

Summary
Efforts to overcome resistance to immune checkpoint blockade therapy have focused on vaccination strategies using neoepitopes, although they cannot be applied on a large scale due to the “private” nature of cancer mutations. Here, we show that infection of tumor cells with Salmonella induces the opening of membrane hemichannels and the extracellular release of proteasome-generated peptides by the exacerbation of endoplasmic reticulum (ER) stress. Peptides released by cancer cells foster an antitumor response in vivo, both in mice bearing B16F10 melanomas and in dogs suffering from osteosarcoma. Mass spectrometry analysis on the supernatant of human melanoma cells revealed 12 peptides capable of priming healthy-donor CD8+ T cells that recognize and kill human melanoma cells in vitro and when xenotransplanted in vivo. Hence, we identified a class of shared tumor antigens that are generated in ER-stressed cells, such as tumor cells, that do not induce tolerance and are not presented by healthy cells.
Keywords: cancer, vaccine, tumor antigens, immunotherapy, Salmonella, ER-stress response
Graphical abstract
Highlights
-
•
Tumor cell infection with Salmonella induces the release of immunogenic peptides
-
•
Peptide-based vaccine boosts a strong antitumor response in dog osteosarcoma
-
•
MS analysis identified shared peptides released by human melanoma cell lines
-
•
Twelve identified peptides are capable of inducing a tumor-specific CD8 response
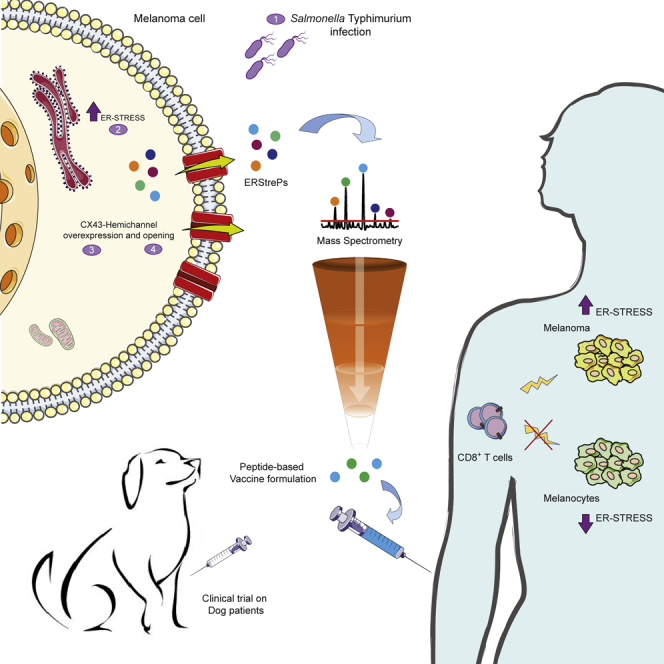
Melacarne et al. demonstrate that Salmonella exacerbates endoplasmic reticulum stress in tumor cells, which induces hemichannel opening and the subsequent release of peptides in the extracellular milieu. Released peptides are immunogenic, shared among tumors, and can be exploited as an anticancer vaccine to prevent metastasis occurrence.
Introduction
Immunotherapy and, in particular, immune checkpoint blockade (ICB) have drastically improved the survival of patients with highly immunogenic solid tumors, such as metastatic melanoma and lung cancer (Vanpouille-Box et al., 2017). However, a large proportion of patients do not respond to therapy (Wolchok et al., 2013). Failure of ICB efficacy is mostly due to the absence of a pre-existing antitumor response that can be boosted or revitalized by ICB (Gros et al., 2014) and/or to the inability of tumor cells to present tumor-derived peptides (Gao et al., 2016; Zaretsky et al., 2016). Combinatorial approaches aimed at both activating the immune response and protecting it from tumor-evasion strategies may represent a valid alternative for treating non-responding patients (Sharma and Allison, 2015). Recently, two clinical trials have shown that therapeutic vaccination based on patient-specific neoantigens can elicit an antitumor response in advanced metastatic melanoma that can be boosted by combining with anti-PD1 ICB (Ott et al., 2017; Sahin et al., 2017). However, the same seems to not be true when analyzing the combination of ICB with only one known melanoma antigenic peptide, such as glycoprotein gp100 (Hodi et al., 2010), suggesting that the nature of the antigen is critical for achieving effective antitumor immunity. Indeed, tumor cells with a high mutational load generate a series of novel antigens that carry somatic mutations that are presented by HLA class I (HLA-I) molecules (Palmieri et al., 2017; Snyder et al., 2014). HLA-I-restricted neoepitopes, i.e., the peptides that carry the somatic mutation and elicit a CD8+ cytotoxic T cell (CTL) response, are generally produced by proteasomes. There are various proteasome isoforms that vary both in their regulatory complexes and in their catalytic subunits of the 20S proteasome core (Mishto and Liepe, 2017). Because neoantigens are the outcome of somatic mutations, they are not presented in the thymus. Hence, there should be limited central tolerance toward neoantigens, and the higher the amount of mutated antigens the lower the probability of inducing peripheral tolerance (Heemskerk et al., 2013). However, a personalized vaccine strategy based on identifying patient-specific neoantigens, although very attractive, is time consuming, expensive, and difficult to translate to all cancer centers and individual patients. Here, we propose a strategy for identifying immunogenic tumor antigens shared among patients to be used in “universal” disease-specific cancer vaccines.
We have previously shown that infection of murine and human tumor cells with the attenuated vaccine strain of Salmonella Typhi leads to the overexpression of Connexin-43 (CX43) (Saccheri et al., 2010), the most abundant and ubiquitous component of plasma membrane hemichannels forming gap junctions (GJs) (Mendoza-Naranjo et al., 2007; Neijssen et al., 2005). These Salmonella-treated tumor cells establish functional GJs with dendritic cells (DCs) by the docking of two plasma membrane hemichannels, allowing the transfer of pre-processed antigens from tumor cells to DCs and leading to the establishment of a strong DC-based antitumor response (Saccheri et al., 2010). However, this strategy would be time consuming and would require the simultaneous generation of a tumor cell line, the differentiation of DCs from peripheral blood monocytes (PBMCs), and the pairing of tumor cells with DCs from patient biomaterial.
It has been shown that, under redox changes and phosphorylation, CX43 undergoes post-translational modifications (PTMs) that lead to the opening of GJ hemichannels without the necessity of full channel formation and to the release of intracellular molecules, including ATP, in the surrounding medium (Pogoda et al., 2016). In addition, Salmonella infection is known to induce a strong oxidative stress response (Suvarnapunya et al., 2003) that leads to the activation of the unfolded protein response (UPR) in infected cells (Antoniou et al., 2019). The UPR is initiated to cope with an ER stress response and has been shown to be linked to antigen presentation (Osorio et al., 2018). Thus, we explored the possibility that infection of tumor cells with Salmonella may foster the UPR response and drive CX43 PTMs, hemichannel opening, and the release of antigenic peptides in the supernatant that could be used for antitumor vaccine formulations. Indeed, we show that infection of tumor cells leads to the opening of GJ hemichannels and subsequent UPR-dependent release of tumor antigenic peptides in the culture supernatant. The released peptides induce a strong antitumor response in both mouse and human models, as well as in a therapeutic clinical trial for sarcoma (SA) and osteosarcoma (OSA) dog patients. We analyzed the nature of these peptides and found that they are shared among different patients affected by the same tumor type and are potently immunogenic but are not presented by melanocytes. Therefore, they could be the core of a potential off-the-shelf peptide-based vaccine.
Results
Salmonella-infected tumor cells release peptides upon plasma membrane hemichannel opening
To test the ability of Salmonella to induce the opening of GJ hemichannels and the release of cytoplasmic material, including antigenic peptides, we monitored ATP release by mouse melanoma B16F10-OVA cells upon infection with Salmonella enterica serovar Typhimurium SL3216AT. Salmonella-infected melanoma cells released a higher amount of ATP than untreated cells (Figure 1A) and the silencing of CX43 (B16F10-CX43KO) impaired ATP release (Figure 1A). Along with ATP, small linear peptides up to 2 kDa (or about 16 amino acids) can diffuse across intercellular GJ (Neijssen et al., 2005, Saccheri et al., 2010). We thus evaluated whether the opening of the hemichannels leads to the release of antigenic peptides in the culture supernatant (secretome) by using B16F10-OVA cells that express the chicken ovalbumin as a model antigen. We tested the release of the OVA epitope with a functional assay by loading murine DCs DC1 (Rotta et al., 2003) with either Salmonella-infected B16F10-OVA or B16F10-OVA-CX43KO-derived secretome and co-culturing them with OVA257-264-specific B3Z hybridoma cells (Rescigno et al., 1998). We observed that only the secretome derived from Salmonella-infected CX43-proficient cells induced mIL-2 production by B3Z cells (Figure 1B), thus indicating the release of OVA257-264 upon Salmonella infection. Secretomes were also tested for their ability to activate OVA257-264-specific CD8 T cells (Figure S1A); DC1 loaded with secretome derived from Salmonella-infected B16F10-OVA cells induced interferon gamma (IFN-γ) production by CD8+ T cells (from OTI mice, Figure S1B). Furthermore, we confirmed the presence of OVA257-264 in the medium by mass spectrometry (MS) (Figure S1C). Together, these results show that tumor cell infection with Salmonella induces not only ATP release but also the release of tumor-derived peptides (such as OVA257-264) by CX43 hemichannels.
Figure 1.

Peptides released by Salmonella-infected tumor cells by plasma membrane hemichannels induce an anti-tumor response in vivo
(A) ATP luminescence test on secretomes of B16F10-OVA and B16F10-OVA CX43KO cells infected with Salmonella (Salm) or left untreated (−).
(B) ELISA quantification of mIL2 secretion by OVA257-264-specific B3Z cells.
(A and B) Data are represented as mean ± SEM using bar plots (n = 3).
(C) Scheme of in vivo immunization experiment; mice were immunized with IFA-Aldara alone (IFA-A), IFA-A combined with secretome derived either from Salmonella-infected B16F10 cells (IFA-A Vax) or untreated cells (IFA-A Ctrl).
(D) Frequency of tumor-reactive CD3+CD8+CD107a+ from PBMCs of immunized mice. Data of 2 pooled experiments are represented as mean ± SD using a scatter dot plot.
(E and F) Tumor growth (E) and Kaplan-Meier survival curves (F) of vaccinated mice. Data are pooled from two independent experiments (n = 5–9).
(G and H) ELISA quantifications of B16F10-specific IgG (G) and Salmonella-specific antibodies (H) in mice sera. Data of 3 pooled experiments are represented as mean ± SEM (n = 6).
(I) Scheme of in vivo CD4 and CD8 depletion. Mice were immunized with IFA-A, IFA-A combined with B16F10 secretome and isotype control antibody (IFA-A Vax Iso), or with B16F10 secretome and depleted of either CD8 (IFA-A Vax CD8) or CD4 (IFA-A Vax CD4) T cells.
(J and K) Tumor growth (J) and Kaplan-Meier survival curves (K) of vaccinated mice (n = 7–9).
(L) Mouse peptides released by B16F10 upon Salmonella infection.
(M) ELISpot of IFNγ spot forming cell (SFC)-splenocytes stimulated ex vivo with the mix of identified peptides. Data of 1 experiment are represented as mean ± SE by using a scatter dot plot (n = 6).
Statistical analysis was evaluated using two-sided Mann-Whitney test (A, B, and D) or ordinary one-way ANOVA with Bonferroni post-test (D, E, G, H, J, and M); log-rank Mantel-Cox test was performed to assess differences among survival curves (F,K). ns, p > 0.05; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. See also Figures S1 and S2 and Table S1.
Peptides released by Salmonella-treated B16F10 cells induce an antitumor response in vivo
We then evaluated if peptides released by Salmonella-infected B16F10, a melanocytic and non-immunogenic melanoma cell line (Overwijk and Restifo 2001), were immunogenic and could be used as an anticancer vaccine. We loaded DC1 cells with secretome derived from Salmonella-infected B16F10 or untreated cells and used them to immunize mice. Mice were vaccinated three times, and 4 weeks later, they were challenged with B16F10 cells (Figure S2A). We found that only mice immunized with DCs loaded with the secretome derived from Salmonella-infected B16F10 cells (DC-Vax) showed significantly delayed tumor growth (Figure S2B) and longer overall survival (OS) (Figure S2C). Interestingly, vaccination with DCs loaded with the combination of two well-known melanoma antigens, namely, rp2180-188 and gp10025-33 (DC Trp2 gp100), induced only a minor tumor growth delay (Figure S2B) and no amelioration in terms of survival (Figure S2D). We then analyzed whether the released peptides could exert an immunogenic effect without the use of DCs, in combination with an adjuvant. The B16F10 cell line secretome was enriched in peptides by chromabond SPE C18 devices and combined with incomplete Freud’s adjuvant (IFA). This combination was used to subcutaneously immunize 6-week-old mice (C57BL/6J) treated topically with the TLR7/8-agonist imiquimod (Aldara, -A) by adopting the schedule described in Figure 1C. Mice were analyzed for development of degranulating (CD107a+) CD8 T cells, antitumor antibody response, tumor growth, and survival. As a control, a group of mice was vaccinated with the secretome derived from untreated cells (IFA-Aldara alone [IFA-A] Ctrl). One week after the last vaccination, only mice immunized with peptides released by Salmonella-infected tumor cells (IFA-A Vax) developed tumor-specific circulating CD8+ T cells (Figure 1D). The detected tumor-specific CD8 response correlated with a delay in tumor growth (Figure 1E) together with a significant improvement in survival (Figure 1F). This response was not observed in mice vaccinated with peptides released by untreated B16F10 cells, indicating that Salmonella treatment was responsible for the release of immunogenic cancer peptides. By analyzing mouse sera taken at the endpoint, we observed that Vax-immunized mice developed immunoglobulins (immunoglobulin Gs [IgGs]) directed to tumor cells (Figure 1G) but not to Salmonella antigens (Figure 1H). The same amplitude of antitumor response was induced by peptides derived from Salmonella-infected B16F10 cells combined with adjuvant TLR9-agonist ODN1826 (ODN Vax, Figures S2E–S2G). To dissect the role of CD4+ and CD8+ T cells in controlling tumor growth, we performed mouse vaccination protocols in the presence of anti-CD4 or -CD8 antibodies (schedules in Figure 1I). The depletion of CD4+ T cells (IFA-A Vax CD4) did not affect the delay in tumor growth and the improved survival of the Vax compared to mice treated with adjuvant alone (IFA-A; Figures 1J and 1K). On the contrary, CD8+ T cell depletion impaired the beneficial effect of the vaccine (IFA-A Vax CD8; Figures 1J and 1K). This finding suggests that although humoral responses were elicited by the vaccine, CD8-mediated immunity plays a major role in the antitumor response.
To dissect the sequences of the peptides that boosted the antitumor response in vivo, we performed a peptidomic analysis. Peptides differentially detected in secretomes derived from untreated versus Salmonella-treated B16F10 cells were selected (Table S1). A further tuning based on the affinity of the peptides for major histocompatibility complex (MHC) class I molecules using in silico prediction tool Net-MHCpan4.1 was applied, and a final list of 10 peptides was selected (Figure 1L).
To assess whether these peptide candidates were responsible for the antitumor response observed in vivo, we synthesized, combined, and used them ex vivo to stimulate splenocytes derived from mice that underwent the vaccination protocol. Splenocytes of both unvaccinated mice (ut) and of mice immunized with adjuvant only (IFA-A) were used as controls. ELISpot analysis showed that the mix of peptides induced T cells of vaccinated, but not IFA-A-treated, mice to produce IFN-γ (Figure 1M), confirming that the selected peptides are a component of the B16F10-secretome vaccine used in vivo and are likely responsible for the observed antitumor response. The MS analysis did not highlight the presence of bacterium-derived peptides nor of danger-associated molecular patterns, thus excluding any adjuvant effect mediated by the vaccine and explaining the absence of a Salmonella-specific antibody response. Consistent with this finding, the secretome-based vaccine did not induce the upregulation of surface activation markers (CD40 and CD86; Figure S2H) in DC1, excluding that the observed in vivo effect was merely due to the capacity of the secretome to activate DCs. Similarly, the exposure of human-monocyte-derived DCs (MoDCs) to the secretome of Salmonella-infected tumor cells did not induce cells activation; no significant changes in the percentage of activated MoDCs (HLA-DR+CD86+CD206−) were observed (Figure S2I).
Secretomes of Salmonella-treated primary tumor cells prime an antitumor response in dogs affected by OSA and high-grade SA
The positive results obtained by our vaccination approach in a poorly immunogenic mouse model of melanoma prompted us to translate this strategy into an immunotherapy clinical setting. As a model system, we treated dog patients affected by OSA, as it shows a similar biological behavior in humans and dogs (Rowell et al., 2011) and melanoma differs profoundly in terms of preferential site of development. The enrolled patients with OSA underwent surgical excision of the primary tumor; we derived OSA cells from the patient’s tumor surgical specimen and expanded them in culture before infecting them with Salmonella. We observed overexpression of the CX43 protein upon Salmonella infection consistent with what was previously shown in human and mouse systems (Figure S3; Saccheri et al., 2010). Secretomes were filtered and lyophilized; for the first 2 immunizations, they were combined with a veterinary adjuvant treatment (leptospirosis vaccine) and were solubilized in water for the following 4 immunizations, all following topically delivered TLR7/8-agonist imiquimod (Aldara cream) at the vaccine-injection site. In total, 6 intradermal vaccinations were performed starting after the 3rd of 4 cycles of carboplatin (schedule in Figure 2A). In some cases, when the primary tumor was not available or tumor cells could not be expanded in vitro, the dog patients were vaccinated using peptides released by tumor cells of another dog suffering of the same tumor type (Vax H, Table 1). To analyze the impact of our vaccination strategy, we compared the survival of patients with OSA to the survival of a control cohort treated with only carboplatin during the same period (2016–2019). The control population was matched with regard to well-known prognostic variables, including clinical stage, serum ALP (alkaline phosphatase level) level, and affected anatomic site, and only dogs without distant metastases were analyzed. Two dogs with humeral OSA (the appendicular site known as the most aggressive) and two dogs with elevated serum ALP level at admission (having a negative prognostic role) were included in each group. Vaccinated dogs received 4 doses of carboplatin, and unvaccinated dogs received 5 to 6 doses. At the writing of this manuscript, 2 patients with OSA that received the vaccine are alive after almost 2 years from diagnosis, and time to metastasis (TTM) as well as OS were significantly longer in vaccinated dogs than those of unvaccinated dogs (TTM of 589 versus 265 days, p = 0.0014; OS of 625 versus 303.5 days, p = 0.0368; Figures 2B and 2C). None of the enrolled patients had a positive reaction to the delayed type hypersensitivity (DTH) test. Erythema or induration after 48–72 h was never detected, thus indicating that none of the peptides included in the vaccine formulation induced a strong and immediate immune response, and also that autoimmune reactions did not occur. Immune-phenotype analysis was performed on samples derived from the 6 patients with OSA that completed the vaccination protocol (Vax state: completed, Table 1). The vaccine induced a B cell response; 4 of 6 patients with OSA developed tumor-specific IgG throughout the vaccination protocol (Figure 2C). Furthermore, in vitro concanavalin-A-expanded PBMCs from 5 of 6 dogs with OSA were activated upon stimulation with vaccine (Vax), confirming the immunogenicity of the Vax (Figure 2D). Importantly, 4 of 5 patients with OSA that responded positively to the vaccine had received a heterologous Vax, suggesting that our immunotherapy strategy enabled the targeting of shared tumor antigens that prompted an immune response. To assess if the vaccination strategy could be extended to other tumor types, 3 dog patients suffering from high-grade SA were enrolled in the clinical trial (Table 1). All patients with SA received an autologous vaccination; 2 of them completed the vaccination protocol and both showed an increased antitumor IgG response over time (Figure 2E), together with a Vax-specific T cell response (Figure 2F). The strong immune response correlated with a long OS (>3 years). These results indicate that the vaccine effectively induces an anticancer immune response that led to objective clinical responses and increased OS in dog patients.
Figure 2.

Secretomes of Salmonella-treated canine primary tumor cells prime an antitumor response in dogs affected by osteosarcoma and high-grade sarcoma
(A) Clinical trial scheme for dog patients with OSA and high-grade SA.
(B and C) Kaplan-Meier of metastasis free (B) and survival curve of (C) patients with OSA treated with carboplatin alone (black) or in combination with Vax (green).
(D and F) Tumor-specific IgG in dog sera shown as frequency of IgG-opsonized tumor cells.
(E and G) ELISA quantification of IFN-γ released by dog patient PBMCs upon stimulation with secretome from untreated (ut) or Salmonella-infected (Vax) SA/OSA cells.
Immunomonitoring experiments were performed once (D and F) or more (n = 2–3; C and E); each condition was tested at least in triplicate. Data are represented as mean ± SEM using a scatter dot plot. Statistical analysis was evaluated using log-rank Mantel-Cox (B), two-way ANOVA Tukey’s multiple comparison (C and E), and two-sided Mann-Whitney test (D and F). ∗p < 0.05,∗∗p < 0.01,∗∗∗p < 0.001,∗∗∗∗p < 0.0001. See also Figure S3.
Table 1.
Dog patients with osteosarcoma and high grade sarcoma enrolled in the clinical trial, see also Figure 3
| Treatment | Dog ID | Breed | Sex | Age (years) | Histotype | Anatomic site | Stage | ALP | Carboplatin (cycles) | Vax (autologous/heterologous) | Vax state | TTM | State (alive/dead) | Death related to cancer (Y/N) | Survival (days) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Osteosarcoma | |||||||||||||||
| Carboplatin + VAX | 0 | Newfoundland | F | 7 | osteoblastic | right distal radius | metastases | Wnl | 4 | H | completed | 653+ | D | N | 653 |
| 1 | American staffordshire terrier | F | 12 | osteoblastic | right proximal humerus | Wnl | 4 | A | 5 out of 6 | 214 | D | Y | 224 | ||
| 8 | mixed breed | M | 12 | osteoblastic | right proximal tibia | increased | 4 | A | 5 out of 6 | 229+ | D | N | 229 | ||
| 17 | mixed breed | M | 10 | osteoblastic | left hemibacin | increased | 6 | A | 4 out of 6 | 246+ | D | Y | 246 | ||
| 19 | Rottweiler | F | 7 | osteoblastic | right proximal tibia | Wnl | 4 | A | completed | 551 | D | Y | 621 | ||
| 23 | Spanish greyhound | F | 9 | chondroblastic | right distal femur | Wnl | 4 | A | 5 out of 6 | 306+ | D | N | 306 | ||
| 42 | border collie | F | 7 | osteoblastic | left proximal tibia | Wnl | 4 | H | completed | 281 | D | Y | 630 | ||
| 47 | boxer | F | 12 | osteoblastic | left proximal humerus | Wnl | 4 | H | completed | 589 | D | Y | 649 | ||
| 51 | Leonberger | M | 3 | osteoblastic | scapula | Wnl | 4 | H | completed | 700+ | A | 700+ | |||
| 52 | mixed breed | F | 8 | osteoblastic | right distal radius | Wnl | 4 | H | completed | 701+ | A | 701+ | |||
| Carboplatin |
mixed breed | F | 7 | osteoblastic | proximal femur | no mets |
Wnl | 4 | no VAX | N/A | 111 | D | Y | 131 | |
| Lagotto | M | 3 | chondroblastic | distal femur | Wnl | 6 | no VAX | N/A | 169 | D | Y | 169 | |||
| German shepherd | M | 8 | osteoblastic | proximal humerus | Wnl | 4 | no VAX | N/A | 375 | D | Y | 375 | |||
| mixed breed | F | 5 | osteoblastic | distal radius | Wnl | 6 | no VAX | N/A | 252 | D | Y | 371 | |||
| mixed breed | F | 13 | osteoblastic | proximal humerus | Wnl | 6 | no VAX | N/A | 298 | D | Y | 329 | |||
| mixed breed | F | 11 | osteoblastic | distal radius | Wnl | 6 | no VAX | N/A | 358 | D | Y | 358 | |||
| Rottweiler | M | 1 | osteoblastic | proximal tibia | increased | 4 | no VAX | N/A | 130 | D | Y | 156 | |||
| mixed breed | F | 10 | osteoblastic | distal tibia | Wnl | 4 | no VAX | N/A | 75 | D | Y | 162 | |||
| pitbull | F | 13 | osteoblastic | distal femur | increased | 4 | no VAX | N/A | 278 | D | Y | 278 | |||
| German shepherd |
M |
9 |
osteoblastic |
distal radius |
Wnl |
6 |
no VAX |
N/A |
351 |
D |
Y |
369 |
|||
| Sarcoma | |||||||||||||||
| Carboplatin +VAX | 5 | pitbull | F | 6 | soft tissue sarcoma | hind limb | no mets | Wnl | 4 | A | completed | 2,224+ | D | N | 2,224 |
| sarcoma relapse | hind limb | A | completed | ||||||||||||
| 32 | beagle | F | 10 | soft tissue sarcoma | abdomen | no mets | Wnl | 4 | A | 4 out of 6 | 244+ | D | Y | 244 | |
| 35 | cocker spaniel | F | 4 | soft tissue sarcoma | gingiva | no mets | Wnl | 4 | A | completed | 1,516+ | A | 1,516+ | ||
Wnl, within normal levels.
Peptides released by 624-38 human melanoma cell line upon Salmonella infection activate tumor-specific CD8+ T cells derived from healthy donor PBMCs
Salmonella induced both mouse and dog tumor cells to release immunogenic peptides that could stimulate an antitumor immune response in vivo. To address whether Salmonella could exert the same effect also on human tumor cells, we used T2 cells to assess the presence of HLA-A∗02:01 binding peptides (Hosken and Bevan, 1990). Melanoma (624-38) and colon adenocarcinoma (HT-29) cell lines were left untreated or infected with an attenuated vaccine strain of Salmonella enterica serovar Typhi (Ty21a); the secretomes were collected and added to the T2 cell line, and the amount of HLA-I molecules at the cell surface was measured by flow cytometry. We observed a higher stabilization of HLA-A∗02:01 at the cell surface when T2 cells were exposed to secretomes of Salmonella-treated cells than that of untreated secretomes (Figure 3A), suggesting that the former was enriched of HLA-binding peptides. Importantly, peptide enrichment was reduced to background levels once the hemichannel blocker heptanol (Hept) (Matsue et al., 2006) was added, thus implying that peptide release was mainly hemichannel mediated.
Figure 3.

Peptides released by 624-38 human melanoma cell line upon Salmonella infection activate tumor-specific CD8+ T cells derived from healthy donor PBMCs
(A) MFI (mean fluorescence intensity) of HLA-A∗02:01 of T2 cells loaded with secretomes of 624-38 and HT-29 cells infected with Salmonella (Salm) with or without Heptanol (Hept) or left untreated (−).
(B) Scheme of CTL-Vax and CTL-Mart126-35 expansion from healthy donor PBMCs.
(C) Tumor cell targets. HLA-A2∗02:01 and Mart1 antigen expression are indicated.
(D and E) Frequency of CD3+CD8+IFN-γ+ (activated) or CD3+CD8+CD107a+ (degranulating) CTL-Vax (D) and CTL-Mart (E) upon co-culture with target cells. Pan HLA-blocker (anti HLA) antibody was added as control, where indicated.
(F) Tumor growth curves of NSG mice injected with 624-38 melanoma cells, adoptively transferred with CTL-Vax (blue) or with CTL-Mart1 (red) at day 7 after tumor injection.
(A, D, and E) Data are pooled from three or more experiments and are expressed as mean ± SE using scatter dot plot. Statistical analysis was evaluated using one way ANOVA (A and F) or two-sided Mann-Whitney test (D and E). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. See also Figures 4, 5, and 6.
To assess the immunogenicity of the released peptides, we tested their capacity to activate antigen-specific-CD8+ T cells starting from PBMCs of HLA-A∗02:01+ healthy donors (Figure 3B). Secretome from Salmonella-infected 624-38 cells was used to stimulate PBMCs (referred to as CTL-Vax). As a positive control, we stimulated PBMCs with the Melanoma antigen recognized by T cells 1 (Mart-126-35) peptide (referred to as CTL-Mart1). CD8+ CTL-Vax recognized antigens presented by the 624-38 cell line, as shown by the intracellular IFN-γ production (Figure 3D) and augmented expression of the degranulation marker CD107a (Figure 3D). Furthermore, CTL-Vax responded to other HLA-A∗02:01+melanoma cell lines (Skmel24, A375, Mewo, and C32) and primary melanoma cells (hMel3) but not to the HLA-A∗02:01-negative (hMel2) primary melanoma cells nor to the colon cancer HLA-A∗02:01+ HT-29 cell line, suggesting that CTL-Vax reacts to HLA-A∗02:01-restricted peptides presented by melanoma cell lines but not by the adenocarcinoma cell line (HLA-A∗02:01 expression on target cells is summarized in Figure 3C). In agreement with the above results, the activation of CD8+ T lymphocytes against melanoma cells was mediated by the recognition of HLA-I-epitope complexes, as these lymphocytes did not degranulate in the presence of a pan-HLA blocker antibody (anti-HLA) and did not recognize the 624-28 cell line, an HLA-A∗02:01-negative 624-MEL cell line caused by aberrant pre-mRNA splicing (Sabatino et al., 2008). As expected, CTL-Mart1 was activated in response to Mart1 expressing 624-38, C32, and MeWo cells (Figure 3E); they expressed CD107a in the presence of the primary hMel3 cell line, but not against SkMel24, Mart-1 negative A375 (Boegel et al., 2014), or the HLA-A∗02:01-negative primary melanoma hMel2 (Figure 3E). To test the fitness of the ex-vivo-expanded CTL-Vax T cells, we assessed their cytotoxic potential in vivo. NOD.Cg-Prkdcscid Il2rgtm1Wjl /SzJ (NSG) mice were subcutaneously inoculated with 624-38 melanoma cells and adoptively transferred with in-vitro-expanded CTL-Vax or CTL-Mart1 cells. Notably, tumor growth was inhibited by CTL-Vax (Figure 3F) that showed a stronger tumor control effect than CTL-Mart1. These results indicate that in vitro priming of Vax-specific cytotoxic T cells effectively induces HLA-dependent lysis of melanoma cell lines and primary melanoma cells in vitro and melanoma cell lines in vivo after adoptive T cell transfer.
Salmonella-induced secretome peptides are released in response to the UPR pathway activation along with hemichannel opening
To sustain cell proliferation and overcome glucose shortage, genomic instability, and high mutational load, tumor cells face several stress stimuli at the level of the ER and are characterized by a high basal level of the UPR, which is now considered a hallmark of cancer (Cerezo et al., 2016; Corazzari et al., 2017; Urra et al., 2016). Three major pathways are activated by the UPR, as follows: (1) the cleavage of ATF6 triggers genes necessary to restore ER homeostasis (i.e., chaperones); (2) the activation of protein kinase RNA-like ER kinase (PERK) increases the translation of activating transcription factor 4 (ATF4), which in turn induces CCAAT-enhancer-binding protein homologous protein (CHOP) that causes the upregulation of the proapoptotic proteins; and (3) the activation of inositol-requiring kinase (IRE) 1-α leads to the splicing of XBP1 (to generate XBP1s) that prompts the transcription of genes involved in cell adaptation. Salmonella infection of human tumor cells exacerbates the activation of 2 of 3 branches of the UPR, as we detected an increased expression of XBP1s, CHOP, and ATF4 both at gene and protein levels (Figures 4A and 4B), without augmenting cell death (Figure 4C). Having observed a stronger activation of XBP1s, we inhibited the upstream regulator of XBP1 protein IRE-1α endoribonuclease in the 624-38 cell line by using 4μ8c; interestingly, the inhibitor had an impact not only on XPB1 splicing but also on ATF4 and CHOP expression both at the mRNA and protein level (Figures S4A and S4B). IRE-1α inhibition significantly impaired peptide release prompted by Salmonella infection (Figure 4D), and these secretomes were less capable of activating CTL-Vax (Figure 4E). We next used CRISPR-Cas9 to knock out Xbp1 in 624-38 cells (XBP1 KO, Figures S5A–S5C) to confirm the involvement of the IRE-1-XBP1 branch of the UPR in peptide release. XBP1 deletion affected the release of peptides upon Salmonella infection (Figure 4D), and accordingly, CTL-Vax was significantly less activated upon stimulation with this secretome (Figure 4E). However, the exacerbation of the UPR alone was not sufficient to allow peptide release. Indeed, the use of brefeldin A (BFA) and thapsigargin (Tg), which induced ER stress but not the upregulation of CX43 (Figures S4C and S4D), did not recapitulate the accumulation of peptides in the secretome (Figure S4E) nor the activation of CTL-Vax (Figure S4F). The importance of hemichannels in peptide release was confirmed by treating melanoma cells with heptanol before Salmonella infection. Secretomes from heptanol-treated Salmonella-infected tumor cells were not enriched in peptides (Figure 4D) and did not activate the CTL-Vax (Figure 4E). Similar results were obtained by parallel studies on murine melanoma cells (Figures S5E–5H). Salmonella-infected B16F10-OVA cells showed an increased expression of ER-stress-related genes (Figure S5E) without inducing cell death (Figure S5F). Inhibition of IRE-1α by 4μ8c impaired OVA257-264 release, whereas neither Tg nor BFA treatment of B16F10-OVA cells could recapitulate OVA257-264 release (Figure S5G). Deleting XBP1 by CRISPR-Cas9 (B16F10-OVA-XBP1KO) significantly decreased OVA257-264 release (Figure S5H).
Figure 4.

Salmonella-induced secretome peptides are mainly tumor proteasome derived and are released in response to UPR pathway activation along with hemichannel opening
(A and B) UPR pathway analysis at gene (A) and protein (B) level in Salmonella-infected 624-38 cells. Data of 3 pooled experiments are normalized to Gapdh (A) or Vinculin (B) and expressed as mean ± SEM of average expression fold change.
(C) Frequency of Annexin−PI− (live), Annexin+PI− (early apoptotic), and Annexin+PI+ (apoptotic) tumor cells Salmonella-infected (Salm) or untreated (−) (n = 2).
(D) MFI of HLA-A∗02:01 on T2 cells loaded with secretomes from untreated (−) or Salmonella-infected (Salm) XBP1WT or XBP1KO cells; from Salmonella-infected XBP1WT in combination with 4μ8c, Heptanol (Hept), MG132 (MG); supernatant of dead cells (C and D).
(E) ELISA of IFN-γ released by CTL-Vax stimulated with secretomes differently derived. (D and E) Data of 3 pooled experiments normalized on (−) are represented as mean ± SEM using a scatter dot plot.
(F) Delfia assay; lysis of target cells by CTL-Vax (effector) at different effector-to-target (E:T) ratios (n = 3).
(G) MFI of HLA-A∗02:01 on 624-38 cells. Data of 3 pooled experiments are represented.
Statistical analysis was evaluated using two-sided Mann-Whitney test (A, B, C, and G), one-way ANOVA followed by multiple comparisons (Dunnett) against XBP1WT-Salm (D and E), or two-way ANOVA followed by multiple comparisons (Tukey) (F). ∗p < 0.05,∗∗p < 0.01,∗∗∗p < 0.001,∗∗∗∗p < 0.0001. See also Figures S4, S5, and S6.
Peptides loaded on HLA-I molecules are mainly generated in the cytoplasm by the proteasome after protein degradation (Groettrup et al., 2010). Therefore, we addressed whether the released peptides were generated through this pathway. We showed that Salmonella infection of tumor cells did not alter the chymotrypsin-like proteasome activity, thus excluding that changes in peptide release were due to an augmented proteasome activity (Figure S4G). However, the inhibition of proteasome proteolytic activity by MG132 (MG, Figure S4G) impaired antigen release of HLA-A∗02:01-binding peptides (Figure 4D) and CTL-Vax activation (Figure 4E), suggesting that released peptides are largely produced by the proteasome of cancer cells. Importantly, none of the tested treatments affected cell viability (Figure S4H); hence, neither the accumulation of peptides nor the CTL-Vax activation were due to cell death. Moreover, induction of cell death with a lethal dose of Tg lead, as expected, to the accumulation of peptides in the cell supernatant (cell death [CD], Figure 4D), but CTL-Vax was poorly activated upon stimulation with CD peptides (Figure 4E). Taken together, our results suggest that UPR activation was necessary, but not sufficient, for peptide release because hemichannel opening had to be induced as well. Moreover, we showed that Salmonella boosts a hemichannel-dependent peptide release that provides higher immunogenic stimuli than a mix of peptides derived from dead cells.
Healthy melanocytes do not present stress-related peptides
The peptides released upon Salmonella infection are presented also by non-infected tumor cells, as shown by the ability of CTL-Vax to degranulate when incubated with untreated tumor cells (Figure 3). We hypothesized that melanoma cells, differently from healthy melanocytes, present stress-related peptides because of their constitutive activation of the UPR response. We first investigated the expression of UPR-related genes in primary melanocytes (mela23, 35, and 41), primary melanoma cells (mel 4, 7, and 10), and melanoma cell lines (SkMel24, 624-28, and 624-38) and monitored the expression of chaperones (grp94, edem, and bip), pro-apoptotic mediators (bbc3, noxa, and bim), and transcription factors (s-xbp1, atf4, and ddit3) at steady state. We observed that the spliced form of the xbp1 transcript was expressed more in melanoma cell lines than that in melanocytes (Figure S6). This observation led us to assess whether the inhibition of XBP1 in tumor cells could affect the presentation of ER-stressed peptides. CTL-Vax showed a high cytotoxicity in response to 624-38 melanoma that was reduced once tumor cells were pre-treated with 4μ8c (Figure 4F) without a concomitant alteration in tumor-HLA expression (Figure 4G). Melanocytes did not present antigens to the CTL-Vax (mela23, Figure 4F). This result suggests that UPR activation at the steady state impacts antigen presentation and indicates that the antigens we identified are presented by ER-stressed cells and not by healthy melanocytes.
Identification and characterization of peptides specifically secreted upon Salmonella infection of human melanoma primary and established cell lines
To determine the nature of the immunogenic peptides, we compared by MS the secretomes of the melanoma cell line 624-38 infected or not with Salmonella. We also searched for peptides with common PTMs. From both datasets (Salmonella-treated and untreated cells), we identified 314 peptides in their native form and/or with PTMs. Consistent with the finding that Salmonella infection fosters the secretion of peptides, we detected a larger number of peptides in the secretome of the Salmonella-infected 624-38 cell line than in the non-infected cell line (Table S2; Figure S7A). We observed an over-representation of peptides derived from the N terminus of the cognate proteins, suggesting that they are generated during protein synthesis (Figure S7B). The peptides identified in both datasets have similar characteristics in terms of length, hydrophobicity, and isoelectric point (Figure S7C), suggesting that they derive from similar generation pathways or from proteins that share common characteristics, or both. Accordingly, proteins represented in the secretomes by these peptides shared similar length, hydrophobicity, and isoelectric point distribution (Figures S7D and S7E). Through the application of the same MS-based pipeline, we evaluated the secretome of primary melanoma cells, which were infected or not with Salmonella. We identified 4,212 peptides in their native form and/or with PTMs derived from the melanoma of 3 human patients (Table S2; Figure 5A). Among the thousands of peptides detected only in the secretomes of Salmonella-infected melanoma primary cell cultures, 28 were present in specimens of all 3 patients. Similar to the 624-38 cell line, all peptides identified and the proteins from which they were derived had similar characteristics in terms of length, hydrophobicity, and isoelectric point, and there was an over-representation of the peptides derived from the N terminus of the cognate proteins (Figures 5B–5E). A Panther pathway enrichment analysis did not show any specific pathway to be over-represented in the cognate proteins from which the peptides were derived (data not shown). Having observed a clear antitumor response also in dogs receiving a heterologous vaccine, we proceeded in the identification of the sequences of the most abundant and shared peptides and in the validation of their immunogenicity; for these purposes, we excluded peptides targeted by PTMs, modifications that are indeed peculiar of tumor cell metabolism and are hardly shared among patients. Out of 135 peptides overrepresented in the 624-38 cell line secretome, we focused on the 95 peptides with no PTM; and to identify shared peptides, we compared them with those released by patient melanoma cells following the pipeline depicted in Figure 6A. As further selection criteria, we considered HLA-binding prediction for the most frequent HLA supertype representatives, namely, HLA-A∗02:01, -A∗03:01, -B∗07:02, -B∗14:02, -B∗40:01, and -B∗58:01. We selected 11 peptides shared with at least 1 patient-derived cell line and containing epitope candidates with a predicted HLA-I-peptide binding affinity (IC50) less than 1,000 nM. Two additional peptides uniquely released by the 624-38 cell line were selected for their efficient HLA binding ability, for a total of 13 candidates (Figure 6B). All 13 peptides were synthesized and, when tested separately, 12 of 13 were able to induce IFN-γ production by CTL-Vax, confirming not only their presence within the Vax but also their immunogenic potential (Figure 6C) that could be further exploited as an anti-cancer vaccine. CTL-Vax was then stimulated twice with irradiated-PBMCs pulsed with a mix of the 13 selected peptides to increase the frequency of peptide-specific CD8+ T cells (CTL-MIX) and eventually were co-cultured with either melanoma cell lines 624-38 (HLA-A∗02:01 proficient) or 624-28 (HLA-A∗02:01-deficient) or primary melanocytes mela23 (HLA-A∗02:01 proficient) and mela41 (HLA-A∗02:01 deficient). CTL-MIX did not express IFN-γ in the presence of primary melanocyte cells, indicating that the selected peptides are not effectively presented by normal melanocytes but are by tumor cells (Figure 6D), further granting their safety and clinical application.
Figure 5.

Characteristics of melanoma primary cell secretome
(A) Frequency of peptides identified only in the secretome of Salmonella-infected (Salm) or untreated (ut) melanoma primary cultures or present in both (common).
(B) Distribution of length, hydrophobicity, and isoelectric point of peptides.
(C) Relative distribution of the secretome’s peptides within the cognate protein sequence.
(D) Frequency of proteins having at least one peptide identified in the secretome.
(E) Distribution of length, hydrophobicity, and isoelectric point of proteins.
In (B)–(E), unique peptides or proteins identified in at least one melanoma patient secretome are reported. In (B) and (E), the violin plots indicate the frequency of the peptides/proteins, with all single peptides/proteins indicated as dots. Red lines indicate the median. See also Figure S7 and Table S2.
Figure 6.

Identification of peptides released by human melanoma cells upon Salmonella infection
(A) Strategy to identify candidate antigens in secretome derived from Salmonella-infected melanoma cells.
(B) List of the selected antigen candidates.
(C) ELISA of IFN-γ released by CTL-Vax upon stimulation with single peptides. Results are representative of 5 independent experiments on CTL-Vax derived from 2 different donors.
(D) Frequency of CD3+CD8+IFN-γ+ CTL-MIX upon co-culture with target cells. Pan HLA-blocker (anti HLA) antibody was added, where indicated.
Data of 2 pooled experiments are represented as mean ± SEM using bar plot. Statistical analysis was evaluated using one-way ANOVA followed by multiple comparisons (Dunnett). ∗p < 0.05,∗∗p < 0.01. See also Table S2.
Discussion
In this work, we provide evidence that Salmonella infection of tumor cells promotes the release of immunogenic peptides capable of inducing antitumor responses in several model systems, providing a rationale for a novel immunotherapy approach.
Starting from a previous finding of our laboratory (Saccheri et al., 2010) that showed that Salmonella infection of mouse melanoma cells promoted a GJ-mediated transfer of immunogenic peptides from tumor cells to adjacent DCs, we proved here that the same stimulus induces the release of immunogenic peptides in the extracellular milieu through plasma membrane hemichannels and that this secretome is effective in inducing a protective antitumor response. When loaded on DCs, peptides released by melanoma cells have a stronger antitumor effect than 2 well-known B16F10-tumor antigens (Trp2180-188 gp10025-33), indicating that our vaccine formulation possibly contains antigens that are more immunogenic than those already known. Released peptides also induced a strong antitumor response in a DC-free vaccine immunotherapy strategy, both when administered in emulsion with IFA and imiquimod (TLR7/8 agonist) and in combination with CpG ODN 1826 (TLR9 agonist). The vaccine prompted both a humoral and a cell-mediated immune response, but only CD8 T cells were necessary for tumor control. The peptides that recapitulated T cell activation in vitro were identified with a peptidomic approach on secretomes derived from Salmonella-infected B16F10, suggesting that the omic approach we have set is a feasible strategy for the selection of immunogenic antigens. This is important when considering translation of this immunotherapeutic approach to a human clinical trial.
We also showed a proof of efficacy of the peptide-based cancer vaccine in dog patients affected by OSA or high-grade SA. Patients with OSA receiving a combination of vaccine and chemotherapy had a significantly longer OS than patients receiving chemotherapy alone. Vaccinated dogs developed both a vaccine-specific T-cell-mediated response and a tumor-directed humoral response. A total of 5 of 10 patients with OSA were vaccinated with peptides released by the tumor cells of another patient with OSA, suggesting the existence of shared tumor antigens between tumor cells of the same histology. Furthermore, 2 of 3 enrolled patients with high-grade SA (SA5 and SA35) had a long OS and SA35 is still alive 3 years from diagnosis. Although the number of patients included in the trial is low (10 vaccinated and 10 control dogs), we indicated that a promising clinical response was induced by the vaccine and that both autologous and heterologous settings were feasible and safe. The inclusion of a higher number of patients in both cohorts will be important to further validate the results of the trial.
The immunogenicity of peptides released by tumor cells upon Salmonella infection was further demonstrated in the human system by their ability to induce the expansion of tumor-specific CD8+ T cells from healthy donor PBMCs. These peptide-specific lymphocytes responded to 5 of 5 melanoma cells, in an HLA-dependent manner, by both producing IFN-γ and recruiting CD107a. CTL specific for a single peptide (CTL-Mart1) could exert a cytotoxic effect to a limited number of melanoma cells (3 of 5), thus showing the advantage of our multi-peptide vaccine strategy. Although our goal is to propose a peptide-based anti-cancer vaccination strategy, we also showed that expanded T cells were fit enough to kill melanoma cells when xenotrasplanted in vivo; this is an important indication of the T cell response that peptides could generate in vivo upon vaccination. Moreover, these tumor-specific CD8+ T cells were activated by 12 of 13 peptides identified as specifically released upon Salmonella infection, thus confirming the immunogenicity of the selected peptides and indicating that there are circulating lymphocytes capable of recognizing these peptides in healthy individuals and implying that T cells are not centrally tolerized to at least 12 of these peptides. It remains to be analyzed whether the 13th peptide is not immunogenic or is tolerized. Even though we showed that peptides could activate an HLA-I-dependent immune response, a peptidomic analysis directly on immune-precipitated HLA-I was never performed to ultimately verify peptide presentation on HLA-I molecules.
The peptides we identified are not patient specific but are shared among several patients. They have been selected for their HLA-binding ability and can be considered as a “signature” of antigens that could be used as a universal melanoma vaccine treatment. They share some characteristics with tumor epitopes associated with impaired peptide processing (TEIPPs; Seidel et al., 2012). For instance, these peptides are derived from housekeeping proteins and are also presented in cells devoid of the transporter associated with antigen processing (TAP) protein (such as the B16F10 melanoma cell line). Furthermore, these peptides, similarly to TEIPPs, are not presented by normal cells and can evade both central and peripheral immune tolerance (Oliveira et al., 2011; Lampen et al., 2010). Indeed, an issue to consider once proposing a peptide-based anticancer vaccine strategy is the autoimmunity that might be induced following immunization. Peptides might boost an aberrant immune response against endogenous HLA-associated peptides instead of inducing an antitumor-specific immune response. None of the mice vaccinated with peptides released by Salmonella-infected B16F10 cells developed vitiligo (white skin patches that are due to the disappearance of melanocytes). Moreover, none of the dog patients developed autoimmunity or serious adverse events. Furthermore, CTLs expanded with the MIX of released human peptides identified by MS did not react against melanocytes, indicating that the selected peptides are expressed by tumor cells only. This is probably related to their presentation in the context of an ER-stressed cell, which is not common in healthy cells, which is a further advantage compared to Mart1-specifc CTLs that are known to lyse melanocytes (Lang et al., 2001).
The peptides released through Salmonella-induced GJ hemichannel opening were generated by the tumor proteasome, as they were not released when tumor cells were treated with a proteasome inhibitor. This has enormous implications. Indeed, because DCs express a different type of proteasome (immunoproteasome), they might present peptides that do not necessarily correspond to the ones presented by tumor cells, thus failing to prompt an effective antitumor response (Chapiro et al., 2006; Dannull et al., 2007). In contrast, peptides generated by the tumor proteasome were also presented by tumor cells, as demonstrated by the ability of CTL-Vax to recognize and kill melanoma cells and by the strong humoral and cellular anti-tumor response elicited in vivo in vaccinated mice and dogs.
The mechanism by which Salmonella infection induced tumor cells to release peptides was elucidated. Indeed, we found that Salmonella exacerbated an already activated UPR response (Cerezo et al., 2016; Corazzari et al., 2017). We showed that either the inhibition of the splicing activity of IRE-1α with 4μ8c or the CRISPR-Cas9-mediated depletion of the XBP1 protein resulted in the inability of tumor cells to release HLA-binding peptides in the secretome and reduced the capacity of restimulating IFN-γ-producing T cells. At the same time, activation of the UPR alone without GJ-hemichannel opening could not recapitulate peptide release, thus suggesting that both pathways need to be prompted. The link between a UPR response and antigen presentation has already been investigated (Osorio et al., 2018). The effect of the UPR response and in particular IRE-1α activation and XBP1 splicing in DCs has been shown to depend on the type of DCs analyzed (Osorio et al., 2014). CD8a+ DCs lose their antigen-presenting capacity in the absence of XBP1, whereas CD11b+ DCs are unaffected (Osorio et al., 2014). The UPR response can reduce the expression of TAP proteins that are required for the translocation of peptides into the ER (Bartoszewski et al., 2011) and can inactivate tumor-infiltrating T cells (Song et al., 2018). In addition, we showed that inhibition of the splicing activity of IRE-1α with 4μ8c in tumor cells at steady state change the profile of HLA-presented antigens. These observations indicate that ER stress and UPR response may be another mechanism of immune evasion adopted by tumor cells to avoid the presentation of immunodominant peptides. However, if correctly exploited, it may be used to activate a potent antitumor response as it generates ER-stressed derived immunogenic peptides (ERstrePs), which are not generally presented by non-ER-stressed healthy cells as shown in melanocytes. We also found that most of the peptides belonged to the N terminus region of the corresponding proteins. A recent report showed that N-terminus-derived peptides may result from prematurely terminated nascent proteins, which are the most frequently presented by highly stressed cells such as cancer cells (Imai et al., 2019).
In conclusion, we demonstrated that infection of tumor cells with Salmonella induced the extracellular release of ERstrePs that could be successfully exploited as cancer vaccines for the prevention or treatment of tumors. The safety, feasibility, and efficacy of the protocol were assessed by the implementation of a vaccine in veterinary clinical practice for the treatment of dogs affected by spontaneous OSA and high-grade SA both in autologous and heterologous regimens. Patients experience a clear clinical benefit, thus paving the way to shared vaccine formulations. An ERstreP-based universal vaccine would lead to an off-the-shelf vaccination treatment that could be administered to patients soon after tumor resection, which would rapidly boost an antitumor response directed against micro-metastases that are often not clinically visible at the moment of primary tumor diagnosis. This may be even more beneficial in a metastatic setting to avoid undergoing long procedures of peptide identification and personalized vaccination. To enhance the efficacy of the cancer vaccine, it will be pivotal to consider a combination with ICB to improve patient clinical benefit.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-human/anti-mouse CX43 | Abcam | Cat #ab11370; AB_297976 |
| anti-human XBP1s | Cell Signaling | Cat #12782S; AB_2687943 |
| anti-human/anti-mouse/anti-canine Vinculin | Sigma | Cat #V9131; AB_477629 |
| anti-canine CX43 | Thermo Fisher Scientific | Cat #13-8300; AB_2533038 |
| Anti-human CHOP | Cell Signaling | Cat # 2895S; AB_2089254 |
| Anti-mouse CD8 (clone YTS 169.4) | BioXcell | Cat # 53-0341-82; RRID:AB_2866440 |
| Anti-mouse CD4 (clone GK1.5) | BioXcell | Cat # 560230; RRID:AB_1645200 |
| rat IgG2a anti-trinitrophenol (clone 2A3) | BioXcell | Cat # ab185230; RRID:AB_2715497 |
| Fixable Viability Stain 510 | BD Biosciences | Cat # 564406; RRID:AB_2869572 |
| Goat anti-canine IgG (H+L) FITC | Invitrogen | Cat # A18764; RRID:AB_2535541 |
| Anti-Human PAN HLA | InVivoMab | Cat #BE0079; RRID:AB_1107730 |
| Anti-Human HLA-A∗02:01 PE | Biolegend | Cat # 343306; RRID:AB_1877227 |
| Anti-human CD3 V450 | BD Biosciences | Cat # 560366; RRID:AB_1645569 |
| Anti-human CD8 PerCP | Biolegend | Cat # 344708; RRID:AB_1967149 |
| Anti-human CD4 FITC | BD Biosciences | Cat # 555346; RRID:AB_395751 |
| Anti-human IFNγ APC | BD Biosciences | Cat # 551385; RRID:AB_398505 |
| Anti-human CD107a PE-Cy7 | Biolegend | Cat # 328618; RRID:AB_11147955 |
| Bacterial and virus strains | ||
| S. typhimurium SL3261AT | This paper | N/A |
| Vivofit® (Thyphoid vaccine live oral Ty21a) | This paper | N/A |
| Biological samples | ||
| PBMC isolation from Buffycoat (approved by the Ethics Committee of Humanitas Clinical and Research Center License No. Humanitas01/2016) | Humanitas Clinical and Research Center | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Heptanol | Sigma-Aldrich | Cat # 51778 |
| MG132 | Sigma-Aldrich | Cat # 474790 |
| 4μ8c | Sigma-Aldrich | Cat # 412512 |
| Brefeldin A | Biolegend | Cat # 420601 |
| Thapsigargin | Sigma-Aldrich | Cat # T9033 |
| Freund’s Incomplete Adjuvant (IFA) | Sigma-Aldrich | Cat # F5506 |
| Aldara 5% Cream (Imiquimod) | Meda AB | N/A |
| ODN1826 | Invivogen | Cat # vac-1826-1 |
| Epoxomycin | Sigma-Aldrich | Cat # E3652 |
| CMV peptide | JPT Peptide Technologies | Cat# PM-PP65-1 |
| Mart126-35 | Iba Lifesciences | Cat# 6-7007-901 |
| IL2 (Proleukin) | Clinigen Healthcare B.V. | N/A |
| RIPA | Cell Signaling | Cat # 9806S |
| Phosphatase Inhibitor | Sigma-Aldrich | Cat # P5726 |
| Protease Inhibiotors | Roche | Cat # 05892988001 |
| Gentamicin Sulfate | Euroclone | Cat# ECM0012B |
| Concanavalin A | Sigma-Aldrich | Cat# C2010 |
| Critical commercial assays | ||
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat # G7572 |
| DuoSet ELISA human IFNg | R&D Systems | Cat # DY285B |
| DuoSet ELISA mouse IL-2 | R&D Systems | Cat # DY402; RRID:AB_2797392 |
| Canine IFNγ ELISA | R&D Systems | Cat # CAIF00 |
| PE Annexin V Apoptosis Detection Kit I | BD PharMingen | Cat # 559763; RRID:AB_2869265 |
| SPE C18 devices | Macherey-Nagel | Cat# MANA730003.250 |
| T Cell TransAct | Miltenyi Biotec | Cat #130-111-160 |
| Delfia-BATDA | Perkin Elmer | Cat # AD0116 |
| fluorometric proteasome 20S assay kit | Sigma-Aldrich | Cat # MAK172-1KT |
| XbpI human gene knockout kit | Origene | Cat# KN401959 |
| XbpI mouse gene knockout kit | Origene | Cat# KN519483 |
| G’NOME DNA isolation kit | MPBiomedicals | Cat # 112010600 |
| Direct-zol RNA MiniPrep Plus Kit | Zymo Research | Cat# R2072 |
| Deposited data | ||
| Original data, Peptidomic Datasets, R-scripts | This paper | Zenodo Repository https://zenodo.org/record/4738409#.YJJEIqHONaQ |
| Experimental models: cell lines | ||
| B16F10 | Saccheri et al., 2010 | N/A |
| B16F10-XBP1KO | This paper | N/A |
| B16F10-OVA | Saccheri et al., 2010 | N/A |
| B16F10-OVA-CX43KO | Saccheri et al., 2010 | N/A |
| B3Z-Hybridoma | Rotta et al., 2003 | N/A |
| DC1 | Rotta et al., 2003 | N/A |
| 624-38 | Sabatino et al., 2008 | N/A |
| 624-38 XBP1KO | This paper | N/A |
| 624-28 | Sabatino et al., 2008 | N/A |
| HT29 | This paper | N/A |
| SkMel24 | Saccheri et al., 2010 | N/A |
| A375 | This paper | ATCC-CRL-1619; RRID:CVCL_0132 |
| MeWo | This paper | ATCC-HTB-65; RRID:CVCL_0445 |
| C32 | This paper | ATCC-CRL-1585; RRID:CVCL_1097 |
| hMel2 | This paper | N/A |
| hMel3 | This paper | N/A |
| T2 | This paper | N/A |
| THP1 | This paper | N/A |
| Mela23, Mela41 | This paper | N/A |
| Experimental models: organisms/strains | ||
| Mouse: C57BL/6J | Charles River | Cat # 027 |
| Mouse: C57BL/6-Tg(TcraTcrb)1100Mjb/Crl (OTI) | Charles River | Cat # 642 |
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl /SzJ (NSG) | Jackson Laboratories | Cat # 005557 |
| Oligonucleotides | ||
| Primers, see Table S3 | This paper | N/A |
| Software and algorithms | ||
| MaxQuant (version 1.5.5.1) | MaxQuant | https://www.maxquant.org/ |
| Perseus (version 1.5.0.31) | Perseus | https://maxquant.net/perseus/ |
| Image Lab | Biorad | http://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
| FlowJo v10.5.3 | TreeStar | https://www.flowjo.com/solutions/flowjo; RRID:SCR_008520 |
| Prism | GraphPad | https://www.graphpad.com:443/ |
| Other | ||
| Count Bright Absolute Counting Beads | Invitrogen | Cat # C36950 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Maria Rescigno (maria.rescigno@hunimed.eu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
Original data, Peptidomic datasets and R-scripts generated during this study have been deposited in Institutional Zenodo community named IRCCS Humanitas Research Hospital & Humanitas University and are available under request https://zenodo.org/record/4738409#.YJJEIqHONaQ. The MS RAW files from which human and murine peptides have been identified (listed in Table S2) are available from the corresponding author on request.
Experimental model and subject details
Dog patient enrollment and vaccination procedure
The study population consisted of 14 treatment-naive dogs, 11 affected by OSA and 3 affected by high-grade (HG) SA. No age, sex, breed or weight limits were applied. Prior to enrollment, dogs underwent diagnostic tests including CT scan or thoracic radiographs and abdominal ultrasound, complete blood count (CBC), serum biochemistry profile and urinalysis. Dogs with histologically confirmed appendicular OSA and HG SA without evidence of metastatic disease were enrolled in this study after undergoing surgery. All dogs were then treated with 4 cycles of single agent carboplatin (300 mg/m2 IV) every three weeks within 14 days of surgery (week 1, 4, 7, 10; schedule described in Figure 2A). Following the third carboplatin administration, dogs were re-evaluated with thoracic radiographs and a first dose of vaccine was administered. A total of 6 intradermal injections were scheduled at week 8, 11, 15, 19, 23, 27. In detail, 2 Control-immunizations (supernatant derived from untreated cells) were performed followed by 4 Vax-immunizations (supernatant derived from Salmonella treated cells). Only for the first and the second immunization lyophilized-vaccine was resuspended in Nobivac (Leptospirosis vaccine chosen as adjuvant) instead of water; all 6 immunizations were injected intradermally following Aldara cream application at the vaccine-injection site. CBC, biochemistry profile, blood draw for both PBMC isolation and serum collection were performed concomitantly with vaccinations I, II, III and VI. Restaging with thoracic radiographs was performed every 8 weeks after completion of vaccine protocol. Delayed type hypersensitivity (DTH) test was performed at week 15. For DTH test, water, Nobivac, Control and Vax were injected intradermally and the diameter of any resulting erythema or induration was measured after 48-72 hours.
Ethics statement
The owners of the 14 dogs recruited for the vaccine treatment were thoroughly informed about the entire procedure and signed an informed consent. Owners also agreed on serial blood collection for PBMC isolation and sera analysis at the time of I, II, III and VI vaccination.
Mice
6-week-old WT C57BL/6J female mice were purchased from Charles River (Calco, Italy). OTI mice (C57BL/6-Tg(TcraTcrb)1100Mjb/Crl) female/male of any age were used to isolate splenocytes. NOD.Cg-Prkdcscid Il2rgtm1Wjl /SzJ (NSG) mice (Jackson Laboratories), 4 week old females, bred in SPF conditions, were used for adoptive transfer experiments. All experiments were performed in accordance with Italian regulations (D.Lgs. 26/2014) and EU guidelines (2010/63/EU).
Cell lines, chemical compounds, and bacterial strains
The murine melanoma B16F10, the murine T cell hybridoma B3Z (whose TCR is restricted for H-2Kb-bound OVA(257–264) peptide) and T2 cells (HLA-A∗02:01 hybrid human cell line lacking TAP-2) were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum South America origin (FBS), 2 mM glutamine, 1x penicillin-streptomycin solution, and 50 mM β-mercaptoethanol (complete RPMI). Murine melanoma B16F10-OVA and B16F10-OVA-Cx43ko were cultured in Iscove’s modified Dulbecco’s medium (IMDM) medium supplemented with 10% FBS, 2 mM glutamine, 1x penicillin-streptomycin solution. Human melanoma cell lines 624-38 (HLA-A∗02:01 proficient), 624-28 (HLA-A∗02:01 deficient), SkMel24, the human colon cancer cell line HT-29 and the human acute monocytic leukemia THP1 cell line were cultured in RPMI 1640 medium supplemented with 10% FBS U.S. origin, 2 mM glutamine, 1x penicillin-streptomycin solution, 1% non-essential amminoacids (NEAA), 1% Sodium Pyruvate (NaPy). The same medium was used to grow the 3-patient-derived melanoma cells (Mel1,4,7). The immature DC line DC1 was cultured in IMDM supplemented with 30% conditioned supernatant derived from murine granulocyte-macrophage colony-stimulating factor (GM-CSF) producing NIH 3T3 cells, containing 10 to 20 ng/ml mouse GM-CSF (Winzler et al., 1997). PBMCs and CTLs were cultured in RPMI supplemented with 5% human serum, 2 mM L-Glutamine, 1x penicillin-streptomycin solution, 1% NEAA, 1% NaPy. Primary melanocytes mela23 and mela41, kindly provided by Giuliana Pelicci (European Institute of Oncology, Milan, IT), were grown in McCoy’s 5A supplemented with 2 mM glutamine, 2% FBS U.S. origin, 1x penicillin-streptomycin solution, 20 pM Cholera Toxin (Sigma), 5 ug/ml Insulin (Roche), 0.5 ug/ml Hydrocortysone (Sigma), 5 ug/ml Transferrin (Sigma), 10 ng/ml stem cell factor (SCF) (Peprotech), 1 ng/ml bFGF (Peprotech), 10 nM Endothelin-1 (Sigma).
Hemichannel blocker Heptanol (1 mM), proteasome inhibitor MG132 (15 μM) and UPR-inhibitor 4μ8c (10 μM), ER-stress response inducer brefeldin A (BFA, 20 nM), Thapsigargin (0.25 μM) were all purchased from Sigma. To generate supernatant from dead cells (CD) a lethal dose of Thapsigargin (40 μM) was used.
S. Typhimurium SL3261AT is an aroA metabolically defective strain on SL1344 background and is grown at 37°C in Luria broth (LB).
Vivofit® (Thyphoid vaccine live oral Ty21a) is a vaccine containing the attenuated strain of Salmonella enterica serovar Typhi Ty21a and is grown at 37°C in Luria broth.
Mice immunization
For preventive vaccination experiment, C57BL/6J mice were injected subcutaneously with a combination of cells’ supernatant and adjuvant for a final volume of 100 μl in their left flank. Supernatants were concentrated by chromabond SPE C18 devices (Macherey-Nagel) and combined with different types of adjuvants according to the in vivo test. IFA-A: Aldara cream (Imiquimod, MEDA) was applied at the immunization site until dried; supernatant emulsified with Incomplete Freud’s Adjuvant (IFA, Sigma) at a 1:1 ratio. ODN1826: 10 μg of ODN1826 (Invivogen) was combined with the supernatant derived by 2 × 106 cells and inoculated. DCs: DC1 cells were incubated for 2 hours with LPS (1 μg/ml) at 37°C and then for another 2 hours either with the supernatant derived by 2 × 106 B16F10 cells or with a mix of Trp2180-188 and gp10025-33 (1 mg/ml each) peptides. 3.5 × 105 DC1 cells were injected per mouse. Preventive vaccination was performed at day 0, 4, 11. 105 B16F10 cells were injected subcutaneously in the right flank of the mice, at day 40 or at day 25. Tumor growth was monitored by measuring the two visible dimensions with a caliper every 2 days. The monoclonal antibodies anti-CD8 (clone YTS 169.4), and anti-CD4 (clone GK1.5) and both rat IgG2a anti-trinitrophenol (clone 2A3) and Rat IgG2b anti-keyhole limpet hemocyanin (clone LTF-2) that were used as isotype controls were all purchased from BioXcell.
Adoptive T cells transfer
7 week old NSG mice were subcutaneously injected with 1 × 106 624-38 cells in their right flank. After one week, tumors became palpable and 8 × 106 antigen-specific CD8+ T cells, expanded in vitro with T Cell TransAct (Miltenyi Biotec) and resuspended in 200 μl PBS, were transferred by intravenous injection. Tumor growth was monitored by measuring the two visible dimensions with a caliper every 2 days.
Method details
In vitro infection with bacteria
Single bacterial colonies were grown overnight and restarted the next day to reach an absorbance at 600 nm of 0.6 corresponding to 0.6 × 109 colony-forming units (CFUs)/ml. Murine and human melanoma cells (2 × 106 cells/ml) were incubated with bacteria for 90 minutes, in tubes, at a cell-to-bacteria ratio of 1:50, in the appropriate medium with L-Glutamine without antibiotics. Cells were washed and incubated in medium supplemented with gentamicin (50 mg/ml), in tissue culture plates, for 18 hours to kill extracellular bacteria. To test proteasome and UPR function, MG132 (MG, 15 μM) and the IRE1-inhibitor 4μ8c were used during the infection protocol, respectively. In detail, cell lines were pre-treated for 1 hour with either MG or 4μ8c; then cells were infected with Salmonella (1:50 cell:bacteria ratio) for 90 minutes in the absence of inhibitors. Afterward, medium was replenished with fresh medium with gentamicin and MG/4μ8c and left for an overnight incubation. At the end of the incubation, cells were harvested and then lysed with RIPA buffer (Sigma) for protein analysis, while supernatant was collected and filtered through 0.22 μm filter to get rid of remaining potentially live bacteria.
ER stress response induction
Murine and human melanoma cells were incubated, in the appropriate medium with L-Glutamine without antibiotics, with Thapsigargin (Tg, 0.25 μM) or with BFA (20 nM) for 90 minutes in tubes. Cells were washed and incubated in plates at a concentration of 2 × 106 cells/ml for 18 hours. At the end of the incubation, cells were harvested and then lysed with RIPA buffer (Sigma) for protein analysis, while supernatant was collected. To induce cell death, tumor cells were treated with Thapsigargin (40 μM) for 4 hours. Cell viability was tested by Annexin/PI staining (BD).
HLA-A∗02:01-peptide binding assay
T2 cells are HLA-A∗02:01 restricted lymphoblastic cells that are deficient for the transporters associated with antigen processing (TAPs) (Durgeau et al., 2011; Leone et al., 2013) leading to the translocation of unstable HLA-I (partially empty) complexes to the cell surface. These can be stabilized in the presence of exogenous peptides (Hosken and Bevan 1990). To assess peptide enrichment inside tumor cells’ supernatant, T2 cells were incubated 2 hours or overnight at 37°C at 2x105 cells/well in serum-free RPMI medium with either 100 μL of supernatant or Mart-126-35 peptide (Iba Lifesciences, 1 μM and 10 μM) as a positive control. After incubation cells were stained with an HLA-A∗02:01 conformation-specific mouse antibody (BD, clone BB7.2).
Antigen specific-CD8+ T cells expansion from healthy donor PBMCs
Total PBMCs isolated from healthy HLA-A∗02:01 donors were loaded with either supernatant derived from 2 × 106 624-38 cells treated with Salmonella, or 20 μM Mart-126-35 (Iba Lifesciences) or CMV peptides (pp65, JPT Peptide Technologies) in rotation at 37°C for 90 minutes. Cells were then plated in complete medium in 24-well plates (2 × 106 cells per well) in a final volume of 2 ml. Starting from day 3, every 2-3 days we added recombinant IL-2 (Proleukin, Novartis) at a final concentration of 20 U/mL. Cells were restimulated every 10 days; expanded lymphocytes were enriched in CD8+ T cells by magnetic column separation (Miltenyi Biotec) and stimulated with irradiated (10 Gy) HLA-A∗02:01 PBMCs that were pulsed with either Mart-1 or CMV peptides-mix, or supernatant from 624-38 cells infected with Salmonella. To pulse, PBMC were incubated for 90 minutes at 37°C in RPMI supplemented with the selected stimulus.
Adenosine5′-triphosfate (ATP) bioluminescent assay
Cell-Titer-Glo Luminescent cell viability Assay (Promega) was used following manufacturer’s instruction. Briefly, supernatants were distributed in 96-well plates and then rapidly mixed with an equivalent volume of MIX-assay solution. Light emission was detected and quantified by a luminometer.
OTI-CD8a+ activation assay
Murine DCs were incubated for 4 hours at 104 cells/well in a 96-well plate with supernatants derived from Salmonella-infected B16F10-OVA cells and from B16F10 cells as control. Cells were then washed twice and incubated for 72 hours with CD8a+ cells isolated from the spleen of OTI mice. CD8a+ cells were obtained using a magnetic column separator device (MACS, Miltenyi Biotec). IFN-γ production in cells’ supernatant was assessed by ELISA (R&D).
B3Z activation assay
Murine DCs were incubated for 4 hours at 104 cells/well in a 96-well plate with supernatants derived from Salmonella-infected B16F10-OVA cells or untreated B16-OVA cells. Cells were then washed twice and incubated for 72 hours with 4 × 104 T hybridoma cells B3Z. mIL2 production in cells’ supernatant was assessed by ELISA (R&D).
CD107a mobilization assay
In this test, human in vitro-expanded CD8 cells or total murine-PBMCs were incubated with target human or mouse tumor cells, respectively. Anti-CD107a monoclonal antibody conjugated with APC (BD), brefeldin A 10 μg/ml (BD) and GolgiStop containing monensin (BD), the HLA-I pan blocker as control (10 ug/ml, Clone W6/32, BioXcell) were added to the cell culture for a final volume of 200 μl and incubated for 5 h at 37°C. Cells were then stained with anti-CD3a, anti-CD8, and anti-CD4 (BD) antibodies. Intracellular staining for IFN-γ (BD) production was also performed in order to further assess T cell activation.
IFN-γ assay for peptide recognition by CTLs
Human acute monocytic leukemia Thp1 cell line was loaded either with antigens at different concentrations (20 uM-1.25 uM) or with conditioned medium and used to stimulate CTL-Vax (ratio 1:1). Cells’ supernatant was collected after 72 hours and IFN-γ production was measured by ELISA (R&D). IFN-γ released by CTLs cocultured with unloaded Thp1 (spontaneous IFN-γ release) was subtracted from IFNγ measured upon antigen stimulation. Activation specificity was further assessed using an HLA-I pan blocker as control (10 μg/ml, Clone W6/32, BioXcell).
IFN-γ ELISpot assay
96-well PVDF membrane plates were pre-treated with 15 μL of 35% EtOH for 30 s, washed 5x with sterile water and coated with 100 μL/well murine IFNγ capture antibody at 12 μg/mL working concentration. After overnight incubation, plates were washed 5x with sterile PBS and blocked for 1 hr in complete culture medium. Murine splenocytes were plated at 4 × 105/well with 1 μM peptide mix. After 24-hour incubation, cells were removed and spots reveal was assessed following standard procedures.
Delfia cytotoxicity test
Target cells (tumor cell lines or melanocytes) were collected, washed once and resuspended at a concentration of 106 cells/ml. Cells were loaded with 3 μl of Delfia-BATDA (DELFIA, Perkin Elmer) reagent and incubated at 37°C for 30 min. Following 4 washes with PBS and 1 wash with medium without serum, 5 × 103 cells were seeded in a v-bottom plate and incubated with effector PBMCs for 90 min at 37°C. Cytotoxicity was then assessed following manufacturer’s instruction (DELFIA, Perkin Elmer).
Proteasome activity
The chymotrypsin-like proteasome activity associated with the proteasome complex was assessed with a fluorometric proteasome 20S assay kit (Sigma). Briefly, after treatment with MG132 and Epoxomycin, cell lines were washed twice and 105 cells/well were plated into a 96-well plate in 100 μl of fresh medium. 100 μl of Proteasome Assay Loading Solution was added to each well and cells were left for 1 hour at room temperature. Acquisition was performed following manufacture’s instruction.
Protein-lysate preparation and immunoblots
Upon treatment, cells were harvested with trypsin; RIPA buffer (Sigma) was used as lysis buffer in the presence of protease inhibitors (cOmplete Mini, EDTA-free, Sigma) and tyrosine protein phosphatases, acid and alkaline-phosphatases inhibitors (Phosphatase Inhibitor Cocktail 2, Sigma). Cell lysates were freshly prepared and equal amounts of proteins, measured using Bradford assay (Bio-Rad), were run on SDS–PAGE followed by western blotting. After 30 min at room temperature in blocking solution (5% milk or 5% bovine serum albumin (BSA) in tris-buffered saline and Tween-20 (TBST) (10 mM Tris, pH 7.5, 150 mM NaCl, 0.1% (v/v) Tween-20)) membranes were probed with primary antibodies in 5% milk (or 5% BSA) in TBST overnight at 4°C, washed in TBST and incubated for 1 h at room temperature with secondary antibodies goat anti-rabbit-HRP (1:10,000, Bio-Rad) or goat anti-mouse-HRP (1:10,000, Bio-Rad). Primary antibodies used are as follows: XBP1s (1:1,000, Cell Signaling), Chop (1:1,000, Cell Signaling), CX43 (1:1,000, Sigma) and Vinculin (1:10,000, V9131, Sigma). Visualization was carried out with enhanced chemiluminescence (Clarity Western ECL substrate, Bio-Rad). Densitometric quantification was performed using ImageJ software.
RNA extraction, RT–PCR and qPCR
Total RNA was isolated using RNeasy Mini Kits (QIAGEN) according to the manufacturer’s instructions, adding a DNase digestion step to eliminate genomic DNA contamination. 1 μg of RNA from each sample was reverse-transcribed using ImProm-II Reverse Transcriptase (Promega) and random primers (Invitrogen), according to the manufacturers’ protocol. Quantitative real-time PCR (qPCR) was carried out with Fast SYBR Green PCR kit using Applied Biosystems 7900HT Fast RT-PCR System (Applied Biosystems). The relative expression level was calculated with the 2[−ΔΔCt] method and expressed as a “fold change”: data were normalized to housekeeping gene (GAPDH) expression and compared to the untreated control. Both human and mouse primer list is reported in Table S3.
Murine humoral response
To assess tumor specific antibodies in mice serum, tumor cells were lysed by freeze and thaw and the lysate corresponding to 10,000 tumor cells was used to coat single wells of PVC microtiter plates (final volume of 50 μl). The coating was an overnight incubation at 37°C. Then, plates were blocked with 5% BSA for 1 hour at 37°C. After removing blocking buffer, mice sera diluted in PBS with 1% BSA was added in each well and incubate for 2 hours at 37°C. Standard ELISA protocol was then performed from this step.
Mass Spectrometry (MS) measurement
Supernatants components were concentrated using chromabond SPE C18 devices (Macherey-Nagel). Once eluted with 80% CH3CN in 0.1% Formic Acid (FA) solution, samples were dried by speed vacuum and resuspended in water. Proteins derived from 4 × 106 cells were sonicated with Bioruptor 30’’ ON + 30’’ OFF (2 cycles) and then loaded on AMICON Ultra 0.5 mL 10K (Millipore). Low molecular weight peptides were analyzed through n-LC-ESI-MS2 Q-Exactive HF. 4 μl of each sample were loaded at max pressure of 900 bar on a LC–ESI–MS-MS quadrupole Orbitrap QExactive-HF mass spectrometer (Thermo Fisher Scientific). Peptides separation was achieved on a linear gradient from 95% solvent A (2% ACN, 0.1% formic acid) to 50% solvent B (80% acetonitrile, 0.1% formic acid) over 33 min and from 50 to 100% solvent B in 2 min at a constant flow rate of 0.25 μl/min on UHPLC Easy-nLC 1000 (Thermo Fisher Scientific) where the LC system was connected to a 23-cm fused-silica emitter of 75 μm inner diameter (New Objective, Inc. Woburn, MA, USA), packed in-house with ReproSil-Pur C18-AQ 1.9 μm beads (Dr Maisch Gmbh, Ammerbuch, Germany) using a high-pressure bomb loader (Proxeon, Odense, Denmark). MS data were acquired using a data-dependent top 15 method for HCD fragmentation. Survey full scan MS spectra (300–1650 Th) were acquired in the Orbitrap with 60,000 resolution, AGC target 3e6, IT20ms. For HCD spectra, resolution was set to 15,000 at m/z 200, AGC target 1e5, IT 80 ms; Normalized Collision energy 28% and isolation with 1.2 m/z. Technical replicates were conducted on the LC-MS-MS part of the analysis.
MS analysis
Data derived from the analysis of both mouse and human-secretomes were controlled by Xcalibur 3.1. Mass spectra were analyzed using MaxQuant software (version 1.5.5.1). Search parameters were set to an initial precursor ion tolerance of 10 ppm and MS/MS tolerance at 20 ppm. Enzyme specificity was set to unspecific and methionine oxidation was set as variable modification. The spectra were searched using the Andromeda search engine in MaxQuant against the Uniprot mouse proteome database (version 2016) or the Uniprot human proteome database including isoforms (version 2016) setting Label-free peptide quantification based on extracted ion chromatograms with min. ratio count 1, PSM FDR and Protein FDR both 0.01 and the minimum required peptide length to 7 amino acids. The reversed sequences of the target database were used as decoy database. Comparative analyses were performed using the Perseus software (version 1.5.0.31). For proteomics analysis, mass spectra of mouse-secretomes and of the secretomes of human melanoma cell lines and patients were analyzed using the Mascot search engine. The precursor tolerance was set to 10ppm and MS/MS tolerance was set to 20ppm. The spectra were searched without enzyme specificity against the Uniprot mouse proteome database (version 2017) and the Uniprot human proteome database including isoforms (version 2016) for mouse-secretomes and human-secretomes, respectively. Methionine oxidation, deamidation (NQ), N-terminal acetylation and phosphorylation (STY) were set as variable PTMs. Although spliced peptides have been identified in cellular proteomes and immunopeptidomes (Faridi et al., 2019; Liepe et al., 2016, 2019; Mishto and Liepe 2017), they were not included in the analysis. Results were filtered for 1% FDR based on peptide spectrum matches (PSMs) using a target decoy approach with a randomized decoy database (Table S2).
Derivation of primary canine osteosarcoma/sarcoma cells
Both primary canine osteosarcoma and sarcoma cells were obtained from dissociation of dog tumor specimens. Tissues were minced with a scalpel in a cell strainer. Cells were washed with complete DMEM and filtered with a cell strainer (100 μm). Cells were counted and cultured at high concentration in complete medium.
Dog patients’ vax-specific T cell response
Dog PBMCs were thawed, counted, resuspended in RPMI complete medium, and plated into a 96-well flat bottom plates in the presence of 1 μg/ml of ConA (Sigma). Every other day IL-2 (100 U/ml, Proleukin, Novartis) was added. After 7-10 days cells were washed twice and plated in a 96-well plate, 2x105cells/well, with or without the Vax (the same used to immunize dog-patient). IFN-γ released by the cells was measured after 72 hours by ELISA (canine IFN-γ, R&D).
Dog patient humoral response
Dog patient tumor cells were plated into a 96-well U-bottom plate at 8x104/well. Blocking step was performed by re-suspending tumor cells in PBS with 5% human serum for 15 minutes at 4°C. After two washes cells were incubated for 2 hours with sera of each patient taken at three different time points. Sera were tested diluted 1:10, 1:40, 1:160. Cells were then washed, stained with a goat anti-canine IgG (H+L) secondary antibody, FITC-conjugated (Invitrogen) and then the percentage of opsonized tumor cells was assessed.
CRISPR/Cas9 protocol
624-38 XBP1KO and B16F10-OVA XBB1KO were obtained using XbpI human and mouse genes knockout kits respectively according to manufacturer’s instructions (Cat# KN401959 and #KN519483, Origene, Rockville, MD, USA). Stable KO cells were selected in complete medium containing 2 μg/ml puromycin for 2 week. Single cell clones were isolated and screened by touchdown PCR. Primers were designed as indicated in Figure S5 and are listed in Table S3. Touchdown PCR were performed using GoTaq G2 Hot Start Taq Polymerase (Promega) and the following cycling conditions: 2 min at 95°C, followed by a touchdown phase of 10 cycles at 95°C for 30 s, 65-55°C (dropping 1°C per cycle) for 30 s and 72°C for 1 min; 25 cycles at 95°C for 30 s, 55°C for 30 s and 72°C for 1 min; 72°C for 5 min. For B16F10-OVA XbpIKO clones, two successive runs of PCR have been performed to obtain enough product to be sequenced. Then PCR products were purified and sequenced. Knockout of protein in both cell lines was confirmed by western blot: Rabbit mAb anti XBP1 s (Cell Signaling, 1:1000).
shRNA-mediated CX43 knockdown
To generate stable clones of B16 and B16-OVA cells silenced for CX43, we infected tumor cells with MISSION Lentiviral Transduction Particles specific for CX43 (Sigma) following the recommended protocol. Four different lentiviruses were tested, of which three successfully knocked down CX43. One of the successful lentiviruses and the unsuccessful lentivirus were cloned by limiting dilution and tested for silencing before and after bacterial infection.
Quantification and statistical analysis
For mice experiments, the sample size was guided by previous murine studies in our laboratory. Results are represented as mean ± standard error mean (SEM). Statistical significance, using GraphPad Prism software, was evaluated with: Mann-Whitney two-sided unpaired t test (for comparison between two groups without Gaussian distribution), two-sided Unpaired t test (for comparison between two groups with Gaussian distribution), Kruskal-Wallis test (for comparison between more than two groups without Gaussian distribution), One-Way ANOVA test (for comparison between more than two groups with Gaussian distribution) followed by Bonferroni’s or Dunn’s post-test and two way ANOVA test followed by Tukey’s post-test using GraphPad Prism software. Log-rank Mantel-Cox test was performed to assess differences among survival curves. Gaussian distribution was evaluated using Column Statistic (D’Agostino & Pearson normality test) in GraphPad Prism software. Following combined experiments, outliers were detected with the Grubbs’ test and excluded from the analysis. Data display normal variance. A probability value of ∗ p < 0.05 was considered to be significant. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessments. All statistical details can be found in the figure legends while P values are shown in the figures.
Acknowledgments
We wish to thank Erica Ghezzi (Ospedale San Michele, Tavazzano [LO], IT) for helping with dog patient information and sample preparation, all dog-patient families that believed in the proposed clinical protocol, and Luisa Lanfrancone (IEO, Milan, IT) that kindly provided primary human melanocytes.
M.R. is supported by Italian Foundation for Cancer Research grants (AIRC 5x1000(2019) “UniCanVax” No. 22757). A.M. is supported by a FIRC-AIRC fellowship (2018). V.F. is supported by an Alan Ghitis Foundation fellowship (2019). M.M. is supported by KCL-Monash Collaborative award 2019, Cancer Research UK (C67500 and A29686), and National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London and/or the NIHR Clinical Research Facility.
Author contributions
Conceptualization, A.M., M.R., and G.P.; investigation, A.M., V.F., L.T., M.L., and M.A.; formal analysis, M.M.; data curation, J.L.; resources, O.Z. and L.M.; writing-original paper, A.M.; writing-review & editing, A.M., V.F., C.P., M.R., M.M., L.M., and M.L.; funding acquisition, A.M., M.M., and M.R.; supervision, A.M. and M.R.
Declaration of interests
The authors declare no competing interests.
Published: July 6, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109312.
Supplemental information
References
- Antoniou A.N., Lenart I., Kriston-Vizi J., Iwawaki T., Turmaine M., McHugh K., Ali S., Blake N., Bowness P., Bajaj-Elliott M. Salmonella exploits HLA-B27 and host unfolded protein responses to promote intracellular replication. Ann. Rheum. Dis. 2019;78:74–82. doi: 10.1136/annrheumdis-2018-213532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoszewski R., Brewer J.W., Rab A., Crossman D.K., Bartoszewska S., Kapoor N., Fuller C., Collawn J.F., Bebok Z. The unfolded protein response (UPR)-activated transcription factor X-box-binding protein 1 (XBP1) induces microRNA-346 expression that targets the human antigen peptide transporter 1 (TAP1) mRNA and governs immune regulatory genes. J. Biol. Chem. 2011;286:41862–41870. doi: 10.1074/jbc.M111.304956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boegel S., Löwer M., Bukur T., Sahin U., Castle J.C. A catalog of HLA type, HLA expression, and neo-epitope candidates in human cancer cell lines. OncoImmunology. 2014;3:e954893. doi: 10.4161/21624011.2014.954893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerezo M., Lehraiki A., Millet A., Rouaud F., Plaisant M., Jaune E., Botton T., Ronco C., Abbe P., Amdouni H. Compounds triggering ER stress exert anti- melanoma effects and overcome BRAF inhibitor resistance. Cancer Cell. 2016;29:805–819. doi: 10.1016/j.ccell.2016.04.013. [DOI] [PubMed] [Google Scholar]
- Chapiro J., Claverol S., Piette F., Ma W., Stroobant V., Guillaume B., Gairin J.E., Morel S., Burlet-Schiltz O., Monsarrat B. Destructive cleavage of antigenic peptides either by the immunoproteasome or by the standard proteasome results in differential antigen presentation. J. Immunol. 2006;176:1053–1061. doi: 10.4049/jimmunol.176.2.1053. [DOI] [PubMed] [Google Scholar]
- Corazzari M., Gagliardi M., Fimia G.M., Piacentini M. Endoplasmic reticulum stress, unfolded protein response, and cancer cell fate. Front. Oncol. 2017;7:78. doi: 10.3389/fonc.2017.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannull J., Lesher D.T., Holzknecht R., Qi W., Hanna G., Seigler H., Tyler D.S., Pruitt S.K. Immunoproteasome down-modulation enhances the ability of dendritic cells to stimulate antitumor immunity. Blood. 2007;110:4341–4350. doi: 10.1182/blood-2007-04-083188. [DOI] [PubMed] [Google Scholar]
- Durgeau A., El Hage F., Vergnon I., Validire P., de Montpréville V., Besse B., Soria J.C., van Hall T., Mami-Chouaib F. Different expression levels of the TAP peptide transporter lead to recognition of different antigenic peptides by tumor-specific CTL. J. Immunol. 2011;187:5532–5539. doi: 10.4049/jimmunol.1102060. [DOI] [PubMed] [Google Scholar]
- Faridi P., Li C., Ramarathinam S.H., Illing P.T., Mifsud N.A., Ayala R., Song J., Gearing L.J., Croft N.P., Purcell A.W. Response to Comment on “A subset of HLA-I peptides are not genomically templated: Evidence for cis- and trans-spliced peptide ligands”. Sci. Immunol. 2019;4:1–12. doi: 10.1126/sciimmunol.aaw8457. [DOI] [PubMed] [Google Scholar]
- Gao J., Shi L.Z., Zhao H., Chen J., Xiong L., He Q., Chen T., Roszik J., Bernatchez C., Woodman S.E. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. 2016;167:397–404.e9. doi: 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groettrup M., Kirk C.J., Basler M. Proteasomes in immune cells: more than peptide producers? Nat. Rev. Immunol. 2010;10:73–78. doi: 10.1038/nri2687. [DOI] [PubMed] [Google Scholar]
- Gros A., Robbins P.F., Yao X., Li Y.F., Turcotte S., Tran E., Wunderlich J.R., Mixon A., Farid S., Dudley M.E. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J. Clin. Invest. 2014;124:2246–2259. doi: 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemskerk B., Kvistborg P., Schumacher T.N.M. The cancer antigenome. EMBO J. 2013;32:194–203. doi: 10.1038/emboj.2012.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi F.S., O’Day S.J., McDermott D.F., Weber R.W., Sosman J.A., Haanen J.B., Gonzalez R., Robert C., Schadendorf D., Hassel J.C. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosken N.A., Bevan M.J. Defective presentation of endogenous antigen by a cell line expressing class I molecules. Science. 1990;248:367–370. doi: 10.1126/science.2326647. [DOI] [PubMed] [Google Scholar]
- Imai J., Otani M., Koike E., Yokoyama Y., Maruya M., Koyasu S., Sakai T. A novel effect of the antigenic peptides position for presentation upon MHC Class I. bioRxiv. 2019 doi: 10.1101/653170. [DOI] [Google Scholar]
- Lampen M.H., Verweij M.C., Querido B., van der Burg S.H., Wiertz E.J.H.J., van Hall T. CD8+ T cell responses against TAP-inhibited cells are readily detected in the human population. J. Immunol. 2010;185:6508–6517. doi: 10.4049/jimmunol.1001774. [DOI] [PubMed] [Google Scholar]
- Lang K.S., Caroli C.C., Muhm A., Wernet D., Moris A., Schittek B., Knauss-Scherwitz E., Stevanovic S., Rammensee H.G., Garbe C. HLA-A2 restricted, melanocyte-specific CD8(+) T lymphocytes detected in vitiligo patients are related to disease activity and are predominantly directed against MelanA/MART1. J. Invest. Dermatol. 2001;116:891–897. doi: 10.1046/j.1523-1747.2001.01363.x. [DOI] [PubMed] [Google Scholar]
- Leone P., Shin E.C., Perosa F., Vacca A., Dammacco F., Racanelli V. MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J. Natl. Cancer Inst. 2013;105:1172–1187. doi: 10.1093/jnci/djt184. [DOI] [PubMed] [Google Scholar]
- Liepe J., Marino F., Sidney J., Jeko A., Bunting D.E., Sette A., Kloetzel P.M., Stumpf M.P.H., Heck A.J.R., Mishto M. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science. 2016;354:354–358. doi: 10.1126/science.aaf4384. [DOI] [PubMed] [Google Scholar]
- Liepe J., Sidney J., Lorenz F.K.M., Sette A., Mishto M. Mapping the MHC Class I – spliced immunopeptidome of cancer cells. Cancer Immunol. Res. 2019;7:62–76. doi: 10.1158/2326-6066.CIR-18-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsue H., Yao J., Matsue K., Nagasaka A., Sugiyama H., Aoki R., Kitamura M., Shimada S. Gap junction-mediated intercellular communication between dendritic cells (DCs) is required for effective activation of DCs. J. Immunol. 2006;176:181–190. doi: 10.4049/jimmunol.176.1.181. [DOI] [PubMed] [Google Scholar]
- Mendoza-Naranjo A., Saéz P.J., Johansson C.C., Ramírez M., Mandakovic D., Pereda C., López M.N., Kiessling R., Sáez J.C., Salazar-Onfray F. Functional gap junctions facilitate melanoma antigen transfer and cross-presentation between human dendritic cells. J. Immunol. 2007;178:6949–6957. doi: 10.4049/jimmunol.178.11.6949. [DOI] [PubMed] [Google Scholar]
- Mishto M., Liepe J. Post-Translational peptide splicing and T Cell responses. Trends Immunol. 2017;38:904–915. doi: 10.1016/j.it.2017.07.011. [DOI] [PubMed] [Google Scholar]
- Neijssen J., Herberts C., Drijfhout J.W., Reits E., Janssen L., Neefjes J. Cross-presentation by intercellular peptide transfer through gap junctions. Nature. 2005;434:83–88. doi: 10.1038/nature03290. [DOI] [PubMed] [Google Scholar]
- Oliveira C.C., Querido B., Sluijter M., Derbinski J., van der Burg S.H., van Hall T. Peptide transporter TAP mediates between competing antigen sources generating distinct surface MHC class I peptide repertoires. Eur. J. Immunol. 2011;41:3114–3124. doi: 10.1002/eji.201141836. [DOI] [PubMed] [Google Scholar]
- Osorio F., Tavernier S.J., Hoffmann E., Saeys Y., Martens L., Vetters J., Delrue I., De Rycke R., Parthoens E., Pouliot P. The unfolded-protein-response sensor IRE-1α regulates the function of CD8α+ dendritic cells. Nat. Immunol. 2014;15:248–257. doi: 10.1038/ni.2808. [DOI] [PubMed] [Google Scholar]
- Osorio F., Lambrecht B.N., Janssens S. Antigen presentation unfolded: identifying convergence points between the UPR and antigen presentation pathways. Curr. Opin. Immunol. 2018;52:100–107. doi: 10.1016/j.coi.2018.04.020. [DOI] [PubMed] [Google Scholar]
- Ott P.A., Hu Z., Keskin D.B., Shukla S.A., Sun J., Bozym D.J., Zhang W., Luoma A., Giobbie-Hurder A., Peter L. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217–221. doi: 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overwijk W.W., Restifo N.P. B16 as a mouse model for human melanoma. Curr. Protoc. Immunol. 2001;Chapter 20:Unit 20.1. doi: 10.1002/0471142735.im2001s39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri G., Colombino M., Cossu A., Marchetti A., Botti G., Ascierto P.A. Genetic instability and increased mutational load: which diagnostic tool best direct patients with cancer to immunotherapy? J. Transl. Med. 2017;15:17. doi: 10.1186/s12967-017-1119-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogoda K., Kameritsch P., Retamal M.A., Vega J.L. Regulation of gap junction channels and hemichannels by phosphorylation and redox changes: a revision. BMC Cell Biol. 2016;17:11. doi: 10.1186/s12860-016-0099-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rescigno M., Citterio S., Thèry C., Rittig M., Medaglini D., Pozzi G., Amigorena S., Ricciardi-Castagnoli P. Bacteria-induced neo-biosynthesis, stabilization, and surface expression of functional class I molecules in mouse dendritic cells. Proc. Natl. Acad. Sci. USA. 1998;95:5229–5234. doi: 10.1073/pnas.95.9.5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotta G., Edwards E.W., Sangaletti S., Bennett C., Ronzoni S., Colombo M.P., Steinman R.M., Randolph G.J., Rescigno M. Lipopolysaccharide or whole bacteria block the conversion of inflammatory monocytes into dendritic cells in vivo. J. Exp. Med. 2003;198:1253–1263. doi: 10.1084/jem.20030335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowell J.L., McCarthy D.O., Alvarez C.E. Dog models of naturally occurring cancer. Trends Mol. Med. 2011;17:380–388. doi: 10.1016/j.molmed.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatino M., Zhao Y., Voiculescu S., Monaco A., Robbins P., Karai L., Nickoloff B.J., Maio M., Selleri S., Marincola F.M., Wang E. Conservation of genetic alterations in recurrent melanoma supports the melanoma stem cell hypothesis. Cancer Res. 2008;68:122–131. doi: 10.1158/0008-5472.CAN-07-1939. [DOI] [PubMed] [Google Scholar]
- Saccheri F., Pozzi C., Avogadri F., Barozzi S., Faretta M., Fusi P., Rescigno M. Bacteria-induced gap junctions in tumors favor antigen cross-presentation and antitumor immunity. Sci. Transl. Med. 2010;2:44ra57. doi: 10.1126/scitranslmed.3000739. [DOI] [PubMed] [Google Scholar]
- Sahin U., Derhovanessian E., Miller M., Kloke B.P., Simon P., Löwer M., Bukur V., Tadmor A.D., Luxemburger U., Schrörs B. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222–226. doi: 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- Seidel U.J.E., Oliveira C.C., Lampen M.H., Hall Tv. A novel category of antigens enabling CTL immunity to tumor escape variants: Cinderella antigens. Cancer Immunol. Immunother. 2012;61:119–125. doi: 10.1007/s00262-011-1160-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P., Allison J.P. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–214. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder A., Makarov V., Merghoub T., Yuan J., Zaretsky J.M., Desrichard A., Walsh L.A., Postow M.A., Wong P., Ho T.S. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014;371:2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M., Sandoval T.A., Chae C.S., Chopra S., Tan C., Rutkowski M.R., Raundhal M., Chaurio R.A., Payne K.K., Konrad C. IRE1α-XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature. 2018;562:423–428. doi: 10.1038/s41586-018-0597-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suvarnapunya A.E., Lagassé H.A.D., Stein M.A. The role of DNA base excision repair in the pathogenesis of Salmonella enterica serovar Typhimurium. Mol. Microbiol. 2003;48:549–559. doi: 10.1046/j.1365-2958.2003.03460.x. [DOI] [PubMed] [Google Scholar]
- Urra H., Dufey E., Avril T., Chevet E., Hetz C. Endoplasmic reticulum stress and the hallmarks of Cancer. Trends Cancer. 2016;2:252–262. doi: 10.1016/j.trecan.2016.03.007. [DOI] [PubMed] [Google Scholar]
- Vanpouille-Box C., Lhuillier C., Bezu L., Aranda F., Yamazaki T., Kepp O., Fucikova J., Spisek R., Demaria S., Formenti S.C. Trial watch: Immune checkpoint blockers for cancer therapy. OncoImmunology. 2017;6:e1373237. doi: 10.1080/2162402X.2017.1373237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzler C., Rovere P., Rescigno M., Granucci F., Penna G., Adorini L., Zimmermann V.S., Davoust J., Ricciardi-Castagnoli P. Maturation stages of mouse dendritic cells in growth factor-dependent long-term cultures. J. Exp. Med. 1997;185:317–328. doi: 10.1084/jem.185.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolchok J.D., Kluger H., Callahan M.K., Postow M.A., Rizvi N.A., Lesokhin A.M., Segal N.H., Ariyan C.E., Gordon R.A., Reed K. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretsky J.M., Garcia-Diaz A., Shin D.S., Escuin-Ordinas H., Hugo W., Hu-Lieskovan S., Torrejon D.Y., Abril-Rodriguez G., Sandoval S., Barthly L. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016;375:819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original data, Peptidomic datasets and R-scripts generated during this study have been deposited in Institutional Zenodo community named IRCCS Humanitas Research Hospital & Humanitas University and are available under request https://zenodo.org/record/4738409#.YJJEIqHONaQ. The MS RAW files from which human and murine peptides have been identified (listed in Table S2) are available from the corresponding author on request.