 weeks. Stable clones with expression of Myc-tagged ABCG2 were verified by Western blot and were maintained in the presence of 0.3 mg/mL G418.
Chemical Cross-Linking, Cell Lysate, and Plasma Membrane Preparations.
Chemical cross-linking using DSS was performed as previously described.9,15 Briefly, confluent cells in 150 mm dishes were washed three times with KCl/Hepes buffer [90 mM KCl and 50 mM Hepes (pH 7.5)] and incubated with 2 mM DSS in the KCl/Hepes buffer for 45 min at room temperature. Tris-HCl (pH 7.4) was then added to a final concentration of 2 mM to quench the reaction. The cells were then collected for plasma membrane preparation as described previously with minor modifications.9,13 Briefly, cells were washed with ice-cold PBS and resuspended in hypotonic buffer [10 mM KCl, 1.5 mM MgCl2, 10 mM Tris (pH 7.4), and 2 mM PMSF] at a density of 106 cells/mL followed by homogenization and centrifugation at 1000g for 10 min. Plasma membrane fractions were prepared by layering the 1000g supernatant on top of a 35% sucrose cushion containing 10 mM Tris (pH 7.4) and 1 mM EDTA, followed by centrifugation at 100000 g for 1 h. The membranes at the interface between the supernatant and the sucrose cushion were collected, mixed with STBS buffer [250 mM sucrose, 150 mM NaCl, and 10 mM Tris (pH 7.5)], and pelleted by centrifugation. The final membrane pellets were resuspended in STBS buffer and stored at –80 °C.
For the preparation of lysates, cells were harvested and lysed in ice-cold lysis buffer [150 mM NaCl, 25 mM Tris (pH 7.4), 1 mM EDTA, and 1% Triton X-100, with freshly added 2 mM PMSF and 1 mM DTT before use] and then passed through 26 gauge needles and incubated on ice for 30 min. The lysates were then centrifuged at 12000g for 15 min. Protein concentrations were determined with the Bio-Rad protein assay kit.
Co-Immunoprecipitation and Western Blot Analyses.
Co-immunoprecipitation (Co-IP) was performed as previously described.9,15 Briefly, 500 μg of fresh cell lysates was mixed with 2 μg of normal mouse IgG, diluted to 1 mL with ice-cold lysis buffer, and incubated for 2 h at 4 °C, followed by addition of 40 μL of protein G-PLUS agarose beads and incubation for 2 h at 4 °C. After centrifugation at 500g for 1 min to preclear the lysate, the supernatants were transferred to new tubes and precipitated with monoclonal anti-Myc or anti-HA antibodies for 4 h at 4 °C. After centrifugation at 10000g for 15 min at 4 °C, the supernatants were transferred to fresh tubes and incubated with 40 μL of protein G-PLUS agarose beads overnight at 4 °C with constant agitation. Agarose beads were then collected by centrifugation and washed five times with lysis buffer before being used for Western blot analysis as previously described.9
Indirect Immunofluorescence Imaging.
Indirect immunofluorescence imaging was performed as previously described.13,16,17 Briefly, HEK293 cells transfected with various ABCG2 constructs were cultured on coverslips and washed with phosphate-buffered saline, fixed with an acetone/methanol mixture, blocked with bovine serum albumin, and probed with an anti-Myc antibody at room temperature for 1 h followed by incubation with an FITC-conjugated secondary antibody at room temperature for 30 min. The coverslips were then mounted on slides before being viewed with a confocal microscope.
MTT and Drug Accumulation Assays.
The MTT assay was performed as previously described9 using different concentrations of adriamycin and mitoxantrone. EC50 is defined as the concentration of the drug required to kill 50% of the cells in the control condition without any drugs. Relative resistance factors were determined by dividing median EC50 values of stable clones with expression of ABCG2Myc-WT by that of cell clones transfected with other constructs.
The drug accumulation assay was performed as previously described11,18 with minor modifications. Briefly, 106 cells were trypsinized, resuspended in 1 mL of DMEM with 20 μM mitoxantrone, and incubated at 37 °C for 30 min. Cells were then collected by centrifugation at 500g and washed three times with ice-cold DMEM. The cells were then resuspended in 1 mL of DMEM and subjected to analysis by flow cytometry using a BD FACSCalibur APC Analyzer. The data were analyzed using Cell Quest Pro (BD Biosciences).
RESULTS
ECL3 Has Oligomerization Activity.
Previously, it was found that the oligomerization activity of ABCG2 is located in its C-terminal domain consisting of three segments, TM5, ECL3, and TM6.11 To determine which segments of this domain have oligomerization activity, we first engineered a construct by replacing ECL1 with ECL3 in the Myc-tagged ABCG2Myc-TM1–2 construct that has previously been shown to have no oligomerization activity11 (Figure 1B). The newly created construct, ABCG2Myc-TM1–2/L1*, was then transfected into HEK293 cells that stably express HA-tagged full-length ABCG2 (ABCG2HA-WT) to determine if it interacts with ABCG2HA-WT using Co-IP with an anti-Myc antibody followed by Western blot analysis. As shown in panels C and D of Figure 1, ABCG2Myc-TM1–2/L1* successfully coprecipitates with ABCG2HA-WT, like the positive control ABCG2Myc-TM5–6, which has been shown previously to interact with ABCG2HA-WT 11 On the other hand, the negative control construct ABCG2Myc-TM1–2 failed to coprecipitate with ABCG2HA-WT as expected (Figure 1C,D). Thus, replacing ECL1 with ECL3 in ABCG2Myc-TM1–2 generated oligomerization activity in this construct. These findings suggest that ECL3 may contain oligomerization activity.
To further confirm the oligomerization activity of ECL3 and to eliminate the potential problem of using truncated constructs in the studies described above, we engineered a new construct by replacing TM5 and TM6 with TM1 and TM2, respectively, in full-length ABCG2 (Figure 2A). As discussed above, the TM1—ECL1—TM2 domain has been shown previously to have no oligomerization activity.11 The new construct, ABCG2Myc-TM5*6*, was then transiently transfected into HEK293 cells with stable expression of ABCG2HA-WT followed by Co-IP using the HA or Myc antibody and Western blot analysis using both Myc and HA antibodies. Panels B—D of Figure 2 show that ABCG2Myc-TM5*6*, like wild-type ABCG2 (ABCG2Myc-WT), coprecipitates with ABCG2HA-WT. However, the negative control ABCG2 without the TM5-ECL3-TM6 domain (ABCG2Myc-TM1–4) does not coprecipitate with ABCG2HA-WT, as we previously demonstrated.11 This finding confirms our conclusion that ECL3 likely contains oligomerization activity.
Figure 2.
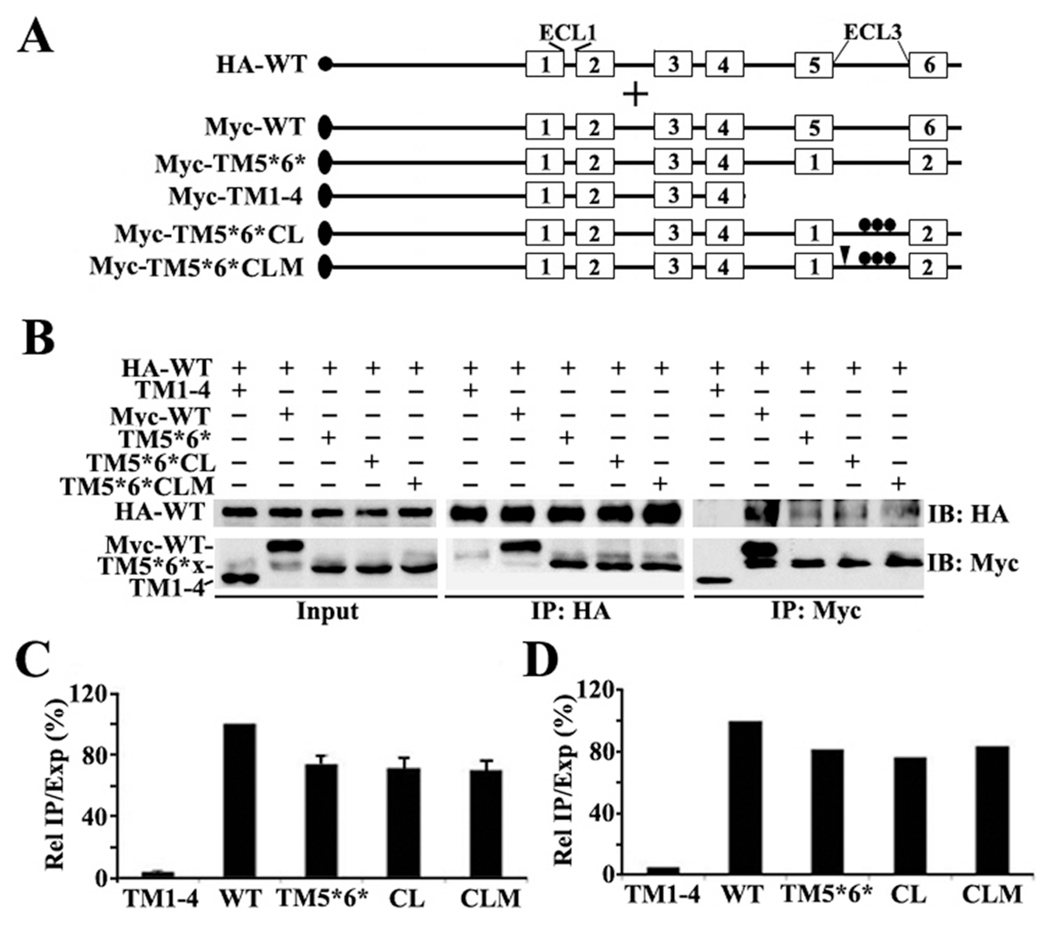
(A) Schematic presentation of HA-tagged full-length and Myc-tagged ABCG2 constructs. The triplet dots represent cysteine mutations, while the arrowheads indicate mutation of the QXXS motif. Other symbols are the same as described for Figure 1. (B) Co-IP. HEK293 cells with stable expression of ABCG2HA-WT were transiently transfected with ABCG2Myc-TM1–4, ABCG2Myc-WT, ABCG2Myc-F-TM5*6*, ABCG2Myc-F-TM5*6*CL, or ABCG2Myc-F-TM5*6*CLM followed by immunoprecipitation with the HA or Myc antibody and Western blot analysis of the precipitates with both HA and Myc antibodies. (C and D) Quantitative analysis of Co-IP. The expression and Co-IP level of constructs from three independent experiments as shown in panel B were quantified using ScnImage and calculation of the relative ratio of Co-IP to the expression level followed by normalization to that of the wild-type positive control construct.
The Conserved Cysteine Residues and the Putative QYFS Motif in ECL3 Are Not Essential for the Oligomerization Activity of ECL3.
It has been shown previously that the conserved cysteine residues in ECL3 of ABCG2, Cys603, Cys592, and Cys608 (Figure S1 of the Supporting Information), are responsible for the formation of inter- and intramolecular disulfide bonds and, thus, possibly dimerization of ABCG2.19,20 To investigate if the oligomerization activity of ECL3 observed using Co-IP in the experiments described above is possibly due to formation of an intermolecular disulfide bond, we created Cys-less ECL3 in the ABCG2Myc-TM5*6* construct by site-directed mutagenesis. This new construct, ABCG2Myc-TM5*6*CL (Figure 2A), was then tested for its interaction with ABCG2HA-WT using the same method that was used for ABCG2Myc-TM5*6*. As shown in Figure 2B–D, ABCG2Myc-TM5*6*CL, compared with ABCG2Myc-TM5*6*, could coprecipitate equally well with ABCG2HA-WT. Thus, the conserved cysteine residues in ECL3 may not contribute to the observed oligomerization activity of ECL3. This finding is consistent with our previous observation that the cysteine residues in ECL3 do not contribute to ABCG2 oligomerization.9
Sequence analysis of ECL3 also shows a conserved QXXS motif [569QYFS (see Figure S1 of the Supporting Information)], which has been shown to facilitate interactions between membrane proteins.21 To determine if this motif is possibly responsible for the oligomerization activity of ECL3, we mutated the 569QYFS motif to 569AYFA in the Cys-less mutant construct ABCG2Myc-TM5*6*CL and created a new one named ABCG2Myc-TM5*6*CLM (Figure 2A). This construct was then tested for its ability to coprecipitate with ABCG2HA-WT as described above. Panels B—D of Figure 2 show that elimination of the motif by mutation had no effect on its ability to oligomerize. Thus, the putative 569QYFS motif is unlikely to be involved in ABCG2 oligomerization.
TM5 and TM6 Also Contain Oligomerization Activity.
Next, we determined if TM5 and TM6 also have oligomerization activity. For this purpose, we first engineered a construct by replacing ECL3 with ECL1 in full-length ABCG2 and created ABCG2Myc-ECL3* (Figure 3A). ABCG2Myc-ECL3*, along with the negative control construct ABCG2Myc-TM1–4 and the positive control construct ABCG2Myc-WT, was then transiently transfected into HEK293 cells with stable expression of ABCG2HA-WT followed by Co-IP and Western blot analysis as described above. As shown in Figure 3B–D, ABCG2Myc-ECL3*, like ABCG2Myc-WT, coprecipitates the ABCG2HA-WT, whereas the negative control construct does not, suggesting that TM5 and TM6 likely also contain oligomerization activity.
Figure 3.
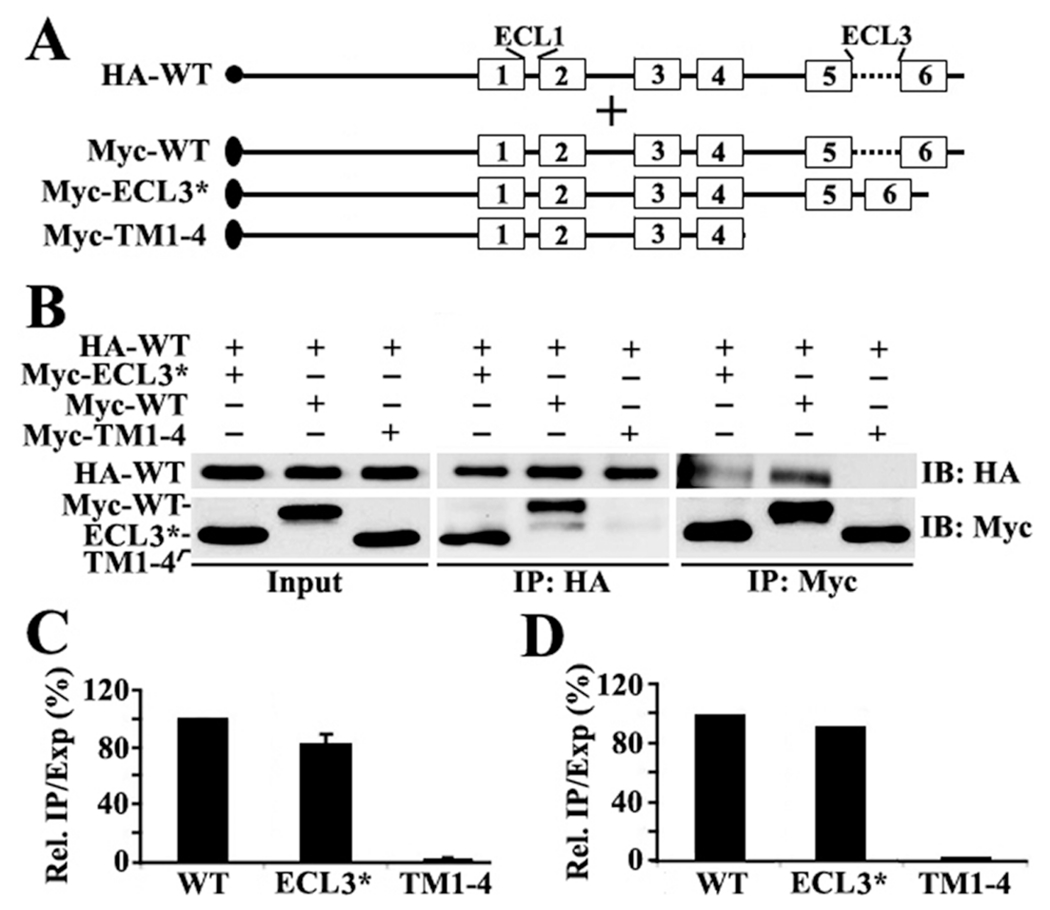
(A) Schematic presentation of HA-tagged full-length and Myc-tagged ABCG2 constructs. The symbols are the same as described for Figure 1. (B) Co-IP. HEK293 cells with stable expression of ABCG2HA-WT were transiently transfected with ABCG2Myc-ECL3*, ABCG2Myc-WT, or ABCG2Myc-TM1–4 followed by immunoprecipitation with the HA or Myc antibody and Western blot analysis of the precipitates with both HA and Myc antibodies. (C and D) Quantitative analysis of Co-IP. The expression and Co-IP level of constructs from three independent experiments as shown in panel B were quantified using ScnImage and calculation of the relative ratio of Co-IP to the expression level followed by normalization to that of the wild-type positive control construct.
We further dissected TM5 and TM6 to determine if both are equally important by creating two more constructs, ABCG2Myc-TM5*L3* and ABCG2TM6*L3* (Figure 4A), by replacing the TM5—ECL3 and ECL3—TM6 domains with TM1—ECL1 and ECL1—TM2 domains, respectively. These constructs along with negative control ABCG2Myc-TM1—4 and positive control ABCG2Myc-WT were tested for their ability to interact with HA-tagged full-length ABCG2 as described above. Panels B—D of Figure 4 show that both constructs have only ~50% of the oligomerization activity of wild-type ABCG2 (ABCG2Myc-WT). Thus, both TM5 and TM6 may each contribute ~50% of the oligomerization activity.
Figure 4.
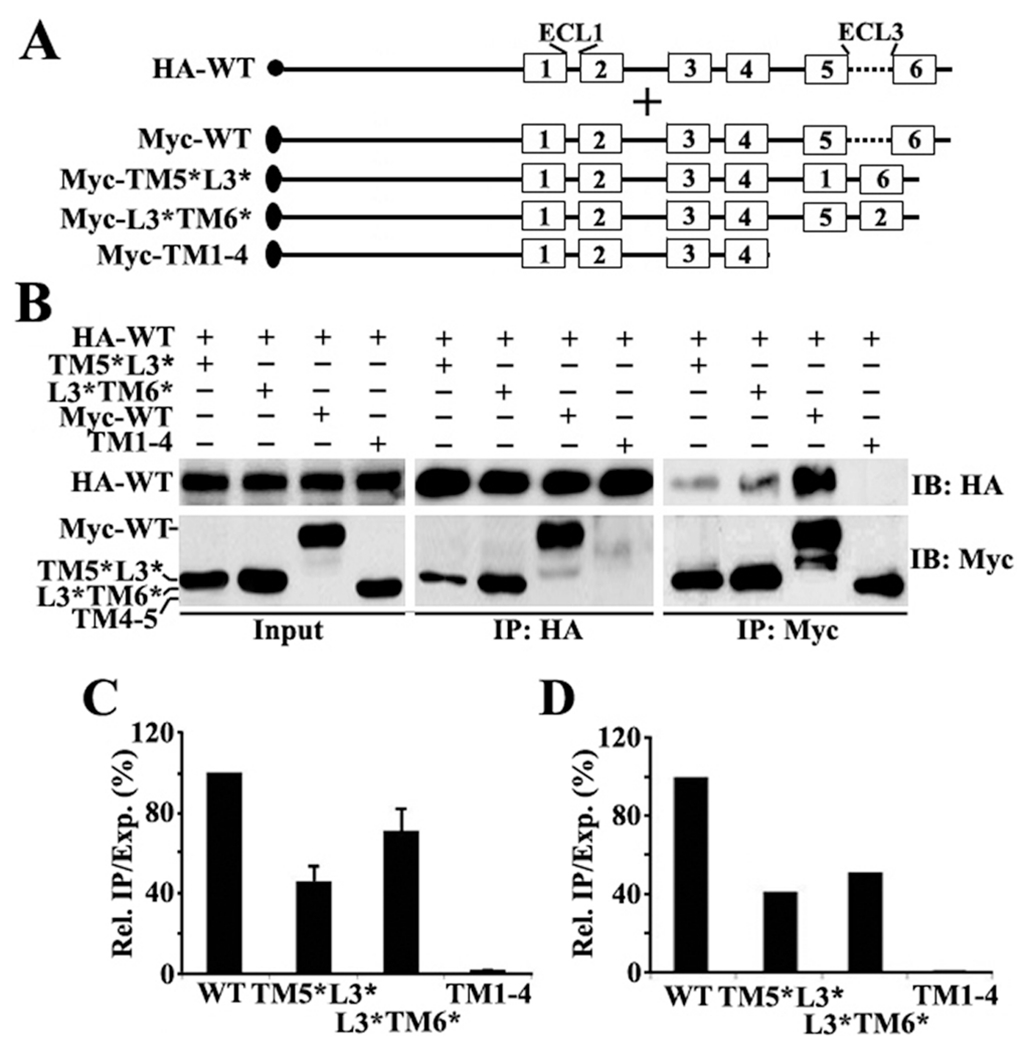
(A) Schematic presentation of HA-tagged full-length and Myc-tagged ABCG2 constructs. The symbols are the same as described for Figure 1. (B) Co-IP. HEK293 cells with stable expression of ABCG2HA-WT were transiently transfected with ABCG2Myc-TM5*L3*, ABCG2Myc-L3*TM6*, ABCG2Myc-WT, or ABCG2Myc-TM1–4 followed by immunoprecipitation with the HA or Myc antibody and Western blot analysis of the precipitates with both HA and Myc antibodies. (C and D) Quantitative analysis of Co-IP. The expression and Co-IP level of constructs from three independent experiments as shown in panel B were quantified using ScnImage and calculation of the relative ratio of Co-IP to the expression level followed by normalization to that of the wild-type positive control construct.
Previously, we found that the TM segments involved in ABCC1 dimerization are hydrophobicity-dependent.22 To determine if the hydrophobicity of TM5 and TM6 is also possibly important for their oligomerization activity and replacing them with TM1 and TM2 may change the hydrophobicity and, thus, oligomerization activity, we performed a sequence analysis of TM5 and TM6 as well as their respective replacements TM1 and TM2. As shown in Table 1, while replacing TM5 with TM1 does not change the hydrophobicity, replacing TM6 with TM2 drastically reduces the hydrophobicity. However, both replacements generated an ~50% reductions in ABCG2 oligomerization activity. Thus, it is possible that the hydrophobicity of TM5 and TM6 may not play important roles in their oligomerization activity.
Table 1.
GRAVY Scores of TM Segments of ABCG2
| TM | no. of amino acids | sequence | GRAVY scorea |
|---|---|---|---|
| TM1 | 21 | IAQIIVTVVLGLVIGAIYFGL | 2.400 |
| TM5 | 21 | VATLLMTICFVFMMIFSGLLV | 2.505 |
| TM2 | 20 | LFFLTTNQCFSSVSAVELFV | 1.260 |
| TM6 | 21 | VALACMIVIFLTIAYLKLLFL | 2.581 |
GRAVY (grand average of hydropathicity) indicates the solubility of the proteins29 (http://www.expasy.ch/tools/protparam.html).
ECL3 Is Not Essential for Supporting ABCG2 Function.
To determine the importance of each segment of the TM5—ECL3—TM6 domain in ABCG2 function, we first established stable cell lines transfected with vector control, ABCG2Myc-WT, ABCG2Myc-TM5*6*, and ABCG2Myc-ECL3* (Figure S2 of the Supporting Information) and tested their ability to resist mitoxantrone and doxorubicin treatments using the MTT assay. All ABCG2 mutants appear to be localized on plasma membranes as determined using immunofluorescence staining (Figure S3 of the Supporting Information). Panels A and B of Figure 5 show that the relative resistance factor of ABCG2Myc-TM5*6* following normalization to its expression level is dramatically reduced compared to that of ABCG2Myc-WT. However, the function of ABCG2Myc-ECL3* did not significantly change. Next, we performed a mitoxantrone accumulation assay using flow cytometry. As shown in Figure 6, ABCG2Myc-ECL3* can significantly reduce the level of mitoxantrone accumulation, like ABCG2Myc-WT, whereas ABCG2Myc-TM5*6* cannot. Hence, it is possible that TM5 and TM6 are functionally important whereas ECL3 is not.
Figure 5.
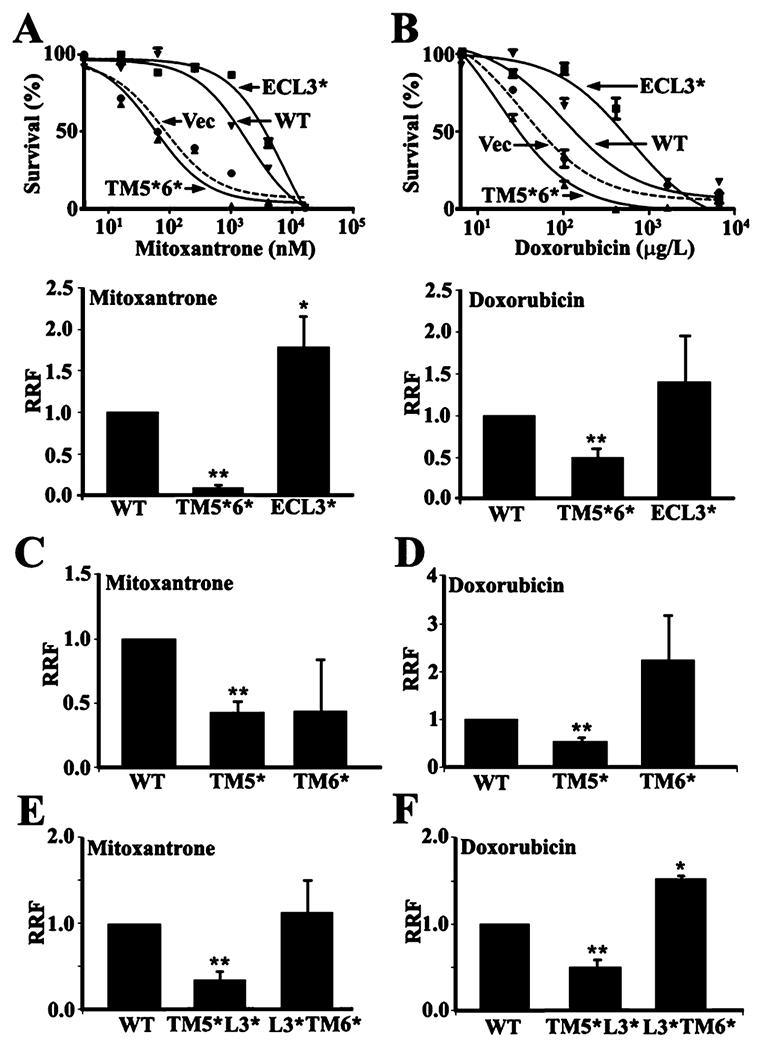
Drug resistance function of wild-type and mutant ABCG2. HEK293 cells with stable expression of ABCG2Myc-WT, ABCG2Myc-ECL3*, ABCG2Myc-TM5*6*, and vector-transfected control (Vec) were subjected to treatment with mitoxantrone (A) and doxorubicin (B) followed by the MTT assay for relative resistance factors (RRF) as described in Materials and Methods. (C—F) Analysis of RRF to mitoxantrone (C and E) and doxorubicin (D and F) of HEK293 cells with stable expression of ABCG2Myc-TM5* and ABCG2Myc-TM6* (C and D) or ABCG2Myc-TM5*L3* and ABCG2Myc-L3*TM6* (E and F) using the MTT assay as shown in panels A and B. **p < 0.01.
Figure 6.
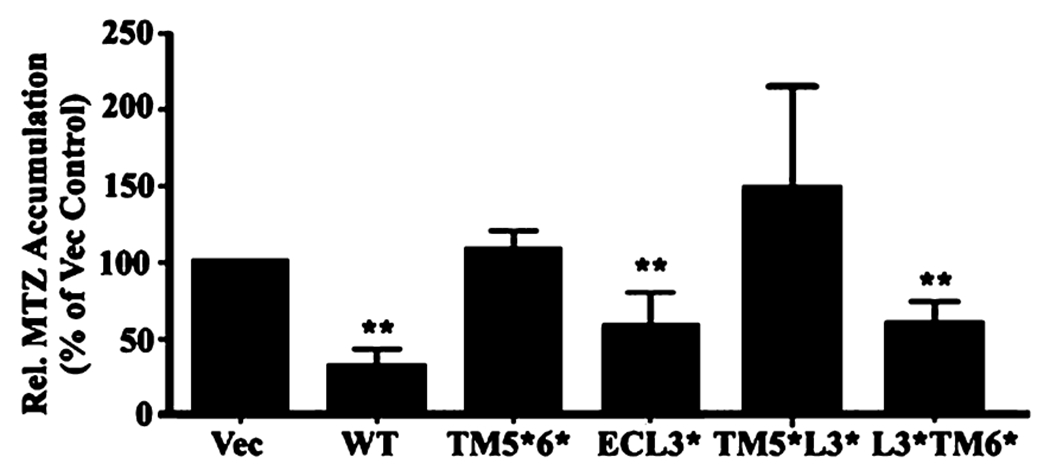
Effect of mutations in ABCG2 on drug efflux function. HEK293 cells with stable expression of ABCG2Myc-WT, ABCG2Myc-TM5*6*, ABCG2Myc-ECL3*, ABCG2Myc-TM5*L3*, ABCG2Myc-L3*TM6*, and vector-transfected control cells (Vec) were subjected to mitoxantrone (MTZ) accumulation analysis using flow cytometry as described in Materials and Methods. The relative accumulation of mitoxantrone was calculated after normalization to the expression level of ABCG2 and then to that of the vector-transfected control from three independent experiments. **p < 0.01.
TM5 Is a Major Contributor of ABCG2 Function.
To further determine the importance of TM5 and TM6 in ABCG2 function and if they make equal contributions, stable HEK293 cell lines transfected with ABCG2Myc-TM5* and ABCG2Myc-TM6* were established and tested for their drug resistance function using mitoxantrone and doxorubicin in the MTT assay. Panels C and D of Figure 5 show that the drug resistance function of ABCG2Myc-TM5* is significantly reduced when compared with that of ABCG2Myc-WT. However, the drug resistance function of ABCG2Myc-TM6* was not significantly reduced versus that of ABCG2Myc-WT. Similar observations were also made with stable cell lines transfected with ABCG2Myc-TM5*L3* and ABCG2Myc-L3*TM6* (Figure 5E,F). Using the drug accumulation assay, we also found that replacing TM5 significantly compromised ABCG2 activity in eliminating the accumulation of mitoxantrone in HEK293 cells (Figure 6). Thus, TM5 may be more important than TM6 in ABCG2 function, although they both play similar roles in ABCG2 oligomerization.
All Mutant Forms of ABCG2 Can Form Homo-Oligomers.
Because replacing TM5 appears to significantly reduce ABCG2 function and reduce its ability to interact with wild-type ABCG2, it is possible that ABCG2 missing the correct TM5 could not form correct homo-oligomeric complexes. To test this possibility, we next determined if the mutant ABCG2 with alterations in TM5, ECL3, and TM6 can form homo-oligomers with itself. For this purpose, we performed chemical cross-linking of live HEK293 cells expressing ABCG2Myc-WT, ABCG2Myc-ECL3*, ABCG2Myc-TM5*6*, ABCG2Myc-TM5*, ABCG2Myc-TM6*, ABCG2Myc-L3*TM6*, or ABCG2Myc-TM5*L3* followed by Western blot analysis of isolated membranes as previously described.9,15 Figure 7 shows that oligomeric ABCG2 molecules can be detected for all wild-type and mutant ABCG2 molecules, although the distribution of cross-linked oligomeric ABCG2 is different between the wild-type and mutant molecules (Table 2). All ABCG2 mutants could successfully form homo-oligomeric complexes, and there is no correlation between formation of oligomers and function of ABCG2 (Table 2). Thus, the loss of function in TM5 mutants may not be due to its loss of formation of homo-oligomeric complexes. These observations also suggest that the existence of any single element in the TM5—ECL3—TM3 domain is enough to support formation of homo-oligomeric ABCG2 complexes with itself.
Figure 7.
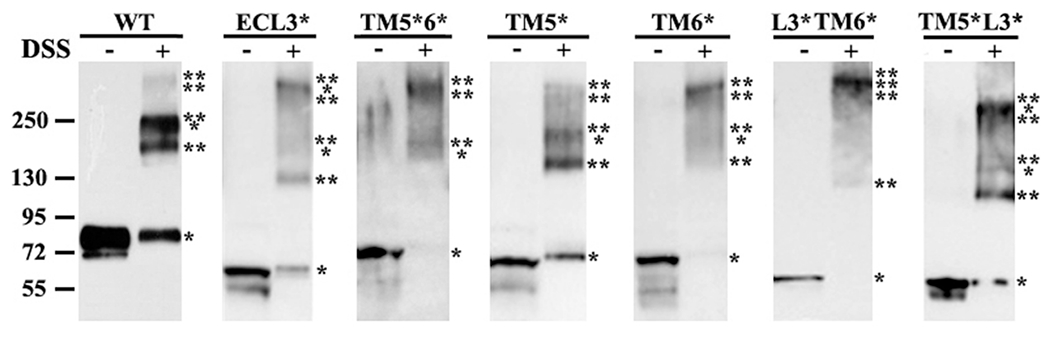
Chemical cross-linking of wild-type and mutant ABCG2. HEK293 cells with expression of ABCG2Myc-WT, ABCG2Myc-ECL3*, ABCG2Myc-TM5*6*, ABCG2Myc-TM5*, ABCG2Myc-TM6*, ABCG2Myc-L3*TM6*, and ABCG2Myc-TM5*L3* were treated without or with DSS followed by isolation of plasma membranes and Western blot analysis of ABCG2 using the Myc antibody. The molecular weight of each cross-linked oligomer was estimated on the basis of linear regression of native protein markers used. The number of asterisks indicates the number of subunits cross-linked.
Table 2.
Correlation between Oligomerization and Function and Distribution of Cross-Linked Oligomeric Complexes
| RRFb |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| ABCG2 construct | Co-IP with WT | monomera Mr (kDa) (%) | dimera Mr (kDa) (%) | trimera Mr (kDa) (%) | tetramera Mr (kDa) (%) | pentamear Mr (kDa) (%) | hexamera Mr (kDa) (%) | MTZc | DOXd |
| WT | 100% | 83 (30%) | 186 (27%) | 232 (37%) | 333 (6%) | 1.0 | 1.0 | ||
| ECL3* | 80–90% | 61 (20%) | 137 (26%) | 181 (11%) | 315 (43%) | 1.9 | 1.4 | ||
| TM5*6* | ~80% | 73 (3%) | 186 (38%) | 307 (59%) | 0.1 | 0.5 | |||
| TM5* | ND | 69 (27%) | 158 (31%) | 202 (32%) | 298 (10%) | 0.4 | 0.6 | ||
| TM6* | ND | 69 (4%) | 158 (18%) | 202 (29%) | 298 (49%) | 0.4 | 2.3 | ||
| L3*TM6* | 50–70% | 57 (0%) | 134 (11%) | 333 (89%) | 1.1 | 1.5 | |||
| TM5*L3* | 40–45% | 55 (12%) | 120 (29%) | 158 (8%) | 260 (51%) | 0.5 | 0.5 | ||
Derived from chemical cross-linking.
RRF is the relative resistance factor.
MTZ is mitoxantrone.
DOX is doxorubicin.
DISCUSSION
ABCG2 has previously been shown to exist as a high-order homo-oligomer, and the oligomerization activity is located in a domain including TM5, ECL3, and TM6.6 In this study, we further dissected this domain and investigated the contribution of each segment in the TM5—ECL3—TM6 domain to ABCG2 oligomerization and function. We found that while ECL3, TM5, and TM6 each have oligomerization activity, each segment alone is not essential for ABCG2 oligomerization. The existence of any single segment is sufficient to support ABCG2 oligomerization. However, TM5 is essential for ABCG2 function, whereas TM6 and ECL3 are replaceable.
ECL3 is the longest extracellular loop of ABCG2 with potential N-linked glycosylation sites and cysteine residues involving formation of potential intra- and intermolecular disulfide bonds.14,23 While ECL3 has full oligomerization activity and its presence is sufficient to support ABCG2 oligomerization, the cysteine residues in ECL3 are not essential for ABCG2 oligomerization. This observation is consistent with our previous finding that the formation of higher-order oligomers is not dependent on disulfide bond formation.9 These cysteine residues have also been shown to be unessential for ABCG2 function.14
The short polar QXXS motif was first identified in the bacterial Tar-1 homodimer TM domain and has been found to be sufficient for inducing stable TM—TM interactions.24 The two polar residues (Q and S) are crucial for the dimerization of the Tar-1 TM domain in vivo, creating symmetric hydrogen bonds that promote and/or stabilize dimeric Tar-1. Substitution of these two polar residues with nonpolar residues markedly impaired the self-association ability of the Tar-1 TM domain. The QXXS motif is common in TMs of bacterial membrane proteins, suggesting a general role for this motif in TM assembly.21 However, the existence of this motif (569QYFS) in ECL3 of ABCG2 does not appear to contribute to the oligomerization activity of ABCG2. This observation may be due to the existence of this motif in the hydrophilic loop but not in the TM segment of ABCG2.
Although both TM5 and TM6 contribute equally to ABCG2 oligomerization in the Co-IP assay with wild-type ABCG2, TM5 and TM6 together have more activity than each does alone. Because any dimerization between the wild-type and mutant ABCG2 could be detected using the Co-IP study, TM5 and TM6 may be important in dimerization as each one contributes to ~50% of their oligomerization activity. On the other hand, ABCG2 mutants with TM5, TM6, or both replaced with TM1 and TM2, respectively, could be efficiently cross-linked, suggesting that the mutant may have a higher affinity for itself than for the wild-type protein and that TM5 and TM6 may not be as important in oligomerization as in dimerization. It is also tempting to speculate that TM5 in one molecule may interact with TM5 in another while TM6 interacts with TM6 (Figure 8). It can be further suggested that ECL3 in one molecule may interact with ECL3 in another. This arrangement of interactions fit well with the dodecameric complex model.9 Currently, it is unknown if the oligomerization activity of TM5 and TM6 is sequence-dependent. However, their oligomerization activities do not appear to depend on the hydrophobicity of these TM segments, which is different from the TM segments involved in ABCC1 dimerization.15
Figure 8.
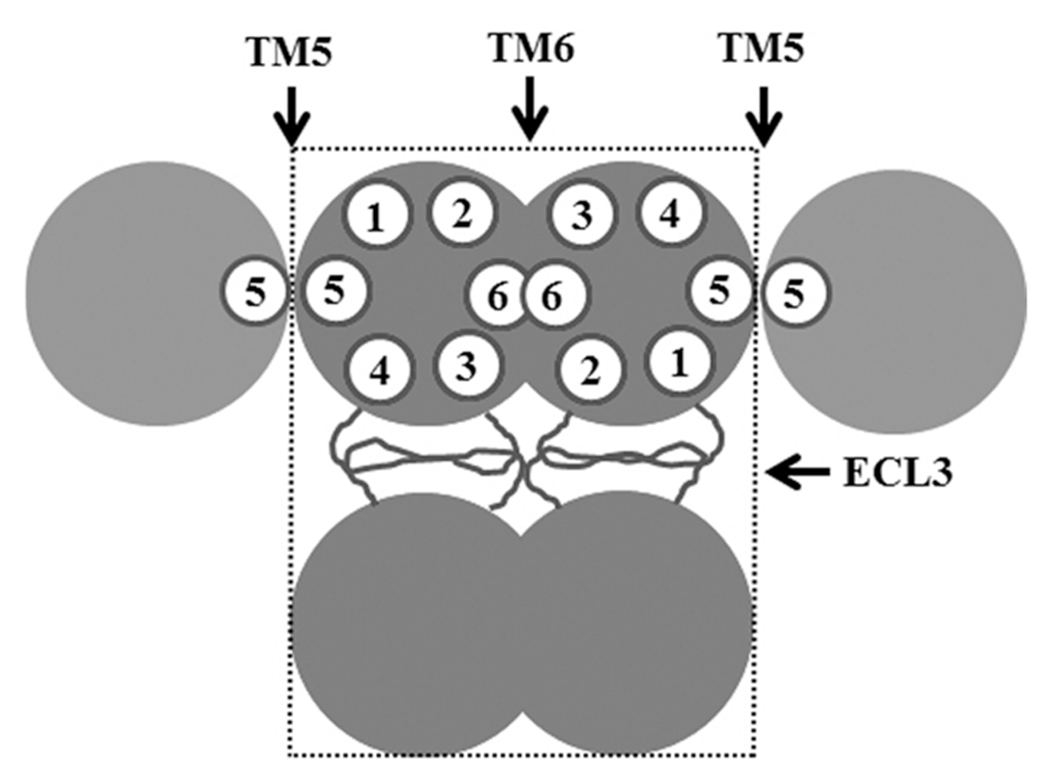
Schematic model of ABCG2 interactions. Three different possible interaction sites contributed by TM5, TM6, and ECL3 are shown. The minimal stable tetrameric unit is shown in a box with dashed lines. The empty circles with numbers represent TM segments.
While the exact drug-binding sites in ABCG2 are unknown, early studies have suggested that the interactions of the substrate with ABCG2 involve multiple binding sites in the protein.25 For example, Arg482 has been shown to affect substrate specificity.26 All our constructs have the same R482G mutation that provides a gain of function in recognition of more substrates. In this study, the only mutant that shows a significant loss of function is replacement of TM5 with TM1, suggesting that TM5 may be important in substrate recognition and binding. This conclusion is consistent with previous reports that TM5 contains several amino acids that are essential for the transport activity of ABCG2. For example, it has been shown that the L554P mutation in TM5 reduces the drug resistance function of ABCG2.27 TM5 has also been found to contain a steroid binding element,28 further suggesting that TM5 may be important in substrate recognition and binding. It is unknown if replacing TM5 with TM1 significantly affects ABCG2 conformation, which results in a loss of function. However, the finding that the mutant can form homo-oligomers with itself in chemical cross-linking studies although replacing TM5 with TM1 weakened the ability of the mutant to oligomerize with wild-type ABCG2, suggests that a major conformational change may not have occurred. Furthermore, replacing TM6 with TM2 did not affect ABCG2 function and also reduced its oligomerization activity with wild-type ABCG2. In addition, replacing ECL3 with ECL1 has no effect on ABCG2 function or oligomerization, which further suggests that the mutations likely have no significant impact on ABCG2 conformation. Thus, we conclude that the functional effect of replacement of TM5 with TM1 is unlikely due to its effect on oligomerization or conformation.
Supplementary Material
Funding
This work was supported in part by National Institutes of Health Grant CA120221 (J.-T.Z.). W.M. was the recipient of a predoctoral fellowship from the Department of Defense Breast Cancer Research Program (W81XWH-08-1-0228). J.Q. was the recipient of a postdoctoral fellowship from the Department of Defense Breast Cancer Research Program (W81XWH-09-1-0489).
Footnotes
ASSOCIATED CONTENT
Supporting Information
Primers for construct engineering, sequence alignment of ECL3 of ABCG2, and Western blot and immunofluorescence analyses of wild-type and mutant ABCG2 in stable cell lines. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).Gottesman MM, Fojo T, and Bates SE (2002) Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2, 48–58. [DOI] [PubMed] [Google Scholar]
- (2).Gottesman MM, Ambudkar SV, Ni B, Aran JM, Sugimoto Y, Cardarelli CO, and Pastan I (1994) Exploiting multidrug resistance to treat cancer. Cold Spring Harbor Symp. Quant. Biol 59, 677–683. [DOI] [PubMed] [Google Scholar]
- (3).Gillet JP, and Gottesman MM (2010) Mechanisms of multidrug resistance in cancer. Methods Mol. Biol 596, 47–76. [DOI] [PubMed] [Google Scholar]
- (4).Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, and Gottesman MM (2006) Targeting multidrug resistance in cancer. Nat. Rev. Drug Discovery 5, 219–234. [DOI] [PubMed] [Google Scholar]
- (5).Zhang JT (2007) Biochemistry and pharmacology of the human multidrug resistance gene product, ABCG2. Zhongnan Daxue Xuebao, Yixueban 32, 531–541. [PubMed] [Google Scholar]
- (6).Xu J, Peng H, and Zhang JT (2007) Human multidrug transporter ABCG2, a target for sensitizing drug resistance in cancer chemotherapy. Curr. Med. Chem. 14, 689–701. [DOI] [PubMed] [Google Scholar]
- (7).Mo W, and Zhang JT (2012) Human ABCG2: Structure, function, and its role in multidrug resistance. Int. J. Biochem. Mol. Biol. 3, 1–27. [PMC free article] [PubMed] [Google Scholar]
- (8).Doyle LA, Yang W, Abruzzo LV, Krogmann T, Gao Y, Rishi AK, and Ross DD (1998) A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. U.S.A 95, 15665–15670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Xu J, Liu Y, Yang Y, Bates S, and Zhang JT (2004) Characterization of oligomeric human half-ABC transporter ATP-binding cassette G2. J. Biol. Chem 279, 19781–19789. [DOI] [PubMed] [Google Scholar]
- (10).McDevitt CA, Collins RF, Conway M, Modok S, Storm J, Kerr ID, Ford RC, and Callaghan R (2006) Purification and 3D Structural Analysis of Oligomeric Human Multidrug Transporter ABCG2. Structure 14, 1623–1632. [DOI] [PubMed] [Google Scholar]
- (11).Xu J, Peng H, Chen Q, Liu Y, Dong Z, and Zhang JT (2007) Oligomerization domain of the multidrug resistance-associated transporter ABCG2 and its dominant inhibitory activity. Cancer Res 67, 4373–4381. [DOI] [PubMed] [Google Scholar]
- (12).Zhang JT, Chen M, Han E, and Wang C (1998) Dissection of de novo membrane insertion activities of internal transmembrane segments of ATP-binding-cassette transporters: Toward understanding topological rules for membrane assembly of polytopic membrane proteins. Mol. Biol. Cell 9, 853–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Yang Y, Chen Q, and Zhang JT (2002) Structural and functional consequences of mutating cysteine residues in the amino terminus of human multidrug resistance-associated protein 1. J. Biol. Chem 277, 44268–44277. [DOI] [PubMed] [Google Scholar]
- (14).Liu Y, Yang Y, Qi J, Peng H, and Zhang JT (2008) Effect of cysteine mutagenesis on the function and disulfide bond formation of human ABCG2. J. Pharmacol. Exp. Ther 326, 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yang Y, Liu Y, Dong Z, Xu J, Peng H, Liu Z, and Zhang JT (2007) Regulation of function by dimerization through the amino-terminal membrane-spanning domain of human ABCC1/MRP1. J. Biol. Chem 282, 8821–8830. [DOI] [PubMed] [Google Scholar]
- (16).Chen Q, Yang Y, Li L, and Zhang JT (2006) The Amino Terminus of the Human Multidrug Resistance Transporter ABCC1 Has a U-shaped Folding with a Gating Function. J. Biol. Chem 281, 31152–31163. [DOI] [PubMed] [Google Scholar]
- (17).Yang Y, Li Z, Mo W, Ambadipudi R, Arnold RJ, Hrncirova P, Novotny MV, Georges E, and Zhang JT (2012) Human ABCC1 Interacts and Colocalizes with ATP Synthase α, Revealed by Interactive Proteomics Analysis. J. Proteome Res 11, 1364–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Peng H, Dong Z, Qi J, Yang Y, Liu Y, Li Z, Xu J, and Zhang JT (2009) A Novel Two Mode-Acting Inhibitor of ABCG2-Mediated Multidrug Transport and Resistance in Cancer Chemotherapy. PLoS One 4, e5676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wakabayashi K, Nakagawa H, Adachi T, Kii I, Kobatake E, Kudo A, and Ishikawa T (2006) Identification of cysteine residues critically involved in homodimer formation and protein expression of human ATP-binding cassette transporter ABCG2: A new approach using the flp recombinase system. J. Exp. Ther. Oncol 5, 205–222. [PubMed] [Google Scholar]
- (20).Kage K, Fujita T, and Sugimoto Y (2005) Role of Cys-603 in dimer/oligomer formation of the breast cancer resistance protein BCRP/ABCG2. Cancer Sci 96, 866–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Sal-Man N, Gerber D, and Shai Y (2005) The identification of a minimal dimerization motif QXXS that enables homo- and hetero-association of transmembrane helices in vivo. J. Biol. Chem 280, 27449–27457. [DOI] [PubMed] [Google Scholar]
- (22).Yang Y, Mo W, and Zhang JT (2010) Role of transmembrane segment 5 and extracellular loop 3 in the homodimerization of human ABCC1. Biochemistry 49, 10854–10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ni Z, Mark ME, Cai X, and Mao Q (2010) Fluorescence resonance energy transfer (FRET) analysis demonstrates dimer/oligomer formation of the human breast cancer resistance protein (BCRP/ABCG2) in intact cells. Int. J. Biochem. Mol. Biol 1, 1–11. [PMC free article] [PubMed] [Google Scholar]
- (24).Sal-Man N, Gerber D, and Shai Y (2004) The composition rather than position of polar residues (QxxS) drives aspartate receptor transmembrane domain dimerization in vivo. Biochemistry 43, 2309–2313. [DOI] [PubMed] [Google Scholar]
- (25).Giri N, Agarwal S, Shaik N, Pan G, Chen Y, and Elmquist WF (2009) Substrate-dependent breast cancer resistance protein (Bcrp1/Abcg2)-mediated interactions: Consideration of multiple binding sites in in vitro assay design. Drug Metab. Dispos. 37, 560–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Honjo Y, Hrycyna CA, Yan QW, Medina-Perez WY, Robey RW, van de Laar A, Litman T, Dean M, and Bates SE (2001) Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells. Cancer Res 61, 6635–6639. [PubMed] [Google Scholar]
- (27).Kage K, Tsukahara S, Sugiyama T, Asada S, Ishikawa E, Tsuruo T, and Sugimoto Y (2002) Dominant-negative inhibition of breast cancer resistance protein as drug efflux pump through the inhibition of S-S dependent homodimerization. Int. J. Cancer 97, 626–630. [DOI] [PubMed] [Google Scholar]
- (28).Velamakanni S, Janvilisri T, Shahi S, and van Veen HW (2008) A functional steroid-binding element in an ATP-binding cassette multidrug transporter. Mol. Pharmacol 73, 12–17. [DOI] [PubMed] [Google Scholar]
- (29).Kyte J, and Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein. J. Mol. Biol 157, 105–132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.