

Summary
Tn7-like transposons have co-opted CRISPR systems, including class 1 type I-F, I-B, and class 2 type V-K. Intriguingly, although these CRISPR-associated transposases (CASTs) undergo robust CRISPR RNA (crRNA)-guided transposition, they are almost never found in sites targeted by the crRNAs encoded by the cognate CRISPR array. To understand this paradox, we investigated CAST V-K and I-B systems and found two distinct modes of transposition: (1) crRNA-guided transposition and (2) CRISPR array-independent homing. We show distinct CAST systems utilize different molecular mechanisms to target their homing site. Type V-K CAST systems use a short, delocalized crRNA for RNA-guided homing, whereas type I-B CAST systems, which contain two distinct target selector proteins, use TniQ for RNA-guided DNA transposition and TnsD for homing to an attachment site. These observations illuminate a key step in the life cycle of CAST systems and highlight the diversity of molecular mechanisms mediating transposon homing.
Introduction
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated proteins (Cas) systems are adaptive immune systems employed by prokaryotes to defend against foreign genetic elements (Barrangou and Horvath, 2017; Marraffini, 2015; Mohanraju et al., 2016). CRISPR-Cas components have been associated with multiple roles beyond adaptive immunity (Faure et al., 2019a, 2019b), however, including guided transposition resulting from the co-option of inactivated Cas effector machinery by transposons (Faure et al., 2019a; Peters et al., 2017). Two distinct subtypes of CRISPR-associated transposon (CAST) systems have been experimentally characterized to date that facilitate RNA-guided targeting of Tn7-like transposons: V-K CAST systems, which utilize Cas12k effectors with naturally inactivated nuclease domains (Strecker et al., 2019), and I-F CAST systems, which utilize Cascade complexes lacking the Cas3 nuclease component (Klompe et al., 2019). In addition, type I-B CAST systems, which also contain components of the Cascade effector in association with Tn7-like transposons, but are extremely rare compared to other CAST systems, have been bioinformatically identified but not yet experimentally characterized (Faure et al., 2019a; Peters et al., 2017).
The prototypical Tn7 transposon from Escherichia coli mediates transposition using the core transposase proteins, TnsA, TnsB, and TnsC. These transposase proteins operate in two modes: (1) homing, where TnsABC interact with TnsD, a site-specific DNA-binding protein, to achieve transposition into the chromosomal glmS attachment site (also referred to as the homing site), or (2) mobile element transposition, where TnsABC interact with TnsE, a protein that preferentially directs the transposon to mobile elements including conjugative plasmids (Peters and Craig, 2001; Waddell and Craig, 1988, 1989). These two pathways allow the transposon to safely and efficiently travel from one host to another: after traveling through a mobile element, the tnsD pathway targets a site conserved across hosts to safely home to bacterial chromosomes. CAST systems, which lack tnsE, rely on spacers within their CRISPR arrays to achieve targeted transposition (Klompe et al., 2019; Strecker et al., 2019), likely to mobile genetic elements (Faure et al., 2019a, 2019b; Peters et al., 2017). The majority of identified CAST systems in bacterial chromosomes, however, are not located at targets programmed by their CRISPR array, but are rather found next to conserved sequences, including IMPDH and SRP RNA in the case of type I-F (Faure et al., 2019a; Peters et al., 2017), suggesting that CAST systems might have an innate homing site. This is also supported by the observation that all known CAST systems identified to date contain tniQ (a homolog of tnsD), and, in the case of type I-B, two tniQ genes. Here, we investigate how CAST systems specifically transpose to bacterial chromosomes and identify two distinct modes of homing in type V-K and type I-B systems involving RNA-mediated and protein-mediated homing, respectively.
Type V-K CAST systems home via a delocalized crRNA
We first conducted a bioinformatic survey to determine where type V-K CAST systems are found in bacterial genomes. Comparison of type V-K CAST loci revealed that the transposons preferentially localize to tRNA genes, as previously noted (Klompe et al., 2019), and that insertions are typically oriented with Cas12k proximal to the tRNA (46 out of 53 analyzed systems, Figure 1A). We also observed two insertions neighboring the DNA mismatch repair genes MutS and MutL, suggesting potential non-random chromosome targets outside of tRNA at a lower frequency. We previously showed that CAST from Scytonema hofmannii (ShCAST) can be reconstituted to achieve RNA-guided transposition in E. coli (Strecker et al., 2019) and we tested whether it can insert next to the tRNA independent of Cas12k. However, expression of tnsB, tnsC, and tniQ along with the introduction of a pDonor plasmid and a pTarget plasmid containing the re-joined flanking chromosomal sequence did not yield any homing insertions into pTarget as assayed by PCR (Figure S1A). Therefore we reasoned that another mechanism or protein might be required to direct the ShCAST transposon to its homing target. In the process of analyzing the left end (LE) sequence of ShCAST, we unexpectedly observed a 17-bp motif that perfectly matches the adjacent tRNA-Leu gene. Further analysis revealed the sequence upstream of this motif has partial homology to the CRISPR direct repeat, indicating that this matching sequence could be a delocalized CRISPR RNA (crRNA) (Figure 1B). The delocalized crRNA has a number of mismatches to the direct repeats in the CRISPR array and is irregularly spaced with respect to the array (which is why it was not previously identified), but it does contain 13 nt that perfectly match the 3′ end of the canonical direct repeat (Figure S1B). The 17-bp tRNA target sequence is flanked by a GGTT PAM sequence, and the LE of ShCAST is positioned 64 bp downstream of the PAM, consistent with target site requirements observed for crRNA-guided transposition (Strecker et al., 2019). Importantly, we note that a similar discovery of delocalized crRNA for type I-F CAST systems was recently uncovered (Petassi et al., 2020) suggesting a potential common mechanism for CAST homing.
Figure 1|. Homing of type V-K ShCAST is guided by a short, delocalized crRNA.
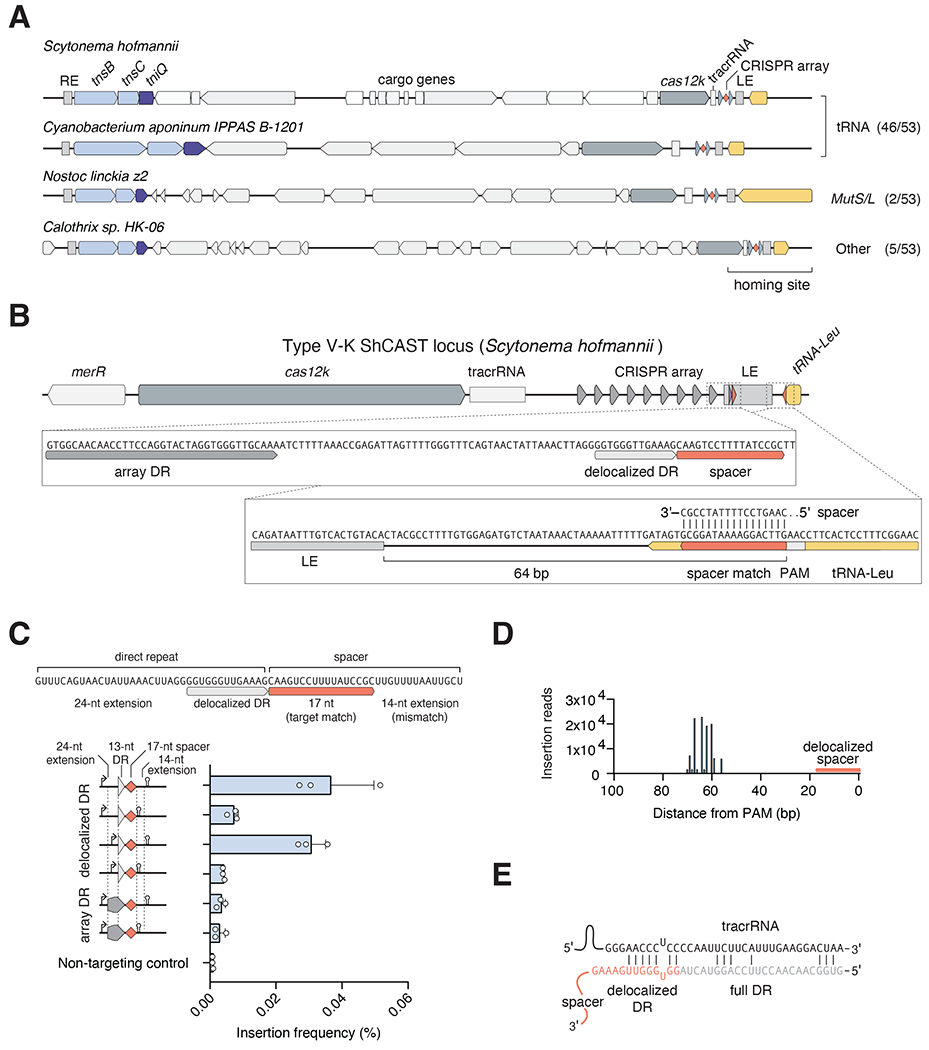
(A) Representative CAST systems from 57 analyzed loci with their respective homing sites in yellow. (B) Diagram of the delocalized crRNA and corresponding spacer match in downstream tRNA-Leu from S. hofmannii CAST. (C) ShCAST homing into a tRNA target plasmid in E. coli. Schematic of the natural delocalized crRNA and insertion activity of crRNA variants; promoters and terminators are indicated with an arrow and hairpin, respectively. Data are represented as mean ± SD. (D) Deep sequencing of the ShCAST homing insertion product. (E) Schematic representation of the ShCAST tracrRNA interacting with a direct repeat from the canonical CRISPR array.
We first tested whether the delocalized crRNA is functional by expressing the natural sequence containing a 37-nt direct repeat and 31-nt spacer (to match the length of crRNA from the adjacent CRISPR array), with only 17 nt of spacer matching the target (Figure 1C), and co-transformed E. coli with pHelper, pDonor, and the tRNA-Leu target plasmid. We observed insertions adjacent to the tRNA, and deep sequencing revealed an insertion product with the LE positioned 60-70 bp downstream of the spacer (Figure 1D), including the most prominent position at 64 bp which matches the natural insertion found in S. hofmannii. We next tested the sequence requirements of the delocalized crRNA by removing 5′ and 3′ sequences. We found the 5′ extension of the 13 nt short direct repeat is dispensable for function whereas loss of the 3′ sequence reduced insertion activity (Figure 1C), even though the additional spacer nucleotides do not match the target site. We observed that a minimal crRNA comprised of 13 nt direct repeat and 17 nt spacer is also capable of homing (Figure 1C). We note that the low insertion activity observed with the delocalized crRNA is likely due to the specific spacer sequence, as targeting PSP1, a previously validated spacer for ShCAST (Strecker et al. 2019), with the same constructs revealed higher activity even though both targets contain the same GGTT PAM (Figure S1C). Interestingly, swapping the delocalized crRNA direct repeat with a canonical direct repeat from the CRISPR array reduced activity between 2-10 fold (Figure 1C), suggesting that the delocalized direct repeat might be more efficient at annealing with the tracrRNA and loading into Cas12k.
Only the final 13 nt of the direct repeat is predicted to bind to the tracrRNA (Figure 1E). To determine if the delocalized crRNA represents the absolute minimal targeting requirements for ShCAST,we performed additional truncation analysis of the delocalized crRNA. We were able to delete an additional 3 nt from the 5′ end of the direct repeat, however, no deletions to the 17 nt homing spacer were tolerated (Figure S1D). We also tested the insertion activity of ShCAST using a synthetic sgRNA containing the 13 nt direct repeat from the delocalized crRNA (Strecker et al. 2019) by deleting different lengths of spacer sequence. We found that 16 nt is both sufficient and near the minimum length required for function (Figure S1E and S1F). Adding double mismatches between the spacer and target revealed that positions 1-16 are sensitive to mutation, while mismatches in positions 17-22 had limited effect on activity (Figure S1G), supporting the idea that only 16 nt of complementary sequence is required for targeted transposition activity. Together these results reveal that the delocalized crRNA is functional for CAST homing and has likely evolved to contain the minimal RNA sequence required for function.
The discovery of a delocalized crRNA in ShCAST led us to examine other type V-K CAST loci, and we were able to identify a delocalized crRNA in 51 out of 53 systems and a matching genomic insertion site in 46 of these 51 systems (Figure 2A, and Table S1). Spacers associated with delocalized crRNAs were 16 or 17 nt in length with only a few observed mismatches to their cognate targets, and all delocalized crRNAs contained the terminal 12-14 nt of the canonical direct repeat (Figure S2A). Analysis of tRNA-targeting spacers revealed several frequent sequences including those which target tRNA-Ala and -Pro (Figure S2B). The location of the delocalized crRNA varied between CAST systems, sometimes occurring as close as 12 bp from the terminal repeat in the array (Figure 2A), and generally with irregular spacing compared to the CRISPR array. This observation suggests that the delocalized crRNAs were not acquired in the same manner as those in the CRISPR array and might be a structural feature of the transposon. Prominent examples of this include the CAST systems from Cyanobacterium aponinum IPPAS B-1201 and Geminocystis sp. NIES-3709, wherein the delocalized crRNAs are fully embedded within the LE sequence and are over 200 bp from the CRISPR array. Interestingly, delocalized crRNAs are not limited to tRNA targets: the genome insertions adjacent to MutS, MutL, and CysH were also found to match a delocalized crRNA within corresponding transposons (Figure 2A). We identified two additional CAST systems that contain a delocalized crRNA targeting tRNA but lack detectable CRISPR arrays (Figure 2B). It is possible that these two transposons might have the ability to home to conserved bacterial sites but have yet to acquire the ability to target diverse mobile genetic elements or have lost their CRISPR array. Finally, although the locations of delocalized crRNA targets varied within the cognate genes, because CAST systems insert downstream of the target site, gene integrity was preserved in all cases, with the closest insertions being only 2 bp from the end of the tRNA gene (Figure 2C).
Figure 2|. Diversity of delocalized crRNA across type V-K CAST systems.
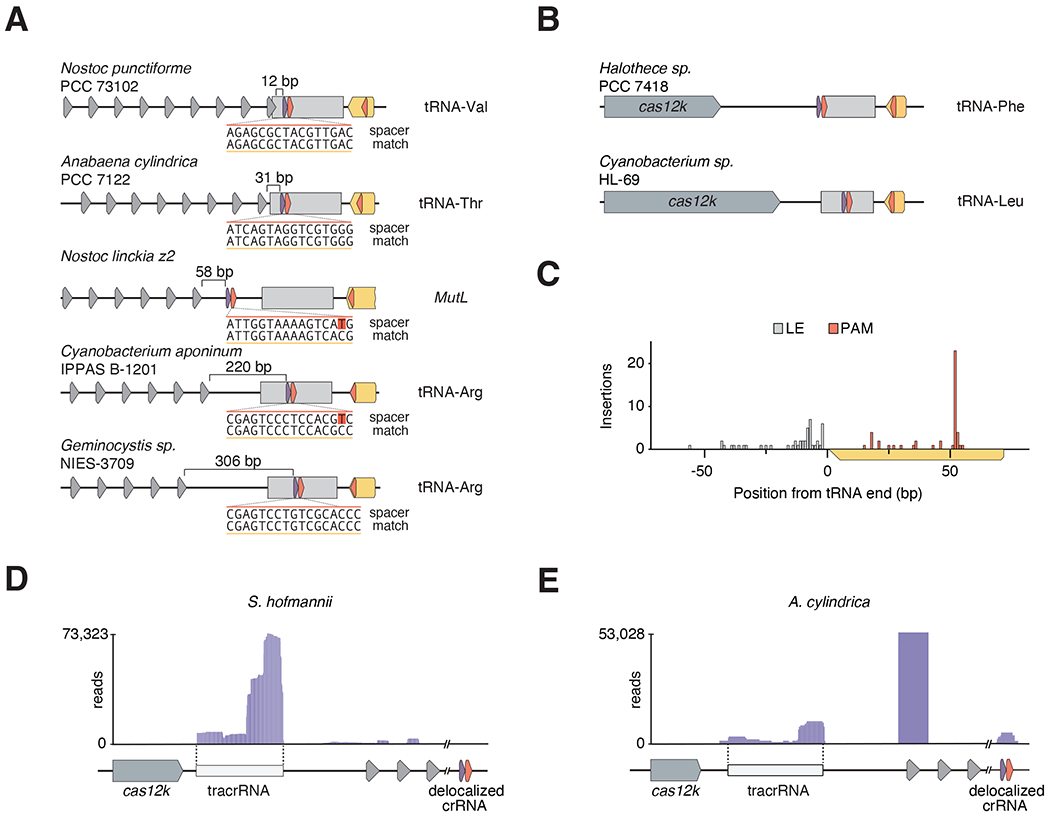
(A) Bioinformatic analysis of sequence and distance from the typical CRISPR array of the delocalized crRNA in five CAST systems (See Supplementary Table 1 for full list). (B) Two type V-K CAST systems (Halothece sp. PCC 7418 and Cyanobacterium sp. HL-69) that contain a short crRNA targeting the adjacent tRNA, but no detectable CRISPR array. (C) Position of the homing target PAM and corresponding left end (LE) for tRNA-homing CAST systems. (D) RNA sequencing reads from S. hofmannii aligned to the delocalized crRNA locus. (E) RNA sequencing reads from A. cylindrica aligned to the delocalized crRNA locus.
To investigate the identity and expression of the delocalized crRNA in natural cyanobacteria, we analyzed small RNA-sequencing data of two CAST system hosts: S. hofmannii and Anabaena cylindrica (Strecker et al., 2019). While we were unable to detect expression of the delocalized crRNA in S. hofmannii (Figure 2D), we could readily detect a small RNA species that matched the predicted delocalized crRNA in A. cylindrica (Figure 2C). The delocalized crRNA from A. cylindrica most commonly contained a 12-nt minimal DR, and ended 2 nt downstream of the 17-nt tRNA spacer (Figure S2C). Based on our experimental characterization of ShCAST, this minimal crRNA contains the required features for RNA-guided transposition and is predicted to be functional. Together, these results suggest that the delocalized crRNA is expressed in some native hosts, but is likely under regulation, and that these delocalized crRNAs function to home type V-K CAST systems to conserved locations on the bacterial chromosomes.
Type I-B systems use two distinct target selector proteins for RNA-guided transposition and homing
To investigate the generality of the RNA-guided homing mechanism to uncharacterized CAST subtypes, we chose to further study the previously identified type I-B CAST system (Faure et al. 2019a; Peters et al. 2017). We searched for delocalized crRNAs in type I-B loci, but we could not detect any crRNAs, canonical or delocalized, that might enable homing. In contrast to type V-K and I-F CAST systems, type I-B subtype1 (hereafter I-B1) CAST systems uniquely encode two TniQ proteins (Figure 3A, and S3A–D), and it was previously suggested that one might function with the Cascade complex to provide RNA-guided targeting while the other might recognize an attachment site (Peters, 2019; Peters et al., 2017). These two TniQ proteins are more similar to prototypical Tn7 TnsD than those found in other CAST systems, suggesting that they likely evolved from a TnsD Tn7 ancestor (Figure 3B, 3C, S3B, and S3C). Although these TniQ proteins are distinct in their C-terminal region, the strong similarity of the longer TniQ to prototypical Tn7 TnsD and the conserved location of type I-B CAST loci near glmS hint that the longer TniQ may facilitate DNA homing as a site-specific DNA-binding protein. Because of the strong similarity with Tn7 TnsD, we named the longer TniQ as TnsD. We therefore sought to investigate the functions of TnsD and TniQ and determine how I-B1 CAST systems achieve both RNA-guided and homing transposition, focusing on the I-B1 system from Anabaena variabilis (ATCC 29413, hereafter AvCAST).
Figure 3|. CAST I-B loci architecture and comparison of the TniQ/TnsD target selectors.

(A) Schematic of the Anabaena variabilis ATCC 29413 CAST I-B subtype 1 (I-B1) locus and ‘Peltigera membranacea cyanobiont’ 210A CAST I-B subtype 2 (I-B2) locus containing both Cascade proteins, Tn7-like proteins including TnsD and TniQ target selectors (Figure S3a, S3b. S3c). (B) A dendrogram showing the similarity of TniQ/TnsD proteins of Tn7 and CAST I-B1 and I-B2, CAST I-F, and CAST V-K systems (Figure S3b, S3d). Dark purple indicates a similar N-terminal region (TniQ/TnsD core). Light purple indicates similarity between the C-terminal regions of Tn7-TnsD, CAST I-B1 TnsD, and remote similarity with CAST I-B2 TnsD (see Figure S4, S12). Dashed lines indicate boundaries of TniQ/TnsD core. Light and dark yellow colors indicate the protein domains annotated from the CAST I-F TniQ structure (see Figure S3d). (C) Phylogenetic tree built with FastTree (WAG model) (Price et al., 2009) from the core region (Figure S3d) of TniQ/TnsD proteins from CAST I-B (TniQ in red; TnsD in purple), Tn7-TnsD (in green), CAST V-K TniQ (in yellow), and CAST I-F TniQ (in cyan). The tree indicates proximity between CAST I-B1 TnsD and Tn7-TnsD and the distinction between CAST I-B1 and CAST I-B2 (Figure S3b, S3d).
We cloned the nine AvCAST-related genes (tnsA, tnsB, tnsC, tniQ, cas6, cas8, cas7, cas5, and tnsD) into a helper plasmid (pHelper) under IPTG-inducible promoters (Figure 4A and Methods). We cloned the predicted transposon right end (RE) and LE sequences (Peters et al., 2017) into a donor plasmid (pDonor) with a 0.5-kbp cargo sequence. Two direct repeats from the AvCAST CRISPR array with appropriate spacers (i.e., the minimal CRISPR array) were further cloned into pHelper, and crRNA was expressed under an IPTG-inducible promoter. Small RNA-sequencing revealed that transcripts from the minimal CRISPR array are processed into 72-nt mature crRNAs (Figure 4B, Table S2).
Figure 4|. Characterization of Type I-B subtype1 AvCAST.
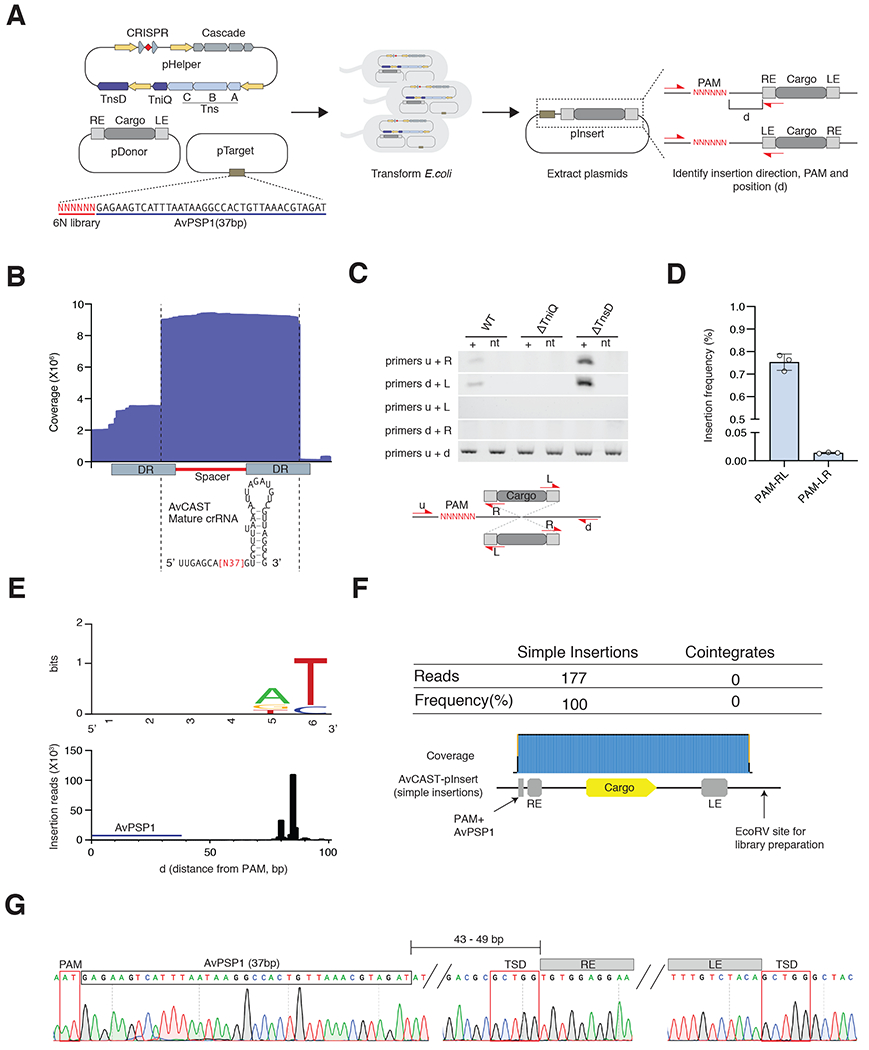
(A) Schematic of experiment to identify insertion direction, PAM, and insertion position by AvCAST in E.coli. (B) RNA sequence reads mapping to the minimal CRISPR array to reveal the mature crRNA sequence of AvCAST. (C) Insertion directionality assayed by diagnostic PCR of pInsert on 6N PAM library plasmid with different primer pairs in ΔTniQ or ΔTnsD conditions. (D) Quantification of insertion frequency in both directions by ddPCR. Data are represented as mean ± SD. (E) Top. PAMs for AvCAST RNA-guided insertions. Bottom. AvCAST RNA-guided insertion positions identified by deep sequencing. (F) Long-read Nanopore sequencing to characterize the structure of pInsert. (G) Sanger sequencing chromatograms of a representative pInsert. PAM, AvPSP1, TSDs, and transposon ends are annotated.
Given that AvCAST likely requires a protospacer adjacent motif (PAM) to recognize target DNA, we first targeted a plasmid (pTarget) library containing AvPSP1 flanked by a 6N motif upstream of the protospacer (Strecker et al., 2019). To clarify the role of AvCAST TniQ and TnsD in RNA-guided transposition, we targeted AvPSP1 with pHelper variants lacking either tniQ or tnsD (Figure 4A). We detected PAM-RE-cargo-LE (PAM-RL) insertion into the target plasmid by PCR when using pHelpers containing tniQ, and furthermore, removal of tnsD from the system increased the efficiency of transposition (Figure 4C). The other PAM-LE-cargo-RE (PAM-LR) direction insertions were confirmed by ddPCR (Figure 4D), indicating that the insertion direction is strongly biased (PAM-RL:PAM-LR=54:1) in contrast to the type I-F system from Vibrio cholerae (Klompe et al., 2019). Deep sequencing analysis of the ΔtnsD condition confirmed the insertion of the RE into pTarget and revealed a preference for 5′ AT PAMs (Figure 4E). We next examined the position of the donor in pInsert products relative to AvPSP1, and we detected insertions mainly within a 80-86-bp window downstream of the PAM (43-49-bp downstream of the spacer) (Figure 4E). To investigate the exact structure of the pInsert, we performed long read, amplification-free nanopore sequencing. We found only simple insertions generated by type I-B1 AvCAST, whereas type V-K systems showed about 20% cointegrate insertions in this experimental context (Figure 4F, (Strecker et al. 2020)). We also confirmed the existence of target site duplications by Sanger sequencing of individual pInserts (Figure 4G). These results show AvCAST can perform RNA-guided transposition and tniQ is necessary and sufficient for this activity.
To test whether AvCAST can target bacterial genomic DNA, we picked 36 PAM-containing targets (labeled AvPSP2 - 37) in E. coli. Two separate helper plasmids, pCascade (bearing cas6, cas8, cas7, cas5, and the minimal CRISPR array) and pTns (bearing tnsA, tnsB, tnsC, tniQ, and tnsD) were co-electroporated into E. coli with pDonor. While clear PCR amplicons were detected on the gel at only 10 out of the 36 sites (27.8%) (Figure S4A), 25 sites showed distinguishable signals compared to the no guide control condition by ddPCR (Figure S4B). Further confirmation by deep sequencing revealed that 33 sites showed insertions 70-100-bp downstream of the PAM (33-63-bp downstream of the spacer), as we observed prominent peaks in a 80-86-bp window downstream of the PAM (43-49-bp downstream of the spacer) in AvPSP1 targeting (Figure S4C). These results suggest that over 90% of guides are functional, with insertion frequencies between 0.02% and 2.45%. For three representative genomic sites (AvPSP2, 7, and 12), we performed Tagmentation-based Tag Integration Site Sequencing (TTISS) (Schmid-Burgk et al., 2020) to determine the specificity of genome targeting by AvCAST. We observed comparable insertion efficiency with newly constructed heat-sensitive pSC101-donor and pDonor (Figure S4D), and confirmed that 98.8%, 99.8%, and 89.2% of the total insertion reads map to each expected target site (AvPSP2, 7, and 12, respectively) by TTISS (Figure S4E). This indicates that AvCAST can be reprogrammed to precisely insert DNA at specific sites in the E.coli genome.
Given that tnsD is dispensable for RNA-guided transposition, we tested the hypothesis that tnsD can mediate homing to the host glmS Tn7 attachment site (attTn7) (Waddell and Craig, 1989). The native AvCAST contig (CP000117) and two other identical contigs (100% of sequence identity at nucleotide level) show insertion 5-bp downstream of glmS gene stop codon, whereas all other homologous loci from diverse contigs (non identical) are constantly inserted about 25-bp downstream from the end of glmS (Figure S5A). To examine whether AvCAST tnsD mediates homing to the A. variabilis glmS gene (A.v. glmS), we generated pTarget(AvCAST) containing the A.v. glmS sequence and AvPSP1 flanked by an AT PAM, which we used to assess the transposition efficiency. As we observed for PAM library plasmid targeting, removal of tnsD from the system increased insertion frequency at the AvPSP1 site, as quantified by ddPCR (3.0-fold, Figure 5A). In contrast to RNA-guided transposition, tniQ is not necessary for Tn7-like transposition to the glmS site. Moreover, removal of tniQ from the system increased insertion frequency at the site (16.8-fold, Figure 5A, and 5B). We found that the prominent insertion site is 24-bp downstream of A.v. glmS gene (Figure. 5C), which, although different from the native locus, is similar to other homologous loci. This insertion site was not affected by co-expression of Cascade and TniQ (Figure 5C). We then cloned the downstream region of glmS from the native contig into pTarget and repeated the transposition assays to determine if there are additional elements that govern insertion sites. We again observed a prominent insertion site at the canonical Tn7 insertion site, 25-bp downstream of glmS (Figure 5C), suggesting there are no additional elements. It is possible the locus found 5-bp downstream glmS in the native contig (CP000117) could result from a 20-bp microdeletion, an idea supported by the lack of target site duplications in this contig.
Figure 5|. Homing of AvCAST is mediated through conserved attTn7 recognition by TnsD.

(A) RNA-guided insertion frequency of AvCAST into the pTarget with AvPSP1 and glmS gene at each ΔTniQ/ΔTnsD condition. Data are represented as mean ± SD. (B) Tn7-like machinery-mediated insertion frequency into the pTarget with AvPSP1 and glmS gene at each ΔTniQ or ΔTnsD condition. Data are represented as mean ± SD. (C) AvCAST-mediated single prominent insertion at glmS Tn7 attachment site on plasmid identified by deep sequencing. Purple bar indicates the position by Tn7-like machinery (tnsA, tnsB, tnsC, and tnsD). Blue bar indicates the position by all AvCAST protein components (Tn7-like machinery + TniQ + Cascade). Light blue bar indicates the insertion position on the plasmid harboring an additional 65-bp downstream sequence of A. variabilis glmS. (D) Tn7-like machinery-mediated insertion frequency of AvCAST into a plasmid bearing the A.variabilis glmS gene, a plasmid bearing mutations in the conserved attTn7 site, and a plasmid bearing the E.coli glmS gene. The 30 bp of the C-terminus of glmS, a part of attTn7 identified by a previous study (Mitra et al.), is shown. Base numbering format follows that of the study (End of glmS = +23), and reported essential positions for TnsD recognition are shown in red. Data are represented as mean ± SD. (E) Schematic of in vitro transposition reactions with purified Tn7-like machinery components of AvCAST. (F) Tn7-like machinery and donor requirements for in vitro transposition on E. coli glmS Tn7 attachment site. pInsert was detected by PCR for LE and RE junctions. (G) Tns protein requirements for in vitro transposition to the E.coli glmS Tn7 attachment site. All reactions contained pDonor and pTarget.
To obtain additional evidence that this homing transposition is mediated by Tn7-like machinery, we focused on the similarity between A. variabilis and E.coli glmS genes and assessed transposition into target plasmids harboring glmS mutations. As expected from the similarity, we observed an insertion 24-bp downstream of the E.coli glmS gene (Figure S5B). Similarly, expression of all Tn7-like machinery proteins with or without the Cascade complex induced transposition 25-bp downstream of glmS in the E. coli genome with TSDs (Figure S5C, S5d, and S5e). Based on previous findings about E.coli glmS attTn7 (Mitra et al., 2010), we introduced single point mutations at 6 positions previously reported to be important for TnsD attachment and that are conserved between A. variabilis and E.coli glmS. We also constructed an additional 3 variants, two with double mutants and one with all six conserved sites mutated. Each of the six single point mutations in the A.v. glmS gene significantly impaired transposition, and both the double point mutants and the sextuple mutant completely abolished it (Figure 5D), suggesting that this homing transposition is achieved through recognition of attTn7 by AvCAST TnsD.
To further validate these observations, we reconstituted Tn7-like transposition of AvCAST in vitro (Bainton et al., 1991) (Figure 5E). As expected, four Tn7 components (tnsA, tnsB, tnsC, and tnsD, Figure S5F) and pDonor resulted in the transposition into a pTarget vector bearing the E. coli glmS Tn7 attachment site (Figure 5F and 5G).
A distinct CAST I-B system homes to ncRNA sites
We sought to investigate the generalizability of this homing mechanism by considering a second subtype of CAST I-B systems, subtype 2 (hereafter I-B2). In contrast to type I-B1, type I-B2 systems are not found at glmS in their native hosts, but are instead found next to tRNA genes (e.g., tRNA-Val and -Ala) (Figure 3A and S3C), similar to type V-K CAST systems. While Type I-B1 and I-B2 share similar Cascade proteins (38% sequence identity, estimated from the concatenation of all Cas protein sequences), their transposon proteins have less than 10% sequence identity (concatenation of all Tn7 protein components). Moreover, although type I-B2 systems contain a TnsC protein and two TniQ proteins (Figure S3C), similar to I-B1 systems, they also harbor a fusion protein of TnsA and TnsB (hereafter TnsAB). Together, these findings suggest Cascade I-B was likely captured by two different transposons in evolutionarily independent events. Given these differences, we sought to determine the mechanisms used by these distinct systems. Among the putative I-B2 CAST systems, we focused on one from Peltigera membranacea cyanobiont 210A (PmcCAST) (Figure 3A) to investigate how it homes to tRNA genes. The PmcCAST locus encodes two TniQ proteins, with the larger TniQ protein having a weak but detectable similarity to Tn7 TnsD in its C-terminal region (HHpred probability is 99.26% with a sequence identity of 14%), indicating this protein (hereafter called TnsD) might be the homing selector target protein.
We cloned the eight PmcCAST genes (tnsAB, tnsC, tniQ, tnsD, cas6, cas8, cas7, and cas5) into a pHelper plasmid with a minimal CRISPR array. Predicted RE and LE sequences with 0.5-kbp cargo were further cloned into pDonor. As described above, we targeted a PmcPSP1-containing 6N PAM library vector to concurrently identify the preferred PAM and insertion position of PmcCAST. We confirmed the expression of a 71-nt mature crRNA by RNA sequencing (Figure S6A, Table S2). Analysis of PmcCAST pInsert plasmids revealed single direction biased insertions (PAM-RL:PAM-LR=1:630) within a 83-92-bp window downstream of a strong ATG PAM (48-57-bp downstream from spacer) (Figure 6A, S6B, and S6C). In contrast to type I-B1 AvCAST, we found cointegrate insertions by this type I-B2 PmcCAST, whose cointegrate ratio (17%) is comparable to that of type V-K ShCAST (20%, (Strecker et al. 2020)) (Figure 6B). We also confirmed the existence of target site duplications of individual pInserts (Figure S6D).
Figure 6|. Type I-B subtype 2 PmcCAST system homes to tRNA-val.
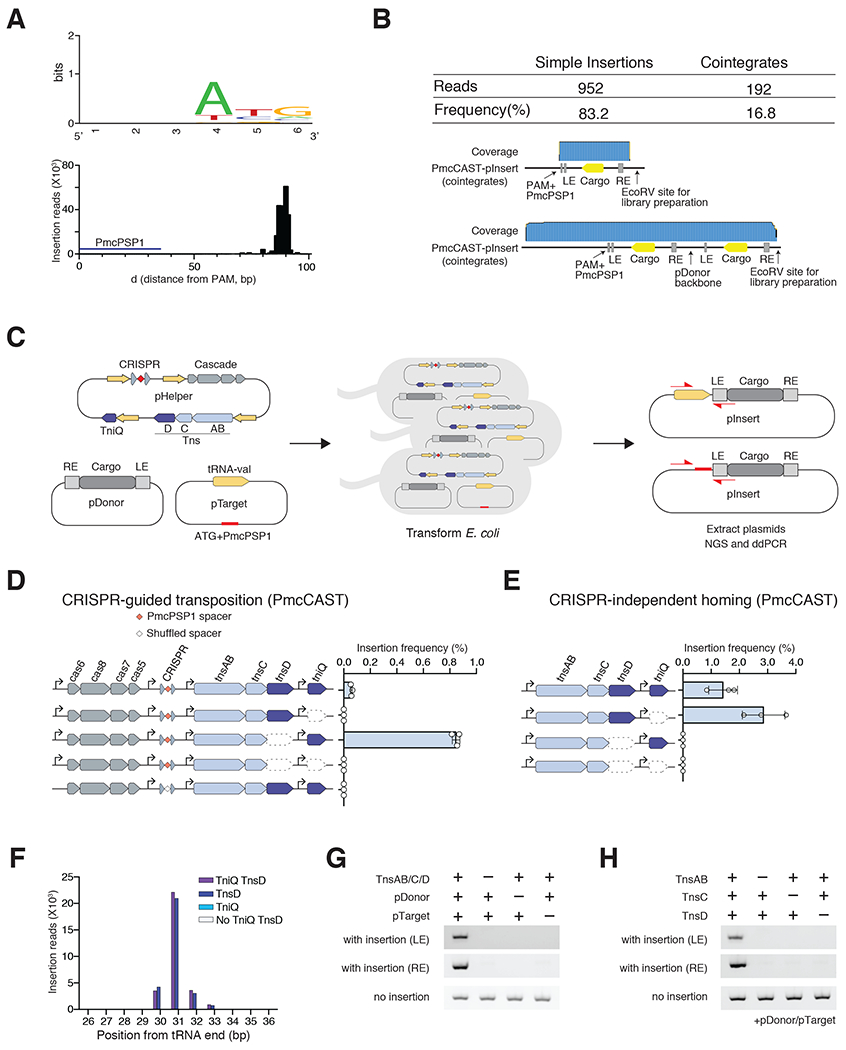
(A) Top. PAMs for PmcCAST RNA-guided insertions. Bottom. AvCAST RNA-guided insertion positions identified by deep sequencing. (B) Long-read Nanopore sequencing to characterize the structure of pInsert. (C) Schematic of experiment to compare RNA-guided transposition and homing to tRNA-val by PmcCAST in E.coli. (D) RNA-guided insertion frequency of PmcCAST into the pTarget with PmcPSP1 and tRNA-val gene at each ΔTniQ or ΔTnsD condition. Data are represented as mean ± SD. (E) Tn7-like machinery-mediated insertion frequency of PmcCAST into pTarget at each ΔTniQ or ΔTnsD condition. Data are represented as mean ± SD. (F) PmcCAST-mediated prominent insertion at tRNA-val gene on target plasmid identified by deep sequencing. Purple bar indicates the position by all Tns proteins (TnsAB, TnsC, TniQ, and TnsD). Blue bar indicates the position at ΔTniQ condition. Light blue bar indicates the position at ΔTnsD condition. White bar indicates the position at ΔTniQΔTnsD conditions. (G) Tn7-like machinery and donor requirements for in vitro transposition on tRNA-val gene. pInsert was detected by PCR for LE and RE junctions. (H) Tns protein requirements for in vitro transposition on tRNA-val gene. All reactions contained pDonor and pTarget.
To examine the role of TniQ and TnsD in PmcCAST RNA-guided transposition and homing, we generated pTarget(PmcCAST) containing the host tRNA-Val gene and PmcPSP1 flanked by an ATG PAM. We targeted PmcPSP1 with pHelper and its variants lacking either tniQ or tnsD (Figure 6C). As with AvCAST, we again found that removal of tnsD from the system increased insertion frequency at the PmcPSP1 site, as quantified by ddPCR (14.5-fold, Figure 6D). Expression of just the Tns proteins by pTns (bearing tnsAB, tnsC, tniQ, and tnsD) also induced transposition on pTarget(PmcCAST), and deep sequencing analysis revealed that the prominent insertion site is 31-bp downstream from the tRNA-val gene, consistent with where PmcCAST is located in the genome (Figure 6E and 6F). For this Tn7-like transposition of PmcCAST to the tRNA-val homing site, TnsD is essential while TniQ is not necessary (Figure 6F). Reconstitution of Tn7-like transposition of PmcCAST in vitro (Figure S6E) validated that three protein components (TnsAB, TnsC, and TnsD) and pDonor resulted in the transposition into pTarget(PmcCAST) (Figure 6G and 6H). Thus, although the homing site is different, type I-B2 achieves RNA-guided transposition through the same mechanism used by type I-B1. The detection of high levels of cointegrate pInsert plasmids suggests that the TnsAB fusion of PmcCAST does not have the ability to process the 5′ ends of the transposon, instead facilitating copy-and-paste transposition into the target site.
Discussion
The non-random occurrence of CAST insertions within bacterial genomes hints at mechanisms for directing transposition into conserved chromosomal sequences, or homing sites (Figure 7A). Here, we uncover two distinct ways by which CAST systems accomplish this task: V-K systems home using a dedicated delocalized crRNA, primarily to tRNA targets, whereas I-B systems utilize a dedicated tniQ/tnsD that mediates homing to glmS via direct DNA motif recognition in subtype 1 (similar to the prototypical Tn7) or to various tRNA genes in subtype 2 (Figure 7B). The use of an atypical crRNA expressed from outside of the CRISPR array in type V-K systems reveals remarkable similarity to type I-F CAST systems, which were discovered to also use a non-canonical crRNA to target homing sites (Petassi et al., 2020). This non-canonical crRNA features a variable repeat that is still able to form a hairpin for Cascade recognition and a full length or truncated spacer that can target homing sites despite numerous mismatches with the target. The similarities in these diverse CAST systems highlights the importance of transposon homing, and that in multiple instances, evolution has selected for an RNA-guided solution (Figure S7). These likely ancient homing spacers have been shaped by evolution from their respective effectors, and shed light on a remarkable ability for target recognition.
Figure 7|. Models of transposition mechanisms of CAST I-B and CAST V-K systems.
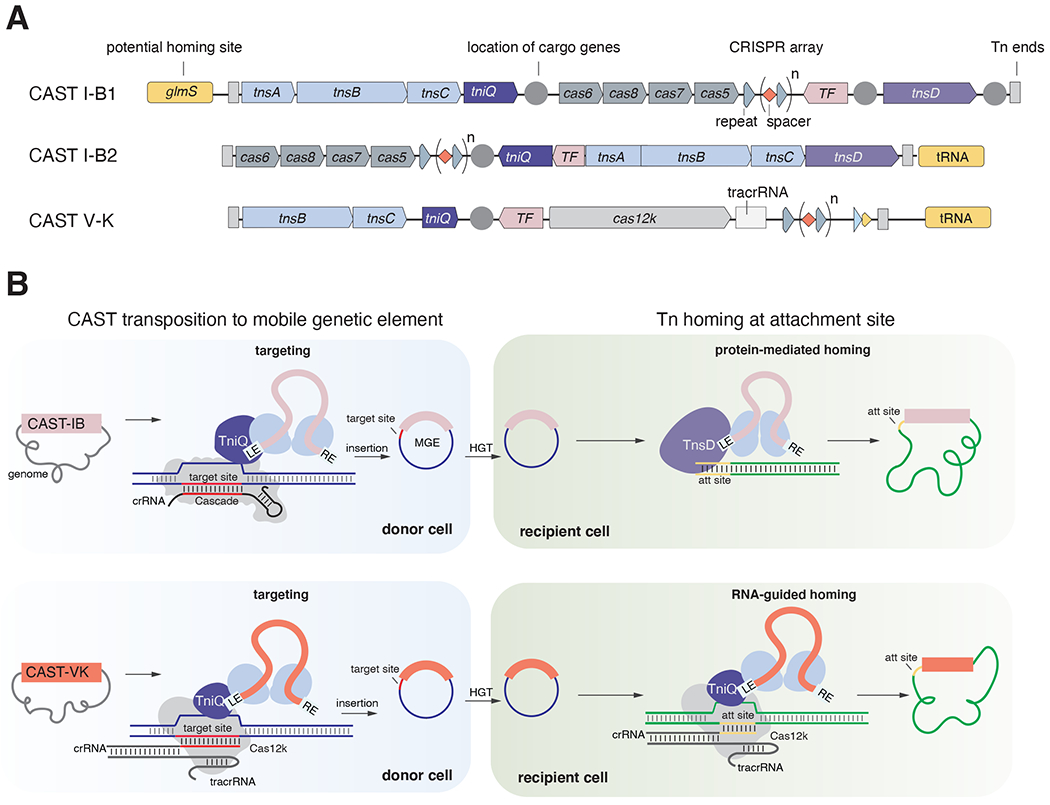
(A) Prototypical loci organization of CAST I-B and CAST V-K systems. The loci are delimited by transposon ends (Tn ends, indicated as light vertical rectangle). Transposon core components (tnsA, tnsB, and tnsC in CAST I-B; tnsB and tnsC in CAST V-K) are colored in light blue while tniQ are indicated in purple with a lighter purple for tnsD. Cas components (Cascade and cas12k) are shown in grey. Conserved transcription factors (TF) are shown in pink. Locations of cargo genes are indicated with dark grey circles. CRISPR arrays are shown as grey triangles for the repeats and red diamonds for the spacers. The delocalized crRNA is indicated by a lighter triangle (partial repeat) and a truncated red diamond (short spacer). Homing target sites are colored in yellow. Horizontal light grey triangle indicates the tracrRNA in the CAST V-K locus. Conserved transcription factors (TF) are shown in light pink. (B) Models for CAST transposition to mobile element (donor cell in blue) and CAST homing transposition to bacterial chromosome (recipient cell in green). CAST transposition to mobile genetic elements is mediated by the Cas effector machinery (IB Cascade and crRNA or Cas12k, tracrRNA, and cRNA; Cas proteins are shown in grey) that recognizes the target site (red) with RNA-guided targeting to insert the transposon using Tn components. TniQ may function as an adaptor between Cas machinery and the transpososome (Tn core machinery bound to the DNA transposon). The mobile genetic element (MGE) where the transposon has inserted can be horizontally transferred (via HGT) to another host where homing transposition can occur. CAST I-B homing transposition is mediated by TnsD (without Cas components), which targets the homing site (light orange) located in the bacterial chromosome (in green). CAST V-K uses a dedicated crRNA to target the homing site.
The identification of a delocalized homing crRNA across almost all diverse type V-K CAST systems suggests that homing is a critical step in the life cycle of these systems, and the minimal functional length of both the direct repeat and spacer reveals strong pressure to retain effective targeting ability during evolution. The irregular spacing, and often separation, of the delocalized homing crRNA relative to the CRISPR array, and its divergent sequence features, imply different evolutionary constraints and possible acquisition mechanisms imparted by the self-targeting homing spacer and mobile element targeting spacers in the array. Consistent with this idea, we identified two CAST systems that have delocalized crRNA, but no detectable CRISPR arrays (Figure 2B). Why RNA-guided homing is programmed from outside the CRISPR array, and with different sequence features, remains an interesting question. One possibility, as identified in the type I-F systems, is that the delocalized spacer is competent for binding and transposition but does not allow for self-cleavage when in contact with nuclease-competent CRISPR systems (Petassi et al., 2020) However, a search of type V-K CAST systems in fully assembled genomes revealed no co-occurrence of other type V CRISPR systems, indicating that preventing crRNA cross-reactivity may not be a necessary function. We note that more comprehensive co-occurrence analysis is needed once complete genomic information is available for incomplete metagenomic type V-K loci to fully rule out this idea. Alternatively, we hypothesize that the sequence features of the delocalized crRNA could enable protection of the type V-K CAST host from self-cleavage by an ancestral Cas12 nuclease during initial co-option of this CRISPR system by the transposon.
The preference for homing to conserved genes and the ability to insert downstream of the target sequence (e.g., tRNA and glmS) allows CAST systems to safely target numerous bacterial species without being deleterious to the host. The use of Cas protein-mediated RNA-guided targeting, which is known to tolerate a small number of mismatches between the RNA guide and its DNA target, of homing sites likely provides some flexibility to recognize variation across bacterial species.
The similarity of CAST I-B1 homing to prototypical Tn7 suggests a close evolutionary relationship and reveals a distinct strategy to integrate safely into chromosomes. Our finding that two separate subtypes of I-B use the same homing mechanism, but target different sites, further supports the notion that type I CRISPR-Cas systems were independently acquired by two distinct Tn7-like transposons.
Our results address a critical gap in our understanding of CAST systems, namely, why they are found at sites not targeted by their CRISPR arrays. We find that while some systems use an RNA-guided homing mechanism, others use a protein-targeted mechanism likely inherited from the transposon. Either one of these mechanisms allows CAST systems to insert themselves into host genomes by targeting well-conserved bacterial genes that serve as safe harbor attachment sites (Figure 7B). Thus, CAST systems seem to utilize two modes of insertion: transposition to mobile elements using RNA-guided targeting and homing using either RNA-guided targeting or the tniQ/tnsD pathway, like Tn7 transposons. How these pathways are regulated remains unclear in type V-K and type I-B CAST systems, but it was recently shown for type I-F that conserved transcription factors are linked to the switch between pathways (Petassi et al., 2020). Consistent with this observation, we can detect conserved transcription factors within CAST V-K and CAST I-B loci (Figure 7A) that appear to be distinct from cargo genes and could be linked to transposon pathway switching. Further analysis remains to be done to confirm they are pathway regulators. We also note our inability to detect expression of the homing crRNA in the cyanobacteria host S. hofmannii hinting at potential regulation. In addition to elucidating homing mechanisms, our work here experimentally characterizes the type I-B CAST systems and demonstrates that both subtypes of I-B CAST systems can transpose via RNA-guided targeting. Importantly, these type I-B CAST systems provide new starting points for developing genome engineering tools for targeted gene insertion in bacterial genomes (Strecker et al. 2019; Klompe et al. 2019; Rubin et al., 2020; Vo et al., 2020) and potential applications in human cells. Collectively, this work advances our understanding of the biology of these systems, highlighting conserved and divergent features, and shedding light on their mechanisms of action.
Limitations of the Study
This study presents two unique targeting mechanisms which enable CAST systems to integrate at their chromosomal attachment sites and reveals two distinct pathways in the CAST lifecycle: mobilization and homing. Further work will elucidate how CAST V-K and I-B systems regulate these two pathways. While we can detect conserved transcription factors in CAST V-K and I-B loci, their role in CAST pathway regulation must be experimentally validated as has been done for CAST I-F systems (Petassi et al., 2020). How spacers are acquired for both mobilization and homing remains a mystery, as the adaptation module is notably absent in CAST loci. Finally, this study adds to the list of potential candidate systems that may be developed for targeted gene insertion applications (along with other CAST V-K and I-F systems) (Strecker et al. 2019; Klompe et al. 2019; Rubin et al., 2020; Vo et al., 2020).
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Feng Zhang (zhang@broadinstitute.org).
Materials Availability
Plasmids generated in this study have been deposited to Addgene.
Data and Code Availability
Expression plasmids are available from Addgene under a uniform biological material transfer agreement; support forums and computational tools are available via the Zhang lab website (https://zhanglab.bio). All CAST loci are available in the Supplementary Dataset as an archive of Genbank files.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacteria strains
For transposition assays, E. coli strains were plated on LB-agar plates supplemented with appropriate antibiotics and IPTG and incubated at 37°C for 17 hours. For recovery of mature crRNAs and Tns proteins of I-B CAST systems, E.coli from a single colony were grown in Terrific Broth media (TB) at 37°C until OD600 reached 0.6 , and further grown at 16°C with IPTG supplementation for expression. Culture for Trichormus variabilis followed the condition suggested by ATCC. Scytonema hofmanni (UTEX B 2349) and Anabaena cylindrica (PCC 7122) were cultured in BG-11 media at 25°C with light periodicity of 14 hours on, 10 hours off.
METHOD DETAILS
Plasmid construction
All plasmids used in this study are described in Table S2. For ShCAST homing analysis, a tRNA target plasmid was cloned in the pACYC background by rejoining the chromosomal sequence up and downstream of the transposon. ShCAST pHelper plasmids (Addgene #127922) with endogenously driven tracrRNA were modified by replacing the previous FnPSP1 spacer with various crRNA driven by the J23117 promoter and flanked with the T7Te terminator using Gibson Assembly (NEB). The 24-nt 5′ extension and 14-nt 3′ extensions of the delocalized crRNA were taken from the natural host to match the length of crRNA derived from the CRISPR array. For sgRNA experiments targeting the E. coli genome, the pHelper_ShCAST_sgRNA plasmid (Addgene #127922) was modified by cloning spacers using Golden Gate assembly and LguI. For all RNA truncations, the length of crRNA or sgRNA spacer was reduced, and mismatches were generated with transversion mutations (A to T, T to A, G to C, C to G). All tested crRNA and sgRNA can be found in Supplementary Table 2. For AvCAST experiments, genes encoding TnsA-TnsB-TnsC-TniQ, TnsD and Cas6-Cas8-Cas7-Cas5 were amplified from the genome of Trichormus variabilis (Anabaena variabilis ATCC 29413). All the Tns genes were cloned into pCOLADuet-1 (Millipore) and the Cascade genes were cloned into pCDFDuet-1 (Millipore), resulting in pTns(AvCAST) and pCascade(AvCAST)_ ΔCRISPR, respectively using Gibson Assembly Master Mix (NEB). A pCascade was generated by inserting a tandem BsaI site flanked by two CRISPR direct repeats into pCascade(AvCAST)_ ΔCRISPR, and specific guides were further inserted by BsaI digestion and ligation. The Tns expression cassette in pTns(AvCAST) was inserted into pCascade(AvCAST) to obtain pHelper(AvCAST). For PmcCAST experiments, genes encoding ‘Peltigera membranacea cyanobiont’ 210A TnsAB-TnsC-TnsD, TniQ, and Cas6-Cas8-Cas7-Cas5 were synthesized (Twist Bioscience) and cloned into pCDFDuet-1 with the appropriate spacer flanked by two CRISPR direct repeats described above. Gene fragments encoding both transposon ends (GENEWIZ) were cloned into pBluescript II SK (+), yielding pDonor(AvCAST) or pDonor(PmcCAST). For PAM screens, a 0.5kb exon fragment amplified from human EMX1 was inserted between the transposon ends as a mock non-functional cargo in E.coli. For other experiments, a chloramphenicol resistance gene (CmR) was used as cargo. To obtain pTns variants, the Q5 Site-Directed Mutagenesis Kit (NEB) was used. For Nanopore sequencing experiments, all CAST components (DR-spacer-DR, Cascade, and Tns proteins) were cloned under a constitutive lac promoter to obtain pHelper2(AvCAST) and pHelper2(PmcCAST). The design of pR6K donors for AvCAST and PmcCAST followed that of pDonor_ShCAST_kanR (Addgene #127924). The glmS locus of BL21(DE3) (3800227-3801226, CP001509) was cloned into pUC19 (NEB). The glmS gene of A. variabilis and tRNA-val gene of ‘Peltigera membranacea cyanobiont’ 210A were cloned into pACYC vector bearing AvPSP1 or PmcPSP1 flanked by an AT or ATG PAM, respectively. These target plasmids were used as pTarget for in vitro transposition assays and also pTarget for assays in bacteria.
PAM and insert position analysis of AvCAST and PmcCAST
To identify the PAM of AvCAST and PmcCAST, 100 ng of randomized 6N sequence-containing pACYC vector libraries (Strecker et al., 2019) was co-electroporated with 100 ng of both pHelper and pDonor into BL21(DE3) electrocompetent cells (Sigma). Cells were recovered for 1 hour and plated on 100 μg/ml carbenicillin, 50 μg/ml spectinomycin, and 50 μg/ml chloramphenicol containing LB-agar plates. After 37°C incubation for 17 hours, all colonies were scraped from the plates, and a portion was re-plated on 0.1 mM IPTG supplemented triple antibiotic LB-agar plates to induce protein expression. Cells were incubated for an additional 17 hours at 37°C. All colonies were scraped and plasmid DNAs were prepared from the colonies using the PureYield Plasmid Midiprep System (Promega). Insertion products containing the 6N sequence were amplified and sequenced using a MiSeq Reagent Kit v2, 300-cycle (Illumina). PAM and insert position were characterized using an established pipeline (Strecker et al., 2019).
E. coli Transposition Assays
For ShCAST, transposition in E.coli was performed by transformation of 20 ng each of pHelper, pTarget, and pDonor into TransforMax EC100 pir+ Electrocompetent E.coli (Lucigen). Cells were recovered for 1 hour and plated on ampicillin, kanamycin, and chloramphenicol-containing plates. Cells were harvested 20 hours after plating, and scraped cells were lysed in 15 uL colony lysis buffer (TE with 0.1% Triton X-100). Genome targeting experiments were performed in a similar manner using pHelper and pDonor plasmids followed by plating on ampicillin and kanamycin. Cells were boiled for 5 min, diluted with 60 uL of water, spun at 4000 x g for 10 min to pellet debris, and the supernatant was used for subsequent analysis. The frequency of genome insertions relative to the concentration of extracted E.coli genomes or to the target plasmid was determined with qPCR as described above with a guide-specific forward primer and a primer/probe set in an unmodified region of the E.coli genome or the pTarget chloramphenicol resistance gene (Table S2). The initial homing test for ShCAST was performed with a modified pHelper plasmid harboring a deletion of cas12k.
For AvCAST, 10 guides with 5′ AT PAMs were chosen in non-coding regions of the E. coli genome (Strecker et al., 2019) and cloned into pCascade(AvCAST) (Table S2). Each 100 ng of pCascade(AvCAST) targeting a specific genomic region was co-electroporated with pTns(AvCAST)_ΔTnsD and pDonor(AvCAST) into BL21(DE3) electrocompetent cells (Sigma) and plated on 100 μg/ml carbenicillin, 50 μg/ml spectinomycin, and 50 μg/ml kanamycin containing LB-agar plates. After 37°C incubation for 17 hours, all colonies were scraped from the plates, and a portion was re-plated on 0.1 mM IPTG supplemented triple antibiotic LB-agar plates to induce protein expression. Cells were incubated for an additional 17 hours at 37°C. All colonies were scraped and the gDNA was purified using the Wizard Genomic DNA Purification Kit (Promega). Insertions were identified by PCR using KAPA HiFi HotStart ReadyMix (Roche). The frequency of genome insertions was determined with droplet digital PCR (ddPCR) as described below with 20 ng template gDNA for 20 μL ddPCR reaction.
Nanopore sequencing to determine constitutive/replicative transposition
Transposition assays for AvCAST and PmcCAST were performed by transformation of 30 ng each of pHelper2, pR6K Donor, and pTarget(AT/ATG+AvPSP1/PmcPSP1) into Pir1 E. coli. Cells were recovered for 1 hour at 37°C and plated on 50 μg/ml kanamycin, 50 μg/ml spectinomycin, and 50 μg/ml chloramphenicol containing LB-agar plates. Cells were harvested 5 days after incubation at room temperature, and subjected to mini-prep by QIAprep Spin Miniprep Kit (Qiagen). Note that we avoided incubation at 37°C to prevent transposed products being actively resolved, which would mask replicative transposition. To recover transposed products, 100 ng mini-prep product was electroporated into Endura Competent Cells (Lucigen). Cells were recovered for 1 hour and plated on 50 μg/ml kanamycin and 50 μg/ml chloramphenicol containing LB-agar plates, and further incubated at room temperature for 5 days. Donor insertion on pTarget was confirmed by Sanger sequencing (position and target site duplication) of mini-prep products for 12 colonies. In parallel, all colonies were harvested and subjected to mini-prep, followed by amplification-free Nanopore sequencing library preparation (Oxford Nanopore Technologies SQK-LSK109). Briefly, mini-prep products were linearized by EcoRV, followed by end-prep and subsequent ligation of sequencing adapters. Resulting libraries were loaded on a MinION R9 flow cell and sequenced. Sequence reads containing 20 bp of AvPSP1/PmcPSP1 and both 20 bp of the outer transposon LE and RE were filtered for further analysis, thereby discarding short, low-quality, and contaminating gDNA reads.
Droplet digital PCR (ddPCR) reactions
Insertion events were quantified using insertion specific primers and a donor specific probe (Table S2). ddPCR Supermix for Probes (No dUTP) (BioRad), primers (900 nM each), a probe (250 nM), and template DNA were combined into 20 μL reactions, and droplets were generated with 70 μL of Droplet Generation Oil for Probes (BioRad) using the QX200 Droplet Generator (BioRad). Thermal cycling conditions for ddPCR reactions were as follows: 1 cycle, 95°C, 10 min; 40 cycles, 94°C, 30 sec, 58°C, 1 min; 1 cycle, 98°C, 10 mins; 4°C hold. PCR products were read with a QX200 Droplet Reader, and absolute concentrations of inserts were determined using QuantaSoft (v1.6.6.0320). Total template (genome or target plasmid) amount was also quantified through this process, and insertion frequency was calculated as inserts/template.
TTISS for AvCAST insertion specificity analysis
AvPSP2, 7, and 12 sites on the E.coli genome were targeted as mentioned above, with the following change: 100 ng of a temperature-sensitive pSC101-donor was used. After incubation on IPTG supplemented triple antibiotic LB-agar plates, cells were re-plated and grown for 12 hours at 43°C to prevent unintended amplification of donor plasmids in the following Tagmentation-based Tag Integration Site Sequencing (TTISS) analysis (Schmid-Burgk et al., 2020). Genomic DNA was extracted from hundreds of colonies on a LB-agar petri dish, and 500 ng genomic DNA (from approximate 108 E. coli cells) was tagmented with Tn5, and re-purified by Wizard SV Gel and PCR Clean-Up System (Promega). Tagmented DNA samples were amplified using two rounds of PCR with KOD Hot Start DNA Polymerase (Millipore) using a Tn5 adapter-specific primer and nested primers within the DNA donor (Table S2). The resulting libraries were sequenced using a NextSeq v2 kit, 75 cycle kit with 45 forward cycles and 30 reverse cycles. Read pairs with R1 containing the terminal 27 bp of the transposon RE sequence were filtered for further analysis and trimmed of the transposon sequence for alignment to the E. coli genome (CP001509). Filtered and trimmed reads were imported into Geneious Prime and aligned using the Geneious aligner with medium-low sensitivity, no fine-tuning, no structural variant, insertion, or deletion discovery enabled, and requiring paired reads to map nearby. The resulting SAM files were exported for further analysis. Aligned R1 reads with length of 18 bp (remaining R1 length after trimming of 27 bp of transposon sequence) and SAM flags 99 (insertion position = POS field) or 147 (insertion position = POS field + trimmed R1 length of 18 bp) were used to determine the transposon insertion position. Reads with an insertion position 50-100-bp downstream of the PAM were considered on-target, whereas remaining reads were counted as off-targets.
qPCR Reactions
Quantitative PCR reactions were performed using TaqMan™ Fast Advanced Master Mix (Applied Biosytems) using 20 uL reactions, 2 uL input of extracted nucleic acid, 900 nM forward primer, 900 nM reverse primer, and 250 nM of probe. Reactions were quantified using the LightCycler 480 Instrument (Roche).
RNA sequencing to recover mature crRNAs
To recover mature RNAs of AvCAST and PmcCAST, each pCascade(AvCAST, AvPSP1 spacer) and pCascade(PmcCAST, PmcPSP1 spacer) was transformed into BL21-CodonPlus (DE3)-RIPL Competent Cells (Agilent 230280). A 10-mL culture was grown in Terrific Broth media (TB) supplemented with 50 μg/ml spectinomycin at 37°C with shaking at 150 rpm until an OD600 of 0.6 was reached. Cascade and crRNA expression was induced by supplementation with IPTG to a final concentration of 0.5 mM. The cells were incubated at 16°C for 16 h , and then harvested by centrifugation for 10 min at 4°C at 4000 rpm. Total RNA was extracted by TRIzol-chloroform (ZYMO RESEARCH) and DNaseI treatment. Ribosomal RNAs were removed by Ribominus Transcriptome Isolation Kit (Invitrogen), and the remaining RNA was treated with 5′ terminator exonuclease, T4 Polynucleotide Kinase, RNA 5′ polyphosphatase, and used as input for NEBNext Small RNA Library Prep Set for Illumina (NEB). Prepared libraries were sequenced using a NextSeq 500/550 High Output Kit v2, 75 cycles. Paired-end reads were aligned to their respective reference expression plasmid sequences using BWA. Resulting transcript sequences were analyzed using Geneious Prime.
Purification of Tn7-like machinery components and in vitro transposition assays
TnsA, B, C, and D of AvCAST and TnsAB, C, and D of PmcCAST were individually cloned into ColE1-based pTwinStrep-SUMO bacterial expression vectors. Each vector was transformed into BL21-CodonPlus (DE3)-RIPL Competent Cells. A 5-mL starter culture was grown in TB supplemented with 100 μg/ml ampicillin for 12 h, which was used to inoculate 4 L of TB for growth at 37°C with shaking at 150 rpm until an OD600 of 0.6 was reached. Protein expression was induced by supplementation with IPTG to a final concentration of 0.5 mM. The cells were incubated at 16°C for 16 h for protein expression, and then harvested by centrifugation for 20 min at 4°C at 4000 rpm (Beckman Coulter Avanti J-E, rotor JLA9.100). Cell pellets were stored at −80°C for later purification. All subsequent steps were performed at 4°C. Cell pellets were resuspended in 200 mL of lysis buffer (50 mM Tris-HCl, 500 mM NaCl, 10% glycerol, 1 mM DTT, pH 8.0) supplemented with cOmplete Protease Inhibitor Cocktail (Millipore Sigma 4693116001). Cells were disrupted using the LM20 Microfluidizer system at 18,000 PSI. Lysate was cleared by centrifugation for 30 min at 4°C at 9500 rpm (Beckman Coulter Avanti J-E, rotor JLA-10.500). The cleared lysate was applied to 2 mL of packed Strep-Tactin Superflow Plus (Qiagen 30004) and incubated with rotation for 1 h, followed by washing of the protein-bound beads in 50 mL of lysis buffer. The bound SUMO-tagged proteins were eluted in 10 mL of lysis buffer supplemented with 2.5 mM desthiobiotin (Sigma). The eluted proteins were further digested by Ulp1 SUMO protease at 4°C for 16 h. Resulting Tns proteins were concentrated using Amicon Ultra Centrifugal Filter Units (Millipore), and protein concentration was estimated by NuPAGE (Invitrogen) and eStain L1 Protein Staining System (GenScript). The concentrated protein was loaded onto a gel filtration column (Superdex 200 Increase 10/300 GL, GE Healthcare) equilibrated with storage buffer (50 mM Tris-HCl, 500 mM NaCl, 5% glycerol, 2 mM DTT, pH 7.5) via FPLC. The resulting fractions from gel filtration were analyzed, and the fractions containing the protein were pooled, snap frozen as 1 mg/mL aliquots, and stored at −80°C. In vitro transposition reactions were carried out with 200 nM of each Tns protein, 100 ng of pDonor(AvCAST)_CmR, and 100 ng of pTarget for AvCAST in vitro (E. coli - glmS) in a final reaction buffer of 26 mM HEPES pH 7.5, 5 mM Tris-HCl pH 8.0, 50 μg/mL BSA, 2 mM ATP, 2.1 mM DTT, 0.05 mM EDTA, 0.2 mM MgCl2, 50 mM NaCl, 21 mM KCl, 1% glycerol supplemented with 15 mM Mg(OAc)2 based on in vitro Tn7 reaction protocol (Bainton et al., 1991). Total reaction volumes were 20 uL, and reactions were incubated for 2 hours at 30°C. For PmcCAST, pDonor(PmcCAST)_CmR and pTarget for PmcCAST (bearing ATG+PmcPSP1 and tRNA-val) were used. The resulting products were purified using Wizard SV Gel and PCR Clean-Up System (Promega), followed by diagnostic PCR.
Comparative genomic analysis of CAST loci and TniQ/TnsD analysis
We searched for new CAST loci using type I-F, I-B, and V-K components in prokaryotic and metagenomic databases (nucleotide sequence available in August 2019 in NCBI database restricted to Prokaryotic sequences WGS and Genbank, and MG-RAST (Keegan et al., 2016)). First, we identified contigs where CAST components (TnsA, TnsB, TnsC, TniQ/TnsD plus Cascade IB, and TnsB, TnsC, TniQ plus Cas12k) are colocalized to form a full CAST locus (CAST-IB: TnsA, TnsB, TnsC, TniQ/TnsD plus Cascade IB; CAST-IV: TnsB, TnsC, TniQ plus Cas12k) within a 50-kb region using tblastn (bitscore superior or equal to 30) (Altschul et al., 1990) from each CAST component extracted from previous literature (Faure et al., 2019a; Peters et al., 2017). Then, we extracted each CAST component from all contigs, clustered them by protein type (TnsA, TnsB, TnsC, TniQ/TnsD, Cas6Ib, Cas7b, Cas8b, Cas5b, Cas12k) and by sequence similarity using mmseqs2(Steinegger and Söding, 2017) (50 percent of sequences identity 50 percent of coverage), aligned members together within each clusters using mafft-einsi (Katoh et al., 2005), and created a hmm profile for each cluster using hmmer (Eddy, 2009; Katoh et al., 2005). From these profiles, we searched for additional contigs where CAST components (TnsA, TnsB, TnsC, TniQ/TnsD plus Cascade IB, and TnsB, TnsC, TniQ plus Cas12k) are colocalized within a 50-kb region using hmmsearch (bitscore superior or equal to 30) against a translated version of our nucleotide database. For each CAST locus, we predicted gene boundaries using prodigal version 2.6.3 (model meta)(Hyatt et al., 2010) and Cas profiles database (Makarova et al., 2020). We predicted CRISPR arrays using minced software (https://github.com/ctSkennerton/minced) (minimum repeat size 16, maximum repeat size 50, minimum spacer size 20, maximum spacer size 50). We predicted tRNA and other ncRNA using tRNA-ScanSE version 2 (Chan and Lowe, 2019) and Infernal version 1.1.3 (Chan and Lowe, 2019; Griffiths-Jones, 2003; Nawrocki and Eddy, 2013) with Rfam database. Transposon ends were determined manually on Geneious framework (Kearse et al., 2012; Price et al., 2009) using the following methodology. We searched for repeat segments of 10 nucleotides with a maximum of 3 mismatched nucleotides and looked for inverted repeats embedding both the transposon components and the Cas components. For every candidate, we used the opposite inverted repeat and ran a similar search. We looked for two blocks of inverted clustered repeats that embed CAST components and validated the ends when possible by searching for the duplication site on both sides of the transposon. If no candidate was found with this repeat search method, we used blastn-short (Altschul et al., 1990) (word side=7) to initially identify repeats, and performed the same downstream strategy to find the transposon’s ends. The structural comparison of TniQ was performed under PyMOL framework version 1.2 (The PyMOL Molecular Graphics System, Schrödinger, LLC) and using Hydrophobic Cluster Analysis (Callebaut et al., 1997; Faure and Callebaut, 2013a, 2013b). Phylogenetic trees were built with FastTree (Price et al., 2009) using the Whelan And Goldman model (WAG model) and visualized with iTOL (Letunic and Bork, 2007).
QUANTIFICATION AND STATISTICAL ANALYSIS
The number and definition of replicates for each experiment can be found in the corresponding figure legend. To quantitatively define PAM preference, insertion read counts stratified by the PAM sequence at the 6N randomized library position were normalized by abundance of the corresponding PAM in the input library. PAMs displaying greater than 16-fold enrichment in abundance compared to the input library were input into WebLogo (https://weblogo.berkeley.edu/) to obtain the corresponding sequence logo. Sequencing data was converted to coverage maps using Geneious Prime and its built-in Geneious aligner. All remaining quantitative data was processed and visualized using GraphPad Prism.
Supplementary Material
Table S1| Type V-K CAST hidden crRNA and targets, Related to Figure 2.
Identification and analysis of Type V-K loci in bacterial metagenomes with annotated homing targets, delocalized crRNA and target sequences, and relevant distances between features.
Table S2| Sequence information related to this study, Related to STAR Methods.
The table contains sequences of plasmids, transposon ends, and crRNA sequences for each CAST system experimentally studied here.
Figure S2| Comparison of delocalized crRNA across Type V-K CAST systems, Related to Figure 2 (A) Alignment of delocalized direct repeats from diverse type V-K CAST systems. (B) Alignment of spacers from delocalized crRNA of type V-K CAST systems, annotated with their corresponding tRNA target. (C) Start and end position of RNA sequencing reads aligned to the predicted delocalized crRNA in A. cylindrica. The predicted delocalized DR and tRNA match are annotated as determined by homology to the canonical DR and downstream tRNA target respectively.
Figure S1| Characterization of Type V-K CAST short, delocalized crRNA, Related to Figure 1 (A) Insertion activity of ShCAST components in E. coli into a CRISPR and homing target plasmid. (B) Alignment of typical direct repeats from the CRISPR array with short direct repeat from ShCAST. (C) Insertion activity of crRNA containing the delocalized direct repeat or the array direct repeat with a PSP1 spacer; promoters and terminators are indicated with an arrow and hairpin, respectively. Both the 17-nt spacer and 14-nt spacer extension match the target sequence. Data are represented as mean ± SD. (D) Insertion activity of ShCAST homing following truncation of its delocalized crRNA. Data are represented as mean ± SD. (E) Effect of sgRNA spacer length on insertion frequency of ShCAST on PSP15 through deletion of 3′ spacer sequence. Data are represented as mean ± SD. (F) Effect of sgRNA spacer length on insertion frequency of ShCAST on PSP42 through deletion of 3′ spacer sequence. Data are represented as mean ± SD. (G) Effect of double mismatches in the sgRNA spacer at the indicated positions on insertion frequency of ShCAST on PSP15. Positions are relative to the 5′ NGTN PAM. Data are represented as mean ± SD.
Figure S4| RNA-guided transposition on E.coli genome by Type I-B subtype1 AvCAST, Related to Figure 4 (A) AvCAST RNA-guided E.coli genomic insertions at 10 targeted loci. Representative data for LE junction amplification are shown. (B) Insertion frequency for 36 targeted loci (AvPSP2 - 37) quantified by ddPCR. Data are represented as mean ± SD. (C) Type I-B subtype1 AvCAST RNA-guided insertion positions on 36 targeted loci by deep sequencing. Both RE and LE junctions were sequenced and representative insertion reads at each position measured by LE junction data are provided. Insertion at PSPs indicated in red were below the limit of detection (few or no insertion reads). (D) Comparison of insertion frequency between pSC101and pBluescript donors. Data are represented as mean ± SD. (E) TTISS analysis of three representative loci (AvPSP2, AvPSP7, and AvPSP12) to test insertion specificity of AvCAST. Reads are mapped on CP001509 E.coli BL21(DE3) complete genome.
Figure S5| Type I-B subtype1 AvCAST system homes to glmS, Related to Figure 5 (A) Alignment of CAST-IB1 loci at the insertion site. Ends of glmS are highlighted in orange while the CAST left end (LE) is shown in grey. Only 2 identical contigs have the transposon inserted 5-bp downstream of glmS while all other transposons are inserted about 24-bp downstream of the homing target site. (B) Deep sequencing reads showing Tn7-like machinery of AvCAST mediates a single prominent insertion at glmS Tn7 attachment site on plasmids. Purple bar indicates the insertion position on the plasmid harboring E. coli glmS. Blue bar indicates the insertion position on the plasmid harboring A. variabilis glmS. (C) Tn7-like machinery-mediated insertion frequency at the glmS gene on the E. coli BL21(DE3) genome in the absence of TniQ, TnsD, or both. Data are represented as mean ± SD. (D) Sanger sequencing chromatograms for RE and LE junctions of insertion at the endogenous glmS Tn7 attachment site on E.coli BL21(DE3) genome. “GCGGG” is target site duplication as a signature of Tn7-like transposition. (E) AvCAST-mediated single prominent insertion at the endogenous glmS Tn7 attachment site on E.coli BL21(DE3) genome identified by deep sequencing. Left: with all Tn7-like machinery of AvCAST; right: with all AvCAST proteins including TniQ and Cascade proteins. (F) Purified Tn7-like machinery components of AvCAST on Coomassie Brilliant Blue stained SDS-PAGE gel.
Figure S7| Models comparing CAST V-K and CAST I-F, Related to Figure 7 Prototypical locus organization of CAST V-K systems and CAST I-F systems and models for CAST transposition to mobile element (donor cell in blue) and CAST homing transposition to bacterial chromosome (recipient cell in green). The loci are delimited by transposon ends (Tn ends, indicated as light vertical rectangles). Transposon core components (tnsA, tnsB, and tnsC in CAST I-F; tnsB and tnsC in CAST V-K) are colored in light blue while tniQ is colored in dark blue. Cas components (Cascade I-F and cas12k) are shown in grey. Conserved transcription factors (TF) are shown in pink. Locations of cargo genes are indicated with dark grey circles. CRISPR arrays are shown as grey triangles for the repeats, and red diamonds for the spacers. In CAST V-K the delocalized crRNA is indicated by a lighter triangle (partial repeat), and a truncated orange diamond (short spacer). In CAST I-F the homing spacer (targeting the homing target site) is indicated as a light orange diamond located at the distal spacer of the CRISPR-array. Homing target genes are colored in large yellow rectangles. Horizontal light grey triangle indicates the tracrRNA in the CAST V-K locus. CAST transposition to mobile genetic elements is mediated by the Cas effector machinery (I-F Cascade and crRNA or Cas12k, tracrRNA, and cRNA) which recognizes the target site (red) with RNA-guided targeting to insert the transposon using Tn components. For homing transposition, CAST V-K uses a dedicated crRNA (spacer in light orange) bearing a truncated spacer to target the homing site while CAST I-F uses a dedicated spacer, either located at the distal region of the CRISPR array or delocalized, to target the homing target site often with numerous mismatches between spacer and target (Petassi et al. 2020).
Figure S6| Characterization of Type I-B subtype2 PmcCAST, Related to Figure 6 (A) RNA sequence reads mapping to the minimal CRISPR array showing the mature crRNA sequence of PmcCAST. (B) Insertion directionality assayed by diagnostic PCR of pInsert on 6N PAM library plasmid with different primer pairs in ΔTniQ or ΔTnsD conditions. (C) Quantification of insertion frequency across the LE and RE junctions by ddPCR. Data are represented as mean ± SD. (D) Sanger sequencing chromatograms of a representative pInsert. PAM, PmcPSP1, TSDs, transposon ends are annotated. (E) Coomassie Brilliant Blue stained SDS-PAGE gel for purified Tn7-like machinery components of PmcCAST.
Figure S3| CAST I-B loci comparison and TnsD/TniQ analysis, Related to Figures 3 and 6 (A) Left, CAST I-B1 loci sharing similar TniQ (non identical and greater than or equal to 50 percent sequence identity). Right, Loci sharing similar CAST I-B1 tnsD (greater than or equal to 50 percent sequence identity). tniQ is colored in red, transposon core genes (tnsA, tnsB, and tnsC) in blue, Cas components in yellow, glmS homing gene in green, CRISPR arrays are shown as thin empty red arrays, and tnsD in pink. Black arrows represent padding to center the tniQ or tnsD genes. Cyan indicates tnsE, a hallmark gene for targeting conjugative plasmids in Tn7. Loci centered on tnsD (right) include Tn7 loci (non CAST) that also home to glmS (green). All loci are available in the Supplementary Dataset (B) Protein sequence alignment of CAST I-B1 TniQ (red), TnsD (purple), and Tn7-TnsD (green) sharing sequence similarity with CAST I-B1 TnsD. Sequences are ordered from a tree built with FastTree (WAG model) (Price et al., 2009). Blue highlights indicate the conserved core of TniQ/TnsD. On top of the alignment sequences are the consensus, the hydrophobicity graph (red), and the conservation graph (green). The alignment is colored using the Rasmol color palette; colored positions indicate conservation in agreement with the consensus sequence. Tn7 TnsD and CAST-IB TnsD are longer than TniQ, and are similar in their C-terminal region. (C) CAST I-B2 loci sharing similar TniQ (non identical and greater than or equal to 50 percent sequence identity). tniQ is colored in red, transposon core genes (tnsAB and tnsC) in blue, Cas components in yellow, tRNA homing gene in green, CRISPR arrays are shown as thin empty red arrays, and tnsD in pink. (D) Protein alignment of TniQ/TnsD core region from CAST I-B TniQ (red), TnsD (purple), and Tn7-TnsD (green) , CAST I-F TniQ (light green), and CAST V-K TniQ (orange). On top of the alignment sequences are the consensus and the conservation graph (green). The alignment is colored using the Rasmol color palette; colored positions indicate conservation in agreement with the consensus sequence. Domain annotation of TniQ dimer mapped on the electron microscopy structure (pdb code: 6pij) (Halpin-Healy et al., 2019) of CAST I-F TniQ. The different colors indicate the potential domains of TniQ, from N-terminal to C-terminal, blue, green, orange, and red. TniQ/TnsD core (red dashed line) is formed by the blue and the green domain (region conserved in CAST TniQ/TnsD and Tn7-TnsD). Black dashed lines indicate interaction with Cascade components found in the structure.
Supplementary Dataset (SupDataset.zip) | CAST loci, Related to STAR Methods.
Annotated CAST loci from V-K, I-B subtype 1, and I-B subtype 2 in Genbank format. Each locus is centered on the tniQ gene, which is annotated as “Center”. All loci have a unique TniQ/TnsD protein: all loci with identical TniQ/TnsD proteins were discarded to keep one representative with longest distance to the edges of the contig. The centered tniQ/tnsD is surrounded by a maximum of 50-kb upstream and downstream (or less if the locus is near the edge of a contig). A padding was used when the centered tniQ/tnsD was near the contig edge. Proteins are annotated from Pfam or Cas profile database (annotated Protein, or CAS_CRISPR). Tn7 components are annotated as Tns_tni. Proteins sharing at least 50% sequence identity have a “Clu” annotation followed by a unique number. tRNA and other ncRNAs are annotated under tRNA and RNA.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial Strains | ||
| One Shot™ PIR1 Chemically Competent E. coli | ThermoFischer | C101010 |
| TransforMax™ EC100D™ pir+ Electrocompetent E.coli | Lucigen | ECP09500 |
| BL21(DE3) Competent E. coli | New England Biolabs | C2527 |
| BL21(DE3) Electrocompetent Cells | Millipore Sigma | CMC0016 |
| Endura™ ElectroCompetent Cells | Lucigen | 60242 |
| One Shot™ Stbl3™ Chemically Competent E. coli | ThermoFischer | C737303 |
| BL21-CodonPlus (DE3)-RIPL Competent Cells | Agilent | 230280 |
| Trichormus variabilis | ATCC | 29413 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| NEBNext® High-Fidelity 2X PCR Master Mix | New England Biolabs | M0541 |
| Q5® High-Fidelity 2X Master Mix | New England Biolabs | M0492 |
| KAPA HiFi HotStart ReadyMix | Roche | KK2602 |
| KOD Hot Start DNA Polymerase | Millipore Sigma | 71086-3 |
| E-Gel™ EX Agarose Gels, 1% | ThermoFischer | G401001 |
| E-Gel™ EX Agarose Gels, 2% | ThermoFischer | G401002 |
| E-Gel™ 48 Agarose Gels, 2% | ThermoFischer | G800802 |
| Wizard® SV Gel and PCR Clean-Up System | Promega | A9282 |
| Wizard® Genomic DNA Purification Kit | Promega | A1120 |
| QIAquick PCR Purification Kit | Qiagen | 28106 |
| QIAprep Spin Miniprep Kit | Qiagen | 27106 |
| PureYield™ Plasmid Midiprep System | Promega | A2495 |
| Gibson Assembly® Master Mix | New England Biolabs | E2611 |
| NEBuilder® HiFi DNA Assembly Master Mix | New England Biolabs | E2621 |
| Q5® Site-Directed Mutagenesis Kit | New England Biolabs | E0554 |
| FastDigest LguI | ThermoFischer | FD1934 |
| BsaI-HF®v2 | New England Biolabs | R3733 |
| BbsI-HF® | New England Biolabs | R3539 |
| T4 DNA Ligase | New England Biolabs | M0202 |
| IPTG | Goldbio | I2481C |
| TRI Reagent | ZYMO RESEARCH | R2050-1-200 |
| RNA Clean & Concentrator-25 | ZYMO RESEARCH | R1017 |
| DNase I (RNase-free) | New England Biolabs | M0303 |
| SUPERase•In™ RNase Inhibitor (20 U/μL) | ThermoFischer | AM2694 |
| Ribominus™ Transcriptome Isolation Kit | ThermoFischer | K155004 |
| Terminator™ 5′-Phosphate-Dependent Exonuclease | Lucigen | TER51020 |
| RiboGuard™ RNase Inhibitor | Lucigen | RG90910K |
| T4 Polynucleotide Kinase | New England Biolabs | M0201 |
| RNA 5′ Polyphosphatase | Lucigen | RP8092H |
| NEBNext® Small RNA Library Prep Set for Illumina® (Multiplex Compatible) | New England Biolabs | E7330 |
| TaqMan Fast Advanced Master Mix | ThermoFischer | 4444963 |
| ddPCR Supermix for Probes (No dUTP) | Bio-Rad | #1863024 |
| Droplet Generation Oil for Probes | Bio-Rad | #1863005 |
| FastDigest Eco32I | ThermoFischer | FD0304 |
| NEBNext® Companion Module for Oxford Nanopore Technologies® Ligation Sequencing | New England Biolabs | E7180S |
| Flow Cell (R9.4) | Oxford Nanopore Technologies | FLO-MIN106 |
| AMPure XP for PCR Purification | Beckman Coulter | A63881 |
| Tn5 | Schmid-Burgk et al., 2020. | N/A |
| cOmplete Protease Inhibitor Cocktail | Millipore sigma | 4693116001 |
| Strep-Tactin Superflow Plus | Qiagen | 30004 |
| D-Desthiobiotin | Millipore sigma | 71610-3 |
| Ulp1 SUMO protease | F. Zhang Lab | N/A |
| Amicon Ultra-15 Centrifugal Filter Units 30kDa NMWL | Millipore sigma | UFC903024 |
| Amicon Ultra-15 Centrifugal Filter Units 50kDa NMWL | Millipore sigma | UFC905024 |
| NuPAGE™ 4-12% Bis-Tris Protein Gels, 1.0 mm, 12-well | ThermoFischer | NP0322BOX |
| NuPAGE™ LDS Sample Buffer (4X) | ThermoFischer | NP0007 |
| Ampicillin, sodium salt | AmericanBio | Ab00115 |
| Carbenicillin disodium salt,89.0-100.5% anhydrous basis | Millipore sigma | C1389 |
| Spectinomycin dihydrochloride pentahydrate | Millipore sigma | S4014 |
| Kanamycin sulfate from Streptomyces kanamyceticus | Sigma | K4000 |
| Chloramphenicol | Sigma | C0378 |
| MiSeq Reagent Kits v2 | Illumina | MS-102 |
| NextSeq 500/550 High Output Kit v2, 75 cycles | Illumina | FC-404-2005 |
| Critical Commercial Assays | ||
| Qubit 1X dsDNA HS (High-Sensitivity) Assay Kit | ThermoFischer | Q33231 |
| eStain L1 Protein Staining System | GenScript | N/A |
| Deposited Data | ||
| Oligonucleotides | ||
| ctgtcgtcggtgacagattaatgtcattgtgac | IDT | Insert Probe |
| aacgctgatgggtcacgacg | IDT | T14 LE Reverse Primer |
| GACTGGAGTTCAGACGTGTGCTCTTCCGATCTtatctgcaaagtcgctgggg | IDT | PSP15 Target Forward Primer |
| GACTGGAGTTCAGACGTGTGCTCTTCCGATCTacgtacttcgccacctgaag | IDT | PSP42 Target Forward Primer |
| cgcggcaactttgtagtaccagc | IDT | E. coli Genome Forward Primer |
| cccttttcagatttctgcccgacgc | IDT | E. coli Genome Reverse Primer |
| acgttcgcgtttgccgtgcgtgtaatgtagtac | IDT | E. coli Genome Probe |
| gagcaagagattacgcgcagac | IDT | PAM library 6N upstream Primer |
| ctgcgtatgtgggcgctgcc | IDT | PAM library 6N downstream Primer |
| tgaaccgaattatatattgttgaacc | IDT | AvCAST RE Primer |
| taatggattcaaacaattgtttaaccg | IDT | AvCAST LE Primer |
| CTTTCCCTACACGACGCTCTTCCGATCTgagcaagagattacgcgcagac | IDT | PAM libarry 6N upstream NGS Primer |
| CTTTCCCTACACGACGCTCTTCCGATCTctgcgtatgtgggcgctgcc | IDT | PAM library 6N downstream NGS Primer |
| GACTGGAGTTCAGACGTGTGCTCTTCCGATCTcttttacttggtacaatcgtactcctttagagg | IDT | AvCAST RE NGS Primer |
| GACTGGAGTTCAGACGTGTGCTCTTCCGATCTcctttatcagtaatggattcaaacaattgtttaaccg | IDT | AvCAST LE NGS Primer |
| cgacagcatcgccagtcactatg | IDT | pTarget control forward Primer for ddPCR |
| caagtagcgaagcgagcaggac | IDT | pTarget contro reverse Primer for ddPCR |
| tgcgttgatgcaatttctatgcgcacccgt | IDT | pTarge control ddPCR Probe |
| agtcatttaataaggccactgttaaacg | IDT | PSP1 insertion site upstream for ddPCR |
| ctaccgcattaaagcttccgcc | IDT | PSP1 insertion site downstream Primer for ddPCR |
| cgacaacggcagattaaatattcgaactgt | IDT | AvCAST LE ddPCR Probe |
| ttcgaactgtaataattgtttaaccg | IDT | AvCAST-TTISS_LE 1st PCR Primer |
| gtctcgtgggctcggagatgtgtataagagacag | IDT | TTISS 1st PCR common Primer |
| AATGATACGGCGACCACCGAGATCTACACAAGTAGAGACACTCTTTCCCTACACGACGctcttccgatctTTGTTTAACCGAATTCTTTGTCTAC | IDT | AvCAST-TTISS_LE 2nd PCR Primer |
| caagcagaagacggcatacgagatcgatcatggtctcgtgggctcggagatgtgt | IDT | TTISS 2nd PCR comon Primer-1 |
| caagcagaagacggcatacgagattagatcctgtctcgtgggctcggagatgtgt | IDT | TTISS 2nd PCR comon Primer-2 |
| caagcagaagacggcatacgagatttactgtcgtctcgtgggctcggagatgtgt | IDT | TTISS 2nd PCR comon Primer-3 |
| caagcagaagacggcatacgagatggcatagggtctcgtgggctcggagatgtgt | IDT | TTISS 2nd PCR comon Primer-4 |
| caagcagaagacggcatacgagatcaaggcgagtctcgtgggctcggagatgtgt | IDT | TTISS 2nd PCR comon Primer-5 |
| caagcagaagacggcatacgagatgacgctatgtctcgtgggctcggagatgtgt | IDT | TTISS 2nd PCR comon Primer-6 |
| gcagaataaataaatcctggtgtc | IDT | pTarget glmS downstream Primer for ddPCR |
| CTTTCCCTACACGACGCTCTTCCGATCTagttggcagcatcacccgacgc | IDT | pTarget glmS downstream NGS primer |
| tggcaggatgtttgattaaaaac | IDT | Non-coding region between pstS and glmS ddPCR Primer |
| agcgataacatgcacatcatc | IDT | glmS Primer |
| CTTTCCCTACACGACGCTCTTCCGATCTgtgattgcaccaatcttctacaccgttc | IDT | glmS NGS Primer |
| GACTGGAGTTCAGACGTGTGCTCTTCCGATCTgcaaagttatggttccagtaacggc | IDT | PmcCAST RE NGS Primer |
| GACTGGAGTTCAGACGTGTGCTCTTCCGATCTgcataagcttgccgttgcgg | IDT | PmcCAST LE NGS Primer |
| gtaagctgttatatgtaaaactagaacagc | IDT | PmcCAST LE primer |
| CTTTCCCTACACGACGCTCTTCCGATCTagcttccttagctcctgaaaatctcg | IDT | pTarget tRNA-val upstream NGS Primer |
| tctggtactgcccagtagtg | IDT | tRNA-val primer for ddPCR |
| acccataactttgccgcaacggcaagctta | IDT | PmcCAST LE ddPCR Probe |
| CACTTTCATATCAGCATACTCCTGCTG | IDT | AvPSP2_Upstream Primer |
| CCCTCACCCCAGTCACTTACTTATG | IDT | AvPSP3_Upstream Primer |
| CTCACCCGGCAATTTCCCATTC | IDT | AvPSP4_Upstream Primer |
| TGTGAAGCTATGCGTTGCTGCC | IDT | AvPSP5_Upstream Primer |
| GATTAAATGTGACGCGACCCGC | IDT | AvPSP6_Upstream Primer |
| GTCCCAGTCCGATCTCTTGC | IDT | AvPSP7_Upstream Primer |
| CGGGCGGGATCGAGTATACAC | IDT | AvPSP8_Upstream Primer |
| TGCCTGACTTTGCTGCGCC | IDT | AvPSP9_Upstream Primer |
| TATCTGATGAGTTCCAAATTATGCCC | IDT | AvPSP10_Upstream Primer |
| ACCGTTGCATCATGTCGCCC | IDT | AvPSP11_Upstream Primer |
| ATTAGAAGGCGCACAATGTTACAGC | IDT | AvPSP12_Upstream Primer |
| CGACAGAACAACGATTGCCAGAG | IDT | AvPSP13_Upstream Primer |
| CTCCATTTTACCCATCCAGGGC | IDT | AvPSP14_Upstream Primer |
| AACGCTGCAACGTCCTGAGC | IDT | AvPSP15_Upstream Primer |
| CAGGTCGCCCGGATTACAGC | IDT | AvPSP16_Upstream Primer |
| CAGCTGGGCATTCGGTTGC | IDT | AvPSP17_Upstream Primer |
| ACGTCAGCATCCTAACAGCACC | IDT | AvPSP18_Upstream Primer |
| TATTAGTCTGAGTTATTTTTGTAGGGC | IDT | AvPSP19_Upstream Primer |
| GGACGTTAAAACGTCACGGCC | IDT | AvPSP20_Upstream Primer |
| GTCTGTGCGCTATGCCTATATTGG | IDT | AvPSP21_Upstream Primer |
| GAATTCGCTTTCTTGGAGAGCTTCTG | IDT | AvPSP22_Upstream Primer |
| ATCTGCAGGCTTTGCAGTCTGC | IDT | AvPSP23_Upstream Primer |
| TGAGTCCGCCCGGAACTCG | IDT | AvPSP24_Upstream Primer |
| GCTTAAGAATCGTAGAACCATCGGC | IDT | AvPSP25_Upstream Primer |
| GAGCCCGTGAACTGAAACCCTC | IDT | AvPSP26_Upstream Primer |
| CGACGGTGGTACGCATAACTTTC | IDT | AvPSP27_Upstream Primer |
| CGGTTTTACCCCTGTTACACGGG | IDT | AvPSP28_Upstream Primer |
| CTGCTAAAAATCGGCGCTAAGAACC | IDT | AvPSP29_Upstream Primer |
| CAGCTGAGACTGGATTTTTTCCAGC | IDT | AvPSP30_Upstream Primer |
| CTGTGCGGTGCCACAATCGG | IDT | AvPSP31_Upstream Primer |
| GCTGTTAGTCATGATTGGCCTGC | IDT | AvPSP32_Upstream Primer |
| CATCCGGCATTAAGCACCAACC | IDT | AvPSP33_Upstream Primer |
| GCAATGAACGATTATCCCTATCAAGC | IDT | AvPSP34_Upstream Primer |
| CAACCAGGCAACATGCCCGAC | IDT | AvPSP35_Upstream Primer |
| TGGCTATTCATTTGAAAGGAGGTCTG | IDT | AvPSP36_Upstream Primer |
| GTCATTCCGCTGCATATCATGAACGC | IDT | AvPSP37_Upstream Primer |
| TTGCATTCTGCGGGAAGGGATATC | IDT | AvPSP2_Downstream Primer |
| AGCGAATAGACAAATCGGTTGCCG | IDT | AvPSP3_Downstream Primer |
| GCCTGTGAACACACAGACAGAG | IDT | AvPSP4_Downstream Primer |
| GACTCTAACCGTCGGCCCC | IDT | AvPSP5_Downstream Primer |
| CTCATCGCCATTTACCTTCATGATAG | IDT | AvPSP6_Downstream Primer |
| GATCTGCGTTTTACAACTCAGATCAC | IDT | AvPSP7_Downstream Primer |
| CGGAAATCATACCATGAGGTTAATCC | IDT | AvPSP8_Downstream Primer |
| TGAACATAACGCCGATAGCAAAAGG | IDT | AvPSP9_Downstream Primer |
| TGGGCAAAATCAAGATACGCGCAG | IDT | AvPSP10_Downstream Primer |
| TAATTGCTAAGGAATAACCCAGTTGC | IDT | AvPSP11_Downstream Primer |
| CCTGGCCGTAAATGAAACACGC | IDT | AvPSP12_Downstream Primer |
| CTGAAAAAGTTTGTCCGCGATGC | IDT | AvPSP13_Downstream Primer |
| GCGCGGGGAACACTCTAAAC | IDT | AvPSP14_Downstream Primer |
| CGTAAGTTTCGCAGCTTATTAACAGC | IDT | AvPSP15_Downstream Primer |
| CCTTATCCGGCCTACAAAGTCG | IDT | AvPSP16_Downstream Primer |
| TTCTACGGGCAGGATGACTGG | IDT | AvPSP17_Downstream Primer |
| CGTGGGAAGGAGGCTATAATGG | IDT | AvPSP18_Downstream Primer |
| GCGTTCTGGTTGAATGGAACGC | IDT | AvPSP19_Downstream Primer |
| GTTGCCAGCAGGTATTATATCGCC | IDT | AvPSP20_Downstream Primer |
| GCAATGTTGCACCGTTTGCTGC | IDT | AvPSP21_Downstream Primer |
| GGTGGTTGACCTAAGGTAGCAG | IDT | AvPSP22_Downstream Primer |
| CCTTTCGCCCTGAATGCAGTC | IDT | AvPSP23_Downstream Primer |
| TCCGACAGTAAGCAAAAATTTGAGAC | IDT | AvPSP24_Downstream Primer |
| AGGTATTTAACCTGTGTTGATTGCTG | IDT | AvPSP25_Downstream Primer |
| ACGCAGCATTAATGCATAGTGGTTAAG | IDT | AvPSP26_Downstream Primer |
| GCGGGGCATTTTTCTTCCTGTTATG | IDT | AvPSP27_Downstream Primer |
| CACTAAACCGGGCCGTTTAGCC | IDT | AvPSP28_Downstream Primer |
| CATATTGCGCATGTTCGCGCAC | IDT | AvPSP29_Downstream Primer |
| GAAAAGCGCGCAAAGTGCGG | IDT | AvPSP30_Downstream Primer |
| GAATGCTTATACTGAAGACCGCGC | IDT | AvPSP31_Downstream Primer |
| CAAGAACAAGGCCATCCCTTTACC | IDT | AvPSP32_Downstream Primer |
| CCACTGTGTCGCTAAAAAGAGACAAC | IDT | AvPSP33_Downstream Primer |
| TTTGAGCCTGGCTTATCGCCG | IDT | AvPSP34_Downstream Primer |
| ATGATGCGGGTTCGATTCCCG | IDT | AvPSP35_Downstream Primer |
| CCGCCAGCTGAAGAAATCGCTAATTC | IDT | AvPSP36_Downstream Primer |
| CATCGGCTACGATGTAAAAATGGGTC | IDT | AvPSP37_Downstream Primer |
| Recombinant DNA | ||
| pHelper_ShCAST | Addgene | #127922 |
| pUC19 Vector | New England Biolabs | N3041 |
| pBluescript II SK (+) | Agilent | 212205 |
| pCDFDuet™-1 DNA | Millipore sigma | 71340 |
| pCOLADuet™-1 DNA | Millipore sigma | 71406 |
| pDonor(pSC101 origin)_ShCAST | F. Zhang Lab | N/A |
| pACYC 6N PAM library | Strecker et al., 2019. | N/A |
| pDonor_ShCAST_kanR | Addgene | #127924 |
| Software and Algorithms | ||
| Geneious | https://www.geneious.com/ | v2020 |
| QuantaSoft™ Software | Bio-Rad | #1864011 |
| Hmmer | http://hmmer.org/ | v3.1 |
| Blast+ | https://blast.ncbi.nlm.nih.gov/ | v2.9.0 |
| iTOL | https://itol.embl.de/ | N/A |
| MAFFT | https://mafft.cbrc.jp/ | V7.407 |
| Minced | https://github.com/ctSkennerton/minced | v0.4.2 |
| Prodigal | https://github.com/hyattpd/Prodigal | v2.6.3 |
| tRNA-ScanSE | http://lowelab.ucsc.edu/tRNAscan-SE/ | v2 |
| Infernal | http://eddylab.org/infernal/ | v1.1.3 |
| PyMOL | https://pymol.org/2/ | v1.2 |
| FastTree | http://www.microbesonline.org/fasttree/ | v2.1 |
| Hydrophobic Cluster Analysis | http://osbornite.impmc.upmc.fr/hca/hca-form.html | N/A |
| Other | ||
| Bench Protocol | (this paper) | STAR Methods |
Acknowledgements
We thank I. Peng, P. Clauer, and J. Kreitz for their assistance with cloning and E.coli transposition assays; S. Kannan for her assistance with RNA sequencing analysis of CAST I-B systems; J. Schmid-Burgk and L. Gao for useful discussions; and the entire Zhang laboratory for support and advice. We also thank A. Farina for preparation of reagents. M.S. was supported by a fellowship from the Swiss National Science Foundation and is currently supported by a Long-Term Fellowship from Human Frontier Science Program. J.S. was supported by a Long-Term Fellowship from the Human Frontier Science Program. F.Z. is supported by the NIH (1DP1-HL141201, R01-HG009761, and RM1-HG006193 grants); G. Harold and Leila Y. Mathers, Edward Malinckrodt Jr., and Gates Foundations; the Howard Hughes Medical Institute; the Poitras Center for Psychiatric Disorders Research, the Open Philanthropy Project; the Hock E. Tan and K. Lisa Yang Center for Autism Research at MIT; and J. and P. Poitras, the Phillips family, and R. Metcalfe.
Footnotes
Competing Interests
The Broad Institute has filed patent applications related to this work. F.Z. is a scientific advisor and cofounder of Editas Medicine, Beam Therapeutics, Pairwise Plants, Arbor Biotechnologies, and Sherlock Biosciences.
References
- Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ (1990). Basic local alignment search tool. J. Mol. Biol 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Bainton R, Gamas P, and Craig NL (1991). Tn7 transposition in vitro proceeds through an excised transposon intermediate generated by staggered breaks in DNA. Cell 65, 805–816. [DOI] [PubMed] [Google Scholar]
- Barrangou R, and Horvath P (2017). A decade of discovery: CRISPR functions and applications. Nature Microbiology 2. [DOI] [PubMed] [Google Scholar]
- Callebaut I, Labesse G, Durand P, Poupon A, Canard L, Chomilier J, Henrissat B, and Mornon JP (1997). Deciphering protein sequence information through hydrophobic cluster analysis (HCA): current status and perspectives. Cell. Mol. Life Sci 53, 621–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PP, and Lowe TM (2019). tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. Methods Mol. Biol 1962, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR (2009). A new generation of homology search tools based on probabilistic inference. Genome Inform. 23, 205–211. [PubMed] [Google Scholar]
- Faure G, and Callebaut I (2013a). Comprehensive repertoire of foldable regions within whole genomes. PLoS Comput. Biol 9, e1003280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure G, and Callebaut I (2013b). Identification of delocalized relationships from the coupling of hydrophobic cluster analysis and domain architecture information. Bioinformatics 29, 1726–1733. [DOI] [PubMed] [Google Scholar]
- Faure G, Shmakov SA, Yan WX, Cheng DR, Scott DA, Peters JE, Makarova KS, and Koonin EV (2019a). CRISPR–Cas in mobile genetic elements: counter-defence and beyond. Nat. Rev. Microbiol 17, 513–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure G, Makarova KS, and Koonin EV (2019b). CRISPR-Cas: Complex Functional Networks and Multiple Roles beyond Adaptive Immunity. J. Mol. Biol 431, 3–20. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S (2003). Rfam: an RNA family database. Nucleic Acids Research 31, 439–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpin-Healy TS, Klompe SE, Sternberg SH, and Fernández IS (2019). Structural basis of DNA targeting by a transposon-encoded CRISPR–Cas system. Nature 577, 271–274. [DOI] [PubMed] [Google Scholar]
- Hyatt D, Chen G-L, Locascio PF, Land ML, Larimer FW, and Hauser LJ (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Kuma K-I, Toh H, and Miyata T (2005). MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan KP, Glass EM, and Meyer F (2016). MG-RAST, a Metagenomics Service for Analysis of Microbial Community Structure and Function. Methods Mol. Biol 1399, 207–233. [DOI] [PubMed] [Google Scholar]
- Klompe SE, Vo PLH, Halpin-Healy TS, and Sternberg SH (2019). Transposon-encoded CRISPR–Cas systems direct RNA-guided DNA integration. Nature 571, 219–225. [DOI] [PubMed] [Google Scholar]
- Letunic I, and Bork P (2007). Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128. [DOI] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, Brouns SJJ, Charpentier E, Cheng D, Haft DH, Horvath P, et al. (2020). Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol 18, 67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA (2015). CRISPR-Cas immunity in prokaryotes. Nature 526, 55–61. [DOI] [PubMed] [Google Scholar]
- Mitra R, McKenzie GJ, Yi L, Lee CA, and Craig NL (2010). Characterization of the TnsD-attTn7 complex that promotes site-specific insertion of Tn7. Mob. DNA 1, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanraju P, Makarova KS, Zetsche B, Zhang F, Koonin EV, and van der Oost J (2016). Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science 353, aad5147. [DOI] [PubMed] [Google Scholar]
- Nawrocki EP, and Eddy SR (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petassi MT, Hsieh S-C, and Peters JE (2020). Guide RNA categorization enables target site choice in Tn7-CRISPR-Cas transposons. Cell, 183, 1757–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JE (2019). Targeted transposition with Tn7 elements: safe sites, mobile plasmids, CRISPR/Cas and beyond. Mol. Microbiol 112, 1635–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JE, and Craig NL (2001). Tn7 recognizes transposition target structures associated with DNA replication using the DNA-binding protein TnsE. Genes Dev. 15, 737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JE, Makarova KS, Shmakov S, and Koonin EV (2017). Recruitment of CRISPR-Cas systems by Tn7-like transposons. Proc. Natl. Acad. Sci. U. S. A 114, E7358–E7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, and Arkin AP (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol 26, 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin BE, Diamond S, Cress BF, Crits-Christoph A, He C, Xu M, Zhou Z, Smock DC, Tang K, Owens TK, et al. (2020). Targeted Genome Editing of Bacteria Within Microbial Communities. 10.1101/2019.12.11.123456v2 [DOI] [Google Scholar]
- Schmid-Burgk JL, Gao L, Li D, Gardner Z, Strecker J, Lash B, and Zhang F (2020). Highly Parallel Profiling of Cas9 Variant Specificity. Mol. Cell 78, 794–800.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinegger M, and Söding J (2017). MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol 35, 1026–1028. [DOI] [PubMed] [Google Scholar]
- Strecker J, Ladha A, Gardner Z, Schmid-Burgk JL, Makarova KS, Koonin EV, and Zhang F (2019). RNA-guided DNA insertion with CRISPR-associated transposases. Science 365, 48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strecker J, Ladha A, Makarova KS, Koonin EV, and Zhang F (2020). Response to Comment on “RNA-guided DNA insertion with CRISPR-associated transposases.” Science 368. [DOI] [PubMed] [Google Scholar]
- Vo PLH, Ronda C, Klompe SE, Chen EE, Acree C, Wang HH, and Sternberg SH (2020). CRISPR RNA-guided integrases for high-efficiency and multiplexed bacterial genome engineering. Nat. Biotechnol 10.1038/s41587-020-00745-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddell CS, and Craig NL (1988). Tn7 transposition: two transposition pathways directed by five Tn7-encoded genes. Genes & Development 2, 137–149. [DOI] [PubMed] [Google Scholar]
- Waddell CS, and Craig NL (1989). Tn7 transposition: recognition of the attTn7 target sequence. Proc. Natl. Acad. Sci. U. S. A 86, 3958–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1| Type V-K CAST hidden crRNA and targets, Related to Figure 2.
Identification and analysis of Type V-K loci in bacterial metagenomes with annotated homing targets, delocalized crRNA and target sequences, and relevant distances between features.
Table S2| Sequence information related to this study, Related to STAR Methods.
The table contains sequences of plasmids, transposon ends, and crRNA sequences for each CAST system experimentally studied here.
Figure S2| Comparison of delocalized crRNA across Type V-K CAST systems, Related to Figure 2 (A) Alignment of delocalized direct repeats from diverse type V-K CAST systems. (B) Alignment of spacers from delocalized crRNA of type V-K CAST systems, annotated with their corresponding tRNA target. (C) Start and end position of RNA sequencing reads aligned to the predicted delocalized crRNA in A. cylindrica. The predicted delocalized DR and tRNA match are annotated as determined by homology to the canonical DR and downstream tRNA target respectively.
Figure S1| Characterization of Type V-K CAST short, delocalized crRNA, Related to Figure 1 (A) Insertion activity of ShCAST components in E. coli into a CRISPR and homing target plasmid. (B) Alignment of typical direct repeats from the CRISPR array with short direct repeat from ShCAST. (C) Insertion activity of crRNA containing the delocalized direct repeat or the array direct repeat with a PSP1 spacer; promoters and terminators are indicated with an arrow and hairpin, respectively. Both the 17-nt spacer and 14-nt spacer extension match the target sequence. Data are represented as mean ± SD. (D) Insertion activity of ShCAST homing following truncation of its delocalized crRNA. Data are represented as mean ± SD. (E) Effect of sgRNA spacer length on insertion frequency of ShCAST on PSP15 through deletion of 3′ spacer sequence. Data are represented as mean ± SD. (F) Effect of sgRNA spacer length on insertion frequency of ShCAST on PSP42 through deletion of 3′ spacer sequence. Data are represented as mean ± SD. (G) Effect of double mismatches in the sgRNA spacer at the indicated positions on insertion frequency of ShCAST on PSP15. Positions are relative to the 5′ NGTN PAM. Data are represented as mean ± SD.
Figure S4| RNA-guided transposition on E.coli genome by Type I-B subtype1 AvCAST, Related to Figure 4 (A) AvCAST RNA-guided E.coli genomic insertions at 10 targeted loci. Representative data for LE junction amplification are shown. (B) Insertion frequency for 36 targeted loci (AvPSP2 - 37) quantified by ddPCR. Data are represented as mean ± SD. (C) Type I-B subtype1 AvCAST RNA-guided insertion positions on 36 targeted loci by deep sequencing. Both RE and LE junctions were sequenced and representative insertion reads at each position measured by LE junction data are provided. Insertion at PSPs indicated in red were below the limit of detection (few or no insertion reads). (D) Comparison of insertion frequency between pSC101and pBluescript donors. Data are represented as mean ± SD. (E) TTISS analysis of three representative loci (AvPSP2, AvPSP7, and AvPSP12) to test insertion specificity of AvCAST. Reads are mapped on CP001509 E.coli BL21(DE3) complete genome.
Figure S5| Type I-B subtype1 AvCAST system homes to glmS, Related to Figure 5 (A) Alignment of CAST-IB1 loci at the insertion site. Ends of glmS are highlighted in orange while the CAST left end (LE) is shown in grey. Only 2 identical contigs have the transposon inserted 5-bp downstream of glmS while all other transposons are inserted about 24-bp downstream of the homing target site. (B) Deep sequencing reads showing Tn7-like machinery of AvCAST mediates a single prominent insertion at glmS Tn7 attachment site on plasmids. Purple bar indicates the insertion position on the plasmid harboring E. coli glmS. Blue bar indicates the insertion position on the plasmid harboring A. variabilis glmS. (C) Tn7-like machinery-mediated insertion frequency at the glmS gene on the E. coli BL21(DE3) genome in the absence of TniQ, TnsD, or both. Data are represented as mean ± SD. (D) Sanger sequencing chromatograms for RE and LE junctions of insertion at the endogenous glmS Tn7 attachment site on E.coli BL21(DE3) genome. “GCGGG” is target site duplication as a signature of Tn7-like transposition. (E) AvCAST-mediated single prominent insertion at the endogenous glmS Tn7 attachment site on E.coli BL21(DE3) genome identified by deep sequencing. Left: with all Tn7-like machinery of AvCAST; right: with all AvCAST proteins including TniQ and Cascade proteins. (F) Purified Tn7-like machinery components of AvCAST on Coomassie Brilliant Blue stained SDS-PAGE gel.
Figure S7| Models comparing CAST V-K and CAST I-F, Related to Figure 7 Prototypical locus organization of CAST V-K systems and CAST I-F systems and models for CAST transposition to mobile element (donor cell in blue) and CAST homing transposition to bacterial chromosome (recipient cell in green). The loci are delimited by transposon ends (Tn ends, indicated as light vertical rectangles). Transposon core components (tnsA, tnsB, and tnsC in CAST I-F; tnsB and tnsC in CAST V-K) are colored in light blue while tniQ is colored in dark blue. Cas components (Cascade I-F and cas12k) are shown in grey. Conserved transcription factors (TF) are shown in pink. Locations of cargo genes are indicated with dark grey circles. CRISPR arrays are shown as grey triangles for the repeats, and red diamonds for the spacers. In CAST V-K the delocalized crRNA is indicated by a lighter triangle (partial repeat), and a truncated orange diamond (short spacer). In CAST I-F the homing spacer (targeting the homing target site) is indicated as a light orange diamond located at the distal spacer of the CRISPR-array. Homing target genes are colored in large yellow rectangles. Horizontal light grey triangle indicates the tracrRNA in the CAST V-K locus. CAST transposition to mobile genetic elements is mediated by the Cas effector machinery (I-F Cascade and crRNA or Cas12k, tracrRNA, and cRNA) which recognizes the target site (red) with RNA-guided targeting to insert the transposon using Tn components. For homing transposition, CAST V-K uses a dedicated crRNA (spacer in light orange) bearing a truncated spacer to target the homing site while CAST I-F uses a dedicated spacer, either located at the distal region of the CRISPR array or delocalized, to target the homing target site often with numerous mismatches between spacer and target (Petassi et al. 2020).
Figure S6| Characterization of Type I-B subtype2 PmcCAST, Related to Figure 6 (A) RNA sequence reads mapping to the minimal CRISPR array showing the mature crRNA sequence of PmcCAST. (B) Insertion directionality assayed by diagnostic PCR of pInsert on 6N PAM library plasmid with different primer pairs in ΔTniQ or ΔTnsD conditions. (C) Quantification of insertion frequency across the LE and RE junctions by ddPCR. Data are represented as mean ± SD. (D) Sanger sequencing chromatograms of a representative pInsert. PAM, PmcPSP1, TSDs, transposon ends are annotated. (E) Coomassie Brilliant Blue stained SDS-PAGE gel for purified Tn7-like machinery components of PmcCAST.
Figure S3| CAST I-B loci comparison and TnsD/TniQ analysis, Related to Figures 3 and 6 (A) Left, CAST I-B1 loci sharing similar TniQ (non identical and greater than or equal to 50 percent sequence identity). Right, Loci sharing similar CAST I-B1 tnsD (greater than or equal to 50 percent sequence identity). tniQ is colored in red, transposon core genes (tnsA, tnsB, and tnsC) in blue, Cas components in yellow, glmS homing gene in green, CRISPR arrays are shown as thin empty red arrays, and tnsD in pink. Black arrows represent padding to center the tniQ or tnsD genes. Cyan indicates tnsE, a hallmark gene for targeting conjugative plasmids in Tn7. Loci centered on tnsD (right) include Tn7 loci (non CAST) that also home to glmS (green). All loci are available in the Supplementary Dataset (B) Protein sequence alignment of CAST I-B1 TniQ (red), TnsD (purple), and Tn7-TnsD (green) sharing sequence similarity with CAST I-B1 TnsD. Sequences are ordered from a tree built with FastTree (WAG model) (Price et al., 2009). Blue highlights indicate the conserved core of TniQ/TnsD. On top of the alignment sequences are the consensus, the hydrophobicity graph (red), and the conservation graph (green). The alignment is colored using the Rasmol color palette; colored positions indicate conservation in agreement with the consensus sequence. Tn7 TnsD and CAST-IB TnsD are longer than TniQ, and are similar in their C-terminal region. (C) CAST I-B2 loci sharing similar TniQ (non identical and greater than or equal to 50 percent sequence identity). tniQ is colored in red, transposon core genes (tnsAB and tnsC) in blue, Cas components in yellow, tRNA homing gene in green, CRISPR arrays are shown as thin empty red arrays, and tnsD in pink. (D) Protein alignment of TniQ/TnsD core region from CAST I-B TniQ (red), TnsD (purple), and Tn7-TnsD (green) , CAST I-F TniQ (light green), and CAST V-K TniQ (orange). On top of the alignment sequences are the consensus and the conservation graph (green). The alignment is colored using the Rasmol color palette; colored positions indicate conservation in agreement with the consensus sequence. Domain annotation of TniQ dimer mapped on the electron microscopy structure (pdb code: 6pij) (Halpin-Healy et al., 2019) of CAST I-F TniQ. The different colors indicate the potential domains of TniQ, from N-terminal to C-terminal, blue, green, orange, and red. TniQ/TnsD core (red dashed line) is formed by the blue and the green domain (region conserved in CAST TniQ/TnsD and Tn7-TnsD). Black dashed lines indicate interaction with Cascade components found in the structure.
Supplementary Dataset (SupDataset.zip) | CAST loci, Related to STAR Methods.
Annotated CAST loci from V-K, I-B subtype 1, and I-B subtype 2 in Genbank format. Each locus is centered on the tniQ gene, which is annotated as “Center”. All loci have a unique TniQ/TnsD protein: all loci with identical TniQ/TnsD proteins were discarded to keep one representative with longest distance to the edges of the contig. The centered tniQ/tnsD is surrounded by a maximum of 50-kb upstream and downstream (or less if the locus is near the edge of a contig). A padding was used when the centered tniQ/tnsD was near the contig edge. Proteins are annotated from Pfam or Cas profile database (annotated Protein, or CAS_CRISPR). Tn7 components are annotated as Tns_tni. Proteins sharing at least 50% sequence identity have a “Clu” annotation followed by a unique number. tRNA and other ncRNAs are annotated under tRNA and RNA.
Data Availability Statement
Expression plasmids are available from Addgene under a uniform biological material transfer agreement; support forums and computational tools are available via the Zhang lab website (https://zhanglab.bio). All CAST loci are available in the Supplementary Dataset as an archive of Genbank files.