

Abstract
Endothelial cells (ECs) are an important target for therapy in a wide range of diseases, most notably atherosclerosis. Developing efficient nanoparticle (NP) systems that deliver RNA interference (RNAi) drugs specifically to dysfunctional ECs in vivo to modulate their gene expression remains a challenge. To date, several lipid-based NPs have been developed and shown to deliver RNAi to ECs, but few of them have been optimized to specifically target dysfunctional endothelium. Here, a novel, targeted poly(β-amino ester) (pBAE) NP is demonstrated. This pBAE NP is conjugated with VHPK peptides that target vascular cell adhesion molecule 1 (VCAM-1) protein, overexpressed on inflamed EC membranes. To test this approach, the novel NPs were used to deliver anti-microRNA-712 (anti-miR-712) specifically to inflamed ECs both in vitro and in vivo, reducing the high expression of pro-atherogenic miR-712. A single administration of anti-miR-712 using the VHPK-conjugated-pBAE NPs in mice significantly reduced miR-712 expression, while preventing the loss of its target gene, tissue inhibitor of metalloproteinase 3 (TIMP3) in inflamed endothelium. miR-712 and TIMP3 expression were unchanged in non-inflamed endothelium. This novel, targeted-delivery platform may be used to deliver RNA therapeutics specifically to dysfunctional endothelium for the treatment of vascular disease.
Keywords: Poly(β-amino ester) (pBAE) nanoparticles, VCAM-1-targeting VHPK peptides, microRNA-712, endothelial inflammation, atherosclerosis
Graphical Abstract
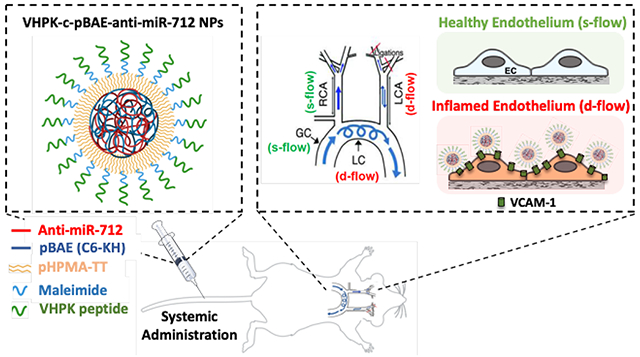
In this study, novel poly(β-amino ester) (pBAE) nanoparticles (NPs) conjugated with VHPK peptides that target vascular cell adhesion molecule 1 (VCAM-1) were developed and used to deliver RNA interference (RNAi) drugs to inflamed endothelial cells (ECs). Specifically, these VHPK-conjugated pBAE NPs successfully delivered anti-microRNA-712 to inflamed ECs both in vitro and in vivo and reduced expression of the pro-atherogenic microRNA-712.
1. Introduction
Atherosclerosis is a critical mechanism underlying myocardial infarction, ischemic stroke, and peripheral artery disease, the leading causes of death worldwide.[1] Atherosclerosis is a chronic inflammatory disease associated with multiple risk factors including hypercholesterolemia, diabetes, hypertension, smoking, age, sex, and physical inactivity. Interestingly, atherosclerosis occurs preferentially in curved and branching arterial regions that are exposed to disturbed blood flow, while straight and non-branching regions exposed to stable flow are protected from the disease. Disturbed flow induces genomic and epigenomic changes in endothelial cells, which lead to endothelial inflammation and the development of atherosclerosis.[2] Therefore, investigators have focused on identifying the pro-inflammatory roles of flow-sensitive endothelial genes and proteins in order to target them for therapeutic intervention in atherosclerosis.[3]
RNA interference (RNAi)-based therapeutics, including microRNAs (miRs) and small interfering RNAs (siRNAs), have the ability to regulate gene expression and show great therapeutic potential.[4] Modulating EC behavior via targeted RNAi-based therapeutics in vivo, however, remains a challenge. This is due to the limited number of efficient, cell-specific nanoparticle (NP) delivery systems, which are required to protect naked RNAi drugs such as siRNAs, miRs and anti-miRs (inhibitor of miRs) from rapid degradation in the bloodstream and to transport them across cell membranes.[5]
Various approaches for NP-based RNAi delivery have been developed, and some have been shown to successfully target vascular ECs in vivo. For example, several different cationic lipids and lipidoids have been reported to deliver RNAi to ECs.[6] However, few of these RNAi delivery platforms are optimized to preferentially target dysfunctional ECs and avoid healthy or non-diseased ECs. Active targeting via nanoparticle conjugation to receptor-binding molecules has been shown to be critical for nanoparticle accumulation in dysfunctional ECs.[7] Recently, we developed coated-cationic lipid nanoparticles (CCLs) conjugated with vascular cell adhesion molecule 1 (VCAM-1)-targeting VHPK peptides, which enabled the targeted-delivery of its cargo (anti-miR-712) to inflamed mouse endothelial cells in vivo.[8] While the lipid NP technologies show potential for EC-specific RNAi delivery, they need further improvement, particularly with regard hepatic accumulation.[9] Here, we demonstrate an alternative, novel delivery system using poly(β-amino ester) (pBAE) NPs.
pBAE NPs are a promising polymeric nanoparticle system for the delivery of RNAi drugs to ECs.[10] pBAEs have low toxicity and high biocompatibility due to their backbone of repeating ester groups that are biodegradable through hydrolysis in the cell cytoplasm. pBAE NPs present fast degradation kinetics once they are internalized by cells.[11] The presence of protonable amines in their structure allows for improved endosomal escape of complexes due to the proton sponge effect once they are internalized.[12] Unlike lipid-based NPs, which hold RNA therapeutics in a liposome or micelle structure, pBAE NPs hold RNA therapeutics within their polymer matrix, allowing for higher encapsulation and RNAi loading efficiencies.[10b] Additionally, pBAE synthesis can be easily modified in order to vary structural components, such as polymer backbones and terminal groups. Alterations in pBAE backbone hydrophobicity have been reported to enhance their stability in physiological media, overcoming the stability concerns of polymeric nanoparticles.[13] By terminal group modification, we have recently developed a wide library of bioinspired oligopeptide-modified pBAE NPs that demonstrate efficient nucleic acid delivery in a highly cell-specific manner.[14]
In the current study, we have developed a novel VHPK-conjugated pBAE NP system that delivers a therapeutic RNA (anti-miR-712) to inflamed ECs both in vitro and in vivo. VHPK peptide is composed of Val-His-Pro-Lys amino acids and targets vascular cell adhesion molecule 1 (VCAM-1) protein, which is overexpressed on the inflamed EC surface.[15] miR-712 is a pro-atherogenic microRNA, which is upregulated in dysfunctional vascular ECs exposed to disturbed blood flow patterns (d-flow) present in curved and branching regions of the arteries that are prone to atherosclerosis development.[2a, 3b, 3c, 16] miR-712 contributes to the development of atherosclerosis by degrading and preventing expression of its target gene, tissue inhibitor of metalloproteinase-3 (TIMP3). TIMP3 is an inhibitor of the pro-atherogenic matrix metalloproteinases (MMPs) and a disintegrin and metalloproteinases (ADAMs), which trigger endothelial inflammation and atherosclerosis.[17] So, loss of TIMP3 by overexpression of miR-712 leads to endothelial dysfunction and atherosclerosis. Here, we demonstrate that anti-miR-712 can be specifically delivered to inflamed ECs to reduce the miR-712 level by using the VHPK-conjugated pBAE NPs both in vitro and in vivo, demonstrating that this novel NP delivery system is a viable option for dysfunctional EC-targeted RNA therapy.
2. Results
2.1. Synthesis of Stable pBAE NPs
Synthesis of C6 polymer was previously reported and is described in detail in the Experimental Section.[13] A library of oligopeptide-modified C6 polymers was generated by reacting terminal acrylate groups from C6 polymers with different natural oligopeptide moieties (Figure 1a). We have previously confirmed that pBAE polymers are able to efficiently complex with nucleic acids and form discrete-sized NPs.[13, 14b] The average NP size was between 50 to 150 nm depending on the pBAE formulation. All NPs had a positive surface charge, with the exception of C6-RD (Figure 1b, c). We previously demonstrated that the oligopeptide composition, cell-specificity and/or nucleic acid delivery efficiency of the NPs can be modulated.[14a-c] Taking that into account, we performed in vitro studies to determine which formulation was the most effective at delivering anti-miRNA to cultured immortalized mouse aortic ECs (iMAECs) (Figure 1d). In general, all pBAE NPs achieved a higher cell internalization than the commercially available control reagent Oligofectamine. Specifically, lysine/histidine oligopeptide modification (C6-KH polymer), containing 50% C6-K and 50% C6-H was the most efficient formulation (Figure 1d, e).
Figure 1: Selection of C6-KH pBAE polymer.

(A) Structure and synthetic scheme of oligopeptide-modified pBAEs. R” terminal can be arginine-, lysine-, histidine-, glutamic acid- or aspartic acid-oligopeptide. (B and C) Biophysical characterization of pBAE NPs. (B) Average hydrodynamic diameter, polydispersity, (C) and zeta potential of pBAE:anti-miR-712 nanoparticles prepared at 75:1 polymer:anti-miR-712 ratio (w/w) with different oligopeptide-modified pBAE polymers are shown. (D) Cellular uptake of oligopeptide-modified pBAEs using fluorescent-labelled anti-miR (anti-miR-Cy3) at a final concentration of 200nM in iMAECs. (E) Fluorescent images of iMAECs using lysine/histidine-modified pBAE (C6-KH). Scale bar: 50 μm. Data are represented as mean ± SEM (n = 3). Multiple comparisons among groups were determined using one-way ANOVA followed by a post-hoc test. P-value: *p < 0.05, **p < 0.01, ***p < 0.001.
2.2. Formulation and Biophysical Characterization of VHPK-c-pBAE NPs
To deliver RNA therapeutic cargo to VCAM-1-expressing, inflamed ECs[2a, 16b], we further modified the C6-KH NPs with VHPK peptides and tested their delivery of a fluorescent payload to inflamed ECs. C6-KH polymer was first complexed with anti-miR-Cy3 at w/w ratio of 100/1, forming pBAE NPs. To conjugate VHPK to the pBAE NPs, a bi-functional, hydrophilic coating polymer (pHPMA-TT) was applied, which contains 2-thiazoline-2-thiol groups (TT groups) that react with primary amines. Optimal coating ratio was determined via anti-miR-Cy3 uptake assay in iMAECs (Figure S12). For VHPK-targeting nanoparticles (VHPK-c-pBAE NPs), 40% of TT groups were pre-modified using N-(2-Aminoethyl)maleimide, which allows for VHPK targeting peptide conjugation via maleimide-thiol reaction at terminal cysteine amino acids. The remaining 60% of TT groups were reacted with lysine residues containing primary amines from pBAE NPs (Figure 2b, c). For non-targeting pBAE NPs (c-pBAE NPs), 100% of TT groups were reacted with pBAE NPs to form a stable coating (Figure 2a).
Figure 2: Preparation of VHPK-conjugated pBAE nanoparticles.

(A) pBAE NPs were coated with pHPMA-TT. The TT groups in the pHPMA-TT copolymer react with amine groups from pBAE NPs. (B) 40% of TT groups were modified using amino-maleimide linker while 60% of TT groups were used to react with pBAE NPs. (C) Maleimide groups were used to conjugate VHPK peptides via their terminal cysteine groups.
To assess pBAE NP coating and targeting, the size and charge of each type of NP (pBAE NP, c-pBAE NP, VHPK-c-pBAE NP) was tracked by dynamic light scattering (DLS) (Figure S11). pBAE NPs have an average size of 70.0 ± 1.3 nm with a positive surface charge of 21.5 ± 1.0 mV. The neutral average charge from the pHPMA-TT polymer decreased the positive surface charge of the pBAE NPs from 21.5 mV to 6.0 mV. This decrease in surface charge indicates successful nanoparticle coating. Addition of VHPK peptide to coated c-pBAE NPs caused the surface charge of the resultant formulation to increase slightly from 6.0 mV to 8.1 mV. In addition, pBAE NPs were further characterized by Nanoparticle Tracking Analysis (NTA) showing a monodisperse nanoparticle size distribution, correlating with the low polydispersity index analyzed by DLS (Figure S11).
2.3. VCAM-1-Dependent In Vitro EC Delivery of Anti-miRNA using VHPK-c-pBAE NPs
To test whether VHPK-c-pBAE NPs can deliver anti-miR-Cy3 to inflamed ECs, iMAEC inflammation and hence VCAM-1 expression was first induced by pre-treating cells with 3 ng/ml tumor necrosis factor α (TNF-α). TNF-α increased VCAM-1 expression by 4-fold compared to the vehicle control (Figure 3a). The internalization efficiencies of VHPK-c-pBAE, c-pBAE, and non-coated pBAE NPs were compared by assessing cellular anti-miR-Cy3 fluorescence. Non-coated pBAE NPs showed the highest uptake efficiency into inflamed iMAECs. As expected, the pHPMA-TT coating decreased c-pBAE NP uptake into inflamed iMAECs. The conjugation of VHPK peptide to the c-pBAE NPs, however, increased internalization to a level similar to that of the pBAE NPs (Figure 3b-c). Furthermore, addition of excess free VHPK peptides (to compete with the conjugated VHPK-c-PBAE NPs) significantly reduced the internalization of anti-miR-Cy3 signal by VHPK-c-pBAE NPs (Figure 3b), demonstrating that VHPK peptide mediates nanoparticle internalization and delivery of the anti-miR cargo to inflamed ECs. Additionally, no significant differences in anti-miR-Cy3 delivery were observed between VHPK-c-pBAE NPs and non-targeted c-pBAE NPs in healthy, non-inflamed iMAECs, indicating that VHPK-c-pBAE NPs specifically target inflamed ECs (Figure S13).
Figure 3: Selective uptake of VHPK-c-pBAE NPs in inflamed iMAECs.

(A) iMAECs were treated with TNF-α (3 ng ml−1) for 2 hours and VCAM1 expression was determined by qPCR. (B) pBAE NPs containing Cy3-anti-miRNA were coated with pHPMA-TT polymer (c-pBAE NP) and conjugated with VHPK peptide (VHPK-c-pBAE NP) to improve their specific delivery to inflamed iMAECs. Internalization efficiency was tested at 2 hours post-transfection using flow cytometry. To assess VHPK-mediated internalization, the VCAM-1 receptor in inflamed iMAECs was blocked using excess VHPK peptide. Internalization of VHPK-c-pBAE NPs was assessed by flow cytometry. (C) VHPK-targeted internalization was validated using fluorescence microscopy. Scale bar: 50 μm. Data are represented as mean ± SEM (n = 3). Pairwise comparisons were determined using Student t-tests. Multiple comparisons among groups were determined using one-way ANOVA followed by a post-hoc test. P-value: *p < 0.05, **p < 0.01, ***p < 0.001.
2.4. In Vitro Anti-miR-712 Delivery to Inflamed ECs
To test their therapeutic efficiency in vitro, VHPK-c-pBAE NPs were used to deliver anti-miR-712 to inflamed iMAECs. An anti-miR-712 concentration curve study was first conducted using pBAE NPs. We found that anti-miR-712 reduced miR-712 level in a concentration-dependent manner, reaching 90% knockdown at 200 nM (Figure 4a). The specificity of anti-mir-712 was confirmed by the finding that miR-15a (a control miR) was not affected (Figure 4b). Additionally, no differences in miR-712 expression were observed when anti-miR-712 was delivered in either the presence or absence of serum proteins, indicating that the NPs likely remain stable in blood, an important criteria for successful in vivo studies.
Figure 4: VHPK-c-pBAE NP delivers anti-miR-712 to inflamed ECs in vitro.
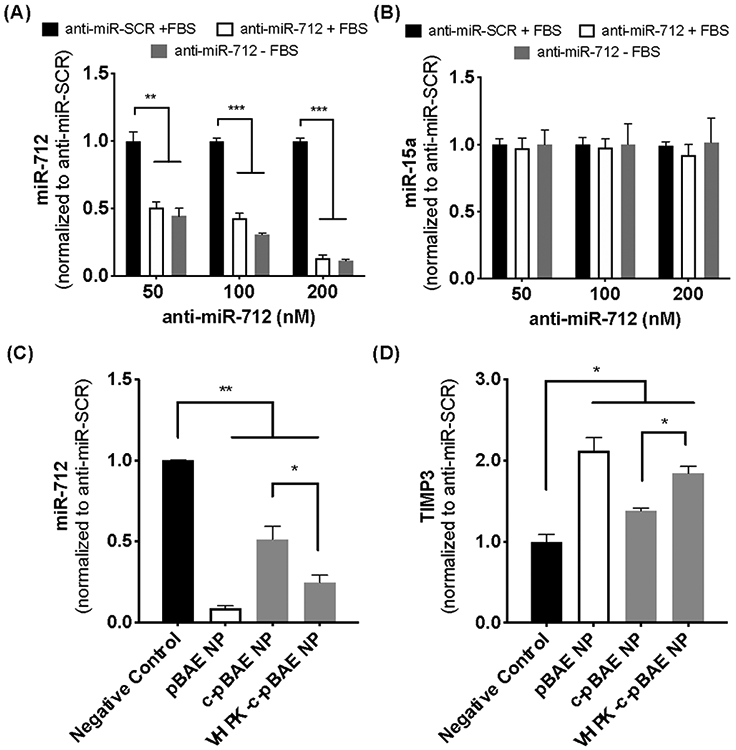
(A, B) iMAECs were transfected with 50-200 nM anti-miR-712 or anti-miR-SCR control in the presence or absence of FBS. miR-712 expression (A) and miR-15a as a control (B) were determined 48 hours post-transfection by qPCR. (C, D) iMAECs treated with TNF-α were treated with anti-miR-712 carried by pBAE, c-pBAE or VHPK-c-pBAE NPs for 48 hours. Anti-miR-SCR carried by VHPK-c-pBAE NPs was used as a negative control (black bar). Levels of miR-712 (C) and TIMP3 (D) were determined by qPCR. Data are represented as mean ± SEM (n = 3). Multiple comparisons among groups were determined using one-way ANOVA followed by a post-hoc test. P-value: *p < 0.05, **p < 0.01, ***p < 0.001.
Delivery of anti-miR-712 using either VHPK-c-pBAE or c-pBAE NPs was next assessed by measuring miR-712 and TIMP3 levels in inflamed iMAECs. Scrambled control anti-miR (anti-miR-SCR) delivered using VHPK-c-pBAE NPs was used as a negative control and pBAE NPs (non-coated and non-targeted NPs) were used as a positive control for transfection (Figure 4c, d). As expected, pBAE NPs induced robust miR-712 knockdown. When pHPMA-TT coating was added to the pBAE NPs to make c-pBAE NPs, anti-miR-712 delivery was reduced. However, when the VHPK peptide was conjugated (VHPK-c-pBAE NPs), an increase in miR-712 knockdown when compared to c-pBAE NPs was observed. Importantly, a similar trend was observed in terms of TIMP3 expression: anti-miR-712 delivered via either pBAE NPs or VHPK-c-pBAE NPs rescued TIMP3 expression, while anti-miR-712 delivered via c-pBAE NPs failed to do so (Figure 4d).
2.5. Anti-miR-712 Delivery to Inflamed Endothelial Cells In Vivo
VHPK-c-pBAE NP delivery of anti-miR-712 to inflamed ECs was next assessed using the partial carotid ligation (PCL) model of atherosclerosis in C57BL/6 mice. PCL surgery induces pro-inflammatory and pro-atherogenic, disturbed flow (d-flow) in the left carotid artery (LCA), while anti-inflammatory and atheroprotective, stable blood flow (s-flow) is present in the right carotid artery (RCA) in the same animal (Figure 5a). 72 hours post-surgery, 25 μg of either anti-miR-712 or anti-miR-SCR control, encapsulated in VHPK-c-pBAE NPs were administered intravenously. NP administration did not affect mouse weight in any of the experiments (Figure S14).
Figure 5: Selective and efficient delivery of anti-miR-712 packaged in VHPK-c-pBAE NPs to inflamed endothelium in mouse carotid arteries in vivo.

(A) Depiction of the partial carotid ligation (PCL) surgery to induce d-flow in LCA and s-flow in the contralateral RCA. The aortic arch regions naturally exposed to s-flow (GC) and d-flow (LC) are shown as comparison. At 3 days following partial carotid ligation, C57BL/6 mice were tail-vein injected with VHPK-c-pBAE NPs carrying either anti-miR-712 or anti-miR-SCR at 1 mg kg−. Two days post-injection, mice were sacrificed, and endothelial-enriched RNA was extracted from the RCA and LCA for qPCR analysis for VCAM-1 (B), markers of EC (PECAM1), smooth muscle cells (SM22a), and leukocytes (CD45) (C), miR-712 (D), and TIMP3 (E). Data are represented as mean ± SEM (n = 5). Pairwise comparisons were determined using Student t-tests. Multiple comparisons among groups were determined using one-way ANOVA followed by a post-hoc test. P-value: *p < 0.05, **p < 0.01, ***p < 0.001.
As we previously demonstrated, VCAM-1 gene expression increased 3-fold in the LCA endothelium compared to the RCA at 3 days post PCL surgery (Figure 5b), confirming that VCAM-1 expression was significantly higher in ECs exposed to d-flow conditions.[18] Two days post anti-miR-712 administration, miR-712 and TIMP3 levels were determined in endothelial enriched RNA samples from RCA and LCA (Figure 5c). A significant 2.5-fold increase of miR-712 expression was observed in the LCA endothelium as compared to the RCA, as expected, when control anti-miR-SCR was injected. However, when anti-miR-712 was administered, a significant decrease of miR-712 levels was observed in the LCA endothelium compared to those in the anti-miR-SCR injected mice, obtaining a similar miR-712 expression level to the s-flow-exposed RCA (Figure 5d). RCA endothelial miR-712 expression was similar in both the scramble control and anti-miR-712 conditions, suggesting anti-miR-712 was specifically targeted to the inflamed LCA endothelium.
Consistent with the miR-712 results, TIMP-3 expression was significantly reduced in the LCA as compared to the RCA after PCL treatment when anti-miR-SCR was injected. In contrast, when anti-miR-712 was delivered using VHPK-c-pBAE NPs, a 2-fold TIMP3 expression increase in the LCA was observed as compared to anti-miR-SCR in LCA. TIMP3 expression was similar in the LCA and RCA endothelium of the anti-miR-712-injected mice. In addition, similar TIMP3 levels were observed using either anti-miR-712 or anti-miR-SCR in RCA (Figure 5e). Importantly, no significant changes of miR-712 or TIMP3 expression were observed in the medial layer composed mostly of vascular smooth muscle cells when anti-miR-712 was administered (Figure S15). Therefore, these results demonstrated that VHPK-c-pBAE NPs can effectively and selectively deliver anti-miR-712 to the inflamed endothelium expressing VCAM-1 in d-flow regions, decreasing miR-712 levels and rescuing TIMP3 expression.
2.6. VHPK-c-pBAE NP Anti-miR-712 Biodistribution Profile
Upon administration, VHPK-c-pBAE NPs containing anti-miR-712 were distributed widely to tissues throughout the mouse tissues. Therefore, anti-miR-712 biodistribution and gene regulation in different tissues were studied using the PCL model of atherosclerosis in C57BL/6 mice. To assess biodistribution, miR-712, miR-15a (negative control), and TIMP3 expression were analyzed 48 hours after delivery of anti-miR-712 using VHPK-c-pBAE NPs. Anti-miR-SCR was used as a negative control. Interestingly, lung tissue presented the highest miR-712 knockdown, reaching 55% miRNA silencing, followed by the spleen and kidneys (Figure 6a). In contrast, no significant changes in miR-712 expression were observed in the thymus, liver or heart. Moreover, the lack of significant changes in miR-15a expression confirm that the anti-miR silencing effects are specific to miR-712 (Figure S16). To confirm that miR-712 downregulation resulted in functional TIMP3 upregulation, its mRNA expression was measured. As expected, a more than 4-fold increase in TIMP3 expression was observed in lung tissue, and no TIMP3 expression changes were observed in the spleen, thymus, or heart. Slight TIMP3 increases were detected in the kidneys and liver (Figure 6b). To further confirm TIMP3 overexpression in lung tissues, TIMP3 protein expression was assessed via Western blot and immunofluorescent imaging. TIMP3 protein expression was significantly increased in the lung tissue of anti-miR-712-injected mice as compared to anti-miR-SCR-injected mice (Figure 6c). In addition, TIMP3 immunofluorescence imaging of pulmonary vasculature cross-sections showed a slight increase in fluorescence when anti-miR-712 was delivered as compared to anti-miR-SCR (Figure 6d).
Figure 6: Biodistribution of anti-miR-712 delivered by VHPK-c-pBAE NPs using PCL model of atherosclerosis in C57BL/6 mice.

Anti-miR-712 biodistribution was analyzed by determining the (A) miR-712 expression and (B) TIMP3 expression by qPCR in tissue samples of the lung, spleen, thymus, kidney, liver and heart obtained from the mouse study described in Figure 5. (C) Anti-miR-712 restores TIMP3 protein expression in lungs. TIMP3 expression was analyzed by Western blot 48 hours post-injection. (D) Frozen lung sections obtained from these mice were used for immunofluorescence staining with antibody specific to TIMP3 shown in purple (n=3 mice). Negative control staining was performed without primary TIMP3 antibody. Scale bar: 20 μm. Data are represented as mean ± SEM (n = 4-6). Pairwise comparisons were determined using Student t-tests. P-value: *p < 0.05, **p < 0.01, ***p < 0.001.
3. Discussion
Here, we have designed a novel polymeric NP system that specifically delivers RNAi drugs to inflamed ECs. We have modified pBAE NPs with the VCAM-1-targeting peptide, VHPK in order to specifically deliver anti-miR-712 to inflamed ECs expressing VCAM-1. First, we developed the VHPK-conjugated pBAE nanoparticles (VHPK-c-pBAE NPs), confirmed their physicochemical proprieties, and demonstrated that they specifically target inflamed ECs in vitro (Figures 1-3). The fact that the VHPK-c-pBAE NPs demonstrated increased EC delivery of anti-miR-Cy3 as compared to the pHPMA-TT-coated nanoparticles (c-pBAE NPs), and that this effect was abolished via competition binding assay with VHPK peptide, indicates that VHPK-conjugated nanoparticles likely enter inflamed ECs via receptor-mediated endocytosis, potentially via the clathrin-mediated endocytosis pathway (Figure 3b, c).[19] Using these VHPK-c-pBAE NPs, anti-miR-712 was delivered to inflamed ECs in vitro and in vivo, causing significant decreases in miR-712 expression, as well as significant increases in downstream TIMP3 expression (Figure 4 and 5). Finally, biodistribution studies demonstrated that the lungs are the most significantly affected organ when anti-miR-712 is delivered using VHPK-c-pBAE NPs (Figure 6).
This novel VHPK-c-pBAE NP formulation shows a unique biodistribution profile of anti-miR-712 as compared to previous studies. Our previous studies showed that subcutaneous injection of naked anti-miR-712 led to significant off-target miR-712 silencing in various cell types across many different organs. Specifically, miR-712 expression was reduced by 89% in the liver, 70% in the spleen and 76% in the thymus.[16a] Our novel VHPK-conjugated pBAE nanoparticle delivery system greatly reduces off-target effects in comparison, specifically in the liver. In prior studies, we have shown that unlike other NP formulations, pBAEs do not accumulate in the liver.[14d] This enhances the therapeutic potential of pBAE NPs due to the fact that the high clearance of NPs in the liver prevents NPs from reaching their target organs in concentrations required for therapeutic effects.[20] Interestingly, with the VHPK-c-pBAE NPs, we observed significant changes in miR-712 and TIMP3 in the lungs. This result is of particular interest as lung tissue is highly vascularized and rich in ECs exposed to pro-inflammatory d-flow patterns.[21] The novel VHPK-conjugated pBAE NPs may thus be of particular importance for RNAi therapies targeting inflamed ECs in diseases of the pulmonary vasculature.
Modified pBAE NPs are a promising delivery system for RNAi therapies. Recently, a new generation of stable pBAE NPs was reported, which have increased backbone hydrophobicity conferring longer stability and better in vivo outcomes.[13] For the current study, we developed a library of oligopeptide-modified pBAEs that are able to self-aggregate with different types of nucleic acids (from small-scale siRNAs to large-scale plasmids) and spontaneously form a wide range of nanoparticles with distinct physicochemical proprieties (Figure 1). By modifying end-oligopeptide moieties, the cell-specific delivery efficiency of pBAE NPs can be enhanced.[14a-c] Oligopeptide formulation screening of our wide family of pBAEs revealed that pBAE polymers with a mixture of 50% lysine and 50% histidine (C6-KH) showed the highest cellular uptake in ECs (Figure 1d). Lysine modifications, which contain primary amines, confer sufficient positive charge to pBAEs for anti-miR complexation. Histidine modifications, which contain ternary amines, confer high buffering capacity to pBAEs for enhanced endosomal escape.[22] Efficient miR-712 knockdown in vitro confirms that C6-KH pBAE NPs are optimized to efficiently deliver anti-miR when internalized by ECs. However, non-specific targeting was observed using C6-KH polymer alone, and the NPs were internalized by all ECs (unpublished data). Therefore, the novel ~100 nm diameter pBAE NPs passively target ECs.[23]
The addition of VHPK peptides to the pBAE NPs via pHPMA-TT coating actively targeted the NPs to VCAM-1-expressing, inflamed ECs. The pHPMA-TT coating plays two roles.[24] Similar to polyethylene glycol (PEG), pHPMA coating protects NPs from the mononuclear phagocyte system and protein interaction, permitting longer NP circulation times.[25] It also links the pBAE formulation to the VHPK peptide. Specifically, TT groups in the coating covalently react with available primary amines from lysine amino acid residues in the pBAE NPs. pHPMA-TT coatings are quite versatile, as they can be tuned to attach different densities of targeting peptides to the pBAE NPs. The conjugated VHPK peptides conferred active targeting of pBAE NPs to VCAM-1-expressing, inflamed ECs. Recent reports demonstrate that active targeting is the main mechanism through which NPs accumulate in cells, and that passive targeting plays a secondary role.[7c, 7d] Therefore, conjugation of receptor-binding peptides such as VHPK is one method of actively targeting nanoparticles to specific cell types. VHPK peptides in particular are well described and known to effectively target NPs to VCAM-1 on inflamed EC membranes, while avoiding healthy, non-inflamed ECs.[8]
4. Conclusion
In the current study, we have described an efficient nanoparticle formulation based on poly(β-amino ester)s that can be used to deliver RNAi drugs in a cell-specific manner in vitro and in vivo. Our novel NP combines previously-described, cell-efficient pBAEs with targeting peptides to enhance cell specificity. We have observed that by packaging the RNAi drug, anti-miR-712 within our pBAE NPs conjugated to the well-described targeting peptide, VHPK, expression of a particular RNA transcript can be modulated in inflamed ECs. Our data shows that anti-miR-712 was specifically internalized in inflamed ECs, and that it effectively silenced miR-712, and rescued the expression of its downstream target gene, TIMP3 both in vitro and in vivo, thus preventing a known mechanism of atherosclerosis. This novel system of active targeting and efficient release of RNAi drugs to inflamed ECs opens up numerous opportunities for the treatment of vascular diseases.
5. Experimental Section
Materials:
Reagents and solvents used for polymer synthesis were purchased from Sigma-Aldrich and Panreac. Oligopeptide moieties used for polymer modification (H-Cys-Arg-Arg-Arg-NH2, H-Cys-Lys-Lys-Lys-NH2, H-Cys-His-His-His-NH2, and H-Cys-Asp-Asp-Asp-NH2) were obtained from GL Biochem (Shanghai) Ltd. with a purity of at least 98%. VHPK peptide (VHPKQHRGGSKGC) was obtained from GenScript with a purity of at least 98%. Labeled anti-miR (Cy3™ Dye-Labeled Anti-miR™ Negative Control #1; AM17011) and Anti-miR™ miRNA Inhibitor Negative Control #1 (AM17010) were used for uptake experiments and purchased from ThermoFisher. Anti-miR-712 and scramble anti-miR (anti-miR-SCR) control were purchased from Qiagen. Oligofectamine™ Transfection Reagent was purchased from Invitrogen and used according to manufacturer instructions.
Synthesis of Oligopeptide-Modified C6 Polymer:
Synthesis of poly(β-amino ester)s (pBAEs) was performed via a two-step procedure, as previously described.[13] First, addition reaction of primary amines to an excess of diacrylates was used to synthesize an acrylate-terminated polymer (termed C6 polymer). Briefly, C6 polymerization was performed by mixing 5-amino-1-pentanol (0.426 g, 4.1 mmol), hexylamine (0.422 g, 4.1 mmol), and 1,4-butanediol diacrylate (2.0 g, 9.1 mmol) via magnetic stirring at 90 °C for 24 hours. Second, C6 polymer was end-capped with different thiol-terminated oligopeptides composed of Cys + 3 amino acids (Arg, Lys, His, Asp, or Glu) at a 1:2.1 molar ratio in dimethyl sulfoxide (DMSO) in an overnight, room temperature reaction. The resulting polymers were collected by precipitation in a mixture of diethyl ether and acetone (7:3). Synthesized structures were dissolved in methanol-d4 and confirmed by 1H-NMR, recorded in a 400 MHz Varian (NMR Instruments, Clarendon Hills, IL) (Supporting Information). Molecular weight (MW), relative to polystyrene standard, was determined by HPLC (HPLC Elite LaChrom system of VWR-Hitech equipped with a GPC Shodex KF-603 column and THF mobile phase).
Synthesis of pHPMA-TT Copolymer:
The syntheses of HPMA and Ma-acap-TT monomers are detailed in the supplementary information (Figure S7 and Figure S8). HPMA (3.0 g, 20.9 mmol), Ma-acap-TT (0.7 g, 2.3 mmol), and AIBN (0.59 g, 3.6 mmol) were dissolved in dry DMSO (25.3 g, 17.1 mL), and allowed to polymerize by reversible addition-fragmentation chain transfer (RAFT) reactions at 60 °C for 6 hours under inert atmosphere(Figure S9).[26] The final product was precipitated in a mixture of acetone/ethanol (1/1) and dried under vacuum until constant weight was achieved, yielding 2.9 g of product (polymerization yield of 78%). The percentage of modified TT groups was determined by absorption (λ=305 nm).
Synthesis of pHPMA-TT/Mal Copolymer:
Synthesis of 1-(2-Aminoethyl)maleimide-modified pHPMA-TT copolymer (termed pHPMA-TT/Mal) was performed by reaction of pHMPA-TT (0.25 g, 1.59 mmol), 1-(2-Aminoethyl)maleimide (15.57 mg, 0.057 mmol), and trimethylamine (0,017g, 0.17 mmol) in DMSO for 12 hours at room temperature (Figure S10). The final product was precipitated in a mixture of acetone/ethanol (1/1) and dried under vacuum. The percentage of modified TT groups was determined by absorption (λ=305 nm).
Targeted pBAE NP Formation and Biophysical Characterization:
pBAE NPs were prepared at 100/1 C6 polymer/anti-miRNA ratio, by mixing equal volumes of anti-miR at 0.05 mg mL−1 with the C6 polymer at 5.0 mg mL−1 in sodium acetate buffer (AcONa) at 12.5 mM. The anti-miR was added to the C6 polymer solution and mixed by pipetting, followed by 10 min incubation at 25°C. For the formation of discrete pBAE NPs, this mixture was nanoprecipitated with the same volume of PBS 1x.
Targeted pBAE NPs were prepared by mixing pHPMA-TT/Mal polymer with pBAE NPs at 6.25/100 w/w ratio. pHPMA-TT/Mal polymer was added to the pBAE NPs and mixed by pipetting, followed by 30 min incubation at 25 °C (termed c-pBAE NPs). After that, VHPK peptide was added at 1:10 Maleimide:VHPK mol/mol ratio. The resulting formulation was incubated for 30 minutes. Excess VHPK peptide was removed by dialysis (10 kDa) (termed VHPK-c-pBAE NP). pHPMA-TT polymer was used to formulate control non-targeted pBAE NPs following the same procedure.
Physicochemical characterization of nanoparticles was performed by ZetaSizer Nano ZS (Malvern Instruments Ltd, United Kingdom, 4 mW laser). Hydrodynamic diameter (nm), polydispersity index (PDI), and surface charge of nanoparticles were measured. Hydrodynamic size was also measured by nanoparticle tracking analysis (NTA), using a NanoSight LM10 from Malvern, equipped with a fluorescent laser (λ = 488 nm).
Cell Lines:
Immortalized mouse aortic endothelial cells (iMAECs) were maintained in Dulbecco’s minimum essential medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units mL−1 penicillin, 100 μg mL−1 streptomycin, 0.1 mM MEM non-essential amino acids (NEAA), 2 mM L-glutamine, and 50 μg mL−1 endothelial cell growth supplement (ECGS).[27]
iMAEC-Specific pBAE NP Formulation Screening:
Screening of the oligopeptide-modified C6 polymer library was carried out using fluorescent anti-miR-Cy3 in iMAECs. Briefly, cells were seeded in 12-well plates at 80,000 cells/well and incubated overnight to roughly 80% confluence prior to performing the transfection experiments. pBAE NPs (without targeting) were synthesized as described previously at a 100:1 w:w ratio, diluted in complete DMEM medium, and added to the cells at a final anti-miR-Cy3 concentration of 200 nM. Cells were incubated for 2 hours at 37 °C in 5% CO2 atmosphere. After incubation, pBAE NP internalization was measured by flow cytometry (BD LSRFortessa cell analyzer). Untreated cells were used as negative control. Oligofectamine reagent was used as a positive control following manufacture instructions.
In Vitro Delivery of Anti-miR-712:
iMAECs were seeded in 12-well plates at 80,000 cells per well, incubated overnight to roughly 80% confluence, and pre-treated using 3 ng mL−1 of mouse TNF-α for 2 hours prior to anti-miR-712 delivery. Two different experiments were performed: (1) Cells were transfected using C6-KH polymer and anti-miR-712 at different concentrations (pBAE NPs), ranging from 50 nM to 200 nM. Transfections were carried out with and without the presence of 10% FBS in the transfection medium. (2) pBAE NPs, c-pBAE NPs, and VHPK-c-pBAE NPs were prepared as described previously and added to the cells at a final concentration of 200 nM for 48 hours. For both experiments, miR-712, miR-15a, and TIMP3 levels were analyzed by qPCR. Anti-miR-SCR was used as a negative control.
Cellular Uptake of Anti-miR-Cy3 Using Targeted pBAE NPs:
iMAECs were seeded in 12-well plates at 80,000 cells per well, incubated overnight to roughly 80% confluence, and pre-treated using 3 ng mL−1 of mouse TNF-α for 2 hours prior to anti-miR-Cy3 delivery. pBAE NPs, c-pBAE NPs, and VHPK-c-pBAE NPs were prepared as described previously and added to the cells at a final anti-miR-Cy3 concentration of 200 nM. After 2 hours, excess nanoparticles were removed, and cells were trypsinized and fixed with 1% paraformaldehyde (PFA) for flow cytometry analysis. Data was acquired and analyzed by FACS (BD LSRFortessa cell analyzer).
pBAE Internalization Studied by Confocal Microscopy:
iMAECs were seeded in 35 mm dishes (P35GCOL-1.5-10-C; from MatTek Corporation) at 150,000 cells/plate, incubated overnight to roughly 50% confluence, and then pre-treated using 3 ng mL-1 of mouse TNF-α for 2 hours prior to anti-miR-Cy3 delivery. c-pBAE NPs and VHPK-c-pBAE NPs were prepared as described previously and added to the cells at a final anti-miR-Cy3 concentration of 200 nM for 2 h at 37 °C and 5% CO2 atmosphere. Subsequently, cells were washed three times with PBS and stained with DAPI following the manufacturer’s protocol. Each fluorescent dye was analyzed with the corresponding filter: DAPI (315–400 nm) in blue and AF555 (510–560 nm) in red.
Lyophilization of pBAE Nanoparticles for In Vivo Studies:
pBAE NPs were generated by pipette mixing an equal volume of anti-miRNA at 0.25 μg μL−1 with C6-KH polymer at 25 μg μL−1 in AcONa buffer solution (12.5 mM, pH 5.5). After 10 minutes of incubation at room temperature, 200 μL of nanoparticles were nanoprecipitated in an equal volume of 10 mM HEPES. Thereafter, 100 μL of pHPMA-TT/Mal polymer at 1.56 μg μL−1 in 10 mM HEPES was added and allowed to react for 30 minutes. VHPK peptide was added at 1:10 Maleimide:VHPK mol/mol ratio, and allowed to react for 30 minutes. Finally, 400 μL of 10 mM HEPES containing 4 wt% sucrose (added as crio and lioprotectors) was added. Then, nanoparticles were freeze-dried. The lyophilized powder was kept at −20 °C until use, when it was reconstituted with DEPC water (120μl).
Mouse Partial Carotid Ligation (PCL) Surgery:
All procedures used in animal studies were conducted at Emory University and were approved by the Emory University Institutional Animal Care and Use Committee (IACUC) (PROTO201700182). Partial ligation of left carotid arteria (LCA) was carried out following a previously described procedure.[28] 8-week-old C57BL/6 male mice were used in each condition.
Isolation of Endothelial-Enriched RNA from Mouse Carotids:
Endothelial-enriched RNA from the carotids was extracted as described previously.[28] First, mice were sacrificed by CO2 inhalation according to Emory University’s IACUC protocol and perfused with saline containing heparin (10 U ml−1) via the left ventricle after severing the inferior abdominal aorta. The left common carotid artery (LCA) and the right common carotid artery (RCA) were then isolated and carefully cleaned of periadventitial fat. Then, the carotid lumen was quickly flushed with 150 μL of QIAzol® lysis reagent using a 29-gauge insulin syringe into an Eppendorf tube. The eluate was snap-frozen in liquid nitrogen for RNA extraction. Additionally, media and adventitia (smooth-muscle cell enriched) were snap-frozen in liquid nitrogen, pulverized by mortar and pestle, and lysed with QIAzol® lysis reagent (300 μL).
Anti-miR-712 Biodistribution Study:
Mice (n = 5) were intravenously injected with either 25 μg (120 μL) of anti-miR-712 or anti-miR-SCR in VHPK-c-pBAE NPs. 48 hours post-injection, mice were sacrificed by CO2 inhalation according to Emory University’s IACUC protocol and perfused with saline solution. The organs were collected and snap-frozen in liquid nitrogen. Then, they were crushed using a mortar and pestle device and lysed with QIAzol for RNA extraction and qPCR analysis. miR-712, TIMP3 and miR-15a expression were analyzed in the lung, liver, spleen, thymus, heart, and kidneys.
RNA Isolation and qPCR Analysis:
Total RNA was extracted and purified using Direct-zol™ RNA Kits according to manufacturer instructions. The amount and purity of RNA was determined by NanoDrop 1000 Spectrophotometer. RNA was reverse-transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). For miRNA transcription, the RNA was reverse-transcribed using the miScript II RT Kit following the manufacturer’s instructions. Gene expression was determined by RT-qPCR using gene-specific primers (Table S1). The expressions of genes of interest were normalized against either 18S or U6 housekeeping genes. All analyses were performed using the 2−ΔΔCT method.
Immunohistochemistry:
Histological sections from the lung (n=3) were stained using anti-TIMP3 antibody (ab39185). Briefly, lung sections were washed with PBS 1x, permeabilizated with 0.25% TritonX in PBS for 20 min, and blocked using 10% Donkey Serum in PBS 1X for 60 minutes. Then, samples were incubated with a 1:100 anti-TIMP3 antibody (ab39185) diluted in 10% Donkey Serum in PBS 1X overnight at 4 °C. After that, samples were washed twice with PBS 1X, and goat anti-rabbit secondary antibody (sc-2004 from Santa Cruz) was added and incubated for 2 hours at 4°C. Finally, samples were washed twice with PBS 1x and imaged using a Zeiss LSM 510 META confocal microscope (Carl Zeiss).
Western Blot:
Lung tissue was incubated with RIPA buffer and disrupted using TissueLyser LT (Qiagen). After tissue disruption, the samples were centrifuged at 4°C for 10 minutes at 10,000 rpm. Cell lysates (20 μg of protein) were loaded into 10% SDS-PAGE for protein separation. Then, proteins were transferred to a polyvinylidene difluoride membrane (Millipore) and probed using primary antibody following the standard western blotting method. TIMP3 (ab39185) and β-actin (A5316; Sigma-Aldrich) primary antibodies were diluted 1:1,000 in TBST with 5% milk. Goat anti-rabbit secondary antibodies (sc-2004 from Santa Cruz) were used, and the membrane staining was revealed via chemiluminescence detection. Blots were analyzed using the NIH IMAGE program.
Statistical Analysis:
Statistical analyses were carried out using Graph-Pad Prism 8 (GraphPad Software). All data are reported as mean ± SEMs. A minimum of n=3 biological replicates were used per condition in each experiment. Pairwise comparisons were performed using Student t-tests. Multiple comparisons among groups were determined using one-way ANOVA followed by a post-hoc test. No specfici pre-processing of data was performed prior to statistical analyses. Differences between groups were considered significant at p-values below 0.05 (* p < 0.05, ** p < 0.01, *** p < 0.001).
Supplementary Material
Acknowledgments
This work was supported by funding from the National Institutes of Health grants HL119798 and HL095070 to HJ. It was also supported by funding from the Spanish Ministerio de Ciencia, Innovación y Universidades for the grant RTI2018-094734-B-C22 to SB. PD received the financial support from AGAUR (Generalitat de Catalunya) 2017FI_B2 00141. IT was supported by NIH training grant T32-GM008602. HJ was supported by John and Jan Portman Professorship and Wallace H. Coulter Distinguished Faculty Chair Professorship. This study was also funded by Grup d’Enginyeria dels Materials (GEMAT). GEMAT would like to acknowledge Agència de Gestió d’ajuts Universitaris i de Recerca, Generalitat de Catalunya (SGR 2017) n° 1559. The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
References
- [1].Herrington W, Lacey B, Sherliker P, Armitage J, Lewington S, Circ Res 2016, 118, 535. [DOI] [PubMed] [Google Scholar]
- [2] a).Davies PF, Nat Clin Pract Cardiovasc Med 2009, 6, 16; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kwak BR, Back M, Bochaton-Piallat ML, Caligiuri G, Daemen MJ, Davies PF, Hoefer IE, Holvoet P, Jo H, Krams R, Lehoux S, Monaco C, Steffens S, Virmani R, Weber C, Wentzel JJ, Evans PC, Eur Heart J 2014, 35, 3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3] a).Kumar S, Kim CW, Son DJ, Ni CW, Jo H, Sci Data 2014, 1, 140039; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kumar S, Williams D, Sur S, Wang JY, Jo H, Vascul Pharmacol 2018, DOI: 10.1016/j.vph.2018.10.001; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kumar S, Kim CW, Simmons RD, Jo H, Arterioscler Thromb Vasc Biol 2014, 34, 2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Martens CR, Bansal SS, Accornero F, J Mol Cell Cardiol 2019, 129, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Reischl D, Zimmer A, Nanomedicine 2009, 5, 8. [DOI] [PubMed] [Google Scholar]
- [6] a).Dahlman JE, Barnes C, Khan O, Thiriot A, Jhunjunwala S, Shaw TE, Xing Y, Sager HB, Sahay G, Speciner L, Bader A, Bogorad RL, Yin H, Racie T, Dong Y, Jiang S, Seedorf D, Dave A, Sandu KS, Webber MJ, Novobrantseva T, Ruda VM, Lytton-Jean AKR, Levins CG, Kalish B, Mudge DK, Perez M, Abezgauz L, Dutta P, Smith L, Charisse K, Kieran MW, Fitzgerald K, Nahrendorf M, Danino D, Tuder RM, von Andrian UH, Akinc A, Schroeder A, Panigrahy D, Kotelianski V, Langer R, Anderson DG, Nat Nanotechnol 2014, 9, 648; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Aleku M, Fisch G, Mopert K, Keil O, Arnold W, Kaufmann J, Santel A, Microvasc Res 2008, 76, 31; [DOI] [PubMed] [Google Scholar]; c) Santel A, Aleku M, Keil O, Endruschat J, Esche V, Fisch G, Dames S, Loffler K, Fechtner M, Arnold W, Giese K, Klippel A, Kaufmann J, Gene Ther 2006, 13, 1222; [DOI] [PubMed] [Google Scholar]; d) Polach KJ, Matar M, Rice J, Slobodkin G, Sparks J, Congo R, Rea-Ramsey A, McClure D, Brunhoeber E, Krampert M, Schuster A, Jahn-Hofmann K, John M, Vornlocher HP, Fewell JG, Anwer K, Geick A, Mol Ther 2012, 20, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7] a).Mansurov A, Ishihara J, Hosseinchi P, Potin L, Marchell TM, Ishihara A, Williford JM, Alpar AT, Raczy MM, Gray LT, Swartz MA, Hubbell JA, Nat Biomed Eng 2020, 4, 531; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Muro S, J Control Release 2012, 164, 125; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Nag OK, Delehanty JB, Pharmaceutics 2019, 11; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Simone E, Ding BS, Muzykantov V, Cell Tissue Res 2009, 335, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kheirolomoom A, Kim CW, Seo JW, Kumar S, Son DJ, Gagnon MK, Ingham ES, Ferrara KW, Jo H, ACS Nano 2015, 9, 8885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9] a).Oberli MA, Reichmuth AM, Dorkin JR, Mitchell MJ, Fenton OS, Jaklenec A, Anderson DG, Langer R, Blankschtein D, Nano Lett 2017, 17, 1326; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wong JKL, Mohseni R, Hamidieh AA, MacLaren RE, Habib N, Seifalian AM, Trends Biotechnol 2017, 35, 1124; [DOI] [PubMed] [Google Scholar]; c) Knudsen KB, Northeved H, Kumar PE, Permin A, Gjetting T, Andresen TL, Larsen S, Wegener KM, Lykkesfeldt J, Jantzen K, Loft S, Moller P, Roursgaard M, Nanomedicine 2015, 11, 467; [DOI] [PubMed] [Google Scholar]; d) Lv H, Zhang S, Wang B, Cui S, Yan J, J Control Release 2006, 114, 100; [DOI] [PubMed] [Google Scholar]; e) Sahay G, Querbes W, Alabi C, Eltoukhy A, Sarkar S, Zurenko C, Karagiannis E, Love K, Chen D, Zoncu R, Buganim Y, Schroeder A, Langer R, Anderson DG, Nat Biotechnol 2013, 31, 653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10] a).Sunshine JC, Sunshine SB, Bhutto I, Handa JT, Green JJ, PLoS One 2012, 7, e37543; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lynn DML, R., J. Am. Chem. Soc. 2000, 122, 10761; [Google Scholar]; c) Gao X, Jin Z, Tan X, Zhang C, Zou C, Zhang W, Ding J, Das BC, Severinov K, Hitzeroth II, Debata PR, He D, Ma X, Tian X, Gao Q, Wu J, Tian R, Cui Z, Fan W, Huang Z, Cao C, Bao Y, Tan S, Hu Z, J Control Release 2020, 321, 654. [DOI] [PubMed] [Google Scholar]
- [11].Riera R, Feiner-Gracia N, Fornaguera C, Cascante A, Borros S, Albertazzi L, Nanoscale 2019, 11, 17869. [DOI] [PubMed] [Google Scholar]
- [12].Behr J, Chim. Int. J. Chem 1997, 51, 34. [Google Scholar]
- [13].Dosta P, Ramos V, Borrós S, Mol. Syst. Des. Eng 2018, 3, 677. [Google Scholar]
- [14] a).Segovia N, Dosta P, Cascante A, Ramos V, Borros S, Acta Biomater 2014, 10, 2147; [DOI] [PubMed] [Google Scholar]; b) Dosta P, Segovia N, Cascante A, Ramos V, Borros S, Acta Biomater 2015, 20, 82; [DOI] [PubMed] [Google Scholar]; c) Nunez-Toldra R, Dosta P, Montori S, Ramos V, Atari M, Borros S, Acta Biomater 2017, 53, 152; [DOI] [PubMed] [Google Scholar]; d) Fornaguera C, Guerra-Rebollo M, Angel Lazaro M, Castells-Sala C, Meca-Cortes O, Ramos-Perez V, Cascante A, Rubio N, Blanco J, Borros S, Adv Healthc Mater 2018, 7, e1800335. [DOI] [PubMed] [Google Scholar]
- [15] a).Bruckman MA, Jiang K, Simpson EJ, Randolph LN, Luyt LG, Yu X, Steinmetz NF, Nano Lett 2014, 14, 1551; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nahrendorf M, Jaffer FA, Kelly KA, Sosnovik DE, Aikawa E, Libby P, Weissleder R, Circulation 2006, 114, 1504. [DOI] [PubMed] [Google Scholar]
- [16] a).Son DJ, Kumar S, Takabe W, Kim CW, Ni CW, Alberts-Grill N, Jang IH, Kim S, Kim W, Won Kang S, Baker AH, Woong Seo J, Ferrara KW, Jo H, Nat Commun 2013, 4, 3000; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Davies PF, Circ Res 2007, 101, 10; [DOI] [PubMed] [Google Scholar]; c) Topper JN, Gimbrone MA Jr., Mol Med Today 1999, 5, 40. [DOI] [PubMed] [Google Scholar]
- [17] a).Stohr R, Cavalera M, Menini S, Mavilio M, Casagrande V, Rossi C, Urbani A, Cardellini M, Pugliese G, Menghini R, Federici M, Atherosclerosis 2014, 235, 438; [DOI] [PubMed] [Google Scholar]; b) Cardellini M, Menghini R, Martelli E, Casagrande V, Marino A, Rizza S, Porzio O, Mauriello A, Solini A, Ippoliti A, Lauro R, Folli F, Federici M, Diabetes 2009, 58, 2396; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Black RA, Nat Genet 2004, 36, 934; [DOI] [PubMed] [Google Scholar]; d) Magid R, Murphy TJ, Galis ZS, J Biol Chem 2003, 278, 32994. [DOI] [PubMed] [Google Scholar]
- [18].Walpola PL, Gotlieb AI, Cybulsky MI, Langille BL, Arterioscler Thromb Vasc Biol 1995, 15, 2. [DOI] [PubMed] [Google Scholar]
- [19] a).Ricard I, Payet MD, Dupuis G, Eur J Immunol 1998, 28, 1708; [DOI] [PubMed] [Google Scholar]; b) Voinea M, Manduteanu I, Dragomir E, Capraru M, Simionescu M, Pharm Res 2005, 22, 1906. [DOI] [PubMed] [Google Scholar]
- [20].Blanco E, Shen H, Ferrari M, Nat Biotechnol 2015, 33, 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21] a).Wu D, Birukov K, Front Bioeng Biotechnol 2019, 7, 172; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dickinson MG, Bartelds B, Borgdorff MA, Berger RM, Am J Physiol Lung Cell Mol Physiol 2013, 305, L1; [DOI] [PubMed] [Google Scholar]; c) Huertas A, Guignabert C, Barbera JA, Bartsch P, Bhattacharya J, Bhattacharya S, Bonsignore MR, Dewachter L, Dinh-Xuan AT, Dorfmuller P, Gladwin MT, Humbert M, Kotsimbos T, Vassilakopoulos T, Sanchez O, Savale L, Testa U, Wilkins MR, Eur Respir J 2018, 51; [DOI] [PubMed] [Google Scholar]; d) Bland RD, Am J Physiol 1990, 259, L30; [DOI] [PubMed] [Google Scholar]; e) Matthay MA, Folkesson HG, Verkman AS, Am J Physiol 1996, 270, L487. [DOI] [PubMed] [Google Scholar]
- [22].Meng Z, Luan L, Kang Z, Feng S, Meng Q, Liu K, J Mater Chem B 2017, 5, 74. [DOI] [PubMed] [Google Scholar]
- [23].Attia MF, Anton N, Wallyn J, Omran Z, Vandamme TF, The Journal of pharmacy and pharmacology 2019, 71, 1185. [DOI] [PubMed] [Google Scholar]
- [24].Ulbrich K, Subr V, Strohalm J, Plocova D, Jelinkova M, Rihova B, J Control Release 2000, 64, 63. [DOI] [PubMed] [Google Scholar]
- [25].Harris JM, Chess RB, Nat Rev Drug Discov 2003, 2, 214. [DOI] [PubMed] [Google Scholar]
- [26].Yanjarappa MJ, Gujraty KV, Joshi A, Saraph A, Kane RS, Biomacromolecules 2006, 7, 1665. [DOI] [PubMed] [Google Scholar]
- [27].Ni CW, Kumar S, Ankeny CJ, Jo H, Vasc Cell 2014, 6, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28] a).Nam D, Ni CW, Rezvan A, Suo J, Budzyn K, Llanos A, Harrison D, Giddens D, Jo H, Am J Physiol Heart Circ Physiol 2009, 297, H1535; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nam D, Ni CW, Rezvan A, Suo J, Budzyn K, Llanos A, Harrison DG, Giddens DP, Jo H, J Vis Exp 2010, DOI: 10.3791/1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.