

Abstract
Objectives
To better understand how immune responses may be harnessed against breast cancer, we investigated which immune cell types and signalling pathways are required for spontaneous control of a mouse model of mammary adenocarcinoma.
Methods
The NOP23 mammary adenocarcinoma cell line expressing epitopes derived from the ovalbumin model antigen is spontaneously controlled when orthotopically engrafted in syngeneic C57BL/6 mice. We combined this breast cancer model with antibody‐mediated depletion of lymphocytes and with mutant mice affected in interferon (IFN) or type 1 conventional dendritic cell (cDC1) responses. We monitored tumor growth and immune infiltration including the activation of cognate ovalbumin‐specific T cells.
Results
Breast cancer immunosurveillance required cDC1, NK/NK T cells, conventional CD4+ T cells and CD8+ cytotoxic T lymphocytes (CTLs). cDC1 were required constitutively, but especially during T‐cell priming. In tumors, cDC1 were interacting simultaneously with CD4+ T cells and tumor‐specific CTLs. cDC1 expression of the XCR1 chemokine receptor and of the T‐cell‐attracting or T‐cell‐activating cytokines CXCL9, IL‐12 and IL‐15 was dispensable for tumor rejection, whereas IFN responses were necessary, including cDC1‐intrinsic signalling by STAT1 and IFN‐γ but not type I IFN (IFN‐I). cDC1 and IFNs promoted CD4+ and CD8+ T‐cell infiltration, terminal differentiation and effector functions. In breast cancer patients, high intratumor expression of genes specific to cDC1, CTLs, CD4+ T cells or IFN responses is associated with a better prognosis.
Conclusion
Interferons and cDC1 are critical for breast cancer immunosurveillance. IFN‐γ plays a prominent role over IFN‐I in licensing cDC1 for efficient T‐cell activation.
Keywords: breast cancer, cancer immunosurveillance, CD4+ T cells, CD8+ T cells, cDC1, IFN‐γ, interferons
Type 1 conventional dendritic cells (cDC1) cross‐present tumor antigens to CD8+ T cells. Understanding the regulation of their antitumor functions is important. In this study, we show that cell‐intrinsic STAT1/IFN‐γ signalling licenses cDC1 for efficient CD4+ and CD8+ T‐cell activation during breast cancer immunosurveillance, as assessed by using a mouse mammary adenocarcinoma orthotopic transplantation model combined with mutant animals enabling cDC1‐specific targeting.
Introduction
Conventional dendritic cells (cDCs) are specialised in antigen (Ag) capture, processing and presentation for T‐cell priming. 1 , 2 cDCs are present in lymphoid organs and peripheral tissues. Lymphoid‐resident cDCs (Res‐cDCs) of the spleen and lymph nodes (LNs) participate in the capture of Ag from blood and lymph respectively. cDC in non‐lymphoid tissues capture Ag at the periphery and migrate to the LNs via the afferent lymphatic vessels. cDC encompass two distinct cell types. 1 Type 1 cDC (cDC1) excel in cytotoxic CD8+ T‐cell (CTL) activation, particularly via cross‐presentation of cell‐associated Ag. 3 , 4 , 5 , 6 , 7 , 8 , 9 Type 2 cDC (cDC2) are particularly effective for helper CD4+ T‐cell activation. 2
Previous studies suggested that cDC1 play a non‐redundant role in antitumor immunity, both for spontaneous control of syngeneic tumor grafts used as a surrogate model for cancer immunosurveillance, and for rejection of established tumors upon immunotherapy. 10 , 11 In human patients suffering from various cancers, several studies have shown that high expression in tumors of genes selectively expressed in cDC1 is of good prognosis, as reviewed in Cancel et al. 10 and exemplified for breast cancer in Bottcher et al. 12 and Hubert et al. 13 Hence, cDC1 represent an attractive target for new generation cancer immunotherapies. 3 , 10 , 11 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 Yet, most of the studies that investigated the antitumor role of cDC1 in vivo used mutant mice whose deficiencies were not affecting exclusively cDC1. Irf8 deficiency in CD11c‐expressing cells also affected the differentiation and functions of plasmacytoid dendritic cells 23 and inflammatory cDC2. 24 Batf3 knockout does not only abrogate cDC1 differentiation 25 but was also recently shown to enhance CD4+ regulatory T‐cell (Treg) induction 26 and to hamper CTL survival and memory 27 , 28 in a cell‐intrinsic manner. Hence, mouse models targeting cDC1 with a higher specificity are mandatory to investigate whether and how they contribute to antitumor immunity. 27 This is the case of mice knocked‐in for the CRE recombinase in the Xcr1 locus. 29
Several key features of cDC1 are proposed to contribute to their critical role in antitumor immunity, beyond their efficiency at cross‐presenting cell‐associated Ag. 10 , 11 , 14 , 17 cDC1 may have the unique ability to simultaneously deliver to CTLs a series of complementary output signals ensuring their optimal response. CXCL9 attracts CXCR3‐expressing memory or effector CTLs to the tumors. IL‐12 production and IL‐15 transpresentation promote CTL IFN‐γ expression and proliferation. cDC1 could also promote delivery to CTLs of help from other immune cells including CD4+ T cells. 10 , 30 , 31 However, which of these output signals are critical for cDC1 antitumor immunity remains to be rigorously investigated. 10 , 14
The antitumor functions of cDC1 have been proposed to depend on their integration of specific input signals, instructing them to deliver the right output signals to CTLs. cDC1 recruitment into the tumor can be promoted by engagement of their chemokine receptor XCR1, 12 whose ligand XCL1 is produced by activated NK cells and CTLs. 8 Triggering of the receptor for type I interferons (IFNAR) on cDC1 is necessary for rejection of immunogenic melanoma and fibrosarcoma by promoting Ag cross‐presentation. 32 , 33 It also boosts their expression of costimulation molecules, induces their transpresentation of IL‐15 and can promote their production of IL‐12. 10 , 34 However, whether these input signals are always required by cDC1 for their antitumor functions is unknown. Moreover, to which extent and how different types of interferons (IFNs) promote the immunogenic maturation of cDC1, not only type I IFNs (IFN‐I) but also type III IFNs (IFN‐III) or IFN‐γ, remains to be formally investigated.
Here, we studied whether cDC1 promote spontaneous immunity to breast cancer, and when, where and how this is achieved. To this aim, we used a mouse model of spontaneous immune control of breast cancer in C57BL/6 female mice, consisting of an orthotopic graft of a clone of the NOP23 syngeneic breast adenocarcinoma cell line expressing epitopes from the ovalbumin (OVA) model Ag. 35 This experimental system enabled us to study the cellular and molecular mechanisms underpinning spontaneous immune control of breast cancer, by harnessing the Karma‐tmt‐hDTR (Karma) 36 , Xcr1Cre / wt ; Rosa26DTA / wt (Xcr1‐DTA), Xcr1Cre and KarmaCre 29 mutant mouse models that we have generated and validated to specifically target cDC1, in combination with other mutant C57BL/6 mice. We showed that cDC1 and IFNs are essential for the spontaneous immune control of breast cancer. We demonstrated that cell‐intrinsic STAT1/IFN‐γ signalling in cDC1 licenses them for efficient CD4+ and CD8+ T‐cell activation during breast cancer immunosurveillance.
Results
CTL, CD4+ Tconv and NK1.1+ cells are instrumental to rejection of NOP23 mammary tumors
We first investigated whether T or NK cells were providing the effector arm of the spontaneous rejection of the NOP23 breast adenocarcinoma cells in C57BL/6J females. Tumor rejection was abolished by continuous depletion of CTLs or of NK1.1+ cells (i.e. NK cells and a fraction of NKT cells) (Figure 1a). However, the anti‐NK1.1 mAb‐mediated depletion showed a delayed and milder effect than CTL depletion. Continuous depletion of all CD4+ T cells also abrogated tumor control (Figure 1b), whereas this was not the case of the selective depletion of intratumor Tregs as achieved with administration of the anti‐CTLA‐4 clone 9D9 37 (Supplementary figure 1). Thus, CTLs, CD4+ conventional T cells (Tconv) and NK/NK T cells are required for NOP23 mammary tumor rejection.
Figure 1.
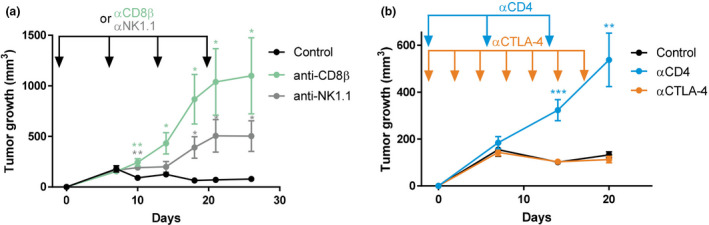
CTL, CD4+ Tconv and NK1.1+ cells are instrumental in spontaneous rejection of breast cancer NOP23. (a) NOP23 tumor growth (mean ± SEM) in the mammary fat pad of female mice treated or not with anti‐CD8β or anti‐NK1.1 mAb at the indicated time (arrows) with the first injection given 1 day before tumor engraftment. One experiment representative of at least 2 independent ones with 5 mice per group is shown. (b) NOP23 tumor growth (mean ± SEM) in female mice treated or not with anti‐CD4 (once a week) or anti‐CTLA‐4 (every 3 days) mAb at the indicated time (arrows) with the first injection given 1 day before tumor engraftment. n = 7 mice per group. *, P < 0.05; **, P < 0.01; and ***, P < 0.001; unpaired t‐test.
cDC1 are critical for breast cancer control, especially during the T‐cell priming phase
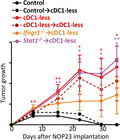
We next examined whether and when cDC1 depletion compromised the immune control of NOP23 growth, by taking advantage of our mutant mouse models specifically targeting cDC1. The Xcr1Cre / wt ; Rosa26DTA / wt (Xcr1‐DTA) model 29 harboured a constitutive and complete lack of all cDC1 in the spleen, inguinal lymph nodes (IngLNs), tumor and tumor‐draining lymph node (TdLN) (Supplementary figures 2a, 3 and 4a). The Xcr1Cre / w t; Rosa26hDTR / wt (Xcr1‐hDTR) mouse model allowed conditional depletion of cDC1 in spleen, and of migratory cDC1 (Mig‐cDC1) and Res‐cDC1 in the IngLNs, for at least 2 days following the administration of a single dose of diphtheria toxin (DT) (Supplementary figure 3a, b), similar to the Karma‐tmt‐hDTR (Karma) mice. 36 None of the other immune cell types examined in the spleen and IngLNs were affected in these mice (Supplementary figures 2 and 3c). Xcr1‐DTA mice harboured a progressive and unabated growth of NOP23 as compared to control WT animals (Figure 2a), demonstrating that cDC1 are necessary to efficiently reject this breast adenocarcinoma. To determine when cDC1 were required to promote this rejection, we conditionally depleted these cells in Karma mice for 10 days during different time windows relative to tumor engraftment (Figure 2a). The earlier the depletions were initiated, the stronger the tumors grew. However, in mice with a transient cDC1 depletion the tumors never grew as strongly as in the Xcr1‐DTA mice that are constitutively devoid of cDC1 (Figure 2a, b). TdLN mig‐cDC1 showed a high expression of the costimulatory molecules CD40 and CD86 at day 4 post‐engraftment, as compared to Res‐cDC1 or to Mig‐cDC1 on any other days (Figure 2c). In summary, cDC1 were required continuously until tumor rejection, but their presence was especially critical during the first 4 days after tumor engraftment, most likely during the T‐cell priming phase.
Figure 2.
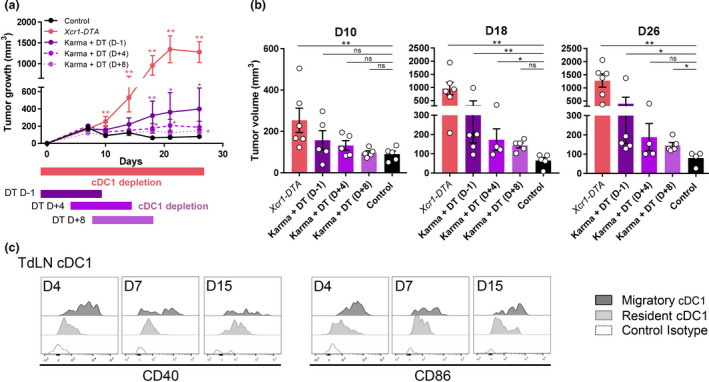
cDC1 are instrumental in spontaneous rejection of breast cancer NOP23, especially during the phase of T‐cell priming. (a) Tumor growth (mean ± SEM) in control (n = 5), in constitutively cDC1‐depleted (Xcr1‐DTA, n = 6) or conditionally cDC1‐depleted (Karma‐tmt‐DTR + DT) female mice. Karma‐tmt‐DTR mice were injected 4 times with DT every 60 h, starting 1 day before engraftment (n = 5, representative of 3 independent datasets), at d + 4 post‐engraftment (n = 5, representative of 2 independent datasets) or at d + 8 post‐engraftment (n = 5, representative of 2 independent datasets). (b) Tumor volumes as measured in a at days 10, 18 and 26. Data are shown as mean ± SEM, with values for individual mice shown as white circles. ns, not significant (P > 0.05); *, P < 0.05; **, P < 0.01; and ***, P < 0.001; non‐parametric Mann–Whitney U‐test. (c) Analysis of CD40 and CD86 expression by flow cytometry on TdLN Mig‐cDC1 and Res‐cDC1. The data shown are from one experiment representative of two independent ones.
CCR7‐ and S1PR1‐dependent immune cell trafficking is critical for NOP23 rejection
At day 7 post‐engraftment, CCR7 was upregulated on cDC1 that had migrated from the tumor site into the TdLN, in addition to being also highly expressed on macrophages in the TdLN and in effector CTLs in the tumor microenvironment (TME) (Figure 3a). We thus investigated whether CCR7‐dependent immune cell trafficking was critical for NOP23 rejection. In Ccr7 −/− mice, the NOP23 tumor grew uncontrolled (Figure 3b). Blocking lymphocyte egress from secondary lymphoid organs with the sphingosine‐1‐phosphate receptor (S1PR1) inhibitor FTY720 38 also abrogated spontaneous tumor control (Figure 3c). It reduced intratumor infiltration of lymphocytes as compared to control animals or to mice depleted of CTLs or NK1.1+ cells (Figure 3d). Taken together, these results suggested that two‐way traffic of immune cells between the tumor site and TdLNs was critical for the establishment of an effective endogenous antitumor immune response within the TME.
Figure 3.
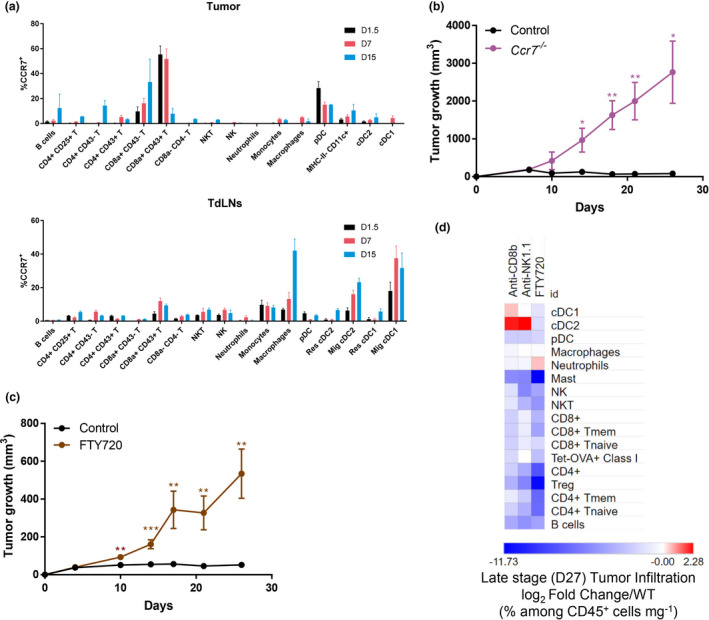
Trafficking of immune cells into and out of the TdLNs is instrumental in breast cancer spontaneous rejection. (a) Kinetics of CCR7 expression amongst immune populations in control tumor and TdLNs (2–6 mice per time point, data are shown as mean ± SEM). T cells were split into different phenotypic populations known to be associated with their activation state, including expression of the glycosylated isoform of CD43 that is specifically expressed on effector T cells, in particular cytotoxic CD8+ T cells. (b) Tumor growth (mean ± SEM) of Ccr7 −/− (n = 3) and control (n = 5) females. One experiment representative of two independent ones is shown. (c) Tumor growth (mean ± SEM) of FTY720‐treated (n = 7) and untreated (control, n = 7) female mice. One experiment representative of two independent ones is shown. (d) Heatmap representing the immune landscapes in d27 tumors in CD8+ T‐cell‐depleted (anti‐CD8β), in NK cell‐depleted (anti‐NK1.1) or in FTY720‐treated females compared with control. The data are shown as log2 fold changes of the %Population/CD45+/mg of tumor in analysed animals compared with WT. n = 3 or 4 mice per group. *, P < 0.05; **, P < 0.01; and ***, P < 0.001; unpaired t‐test.
cDC1 interact with CD4+ T cells and tumor‐specific CTLs in the TME
Because cDC1, CTLs and CD4+ Tconv were all critical to trigger NOP23 rejection, we wondered whether cDC1 interacted with T cells in the TME. To follow the behaviour of Ag‐specific CTLs in our experimental models, 1 day before tumor engraftment we adoptively transferred low numbers (1000) of naïve GFP‐expressing OT‐I cells into Xcr1Cre / wt ; Rosa26tdRFP / wt mice that allow in situ visualisation of cDC1 by microscopy because of their specific expression of the RFP fluorescent reporter protein (Supplementary figure 5). At d7 in the tumor bed, RFP+ cDC1 were engaged in cell–cell contacts with both CD4+ T cells and antitumor CTLs, in close proximity to the HER2+ NOP23 cells (Figure 4). This suggested that, in the tumor stroma, cDC1 might have been simultaneously cross‐presenting tumor Ags to CTLs and relaying them the CD4+ Tconv help.
Figure 4.
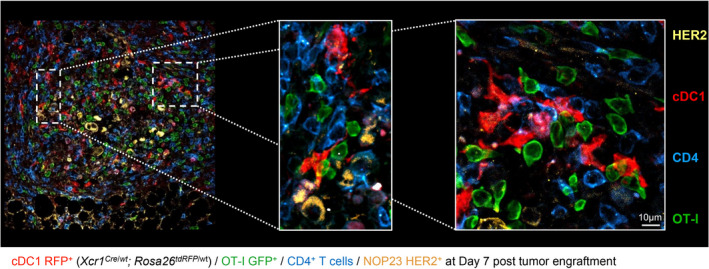
cDC1 interact with CD4+ T and tumor‐specific CD8+ T cells together in the tumor microenvironment. Xcr1Cre / wt ; Rosa26tdRFP / wt mice were adoptively transferred with 1000 GFP+ OT‐I cells 1 day prior to tumor engraftment. 7d post‐engraftment, tumors sections were stained for RFP expression (cDC1), GFP (OT‐I), CD4 (CD4+ T cells) and HER2 (NOP23 cells). This image is representative of 5 individual mice.
CXCL9 or IL‐12 production by cDC1 and their transpresentation of IL‐15 are individually dispensable for the immune control of NOP23 tumors
To dissect how cDC1 promoted spontaneous immune control of NOP23, we first tested the candidate molecules CXCL9, IL‐12 and IL‐15, which had been proposed to be key output signals delivered by cDC1 to T or NK/NK T cells for their recruitment into the tumor or the activation of their effector functions. 10 The NOP23 tumors were controlled in Cxcl9 −/−, Il12b −/− and Il15ra −/− mice as efficiently as in control mice (Figure 5a). Consistent with these results, conditional inactivation of Cxcl9 or Il15ra in cDC1 had no impact on tumor growth (Supplementary figure 6). Thus, CXCL9 or IL‐12 production and IL‐15 transpresentation by cDC1 were not required individually for breast cancer rejection in our experimental settings.
Figure 5.
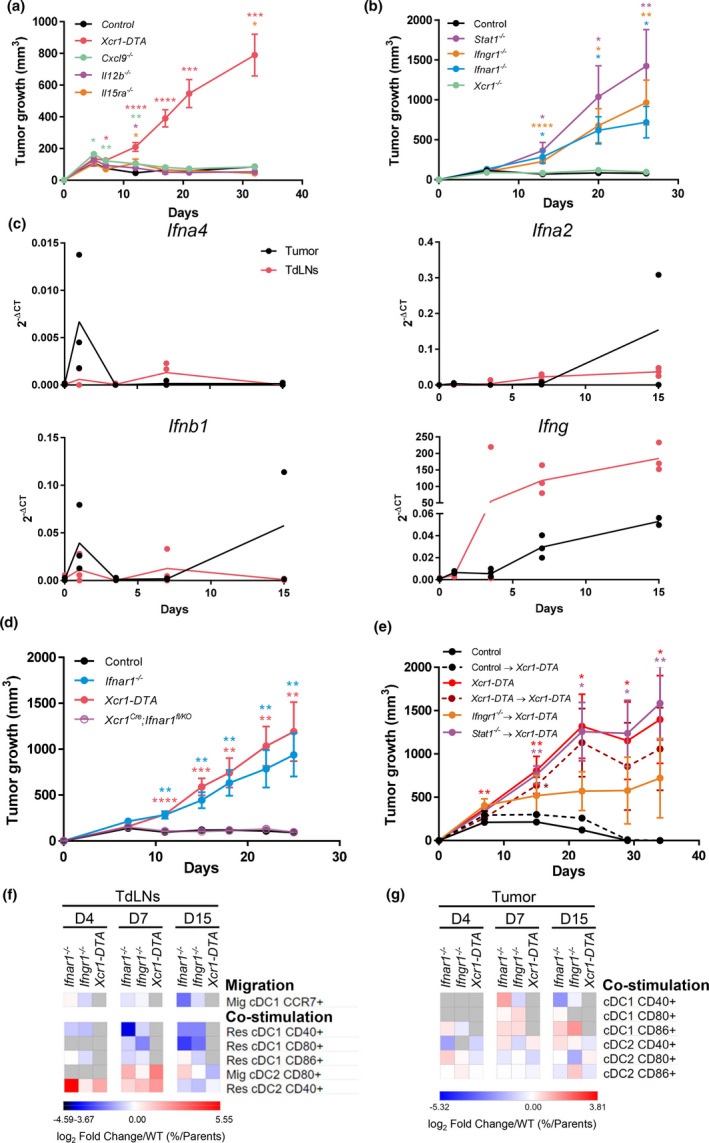
IFNs and cDC1‐intrinsic IFN‐γ and STAT1 signalling are necessary for breast cancer spontaneous rejection. (a) Tumor growth (mean ± SEM) in Xcr1‐DTA (n = 10), Cxcl9 −/− (n = 9), Il12b −/− (n = 6), Il15ra −/− (n = 5) and control (n = 8) female mice. One experiment representative of two independent ones is shown. (b) Tumor growth (mean ± SEM) in Xcr1 −/− (n = 4), Ifnar1 −/− (n = 7), Ifngr1 −/− (n = 5), Stat1 −/− (n = 5) and control (n = 6) female mice. One experiment representative of at least 2 independent ones is shown. (c) Expression analysis of the Ifna4, Ifna2, Ifnb and Ifng genes in control tumors and their TdLNs (n = 2‐4) by qPCR. (d) Tumor growth (mean ± SEM) in Xcr1‐DTA (n = 7), Ifnar1 −/− (n = 8), Xcr1cre ; Ifnar1fl / KO (n = 13) and control (n = 7) female mice. One experiment representative of two independent ones is shown. (e) Tumor growth (mean ± SEM) in different types of shield bone marrow chimeric female mice, control→Xcr1‐DTA (n = 4), Xcr1‐DTA→Xcr1‐DTA (n = 4), Ifngr1 −/−→Xcr1‐DTA (n = 4) and Stat1 −/−→Xcr1‐DTA (n = 4). Xcr1‐DTA (n = 5) and control (n = 5) female mice were used as controls. One experiment representative of two independent ones is shown. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001; unpaired t‐test. (f, g) Heatmaps representing the expression of costimulatory receptors on lymphoid‐resident (Res) and migratory (Mig) cDC1 and cDC2 in the TdLNs (f) and tumors (g) at d4, d7 and d15 after engraftment in Ifnar1 −/−, Ifngr1 −/− and Xcr1‐DTA compared with control mice. The data are shown as log2 fold changes in the ratio of % Population/Parent population from mutant animals to WT (n = 3–6 mice per group). The data shown are from two independent experiments pooled together.
IFN‐I and IFN‐γ responses, but not XCR1, are necessary for NOP23 tumor control
We next sought to identify the input signals received by cDC1 and promoting their antitumor functions. Xcr1 −/− mice efficiently controlled the NOP23 tumor cells (Figure 5b). Hence, XCR1‐dependent recruitment of cDC1 to the tumor 12 or micro‐anatomical attraction to XCL1‐producing effector lymphocytes within the tissue 39 was not necessary for breast tumor elimination in our experimental settings. To assess functionally the importance of IFN‐I and IFN‐γ signalling in NOP23 control, we compared tumor growth between WT animals and Ifnar1 −/−, Ifngr1 −/− or Stat1 −/− mice, respectively, lacking the ability to respond to IFN‐I, to IFN‐γ or to all types of IFNs including type III IFNs (IFN‐III). All three mutant mice failed to control tumor growth, with a more pronounced effect in Stat1 −/− mice (Figure 5b). Thus, both IFN‐I and IFN‐γ were promoting antitumor immunity in the NOP23 breast cancer model. The analysis of the kinetics of induction of the Ifna4, Ifna2 and Ifnb1 genes and of interferon‐stimulated genes (ISGs) showed that IFN‐I responses were induced in the tumor within the first day after engraftment before rapidly decreasing, while they remained generally low in the TdLNs (Figure 5c and Supplementary figure 7). Conversely, Ifng expression increased gradually over time and was higher in the TdLNs than in the tumors (Figure 5c). These results suggested that distinct IFNs and ISGs could have complementary roles at different times and locations to initiate and maintain protective antitumor immune responses.
The antitumor protective effects of IFN‐γ and STAT1 occur at least in part in cDC1, whereas cDC1‐intrinsic signalling by IFN‐I is dispensable for NOP23 control
We next investigated whether the protective antitumor roles of IFNs were at least in part because of cell‐intrinsic effects on cDC1. NOP23 cells were efficiently rejected in Xcr1Cre ; Ifnar1fl / KO and KarmaCre / wt ; Ifnar1fl / fl mice that are deficient for IFN‐I responsiveness selectively in cDC1 (Figure 5d and Supplementary figure 8a). Moreover, cDC1 maturation and tumor‐specific CTL activation in tumors of Xcr1Cre ; Ifnar1fl / KO mice were similar to those in control tumors (Supplementary figures 4 and 8b, c). Thus, in our experimental settings, cDC1‐intrinsic signalling by IFN‐I was dispensable for NOP23 control.
To determine whether IFN‐γ or overall IFN responses in cDC1 were critical for their promotion of NOP23 rejection, we generated shield bone marrow chimeric (SBMC) mice deficient selectively in cDC1 for key components of the corresponding signalling pathways, namely Ifngr1 −/−→Xcr1‐DTA and Stat1 −/−→Xcr1‐DTA animals. Tumors grew progressively in Ifngr1 −/−→Xcr1‐DTA SBMC mice, but to a lower extent than in Stat1 −/−→Xcr1‐DTA SBMC mice that harboured a progressive and unabated tumor growth, similar to Xcr1‐DTA animals and Xcr1‐DTA→Xcr1‐DTA SBMC mice (Figure 5e). Thus, the beneficial antitumor effects of IFN‐γ and STAT1 signalling occurred at least in part in cDC1.
Compared with controls, Mig‐cDC1 from Ifnar1 −/− and Ifngr1 −/− TdLNs expressed less CCR7, and their Res‐cDC1 were less mature with a decrease in CD40, CD80 and CD86 expression (Figure 5f), contrasting with a higher expression of CD40 on Res‐cDC2 at d4‐7, and of CD80 on Mig‐cDC2 at d7 (Figure 5f). cDC1 expressed less CD40 in the tumors from Ifnar1 −/− and Ifngr1 −/− mice at d15 (Figure 5g). This suggested that loss of IFN responses led to a defective cDC1 maturation in the tumor and TdLNs, with a compensatory increase in cDC2 maturation that was not sufficient to maintain protective antitumor immunity.
cDC1 and IFNs promote the infiltration of the tumor and its draining lymph node by protective over putatively deleterious immune cell types
To better understand the respective roles of cDC1 and IFNs in the antitumor response, we quantified different immune populations in control, Ifnar1 −/−, Ifngr1 −/− and Xcr1‐DTA tumor at d4, d7 and d15 (Figure 6, Supplementary figures 4, 9a). The overall immune cell infiltration in the tumors increased between d4 and d7, the highest in WT mice as compared to mutant animals, and later decreased in all mice (Figure 6a). At d4, the major difference observed between mutant mice and WT controls was a decrease in cDC1 (Figure 6b). At this stage, the tumor was mainly infiltrated in all mouse strains by neutrophils, macrophages and γδ T lymphocytes identified as CD8− CD4− CD3ε+ cells (Supplementary figure 9a). At day 7, the proportion of cDC1 and NK cells within immune cells remained lower in mutant animals as compared to WT mice (Figure 6b, c and Supplementary figure 9a). In contrast, the immune infiltrate from the tumors of mutant animals harboured increased proportion of Lin− Siglec‐H− CD64− CD11c+ MHC‐II− cells (Figure 6d and Supplementary figure 9a), corresponding most likely to DC precursors 40 or immature DC. As compared to WT controls, the mutant mice showed a slight decrease in CD8+ T‐cell proportion at d7 (Figure 6d, e and Supplementary figure 9) and a marked decrease in the proportions of CD4+ T cells mostly at d7 (Figure 6d, f and Supplementary figure 9). The delayed tumor growth observed upon NK1.1+ cell depletion as compared to CTL depletion (Figure 1a) was consistent with the late infiltration of the tumor by NK cells (Figure 6c) and NK T cells (Figure 6g) as compared to T cells (Figure 6e, f). The fraction of activated (CD44+) CTLs in the immune infiltrates remained lower in all mutant animals (Figure 6d and Supplementary figure 9). This was also the case for the fraction of CD4+ T cells in Xcr1‐DTA mice (Figure 6d, f and Supplementary figure 9) and for the fraction of NK cells in Ifngr1 −/− animals (Figure 6c, d and Supplementary figure 9). Conversely, at d15, the proportion of neutrophils in the immune infiltrates was much higher in mutant mice than in WT controls (Figure 6d), even though the absolute numbers of neutrophils in tumors decreased sharply over time (Supplementary figure 9a).
Figure 6.
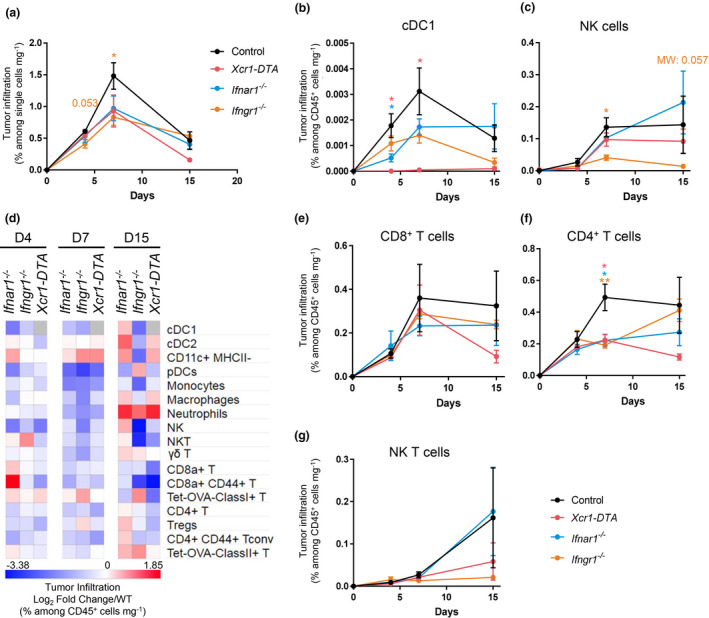
cDC1, type I IFN and type II IFN signalling shape the tumor immune landscape. (a) Kinetics of the tumor infiltration by CD45+ cells at d4, d7 and d15, in control, Ifnar1 −/−, Ifngr1 −/− and Xcr1‐DTA mice, as assessed by flow cytometry. (b, c) Kinetics of the tumor infiltration by cDC1 (b) and NK cells (c) at d4, d7 and d15, in control, Ifnar1 −/−, Ifngr1 −/− and Xcr1‐DTA mice. (d) Heatmap representing the tumor immune landscapes in Ifnar1 −/−, Ifngr1 −/− and Xcr1‐DTA mice at d4, d7 and d15. The data are shown as log2 fold changes calculated as the ratio of % Population/CD45+/mg of tumor from mutant animals to WT (n = 3–6 mice per group). The data shown are from two independent experiments pooled together. (e–g) Kinetics of the tumor infiltration by CD8+ T cells (e), CD4+ T cells (f) and NK T cells (g) in control, Ifnar1 −/−, Ifngr1 −/− and Xcr1‐DTA mice. For a–c, e–g, the data shown (mean ± SEM) are from two independent experiments pooled together (n = 3‐6 mice per group). For a–c, e–g, data are shown as mean ± SEM. ns, not significant (P > 0.05); *, P < 0.05; **, P < 0.01; and ***, P < 0.001 according to the unpaired t‐test or non‐parametric Mann–Whitney U‐test (MW) when specified.
In the TdLNs (Supplementary figure 2), the proportion of activated (CD44+) CTL was lower at all times in Xcr1‐DTA and Ifnar1 −/− mice (Supplementary figure 9b). Neutrophils were increased over time in all mutant mice, and monocytes or macrophages in Xcr1‐DTA and Ifngr1 −/− mice, as compared to WT animals (Supplementary figure 9b). Starting at d4, Mig‐cDC1 accumulated less in Ifnar1 −/− and Ifngr1 −/− mice than in WT animals (Supplementary figure 9b). Conversely, the proportion of migratory cDC2 (Mig‐cDC2) increased over time (Supplementary figure 9b). This suggested that the early migration of cDC1 from the tumor to the TdLNs is IFN‐dependent and that its absence leads to a compensatory phenomenon of increased cDC2 migration from the tumor to the TdLN.
Altogether, these results showed that IFNs and cDC1 contributed to sculpt the immune composition of the tumor and TdLN by promoting a higher ratio of protective immune cells, not only NK cells and CTLs but also CD4+ Tconv, over potentially deleterious myeloid cells including subtypes of macrophages 41 and neutrophils. IFN effects on the tumor immune infiltration may have occurred in part indirectly through promoting early cDC1 recruitment.
cDC1 and IFNs are necessary for CD4+ Tconv and CTL terminal activation and effector functions in the TME
We then focused on tumor‐specific (Tetramer+) CD4+ T cells and CTL responses. Their proportions within tumor‐infiltrating immune cells did not differ between experimental groups (Figure 7a). As compared to total T cells, tumor‐infiltrating Tetramer+ T cells expressed higher levels of the checkpoint receptors PD‐1, Tim‐3 or LAG3 (Figure 7b, Supplementary figures 4b, 10), whose individual expression has been shown to peak at maximal effector phase and reflects CTL activation rather than exhaustion. 42 , 43 Tumor‐infiltrating Tetramer+ CD4+ T cells or CTLs harboured decreased percentages of PD‐1+ or Tim‐3+ cells in mutant mice as compared to control animals, suggesting an incomplete effector differentiation (Figure 7b and Supplementary figure 10). This was also the case for Tetramer+ CTLs in TdLN (Supplementary figure 11). The coexpression on the same cell of multiple checkpoint receptors, such as PD‐1, Tim‐3 and LAG3, has been proposed to define functionally exhausted or dysfunctional lymphocytes. 42 The proportion of triple‐positive cells were very low on both total and Tetramer+ T lymphocytes not only in WT mice but also in mutant animals, suggesting that the vast majority of antitumor T cells were not exhausted even in the absence of cDC1 or of IFN responses (Supplementary figure 12). Altogether, these results showed that cDC1 and IFNs were essential to promote CTL and CD4+ T‐cell effector differentiation in the tumor and TdLNs.
Figure 7.
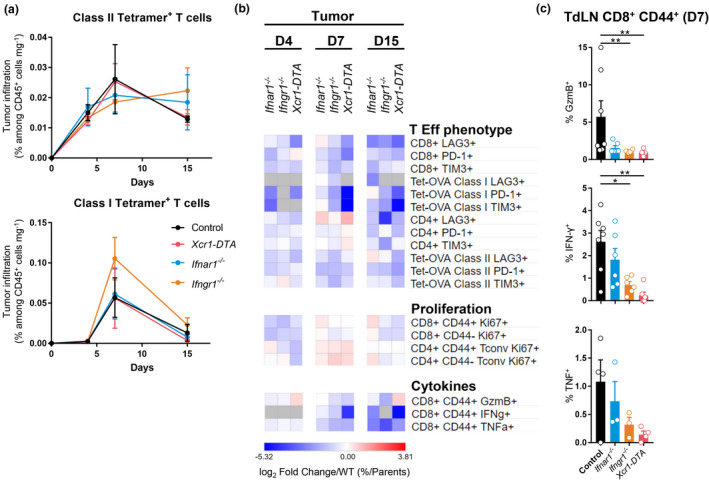
cDC1, type I IFN and type II IFN signalling are necessary for CD4+ and CD8+ T‐cell terminal activation and effector functions in the TME. (a) Kinetics of the tumor infiltration by Ag‐specific CD4+ (up) and CD8+ (bottom) T cells at d4, d7 and d15 in Ifnar1 −/−, Ifngr1 −/− Xcr1‐DTA and control mice. The data shown (mean ± SEM) are from two independent experiments pooled together (n = 3–6 mice per group). (b) Heatmap representing T‐cell effector phenotype and proliferation, and CTL cytokine production. The data are shown as log2 fold changes in the ratio of % Population/Parent population from mutant animals to WT (n = 3–6 mice per group). (c) Expression of GzmB, IFN‐γ and TNF by OVA‐specific CD44+ CD8+ T cells in the TdLN of Ifnar1 −/−, Ifngr1 −/−, Xcr1‐DTA and control mice day 7 post‐engraftment. The data shown are from two independent experiments pooled together. For a and c, data are shown as mean ± SEM, with values for individual mice shown as white circles in (c). *, P < 0.05; **, P < 0.01; and ***, P < 0.001; non‐parametric Mann–Whitney U‐test.
We next compared mouse strains for the ability of their tumor‐ or TdLN‐associated T cells to expand and to produce cytokines upon ex vivo Ag‐specific restimulation. Tumor‐infiltrating naïve (CD44−) and activated (CD44+) Tconv and CTLs proliferated less in mutant mice than in control animals (Figure 7b). Activated (CD44+) CTLs expressed less granzyme B, IFN‐γ and TNF in mutant mice than in control animals, at all time points examined in the tumor (Figure 7b) and at day 7 in TdLN (Figure 7c). Altogether, these results showed that IFNs and cDC1 in tumor and TdLN contributed to promote the terminal differentiation of antitumor CTLs and CD4+ Tconv for the acquisition of protective effector functions.
Genes associated with cDC1, CTLs, helper T cells, IFN‐I and IFN‐γ are associated with a better prognosis in human breast cancer patients
We wanted to know whether the immune cells and signalling pathways associated with the immune control of the NOP23 mouse breast adenocarcinoma model were of good prognosis in breast cancer patients. To infer the degree of infiltration of human mammary tumors by cDC1 and its association with the clinical outcome, we analysed a compendium of breast cancer microarrays and the TCGA database, via the KM plotter online resource. 44 , 45 The expression of the genes CLEC9A, 46 XCR1 8 , 9 and CLNK 47 is highly specific for cDC1 as compared to all other cell types in humans (Supplementary figure 13a). Therefore, we used the mean expression for the aggregated expression of these three genes in the tumors for TCGA breast cancer patients as a measurement of their infiltration by cDC1. We compared the overall survival between two patient groups harbouring a higher versus lower cDC1 infiltration in their tumors, respectively, based on automatic selection of the best‐performing cDC1 signature threshold value amongst all those possible between the lower and upper quartile. The patients whose tumors expressed the cDC1 gene signatures to higher levels had a significantly better overall survival (Figure 8a), showing that a higher cDC1 infiltration in breast cancer was associated with a better prognosis. Similar results were obtained with the other independent data set analysed, the breast cancer microarray compendium (Figure 8b), using the best ProbeSets for XCR1 (1561226_at) and CLNK (241483_at) (Supplementary figure 13a), without the possibility to take CLEC9A expression into account because of the lack of ProbeSet for this gene on the microarrays used. Patients whose tumor harboured a higher expression of CCR7 or of its ligand CCL19 also had a significantly better overall survival (Figure 8b), suggesting that activation of the CCR7/CCL19 axis in human breast tumors promotes more efficient immune responses as shown in the NOP23 mouse mammary tumor model. Finally, we used the Human Protein Atlas Web interface to retrieve the gene lists associated with a good (n = 367) or bad (n = 210) prognosis in TCGA breast cancer patients and queried their associations with biological processes and signalling pathways by using gene ontology. The GO terms enriched in the gene list associated with a good prognosis were linked to the activation and helper or cytotoxic functions of T‐cell responses and to IFN‐I and IFN‐γ signalling pathways (Figure 8e). Conversely, the GO terms associated with the bad prognosis gene list were linked to mitochondria and translation, likely reflecting active metabolism and proliferation of tumor cells (Supplementary figure 13b). In conclusion, these analyses strongly suggested that cDC1, CD4+ T cells, CTLs, NK cells and IFNs play together a crucial role in the immune control of breast cancer, not only in the NOP23 mouse model but also in human patients.
Figure 8.
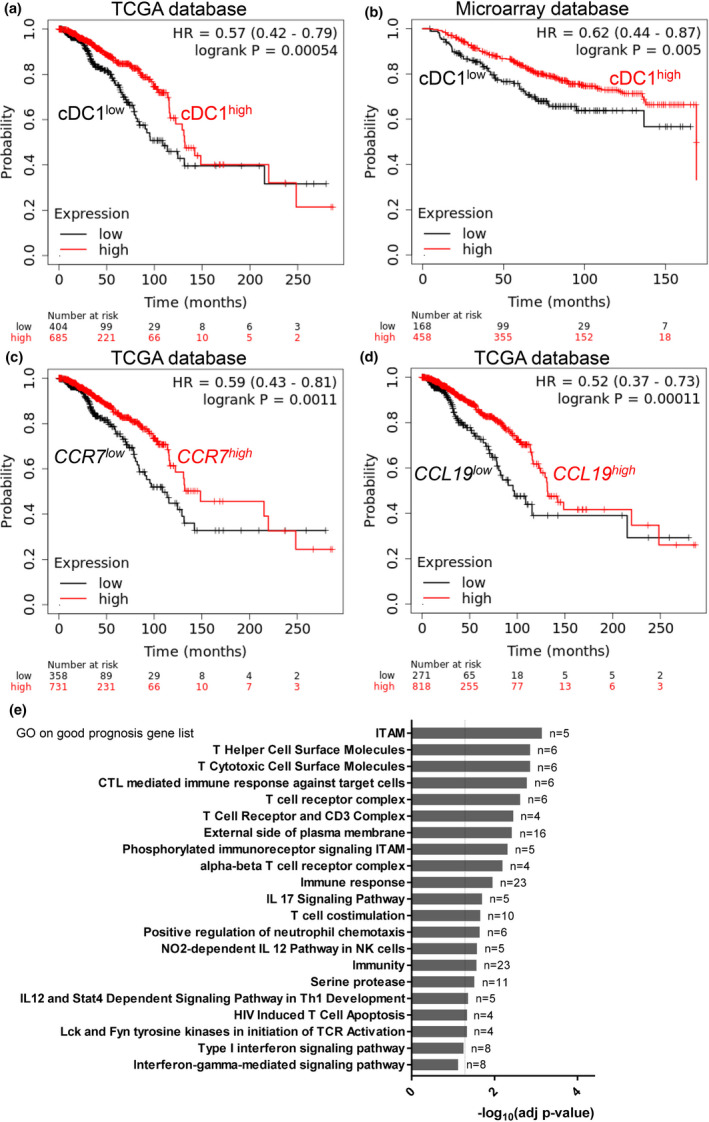
Intratumor expression of a cDC1 transcriptomic fingerprint and gene ontology annotations linked to CTL, helper T‐cell and IFN‐I/II signalling are all associated with a better prognosis in human breast cancer patients. (a, b) The Kaplan–Meier plot for human breast cancer patients based on the expression levels of a cDC1 gene signature in their tumor, for the TCGA data set (a) and for an independent compendium of breast cancer microarrays (b). (c, d) The Kaplan–Meier plot for the TCGA human breast cancer patients based on the expression levels of CCR7 (c) or CCL19 (d) in their tumor. (e) Gene Ontology analysis of a breast cancer good prognosis gene list. a–d were generated by using the KM plotter Web resource; e was retrieved from the Human Protein Atlas Web resource.
Discussion
Here, we investigated whether and how cDC1 promote the spontaneous control of breast cancer in mice, by combining mutant animals enabling specific targeting of cDC1 in vivo with an orthotopic model of engraftment of the syngeneic breast adenocarcinoma cell line NOP23. Specifically, we investigated how tumor growth and the nature of the immune infiltrate in the tumor or TdLN were affected by a specific, constitutive or conditional, depletion of cDC1, or by the genetic inactivation of candidate input or output signals specifically in cDC1. We demonstrated the non‐dispensable role of cDC1 in the containment of the breast cancer NOP23. By harnessing sophisticated genetically engineered mice, we identified the signals that were redundant and those that were essential in the immunological control of tumor growth. Whereas CXCL9, IL‐12 and IL‐15 were not required for tumor rejection, IFN responses were critical including cDC1‐intrinsic signalling via STAT1 and IFN‐γ. cDC1 and IFN promoted CD4+ and CD8+ T‐cell infiltration into the tumor, their differentiation and effector functions. Importantly, the attributes associated with tumor control in the mouse were also associated with better prognosis in breast cancer patients.
We showed unequivocally that cDC1 were required for the CTL‐dependent control of the NOP23 breast adenocarcinoma, throughout the entire antitumor immune response, but especially very early on at the time when antitumor T cells are primed. However, surprisingly, in mice lacking cDC1 the numbers of OVA‐specific CD8+ T cells were not significantly decreased, raising the possibility that other Ag‐presenting cells could cross‐prime them, whereas cDC1 were non‐redundant for their proper functional polarisation and sustained activation. We then wondered which molecular mechanisms promoted cDC1 infiltration into the tumor, and their later migration to the draining lymph node. CXCL9 has been proposed to promote infiltration of pre‐cDC1 in the tumor. 48 XCR1 expression by cDC1 can contribute to their recruitment by XCL1‐producing NK/NK T cells and CTLs in infected tissues. 39 , 49 However, neither XCR1 nor CXCL9 were individually required for this function in the NOP23 model. Hence, different chemokine receptors may redundantly promote cDC1 recruitment into the tumor bed, close to effector lymphocytes, as observed for XCR1 and CCR5 in melanoma or colorectal tumors in mice. 12 The lack of tumor control in Ccr7 −/− mice might in part result from a strict requirement of this chemokine receptor for cDC1 migration from the tumor to the TdLNs, as previously suggested in a model of melanoma. 50
We then investigated the role of candidate output signals delivered by cDC1 for the promotion of the recruitment of cytotoxic lymphocytes to the tumor and for the activation of their antitumor functions. In microbial infections, cDC1 production of high levels of CXCL9 promotes the recruitment of effector and memory CTLs expressing CXCR3 in secondary lymphoid organs. 36 This has been proposed to be also the case in tumors. 51 , 52 , 53 cDC1 are also a major source of IL‐12 and IL‐15 that promote activation, survival and cytotoxic functions of NK cells and CTLs. 36 , 52 , 53 , 54 , 55 , 56 , 57 , 58 However, CXCL9 and IL‐12 production and IL‐15 transpresentation by cDC1 were individually dispensable for the immune control of the NOP23 breast adenocarcinoma. In contrast to our observation with the NOP23 mammary tumor, IL‐12 activity was critical for the CD8+ T‐cell‐mediated tumor rejection in another mouse breast cancer model, the MMTV‐PyMT transgenic mice treated with paclitaxel and an anti‐IL‐10‐neutralising antibody. 58 More generally, in various cancer models, tumor rejection by NK, NK T, CD8+ T cells or CD4+ T cells was shown to depend partially 55 , 59 , 60 , 61 , 62 , 63 or critically 57 , 64 , 65 , 66 , 67 , 68 , 69 , 70 , 71 , 72 , 73 , 74 , 75 , 76 on endogenous IL‐12 activity. Yet, host IL‐12 responses were found to be dispensable for immune‐mediated tumor control in several other mouse cancer models, including under conditions requiring CD8+ T‐cell, CD4+ T‐cell, CD40 or IFN‐γ responses 77 , 78 , 79 , 80 , 81 , 82 . In some of these studies, loss of IL‐12 activity might have been compensated with other innate cytokines with overlapping functions such as IL‐18/IL‐2, 79 IL‐1 77 or IFN‐I. 80 This would be consistent with observations during certain microbial infections where IL‐12, IL‐15, IL‐18 and IFN‐I/III or other cytokines can exert overlapping effects for promoting the proliferation, IFN‐γ production and cytotoxic activity of NK and CTLs. 83 , 84 Similarly, our results suggest a level of redundancy higher than expected between different types of output signals delivered by cDC1 for the control of the NOP23 mammary tumor.
We sought for key input signals required for cDC1 to mediate protective antitumor effects. During microbial infections, the induction of protective NK and T‐cell responses critically depends on the immunogenic maturation of cDC1, which is driven at least in part by their responses to IFN‐I 34 or IFN‐γ. 36 , 85 , 86 Here, we showed that overall IFN‐I, IFN‐γ and STAT1 responses were critical for rejection of the NOP23 breast adenocarcinoma. Only IFN‐γ and STAT1 responses were required to occur in cDC1, whereas cell‐intrinsic response to IFN‐I in cDC1 was dispensable for tumor rejection. Hence, we show here that IFN‐I is not always critical for enhancing cDC1 cross‐presentation and more generally for licensing them to promote tumor control. This raises the question of the extent to which cDC1‐intrinsic IFN‐I signalling is critical for effective immunity against cancer, besides for immune‐mediated rejection of syngeneic and immunogenic fibrosarcoma and melanoma in mice. 32 , 33 However, cDC1‐specific inactivation of Stat1 led to a higher tumor growth than the loss of IFN‐γ signalling alone, suggesting some level of redundancy between these two activation pathways for the promotion of cDC1 immunogenic maturation. STAT1 is also key in transducing the signal of IFN‐III, whose expression is associated with a better prognosis for breast cancer patients, together with the level of cDC1 infiltration in the tumor. 13 Moreover, cDC1 are a main source of IFN‐III in human breast tumors. 13 Hence, it would be interesting in future studies to investigate whether cDC1 produce, and respond to, IFN‐III in the NOP23 breast adenocarcinoma model, and how this may contribute to the spontaneous rejection of the tumor. It will also be interesting to investigate whether further boosting cDC1 development and IFN‐III production could help improve immune responses against triple‐negative breast cancer, similar to what was recently reported in a therapeutic vaccination trial in human melanoma patients. 22 Since overall IFN‐I responses but not cDC1 responses to IFN‐I were essential for the immune control of the NOP23 breast adenocarcinoma, IFN‐I must exert critical effect on other immune cells, likely CTL themselves as was reported in microbial infections. 84 , 87 , 88
Finally, to attempt to better understand how IFNs and cDC1 were promoting immune rejection of the NOP23 breast adenocarcinoma, we examined how their loss affected the immune landscape of the tumor and of the TdLN. We observed that cDC1 and IFNs shaped the tumor immune landscape by promoting CD4+ Tconv and CTL infiltration and their terminal differentiation with enhanced effector functions. Conversely, cDC1 and IFN responses limited the numbers of putatively deleterious myeloid cell types in the tumor and in TdLN, including subtypes of macrophages and neutrophils. In the tumors from Ifnar1 −/− and Ifngr1 −/− mice, cDC1 expressed less CD40. Together with our preliminary observation of the simultaneous interactions of cDC1 with CD4+ T cells and CTLs in the tumor bed, this suggested that cell‐intrinsic responses of cDC1 to IFN may be critical to promote their ability to deliver to CTL the help from CD4+ Tconv in a manner depending on their interactions via CD40/CD40L. This hypothesis is consistent with the demonstration that simultaneous presentation of viral Ags by cDC1 to CTLs and CD4+ T cells is key for robust antiviral cellular immunity 89 , 90 and with publications linking CD40 expression on cDC1, their ability to activate CD4+ Tconv and the CTL‐dependent rejection of tumors. 30 , 91
To the best of our knowledge, our preliminary confocal microscopy data are the first direct evidence showing that cDC1 in tumors might act as a unique cellular platform docking simultaneously CD4+ Tconv and CTLs, which is likely key to their ability to relay CD4+ T‐cell help to CTLs in situ in tumor, akin to what had been previously shown in infectious settings. 89 , 90 However, further experiments including dynamic data and spatial distribution assessments will be required to robustly determine whether cDC1 simultaneously interact with cognate CD4+ and CD8+ T cells in the tumor bed, and more efficiently than other Ag‐presenting cells. We also rigorously demonstrate for the first time that cell‐intrinsic signalling by IFN‐γ and STAT1 in cDC1 is critical to their antitumor functions. Although this had been suggested before, 33 , 76 it was never formally proven because of the lack of adequate models to specifically inactivate IFN‐γ signalling in cDC1 without also affecting it in other CD11c+ cells. Moreover, in our experimental settings, the licensing of cDC1 by IFN‐γ for promoting the control of the NOP23 breast cancer model does not require IL‐12 contrary to what has been previously observed or proposed. 54 , 58 , 76 Beside tumor Ag cross‐presentation, the precise nature of the output signals delivered by cDC1 that are critical to induce and maintain protective functions of antitumor CD4+ Tconv and CTLs remains to be identified, but may encompass IFN‐I/III production and CD40 expression. Future studies based on comparative gene expression profiling of WT versus Ifngr1 −/− or Stat1 −/− cDC1 infiltrating the tumor or having migrating to the TdLN should help to identify the output signals whose delivery from cDC1 to effector antitumor lymphocytes is critical for the spontaneous rejection of the NOP23 cancer adenocarcinoma model.
Consistent with our experimental results in mice, we confirmed that human breast cancer patients harbouring a high expression in their tumor of genes specific to cDC1 have a significantly better clinical outcome, in line with previous studies. 10 , 12 , 13 We showed that the genes associated with a better prognosis were enriched for annotations linked to CTLs, helper T cells or IFN responses. Therefore, we propose the following model of how cDC1 promote tumor immunosurveillance (Supplementary figure 14). Following tumor cell immunogenic death, cDC1 capture tumor Ag, undergo immunogenic maturation and likely migrate to the TdLNs in a CCR7‐dependent manner to prime CD4+ Tconv towards TH1 and CD8+ T cells towards multipotent protective CTLs. In turn, activated antitumor T cells infiltrate the tumor, contributing (i) to enhance local recruitment of cDC1, by producing redundant chemokines such as XCL1 and CCL5, 12 and (ii) to induce their immunogenic maturation via IFN‐γ. This leads to quadripartite interactions in the tumor between cDC1, tumor cells, CD4+ Tconv and CTLs, ensuring local amplification and maintenance of the effector functions of antitumor T‐cell responses, leading to tumor eradication. IFN‐I and IFN‐III produced by the cDC1 themselves or other cells might also redundantly contribute to induce cDC1 immunogenic maturation, together with IFN‐γ.
In conclusion, we showed that cDC1 and their cell‐intrinsic response to STAT1 signalling are critical for adequate activation of the effector functions of CD8+ and CD4+ T cells and for spontaneous immune control of a mouse model of regressive breast cancer. In future studies, it will be important to determine the precise nature of the protective cDC1 functions induced by STAT1 signalling, and to investigate whether they can be mobilised in preclinical mouse models of progressive triple‐negative breast cancer upon specific chemotherapy 92 or immunotherapy. 93 This should then help further optimising new generation immunotherapeutic strategies aiming at mobilising human cDC1 to treat various cancers. 14 , 15 , 16 , 17
Methods
Mice and in vivo treatments
Mice were bred and maintained in the CIPHE pathogen‐free animal facility. All experiments were performed with female littermate mice at 7–15 weeks of age. The mouse strains used are all on the C57BL/6J background and listed in Supplementary table 1. For a sustained and efficient cDC1 conditional depletion for at least 10 consecutive days, Karma‐tmt‐hDTR and Xcr1Cre / wt ; Rosa26hDTR / wt mice received a first dose of 32 ng per g of body weight of DT (Merck, Darmstadt, Germany), followed by one injection of 16 ng per g every 60 h. For in vivo antibody‐mediated cell depletion, C57BL/6 mice were injected i.p. with the antibody and doses indicated in Supplementary table 2, starting 1 day before tumor engraftment and then as indicated in the figures. To block lymphoid cell egress from peripheral lymphoid organs, mice received 20 µg of FTY720 (Cayman Chemical, Ann Arbor, USA) starting 1 day before tumor engraftment, then every 2d. The study was carried out in accordance with institutional guidelines and with protocols (#1221 and ROXinAIR #16555) approved by the Comité National de Réflexion Ethique sur l'Expérimentation Animale #14 and the Ministère de l’Enseignement Supérieur, de la Recherche et de l’Innovation.
Tumor experiments
We used a breast adenocarcinoma tumor cell line that is spontaneously rejected when orthotopically implanted in C57BL/6 females. It was derived from the NOP23 cell line, which was established from a spontaneous mammary tumor of a transgenic mouse expressing a dominant negative version of p53 and the rat NEU (HER2) oncogene fused at its COOH terminus to class I and II OVA peptide sequences. 35 NOP23 cells were grown in DMEM, 10% FCS, supplemented with 10 mg mL−1 of insulin–transferrin–sodium selenite media supplement (Sigma‐Aldrich, St. Louis, USA). 5 × 106 NOP23 cells were injected in the mammary inguinal fat pad, under isoflurane anaesthesia. Tumor volume was calculated as , where L is the greatest length, and W is the width of the tumor, measured with a calliper. Tumor volumes in graphs are represented as mean ± SEM.
Shield bone marrow chimeric mice
To generate Ifngr1 −/−→Xcr1‐DTA and Stat1 −/−→Xcr1‐DTA shield bone marrow chimeric (SBMC) mice, the hind legs of Xcr1‐DTA recipient mice were irradiated with one dose of 8 Gy, to preserve most of their haematopoietic system that remained WT. The cDC1 empty niche of these recipient mice was then reconstituted upon injection of 30 × 106 bone marrow cells from Ifngr1 −/− or Stat1 −/− donor mice. Xcr1‐DTA→Xcr1‐DTA and WT→Xcr1‐DTA SBMC mice were used as controls. NOP23 cells were engrafted 8–14 weeks later.
Preparation of cell suspensions from tumors and TdLNs for flow cytometry
Tumors and inguinal TdLNs were cut into small pieces with a scalpel and incubated for 30 min at 37°C in an enzymatic cocktail consisting in RPMI with Collagenase D at 1 mg mL−1 and DNase I at 70 μg mL−1 (Roche, Basel, Switzerland). Ice‐cold PBS/EDTA (2 mM) was added for 5 min. Digested tissues were crushed through a 70‐μm nylon sieve. For flow cytometry, cells were pre‐incubated with 2.4G2 mAb to block Fc‐receptors, then stained with the mAb listed in Supplementary table 2 for 25 min at 4°C. Class I OVA Tetramer (iTAg Tetramer/PE ‐ H‐2 Kb OVA [SIINFEKL], MBL International, Woburn, USA) was incubated at 1:100 at 4°C for 1 hour, and Class II OVA Tetramer (T‐Select I‐Ab OVA323‐339 [ISQAVHAAHAEINEAGR] Tetramer‐APC, MBL International), at 1:12.5 at room temperature for 1 h, before proceeding to staining with mAb. For CCR7 staining, cells were incubated for 30 min at 37°C. For intracellular staining, cells were re‐stimulated ex vivo for 4h at 37°C with 0.5 μg mL−1 of SIINFEKL peptide and 10 μg mL−1 of Brefeldin A (Sigma‐Aldrich) in complete RPMI. Cytokines, Ki67 and Foxp3, were stained after fixation/permeabilisation with Foxp3/Transcription Factor Staining Buffer Set (eBioscience, San Diego, USA). Data were acquired on a LSRFortessa X‐20 flow cytometer (BD Biosciences, Franklin Lakes, USA) and analysed using FlowJo (Tree Star, Inc., Ashland, USA). Flow cytometry heatmaps were performed using the Morpheus website from the Broad Institute (https://software.broadinstitute.org/morpheus/).
Immunohistofluorescence
1000 naïve GFP‐expressing OT‐I cells were purified from a TgTcraTcrb1100Mjb ; Rag2 −/−; Ubc‐GFP +/+ spleen with the Dynabeads Untouched Mouse CD8 Cell Kit (Thermo Fisher Scientific, Waltham, USA) and transferred i.v. in Xcr1Cre / wt ; Rosa26tdRFP / wt mice 1 day before tumor engraftment. 7d after, tumors were harvested and 12‐μm frozen sections were stained with the antibodies listed in Supplementary table 2, as described previously. 39
Quantitative PCR analysis
Total RNA from tumors and TdLNs were prepared with the RNeasy Plus Mini Kit (Qiagen, Venlo, the Netherlands). RNA was reverse‐transcribed into cDNA using the QuantiTect Reverse Transcription Kit (Qiagen). qPCR was performed with the SYBR Green Kit (Takara, Kusatsu, Japan) and specific primers (Supplementary table 3), and run on a 7500 Real Time PCR System apparatus (Applied Biosystems, Waltham, USA). Relative gene expression was calculated using the ΔΔCt method with Hprt as housekeeping control gene.
Transcriptomic data from breast cancer patients
The Kaplan–Meier plots were obtained from the KM plotter database (https://kmplot.com/analysis/). 44 , 45 Transcripts for which expression levels in the tumors of TCGA BC patients were correlated with a better overall survival (367 genes) or with a poor prognosis (210 genes) were extracted through the Human Protein Atlas Web resource (https://www.proteinatlas.org/humanproteome/pathology). 94 A gene was considered prognostic if correlation analysis of gene expression and clinical outcome resulted in the Kaplan–Meier plots with high significance (P < 0.001). The gene ontology analyses on the good and bad prognosis gene lists were performed with DAVID 6.8 (https://david.ncifcrf.gov/). 95
Statistical analyses
Statistical analyses were performed using unpaired Student’s t‐tests or the non‐parametric Mann‐Whitney U‐tests (MW) when specified. N.S., non‐significant (P > 0.05); *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; and ****, P ≤ 0.0001.
Conflict of interest
The authors declare that they have no competing interests.
Author contributions
Raphaël Mattiuz: Conceptualization; Formal analysis; Investigation; Methodology; Visualization; Writing‐original draft; Writing‐review & editing. Carine Brousse: Formal analysis; Investigation. Marc Ambrosini: Formal analysis; Investigation; Visualization. Jean‐Charles Cancel: Investigation. Gilles Bessou: Investigation; Methodology; Resources. Julie Mussard: Investigation. Amélien Sanlaville: Methodology. Christophe Caux: Funding acquisition; Methodology; Writing‐review & editing. Nathalie Bendriss‐Vermare: Methodology; Resources; Writing‐review & editing. Jenny Valladeau‐Guilemond: Resources; Writing‐review & editing. Marc Dalod: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Supervision; Validation; Visualization; Writing‐original draft; Writing‐review & editing. Karine Crozat: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Supervision; Validation; Visualization; Writing‐original draft; Writing‐review & editing.
Supporting information
Acknowledgments
This work was supported by grants from the ANR (XCR1‐DirectingCells to KC), the Fondation pour la Recherche Médicale (label Equipe FRM 2011, project number DEQ20110421284 to MD), the Fondation ARC (to KC), the Institut National du Cancer (INCa PLBIO 2018‐152 to CC and MD) and the European Research Council under the European Community's Seventh Framework Program (FP7/2007–2013 grant agreement number 281225 to MD). This work also benefited from institutional funding by CNRS and INSERM. RM was supported by doctoral fellowships from the Biotrail Ph.D. programme (Fondation A*MIDEX) and from Fondation ARC. JCC had a doctoral fellowship from LNCC. We thank Dr. Brad Nelson (Director, Deeley Research Centre, BC Cancer Agency, Victoria BC, Canada) for authorisation to use the NOP23 cell line. We thank members of the CIML cytometry, ImagImm and histology facilities and members from the CIML/CIPHE mouse houses. We acknowledge France Bio lmaging infrastructure supported by the Agence Nationale de la Recherche (ANR‐10‐INBS‐04‐01, call ‘Investissement d’Avenir’). We thank Hugues Lelouard (CIML) for sharing mice, and Rebecca Gentek and Pierre Golstein (CIML) for helpful discussions and protocols. This work benefited from data assembled by the TCGA, The Human Protein Atlas, and Kaplan–Meier plotter databases. The model figure has been created with BioRender (https://biorender.com/) under academic licence.
Contributor Information
Marc Dalod, Email: dalod@ciml.univ-mrs.fr.
Karine Crozat, Email: crozat@ciml.univ-mrs.fr.
REFERENCES
- 1. Guilliams M, Ginhoux F, Jakubzick C et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol 2014; 14: 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vu Manh TP, Bertho N, Hosmalin A, Schwartz‐Cornil I, Dalod M. Investigating evolutionary conservation of dendritic cell subset identity and functions. Front Immunol 2015; 6: 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sancho D, Joffre OP, Keller AM et al. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 2009; 458: 899–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Theisen DJ, Davidson JT, Briseño CG et al. WDFY4 is required for cross‐presentation in response to viral and tumor antigens. Science 2018; 362: 694–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Laoui D, Keirsse J, Morias Y et al. The tumour microenvironment harbours ontogenically distinct dendritic cell populations with opposing effects on tumour immunity. Nat Commun 2016; 7: 13720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jongbloed SL, Kassianos AJ, McDonald KJ et al. Human CD141+ (BDCA‐3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross‐presents necrotic cell antigens. J Exp Med 2010; 207: 1247–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chiang MC, Tullett KM, Lee YS et al. Differential uptake and cross‐presentation of soluble and necrotic cell antigen by human DC subsets. Eur J Immunol 2016; 46: 329–339. [DOI] [PubMed] [Google Scholar]
- 8. Crozat K, Guiton R, Contreras V et al. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8α+ dendritic cells. J Exp Med 2010; 207: 1283–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bachem A, Guttler S, Hartung E et al. Superior antigen cross‐presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med 2010; 207: 1273–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cancel JC, Crozat K, Dalod M, Mattiuz R. Are conventional type 1 dendritic cells critical for protective antitumor immunity and how? Front Immunol 2019; 10: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol 2020; 20: 7–24. [DOI] [PubMed] [Google Scholar]
- 12. Bottcher JP, Bonavita E, Chakravarty P et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell 2018; 172: 1022–1037 e1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hubert M, Gobbini E, Couillault C et al. IFN‐III is selectively produced by cDC1 and predicts good clinical outcome in breast cancer. Sci Immunol 2020; 5: eaav3942. [DOI] [PubMed] [Google Scholar]
- 14. Balan S, Radford KJ, Bhardwaj N. Unexplored horizons of cDC1 in immunity and tolerance. Adv Immunol 2020; 148: 49–91. [DOI] [PubMed] [Google Scholar]
- 15. Pearson FE, Tullett KM, Leal‐Rojas IM et al. Human CLEC9A antibodies deliver Wilms' tumor 1 (WT1) antigen to CD141+ dendritic cells to activate naive and memory WT1‐specific CD8+ T cells. Clin Transl Immunology 2020; 9: e1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Masterman KA, Haigh OL, Tullett KM et al. Human CLEC9A antibodies deliver NY‐ESO‐1 antigen to CD141+ dendritic cells to activate naive and memory NY‐ESO‐1‐specific CD8+ T cells. J Immunother Cancer 2020; 8: e000691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Radford KJ, Tullett KM, Lahoud MH. Dendritic cells and cancer immunotherapy. Curr Opin Immunol 2014; 27: 26–32. [DOI] [PubMed] [Google Scholar]
- 18. Chan JD, von Scheidt B , Zeng B et al. Enhancing chimeric antigen receptor T‐cell immunotherapy against cancer using a nanoemulsion‐based vaccine targeting cross‐presenting dendritic cells. Clin Transl Immunology 2020; 9: e1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sosa Cuevas E, Ouaguia L, Mouret S et al. BDCA1+ cDC2s, BDCA2+ pDCs and BDCA3+ cDC1s reveal distinct pathophysiologic features and impact on clinical outcomes in melanoma patients. Clin Transl Immunol 2020; 9: e1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Salmon H, Idoyaga J, Rahman A et al. Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD‐L1 and BRAF inhibition. Immunity 2016; 44: 924–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hammerich L, Marron TU, Upadhyay R et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat Med 2019; 25: 814–824. [DOI] [PubMed] [Google Scholar]
- 22. Bhardwaj N, Friedlander PA, Pavlick AC et al. Flt3 ligand augments immune responses to anti‐DEC‐205‐NY‐ESO‐1 vaccine through expansion of dendritic cell subsets. Nat Cancer 2020; 1: 1204–1217. [DOI] [PubMed] [Google Scholar]
- 23. Sichien D, Scott CL, Martens L et al. IRF8 transcription factor controls survival and function of terminally differentiated conventional and plasmacytoid dendritic cells, respectively. Immunity 2016; 45: 626–640. [DOI] [PubMed] [Google Scholar]
- 24. Bosteels C, Neyt K, Vanheerswynghels M et al. Inflammatory type 2 cDCs acquire features of cDC1s and macrophages to orchestrate immunity to respiratory virus infection. Immunity 2020; 52: 1039–1056 e1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hildner K, Edelson BT, Purtha WE et al. Batf3 deficiency reveals a critical role for CD8α+ dendritic cells in cytotoxic T cell immunity. Science 2008; 322: 1097–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee W, Kim HS, Hwang SS, Lee GR. The transcription factor Batf3 inhibits the differentiation of regulatory T cells in the periphery. Exp Mol Med 2017; 49: e393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ataide MA, Komander K, Knopper K et al. BATF3 programs CD8+ T cell memory. Nat Immunol 2020; 21: 1397–1407. [DOI] [PubMed] [Google Scholar]
- 28. Qiu Z, Khairallah C, Romanov G, Sheridan BS. Cutting edge: Batf3 expression by CD8 T cells critically regulates the development of memory populations. J Immunol 2020; 205: 901–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mattiuz R, Wohn C, Ghilas S et al. Novel cre‐expressing mouse strains permitting to selectively track and edit type 1 conventional dendritic cells facilitate disentangling their complexity in vivo . Front Immunol 2018; 9: 2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ferris ST, Durai V, Wu R et al. cDC1 prime and are licensed by CD4+ T cells to induce anti‐tumour immunity. Nature 2020; 584: 624–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tullett KM, Leal Rojas IM, Minoda Y et al. Targeting CLEC9A delivers antigen to human CD141+ DC for CD4+ and CD8+T cell recognition. JCI Insight 2016; 1: e87102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Diamond MS, Kinder M, Matsushita H et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 2011; 208: 1989–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fuertes MB, Kacha AK, Kline J et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8α+ dendritic cells. J Exp Med 2011; 208: 2005–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tomasello E, Pollet E, Vu Manh TP, Uze G, Dalod M. Harnessing mechanistic knowledge on beneficial versus deleterious IFN‐I effects to design innovative immunotherapies targeting cytokine activity to specific cell types. Front Immunol 2014; 5: 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Martin ML, Wall EM, Sandwith E et al. Density of tumour stroma is correlated to outcome after adoptive transfer of CD4+ and CD8+ T cells in a murine mammary carcinoma model. Breast Cancer Res Treat 2010; 121: 753–763. [DOI] [PubMed] [Google Scholar]
- 36. Alexandre YO, Ghilas S, Sanchez C, Le Bon A, Crozat K, Dalod M. XCR1+ dendritic cells promote memory CD8+ T cell recall upon secondary infections with Listeria monocytogenes or certain viruses. J Exp Med 2016; 213: 75–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Simpson TR, Li F, Montalvo‐Ortiz W et al. Fc‐dependent depletion of tumor‐infiltrating regulatory T cells co‐defines the efficacy of anti‐CTLA‐4 therapy against melanoma. J Exp Med 2013; 210: 1695–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Matloubian M, Lo CG, Cinamon G et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004; 427: 355–360. [DOI] [PubMed] [Google Scholar]
- 39. Ghilas S, Ambrosini M, Cancel J‐C et al. NK cells orchestrate splenic cDC1 migration to potentiate antiviral protective CD8+ T cell responses. bioRxiv 2020: 10.1101/2020.1104.1123.057463. [DOI]
- 40. Schlitzer A, Sivakamasundari V, Chen J et al. Identification of cDC1‐ and cDC2‐committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat Immunol 2015; 16: 718–728. [DOI] [PubMed] [Google Scholar]
- 41. Ramos RN, Rodriguez C, Hubert M et al. CD163+ tumor‐associated macrophage accumulation in breast cancer patients reflects both local differentiation signals and systemic skewing of monocytes. Clin Transl Immunol 2020; 9: e1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Blank CU, Haining WN, Held W et al. Defining 'T cell exhaustion'. Nat Rev Immunol 2019; 19: 665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Singer M, Wang C, Cong L et al. A distinct gene module for dysfunction uncoupled from activation in tumor‐infiltrating T cells. Cell 2016; 166: 1500–1511 e1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gyorffy B, Lanczky A, Eklund AC et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat 2010; 123: 725–731. [DOI] [PubMed] [Google Scholar]
- 45. Nagy A, Munkacsy G, Gyorffy B. Pancancer survival analysis of cancer hallmark genes. Sci Rep 2021; 11: 6047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Poulin LF, Reyal Y, Uronen‐Hansson H et al. DNGR‐1 is a specific and universal marker of mouse and human Batf3‐dependent dendritic cells in lymphoid and nonlymphoid tissues. Blood 2012; 119: 6052–6062. [DOI] [PubMed] [Google Scholar]
- 47. Robbins SH, Walzer T, Dembele D et al. Novel insights into the relationships between dendritic cell subsets in human and mouse revealed by genome‐wide expression profiling. Genome Biol 2008; 9: R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cook SJ, Lee Q, Wong AC et al. Differential chemokine receptor expression and usage by pre‐cDC1 and pre‐cDC2. Immunol Cell Biol 2018; 96: 1131–1139. [DOI] [PubMed] [Google Scholar]
- 49. Brewitz A, Eickhoff S, Dahling S et al. CD8+ T cells orchestrate pDC‐XCR1+ dendritic cell spatial and functional cooperativity to optimize priming. Immunity 2017; 46: 205–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Roberts EW, Broz ML, Binnewies M et al. Critical role for CD103+/CD141+ dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell 2016; 30: 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Spranger S, Dai D, Horton B, Gajewski TF. Tumor‐Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017; 31: 711–723 e714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. de Mingo Pulido A, Gardner A, Hiebler S et al. TIM‐3 regulates CD103+ dendritic cell function and response to chemotherapy in breast cancer. Cancer Cell 2018; 33: 60–74 e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bergamaschi C, Pandit H, Nagy BA et al. Heterodimeric IL‐15 delays tumor growth and promotes intratumoral CTL and dendritic cell accumulation by a cytokine network involving XCL1, IFN‐γ, CXCL9 and CXCL10. J Immunother Cancer 2020; 8: e000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Broz ML, Binnewies M, Boldajipour B et al. Dissecting the tumor myeloid compartment reveals rare activating antigen‐presenting cells critical for T cell immunity. Cancer Cell 2014; 26: 638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Beavis PA, Henderson MA, Giuffrida L et al. Dual PD‐1 and CTLA‐4 checkpoint blockade promotes antitumor immune responses through CD4+Foxp3‐ cell‐mediated modulation of CD103+ dendritic cells. Cancer Immunol Res 2018; 6: 1069–1081. [DOI] [PubMed] [Google Scholar]
- 56. Greyer M, Whitney PG, Stock AT et al. T cell help amplifies innate signals in CD8+ DCs for optimal CD8+ T cell priming. Cell Rep 2016; 14: 586–597. [DOI] [PubMed] [Google Scholar]
- 57. Mittal D, Vijayan D, Putz EM et al. Interleukin‐12 from CD103+ Batf3‐dependent dendritic cells required for NK‐cell suppression of metastasis. Cancer Immunol Res 2017; 5: 1098–1108. [DOI] [PubMed] [Google Scholar]
- 58. Ruffell B, Chang‐Strachan D, Chan V et al. Macrophage IL‐10 blocks CD8+ T cell‐dependent responses to chemotherapy by suppressing IL‐12 expression in intratumoral dendritic cells. Cancer Cell 2014; 26: 623–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liu J, Xiang Z, Ma X. Role of IFN regulatory factor‐1 and IL‐12 in immunological resistance to pathogenesis of N‐methyl‐N‐nitrosourea‐induced T lymphoma. J Immunol 2004; 173: 1184–1193. [DOI] [PubMed] [Google Scholar]
- 60. Smyth MJ, Thia KY, Street SE et al. Differential tumor surveillance by natural killer (NK) and NKT cells. J Exp Med 2000; 191: 661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Street SE, Cretney E, Smyth MJ. Perforin and interferon‐γ activities independently control tumor initiation, growth, and metastasis. Blood 2001; 97: 192–197. [DOI] [PubMed] [Google Scholar]
- 62. He D, Li H, Yusuf N et al. IL‐17 mediated inflammation promotes tumor growth and progression in the skin. PLoS One 2012; 7: e32126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Blake SJ, Stannard K, Liu J et al. Suppression of metastases using a new lymphocyte checkpoint target for cancer immunotherapy. Cancer Discov 2016; 6: 446–459. [DOI] [PubMed] [Google Scholar]
- 64. Airoldi I, Di Carlo E, Cocco C et al. Lack of Il12rb2 signaling predisposes to spontaneous autoimmunity and malignancy. Blood 2005; 106: 3846–3853. [DOI] [PubMed] [Google Scholar]
- 65. Esche C, Gambotto A, Satoh Y et al. CD154 inhibits tumor‐induced apoptosis in dendritic cells and tumor growth. Eur J Immunol 1999; 29: 2148–2155. [DOI] [PubMed] [Google Scholar]
- 66. Evel‐Kabler K, Song XT, Aldrich M, Huang XF, Chen SY. SOCS1 restricts dendritic cells' ability to break self tolerance and induce antitumor immunity by regulating IL‐12 production and signaling. J Clin Invest 2006; 116: 90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Maeda A, Schneider SW, Kojima M, Beissert S, Schwarz T, Schwarz A. Enhanced photocarcinogenesis in interleukin‐12‐deficient mice. Cancer Res 2006; 66: 2962–2969. [DOI] [PubMed] [Google Scholar]
- 68. Meeran SM, Mantena SK, Meleth S, Elmets CA, Katiyar SK. Interleukin‐12‐deficient mice are at greater risk of UV radiation‐induced skin tumors and malignant transformation of papillomas to carcinomas. Mol Cancer Ther 2006; 5: 825–832. [DOI] [PubMed] [Google Scholar]
- 69. Swann JB, Vesely MD, Silva A et al. Demonstration of inflammation‐induced cancer and cancer immunoediting during primary tumorigenesis. Proc Natl Acad Sci USA 2008; 105: 652–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fujita M, Zhu X, Ueda R et al. Effective immunotherapy against murine gliomas using type 1 polarizing dendritic cells–significant roles of CXCL10. Cancer Res 2009; 69: 1587–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nunez NG, Andreani V, Crespo MI et al. IFN‐β produced by TLR4‐activated tumor cells is involved in improving the antitumoral immune response. Cancer Res 2012; 72: 592–603. [DOI] [PubMed] [Google Scholar]
- 72. Mattarollo SR, West AC, Steegh K et al. NKT cell adjuvant‐based tumor vaccine for treatment of myc oncogene‐driven mouse B‐cell lymphoma. Blood 2012; 120: 3019–3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Baird JR, Byrne KT, Lizotte PH et al. Immune‐mediated regression of established B16F10 melanoma by intratumoral injection of attenuated Toxoplasma gondii protects against rechallenge. J Immunol 2013; 190: 469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Baird JR, Fox BA, Sanders KL et al. Avirulent Toxoplasma gondii generates therapeutic antitumor immunity by reversing immunosuppression in the ovarian cancer microenvironment. Cancer Res 2013; 73: 3842–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mittal D, Lepletier A, Madore J et al. CD96 is an immune checkpoint that regulates CD8+ T‐cell antitumor function. Cancer Immunol Res 2019; 7: 559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Garris CS, Arlauckas SP, Kohler RH et al. Successful anti‐PD‐1 cancer immunotherapy requires T cell‐dendritic cell crosstalk involving the cytokines IFN‐γ and IL‐12. Immunity 2018; 49: 1148–1161. e1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ghiringhelli F, Apetoh L, Tesniere A et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL‐1β‐dependent adaptive immunity against tumors. Nat Med 2009; 15: 1170–1178. [DOI] [PubMed] [Google Scholar]
- 78. VanLith ML, Kohlgraf KG, Sivinski CL, Tempero RM, Hollingsworth MA. MUC1‐specific anti‐tumor responses: molecular requirements for CD4‐mediated responses. Int Immunol 2002; 14: 873–882. [DOI] [PubMed] [Google Scholar]
- 79. Wigginton JM, Lee JK, Wiltrout TA et al. Synergistic engagement of an ineffective endogenous anti‐tumor immune response and induction of IFN‐γ and Fas‐ligand‐dependent tumor eradication by combined administration of IL‐18 and IL‐2. J Immunol 2002; 169: 4467–4474. [DOI] [PubMed] [Google Scholar]
- 80. Simma O, Zebedin E, Neugebauer N et al. Identification of an indispensable role for tyrosine kinase 2 in CTL‐mediated tumor surveillance. Cancer Res 2009; 69: 203–211. [DOI] [PubMed] [Google Scholar]
- 81. Zhang S, Bernard D, Khan WI, Kaplan MH, Bramson JL, Wan Y. CD4+ T‐cell‐mediated anti‐tumor immunity can be uncoupled from autoimmunity via the STAT4/STAT6 signaling axis. Eur J Immunol 2009; 39: 1252–1259. [DOI] [PubMed] [Google Scholar]
- 82. Byrne KT, Vonderheide RH. CD40 stimulation obviates innate sensors and drives T cell immunity in cancer. Cell Rep 2016; 15: 2719–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Cousens LP, Peterson R, Hsu S et al. Two roads diverged: interferon‐α/β‐ and interleukin 12‐mediated pathways in promoting T cell interferon‐γ responses during viral infection. J Exp Med 1999; 189: 1315–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Thompson LJ, Kolumam GA, Thomas S, Murali‐Krishna K. Innate inflammatory signals induced by various pathogens differentially dictate the IFN‐I dependence of CD8 T cells for clonal expansion and memory formation. J Immunol 2006; 177: 1746–1754. [DOI] [PubMed] [Google Scholar]
- 85. Lee SH, Carrero JA, Uppaluri R et al. Identifying the initiating events of anti‐Listeria responses using mice with conditional loss of IFN‐γ receptor subunit 1 (IFNGR1). J Immunol 2013; 191: 4223–4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Deauvieau F, Ollion V, Doffin AC et al. Human natural killer cells promote cross‐presentation of tumor cell‐derived antigens by dendritic cells. Int J Cancer 2015; 136: 1085–1094. [DOI] [PubMed] [Google Scholar]
- 87. Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali‐Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med 2005; 202: 637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Havenar‐Daughton C, Kolumam GA, Murali‐Krishna K. Cutting Edge: The direct action of type I IFN on CD4 T cells is critical for sustaining clonal expansion in response to a viral but not a bacterial infection. J Immunol 2006; 176: 3315–3319. [DOI] [PubMed] [Google Scholar]
- 89. Eickhoff S, Brewitz A, Gerner MY et al. Robust anti‐viral immunity requires multiple distinct T cell‐dendritic cell interactions. Cell 2015; 162: 1322–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hor JL, Whitney PG, Zaid A, Brooks AG, Heath WR, Mueller SN. Spatiotemporally distinct interactions with dendritic cell subsets facilitates CD4+ and CD8+ T cell activation to localized viral infection. Immunity 2015; 43: 554–565. [DOI] [PubMed] [Google Scholar]
- 91. Yamauchi T, Hoki T, Oba T, Attwood K, Cao X, Ito F. CD40 and CD80/86 signaling in cDC1s mediate effective neoantigen vaccination and generation of antigen‐specific CX3CR1+ CD8+ T cells in mice. bioRxiv 2020: 10.1101/2020.1106.1115.151787. [DOI] [PMC free article] [PubMed]
- 92. Lamballe F, Ahmad F, Vinik Y et al. Modeling heterogeneity of triple‐negative breast cancer uncovers a novel combinatorial treatment overcoming primary drug resistance. Adv Sci 2020; 8: e2003049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zanker DJ, Spurling AJ, Brockwell NK et al. Intratumoral administration of the Toll‐like receptor 7/8 agonist 3M–052 enhances interferon‐driven tumor immunogenicity and suppresses metastatic spread in preclinical triple‐negative breast cancer. Clin Transl Immunol 2020; 9: e1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Uhlen M, Zhang C, Lee S et al. A pathology atlas of the human cancer transcriptome. Science 2017; 357: eaan2507. [DOI] [PubMed] [Google Scholar]
- 95. da Huang W , Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4: 44–57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials