

1. INTRODUCTION
Physiological pain is a protective mechanism that involves the detection of stimuli that have a potentially damaging effect on an organism [1]. Pain-producing noxious stimuli are commonly mechanical, chemical, or thermal in origin and activate the peripheral nerve terminals of specialized sensory neurons that innervate tissues and whose cell bodies are localized in the dorsal root and trigeminal ganglia. These pain-sensing afferents are called nociceptors, and depolarization of their nerve terminals initiates the firing of action potentials that invade the dorsal root ganglia (DRG) [2]. Studies of the electrophysiological properties of nociceptors have demonstrated that electrogenesis in these neurons requires the activation of specific types of voltage-gated sodium channels (NaV), NaV1.7, NaV1.8, and NaV1.9. In humans, both gain-of-function and loss-of-function mutations in these genes have been described, and these result in marked pain phenotypes including congenital insensitivity to pain or erythromelalgia (a rare syndrome characterized by episodes of severe, burning pain, redness and increased skin temperature, mostly in the feet) among other syndromes (reviewed in [3] [4]).
The normal processing of pain is substantively altered in response to pathology. In many instances, such as in inflammation or tissue injury, pain pathways become sensitized so that stimuli that were previously innocuous now trigger painful responses [2]. In some cases, these pain responses become dissociated from their protective functions and ongoing pain is experienced in the absence of physiologically appropriate stimuli. The molecular and cellular mechanisms that mediate pain in association with pathology involve changes to the properties of neurons at all levels of the neuraxis, both in the periphery and in the central nervous system (CNS) [2, 5].
Osteoarthritis (OA) is one of the major causes of chronic pain in the world [6]. Pain in OA is typically mechanical in nature – for example, patients with knee OA complain that it hurts to climb stairs [7]. In addition to the symptom of pain, OA patients show signs of peripheral and central sensitization, which can be detected by quantitative sensory testing (QST), including reduced pressure pain thresholds and increased temporal summation, both at the OA joint as well as at anatomical sites away from the joint [8] [9]. In the majority of patients, OA joint pain is relieved by joint replacement surgery, strongly suggesting that pain signaling in OA continues to be dependent on peripheral triggers throughout the disease process. Mechanistically, this involves the detection of painful stimuli by joint-innervating nociceptors. In mice, chemogenetic methods involving the expression of “Designer receptors exclusively activated by designer drugs” (DREADDs) in specific nerve populations, can be used to selectively stimulate Gi/o protein signaling in order to reduce peripheral sensory nerve activity [10], and this results in inhibition of pain behaviors in experimental OA models, particularly in early phases of disease [11, 12]. On the other hand, acute chemogenetic inhibition of nociceptor function has been reported to be ineffective in ameliorating pain behaviors at late stages of murine experimental OA in one study [11], indicating that other factors that affect pain signaling may be of overriding importance in late-stage disease. This may be important for understanding underlying mechanisms for those patients who experience little pain relief from joint replacement (around 20% of patients with knee OA, fewer for hip OA [13]), suggesting that central sensitization in those patients is no longer dependent on peripheral input [14].
Understanding the mechanisms that underlie OA pain requires, first and foremost, a complete description of the properties of the neuronal pathways that mediate pain signaling in the joint. In the course of chronic progressive disease, these pathways are altered and modified by interaction with the degenerating and remodelling tissues they innervate, and with immune and inflammatory cells that react to the disease [15] [16] [17]. The specific changes in the peripheral neuronal pathways underlying joint pain in OA are the focus of this narrative review (for discussion on central mechanisms, we refer the reader to other reviews [18] [19] [8]. We searched PubMed for the following terms: “osteoarthritis”, “pain”, “sensitization”, “DRG neurons”, “nociceptors”, “mechanosensation”, “animal models”, “NGF”, “DAMPs”, “TLR”. First, we describe the sensory nervous system in OA with a focus on recent discoveries, driven by new molecular technologies such as single cell RNA sequencing (scRNAseq) that are revealing that DRG neurons can be classified into distinct functional classes characterized by expression of selected marker molecules. We discuss how these discoveries may be relevant for joint pain in OA. Then, we briefly summarize how the sensory nervous system interacts with its environment in the OA joint, and how this modulates pain processing in OA.
2. THE NERVOUS SYSTEM IN OSTEOARTHRITIS.
It should be realized -and this cannot be stressed too strongly- that because of the chronic nature of OA, the anatomy and physiology of nociception undergoes considerable plasticity during the course of the disease. Hence, it is important to understand the pharmacology and physiology of pain as it is manifest during progressive disease rather than in unaffected animals or human subjects. As we shall discuss, the plasticity of the nociceptive system in OA involves changes in the structural, physiological and genetic properties of neurons in pain pathways. Understanding the exact nature of this plasticity, and how it comes about, are key issues if we are to progress in the treatment of OA-associated pain.
2.1. Types of DRG neurons.
Primary sensory afferent neurons, including nociceptors, are pseudo-unipolar in structure, with one process attached to the cell body that bifurcates into two branches. The peripheral or distal processes of these neurons project to the cutaneous or deep peripheral tissues they innervate, and the other branch, referred to as the proximal process, terminates in the dorsal horn of the spinal cord or sensory nuclei of the brain stem. These sensory neurons are responsible for mediating diverse aspects of somatosensory physiology and are quite heterogeneous anatomically, physiologically, and biochemically. There are clearly distinct types of sensory neurons with different response profiles that underlie their ability to discriminate between different types of sensations, such as heat, cold, pain, itch, as well as mechanical sensations like touch and proprioception [20]. Numerous investigations have sought to answer the question as to the precise properties of each set of neurons that mediate particular somatosensory functions, including nociception, and how the properties of these neurons change in the face of pathology.
How then do we describe the class of sensory neurons that may mediate OA pain? The first way that sensory neuron subtypes were categorized was according to their degree of myelination and associated electrophysiological properties such as conduction velocity. Sensory neurons were shown to be heavily and moderately myelinated Aα and Aβ fibers, thinly myelinated Aδ-fibers and unmyelinated C-fibers. The degree of myelination has functional consequences determining the speed of action potential conduction, with heavily myelinated A-fibers conducting action potentials the most rapidly [21]. These neurophysiological differences in conduction velocity have also been associated with discrete functions. Heavily and moderately myelinated A-fibers are thought to be generally associated with light touch and proprioception, while thinly myelinated Aδ-fibers detect innocuous and noxious stimuli. C-fibers are unmyelinated and mostly involved in detecting painful stimuli, although some C-fibers, termed low threshold mechanoreceptors (C-LTMR), are also responsible for relaying innocuous light touch stimulation (such as pleasant touch elicited by the stoking of a soft brush). Nociceptors, the C and Aδ-fibers which detect noxious stimuli, can be either polymodal, responding to a variety of noxious stimuli (i.e., mechanical and thermal), or unimodal, responding to only a single type of noxious stimulation [22].
It has, however, become clear that fiber classification alone does not provide a sufficient means of predicting neuronal function [23]. This has led to other complementary methods of sensory neuron classification that depend on the expression of selected marker molecules such as peptidergic neurotransmitters, ion channels, calcium binding proteins and receptors, which indicate some specific function associated with each neuron type. Such markers also allow the identification of sensory neuron types using histological methods such as in situ hybridization or immunohistochemistry. From the histological perspective, large light and the small dark neurons were originally recognized. Large light neurons were shown to stain for neurofilament (NF) 200 and are mostly Aα/β, whereas the small dark neurons are neurofilament 200 negative and mostly correspond to unmyelinated nociceptive C fibers [24]. Additionally, nociceptors can be subdivided based on a limited number of neurochemical features particularly a division between peptidergic and non-peptidergic classes, the former being defined by containing neuropeptides such as substance P and calcitonin-gene related peptide (CGRP), while non-peptidergics traditionally do not contain these neuropeptides, but bind the plant lectin Griffonia simplicifolia I-B4 (IB4). The peptidergic neurons are either C- or Aδ-fiber neurons, while non-peptidergic neurons are all C-fibers. Of importance for a discussion of OA pain, it was also originally proposed that the majority of peptidergic neurons express tropomyosin receptor kinase A (TrkA), the high affinity receptor for the pain-producing neurotrophin, nerve growth factor (NGF), while non-peptidergic neurons do not [25], although this conclusion may have to be revised based on single cell RNA sequencing data (see below). In recent years, more advanced molecular biology techniques have led to the discovery of an increased number of marker molecules, resulting in the further devolution of these categories into ever more diverse groups. For example, subpopulations of nonpeptidergic neurons express the Mas-related G protein–coupled receptors, which define at least three subgroups of IB4-binding and Ret-expressing nonpeptidergic neurons, Mrgprd (Mas1-related G protein–coupled receptor D), MrgprA3/MrgprC11, and MrgprB4 [26]. These genetically defined subpopulations also appear functionally relevant, as Mrgprd+ neurons are mechanically responsive [27], while Mrgpra3+ neurons have been linked to response to itch [28], and Mrgprb4+ neurons appear to play a role in response to light touch of hairy skin [29].
Although the traditional classification of nociceptors has been an important aid to our understanding of pain mechanisms, sensory neuron biology has recently entered the age of microanatomy and function based on the revolutionary use of powerful nucleic sequencing techniques. Single-cell RNA sequencing of large numbers of cells and their transcripts together with bioinformatic procedures enable the unbiased identification of categories of neurons based on their transcriptome profiles [30, 31]. These techniques can provide extremely valuable data as to the precise molecular identity of DRG neurons, and their specific somatosensory specialization. There is not yet a complete consensus as to the number of DRG neuron classes and their functional specializations, but according to a recent review that parsed the currently published data sets, there may be a total of 18 somatosensory DRG neuron subtypes [23], which define functional specificity (Figure 1). It has also become clear that all nociceptors, except NP1.1 and NP1.2 can express neuropeptides [23]. Suffice it to say that the precise number of types of DRG neurons and their respective functions are likely to be much greater than originally anticipated. Importantly, these current classifications are based on studies in young adult healthy mice. Now, these advanced molecular and genetic techniques can be used to explore how the molecular and functional identity of DRG neurons is altered during the course of chronic painful diseases such as OA. Such studies will provide a basis for rational experiments to explore how these different subsets of nociceptors relate to pain in joints affected OA, for example through functional manipulation of subsets of sensory neurons. It can be expected that this will lead to the identification of specific neuronal targets and development of novel targeted analgesic therapies.
Fig 1.
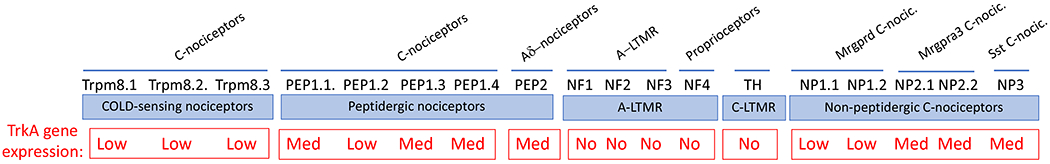
Classification of DRG sensory neurons, based on reference 23. We used http://mousebrain.org/genesearch.html, which shows gene expression throughout the mouse nervous system based on single-cell RNA-seq profiling, to determine Ntrk1 (TrkA) expression in each subtype. High expression of Ntrk1 is found in sympathetic neurons, while C and Aδ nociceptors show low to medium expression. A-LTMR neurons and TH C-LTMRs do not express Ntrk1.
LTMR= low threshold mechanoreceptor; PEP=peptidergic; NF= neurofilament; NP= non peptidergic; Mrgpr= Mas-related g-protein receptor; Sst= somatostatin; TH, tyrosine hydroxylase.
2.2. Joint innervation in OA
A critical step in unravelling the contribution of sensory neurons to joint pain in OA is to precisely describe where different relevant subsets of neurons are located in the joint. OA pathology affects all joint tissues, including articular cartilage, subchondral bone, synovium, menisci, and ligaments and many of these tissues are innervated by free nerve endings of nociceptors. Indeed, in healthy joints, the capsule, ligaments, menisci, periosteum and subchondral bone – but not articular cartilage – have all been shown to be richly innervated by sensory and sympathetic neurons, with the vast majority of sensory afferents being nociceptors [32] [33] [34]. It has also emerged that in the course of OA, the innervation may change - for example, anatomical studies in human OA knee joints and in rat models have revealed vascular penetration and nerve growth in the menisci, in osteophytes, and in the subchondral bone [35, 36]. Importantly, the appearance of osteochondral channels that breach the tidemark between the subchondral bone and the articular cartilage was described many years ago in human subjects with OA, as well as in rat models of OA, with evidence that these channels contain neurons (PGP9.5 staining) and blood vessels [35] [37] [38]. More recently, the authors expanded their work to show that the presence of CGRP-immunoreactive nociceptors in these osteochondral channels was associated with pain in human OA knees, as well as in a rat meniscal transection model [39].
Recent identification of specific molecular markers for different types of DRG neurons, as described above, enables the selective genetic labeling of these neurons in mice using appropriate Cre-recombinase driver lines to produce the expression of fluorescent markers in selected types of neurons. The resulting mice can be used for examining the precise innervation of the knee joint and whether this is altered during the development of OA in mouse models, something that has never previously been examined. In order to define changes in joint innervation that might contribute to OA pain, we employed fluorescently labeled NaV1.8 expressing neurons to investigate the nociceptive innervation of the mouse knee [40]. NaV1.8, in particular, is highly localized to nociceptors [41] and, therefore, NaV1.8-Cre can be used to label most of these neurons in mice. In young male mice (10 weeks old), innervation was observed in the lateral synovium, the connective tissue layer (the epiligament) surrounding the cruciate ligaments, and the insertion sites of the cruciate ligaments, as well as dense innervation of bone marrow cavities. These neurons are likely to be mostly Aδ/C-type nociceptors as reported previously in the literature [41]. Important changes in this pattern were observed 16 weeks after destabilization of the medial meniscus (DMM) surgery, a procedure that produces slowly developing OA-like joint damage and accompanying pain behaviors [42]. In association with joint damage in the medial compartment of the operated knee, fluorescently labelled nerves were noted in the synovium, as well as increased innervation of the meniscus and major changes in the innervation of the subchondral bone. In particular, remodeling in the sclerotic subchondral bone was accompanied by densely innervated subchondral bone channels in the tibial plateaux and femoral condyles (for an example, see Figure 2). Thus, these studies confirm immunohistochemical studies in human knees with end-stage knee OA, and show that genetically labelled mice such as these can be used to document changes in joint innervation over time.
Fig. 2.
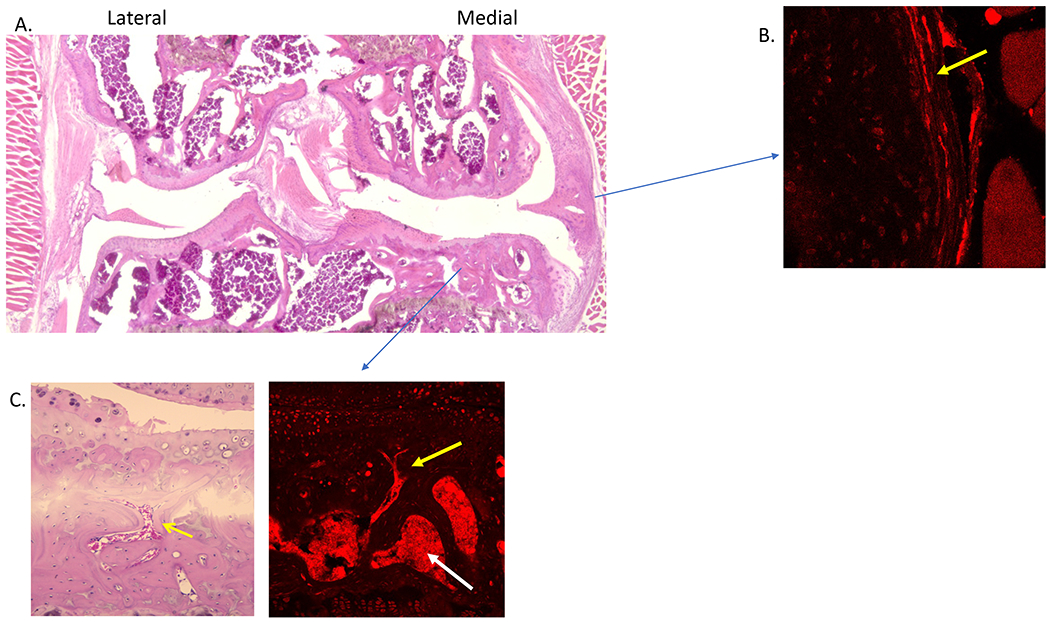
A. Using NaV1.8 tdTomato reporter mice, H&E staining shows osteoarthritis joint damage in the medial compartment 16 weeks after destabilization of the medial meniscus; B. New nociceptors are present in the medial synovium; C. Subchondral bone channels (left, yellow arrow) contain NaV1.8 nociceptors (right, yellow arrow). The white arrow points to bone marrow cavities, which are also innervated.
(Data from Obeidat AM, Miller RE, Miller RJ, Malfait AM: The nociceptive innervation of the normal and osteoarthritic mouse knee. Osteoarthritis Cartilage 2019, 27(11):1669-1679.)
In recent years, Ca2+ imaging techniques of DRGs in live mice have been developed [43], enabling visualization as a marker of electrical activity of L4-DRG neurons (where the majority of cell bodies of afferents coming from the knee and paw are located) in response to mechanical stimuli applied to the knee [44]. It is interesting to note that when the activity of knee-innervating DRG neurons is examined using this in vivo Ca2+ imaging technique, a noxious knee twist or knee pinch produces an increased number of activated neurons 8 weeks following DMM in comparison to sham surgery, indicating that this stimulus now recruits a population of newly sensitized neurons - possibly representing a category of previously silent nociceptors [45] [46] [44]. Increased responses to knee pinch have also been observed using this technique in the mouse anterior cruciate ligament transection (ACLT) model [47]. An important question is whether any of the plastic changes in knee innervation observed in OA, such as the neurons present in the subchondral bone channels, or in the synovium and meniscus, correspond to these newly recruited previously “silent” nociceptors. If this is the case, they would provide a defined target for therapeutic intervention in OA pain, and their subtype could be molecularly defined, as above, in order to identify druggable targets expressed by these newly recruited neurons.
3. THE ROLE OF NGF IN OSTEOARTHRITIS PAIN
An important development that has illuminated mechanisms underlying OA pain has been the discovery of the involvement of nerve growth factor (NGF) signaling. The neurotrophin, NGF, was first identified as an important factor that regulates the growth and survival of both sympathetic and sensory neurons [48]. Mice where the Ngf gene is deleted show an extensive loss of sensory and sympathetic innervation, and diminished response to noxious stimuli [49]. In humans, several mutations have been described in the NGF gene, as well as in the gene encoding TrkA (NTRK1), and these mutations are associated with congenital insensitivity to pain, complicated by severe musculoskeletal manifestations such as fractures and neuropathic joints (reviewed in [3]). After development, NGF has been shown to have a number of important functions, both within and outside of the nervous system; most notably, it is a key protein for producing pain (reviewed in [50, 51]). NGF can exert its pro-algesic actions through a number of different mechanisms. For example, it produces rapid excitatory effects in a subset of nociceptors that express its high-affinity receptor, TrkA. Activation of this receptor produces several rapid excitatory signaling events such as transactivation and increased expression of a variety of ion channels and receptors including the transient receptor vanilloid 1 (TRPV1), voltage-gated sodium and calcium channels, acid-sensing ion channels and mechanosensitive channels (see below).
Consistent with these effects, local injection of NGF produces immediate and often long-lasting pain responses in animals and humans (reviewed in [52]). In healthy adults, for example, a single subcutaneous injection of recombinant NGF has been shown to elicit rapid onset local injection-site hyperalgesia. Interestingly, this effect persists for up to 7 weeks [53]. Likewise, intradermal injection of NGF produces long-lasting local pressure allodynia [54]. The long-lasting effects suggest that they might be mediated by new nerve growth, and it was hypothesized that NGF-induced nerve growth might produce beneficial effects in certain neurodegenerative diseases, for example in diabetic neuropathy, in which there is a marked degeneration of the cutaneous innervation [55]. However, the painful effects of NGF following injection in patients resulted in cancellation of clinical trials that explored the use of NGF for this indication [56]. NGF produces pain upon injection into the cervical facet joints of the spine [57]. When injected intra-articularly into the knee joints of healthy rats, it rapidly causes acute swelling and pain behaviors, including mechanical allodynia and weightbearing deficits [58] [59]. Interestingly, OA knees are more sensitive to the pain-inducing effects of NGF than healthy knees [58].
Because of the clear ability of NGF to produce pain behaviors it has been considered to be a good candidate as an essential algogenic mediator involved in several chronic painful diseases in humans. Consequently, neutralizing antibodies that sequester NGF and prevent it from binding to TrkA were developed, and clinical trials testing these antibodies have been going on for over 20 years in conditions such as chronic low back pain and OA (reviewed in [52]). Of all these efforts, the beneficial effects of anti-NGF in OA have proved to be particularly marked. The anti-NGF antibody, tanezumab, is expected to be approved by the US Food and Drug Administration (FDA) for the treatment of OA pain in the very near future, in spite of the occurrence of rapidly progressive OA as a poorly understood side-effect in a small percentage of people receiving the antibody (discussed in [52]). Very recently (October 1 2020), anti-NGF Abs were approved in Europe for alleviation of OA pain in dogs (https://www.ema.europa.eu/en/medicines/veterinary/summaries-opinion/librela). Blockade of NGF-TrkAs signaling has also shown analgesic effects in several rodent models of OA, with varying effects on joint health - although in depth studies are lacking (reviewed in [52] [60]). Irrespective of anything else, these observations clearly indicate that NGF signaling does play a role in enabling chronic OA pain. In addition, since these antibodies do not cross the blood-brain barrier, these findings further strongly support the idea that peripheral drive maintains pain in this disease. Consequently, elucidating how exactly NGF contributes to OA pain, and how inhibiting its biological effects modifies pain pathways in OA- and may also affect joint integrity - will be of great importance for understanding the entire disease.
Clearly, NGF is present in joints affected by OA – for example, levels are higher in the synovial fluid in patients with knee OA than in controls [61], and synovial fluid levels have been correlated with poor knee function [62]. However, while it is known that many cell types – including joint cells such as chondrocytes, fibroblasts, and innate immune cells can produce NGF in response to cytokine stimulation [63] (reviewed in [50]), it is not clear exactly which cells in osteoarthritic joints produce the neurotrophin. In surgical mouse models of OA, Ngf mRNA is upregulated in the whole joint [64], specifically in articular cartilage [65] immediately after surgery and also in the late stage of the model, when mice show chronic pain. Recent work in human OA knee joints has reported NGF-immunoreactivity in specific localizations to be associated with knee pain. Specifically, NGF-immunoreactivity in the synovium, mostly in synovial fibroblasts and to a lesser extent in macrophages was associated with pain [66]. Importantly, NGF expression in osteochondral channels as well as increased osteoclast density were associated with symptomatic knee OA, independently of synovitis or chondropathy [67]. NGF-like immunoreactivity in the subchondral bone was predominantly found associated with osteoclasts and mononuclear cells [67].
There are several mechanisms through which anti-NGF antibodies could produce relief of OA pain. It is clear from studies using in vivo Ca2+ imaging to monitor the activity of NaV1.8 expressing nociceptors that injection of NGF into the mouse knee produces immediate nociceptor excitation [68], presumably due to its ability to depolarize these neurons through mechanisms such as the transactivation of TRPV1 (see above). In addition, NGF signaling modifies the properties of nociceptor ion channels resulting in sensitization so that nerves are more likely to fire in response to normal mechanically driven stimuli associated with joint usage. Hence, ongoing release of NGF in OA can be expected to produce pain and inhibiting these actions will block these effects. During the past two decades, numerous ion channels, including ASIC channels, TRPA1, TRPC3, TRPC6, TACAN (aka Tmem120A), and PIEZO2, have been proposed to contribute to mechanosensitivity of sensory neurons. Interestingly, the biophysical properties of PIEZO2 appear well suited for mediating light touch (low threshold of activation, rapidly inactivating) and proprioception [69] rather than pain, whereas a channel with a high activation threshold which exhibits slow inactivation such as TACAN would seem more appropriate [70]. However, PIEZO2 has been shown to be expressed by nociceptors and to play a role in mediating mechanical pain associated with models of inflammation and nerve injury [46, 71–73], and so how it may contribute to mechanical pain, particularly in disease states, is an area of active investigation. Indeed, at this time it is unclear which channels mediate mechanical depolarization of the nociceptor population involved in OA and how these may be related to the effects of NGF. An interesting set of observations demonstrated that application of NGF to DRG neurons in culture greatly increased their mechanosensitivity by sensitizing the activity of PIEZO2 mechanoreceptors in neurons [46]. These observations might therefore correspond to the increased activity of previously silent nociceptors observed in in vivo Ca2+ imaging studies referred to above. A precise description of the cellular populations of DRG neurons that express mechanosensitive channels such as PIEZO2 and TACAN as well as their co-localization with TrkA would clearly be of interest, in order to increase our understanding of the role NGF plays in the mechanical pain that is a feature of OA.
In addition to its acute actions that result in the functional sensitization of nociceptor populations, the continuous action of NGF in OA might well be responsible for the remodeling of the joint innervation observed in OA – although this has not yet directly been investigated. We recently found that injection of NGF into the mouse knee produces marked NaV1.8-hyper-innervation of joint tissues, most notably the synovium and the subchondral bone (Obeidat et al., manuscript in preparation) - effects that are not surprising considering the well known ability of NGF to support growth and survival of sensory neurons. The purpose of the anatomical remodeling of joint nociceptors in the OA joint, as described above, is unclear. It may represent an attempt to produce neurogenic repair of the joint in the face of ongoing bone and cartilage degradation. At the same time, this enhanced nerve growth may provide a substrate for increased nociceptor activation and pain, which may be regarded as an injury-related signal for preventing joint usage and thus promote healing. If anti-NGF treatment also reduces enhanced nerve innervation of the joint then this may well contribute to its antinociceptive actions, particularly with regard to the long-lasting nature of its effects in humans. Interestingly, it has been reported that administration of the small molecule TrkA inhibitor, AR786, reversed the appearance of CGRP-immunoreactive nerves in the osteochondral channels, and reduced pain behaviors in rats with surgically induced OA [39], an observation that further suggests the importance of these structures in OA pain.
It is not known at this point which of these NGF actions are most important for the observed analgesic effects of anti-NGF in OA, and it is conceivable that they are both involved during the progression of the disease. Furthermore, it is also possible that NGF sensitizes nociceptors indirectly through actions on non-neuronal cells in the joint. Not only do some populations of immune cells produce NGF – for example, in OA knee joints, NGF immunoreactive macrophages have been detected in the synovium, and this was correlated with pain [66]- but others express TrkA and respond to NGF by releasing inflammatory cytokines, which can then further enhance nociceptor excitability by directly activating these neurons, or through an action on other non-neuronal cells further elaborating an ever expanding excitatory cytokine cascade. For example, in response to NGF, mast cells produce prostaglandin D2 that can then act on nociceptors, a mechanism which may contribute to mechanical allodynia in a mouse model of OA [74]. NGF has also been found to be present in, and released from, human CD14+ T cell clones and human monocytes. NGF has been shown to increase the release of mediators including IL-1β, bradykinin, histamine, ATP, serotonin, and protons from inflammatory cells. It is interesting to note that many non-neuronal cells, including bone and chondrocytes also express mechanosensitive PIEZO channels whose activation may be linked to the synthesis and release of inflammatory cytokines [75–77]. Whether these PIEZO channels can also be sensitized by NGF as in sensory neurons (see above) is not yet known, but this should be an important question to address in the context of a mechanically driven diseases such as OA.
Study of the current literature therefore reveals that knowledge of the expression patterns and precise biological effects of NGF-TrkA signaling in the OA joint is rather limited. Importantly, the clinical trials with anti-NGF revealed that a small percentage of patients treated with the antibody developed rapidly progressive OA, which resulted in the need for joint replacement [78] [52]. In view of this unexplained side effect, going forward it will clearly be important to paint an accurate picture of the entire NGF-TrkA “interactome” in the OA joint, detailing which cells express NGF and TrkA at which points in time during the development of the disease. Because the pain-relieving effects of anti-NGF are not in doubt, a detailed understanding of its mechanism of action will certainly help to understand the basis of OA pain, and may also provide insight into the relationship between pain and joint damage. Furthermore, the observed analgesic effects with targeting NGF-TrkA may also suggest novel strategies for targeting OA pain. Indeed, if the precise identity of the neuronal subtypes involved in OA pain can be defined - and these may well be TrkA expressing neurons (Figure1)- then, because single cell RNA sequencing can identify the receptome of these DRG neurons, targeting receptors that are particularly associated with these specific neurons may enable the selective inhibition of OA pain.
4. NEUROIMMUNE INVOLVEMENT IN OSTEOARTHRITIS PAIN
As described above, the sensory nervous system is designed to perform its function of detecting potentially noxious stimuli, and producing pain in order to help maintain homeostasis in the face of danger. One aspect of this response is that the nervous system is in constant communication not only with innervated tissues but also with the immune system - which also plays a vital role in the protection of organisms and the maintenance of homeostasis. The bi-directional crosstalk between the nervous system and the immune system is currently the focus of intense investigation as a major pathogenic contributor to the establishment and maintenance of chronic pain [15] [79]. In the case of OA, emphasis is on the innate immune system, which is increasingly appreciated to play a role in driving joint damage and pain, as discussed in depth in several recent reviews [80] [17]. One particular pathway that is shared by the innate immune and the sensory nervous system is mediated by Pattern Recognition Receptors (PRRs) that can respond to signals provided by pathogens (Pathogen Associated Molecular Patterns or PAMPs, such as microbial nucleic acids, lipoproteins, and carbohydrates) or Damage-Associated Molecular Patterns (DAMPs, a.k.a. “alarmins”, such as S100A8/9). These molecular patterns bind PRRs such as toll-like receptors (TLRs), NOD-like receptors (NLRs), and the intracellular RNA-sensing retinoic acid-inducible gene I (RIG-I)-like receptors, resulting in cellular responses that try to re-establish tissue homeostasis through triggering the synthesis of inflammatory cytokines and chemokines [81]. In the context of OA, it has been proposed that molecules arising from the degradation of joint tissues may activate TLRs expressed by nociceptors, resulting in direct excitation, and pain. This has been shown for the TLR4 ligand, S100A8/9 [82] [83], as well as for a 32-amino-acid aggrecan fragment that is released from cartilage when ADAMTS4/5 and MMPs degrade aggrecan in the interglobular domain [84]. This 32-mer aggrecan fragment can activate nociceptors through TLR2, and produce hyperalgesia following its injection into the knee cavity of naive mice. In vivo studies in mice have revealed that Tlr4 null or Tlr2 null mice develop joint damage after DMM surgery, but they are protected from knee hyperalgesia ([84]and unpublished data). These data therefore exemplify how a DAMP that is specific to OA cartilage damage can produce pain behavior through direct activation of DRG neurons. In contrast, Tlr2 and Tlr4 null mice are not protected from developing persistent mechanical allodynia, suggesting that many different pathways further contribute to the chronification of pain. Neuroimmune crosstalk at the level of the DRGs is thought to be key in maintenance of pain [85], and in the context of the DMM model, we recently reported a microarray study of the knee-innervating DRGs, where pathway analysis suggested a role for immune cell recruitment in this model [86].
Neuroimmune pathways of pain generation can be further amplified when inflammatory cytokines synthesized as a result of TLR-activation in resident and infiltrating joint cells, such as chondrocytes and macrophages, act upon joint nociceptors directly - or on other joint tissues and immune cells, triggering further elements of an inflammatory cascade. Indeed, it is quite clear that cytokines like interleukin (IL)-1, IL-6, IL-17, TNF-α, and chemokines such as CCL2 can engage receptors expressed by nociceptors and trigger excitation and sensitization through variety of mechanisms, including TRP channel and voltage gated sodium channel transactivation [87].In the case of CCL2 and its receptor CCR2, a series of studies have demonstrated that these molecules are upregulated following DMM and contribute to OA pain at defined points in the progression of the disease in this model [88–90].
The possible role of DAMPs as mediators of OA pain is clearly an attractive possibility but needs further confirmation in both mice and humans. Furthermore, it is important to try and understand how such DAMP mediated effects might be integrated into the developing narrative of NGF signaling in OA pain. In fact, this is generally true of all investigations directed at understanding the mechanisms of OA pain. Traditionally, the pharmacology of OA pain has been envisaged as developing particular “targets” which can be modified by highly specific drugs and clearly this is part of the process. However, it is important to understand that all of these targets operate in the context of constantly shifting joint biology. Observations from human subjects and from animals using models such as the DMM and MIA models, have clearly indicated that the establishment of OA pain is a time-dependent process and that different biological processes, involving different molecular targets are operating during different phases of the disease (these temporal considerations were recently elegantly reviewed in [91]). Drugs that modify NGF-dependent joint innervation and drugs that modify neuroimmune or other influences may be appropriate at different stages of the disease in different patient populations. Future treatment of OA pain may constitute an array of targeted interventions designed to treat specific populations of patients. In order to develop these effective therapeutic approaches to OA pain we must continue to establish an accurate description of exactly how the disease develops from a cellular, molecular and genetic perspective and how different populations of joint cells communicate with one another across space and time.
Synopsis:
The specific changes in the peripheral neuronal pathways underlying joint pain in osteoarthritis are the focus of this narrative review. We discuss the plasticity of the nociceptive system in osteoarthritis, and how this involves changes in the structural, physiological and genetic properties of neurons in pain pathways. We discuss the role of the neurotrophin, nerve growth factor, in these pathogenic processes. Finally, we briefly consider how neuronal pathways are modified by interaction with the degenerating joint tissues they innervate, and with the innate immune system. These extensive cellular interactions will provide a substrate for identification of targets for osteoarthritis pain.
Clinics Care Points (evidence-based pearls and pitfalls relevant to the point of care).
Osteoarthritis (OA) is worldwide one of the major causes of chronic pain. Currently available analgesic drugs fall short of patients’ needs.
Neutralizing antibodies targeting the neurotrophin, NGF, are in clinical development for OA pain. Trial results indicate that this is a highly promising biological therapy for OA pain, but a small proportion of patients treated with the antibodies develop rapidly progressive OA. The mechanism underlying this side effect is not understood.
Neuroimmune interactions provide a rich substrate of potential targets for the development of new drugs for OA pain.
Future treatment of OA pain may constitute an array of targeted interventions designed to treat specific populations of patients with OA.
Acknowledgements
The authors thank the National Institutes of Health (National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), grant numbers R01AR060364 to AMM, and R01AR064251 to AMM and RJM, and K01AR070328 to REM. AMM is supported by the George W. Stuppy, MD, Chair of Arthritis at Rush University.
REFERENCES
- 1.Raja SN, Carr DB, Cohen M, Finnerup NB, Flor H, Gibson S, Keefe FJ, Mogil JS, Ringkamp M, Sluka KA et al. : The revised International Association for the Study of Pain definition of pain: concepts, challenges, and compromises. PAIN 2020, 161(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woller SA, Eddinger KA, Corr M, Yaksh TL: An overview of pathways encoding nociception. Clinical and experimental rheumatology 2018, 36(1):172. [PubMed] [Google Scholar]
- 3.Drissi I, Woods WA, Woods CG: Understanding the genetic basis of congenital insensitivity to pain. Br Med Bull 2020, 133(1):65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Emery EC, Luiz AP, Wood JN: Nav1.7 and other voltage-gated sodium channels as drug targets for pain relief. Expert Opin Ther Targets 2016, 20(8):975–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woolf CJ: Central sensitization: implications for the diagnosis and treatment of pain. Pain 2011, 152(3 Suppl):S2–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collaborators G: Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392(10159):1789–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malfait AM, Schnitzer TJ: Towards a mechanism-based approach to pain management in osteoarthritis. Nat Rev Rheumatol 2013, 9(11):654–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arendt-Nielsen L: Pain sensitisation in osteoarthritis. Clinical and experimental rheumatology 2017, 35 Suppl 107(5):68–74. [PubMed] [Google Scholar]
- 9.Carlesso LC, Neogi T: Identifying pain susceptibility phenotypes in knee osteoarthritis. Clinical and experimental rheumatology 2019, 37 Suppl 120(5):96–99. [PMC free article] [PubMed] [Google Scholar]
- 10.Roth BL: DREADDs for Neuroscientists. Neuron 2016, 89(4):683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller RE, Ishihara S, Bhattacharyya B, Delaney A, Menichella DM, Miller RJ, Malfait AM: Chemogenetic Inhibition of Pain Neurons in a Mouse Model of Osteoarthritis. Arthritis Rheumatol 2017, 69(7):1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chakrabarti S, Pattison LA, Doleschall B, Rickman RH, Blake H, Callejo G, Heppenstall PA, Smith ESJ: Intraarticular Adeno-Associated Virus Serotype AAV-PHP.S-Mediated Chemogenetic Targeting of Knee-Innervating Dorsal Root Ganglion Neurons Alleviates Inflammatory Pain in Mice. Arthritis Rheumatol 2020. [DOI] [PubMed] [Google Scholar]
- 13.Beswick AD, Wylde V, Gooberman-Hill R, Blom A, Dieppe P: What proportion of patients report long-term pain after total hip or knee replacement for osteoarthritis? A systematic review of prospective studies in unselected patients. BMJ Open 2012, 2(1):e000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walsh DA: Editorial: Arthritis Pain: Moving Between Early- and Late-Stage Disease. Arthritis Rheumatol 2017, 69(7):1343–1345. [DOI] [PubMed] [Google Scholar]
- 15.Hore Z, Denk F: Neuroimmune interactions in chronic pain - An interdisciplinary perspective. Brain Behav Immun 2019, 79:56–62. [DOI] [PubMed] [Google Scholar]
- 16.Chu C, Artis D, Chiu IM: Neuro-immune Interactions in the Tissues. Immunity 2020, 52(3):464–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller RJ, Malfait AM, Miller RE: The innate immune response as a mediator of osteoarthritis pain. Osteoarthritis Cartilage 2020, 28(5):562–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lluch E, Torres R, Nijs J, Van Oosterwijck J: Evidence for central sensitization in patients with osteoarthritis pain: a systematic literature review. Eur J Pain 2014, 18(10):1367–1375. [DOI] [PubMed] [Google Scholar]
- 19.Walsh DA, Stocks J: New Therapeutic Targets for Osteoarthritis Pain. SLAS Discov 2017, 22(8):931–949. [DOI] [PubMed] [Google Scholar]
- 20.Basbaum AI, Bautista DM, Scherrer G, Julius D: Cellular and molecular mechanisms of pain. Cell 2009, 139(2):267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raja SN, Meyer RA, Campbell JN: Peripheral mechanisms of somatic pain. Anesthesiology 1988, 68(4):571–590. [DOI] [PubMed] [Google Scholar]
- 22.Woolf CJ, Ma Q: Nociceptors--noxious stimulus detectors. Neuron 2007, 55(3):353–364. [DOI] [PubMed] [Google Scholar]
- 23.Emery EC, Ernfors P: Dorsal Root Ganglion Neuron Types and Their Functional Specialization. The Oxford Handbook of the Neurobiology of Pain Edited by John N Wood 2019. [Google Scholar]
- 24.Lawson SN, Waddell PJ: Soma neurofilament immunoreactivity is related to cell size and fibre conduction velocity in rat primary sensory neurons. J Physiol 1991, 435:41–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Averill S, McMahon SB, Clary DO, Reichardt LF, Priestley JV: Immunocytochemical localization of trkA receptors in chemically identified subgroups of adult rat sensory neurons. Eur J Neurosci 1995, 7(7):1484–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McNeil B, Dong X: Mrgprs as Itch Receptors. In: Itch: Mechanisms and Treatment. Edited by Carstens E, Akiyama T. Boca Raton, FL: CRC Press/Taylor & Francis; 2014. [PubMed] [Google Scholar]
- 27.Zylka MJ, Rice FL, Anderson DJ: Topographically distinct epidermal nociceptive circuits revealed by axonal tracers targeted to Mrgprd. Neuron 2005, 45(1):17–25. [DOI] [PubMed] [Google Scholar]
- 28.Liu Q, Tang Z, Surdenikova L, Kim S, Patel KN, Kim A, Ru F, Guan Y, Weng H-J, Geng Y et al. : Sensory Neuron-Specific GPCR Mrgprs Are Itch Receptors Mediating Chloroquine-Induced Pruritus. Cell 2009, 139(7):1353–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Q, Vrontou S, Rice FL, Zylka MJ, Dong X, Anderson DJ: Molecular genetic visualization of a rare subset of unmyelinated sensory neurons that may detect gentle touch. Nature Neuroscience 2007, 10(8):946–948. [DOI] [PubMed] [Google Scholar]
- 30.Usoskin D, Furlan A, Islam S, Abdo H, Lonnerberg P, Lou D, Hjerling-Leffler J, Haeggstrom J, Kharchenko O, Kharchenko PV et al. : Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci 2015, 18(1):145–153. [DOI] [PubMed] [Google Scholar]
- 31.Zeisel A, Hochgerner H, Lonnerberg P, Johnsson A, Memic F, van der Zwan J, Haring M, Braun E, Borm LE, La Manno G et al. : Molecular Architecture of the Mouse Nervous System. Cell 2018, 174(4):999–1014 e1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skoglund S: Anatomical and physiological studies of knee joint innervation in the cat. Acta Physiol Scand Suppl 1956, 36(124):1–101. [PubMed] [Google Scholar]
- 33.Heppelmann B: Anatomy and histology of joint innervation. J Peripher Nerv Syst 1997, 2(1):5–16. [PubMed] [Google Scholar]
- 34.McDougall JJ, Bray RC, Sharkey KA: Morphological and immunohistochemical examination of nerves in normal and injured collateral ligaments of rat, rabbit, and human knee joints. Anat Rec 1997, 248(1):29–39. [DOI] [PubMed] [Google Scholar]
- 35.Suri S, Gill SE, Massena de Camin S, Wilson D, McWilliams DF, Walsh DA: Neurovascular invasion at the osteochondral junction and in osteophytes in osteoarthritis. Ann Rheum Dis 2007, 66(11):1423–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ashraf S, Wibberley H, Mapp PI, Hill R, Wilson D, Walsh DA: Increased vascular penetration and nerve growth in the meniscus: a potential source of pain in osteoarthritis. Ann Rheum Dis 2011, 70(3):523–529. [DOI] [PubMed] [Google Scholar]
- 37.Mapp PI, Avery PS, McWilliams DF, Bowyer J, Day C, Moores S, Webster R, Walsh DA: Angiogenesis in two animal models of osteoarthritis. Osteoarthritis Cartilage 2008, 16(1):61–69. [DOI] [PubMed] [Google Scholar]
- 38.Mapp PI, Sagar DR, Ashraf S, Burston JJ, Suri S, Chapman V, Walsh DA: Differences in structural and pain phenotypes in the sodium monoiodoacetate and meniscal transection models of osteoarthritis. Osteoarthritis Cartilage 2013, 21(9):1336–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aso K, Shahtaheri SM, Hill R, Wilson D, McWilliams DF, Nwosu LN, Chapman V, Walsh DA: Contribution of nerves within osteochondral channels to osteoarthritis knee pain in humans and rats. Osteoarthritis Cartilage 2020, 28(9):1245–1254. [DOI] [PubMed] [Google Scholar]
- 40.Obeidat AM, Miller RE, Miller RJ, Malfait AM: The nociceptive innervation of the normal and osteoarthritic mouse knee. Osteoarthritis Cartilage 2019, 27(11):1669–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shields SD, Ahn HS, Yang Y, Han C, Seal RP, Wood JN, Waxman SG, Dib-Hajj SD: Nav1.8 expression is not restricted to nociceptors in mouse peripheral nervous system. Pain 2012, 153(10):2017–2030. [DOI] [PubMed] [Google Scholar]
- 42.Miller RE, Malfait AM: Osteoarthritis pain: What are we learning from animal models? Best Pract Res Clin Rheumatol 2017, 31(5):676–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim YS, Anderson M, Park K, Zheng Q, Agarwal A, Gong C, Saijilafu, Young L, He S, LaVinka PC et al. : Coupled Activation of Primary Sensory Neurons Contributes to Chronic Pain. Neuron 2016, 91(5):1085–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller RE, Kim YS, Tran PB, Ishihara S, Dong X, Miller RJ, Malfait AM: Visualization of Peripheral Neuron Sensitization in a Surgical Mouse Model of Osteoarthritis by In Vivo Calcium Imaging. Arthritis Rheumatol 2018, 70(1):88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmidt R, Schmelz M, Forster C, Ringkamp M, Torebjork E, Handwerker H: Novel classes of responsive and unresponsive C nociceptors in human skin. J Neurosci 1995, 15(1 Pt 1):333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prato V, Taberner FJ, Hockley JRF, Callejo G, Arcourt A, Tazir B, Hammer L, Schad P, Heppenstall PA, Smith ES et al. : Functional and Molecular Characterization of Mechanoinsensitive “Silent” Nociceptors. Cell Rep 2017, 21(11):3102–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu S, Zhu J, Zhen G, Hu Y, An S, Li Y, Zheng Q, Chen Z, Yang Y, Wan M et al. : Subchondral bone osteoclasts induce sensory innervation and osteoarthritis pain. J Clin Invest 2019, 129(3):1076–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cohen S, Levi-Montalcini R: Purification and properties of a nerve growth-promoting factor isolated from mouse sarcoma 180. Cancer Res 1957, 17(1):15–20. [PubMed] [Google Scholar]
- 49.Crowley C, Spencer SD, Nishimura MC, Chen KS, Pitts-Meek S, Armanini MP, Ling LH, McMahon SB, Shelton DL, Levinson AD et al. : Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell 1994, 76(6):1001–1011. [DOI] [PubMed] [Google Scholar]
- 50.Denk F, Bennett DL, McMahon SB: Nerve Growth Factor and Pain Mechanisms. Annu Rev Neurosci 2017, 40:307–325. [DOI] [PubMed] [Google Scholar]
- 51.Barker PA, Mantyh P, Arendt-Nielsen L, Viktrup L, Tive L: Nerve Growth Factor Signaling and Its Contribution to Pain. Journal of pain research 2020, 13:1223–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Malfait AM, Miller RE, Block JA: Targeting neurotrophic factors: Novel approaches to musculoskeletal pain. Pharmacol Ther 2020, 211:107553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Petty BG, Cornblath DR, Adornato BT, Chaudhry V, Flexner C, Wachsman M, Sinicropi D, Burton LE, Peroutka SJ: The effect of systemically administered recombinant human nerve growth factor in healthy human subjects. Ann Neurol 1994, 36(2):244–246. [DOI] [PubMed] [Google Scholar]
- 54.Dyck PJ, Peroutka S, Rask C, Burton E, Baker MK, Lehman KA, Gillen DA, Hokanson JL, O’Brien PC: Intradermal recombinant human nerve growth factor induces pressure allodynia and lowered heat-pain threshold in humans. Neurology 1997, 48(2):501–505. [DOI] [PubMed] [Google Scholar]
- 55.Hefti F, Hartikka J, Knusel B: Function of neurotrophic factors in the adult and aging brain and their possible use in the treatment of neurodegenerative diseases. Neurobiol Aging 1989, 10(5):515–533. [DOI] [PubMed] [Google Scholar]
- 56.Apfel SC: Nerve growth factor for the treatment of diabetic neuropathy: what went wrong, what went right, and what does the future hold? Int Rev Neurobiol 2002, 50:393–413. [DOI] [PubMed] [Google Scholar]
- 57.Kras JV, Weisshaar CL, Pall PS, Winkelstein BA: Pain from intra-articular NGF or joint injury in the rat requires contributions from peptidergic joint afferents. Neurosci Lett 2015, 604:193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ashraf S, Mapp PI, Burston J, Bennett AJ, Chapman V, Walsh DA: Augmented pain behavioural responses to intra-articular injection of nerve growth factor in two animal models of osteoarthritis. Ann Rheum Dis 2014, 73(9):1710–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haywood AR, Hathway GJ, Chapman V: Differential contributions of peripheral and central mechanisms to pain in a rodent model of osteoarthritis. Sci Rep 2018, 8(1):7122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vincent TL: Of mice and men: converging on a common molecular understanding of osteoarthritis. Lancet Rheumatol 2020, 2(10):e633–e645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Montagnoli C, Tiribuzi R, Crispoltoni L, Pistilli A, Stabile AM, Manfreda F, Placella G, Rende M, Cerulli GG: beta-NGF and beta-NGF receptor upregulation in blood and synovial fluid in osteoarthritis. Biol Chem 2017, 398(9):1045–1054. [DOI] [PubMed] [Google Scholar]
- 62.Nees TA, Rosshirt N, Zhang JA, Reiner T, Sorbi R, Tripel E, Walker T, Schiltenwolf M, Hagmann S, Moradi B: Synovial Cytokines Significantly Correlate with Osteoarthritis-Related Knee Pain and Disability: Inflammatory Mediators of Potential Clinical Relevance. J Clin Med 2019, 8(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pecchi E, Priam S, Gosset M, Pigenet A, Sudre L, Laiguillon MC, Berenbaum F, Houard X: Induction of nerve growth factor expression and release by mechanical and inflammatory stimuli in chondrocytes: possible involvement in osteoarthritis pain. Arthritis Res Ther 2014, 16(1):R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McNamee KE, Burleigh A, Gompels LL, Feldmann M, Allen SJ, Williams RO, Dawbarn D, Vincent TL, Inglis JJ: Treatment of murine osteoarthritis with TrkAd5 reveals a pivotal role for nerve growth factor in non-inflammatory joint pain. Pain 2010, 149(2):386–392. [DOI] [PubMed] [Google Scholar]
- 65.Driscoll C, Chanalaris A, Knights C, Ismail H, Sacitharan PK, Gentry C, Bevan S, Vincent TL: Nociceptive Sensitizers Are Regulated in Damaged Joint Tissues, Including Articular Cartilage, When Osteoarthritic Mice Display Pain Behavior. Arthritis Rheumatol 2016, 68(4):857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stoppiello LA, Mapp PI, Wilson D, Hill R, Scammell BE, Walsh DA: Structural associations of symptomatic knee osteoarthritis. Arthritis Rheumatol 2014, 66(11):3018–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aso K, Shahtaheri SM, Hill R, Wilson D, McWilliams DF, Walsh DA: Associations of Symptomatic Knee Osteoarthritis With Histopathologic Features in Subchondral Bone. Arthritis Rheumatol 2019, 71(6):916–924. [DOI] [PubMed] [Google Scholar]
- 68.Ren D, Miller R, Malfait A, Miller R: Developing a Functional Imaging Method for Pharmacologically Characterizing Intra-articular Sensory Neurons. Orthopedic Research Society annual meeting (abstract) 2020. [Google Scholar]
- 69.Murthy SE, Dubin AE, Patapoutian A: Piezos thrive under pressure: mechanically activated ion channels in health and disease. Nat Rev Mol Cell Biol 2017, 18(12):771–783. [DOI] [PubMed] [Google Scholar]
- 70.Beaulieu-Laroche L, Christin M, Donoghue A, Agosti F, Yousefpour N, Petitjean H, Davidova A, Stanton C, Khan U, Dietz C et al. : TACAN Is an Ion Channel Involved in Sensing Mechanical Pain. Cell 2020, 180(5):956–967.e917. [DOI] [PubMed] [Google Scholar]
- 71.Murthy SE, Loud MC, Daou I, Marshall KL, Schwaller F, Kuhnemund J, Francisco AG, Keenan WT, Dubin AE, Lewin GR et al. : The mechanosensitive ion channel Piezo2 mediates sensitivity to mechanical pain in mice. Sci Transl Med 2018, 10(462). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Szczot M, Liljencrantz J, Ghitani N, Barik A, Lam R, Thompson JH, Bharucha-Goebel D, Saade D, Necaise A, Donkervoort S et al. : PIEZO2 mediates injury-induced tactile pain in mice and humans. Sci Transl Med 2018, 10(462). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Szczot M, Pogorzala LA, Solinski HJ, Young L, Yee P, Le Pichon CE, Chesler AT, Hoon MA: Cell-Type-Specific Splicing of Piezo2 Regulates Mechanotransduction. Cell Rep 2017, 21(10):2760–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sousa-Valente J, Calvo L, Vacca V, Simeoli R, Arevalo JC, Malcangio M: Role of TrkA signalling and mast cells in the initiation of osteoarthritis pain in the monoiodoacetate model. Osteoarthritis Cartilage 2018, 26(1):84–94. [DOI] [PubMed] [Google Scholar]
- 75.Lee W, Leddy HA, Chen Y, Lee SH, Zelenski NA, McNulty AL, Wu J, Beicker KN, Coles J, Zauscher S et al. : Synergy between Piezo1 and Piezo2 channels confers high-strain mechanosensitivity to articular cartilage. Proc Natl Acad Sci U S A 2014, 111(47):E5114–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Servin-Vences MR, Moroni M, Lewin GR, Poole K: Direct measurement of TRPV4 and PIEZO1 activity reveals multiple mechanotransduction pathways in chondrocytes. Elife 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li X, Han L, Nookaew I, Mannen E, Silva MJ, Almeida M, Xiong J: Stimulation of Piezo1 by mechanical signals promotes bone anabolism. Elife 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hochberg MC, Tive LA, Abramson SB, Vignon E, Verburg KM, West CR, Smith MD, Hungerford DS: When Is Osteonecrosis Not Osteonecrosis?: Adjudication of Reported Serious Adverse Joint Events in the Tanezumab Clinical Development Program. Arthritis Rheumatol 2016, 68(2):382–391. [DOI] [PubMed] [Google Scholar]
- 79.Jain A, Hakim S, Woolf CJ: Unraveling the Plastic Peripheral Neuroimmune Interactome. J Immunol 2020, 204(2):257–263. [DOI] [PubMed] [Google Scholar]
- 80.van den Bosch MHJ, van Lent P, van der Kraan PM: Identifying effector molecules, cells, and cytokines of innate immunity in OA. Osteoarthritis Cartilage 2020, 28(5):532–543. [DOI] [PubMed] [Google Scholar]
- 81.Newton K, Dixit VM: Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol 2012, 4(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Miller RE, Belmadani A, Ishihara S, Tran PB, Ren D, Miller RJ, Malfait AM: Damage-associated molecular patterns generated in osteoarthritis directly excite murine nociceptive neurons through Toll-like receptor 4. Arthritis Rheumatol 2015, 67(11):2933–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Blom AB, van den Bosch MH, Blaney Davidson EN, Roth J, Vogl T, van de Loo FA, Koenders M, van der Kraan PM, Geven EJ, van Lent PL: The alarmins S100A8 and S100A9 mediate acute pain in experimental synovitis. Arthritis Res Ther 2020, 22(1):199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Miller RE, Ishihara S, Tran PB, Golub SB, Last K, Miller RJ, Fosang AJ, Malfait AM: An aggrecan fragment drives osteoarthritis pain through Toll-like receptor 2. JCI Insight 2018, 3(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Raoof R, Willemen H, Eijkelkamp N: Divergent roles of immune cells and their mediators in pain. Rheumatology (Oxford, England) 2018, 57(3):429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Miller RE, Tran PB, Ishihara S, Syx D, Ren D, Miller RJ, Valdes AM, Malfait AM: Microarray analyses of the dorsal root ganglia support a role for innate neuro-immune pathways in persistent pain in experimental osteoarthritis. Osteoarthritis Cartilage 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Miller RJ, Jung H, Bhangoo SK, White FA: Cytokine and chemokine regulation of sensory neuron function. Handb Exp Pharmacol 2009(194):417–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Miller RE, Tran PB, Das R, Ghoreishi-Haack N, Ren D, Miller RJ, Malfait AM: CCR2 chemokine receptor signaling mediates pain in experimental osteoarthritis. Proc Natl Acad Sci U S A 2012, 109(50):20602–20607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Longobardi L, Temple JD, Tagliafierro L, Willcockson H, Esposito A, D’Onofrio N, Stein E, Li T, Myers TJ, Ozkan H et al. : Role of the C-C chemokine receptor-2 in a murine model of injury-induced osteoarthritis. Osteoarthritis Cartilage 2017, 25(6):914–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Miotla Zarebska J, Chanalaris A, Driscoll C, Burleigh A, Miller RE, Malfait AM, Stott B, Vincent TL: CCL2 and CCR2 regulate pain-related behaviour and early gene expression in post-traumatic murine osteoarthritis but contribute little to chondropathy. Osteoarthritis Cartilage 2017, 25(3):406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Vincent TL: Peripheral pain mechanisms in osteoarthritis, PAIN: September 2020 - Volume 161 - Issue - p Pain 2020, 161(doi: 10.1097/j.pain.0000000000001923):S138–S146 [DOI] [PMC free article] [PubMed] [Google Scholar]