

Abstract
The COVID-19 pandemic, caused by severe acute respiratory coronavirus 2 (SARS-CoV-2), has become a public health emergency and widely spread around the world. Rapid, accurate and early diagnosis of COVID-19 infection plays a crucial role in breaking this pandemic. However, the detection accuracy is limited for current single-gene diagnosis of SARS-CoV-2. Herein, we develop an autonomous lab-on-paper platform for multiplex gene diagnosis of SARS-CoV-2 by combining reverse transcription recombinase polymerase amplification (RT-RPA) and CRISPR-Cas12a detection. The autonomous lab-on-paper is capable of simultaneously detecting nucleoprotein (N) gene and spike (S) gene of SARS-CoV-2 virus as well as Human housekeeping RNAse P gene (an internal control) in a single clinical sample. With the developed platform, 102 copies viral RNA per test can be detected within one hour. Also, the lab-on-paper platform has been used to detect 21 swab clinical samples and obtains a comparable performance to the conventional RT-PCR method. Thus, the developed lab-on-paper platform holds great potential for rapid, sensitive, reliable, multiple molecular diagnostics of COVID-19 and other infectious diseases in resource-limited settings.
Keywords: Autonomous lab-on-paper, Multiplex gene diagnosis, CRISPR-Cas12a, RPA amplification, COVID-19 screening
Graphical Abstract
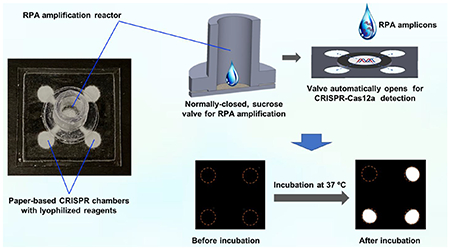
Autonomous lab-on-paper platform for simple, rapid, low-cost, and multiplex gene diagnosis of SARS-CoV-2 in clinical samples.
Introduction
Recently, the novel coronavirus (COVID-19) pandemic, caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has spread worldwide as a public health emergency.1 According to the World Health Organization (WHO), millions of people lost their lives after COVID-19 infection. The SARS-CoV-2 is primarily transmitted between people via contact and respiratory droplets. Due to the lack of an effective therapeutics strategy, rapid screening of symptomatic and asymptomatic infected individuals plays a critical role in reducing the spread of SARS-CoV-2 virus and saving lives.2,3 Most of the governments are exercising this practice, but mainly focus on the symptomatic patients due to shortage of testing capacity. However, the delay of detection and isolation of asymptomatic carriers increase the risk of the worldwide spread of COVID-19 pandemic.4,5
Nucleic acid-based molecular diagnostics provides a sensitive and reliable approach for pathogen detection. Among nucleic acid testing, quantitative reverse transcription polymerase chain reaction (RT-qPCR) is considered as the “gold-standard” method for SARS-CoV-2 detection and has been widely used for COVID-19 early diagnostics in clinical laboratory.6,7 However, The RT-qPCR method typically requires expensive equipment, well-trained operator, and relatively long turnaround time, which is not ideal for rapid on-site testing, at small clinics, and especially in resource-limited settings.8–10 In addition, the efficient SARS-CoV-2 surveillance requires the frequent testing with a fast turnaround time to prevent current pandemic.11,12 Therefore, a simple, rapid, reliable, sensitive, and cost-effective diagnostics method is still essential and meaningful.
In recent years, a series of novel CRISPR-based diagnosis methods have been developed to detect pathogens as an attractive alternative to conventional PCR method and shown great promise as the next-generation of nucleic acid detection.8,13–15 For instance, Cas12a is one of RNA-guided nucleases that owns collateral cleavage activities on single stranded DNA (ssDNA) under the guidance of a CRISPR crRNA.16 After recognizing its specific DNA targets, the cleavage activity of Cas12a can be activated, cleave the collateral ssDNA reporter and generate fluorescent signal.17 By combining with nucleic acid pre-amplification, a highly sensitive CRISPR-based nucleic acid detection can be achieved with an excellent specificity. However, most CRISPR-based molecular detections require multiple, separate manual operations to transfer amplification products, which complicates the testing procedures and increases the risk of carry-over contamination.16,18–20 Although the paper-based lateral flow strips provide a simple and visual detection for CRISPR-based nucleic acid diagnostics, it typically carries out in the open environment and potentially causes false-positive results.17 Additionally, due to the nature of non-specific collateral cleavage of CRISPR-Cas12a enzyme, most CRISPR-Cas12a-based diagnostic systems lack the ability of multiple detection and are able to only detect single gene of SARS-CoV-2, which potentially affects the detection accuracy.8,21 Therefore, a simple testing method for multiplex gene diagnosis of SARS-CoV-2 is of vital importance to improve the detection efficiency, accuracy, and clinical applicability.22
Herein, we have developed an autonomous lab-on-paper platform for simple, sensitive, reliable, simultaneous, multiplex-gene diagnosis of SARS-CoV-2 by CRISPR-Cas12a technology. The paper-based CRISPR detection chambers were designed and fabricated by wax printing technology. CRISPR-Cas12a detection reagent with specific crRNAs was pre-loaded and lyophilized in the detection chambers. The 3D-printed, recombinase polymerase amplification (RPA) reactor was physically separated from the paper-based CRISPR detection chambers by a programmable, normally closed, paper-based sucrose valve.23 After RPA amplification at pre-set time, the sucrose valve automatically opens and RPA amplicons migrate to the CRISPR-based detection chambers, enabling multiple gene detection of SARS-CoV-2. For clinical applications, the developed platform was demonstrated to simultaneously detect the nucleoprotein (N) gene and spike (S) gene of SARS-CoV-2 as well as human RNAse P gene (an housekeeping gene as an internal control) in a single clinical swab sample.
Experimental Methods
Reagents and samples
Cas12a (100 μM), deoxynucleotide (dNTP) mix (10 mM of each), AMV reverse transcriptase (10,000 U/mL) and nuclease-free water were purchased from New England BioLabs (Ipswich, MA). The crRNAs for the N and S genes of SARS-CoV-2 as well as the Human RNAse P gene were synthesized from Integrated DNA Technologies (IDT) (Coralville, IA). TEMED, (NH4)2S2O8, 30% acrylamide/bis-acrylamide solution, and 10×TBE Buffer were purchased from Bio-Rad Laboratories (Hercules, CA). Clear resin (FLGPCL02) was purchased from Formlabs. The quantitative PCR (qPCR) control RNA from heat inactivated SARS-CoV-2 (NR-52347) was obtained from the BEI Resources. TwistAmp Basic Kit was purchased from TwistDx Limited (Maidenhead, UK). The clinical swab samples were de-identified and in compliance with ethical regulations and the approval of Institutional Review Board of the University of Health Center (protocol #: P61067). All chemicals used were analytical reagent grade or better.
SARS-CoV-2 detection by RT-RPA/CRISPR-Cas12a assay
RT-RPA/CRISPR-Cas12a detection of the N and S gene of SARS-CoV-2 in reaction tubes was carried out as described in previous study.16 First, multiple RPA amplification reaction was carried out at 37 °C for 15 min with 0.48 μM RPA forward and reverse primer of the N gene of SARS-CoV-2, 0.48 μM RPA forward and reverse primer of the S gene of SARS-CoV-2, 14 mM magnesium acetate, 2 U of AMV reverse transcriptase, 1 μL SARS-CoV-2 RNA target and TwistAmp Basic reagent. Next, 2 μL RPA amplicons was distributed into individual reaction tube containing 13 μL CRISPR-Cas12a detection solution (100 nM Cas12a, 62.5 nM specific crRNA, 50 nM ssDNA-FQ reporter in reaction buffer) for CRISPR-based fluorescence detection. At the end of 25-min incubation reaction at 37 °C, the endpoint images were taken by either ChemiDocTM MP Imaging System or LED blue light illuminator (Maestrogen UltraSlim). The fluorescence intensity data was analyzed by Image J.
Fabrication of autonomous lab-on-paper platform
The RPA amplification reactor was designed by using SolidWorks software and fabricated on a Form 2 3D printer (Formlabs) with clear methacrylate-based resin (Formlabs, FLGPCL02). The paper-based CRISPR detection chambers and paper-based sucrose valve were designed by using SolidWorks software and printed on the Whatman Grade 1 paper using black wax by Xerox Colorqube 8870 printer. After printing, the paper-based CRISPR detection chambers were put on one hot plate for 30 seconds at 120 °C, allowing the printed wax to melt and penetrate through the paper-based cellulose membrane. For paper-based CRISPR detection chambers, each chamber was added with 2 μL CRISPR reaction solution. For the fabrication of the sucrose valve, sucrose solution at different concentrations (5-15%) was added on the paper-based valve, and dried at room temperature for 24 h. Then, the sucrose solution was added again on another side. Lastly, the 3D printed RPA amplification reactor, paper-based sucrose valve and paper-based CRISPR detection chambers were assembled by the 3M double-sided tape (9500 PC) and PCR Sealers tape (Microseal® ‘B’ Film) (Bio-Rad).
Lyophilization of CRISPR-Cas12a reagents on paper-based CRISPR chambers
The CRISPR-Cas12a reaction reagents contain 1 μM Cas12a, 500 nM ssDNA-FQ reporter, 10% trehalose solution. In our lab-on-paper platform, 1 μL CRISPR-Cas12a reaction solution was pre-loaded on each reaction chamber of the paper-based microfluidics, and lyophilized at −80 °C for 1 h using freeze-drying system (FreeZone 2.5 liter benchtop, Labconco). For SARS-CoV-2 detection, 0.625 μM crRNAs for SARS-CoV-2 N gene and S gene were, respectively added into CRISPR-Cas12a reaction solution. For housekeeping gene detection, 0.625 μM crRNA of human RNAse P gene was introduced into CRISPR-Cas12a reaction solution and lyophilized on the RNAse P gene detection chamber. For blank control, no crRNA was added into CRISPR-Cas12a reaction reagents.
Operation and sensitivity analysis of autonomous lab-on-paper platform
To investigate the sensitivity of SARS-CoV-2 detection on the lab-on-paper platform, ten-fold serial dilutions of SARS-CoV-2 RNA from 0 to 104 copies per test were first mixed with 25 μL RPA reaction solutions containing specific forward and reverse primers. After the mixture was added into the RPA amplification reactor, the reactor was sealed by the PCR Sealers tape (Microseal® ‘B’ Film) to avoid the aerosol contamination. Next, the autonomous lab-on-paper platform was incubated on the heating plate at 37 °C for 40 min. After the incubation reaction, the endpoint images of the lab-on-paper platform were taken by ChemiDoc™ MP Imaging System. The fluorescence intensity data of the CRISPR detection chambers was analyzed by Image J. The highest fluorescence intensity of positive signal collected was applied as the standard for the normalized fluorescence calculation. The test result was defined as positive if the normalized fluorescence was three standard deviation above the mean normalized fluorescence of the negative groups.
Multiple gene detection of SARS-CoV-2 in clinical swab samples
To investigate the clinical utility of the autonomous lab-on-paper platform for multiplex-gene detection, 21 de-identified clinical swab samples (including eight COVID-19 positive samples) were detected. Their viral RNAs were extracted by QIAamp DSP Viral RNA Mini Kit (QIAGEN N.V., Venlo) according to its instruction. 1 μL of RNA extracts was used for multiple detection in the autonomous lab-on-paper platform. After incubation at 37 °C for 40 min, the images of the lab-on-chip platform were taken by the ChemiDoc™ MP Imaging System.
Result and Discussion
Working principle of autonomous lab-on-paper platform
Nowadays, the novel CRISPR-Cas detection system combing target nucleic acid pre-amplification and CRISPR-Cas-based signal generation has emerged as a next-generation of nucleic acid-based molecular diagnosis technique.24 However, these detection methods typically require separate and multiple manual operations, such as amplification products transferring, which not only complicates the testing procedures but increases the risk of carry-over contaminations. To overcome these challenges, an autonomous lab-on-paper has been established for simple, rapid, automated, and multiplex gene diagnosis of SARS-CoV-2. As shown in Fig. 1A, the autonomous lab-on-paper platform mainly consists of: i) 3D printed RPA amplification reactor for multiple RPA amplification, ii) paper-based sucrose valve, and iii) paper-based CRISPR-Cas12a detection chambers. The 3D printed RPA amplification reactor and paper-based CRISPR detection chambers are physically separated by the sucrose valve, which is normally closed and automatically opens after RPA amplification at a pre-set time (e.g., 15 min) due to dissolving of sucrose in the paper-based valve. As shown in Fig. 1B, the CRISPR-Cas12a reaction solution was pre-loaded and lyophilized on the paper-based CRISPR detection chambers. After the valve opens, RPA amplicons automatically migrates to the CRISPR detection chambers. The migrated amplicons specifically trigger the non-specific cleavage activity of CRISPR-Cas12a, which further cleaves the fluorophore quencher (FQ)-labeled ssDNA probe and generates strong fluorescent signals for detection. As shown in Fig. 1C, the fluorescent signal can be directly read by the naked eye or recorded by smartphone camera. As a low-cost diagnostics technology, the developed lab-on-chip platform provides a simple, sensitive and accurate approach for comprehensive COVID-19 screening, especially in resource-limited settings.
Figure 1.
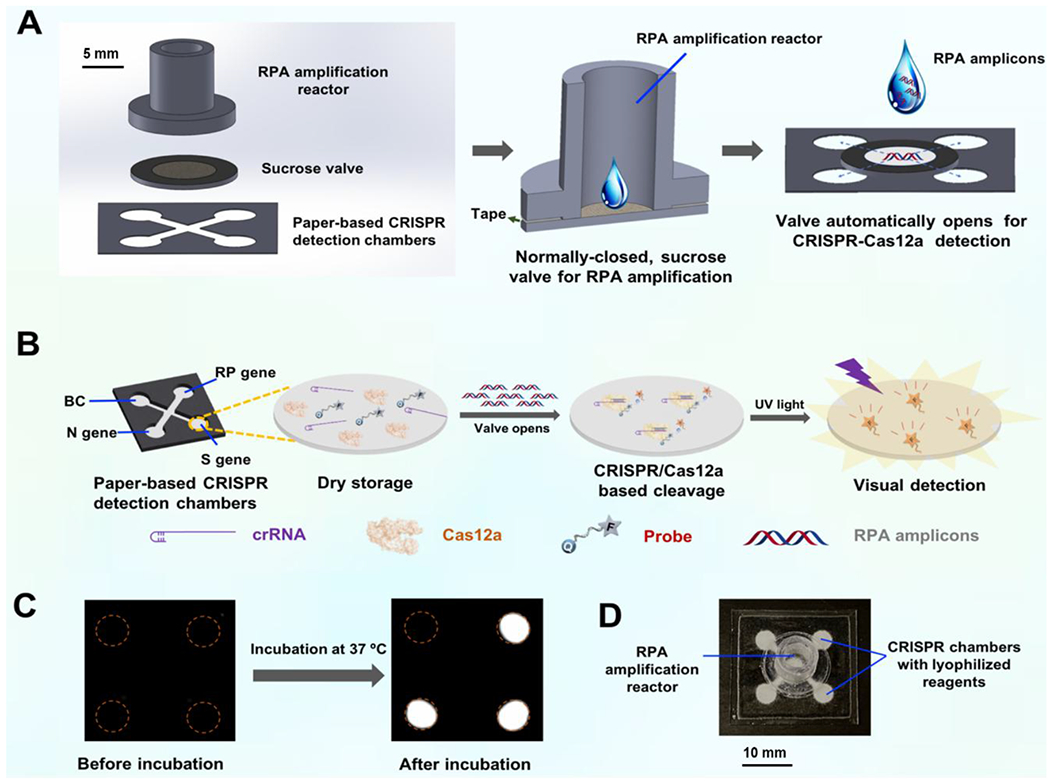
Scheme of the autonomous lab-on-paper platform for multiple, CRISPR-based diagnostics of SARS-CoV-2. A. Schematic illustration of the device configuration and working mechanism. The multiple RPA amplification reactor and CRISPR detection chambers were initially separated by a normally-closed, sucrose valve. B. Paper-based CRISPR chambers for multiplex gene diagnosis. The CRISPR-Cas12a detection reagents were pre-stored on the CRISPR detection chambers through lyophilization. C. Fluorescence image of multiple gene diagnostics of SARS-CoV-2 on the lab-on-paper platform. D. Photograph of the autonomous lab-on-paper platform.
SARS-CoV-2 detection by RT-RPA/CRISPR-Cas12a assay
The COVID-19 pandemic caused by the SARS-CoV-2, has seriously threatened human health. The early screening based on molecular diagnosis technology is an effective approach to prevent the rapid spread of SARS-CoV-2 virus. Here, RT-RPA/CRISPR-Cas12a system was applied for rapid, sensitive detection of SARS-CoV-2 by targeting its N and S genes (Fig. 2A). Before adapting the RT-RPA/CRISPR-Cas12a assay to our lab-on-paper platform, we first investigated the detection performance of the RT-RPA/CRISPR-Cas12a assay for SARS-CoV-2 detection in the reaction tubes. The Cas12 guide RNAs (gRNAs) to the SARS-CoV-2 were designed following the previous studies (Table S1).25,26 Because CRISPR-based fluorescence detection itself lacks multiple detection ability, the RPA amplicons were separately added into individual reaction tubes for CRISPR-based fluorescence detection after 15-min RPA amplification incubation. As shown in Fig. 2B, we could detect N gene and S gene of 10 copies SARS-CoV-2 RNA per test (n=3). In addition, the fluorescent signal can be collected by the fluorescence imaging system and be directly recognized by the naked eye under the LED blue light illuminator.
Figure 2.
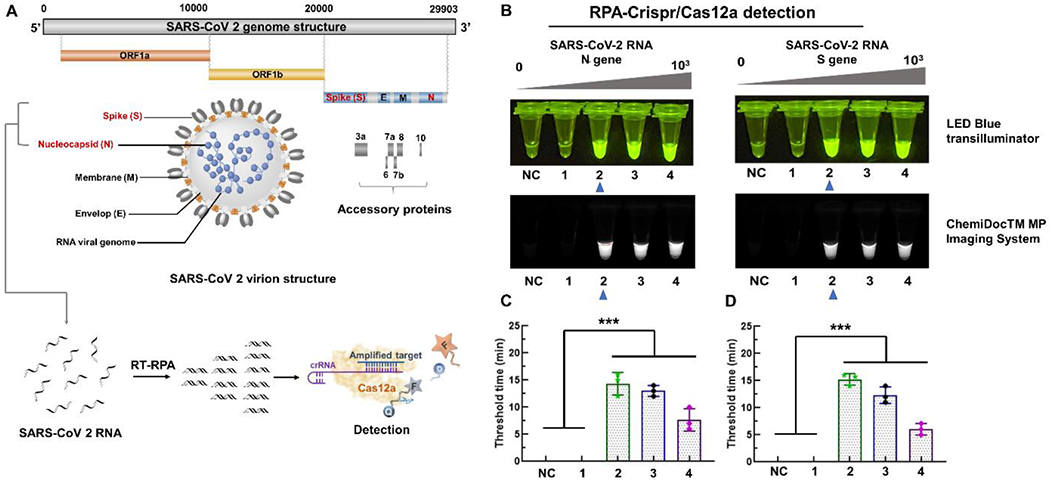
Detection of SARS-CoV-2 by RT-RPA/CRISPR-Cas12a assay in reaction tubes. A. The target genes and detection strategy of SARS-CoV-2. B. Fluorescence detection of N and S genes of SARS-CoV-2 by RT-RPA/CRISPR-Cas12a assay. NC, negative control without SARS-Cov-2 RNA. Tubes 1-4 contain, respectively, 1, 10, 102, 103 copies SARS-CoV-2 RNA spiked in the reaction solution. The images were taken under the LED blue transilluminator and ChemiDocTM MP Imaging System, respectively. C. Threshold time of the N gene detection of SARS-CoV-2 at different concentrations (0, 1, 10, 102, 103 copies). D. Threshold time of the S gene detection of SARS-CoV-2 at different concentrations (0, 1, 10, 102, 103 copies). *** indicate a significant difference of the NC and 1 copy with 10 to 103 copies SARS-CoV-2 in the reaction solution (p < 0.001, t-test). Error bars denote s.d. (n =3).
Optimization of autonomous lab-on-paper platform
To achieve simple and integrated detection of SARS-CoV-2, an autonomous lab-on-paper platform was established for multiple gene diagnosis. In our lab-on-paper platform, multiple RPA amplification reaction was firstly carried out in the RPA amplification reactor when the sucrose valve was initially closed. When the sucrose on the paper-based valve was totally dissolved, the paper-based sucrose valve opened, which brought the RPA amplification reactor and CRISPR detection chambers connected with each other. To optimize the opening time of the paper-based sucrose valve, different sucrose concentrations ranging from 5 to 15% were investigated in our lab-on-paper platform. As shown in Fig. 3A, the higher the sucrose concentration, the longer the valve’s opening time. Also, the sucrose valves exhibited satisfactory reliability. In our assay, 15-minute RPA incubation provides enough RPA amplicons for downstream CRISPR-based fluorescence detection. Thus, 10% sucrose solution was used to fabricate the paper-based sucrose valve of our lab-on-paper platform. To improve the lyophilized Cas12a’s activity, trehalose was added into the CRISPR reaction solution before lyophilization on the paper-based CRISPR detection chambers. As shown in Fig. S1, 10% trehalose solution showed the best performance in our lab-on-paper platform. To demonstrate the feasibility of our lab-on-paper platform for diagnostics without need for a cold chain, we evaluated and tested the lyophilized CRISPR reagents on the CRISPR detection chamber at room temperature for at least 30 days. As shown in Fig. S2, there was no significant decrease of fluorescence signals over a 30-day period at room temperature. Therefore, the experimental results show that our lab-on-paper platform can be transported at room temperature and has a potential for diagnostic applications in resource-limited settings.
Figure 3.
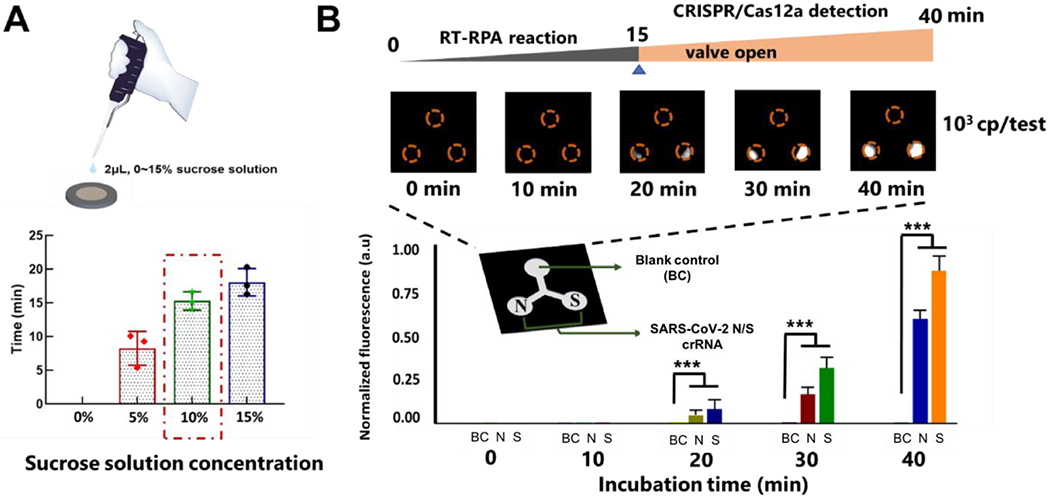
A. Optimization of the concentration of sucrose solution used for the paper-based sucrose valve. ddH2O (0%), 5%, 10% and 15% sucrose solution were dropped and dried on the both sides of the paper-based valve, respectively. B. Fluorescence detection of SARS-CoV-2 on the autonomous lab-on-paper platform at different incubation times. *** indicate a significant difference (p < 0.001, t-test). Error bars denote s.d. (n=3).
To optimize the detection time of the autonomous lab-on-paper platform, 103 copies SARS-CoV-2 RNA per test was added into the RPA amplification reactor and incubated at 37 °C for different time (e.g., 10, 20, 30 and 40 min). As shown in Fig. 3B, no fluorescence signal was observed within 15 min-incubation, which should be attributed to the initial closing of the sucrose valve. With the increasing of the incubation time, the fluorescence signals of the CRISPR detection chambers become stronger and stronger. After 40 min incubation, the strong fluorescent signals can be obviously observed on the positive CRISPR chambers, which indicates that 40 min-incubation is enough for our lab-on-paper platform. To further investigate the sensitivity of the developed lab-on-paper platform for SARS-CoV-2 detection, tenfold serial dilutions of SARS-CoV-2 RNA from 0 to 104 copies per test were added into the RPA amplification reactor of the lab-on-paper platform. As shown in Fig. 4, 102 copies SARS-CoV-2 RNA per test could be detected by targeting both N and S genes on the lab-on-paper platform. Additionally, we observed 0 occurrences of nonspecific amplification out of 5 no-template controls for both N and S genes detection, obtaining a specificity of 100%. Further, the fluorescent intensity of the CRISPR detection chambers was analyzed by the Image J. As shown in Fig. 4, the SARS-CoV-2 RNA concentration from 102 to 104 copies per test showed a significant difference with blank control groups.
Figure 4.
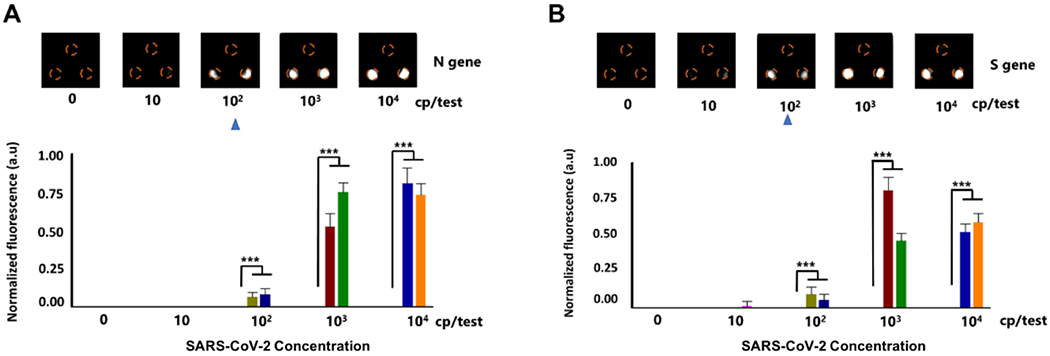
SARS-CoV-2 detection on the autonomous lab-on-paper platform. A & B. Detection sensitivity for N and S genes of SARS-CoV-2 on the lab-on-paper platform. *** indicates a significant difference in the fluorescent intensity (p < 0.001, t-test). Error bars denote s.d. (n=5).
Multiple gene detection of SARS-CoV-2 from clinical swab samples
To evaluate the clinical feasibility of the developed autonomous lab-on-paper platform for multiple gene detection of SARS-CoV-2, the RNA samples extracted from 21 nasopharyngeal swab clinical samples were tested. Among them, there were 8 SARS-CoV-2 positive samples and 13 SARS-CoV-2 negative samples which have been confirmed by RT-PCR method (Table S2).25 The detection workflow of the lab-on-paper platform for clinical swab samples is shown in Fig. 5A. Total turnaround time including nucleic acid preparation is less than 1 hour. To achieve an accurate nucleic acid-based molecular detection, it is crucial to simultaneously detect the housekeeping gene as an internal control to verify and monitor the test performance. The internal control can validate whole diagnostic workflows, including nucleic acid sample preparation, multiplex RT-RPA reaction, lyophilized reagent quality and CRISPR-based fluorescence detection. We detected RNA extracts of 21 nasopharyngeal swab samples using our lab-on-paper platform. As shown in Fig. 5B, for all 8 positive samples, both N and S genes of SARS-CoV-2, and housekeeping RNAse P gene were simultaneously detected on the lab-on-paper platform, which is comparable with that of RT-PCR method. On the contrary, for 13 negative samples, only human RNase P gene was detected. We did not observe nonspecific signals of 13 negative clinical samples, which may be attributed to high specificity of CRISPR detection. The detection of the housekeeping RNAse P gene in clinical samples indicates that high quality nucleic acid samples were extracted from the clinical samples. Therefore, these results indicated that the established lab-on-paper platform could simultaneously detect multiple genes for SARS-CoV-2 diagnostics in a single clinical sample, which further improved detection accuracy and reliability. To our best knowledge, it is the first time to achieve CRISPR-based multi-gene detection of SARS-CoV-2 on paper-based microfluidics platform.
Figure 5.
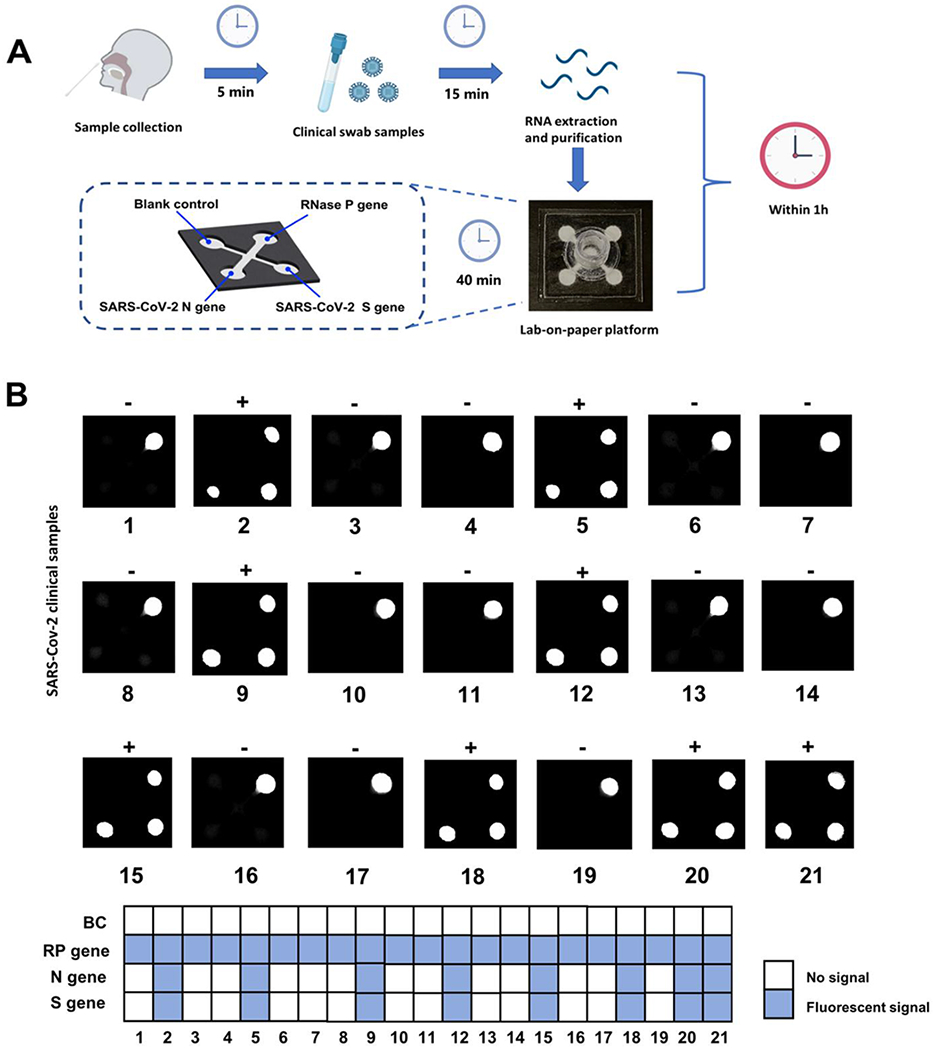
Multiple gene diagnosis of SARS-CoV-2 from clinical human swab samples. A. Workflow and testing time for SARS-CoV-2 detection in clinical swab samples by using the autonomous lab-on-paper platform. B. Multiple gene diagnosis results of SARS-CoV-2 from 21 clinical swab samples on the autonomous lab-on-paper platform. BC, blank control. RP gene, RNase P gene.
Conclusions
In summary, we developed an integrated, autonomous lab-on-paper platform for simultaneous detection of multiple genes for SARS-CoV-2 diagnostics. A programmable sucrose valve was designed to initially separate multiple RT-RPA amplification reactions and CRISPR-Cas12a detection, enabling simple, low-cost, automated, and multiple gene detection. With the autonomous lab-on-paper platform, we achieved sensitivities of 102 copies per test for SARS-CoV-2 detection. Further, the lab-on-paper platform has been successfully used to detect clinical sample within 1 hour, obtaining comparable results with the RT-PCR method. Therefore, the developed lab-on-paper platform can significantly improve the accessibility of on-site testing of SARS-CoV-2 infection and attribute to the comprehensive screening for COVID-19 pandemic control. Moreover, the novel platform also provides an accurate and convenient pathway for the surveillance and control of other infectious diseases (e.g., influenza virus), especially in the resource-limited settings.
Supplementary Material
Acknowledgments
The work was supported, in part, by R01EB023607, R61AI154642, R01CA214072, and UConn COVID-19 Rapid Start Funding (COVID-RSF) (G401911).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- (1).Schroöder I ACS Chemical Health & Safety 2020.
- (2).Wang CJ; Ng CY; Brook RH Jama 2020, 323, 1341–1342. [DOI] [PubMed] [Google Scholar]
- (3).Ferretti L; Wymant C; Kendall M; Zhao L; Nurtay A; Abeler-Dörner L; Parker M; Bonsall D; Fraser C Science 2020, 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Lavezzo E; Franchin E; Ciavarella C; Cuomo-Dannenburg G; Barzon L; Del Vecchio C; Rossi L; Manganelli R; Loregian A; Navarin N Nature 2020, 584, 425–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Bai Y; Yao L; Wei T; Tian F; Jin D-Y; Chen L; Wang M Jama 2020, 323, 1406–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Vogels CB; Brito AF; Wyllie AL; Fauver JR; Ott IM; Kalinich CC; Petrone ME; Casanovas-Massana A; Muenker MC; Moore AJ Nature microbiology 2020, 5, 1299–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lan L; Xu D; Ye G; Xia C; Wang S; Li Y; Xu H Jama 2020, 323, 1502–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Broughton JP; Deng X; Yu G; Fasching CL; Servellita V; Singh J; Miao X; Streithorst JA; Granados A; Sotomayor-Gonzalez A Nature Biotechnology 2020, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Niemz A; Ferguson TM; Boyle DS Trends in biotechnology 2011, 29, 240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Woo CH; Jang S; Shin G; Jung GY; Lee JW Nature biomedical engineering 2020, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Larremore DB; Wilder B; Lester E; Shehata S; Burke JM; Hay JA; Tambe M; Mina MJ; Parker R MedRxiv 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Tahamtan A; Ardebili A; Taylor & Francis, 2020. [Google Scholar]
- (13).Chertow DS Science 2018, 360, 381–382. [DOI] [PubMed] [Google Scholar]
- (14).Yin K; Ding X; Li Z; Zhao H; Cooper K; Liu C Analytical Chemistry 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Caliendo AM; Hodinka RL New England Journal of Medicine 2017, 377, 1685–1687. [DOI] [PubMed] [Google Scholar]
- (16).Chen JS; Ma E; Harrington LB; Da Costa M; Tian X; Palefsky JM; Doudna JA Science 2018, 360, 436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Gootenberg JS; Abudayyeh OO; Kellner MJ; Joung J; Collins JJ; Zhang F Science 2018, 360, 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Gootenberg JS; Abudayyeh OO; Lee JW; Essletzbichler P; Dy AJ; Joung J; Verdine V; Donghia N; Daringer NM; Freije CA Science 2017, 356, 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Li S-Y; Cheng Q-X; Wang J-M; Li X-Y; Zhang Z-L; Gao S; Cao R-B; Zhao G-P; Wang J Cell discovery 2018, 4, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wang B; Wang R; Wang D; Wu J; Li J; Wang J; Liu H; Wang Y Analytical chemistry 2019, 91, 12156–12161. [DOI] [PubMed] [Google Scholar]
- (21).Thi VLD; Herbst K; Boerner K; Meurer M; Kremer LP; Kirrmaier D; Freistaedter A; Papagiannidis D; Galmozzi C; Stanifer ML Science translational medicine 2020, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Xiong E; Jiang L; Tian T; Hu M; Yue H; Huang M; Lin W; Jiang Y; Zhu D; Zhou X Angewandte Chemie 2020. [DOI] [PubMed] [Google Scholar]
- (23).Houghtaling J, Liang T, Thiessen G and Fu E, Analytical chemistry, 2013, 85, 11201–11204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Li Y; Li S; Wang J; Liu G Trends in biotechnology 2019, 37, 730–743. [DOI] [PubMed] [Google Scholar]
- (25).Ding X; Yin K; Li Z; Lalla RV; Ballesteros E; Sfeir MM; Liu C Nature communications 2020, 11, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wu F; Xiao A; Zhang J; Gu X; Lee WL; Kauffman K; Hanage W; Matus M; Ghaeli N; Endo N medRxiv 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Liu J; Liao X; Qian S; Yuan J; Wang F; Liu Y; Wang Z; Wang F-S; Liu L; Zhang Z 2020.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.