

Abstract
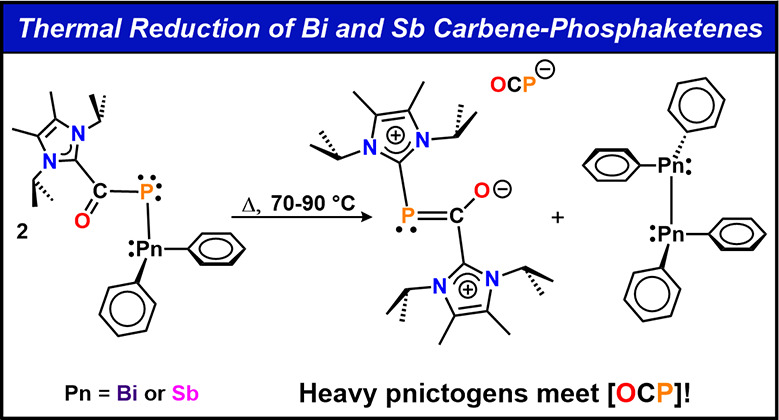
The synthesis and thermal redox chemistry of the first antimony (Sb)– and bismuth (Bi)–phosphaketene adducts are described. When diphenylpnictogen chloride [Ph2PnCl (Pn = Sb or Bi)] is reacted with sodium 2-phosphaethynolate [Na[OCP]·(dioxane)x], tetraphenyldipnictogen (Ph2Pn–PnPh2) compounds are produced, and an insoluble precipitate forms from solution. In contrast, when the N-heterocyclic carbene adduct (NHC)–PnPh2Cl is combined with [Na[OCP]·(dioxane)x], Sb– and Bi–phosphaketene complexes are isolated. Thus, NHC serves as an essential mediator for the reaction. Immediately after the formation of an intermediary pnictogen–phosphaketene NHC adduct [NHC–PnPh2(PCO)], the NHC ligand transfers from the Pn center to the phosphaketene carbon atom, forming NHC–C(O)P-PnPh2 [Pn = Sb (3) or Bi (4)]. In the solid state, 3 and 4 are dimeric with short intermolecular Pn–Pn interactions. When compounds 3 and 4 are heated in THF at 90 and 70 °C, respectively, the pnictogen center PnIII is thermally reduced to PnII to form tetraphenyldipnictines (Ph2Pn–PnPh2) and an unusual bis-carbene-supported OCP salt, [(NHC)2OCP][OCP] (5). The formation of compound 5 and Ph2Pn–PnPh2 from 3 or 4 is unique in comparison to the known thermal reactivity for group 14 carbene–phosphaketene complexes, further highlighting the diverse reactivity of [OCP]− with main-group elements. All new compounds have been fully characterized by single-crystal X-ray diffraction, multinuclear NMR spectroscopy (1H, 13C, and 31P), infrared spectroscopy, and elemental analysis (1, 2, and 5). The electronic structure of 5 and the mechanism of formation were investigated using density functional theory (DFT).
Short abstract
An N-heterocyclic carbene (NHC) was used to support the otherwise unstable Ph2Sb—P=C=O and Ph2Bi—P=C=O moieties. Exploration of the thermal chemistry of these NHC−phosphaketene adducts reveals the formation of the salt [NHC2OCP][OCP]. This present work demonstrates the thermal chemistry of the 2-phospaethynolate anion with heavier pnictogens (Sb and Bi).
Introduction
Due to their unique electron distribution, heteroketenes show versatile and fascinating chemistry. Phosphorus-containing members of this family are phosphaketenes, R—P=C=O.1 Although the first stable phosphaketene was reported nearly four decades ago,2 the synthetic chemistry was experimentally challenging, and various products were thermally unstable. However, in the past decade, simple synthetic routes toward such compounds have emerged, which has resulted in the rapid development of the field. The utilization of the 2-phosphaethynolate anion,3 [OCP]−, as a synthon has proved to be an effective way to access phosphaketenes via nucleophilic substitution.4 However, these synthetic processes are not always straightforward, and phosphaketene stability and reactivity may be hampered by a number of complications. The most important of these are summarized as follows. (i) Dimer formation: the P=C bond of phosphaketenes is prone to cycloaddition, which results in 4-membered rings; however, this process can be minimized with the incorporation of bulky substituents, or with heteroatoms.5 (ii) Formation of constitutional isomers: due to its ambident reactivity, the [OCP]− anion may bind through the P or the O center. Highly oxophilic species, such as s-3c,6 or f-block elements,7 favor the oxyphosphaalkyne isomer, while soft Lewis acidic elements, for example, the heavy group 14 elements (Ge, Sn, Pb),4b,8 and Ga4c,9 favor the phosphaketene isomer. Both O- and P-bound isomers are known for B4a,10 and Si,4b illustrating the ambident nature of the [OCP]− anion. (iii) Redox chemistry: the OCP anion is prone to oxidation by many metals due to its reductive nature and the electrophilic character of the metals.3a,11 While point (i) can be circumvented using sterically demanding substituents to stabilize the phosphaketene, points (ii) and (iii) are more challenging to avoid because the inherent electrophilic properties of the main-group elements differ widely across the periodic table. Nevertheless, phosphaketene isomers R—P=C=O are usually more stable than their oxyphosphaalkyne R—O—C≡P analogues; thus (ii) is a less common problem in synthetic routes. In this Article, we aim to offer a solution to the problem described in point (iii). Since the heavier pnictogens are easily reduced, neutral donor ligands such as N-heterocyclic carbenes (NHC) can be employed to stabilize the phosphaketene motif, thereby preventing reduction at the pnictogen center.
Phosphaketenes show rich and often unprecedented chemistry. In recent years, the phosphanyl- and tetrel-substituted phosphaketenes have attracted special interest. Bertrand, Su, and Grützmacher discovered a unique reaction where OCP rearranges to OPC when an N-heterocyclic phosphane (NHP)–phosphaketene adduct is reacted with NHC (Figure 1A).4e Nucleophilic attack on the OCP carbon atom by the NHC results in a zwitterionic intermediate, which is followed by migration of the NHP unit to oxygen. Grützmacher et al. showed that the CO unit of the phosphaketene can be substituted by a carbene, demonstrating similar phosphaketene reactivity with NHCs (Figure 1B).4d Addition of NHC to a triphenylgermanium– or tin–phosphaketene led to the formation of NHC–phosphaketene adducts. When heated, the NHC transfers to phosphorus to release CO, thereby forming NHC–phosphinidene germanium and tin complexes. Similarly, the C≡O unit of a phosphaketene can be exchanged by another donor. Bertrand observed loss of CO from NHP–phosphaketenes when a Lewis basic phosphine was introduced with moderate heating (Figure 1C).12 It is noteworthy that the loss of CO from P–CO-containing molecules has been explored computationally.13 The reaction proceeds via an associative mechanism, whereby the phosphine binds to the −PCO unit first, followed by loss of CO.
Figure 1.
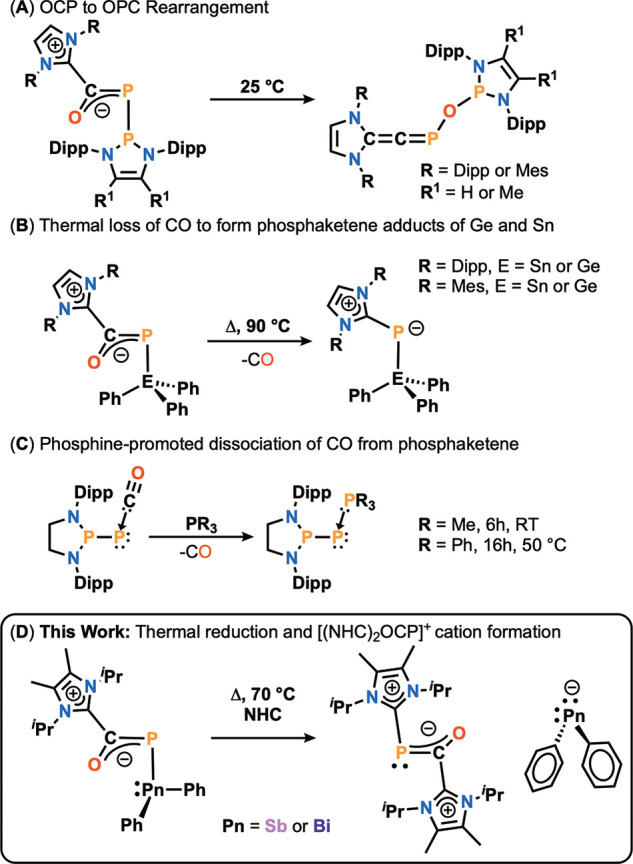
(A) OCP to OPC rearrangement; (B) thermal loss of CO from NHC−phosphaketene adducts of triphenyl-germanium or -tin to form NHC−phosphinidenes; (C) phosphine-promoted CO dissociation; (D) This work: thermal reduction involving Sb and Bi phosphaketene.
Within the realm of main-group elements, the reactivity of Na[OCP] has been established for group 2,6a,6b group 13,4c,9,10 group 14,4b,4e,8,14 and group 15.4e,15 For the lattermost, these examples are limited to phosphorus and arscenic, with no current reports describing reactions of Na[OCP] with the heavier pnictogens (Sb and Bi). Nevertheless, the chemistry of the heavier two Pn elements (Sb, Bi) has seen a substantial increase in interest recently as novel bonding motifs, and new applications in catalysis continue to be discovered.16
Herein, we report the first reactions of Na[OCP] with antimony and bismuth compounds, [NHC–Sb(Ph)2Cl]2 (1) and [NHC–Bi(Ph)2Cl]2 (2). [NHC–PnPh2Cl]2 was combined with [Na[OCP]·(dioxane)x] to afford Sb– and Bi–phosphaketene complexes (3 and 4, respectively). Notably, compounds 3 and 4 were susceptible to a thermal reduction process where the PnIII center is reduced to PnII to form either tetraphenyldistibine or tetraphenyldibismuthine and the [(NHC)2OCP][OCP] salt (5). Compound 5 is a unique example of a salt with an [OCP] moiety imbodying both the cation and the anion. DFT calculations demonstrate that the formation of the cationic unit in 5 occurs in a mechanistic step where nucleophilic attack of a dissociated NHC on one unit of 4 leads to the loss of [Ph2Bi]− (Figure 1D).
Results and Discussion
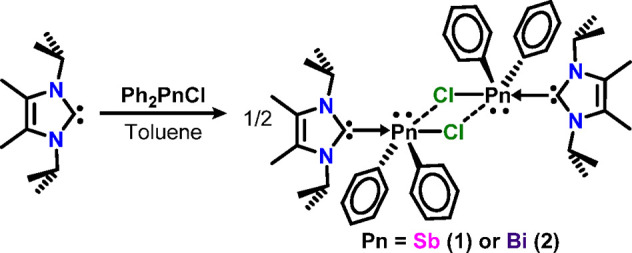
We initially performed the reaction of Na[OCP] with Ph2PnCl (Pn = Sb or Bi) and observed the formation of tetraphenyldipnictine and an insoluble unidentified precipitate. Extending the scope of this reaction, NHC ligand 4,5-dimethyl-1,3-diisopropylimidazolin-2-ylidene was reacted with diphenylantimony chloride (Ph2SbCl) or diphenylbismuth chloride (Ph2BiCl) in THF for 1 h at room temperature (Scheme 1). Compounds 1 (Sb) and 2 (Bi) were obtained as white solids in 94% and 85% yield, respectively. The 1H NMR spectrum of 1 in C6D6 shows a broad heptet at 4.69 ppm, attributed to the NHC methine proton. This is shifted downfield from the methine of the NHC ligand (3.96 ppm). Due to poor solubility in C6D6, the 1H NMR spectrum of compound 2 was recorded in THF-d8, which showed a broadened heptet at 4.51 ppm, attributed to the methine protons of coordinated NHC.
Scheme 1. Synthesis of Diphenylpnictogen Halide N-Heterocyclic Carbene Complexes.
Colorless crystals suitable for X-ray diffraction of both 1 and 2 were obtained from toluene/hexane (10:1) mixtures at −37 °C. The molecular structures of compounds 1 and 2 are dimeric with distorted square pyramidal geometry around the metal center (Figure 2). The C1–Sb1 bond distance in compound 1 [2.356(3) Å] is outside the range of other NHCC–Sb bonds (2.144–2.268 Å);16o,16q,16r,17 likewise, the C1–Bi1 bond in compound 2 [2.489(6)] is slightly longer than the known range for NHCC–Bi bonds (2.339–2.428 Å).17b,18 The Pn–Cl bond lengths in 1 (2.8006(8) Å) and 2 (2.8696(16) Å) are also significantly longer than those in reported complexes containing Sb–Cl (2.332–2.402 Å)17a−17c and Bi–Cl (2.437–2.705 Å)18a,18b bonds. The longer NHCC–Pn bonds results from the weak Lewis acidity of Ph2PnCl compared to PhBiCl2 and BiCl3. The intermolecular Pn–Cl distances in 1 [3.9544(10) Å] are longer than those in 2 [3.7211(17) Å], which is due to the pronounced Lewis acidity at the Bi center.
Figure 2.
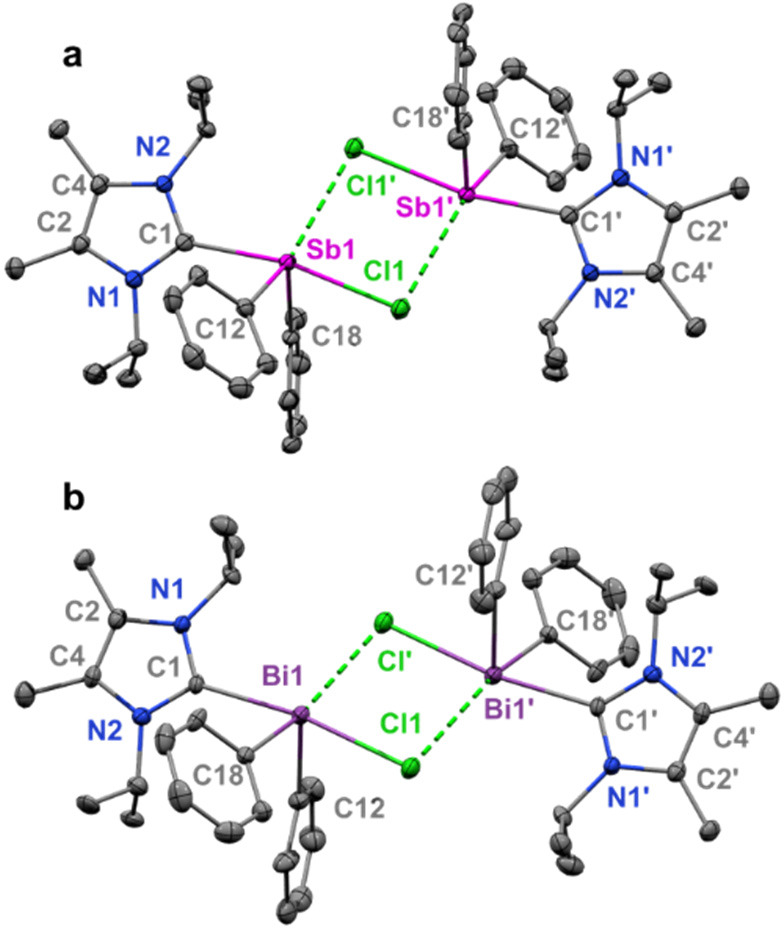
Molecular structure of 1 (a): Thermal ellipsoids at 50% probability; H atoms omitted for clarity. Selected bond distances (Å) and angles (deg): Sb1–C1 2.356(3); Sb1–Cl1 2.8006(8); Sb1–Cl1′ 3.9544(10); Sb1–C18 2.168(3); Sb1–C12 2.171(4). C18–Sb1–C12 102.19(14); C18–Sb1–C1 87.58(12); C12–Sb1–C1 86.33(12); C18–Sb1–Cl1 87.00(8); C12–Sb1–Cl1 85.24(9); C1–Sb1–Cl1 168.80(9). Molecular structure of 2 (b): Thermal ellipsoids at 50% probability; H atoms were omitted for clarity. Selected bond distances (Å) and angles (deg): Bi1–C1 2.489(6); Bi1–Cl1 2.8696(16); Bi1–Cl1′ 3.7211(17); Bi1–C18 2.257(6); Bi1–C12 2.267(6). C18–Bi1–C12 99.0(2); C18–Bi1–C1 86.3(2); C12–Bi1–C1 88.1(2); C18–Bi1–Cl1 86.26(16); C12–Bi1–Cl1 88.26(16); C1–Bi1–Cl1 171.05(15).
For both compounds 1 and 2, we hypothesized that a combination of electronic stabilization from the coordinated NHC and steric protection from the two phenyl groups may stabilize their OCP adducts. Based on the reactivity known for [OCP]− with other main group elements,3a we predicted the formation of a pnictogen–phosphaketene adduct, Pn–PCO. Therefore, we reacted compounds 1 and 2 with Na[OCP]·(dioxane)x at −37 °C in THF (Scheme 2). The 31P NMR spectra of the isolated complexes revealed shifts at 58.2 ppm (Sb) and 82.2 ppm (Bi), which are downfield from known main-group element Pn–PCO compounds (−441 to −225.8 ppm).3a Two doublets were observed in the 13C NMR spectra for both the antimony [203.1 ppm (1JCP = 76.0 Hz) and 148.8 ppm (2JCP = 52.8 Hz)] and bismuth [203.6 ppm (2JCP = 81.7) and 152.0 ppm (1JCP = 49.6 Hz)] complexes.
Scheme 2. Synthesis of Antimony– and Bismuth–Phosphaketene Adducts.

Single crystals of compounds 3 and 4 were obtained by layering the original THF filtrate with hexanes in a 1:1 ratio at −37 °C. Interestingly, the molecular structure revealed that the NHC transferred from the pnictogen center to the phosphaketene (Figure 3). Compounds 3 and 4 are unstable at room temperature and −37 °C, respectively, and decompose slowly in the solid state after a few days. The formation of these products is consistent with the 13C NMR spectra. A stretching frequency was not observed for the carbonyl group in the IR spectrum; however, this is consistent with reported NHC–phosphaketenyl species.4d The solid-state structures of 3 and 4 reveal pnictogen centers in a seesaw environment with intermolecular Pn–Pn interactions at 3.9619(17) Å and 3.8204(6) Å, respectively. The Pn–P bond lengths for both 3 (2.5042(16) Å) and 4 (2.589(2)–2.594(2) Å) are close to the sum of covalent radii for Sb and P (RSbP = 2.50 Å), as well as for Bi and P (RBiP = 2.61 Å).19
Figure 3.
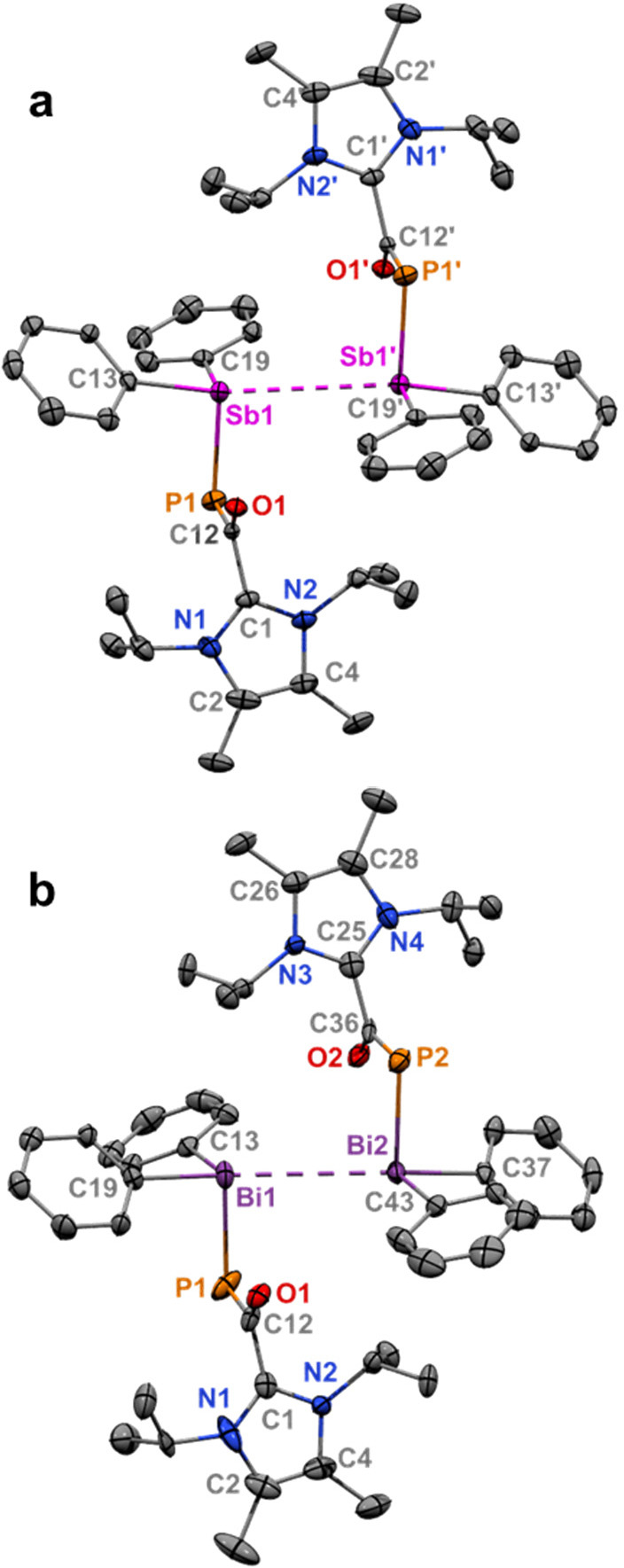
Molecular structure for 3 (a): Thermal ellipsoids at 50% probability; H atoms omitted for clarity. Selected bond distances (Å) and angles (deg): Sb1–C13 2.154(6); Sb1–C19 2.155(6); Sb1–P1 2.5042(16); Sb1–Sb1′ 3.9619(17); P1–C12 1.748(6); O1–C12 1.264(7); C1–C12 1.529(7). C13–Sb1–C19 97.7(2); C13–Sb1–P1 99.79(15); C19–Sb1–P1 91.77(15); Sb1′–Sb1–P1 94.602(40); Sb1′–Sb1–C19 97.237(158); Sb1′–Sb1–C13 158.899(168). Molecular structure for 4 (b): Thermal ellipsoids at 50% probability; H atoms were omitted for clarity. Selected bond distances (Å) and angles (deg): Bi1–C19 2.229(6); Bi1–C13 2.272(7); Bi1–P1 2.589(2); Bi1–Bi2 3.8204(6); Bi2–C37 2.251(7); Bi2–C43 2.257(8); Bi2–P2 2.594(2); P1–C12 1.736(7); P2–C36 1.746(8); O1–C12 1.255(8); O2–C36 1.258(8); C1–C12 1.525(11); C25–C36 1.515(11). C19–Bi1–C13 94.2(2); C19–Bi1–P1 97.38(19); C13–Bi1–P1 87.26(19); C37–Bi2–C43 94.8(3); C37–Bi2–P2 98.56(19); C43–Bi2–P2 90.99(19); Bi2–Bi1–P1 86.913(49); Bi2–Bi1–C13 101.940(176); Bi2–Bi1–C19 163.528(178); Bi1–Bi2–P2 89.365(43); Bi1–Bi2–C43 104.176(179); Bi1–Bi2–C37 159.346(177).
Recently, Grützmacher and co-workers demonstrated that N-heterocyclic carbene (NHC)–phosphaketene adducts of Ph3Sn—P=C=O and Ph3Ge—P=C=O undergo a decarbonylation reaction when heated to form the phosphenidinyl complexes NHC–P–SnPh3 and NHC–P–GePh3.4d We were therefore interested in probing the thermal reactivity of compounds 3 and 4 (Scheme 3), which can be considered group 15 analogues of the aforementioned Sn and Ge phosphaketene complexes. Compound 3 was heated to 90 °C for 24 h in a J-Young NMR tube. The peaks in this 1H NMR spectrum matched those reported in the literature for tetraphenyldistibine (Figure S14).16s Compound 4, being less stable than 3, was heated at 70 °C for 3 h in C6D6. Free NHC emerged along with 100% conversion to tetraphenyldibismuthine (Figure S15). Single crystals suitable for X-ray diffraction were grown from C6D6 inside the NMR tube. The solid-state structure revealed a new polymorph of tetraphenyldibismuthine (6) (Figure S19). It is noteworthy that compounds 3 and 4 slowly covert to 5 and tetraphenyldipnictogen at room temperature; therefore, heat was applied to escalate the reactions as described.
Scheme 3. Thermal Reduction at Sb or Bi Center to Tetraphenyldipnictines and [(NHC)2OCP][OCP].

In addition to the formation of tetraphenyldipnictine, an orange solid precipitated from the C6D6 solution. The orange solid is insoluble in most common organic solvents except for dichloromethane but decomposes within an hour after dissolution. The 1H NMR spectrum of the orange solid revealed one broad and one well-defined heptet, suggesting two distinct NHC ligand environments. The 31P NMR showed a broad singlet at 22.1 ppm and a sharp singlet at −395.1 ppm. The latter shift closely resembles the resonance of 2-phosphaethynolate in D2O (−396.4 ppm).3b Four doublets (δ = 200.7, 170.2, 150.2, 146.0) were observed in the 13C NMR spectrum. Further supporting our assignment, the signal at 170.2 ppm (1JCP = 63.4 Hz) agrees well with known 13C NMR shifts for 2-phosphaethynolate, while the other signals are attributed to three new 13C–31P coupling environments. Similar to compounds 3 and 4, no signals were observed in the IR spectrum for the CO stretch in the cationic unit of 5. Two different stretches were observed for the phosphaalkyne at 1788 and 1768 cm–1, resulting from different orientations of [OCP]− in the solid-state structure.
Orange single crystals of compound 5 suitable for X-ray diffraction were obtained by heating a THF solution of 3 at 55 °C overnight. The crystal structure shows a cation containing two NHCs coordinated to a [OCP] core with an [OCP]− counteranion (Figure 4). A 2-fold rotation axis perpendicular to the P1–C12 bond in the cation causes the two halves of the molecule to be disordered by symmetry in the solid state. This symmetry results in identical bond lengths and angles for both NHC ligands. A similar disorder exists in the anion. There are currently eight other molecular structures containing uncoordinated [OCP]− counter-anions reported in the CSD database.4c,6c,6d,20 The C1–P1 bond (1.890(6) Å) is longer than those in neutral NHC2P2 complexes (1.750–1.754 Å)21 and cationic [NHC2P2]+ complexes (1.795–1.841 Å).17c,21a,22
Figure 4.
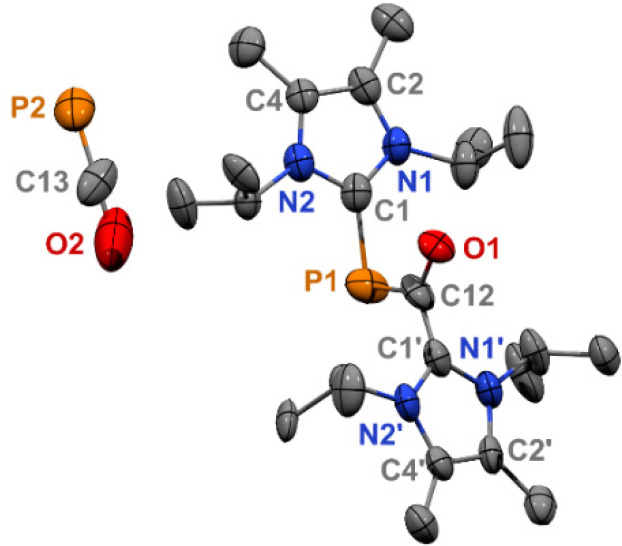
Molecular structure of 5: Thermal ellipsoids at 50% probability; H atoms omitted for clarity. Only one orientation of the symmetry disordered [OCP]− anion is shown. Selected bond distances (Å) and angles (deg): C1–P1 1.890(6); P1–C12 1.755(14); C12–O1 1.268(12); C12–C1′ 1.421(16). C1–P1–C12 98.3(5); O1–C12–C1′ 117.0(14).
To gain insights into the formation mechanism leading to the new compounds and the bonding situation thereof, we carried out DFT calculations employing the ωB97XD range separated functional with the def2-SVP and def2-TZVP basis sets, which is similar to the level of theory used previously to describe the bonding in carbene complexes of bismuth.17b Relevant energies and structural parameters are shown in Table 1.
Table 1. Complex Formation Energies (ΔE) and Gibbs Free Energies (ΔG)a, Geometrical Parametersb, NPA Partial Charges of Pn (q) in Electrons, and Net Charge Transfer in Electrons (Δq) at the ωB97XD/def2-TZVP Level.
| compound | 1 | 2 | NHC–SbPh2PCO | NHC–BiPh2PCO | 3 | 4 |
|---|---|---|---|---|---|---|
| ΔE | –23.9 | –26.2 | –19.2 | –21.5 | –29.7 | –29.2 |
| ΔG | –9.4 | –11.7 | –3.0 | –7.1 | –12.4 | –12.0 |
| d(Pn–Ccarbene/C–Ccarbene) | 2.572 | 2.715 | 2.638 | 2.801 | 1.513 | 1.513 |
| WBI(Pn–Ccarbene/C–Ccarbene) | 0.37 | 0.31 | 0.32 | 0.26 | 0.93 | 0.93 |
| q(Pn) | 1.182 | 1.246 | 1.058 | 1.121 | 0.899 | 0.936 |
| Δq | 0.235 | 0.202 | 0.226 | 0.186 | 0.755 | 0.751 |
In kcal/mol.
Bond length in Å/Wiberg bond indicies.
The complex formation energy leading to adduct 2 is −26.2 kcal/mol (calculated with respect to the isolated carbene and diphenyl bismuth chloride). This value is greater than the values of −35.9 to −44.6 kcal/mol reported for NHC and CAAC complexes of PhBiCl2,18b a stronger Lewis acid owing to the presence of two chlorine atoms instead of one in Ph2BiCl. This agrees nicely with the observations above on the solid-state structures, which revealed rather long NHCC–Pn bonds as a result of a weaker interaction. Compared to 2, the antimony analogue 1 is slightly less stable (ΔE = −23.9 kcal/mol), explainable by the weaker electron pair accepting property of antimony than that of bismuth. The same phenomenon is observed for the NHC–Ph2PnPCO complexes, which are assumed as possible intermediates during the replacement of the chlorides of 1 and 2 by phosphaethynolate anion. However, the phosphaketene complexes are destabilized compared to their chloro-analogues, due to the lower electronegativity of P compared to Cl. Indeed, the partial charge at the Bi center in the uncomplexed Ph2BiPCO and Ph2BiCl is +1.010 and +1.226 e, respectively, in line with the lower Lewis acidity of the former compared to the latter. The reduced stability of the phosphaketene complexes compared to analogous chloro-complexes is accompanied by the weakening of the NHCC–Pn bonds; these bonds are longer and their Wiberg bond indices (WBI), accounting for the covalent character, are lower. Thus, the net charge transfer is smaller. The LUMO of Ph2BiPCO (Figure 5) shows main contributions both at the Bi center and the carbon atom of the PCO moiety, explaining why this species may be complexed either at Bi or on the phosphaketenyl carbon center. The rearranged phosphaketene carbene adducts 3 and 4, in which the carbene is coordinated to the PCO carbon atom, are significantly more stable than the Pn-coordinated analogues. Thus, the driving force for the carbene migration is the formation of a stronger C–C bond instead of a dative C–Pn bond. These C–C bonds show a high covalent character (WBI: 0.93) and remarkable net charge transfer from the carbene to the PCO moiety of Δq = 0.751 and 0.755, meaning that the carbenic unit possesses a large partial positive charge. Furthermore, the WBIs of PC/CO bonds (1.41/1.48 and 1.42/1.47 for 3 and 4, respectively) indicate delocalization in the PCO fragment. Hence, the structure of the C-coordinated Ph2PnPCO adducts 3 and 4 can be best described as a superposition of two zwitterionic resonance structures (Figure 6A). We also studied the electronic structure and bonding of the cationic fragment of compound 5. Even though [(NHC)2OCP]+ can be regarded formally as an adduct of a cationic OCP+ unit and two carbenes, the NPA charges and WBI values suggest the positive charge is localized on the NHC ligands (Figure 6B). While the sum of charges in the OCP core is −0.375e, both NHC fragments possess high partial charges of 0.804e and 0.571e. The WBI of the P–C(carbene) and C–C(carbene) bonds of 0.93 indicate covalent character, and the PC/CO bonds show a delocalization in the OCP moiety. The bis-zwitterionic charge distribution of the [(NHC)2OCP]+ cation is also visible on the molecular electrostatic potential (Figure 7).
Figure 5.
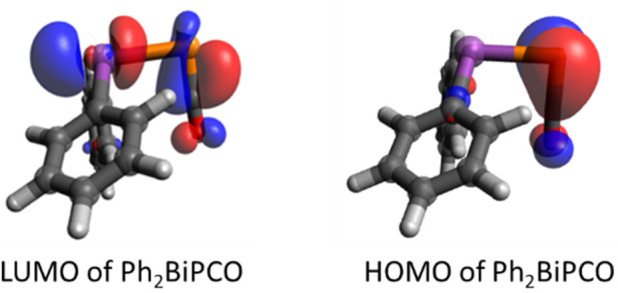
LUMO and HOMO of Ph2BiPCO.
Figure 6.
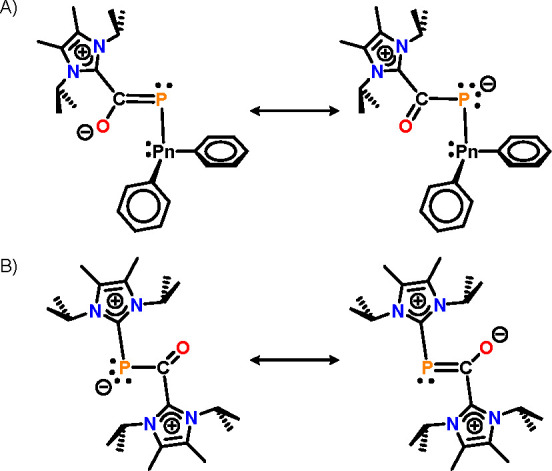
Resonance structures for compounds 3/4 (A) and cation 5 (B).
Figure 7.
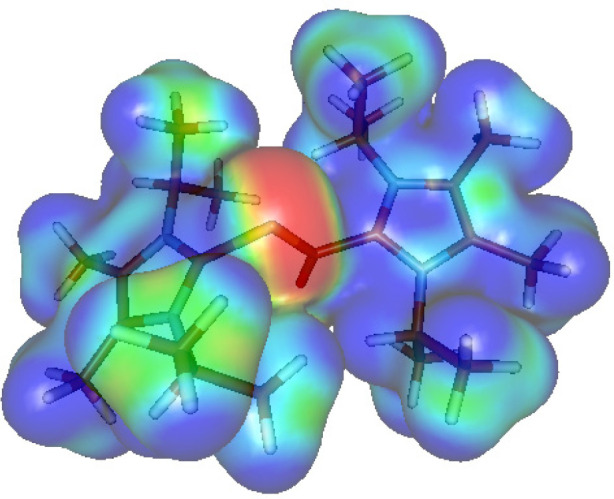
Molecular electrostatic potential for the cationic moiety of 5.
We also aimed to understand the formation of the Ph2PnPnPh2 dimers and compound 5; therefore, we investigated possible reaction mechanisms by means of computations. As the reactivity of 3 and 4 are rather similar, we focused on the bismuth analogue. Because this reaction proceeds in C6D6, the gas phase approximation seems to be appropriate without solvent effects. In the following, we discuss the energies obtained at the ωB97XD/def2-SVP level.
The formation of the tetraphenyldibismuthine may indicate a radical mechanism, in which the first step would be the homolytic dissociation at the P–Bi bond of adduct 4, or alternatively, the free Ph2BiPCO. However, both reactions are highly endothermic (ΔE = 53.0 and 51.7 kcal/mol, respectively); thus, they are unlikely to happen even at higher temperature. We considered further alternative pathways and a plausible mechanism (Figure 8). The first step of the reaction is the partial dissociation of adduct 4, resulting in the free carbene and Ph2BiPCO. This reaction is rather endothermic and proceeds via an activation barrier of 27.9 kcal/mol (Figure 9), resulting in a weakly bound complex of NHC and Ph2BiPCO at the energy of 27.2 kcal/mol. Even though this reaction is likely shifted toward the side of the starting adduct, the formation of small amounts of free carbene is expected, especially if the entropy factor is taken into account (dissociation Gibbs free energy: 18.9 kcal/mol). This is further supported by the experimental observation of uncoordinated NHC during the reaction. The second step of the reaction is an attack of the free carbene onto the P center of adduct 4, delivering the contact ion pair of the [(NHC)2OCP]+ cation with a diphenyl bismuthide ([BiPh2]−) counteranion. The nucleophilic substitution at the phosphaketene P center is known in the literature, and it has been shown that the attack of Lewis bases (L) on the phosphorus center of phosphanyl phosphaketenes R—P=C=O results in the adduct R—P=L and carbon monoxide. In our case, however, the C of the PCO unit is occupied by the carbene fragments; thus, the decarbonylation is hampered. Instead, the bismuthide anion is released in a slightly exothermic reaction (ΔE = −10.1 kcal/mol). Since all of our attempts to locate the transition state of step 2 failed, we performed a relaxed optimization scan connecting the structures at the two sides of the equation and estimated a barrier of 1.4 kcal/mol via this approach. The thermodynamic sink is obtained in reaction step 3, which is strongly exothermic with a reaction energy of ΔE = −31.7 kcal/mol. Since we could not locate any transition states for this step, we performed a relaxed scan computation which showed a continuous decrease in the energy; therefore, this reaction step is assumed to proceed without barrier. In this final step, the attack of the [BiPh2]− anion at the Bi center of Ph2BiPCO formed in step 1 delivers the dibismuthine Ph2BiBiPh2 as well as the [OCP]− anion for compound 5.
Figure 8.

Proposed mechanism for the formation of [(NHC)2OCP]+[OCP]−.
Figure 9.

Energy profile for the decomposition of 4 leading to 5. The three mechanistic steps illustrated in Figure 8 proceed on different potential energy surfaces. For easier understanding, the energy levels of each initial mechanistic step are shifted to the energy of the previous step. (Atom colors: C, black; O, red; N, blue; P, orange; Bi, purple). In step 3 the countercation is not shown for clarity but was included in the computations.
Conclusion
The reaction of Ph2PnCl with Na[OCP] results in the formation of tetraphenyldipnictine and an insoluble unidentifiable product. Therefore, we prepared the respective NHC-supported Ph2PnCl compounds (1 and 2) and explored their reactivity with Na[OCP]. In both cases, the NHC transfers from the pnictogen center to the phosphaketene carbon atom. The crystal structures of these two OCP complexes reveal significant metal–metal interactions. Heating the NHC–phosphaketene adducts 3 and 4 results in a formal reduction at the pnictogen center, PnIII to PnII, resulting in the formation of tetraphenyldipnictine and [(NHC)2OCP]+[OCP]− (5). Notably, compound 5 represents the first example of an ionic compound where the cation and anion each possess an OCP unit. These results further demonstrate the utility of the 2-phosphaethynolate ion as a reductant and contrast with the chemistry observed for the group 14 (Sn and Ge) analogues, which undergo decarbonylation to yield phosphinidenyl species.
Experimental Section
General Considerations
All reactions were carried out under an atmosphere of purified argon in a MBRAUN LABmaster glovebox equipped with a −37 °C freezer. All solvents were distilled over sodium/benzophenone. Glassware was oven-dried at 190 °C overnight. Deuterated solvents were purchased from Acros Organics and Cambridge Isotope Laboratories and were dried the same way as their protic analogues. The NMR spectra were recorded at room temperature on a Varian Inova 500 MHz (1H: 500.13 MHz and 31P: 202.46 MHz) and a Bruker Avance 800 MHz spectrometer (1H: 800.13 MHz, 13C: 201.19 MHz). 1H and 13C chemical shifts are reported in parts per million (ppm) and are referenced using the residual proton and carbon signals of the deuterated solvent (1H: C6D6, δ 7.16; 13C: C6D6, δ 128.06; 1H: THF-d8, δ 3.58, 1.72; 13C: THF-d8, δ 67.21, 25.31). 31P NMR chemical shifts are reported in ppm and are referenced externally to an 85% H3PO4 solution. Elemental analyses were performed at the University of Virginia and Midwest Microlab, 7212 North Shadeland Avenue, Suite 110, Indianapolis, IN 46250, USA. Single-crystal X-ray diffraction data were collected on a Bruker Kappa APEXII Duo system. An Incoatec Microfocus IμS (Cu Kα, λ = 1.54178 Å) and a multilayer mirror monochromator were used for 1, 3, and 4, and a fine-focus sealed tube (Mo Kα, λ = 0.71073 Å) and a graphite monochromator were used for 2, 5, and 6. The frames were integrated with the Bruker SAINT software package21 using a narrow-frame algorithm. Data were corrected for absorption effects using the Multi-Scan method.21 The structures were solved and refined using the Bruker SHELXTL software package22 within APEX321 and OLEX2.23 Non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed in geometrically calculated positions with Uiso = 1.2Uequiv of the parent atom (Uiso = 1.5Uequiv for methyl). For 3, CELL_NOW24 was used to identify a two-component twin. Starting with 1058 reflections, 889 reflections were fit to the first domain and 441 to the second domain (165 exclusively), with 4 unindexed reflections remaining. The twin domain was oriented at a 179.9° rotation about the reciprocal axis 0.003 0.500 1.000. The twin law was −0.994 0.009 0.004/0.622 −0.318 0.657/1.257 1.364 0.311. The structure was refined as a two-component twin on HKLF5 data, with the BASF for the twin domain refining to 0.12525. One isopropyl group was found to be disordered over two positions. The relative occupancy was freely refined, and constraints were used on the anisotropic displacement parameters of one pair of disordered atoms. For 4, the relative occupancies of the disordered isopropyl groups were freely refined. Constraints were used on the anisotropic displacement parameters of the disordered C6/C6a pair. For 5, one isopropyl group was disordered over two positions. The relative occupancy was freely refined and restraints were used on the anisotropic displacement parameters of the disordered atoms. The OCP unit connecting the two carbenes was disordered by symmetry and was therefore modeled at 50% occupancy. The outer-sphere [OCP]− anion was disordered over two positions, each of which was located on a symmetry element. The relative occupancies of the different orientations were freely refined, at 50% occupancy to account for the symmetry and then with restraints on the anisotropic displacement parameters and bond lengths of the disordered atoms.
Synthesis of (NHC)SbPh2Cl (1)
To a 20 mL scintillation vial, Ph2SbCl (690 mg, 2.22 mmol) was added and stirred in toluene (5 mL). A toluene solution (5 mL) of NHC (400 mg, 2.22 mmol) was added, and then, the reaction was allowed to stir for 1 h. After the filtration, the crude solid was washed with hexanes and then dried in vacuo. Compound 1 was obtained as a white solid (925 mg, 85%). Crystals suitable for X-ray diffraction studies were obtained from a toluene/hexane mixture at −37 °C. 1H NMR (C6D6, 500.13 MHz): δ 8.17 (t, 4H, CHortho), 7.22 (t, 4H, CHmeta), 7.12 (t, 2H, CHpara), 4.69 (Br, 2H, CH(CH3)2), 1.56 (s, 6H, C(backbone)–CH3), 0.81 (s, 12H, CH(CH3)2). 13C{1H} NMR (THF-d8, 201.193 MHz): δ 146.53 (CPh-i), 136.50 (CPh-o), 128.51 (CPh-m), 128.23 (CPh-p), 125.33 (Cvinyl), 52.34 (N–CH–(CH3)2), 21.23 (N–CH–(CH3)2), 9.74 (Cvinyl–CH3). Anal. calcd for C23H30N2SbCl: C, 56.18; H, 6.15; N, 5.70%. Found: C, 55.95; H, 6.22; N, 5.68%.
Synthesis of (NHC)BiPh2Cl (2)
To a 20 mL scintillation vial, Ph2BiCl (1.111 g, 2.77 mmol) was added and stirred in toluene (5 mL). A toluene solution (5 mL) of NHC (500 mg, 2.77 mmol) was added, and then, the reaction was allowed to stir for 1 h. After the filtration, the crude solid was washed with hexanes and then dried in vacuo. Compound 2 was obtained as a white solid (1.51 g, 94%). Colorless crystals suitable for X-ray diffraction studies were obtained from a toluene/hexane mixture at −37 °C. 1H NMR (THF-d8, 500.13 MHz): δ 8.35 (br, 4H, CHortho), 7.41 (t, J = 7.6 Hz, 4H, CHmeta), 7.21 (t, J = 7.3 Hz, 2H, CHpara), 4.51 (hept, J = 6.7 Hz, 2H, CH(CH3)2), 2.15 (s, 6H, C(backbone)–CH3), 1.17 (d, J = 7 Hz, 12H, CH(CH3)2). 13C{1H} NMR (THF-d8, 201.19 MHz): δ 139.35 (CPh-o), 131.45 (CPh-m), 128.08 (CPh-p), 126.48 (Cvinyl), 54.12 (N–CH–(CH3)2), 22.75 (N–CH–(CH3)2), 10.28 (Cvinyl–CH3). Anal. calcd for C23H30N2BiCl: C, 47.72; H, 5.22; N, 4.84%. Found: C, 47.37; H, 5.41; N, 4.77%.
Synthesis of NHC–C(O)P-SbPh2 (3)
To a 20 mL vial, (NHC)BiPh2Cl (97 mg, 0.197 mmol) was added and stirred in THF. Na[OCP]•(dioxane)x (65 mg, 0.217 mmol) was added to the stirring solution. upon addition, the solution immediately turned yellow. After stirring for 5 min at room temperature, insoluble NaCl was removed by filtration, and the yellow THF solution was layered with hexanes in a 1:1 ratio and allowed to sit for 1 day at −37 °C. After removal of the solvent and drying in vacuo, the product was obtained as a yellow crystalline solid (50 mg, 49% yield). Note: compound 3 decomposes to 5 and Ph4Bi2 at room temperature. Note: compound 3 decomposes to 5 and Ph4Bi2 at −37 °C. 1H NMR (C6D6, 500.13 MHz) δ 8.18 (d, J = 7.9 Hz, 4H, CHortho), 7.21–7.15 (m, 4H, CHmeta), 7.12 (t, J = 7.3 Hz, 2H CHpara), 5.10 (hept, J = 6.9 Hz, 2H, CH(CH3)2), 1.34 (s, 6H, C(backbone)–CH3), 1.06 (d, J = 7.1 Hz, 12H, CH(CH3)2). 13C{1H} NMR (201.19 MHz, C6D6) δ 203.13 (d, J = 76.0 Hz, C=O), 148.84 (d, J = 52.8 Hz, CNHC), 141.01 (CPh-i), 137.73 (CPh-o), 128.28 (CPh-m), 127.28 (CPh-p), 123.30 (Cvinyl), 51.16 (N-CH-(CH3)2), 21.08 (N–CH-(CH3)2), 9.25 (Cvinyl-CH3). 31P{1H} NMR (202.46 MHz, C6D6) δ 58.18 (s, 1P). IR: ν = 3040, 2971, 2934, 2854, 1625, 1574, 1427, 1371, 1310, 1217, 1105, 1051, 928, 729, 697 cm–1. Suitable elemental analysis could not be obtained due to solid-state instability. Thus, purity was assessed by immediately collecting the 1H, 13C, and 31P NMR data of a freshly made sample of 3.
Synthesis of NHC–C(O)P-BiPh2 (4)
To a 20 mL vial, (NHC)BiPh2Cl (200 mg, 0.344 mmol) was added and stirred in THF. Na[OCP]·(dioxane)x (302 mg, 0.344 mmol) was added to the stirring solution. Immediately upon addition, the solution turned yellow. After it stirred for 5 min at room temperature, insoluble NaCl was removed by filtration and the yellow THF solution was layered with hexanes in a 1:1 ratio and allowed to sit for 1 day at −37 °C. After removal of the solvent and drying in vacuo, the product was obtained as a yellow crystalline solid (92 mg, 48% yield). Note: Compound 4 decomposes to 5 and Ph4Bi2 at −37 °C. 1H NMR (C6D6, 500.13 MHz): δ 8.51 (d, J = 7.6 Hz, 4H, CHortho), 7.25 (t, J = 7.5 Hz, 4H, CHmeta), 7.20–7.13 (m, 2H, CHpara), 5.13 (hept, J = 6.9 Hz, 2H, CH(CH3)2), 1.33 (s, 6H, C(backbone)–CH3), 1.06 (d, J = 7.1 Hz, 12H, CH(CH3)2). 13C{1H} NMR (201.19 MHz, C6D6): δ 203.58 (d, J = 81.7 Hz, C=O), 152.03 (d, J = 49.6 Hz, CNHC), 151.04 (CPh-i), 140.05 (CPh-o), 130.15 (CPh-m), 126.75 (CPh-p), 123.45 (Cvinyl), 51.36 (N–CH–(CH3)2), 21.37 (N–CH–(CH3)2), 9.46 (Cvinyl–CH3). 31P{1H} NMR (202.46 MHz, C6D6): δ 82.17 (s, 1P). IR: ν = 3033, 2975, 2932, 2869, 1625, 1569, 1418, 1371, 1312, 1217, 1110, 995, 928, 723, 697 cm–1. Suitable elemental analysis could not be obtained due to solid-state instability. Thus, purity was assessed by immediately collecting the 1H, 13C, and 31P NMR data of a freshly made sample of 4.
Synthesis of [NHC–PC(=O)–(NHC)][OCP] (5)
To a 20 mL scintillation vial, (NHC)BiPh2Cl (505 mg, 869 μmol) was added and suspended in 10 mL of dry THF. Na[OCP]·(dioxane)x (262 mg, 869 μmol) was added to the suspension, and the suspension was shaken vigorously for 1 min. The reaction mixture was then extracted into a 100 mL Schlenk tube. Orange crystals of [(NHC)2OCP][OCP] formed from the solution after sitting undisturbed at 55 °C overnight (106 mg, 51%). 1H NMR (500.13 MHz, CD2Cl2): δ 5.35 (br, 4.99, 2H, CH(CH3)2) (hept, J = 7.0 Hz, 2H, CH(CH3)2), 2.38 (s, 6H, C(backbone)–CH3), 2.35 (s, 6H, C(backbone)–CH3), 1.62 (d, J = 7.1 Hz, 12H, CH(CH3)2), 1.58 (d, J = 7.1 Hz, 12H, CH(CH3)2). 13C{1H} NMR (201.19 MHz, CD2Cl2): δ 200.71 (d, J = 64.2 Hz, C=O), 170.20 (d, J = 63.0 Hz, OCP), 150.24 (d, J = 86.9 Hz, CNHC–P), 146.04 (d, J = 67.5 Hz CNHC–C=O), 128.8 (Cvinyl), 126.5 (Cvinyl), 53.9 (N–CH–(CH3)2), 52.5 (N–CH–(CH3)2), 21.9 (N–CH–(CH3)2), 21.8 (N–CH–(CH3)2), 11.0 (Cvinyl–CH3), 10.7 (Cvinyl–CH3). 31P{1H} NMR (242.94 MHz, CD2Cl2): δ 22.72 (br, 1P, [OCP]+), −395.09 (s, 1P, [OCP]−). IR: ν = 3044, 2973, 2934, 2873, 1788, 1768, 1634, 1574, 1429, 1371, 1312, 1217, 1051, 930, 729, 729, 699 cm–1. Anal. calcd for C24H40N4O2P2: C, 60.24; H, 8.43; N, 11.71%. Found: C, 60.09; H, 8.52; N, 11.70%.
Computational Details
The computations were carried out with the Gaussian 09 suite of programs.23 The structures were optimized using the ωB97XD functional in combination with the def2-SVP and the def2-TZVP basis sets. At each of the optimized structures vibrational analysis was accomplished to check whether the stationary point located is a minimum or a saddle point of the potential energy hypersurface. We neglected the solvent effect because toluene was used as the solvent. For Wiberg Bond Indexes and NPA charges, the NBO program version 5.0 was employed.24 The plotting of the orbitals was carried out with the AVOGADRO program (www.avogadro.cc).
Acknowledgments
The authors are grateful to the University of Virginia for support of this work. Further support came from NKFIH PD 116329, the Bolyai Research Fellowship of MTA, an UNKP grant (UNKP-20-5-BME-317) and a BME Nanotechnology and Materials Science TKP2020 IE grant from NKFIH Hungary (BME IE-NAT TKP2020).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.0c03683.
NMR spectral data; IR spectral data; crystal structure of compound 6; crystallographic refinement details; and computational details (PDF)
Accession Codes
CCDC 1962839–1962843 and 1976639 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- a Appel R.; Knoll F., Double Bonds Between Phosphorus and Carbon. In Adv. Inorg. Chem., Sykes A. G., Ed.; Academic Press: 1989; Vol. 33, pp 259–361. [Google Scholar]; b Escudié J.; Ranaivonjatovo H.; Rigon L. Heavy Allenes and Cumulenes ECE′ and ECCE′ (E = P, As, Si, Ge, Sn; E′ = C, N, P, As, O, S). Chem. Rev. 2000, 100, 3639–3696. 10.1021/cr990013z. [DOI] [PubMed] [Google Scholar]; c Rey A.; Espinosa Ferao A.; Streubel R. Quantum Chemical Calculations on CHOP Derivatives-Spanning the Chemical Space of Phosphinidenes, Phosphaketenes, Oxaphosphirenes, and COP– Isomers. Molecules 2018, 23, 3341. 10.3390/molecules23123341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel R.; Paulen W. The First Stable Phosphaketene. Angew. Chem., Int. Ed. Engl. 1983, 22, 785–786. 10.1002/anie.198307852. [DOI] [Google Scholar]
- a Goicoechea J. M.; Grützmacher H. The Chemistry of the 2-Phosphaethynolate Anion. Angew. Chem., Int. Ed. 2018, 57, 16968–16994. 10.1002/anie.201803888. [DOI] [PubMed] [Google Scholar]; b Puschmann F. F.; Stein D.; Heift D.; Hendriksen C.; Gal Z. A.; Grützmacher H.-F.; Grützmacher H. Phosphination of Carbon Monoxide: A Simple Synthesis of Sodium Phosphaethynolate (NaOCP). Angew. Chem., Int. Ed. 2011, 50, 8420–8423. 10.1002/anie.201102930. [DOI] [PubMed] [Google Scholar]; c Becker G.; Schwarz W.; Seidler N.; Westerhausen M. Acyl- und Alkylidenphosphane. XXXIII. Lithoxy-methylidenphosphan DME und -methylidinphosphan 2 DME ? Synthese und Struktur. Z. Anorg. Allg. Chem. 1992, 612, 72–82. 10.1002/zaac.19926120113. [DOI] [Google Scholar]
- a Yang W.; Krantz K. E.; Dickie D. A.; Molino A.; Wilson D. J. D.; Gilliard R. J. Jr. Crystalline BP-Doped Phenanthryne via Photolysis of The Elusive Boraphosphaketene. Angew. Chem., Int. Ed. 2020, 59, 3971–3975. 10.1002/anie.201916362. [DOI] [PubMed] [Google Scholar]; b Heift D.; Benkő Z.; Grützmacher H. Is the phosphaethynolate anion, (OCP)–, an ambident nucleophile? A spectroscopic and computational study. Dalton Trans. 2014, 43, 5920–5928. 10.1039/c3dt53569j. [DOI] [PubMed] [Google Scholar]; c Mei Y.; Borger J. E.; Wu D.-J.; Grützmacher H. Salen supported Al–O–C–P and Ga–P–C–O complexes. Dalton Trans. 2019, 48, 4370–4374. 10.1039/C9DT00485H. [DOI] [PubMed] [Google Scholar]; d Li Z.; Chen X.; Li Y.; Su C.-Y.; Grützmacher H. N-Heterocyclic carbene phosphaketene adducts as precursors to carbene–phosphinidene adducts and a rearranged π-system. Chem. Commun. 2016, 52, 11343–11346. 10.1039/C6CC05916C. [DOI] [PubMed] [Google Scholar]; e Li Z.; Chen X.; Benkő Z.; Liu L.; Ruiz D. A.; Peltier J. L.; Bertrand G.; Su C.-Y.; Grützmacher H. N-Heterocyclic Carbenes as Promotors for the Rearrangement of Phosphaketenes to Phosphaheteroallenes: A Case Study for OCP to OPC Constitutional Isomerism. Angew. Chem., Int. Ed. 2016, 55, 6018–6022. 10.1002/anie.201600903. [DOI] [PubMed] [Google Scholar]; f Weber L. 2-Phospha- and 2-Arsaethynolates – Versatile Building Blocks in Modern Synthetic Chemistry. Eur. J. Inorg. Chem. 2018, 2018, 2175–2227. 10.1002/ejic.201800179. [DOI] [Google Scholar]
- a Appel R.; Laubach B.; Siray M. Einschiebungsreaktion von kohlendioxid in disilylierte phosphane. Tetrahedron Lett. 1984, 25, 4447–4448. 10.1016/S0040-4039(01)81462-3. [DOI] [Google Scholar]; b Heift D.; Benkő Z.; Grützmacher H. Coulomb repulsion versus cycloaddition: formation of anionic four-membered rings from sodium phosphaethynolate, Na(OCP). Dalton Trans. 2014, 43, 831–840. 10.1039/C3DT52359D. [DOI] [PubMed] [Google Scholar]
- a Gilliard R. J.; Heift D.; Benkő Z.; Keiser J. M.; Rheingold A. L.; Grützmacher H.; Protasiewicz J. D. An isolable magnesium diphosphaethynolate complex. Dalton Trans. 2018, 47, 666–669. 10.1039/C7DT04539E. [DOI] [PubMed] [Google Scholar]; b Westerhausen M.; Schneiderbauer S.; Piotrowski H.; Suter M.; Nöth H. Synthesis of alkaline earth metal bis(2-phosphaethynolates). J. Organomet. Chem. 2002, 643–644, 189–193. 10.1016/S0022-328X(01)01267-0. [DOI] [Google Scholar]; c Jupp A. R.; Goicoechea J. M. The 2-Phosphaethynolate Anion: A Convenient Synthesis and [2 + 2] Cycloaddition Chemistry. Angew. Chem., Int. Ed. 2013, 52, 10064–10067. 10.1002/anie.201305235. [DOI] [PubMed] [Google Scholar]; d Hennersdorf F.; Frötschel J.; Weigand J. J. Selective Derivatization of a Hexaphosphane from Functionalization of White Phosphorus. J. Am. Chem. Soc. 2017, 139, 14592–14604. 10.1021/jacs.7b07704. [DOI] [PubMed] [Google Scholar]
- a Hoerger C. J.; Heinemann F. W.; Louyriac E.; Maron L.; Grützmacher H.; Meyer K. Formation of a Uranium-Bound η1-Cyaphide (CP−) Ligand via Activation and C–O Bond Cleavage of Phosphaethynolate (OCP−). Organometallics 2017, 36, 4351–4354. 10.1021/acs.organomet.7b00590. [DOI] [Google Scholar]; b Camp C.; Settineri N.; Lefèvre J.; Jupp A. R.; Goicoechea J. M.; Maron L.; Arnold J. Uranium and thorium complexes of the phosphaethynolate ion. Chem. Sci. 2015, 6, 6379–6384. 10.1039/C5SC02150B. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bestgen S.; Chen Q.; Rees N. H.; Goicoechea J. M. Synthesis and reactivity of rare-earth metal phosphaethynolates. Dalton Trans. 2018, 47, 13016–13024. 10.1039/C8DT03427C. [DOI] [PubMed] [Google Scholar]
- a Yao S.; Xiong Y.; Szilvási T.; Grützmacher H.; Driess M. From a Phosphaketenyl-Functionalized Germylene to 1,3-Digerma-2,4-diphosphacyclobutadiene. Angew. Chem., Int. Ed. 2016, 55, 4781–4785. 10.1002/anie.201600701. [DOI] [PubMed] [Google Scholar]; b Wu Y.; Liu L.; Su J.; Zhu J.; Ji Z.; Zhao Y. Isolation of a Heavier Cyclobutadiene Analogue: 2,4-Digerma-1,3-diphosphacyclobutadiene. Organometallics 2016, 35, 1593–1596. 10.1021/acs.organomet.6b00187. [DOI] [Google Scholar]; c Hinz A.; Goicoechea J. M. Limitations of Steric Bulk: Towards Phospha-germynes and Phospha-stannynes. Chem. - Eur. J. 2018, 24, 7358–7363. 10.1002/chem.201801329. [DOI] [PubMed] [Google Scholar]; d Xiong Y.; Yao S.; Szilvási T.; Ballestero-Martínez E.; Grützmacher H.; Driess M. Unexpected Photodegradation of a Phosphaketenyl-Substituted Germyliumylidene Borate Complex. Angew. Chem., Int. Ed. 2017, 56, 4333–4336. 10.1002/anie.201701337. [DOI] [PubMed] [Google Scholar]; e Del Rio N.; Baceiredo A.; Saffon-Merceron N.; Hashizume D.; Lutters D.; Müller T.; Kato T. A Stable Heterocyclic Amino(phosphanylidene-σ4-phosphorane) Germylene. Angew. Chem., Int. Ed. 2016, 55, 4753–4758. 10.1002/anie.201511956. [DOI] [PubMed] [Google Scholar]
- Wilson D. W. N.; Myers W. K.; Goicoechea J. M. Synthesis and decarbonylation chemistry of gallium phosphaketenes. Dalton Trans. 2020, 49, 15249–15255. 10.1039/D0DT03174G. [DOI] [PubMed] [Google Scholar]
- a Wilson D. W. N.; Hinz A.; Goicoechea J. M. An Isolable Phosphaethynolatoborane and Its Reactivity. Angew. Chem., Int. Ed. 2018, 57, 2188–2193. 10.1002/anie.201712624. [DOI] [PubMed] [Google Scholar]; b Wilson D. W. N.; Franco M. P.; Myers W. K.; McGrady J. E.; Goicoechea J. M. Base induced isomerisation of a phosphaethynolato-borane: mechanistic insights into boryl migration and decarbonylation to afford a triplet phosphinidene. Chem. Sci. 2020, 11, 862–869. 10.1039/C9SC05969E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Alidori S.; Heift D.; Santiso-Quinones G.; Benkő Z.; Grützmacher H.; Caporali M.; Gonsalvi L.; Rossin A.; Peruzzini M. Synthesis and Characterization of Terminal [Re(XCO)(CO)2(triphos)] (X=N, P): Isocyanate versus Phosphaethynolate Complexes. Chem. - Eur. J. 2012, 18, 14805–14811. 10.1002/chem.201202590. [DOI] [PubMed] [Google Scholar]; b Mielke Z.; Andrews L. Infrared detection of the PCO radical and HPCO molecule. Chem. Phys. Lett. 1991, 181, 355–360. 10.1016/0009-2614(91)80084-B. [DOI] [Google Scholar]; c Hou G.-L.; Chen B.; Transue W. J.; Yang Z.; Grützmacher H.; Driess M.; Cummins C. C.; Borden W. T.; Wang X.-B. Spectroscopic Characterization, Computational Investigation, and Comparisons of ECX– (E = As, P, and N; X = S and O) Anions. J. Am. Chem. Soc. 2017, 139, 8922–8930. 10.1021/jacs.7b02984. [DOI] [PubMed] [Google Scholar]
- Hansmann M. M.; Bertrand G. Transition-Metal-like Behavior of Main Group Elements: Ligand Exchange at a Phosphinidene. J. Am. Chem. Soc. 2016, 138, 15885–15888. 10.1021/jacs.6b11496. [DOI] [PubMed] [Google Scholar]
- von Frantzius G.; Espinosa Ferao A.; Streubel R. Coordination of CO to low-valent phosphorus centres and other related P–C bonding situations. A theoretical case study. Chem. Sci. 2013, 4, 4309–4322. 10.1039/c3sc52027g. [DOI] [Google Scholar]
- a Do D. C. H.; Protchenko A. V.; Vasko P.; Campos J.; Kolychev E. L.; Aldridge S. N-nacnac Stabilized Tetrelenes: Formation of an N,P-Heterocyclic Germylene via C–C Bond Insertion. Z. Anorg. Allg. Chem. 2018, 644, 1238–1242. 10.1002/zaac.201800259. [DOI] [Google Scholar]; b Szkop K. M.; Jupp A. R.; Stephan D. W. P,P-Dimethylformylphosphine: The Phosphorus Analogue of N,N-Dimethylformamide. J. Am. Chem. Soc. 2018, 140, 12751–12755. 10.1021/jacs.8b09266. [DOI] [PubMed] [Google Scholar]; c Paparo A.; Jones C. Beryllium Halide Complexes Incorporating Neutral or Anionic Ligands: Potential Precursors for Beryllium Chemistry. Chem. - Asian J. 2019, 14, 486–490. 10.1002/asia.201801800. [DOI] [PubMed] [Google Scholar]
- a Li Z.; Chen X.; Bergeler M.; Reiher M.; Su C.-Y.; Grützmacher H. A stable phosphanyl phosphaketene and its reactivity. Dalton Trans. 2015, 44, 6431–6438. 10.1039/C4DT04012K. [DOI] [PubMed] [Google Scholar]; b Mehta M.; McGrady J. E.; Goicoechea J. M. B(C6F5)3-Enabled Synthesis of a Cyclic cis-Arsaphosphene. Chem. - Eur. J. 2019, 25, 5445–5450. 10.1002/chem.201901022. [DOI] [PubMed] [Google Scholar]
- a Kindervater M. B.; Marczenko K. M.; Werner-Zwanziger U.; Chitnis S. S. A Redox-Confused Bismuth(I/III) Triamide with a T-Shaped Planar Ground State. Angew. Chem., Int. Ed. 2019, 58, 7850–7855. 10.1002/anie.201903354. [DOI] [PubMed] [Google Scholar]; b Calderazzo F.; Morvillo A.; Pelizzi G.; Poli R. Synthesis and crystal and molecular structure of tetraphenyldibismuthine, Bi2Ph4, the first crystallographically characterized tetraorganyl derivative of bismuth(II). J. Chem. Soc., Chem. Commun. 1983, 507–508. 10.1039/c39830000507. [DOI] [Google Scholar]; c Yasui M.; Kikuchi T.; Iwasaki F.; Suzuki H.; Murafuji T.; Ogawa T. First X-ray structure determination of a bismuthio ylide: 4,4-dimethyl-2,6-dioxo-1-triphenylbismuthiocyclohexanide. J. Chem. Soc., Perkin Trans. 1 1990, 3367–3368. 10.1039/p19900003367. [DOI] [Google Scholar]; d Sasamori T.; Takeda N.; Fujio M.; Kimura M.; Nagase S.; Tokitoh N. Synthesis and Structure of the First Stable Phosphabismuthene. Angew. Chem., Int. Ed. 2002, 41, 139–141. 10.1002/1521-3773(20020104)41:1<139::AID-ANIE139>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]; e Vehkamäki M.; Hatanpää T.; Ritala M.; Leskelä M. Bismuth precursors for atomic layer deposition of bismuth-containing oxide films. J. Mater. Chem. 2004, 14, 3191–3197. 10.1039/B405891G. [DOI] [Google Scholar]; f Soran A. P.; Silvestru C.; Breunig H. J.; Balázs G.; Green J. C. Organobismuth(III) Dihalides with T-Shaped Geometry Stabilized by Intramolecular N→Bi Interactions and Related Diorganobismuth(III) Halides. Organometallics 2007, 26, 1196–1203. 10.1021/om0611608. [DOI] [Google Scholar]; g Conrad E.; Burford N.; McDonald R.; Ferguson M. J. Bismuthenium-pnictonium dications [R′BiPnR3]2+ (Pn = As, Sb) containing carbenoid bismuth centers and rare Bi–Sb bonds. Chem. Commun. 2010, 46, 4598–4600. 10.1039/c0cc00111b. [DOI] [PubMed] [Google Scholar]; h Zhang X.-P.; Tian H.-R.; Yan G.-F.; Su Y.; Feng Y.-L.; Cheng J.-W. Incorporating different secondary building units of {Bi2}, {Bi8} and {Bi10} to construct diversity of luminescent bismuth–organic frameworks. Dalton Trans. 2013, 42, 1088–1093. 10.1039/C2DT31821K. [DOI] [PubMed] [Google Scholar]; i Balasanthiran V.; Chisholm M. H.; Durr C. B.; Gallucci J. C. Single-site bismuth alkoxide catalysts for the ring-opening polymerization of lactide. Dalton Trans. 2013, 42, 11234–11241. 10.1039/c3dt50629k. [DOI] [PubMed] [Google Scholar]; j Schwamm R. J.; Day B. M.; Coles M. P.; Fitchett C. M. Low-Coordinate Bismuth Cations. Inorg. Chem. 2014, 53, 3778–3787. 10.1021/ic500113g. [DOI] [PubMed] [Google Scholar]; k Solyntjes S.; Bader J.; Neumann B.; Stammler H.-G.; Ignat’ev N.; Hoge B. Pentafluoroethyl Bismuth Compounds. Chem. - Eur. J. 2017, 23, 1557–1567. 10.1002/chem.201604910. [DOI] [PubMed] [Google Scholar]; l Turner Z. R. Bismuth Pyridine Dipyrrolide Complexes: a Transient Bi(II) Species Which Ring Opens Cyclic Ethers. Inorg. Chem. 2019, 58, 14212–14227. 10.1021/acs.inorgchem.9b02314. [DOI] [PubMed] [Google Scholar]; m Wang F.; Planas O.; Cornella J. Bi(I)-Catalyzed Transfer-Hydrogenation with Ammonia-Borane. J. Am. Chem. Soc. 2019, 141, 4235–4240. 10.1021/jacs.9b00594. [DOI] [PMC free article] [PubMed] [Google Scholar]; n Lo Y.-H.; Gabbaï F. P. An Antimony(V) Dication as a Z-Type Ligand: Turning on Styrene Activation at Gold. Angew. Chem., Int. Ed. 2019, 58, 10194–10197. 10.1002/anie.201903964. [DOI] [PubMed] [Google Scholar]; o Alič B.; Štefančič A.; Tavčar G. Small molecule activation: SbF3 auto-ionization supported by transfer and mesoionic NHC rearrangement. Dalton Trans. 2017, 46, 3338–3346. 10.1039/C6DT04909E. [DOI] [PubMed] [Google Scholar]; p Dorsey C. L.; Mushinski R. M.; Hudnall T. W. Metal-Free Stabilization of Monomeric Antimony(I): A Carbene-Supported Stibinidene. Chem. - Eur. J. 2014, 20 (29), 8914–8917. 10.1002/chem.201403578. [DOI] [PubMed] [Google Scholar]; q Arduengo A. J. III; Davidson F.; Krafczyk R.; Marshall W. J.; Schmutzler R. Carbene Complexes of Pnictogen Pentafluorides and Boron Trifluoride. Monatsh. Chem. 2000, 131, 0251–0265. 10.1007/s007060070101. [DOI] [Google Scholar]; r Krüger J.; Wölper C.; John L.; Song L.; Schreiner P. R.; Schulz S. Syntheses, Structures, and Bonding Analyses of Carbene-Stabilized Stibinidenes. Eur. J. Inorg. Chem. 2019, 2019, 1669–1678. 10.1002/ejic.201900167. [DOI] [Google Scholar]; s Li Y.-Z.; Ganguly R.; Leong W. K. Oxidative Addition across Sb–H and Sb–Sb Bonds by an Osmium Carbonyl Cluster: Trapping the Intermediate. Organometallics 2014, 33, 823–828. 10.1021/om401206d. [DOI] [Google Scholar]
- a Henne F. D.; Dickschat A. T.; Hennersdorf F.; Feldmann K. O.; Weigand J. J. Synthesis of Selected Cationic Pnictanes [LnPnX3–n]n+ (L = Imidazolium-2-yl; Pn = P, As; n = 1–3) and Replacement Reactions with Pseudohalogens. Inorg. Chem. 2015, 54, 6849–6861. 10.1021/acs.inorgchem.5b00765. [DOI] [PubMed] [Google Scholar]; b Waters J. B.; Chen Q.; Everitt T. A.; Goicoechea J. M. N-Heterocyclic carbene adducts of the heavier group 15 tribromides. Normal to abnormal isomerism and bromide ion abstraction. Dalton Trans. 2017, 46, 12053–12066. 10.1039/C7DT02431B. [DOI] [PubMed] [Google Scholar]; c Ho L. P.; Nasr A.; Jones P. G.; Altun A.; Neese F.; Bistoni G.; Tamm M. London Dispersion Interactions in Pnictogen Cations [ECl2]+ and [E = E]2+ (E = P, As, Sb) Supported by Anionic N-Heterocyclic Carbenes. Chem. - Eur. J. 2018, 24, 18922–18932. 10.1002/chem.201804714. [DOI] [PubMed] [Google Scholar]
- a Aprile A.; Corbo R.; Vin Tan K.; Wilson D. J. D.; Dutton J. L. The first bismuth–NHC complexes. Dalton Trans. 2014, 43, 764–768. 10.1039/C3DT52715H. [DOI] [PubMed] [Google Scholar]; b Wang G.; Freeman L. A.; Dickie D. A.; Mokrai R.; Benkő Z.; Gilliard R. J. Highly Reactive Cyclic(alkyl)(amino) Carbene- and N-Heterocyclic Carbene-Bismuth(III) Complexes: Synthesis, Structure, and Computations. Inorg. Chem. 2018, 57, 11687–11695. 10.1021/acs.inorgchem.8b01813. [DOI] [PubMed] [Google Scholar]; c Walley J.; Warring L.; Wang G.; Dickie D. A.; Pan S.; Frenking G.; Gilliard R. J., Carbodicarbene Bismaalkene Cations: Unravelling the Complexities of Carbene versus Carbone in Heavy Pnictogen Chemistry. Angew. Chem., Int. Ed. 2020, DOI: 10.1002/anie.202014398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom R.; Haaland A. A modification of the Schomaker—Stevenson rule for prediction of single bond distances. J. Mol. Struct. 1985, 128, 21–27. 10.1016/0022-2860(85)85036-5. [DOI] [Google Scholar]
- a Jost M.; Finger L. H.; Sundermeyer J.; von Hänisch C. Simple access to ionic liquids and organic salts containing the phosphaethynolate (PCO−) and Zintl (Sb113−) anions. Chem. Commun. 2016, 52, 11646–11648. 10.1039/C6CC06620H. [DOI] [PubMed] [Google Scholar]; b Liu L.; Ruiz D. A.; Dahcheh F.; Bertrand G.; Suter R.; Tondreau A. M.; Grützmacher H. Isolation of Au-, Co-η1PCO and Cu-η2PCO complexes, conversion of an Ir−η1PCO complex into a dimetalladiphosphene, and an interaction-free PCO anion. Chem. Sci. 2016, 7, 2335–2341. 10.1039/C5SC04504E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Doddi A.; Bockfeld D.; Zaretzke M.-K.; Kleeberg C.; Bannenberg T.; Tamm M. A modular approach to carbene-stabilized diphosphorus species. Dalton Trans. 2017, 46, 15859–15864. 10.1039/C7DT03436A. [DOI] [PubMed] [Google Scholar]; b Wang Y.; Xie Y.; Wei P.; King R. B.; Schaefer H. F.; Schleyer P. v. R.; Robinson G. H. Carbene-Stabilized Diphosphorus. J. Am. Chem. Soc. 2008, 130, 14970–14971. 10.1021/ja807828t. [DOI] [PubMed] [Google Scholar]; c Wang Y.; Robinson G. H. Carbene Stabilization of Highly Reactive Main-Group Molecules. Inorg. Chem. 2011, 50, 12326–12337. 10.1021/ic200675u. [DOI] [PubMed] [Google Scholar]
- a Waters J. B.; Everitt T. A.; Myers W. K.; Goicoechea J. M. N-heterocyclic carbene induced reductive coupling of phosphorus tribromide. Isolation of a bromine bridged P–P bond and its subsequent reactivity. Chem. Sci. 2016, 7, 6981–6987. 10.1039/C6SC02343F. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Back O.; Donnadieu B.; Parameswaran P.; Frenking G.; Bertrand G. Isolation of crystalline carbene-stabilized P2-radical cations and P2-dications. Nat. Chem. 2010, 2, 369–373. 10.1038/nchem.617. [DOI] [PubMed] [Google Scholar]; c Beil A.; Gilliard R. J.; Grützmacher H. From the parent phosphinidene–carbene adduct NHC–PH to cationic P4-rings and P2-cycloaddition products. Dalton Trans. 2016, 45, 2044–2052. 10.1039/C5DT03014E. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; et al. Gaussian 09, rev A.02; Gaussian, Inc.: Wallingford, CT, 2009.
- Glendening E. D.; Badenhoop J. K.; Reed A. E.; Carpenter J. E.; Bohmann J. A.; Morales C. M.; Weinhold F. In NBO 5.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, 2001.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.