

Abstract
BACKGROUND:
Recent advances in medical care have increased life expectancy and improved the quality of life for people with Down syndrome (DS). These advances are the result of both pre-clinical and clinical research but much about DS is still poorly understood. In 2020, the NIH announced their plan to update their DS research plan and requested input from the scientific and advocacy community.
OBJECTIVE:
The National Down Syndrome Society (NDSS) and the LuMind IDSC Foundation worked together with scientific and medical experts to develop recommendations for the NIH research plan.
METHODS:
NDSS and LuMind IDSC assembled over 50 experts across multiple disciplines and organized them in eleven working groups focused on specific issues for people with DS.
RESULTS:
This review article summarizes the research gaps and recommendations that have the potential to improve the health and quality of life for people with DS within the next decade.
CONCLUSIONS:
This review highlights many of the scientific gaps that exist in DS research. Based on these gaps, a multidisciplinary group of DS experts has made recommendations to advance DS research. This paper may also aid policymakers and the DS community to build a comprehensive national DS research strategy.
Keywords: Down syndrome, intellectual disability, cognitive development, autism spectrum disorder, obstructive sleep apnea, congenital heart disease, obesity, muscle hypotonia, leukemia, autoimmune disease, periodontitis, Alzheimer’s disease
1. Introduction
Down syndrome (DS) is the most common chromosomal disorder in humans, affecting about one of every 675 births [1]. Underlying the diverse spectrum of phenotypes seen in people with DS is an extra copy of chromosome 21 (Chr21), or trisomy 21 (T21), which results in overexpression of many genes and changes in the proteome [2]. It is associated with intellectual disability, facial dysmorphism, short stature, and poor muscle tone or loose joints. There is also an increased risk of Alzheimer’s disease (AD), childhood leukemia, congenital heart disease, sleep dysfunction, metabolic disorders, autoimmune disorders such as thyroid disease, Type 1 diabetes, celiac disease, rheumatoid arthritis, and developmental disorders such as autism spectrum disorder (ASD) [3]. At the same time, however, individuals with DS have a decreased risk of developing atherosclerosis and certain adult cancers [4, 5]. With improved healthcare, people with DS are now living longer, with a life expectancy of > 55 years of age compared to just 25 years of age in the 1980’s. It is estimated that there are 210,000 people with DS in the United States and 40% are over the age of 30 years old [6].
Research to better understand DS across clinical, pathological, genetic, cellular, and molecular domains is essential to develop appropriate interventions to promote health and wellness across the lifespan. Such research has flourished in recent years, driven by technological innovations, emerging international collaborations, increased funding, and worldwide advocacy efforts. With the hope of leveraging the growing momentum to support DS research, two major DS organization, the National Down Syndrome Society (NDSS) and the LuMind IDSC Foundation crafted recommendations for a research strategy focused on the quality of life and care priorities of people with DS by 2030. The impetus for this effort was the US National Institutes of Health (NIH) RFI (Request for Information, Notice Number: NOT-HD-20–013) to update NIH research plans on Down syndrome in 2020. The NDSS and LuMind IDSC worked together with the scientific community to develop recommendations for the NIH. In addition, other DS organizations contributed, including the Jérôme Lejeune Foundation and the National Task Group on Intellectual Disabilities and Dementia Practices.
2. Methods
DS research is broad in scope and engages a wide spectrum of scientific and health care disciplines. To accomplish the goals of the project, eleven working groups made up of experts across multiple disciplines were established to focus on specific issues of special concern to people with DS. These topical groups addressed cognitive development; autism spectrum and behavioral disorders; impaired speech, language, hearing, and vision; heart and vascular disorders; sleep and respiratory problems; obesity, metabolic and musculoskeletal problems; cancer; immune system disorders; dental and oral health; and AD and aging. The eleventh working group focused on the importance of community engagement in all aspects of DS research to ensure that the strategy developed through this process is driven by and resonates with the DS community. Members of the community engagement working group came from a variety of organizations including LuMind IDSC, NDSS, local Down syndrome affiliates, and GiGi’s Playhouse. Important contributions were also made by caregivers and self-advocates to ensure that the DS community had input into the recommendations.
Workgroup members met via teleconference for over four months to discuss the state of the science in their respective topic areas, with plans to distill their findings into a research agenda for the next decade. In April 2020, a two-day meeting was held in a virtual format due to the COVID-19 pandemic. The virtual meeting stimulated discussion that enabled top researchers in the field to share information, look for areas of overlap across workgroup topics, and identify unaddressed needs and research gaps. This paper summarizes areas of medical need for people with DS that have been understudied, unfunded or underfunded historically by federal and state agencies and foundations. The article is intended to serve as a call to action to the entire biomedical research community, to act as a catalyst for advocacy in research on DS, and to give research on DS the attention and funding that people with DS deserve.
3. Cognitive development and independence
To understand the effects of T21 on the development and function of the brain, three main questions were identified as areas to prioritize: 1) What brain regions and cell types are affected and what is the relationship of these effects to cognitive phenotypes? 2) When do deficits arise; are they the consequence of neurodevelopmental changes or functional changes in neural cells; and are they preventable or potentially correctable? 3) What are the underlying mechanisms by which T21 causes cognitive deficits? By understanding the answers to these questions at cellular and molecular levels, it may be possible to identify therapeutic targets and at what point targeting them might prove useful to positively affect cognition.
Human studies provide information about how brain anatomy and structure change across the lifespan and are also critical for genomic analyses. Anatomical studies indicate that DS is associated with a smaller cortex and cerebellum, with reductions in the number of neurons but increases in numbers of glia [7–9]. Neurons have dendritic spine defects and altered synaptic plasticity while glial defects indicate altered myelination [10, 11].
Deficits have also been reported in the function of synapses, mitochondria, and endosomes; and cell stress pathways appear to be activated. Several human Chr21 (HSA21) genes have been implicated in development, including some that regulate neural development and developmental signaling pathways and others involved in multiple stress mechanisms. Many more HSA21 genes have not been well studied and gene interaction effects must also be considered. In addition, T21 affects the expression of genes throughout the genome and so consideration of molecular and cellular pathways, metabolic and immune defects, and environmental factors may also impact brain function in DS.
Translating these anatomical and neural findings has helped refine what aspects of cognition are targeted in studies of individuals with Down syndrome. Specific constructs of executive functioning, learning and memory, and working memory are being targeted with an emphasis on determining the best measures for assessing these constructs and change in these constructs [12–16]. Further attention to the measurement of cognitive constructs has focused on social cognition, emotion recognition, and evaluating cognitive batteries [17–28]. The downstream impact of these cognitive constructs on functional outcomes has primarily considered children with DS, understanding the impact on formal education, and adaptive daily living skills [29–35]. Given a pattern of findings linking cognitive skills to fine and gross motor control [36–41], preliminary small pilot trials to improve cognitive outcomes have more recently focused on the impact of physical activity interventions [42–44]. Both behavioral and pharmaceutical interventions are also being piloted to improve cognitive outcomes in individuals with DS [44–51]. Research on individuals with DS has frequently focused on between-group differences in comparison to other individuals with intellectual disability [52–57]. This focus has helped identify the unique cognitive phenotype common in individuals with DS. Group differences have also elucidated the impact of comorbid medical conditions on cognitive outcomes for individuals with DS, specifically cognitive outcomes for children with DS with or without comorbid congenital heart defects, sleep challenges including obstructive sleep apnea (OSA), and autism spectrum disorder [58–75].
Cognition may have impacts on other aspects of life such as independence. For individuals with DS, the degree of intellectual disability is one possible factor that may restrict independence. Studies show that speech ability and training, and access to a medical home can improve independence for individuals with DS [76, 77]. Measures of adaptive skills and surveys show that independence decreases with age in DS, which is likely multifactorial and presumably related to increasing risk for dementia [78, 79]. Drawing from studies of individuals with intellectual disability, speech independence, autonomy, and self-management predict independence, while low physical fitness and changes in ADLs may indicate decreasing independence [80–82]. Importantly, independence for individuals with intellectual disability can be promoted through interventions including video prompts, video self-modeling, staff training, and the use of technology and remote support services [81, 83–85].
While studies in humans are the gold standard, two types of research models are valuable to address cellular and molecular aspects of T21 in the brain: trisomic mouse models, and human T21 induced pluripotent stem cells (iPSCs) [86]. Human stem cell research enables investigations of T21 in the human genetic background at the molecular and cellular levels and enables mechanistic investigation. Current understanding of DS neurobiology is derived largely from mouse studies (e.g. Ts65Dn, Tc1) [87, 88]. Support for new mouse models that will minimize non-HSA21 genetic changes are needed [89]. In addition, it is becoming clear that aspects of gene regulation and brain development and function differ between mice and humans, so integrating information from these complementary models is essential to understand trisomy neurobiology and development [90–92].
Many drug candidates tested are reported to improve learning and memory in DS mouse models; however, the relevance of these findings to humans remains uncertain. Human cell and organoid models may be useful for testing drugs that target cell-based pathologies. Biomarkers – including functional measures such as functional magnetic resonance imaging (fMRI), electroencephalography (EEG), and functional near-infrared spectroscopy (fNIRS) - are clearly needed to advance drug development.
4. Cognitive development and independence research gaps
To gain a better understanding of cognitive deficits associated with DS, new preclinical models of cognition are needed to help establish the stage of cognitive development that could be amenable to improvement. Therefore, inducible models of trisomy silencing or specific gene silencing should be developed. In addition, mouse studies are needed that can establish a correlation between changes in synaptic plasticity with learning and memory deficits. Finally, new DS models should reflect human physiology and genetic heterogeneity (e.g. outbred backgrounds).
More support of human cognition research in DS is also needed. To establish the link between cognitive performance and underlying brain structure, neurocognitive tools such as fMRI, EEG, and transcranial magnetic stimulation (TMS) should be utilized. Reliable and valid clinical outcome measures across the lifespan should be developed, as well as studies on the natural development of these associated constructs, to enable longitudinal studies and to advance clinical trials [12, 15]. It is important to support research focused on the wide heterogeneity of cognitive function across the DS population, including the effects of medical conditions (e.g. Attention-Deficit Hyperactivity Disorder (ADHD), ASD, anxiety, Acute Myelocytic Leukemia (AML), Acute Lymphocytic Leukemia (ALL)) in order to identify those individuals who will benefit from a particular intervention and who would be appropriate for inclusion in a clinical trial. To better understand all the factors that impact cognitive skills and meaningful functional outcomes, it is important to establish strong collaboration across behavioral disciplines.
There is need to include assessments of independence when studying human cognition. To understand the factors that contribute most to independence, additional research on the natural history of independence in people with DS is needed. Specifically, consideration should be given to what aspects are most meaningful to an individual with DS and caregivers, how to measure independence reliably and validly, and how to modify those factors. Successful interventions used in individuals with ID can guide implementation and study in individuals with DS. Large, multi-site, longitudinal studies could evaluate human cognition and independence simultaneously to connect the lessons learned from basic science to clinical significance; with the ultimate goal of improving independence.
5. Behavior and autism spectrum disorder
The prevalence of coexisting psychiatric or serious maladaptive behaviors in children and adolescents with DS is high and the consequences on education, family functioning, and socialization substantial. It has been reported that coexisting neurobehavioral and psychiatric disorders in those with DS range from 18 to 38% [93]. In a Swedish population-based study of children and adolescents with DS, 42% were diagnosed with ASD and 34% with ADHD [94], with the severity of ASD but not ADHD positively associated with the level of intellectual disability [95]. Other studies have found similar rates of ASD and behavioral symptoms in children with DS [96].
Some adolescents and young adults with DS also present with progressive and sometimes rapid cognitive deterioration or regression, which may dramatically impair independence and autonomy [97, 98]. A minority of individuals with DS experience worsening autistic characteristics that progress to a dementia-like state in what has been called Down syndrome disintegrative disorder (DSDD) [99, 100]. The causes of regression and DSDD remain unclear, but may be associated with depression, hypothyroidism, autoimmune disorders, OSA, and/or subclinical epilepsy. One small study, which hypothesized an immune-related etiology, demonstrated significant improvements following treatment with immunotherapy [101].
6. Gaps in behavior and ASD research
Population-based normative data on maladaptive behaviors and psychiatric syndromes across the lifespan in DS are needed from studies with large and diverse cohorts. In addition, population-based studies are needed to evaluate the associations between ADHD, ASD, anxiety, depression, movement disorders, seizure disorders, and sleep disturbances. People with DS should be included in clinical trials of emerging therapies (e.g. novel biologics, anti-immune, anti-inflammatory, GABA/glutamate, and cell-based therapies) for autism and behavioral disorders. However, validated diagnostic and outcome measures need to be developed in order to assess behavioral disorders and autism for the DS population. In addition, neuroimaging and neurophysiology studies of behavioral disorders and ASD should be conducted in people with DS. Finally, there is a shortage of trained providers and researchers knowledgeable about DS. This has had a negative impact on advancing understanding of behavioral and psychiatric disorders in DS. Additional support for training fellowships in DS is needed.
7. Communication, vision, and hearing
A variety of structural and functional vision, hearing, and communication deficits are associated with DS, as well as other anomalies of the auditory/vestibular system. Variation in these structures and functions can be influenced by other aspects of the behavioral phenotype associated with DS. Taken together, these problems can interfere with the development and maintenance of communication skills critical to reading and oral language.
7.1. Communication disorders in DS
Difficulties with communication often occur in individuals with intellectual disability, but people with DS are 2.6 times more likely to have moderate communication difficulties and 1.9 times more likely to have severe communication difficulties than people with intellectual disability not associated with this syndrome [102]. It is important to understand the reasons why many individuals with DS have lifelong problems with oral and written communication. Individuals with DS often exhibit delays in language development, with greater delays in expressive than receptive language [103]. Difficulties with language development are demonstrated from very early in development and continue into adulthood, with delays observed in the earliest stages of prelinguistic communication. These delays continue into the adolescent and young adult years, including the use of complex language for participation and social interaction in everyday life [104]. Speech is the main form of communication in 97% of all people with DS and therefore is critical to social interactions of all kinds. Reduced speech intelligibility is common in children and adults with DS [105, 106], resulting from a combination of motor impairments, phonological delay or disorder, hearing loss, and craniofacial and laryngeal dysmorphology [107]. Speech intelligibility and language delays are major concerns for children with DS and their families. But many adults with DS also experience lifelong difficulty with the intelligibility of their speech [108]. Nonetheless, children with DS do demonstrate gains in language development and a pattern of relative strengths and weaknesses in this domain, including more substantial delays in expressive syntax than in expressive vocabulary, for example [109]. Recent studies have also shown that verb usage is particularly limited in people with DS, leading to less language complexity and poorer discourse skills [110]. However, there are developmental changes in this profile over time [103, 104, 111]. For example, receptive and expressive vocabulary have been shown to improve during early adolescence but to decline in late adolescence and early adulthood, perhaps foreshadowing the later more dramatic declines associated with early onset Alzheimer’s disease. Given nuances in language for people with DS, treatment and intervention for language impairments in this population must consider both the language phenotype and overall behavioral phenotype [103, 104, 112], leveraging language strengths to support treatment and intervention efforts.
7.2. Gaps in communication research
Research focused on the development of assessments and new interventions for improved communication for individuals with DS across the lifespan is needed. Validated, standardized measures of language may be used but may need modification for extensive testing in the DS population. The development of DS-specific language norms is needed, with the incorporation of other psychometric features, especially those relevant for use as outcome measures in treatment studies, including clinical trials. Studies that can distinguish the effects of motor speech disorders from effects caused by anatomical dysmorphology are needed to develop tools for clinical assessments and to facilitate the development of effective interventions across the lifespan. More and larger studies assessing the efficacy of high-intensity speech interventions in young children are needed. There is also a need to more fully understand the ways in which the social and linguistic environment supports or hinders language development in order to identify potential pathways and mechanisms for intervention. Lastly, there is a need for studies on language and communication in adulthood to understand the trajectory of improvement and possible age-related decline.
7.3. Vision disorders in DS
Ocular disorders are common in DS with reduced visual acuity observed even with refractive corrections. Specific impairments in vision include elevated refractive errors, strabismus, nystagmus, and corneal abnormalities all of which may contribute in part to the reduced visual acuity observed in DS [113]. These disorders are associated with abnormal visual developmental and possibly structural differences in the retina, cornea, and optic nerve [114–117]. Improved clinical strategies are needed to address visual deficits in this group, as most individuals do not achieve normal visual performance with standard refractions. Early intervention is likely to aid visual development for children with DS, but structural deficits in ocular tissues, such as those commonly observed in the cornea, may progressively worsen in later years, leading to additional losses in vision quality.
7.4. Gaps in vision research
A greater understanding is needed regarding the causes of reduced visual acuity, e.g., the contributions of retinal, corneal, and neural abnormalities. There is also a need to expand studies of the impact of optimized refractive correction on visual outcomes to younger children. Longitudinal studies with cohorts large enough to achieve statically meaningful results are needed to understand the progression of structural changes in the ocular system and correlations with visual outcomes across the lifespan.
7.5. Hearing loss and vestibular problems in DS
Hearing loss is common in children and adults with DS. The incidence of hearing loss in neonates and infants with DS is between 15% and 30% [118, 119]; between 25% and 85% in children and adolescents [120, 121]; and between 50% and 75% in adults [122, 123]. While most hearing loss in DS is associated with otitis media with effusion, mixed conductive-sensorineural loss is also common and is usually secondary to inner ear malformations [124], which may also cause symptoms of vestibular dysfunction such as dizziness, vertigo, and balance disorders. Anomalies in the auditory system in DS have been reported for the outer ear, middle ear, and inner ear [125–131]. Other factors that may contribute to vestibular dysfunction in DS include: hypotonicity, joint laxity, decreased deep tendon reflexes and delays in reaction timing, and equilibrium reactions. The vestibular system works with other senses such as vision and proprioception to maintain balance and motor coordination.
7.6. Gaps in hearing loss and vestibular function research
More research on the detection and treatment of hearing loss across the lifespan of individuals with DS is needed. Assessment and detection measures are needed that account for developmental and aging effects as well as structural differences that may be present. Research on strategies to increase the use of hearing aids among adults with DS who has hearing loss also is needed. There is evidence that DS-AD is sometimes misdiagnosed since the symptoms observed may actually be due to hearing impairment or other sensory deficits [132]. Hearing loss may also be a contributing factor given that it is more frequent among adults with DS and co-morbid dementia [133]. More research on the diagnosis of hearing impairment in adults with DS is needed.
8. Heart and vascular
Congenital heart disease (CHD) represents one of the cardinal features of DS, affecting more than 40 percent of infants with T21 [134]. Multiple subtypes are seen, including complete atrioventricular septal defects (AVSD), ventricular septal defect, atrial septal defect, partial atrioventricular septal defect, Tetralogy of Fallot, and patent ductus arteriosus; and the presence of multiple anomalies is common [135–139]. Early diagnosis improved surgical outcomes, and better perioperative care have resulted in significant increases in survival over the past two decades, contributing to the improved longevity for people with DS.
Trisomy 21 clearly significantly increases the risk for AVSD and other heart defects but is not sufficient to cause CHD. It was originally hypothesized that T21 unmasks a common susceptibility variant that explained the hugely increased in risk for AVSD observed in DS. Using a genome-wide association study approach, no evidence for this hypothesis was obtained [140–142]. Instead, current findings suggest an increased burden of rare variants as contributing to risk for AVSD in DS, some of which overlap with those found for non-syndromic CHD (nsCHD). Such variants (including single-nucleotide variants, microRNAs, and copy number variants) are not restricted to chromosome 21, but found throughout the genome [140, 141, 143–145]. Thus, the modified hypothesis states that having an extra Chr21 predisposes to abnormal heart development, but additional rare variants or environmental triggers are required to exceed a susceptibilty threshold to lead to CHD. Support for this hypothesis and proof-of-principle has been provided by the Ts65Dn mouse model [146, 147]. Evidence for altered pathways include the ciliome, Notch signaling, VEGF-A and folate/homocysteine metabolism [144, 145, 148].
In addition to CHD, individuals with DS often experience other cardiovascular problems including cardiac arrhythmia, pulmonary hypertension, and sleep apnea. A higher risk of cerebrovascular events including stroke and transient ischemic attack is also seen, particularly in women [149]. Immune dysfunction and thyroid dysfunction – both prominent in DS –may also affect cardiovascular function; and extra-cardiac comorbidities such as differences in vascular resistance and arterial stiffness may contribute to cardiovascular dysfunction. Alterations in heart rate variability and blood pressure in the low normal range are also relatively common in DS and may reflect autonomic dysfunction [150]. The consequences of lower blood pressure and heart rate in DS over the lifespan are poorly understood, as is the effect of exercise on autonomic function. The Ts65Dn mouse model also demonstrates reduced blood pressure and heart rate variability alterations from wild-type mice, suggesting this model could be used to explore mechanistic changes in cardiovascular function across the lifespan in DS [151]. Moyamoya disease may also occur in people with DS [152] and contributes to increased stroke risk over the lifespan, particularly with those who have blood pressure in a higher, but normal range [153].
9. Heart and vascular disease research gaps
The genetic factors and dysregulated pathways in DS-associated CHD are just beginning to be identified. The roles of nuclear and mitochondrial genes and their possible interactions with HSA21 genes have yet to be determined. Research integrating transcriptomic, metabolomic, proteomic and other -omic approaches should be applied to these knowledge gaps. In addition, epigenomic approaches could facilitate identification of environmental exposures associated with CHD. The integration of -omics data could help identify underlying molecular mechanisms, help define genotypes and phenotypes which could lead to prevention and novel treatments. From the clinical and epidemiological research perspective, larger sample cohorts with well-defined heart phenotypes (cardiac and extra-cardiac) are needed to define genotype / phenotype correlations in DS-associated CHD. Complementing these studies with model systems is essential to understand genotype functions and their interactions with the environment.
More clinical research is needed to better understand cardiovascular disease in DS. For example, there is a need to advance understanding of the surgical and postoperative care needs of children with DS and CHD. The early detection of Moyamoya disease is essential for better outcomes but this will require a better understanding of early risk factors and the development of novel biomarkers. The role of lifestyle factors such as sedentary lifestyle and exercise on cardiovascular function in individuals with DS is not well understood. There is early evidence that increasing exercise, even passive exercise, may also have positive effects on cognition and learning [44, 154]. One issue that needs to be addressed to advance clinical research in DS is the issue of control groups. Proper controls are essential for cardiovascular research in DS, however, there are many different types of controls utilized (heart rate controls, intellectual disability without DS controls, BMI controls, activity level controls), which complicates interpretation of data from these studies. The field should address this issue and build consensus around a single type of DS control.
10. Sleep and respiratory
Individuals with DS are prone to develop OSA due to a combination of anatomic and neuromotor factors, such as midfacial hypoplasia, macroglossia, tracheal and laryngeal abnormalities, and hypotonia [155, 156]. With aging, the prevalence of OSA increases but it remains frequently underdiagnosed. Untreated sleep disorders such as OSA may contribute to worse cognitive function and accelerate cognitive decline [45, 46, 47]. Other respiratory problems including increased risk of infection are common in children and adults with DS due to a combination of the mentioned anatomical and functional abnormalities, immune dysfunction, and cardiac problems [157, 158]. For example, respiratory syncytial virus (RSV) infection is particularly prevalent and associated with high morbidity and mortality in infants and children with DS [159]. Dysphagia, also common in DS, increases the risk of aspiration and recurrent pneumonia [160].
Children with DS also have an increased risk for developing multifactorial pulmonary hypertension [161] associated with hypoxemia, OSA, pulmonary hypoplasia, increased pulmonary vascular resistance, and increased hemodynamic stress. The sub-group of children with DS and CHD are at particularly high risk of developing pulmonary hypertension, but it can also be a consequence of respiratory distress syndrome [162]. Lower airway anomalies such as hypoplasia of the alveoli and other more distal structures have also been reported [163].
11. Gaps in sleep and respiratory research
Despite previous sleep research, normative data on sleep patterns in DS are still lacking. In children and adults with DS, research is needed to evaluate the association of a characteristic DS sleep phenotypes, i.e. reduced sleep efficiency, decreased rapid-eye movement (REM) sleep, and increased slow-wave sleep (SWS, also called non-rapid-eye-movement 3 (NREM3 or N3) sleep), with learning and behavioral difficulties in the context of baseline neurocognitive impairment.
Neurocognitive studies to evaluate the impact of sleep disturbances in adults with DS and its relationship to cognition, behavior, and quality of life are needed. Indeed, it is important to prospectively assess the impact of sleep disturbances on cognitive impairment and progression to dementia in adults with DS. Objective sleep evaluation methods and a DS-specific cognitive battery are needed to properly evaluate baseline relationships and intervention outcomes. More research is required on circadian rhythm disruptions in people with DS. Finally, the design and validation of new sleep questionnaires and simple at-home devices to screen for sleep disorders are needed.
Pulmonary hypertension is a significant cause of morbidity in children and infants with DS. More research is needed to distinguish the clinical and molecular pulmonary hypertension phenotypes in DS compared to children without DS. In addition, non-invasive airway evaluation guidelines for children with DS that incorporate multidisciplinary aerodigestive programs are needed.
12. Musculoskeletal, metabolic factors, and obesity
Past research findings indicate that individuals with DS across the lifespan are at high risk for musculoskeletal issues and metabolic disorders such as obesity and Type 2 diabetes [164, 165]. The genetic and molecular mechanisms that underlie musculoskeletal and metabolic dysfunction in DS remain unclear. T21 has been associated with altered insulin secretion, impaired hepatic glucose metabolism, and altered insulin sensitivity in muscle. Autonomic dysfunction with altered peripheral blood flow to muscle may affect blood distribution to the muscle and build-up of toxins, which may lower the pain threshold and tolerance for physical activity. Research in both humans with DS and DS mouse models has identified changes in metabolism related to obesity, inflammation, mitochondrial function, immunity, insulin resistance, and glucose tolerance [166].
Individuals with DS across the lifespan are at higher risk to be overweight and obese compared to the general population [167–170]. The possible determinants of increased weight and obesity in individuals with DS include low resting metabolic rate, low physical activity levels, and unhealthy dietary behaviors [168, 171–173]. Obesity appears to have adverse health outcomes among individuals with DS such as dyslipidemia, hyperinsulinemia, OSA, and gait problems [168, 174–178].
Musculoskeletal problems associated with DS include muscle hypotonia and joint laxity, both of which can cause ambulation difficulties and other functional impairments [164]. Moreover, these problems may contribute to reduced physical activity, increased levels of obesity [168], and an elevated risk of many disorders including heart disease, Type 2 diabetes, cancer, and osteoporosis [179]. Individuals with DS across the lifespan have very low physical fitness and physical activity levels [180–184]; these attributes may be either causes or outcomes of musculoskeletal and metabolic issues or bidirectional relationships may be at play. Certainly, improving the physical activity and physical fitness levels of individuals with DS may positively impact their health and functional profiles.
There are ongoing international efforts to better understand metabolic dysregulation in DS. The Gene Overdosage and comorbidities during the early lifetime in Down syndrome (GO-DS21) project recently launched in the European Union to explore metabolism and metabolomics in DS.
13. Gaps in musculoskeletal and metabolic research
Longitudinal studies on muscle development and weight changes across the lifespan in DS are needed to explore differences between people with DS and the general population. These studies should evaluate the development of obesity across the lifespan, delineate its causes and clinical impact, and test interventions for reducing obesity in individuals with DS. Hypotonia is very common in DS but it is not well understood. Research on the genetic and biochemical basis of hypotonia including the role of mitochondrial alterations is needed to clarify the etiology of the condition. In addition, the effect of impaired autonomic function on cardiovascular fitness is not well understood and needs more research. There have been a few small physical activity intervention trials in DS [185–187]. More and larger trials are needed to test the effects of physical activity as an intervention on a range of health outcomes and the modifications that may be needed to implement such interventions in the DS population.
14. Cancer
Although there is no difference in the overall incidence of cancer among the DS population compared to the non-DS population, DS is associated with a decreased incidence of solid tumors and an increased incidence of hematological malignancies, specifically acute leukemias [5, 188].
Compared to the non-DS population, individuals with DS are 500 times more likely to be diagnosed with acute myeloid leukemia (ML-DS) and 20 times more likely to be diagnosed with acute lymphoblastic leukemia (DS-ALL). Survival outcomes for ML-DS are superior to the general population, with event-free survival rates ranging from 80–100%. Clinical trials are focused on optimizing chemotherapy to reduce toxicity while maintaining survival outcomes [189, 190]. Outcomes for patients with DS-ALL are inferior to the general population and patients with DS treated for ALL have excess treatment toxicity and treatment morbidity. Novel approaches to treat DS-ALL are needed to improve survival and decrease treatment morbidity [191, 192]. Factors that contribute to variability in treatment response and outcomes are not fully understood but may include differences in the genetics and biology of DS-leukemia as well as those related to the DS-phenotype (i.e., gene dose effect from T21).
The risk for DS-leukemia (ML-DS and DS-ALL) is greatest during early childhood (ages 1–4), a critical neurodevelopmental period. Treatment for DS-leukemia (DS-ALL in particular) involves central nervous system (CNS)-directed chemotherapy. Treatment for DS-ALL is 2.5 – 3 years in duration with extended periods of immunocompromise resulting in missed opportunities for social interaction, early intervention and education services that support developmental gains throughout childhood. It has been well-established that survivors of childhood ALL without DS have a higher risk of neurocognitive deficits [193, 194]. Given the preexisting cognitive vulnerability in DS, the cumulative impact of CNS-directed treatment and missed community participation may add to neurocognitive deficits. Compared to survivors of leukemia without DS, survivors of DS-leukemia may be at increased risk for treatment late effects and poorer quality of life [195–197]. Improved understanding of the impact of leukemia and its therapy on neurodevelopmental, health, and quality of life outcomes has the potential to inform modifications to treatment, approaches to supportive care during therapy, and interventions to ameliorate side effects.
15. Gaps in cancer
Improved characterization of clinical, biological, and genetic phenotype in DS-leukemia is needed to identify therapeutic targets and further refine treatment. A better understanding of DS-leukemia genomics may also have implications for surveillance and diagnosis. Evidence-based approaches to supportive care, improved assessment and management of side effects and toxicities in DS leukemia needs to be developed. Longitudinal studies of neurodevelopmental, health, and quality of life outcomes in cancer survivors beginning during therapy, in order to inform supportive care and interventions to ameliorate problems are also needed.
15.1. Immune system disorders
The architecture of the immune system is significantly altered in people with DS, with common findings of leukopenia, lower B cell frequencies, and pro-inflammatory shifts including increased proportion of memory T cells, pro-inflammatory T cell subsets, and pro-inflammatory cytokine-producing cells [198, 199]. Consistent with this pro-inflammatory milieu, people with DS exhibit a highly increased risk of developing autoimmune diseases including thyroid disease, Type 1 diabetes, celiac disease, rheumatoid arthritis, systemic lupus erythematosus, and atopy. Paradoxically despite this augmented immune response, individuals with DS are more susceptible to infectious disease, which account for 50% of deaths in individuals with DS [3]. This includes increased mortality secondary to RSV and now to COVID-19 infections. [198–204]. Additionally, these diseases may manifest at earlier ages and greater severity in people with DS.
16. Gaps in immune system disorders research
Critical gaps exist in our understanding of the specific facets of immune dysregulation and the underlying mechanistic pathways, which drive predisposition to autoimmunity and poor outcomes with infection. Research is needed to clarify which existing therapeutics are most effective for people with DS and to prioritize targets for novel therapeutics. Although candidate genes on Chr21, including IFNAR1/2 and DYRK1A, have been identified, the role of most HSA21 genes is unclear. Additionally, studies showing that T21 can impact transcription on other chromosomes suggests that the scope of this research will likely extend beyond Chr21. Because the risk of autoimmunity increases with age, longitudinal studies are also needed with genomic confirmation of full or partial trisomy of genes associated with immunity. Sampling prior to developing autoimmunity is necessary to allow us to better understand who is at higher risk, and how that risk might be mitigated. Genotyping for known autoimmunity-associated polymorphisms will help us better understand how T21 modulates single nucleotide polymorphisms (SNP)-associated risk. Similar SNPs on T21 that are normally rendered silent might exert their effect due to gene dosage, but also perhaps due to overall perturbed clinical state. Linked clinical metadata, particularly the severity of autoimmunity and response to specific therapeutics, will enhance translational significance of this work. To better understand the role of thymic dysfunction in autoimmunity, studies should assess the consequences of thymectomy (e.g. on the differentiation of T cells into subsets and the production of self-reactive T cells) in children with CHD who undergo this procedure to provide surgical access as part of their cardiac surgery. As the immune system penetrates all organ systems, it is important to better understand how the immune system connects to dermatological, neurological, and gastrointestinal systems and how it may influence the course of disease in cancer and AD alike. Finally, the emergence of the COVID-19 pandemic makes research in the DS population a priority including for the development of potential therapies.
17. Dental & oral health
Issues with dental and oral health are very common in DS and often directly impact the quality of life. There are many differences between individuals with DS and the general population [205]. Differences include delayed eruption in babies and children, differences in eruption sequences for primary teeth, microdontia, and hypodontia [206, 207]. People with DS may have large tongues (macroglossia), or they may have an average size tongue but a small upper jaw that makes their tongue too large for their mouth (relative macroglossia) causing difficulties with speech and breathing and contributing to OSA. Small jaws and microdontia can cause tooth crowding and problems with spacing where the teeth of the upper and lower jaws do not touch affecting the bite. Orthodontics may be able to improve some of these issues, but this may present a particular challenge for children with DS. Finally, there are reports that people with DS have a lower risk for cavities; however, much of that research was done when people with DS lived in institutions and had very restricted diets. As such, more research is needed on the caries prevalence in people with DS [208, 209].
People with DS may have an aggressive form of periodontitis characterized by rapid progression, significant bacterial and inflammatory burden, and an onset as early as 6 years of age, which could contribute to other systemic diseases as well [210–213]. However, studies have shown that periodontal therapies may be effective [214]. It has been observed that in elderly cognitively normal people, measures of periodontal destruction and periodontal dysbiosis is associated with brain amyloidosis [215]. Given the high prevalence of both AD and periodontal disease in DS, more research is needed to determine if periodontitis contributes to AD pathology [216].
Dental and oral health has a broad impact on overall health and quality of life. Thus, more research is needed to understand the impact of dental and oral health in DS and the association with development, sleep disorders, the immune system, and other common co-occurring conditions. This research could lead to greater insights in the role of oral health on the overall health of people with DS and lead to better treatment options.
18. Dental and oral health research gaps
The development and validation of cellular and animal models are needed to better understand the characteristics of dental and oral health in individuals with DS. More research is needed to better characterize the differences between DS and the general population in dental development and oral health to help inform medical guidelines. Furthermore, the impact of periodontal disease on the overall health of individuals with DS is not well understood. DS mouse models should be employed to study the possible association between periodontal disease and AD related pathology such as Aβ plaques, neurofibrillary tangles, and neurodegeneration. Longitudinal and interventional studies should evaluate the role of periodontal inflammation and bacterial dysbiosis on AD progression [217].
19. Alzheimer’s disease and aging
With improved care and extended longevity for people with DS, there has been an increase in age-related disorders, especially AD, which typically occurs at an earlier age than that observed in the general population [218] (Fig. 1). Indeed, compared to people without triplication of Chr21, individuals with DS are at a markedly increased risk of developing AD. By age 40 years, β-amyloid plaque pathology and neurofibrillary tangles are present in almost all people with full trisomy [219]. It is estimated that the lifetime risk of AD is > 90% and that AD is the leading cause of death for adults with DS [220, 221]. However, few adults with DS associated AD (DS-AD) or with other age-related disorders receive appropriate care for a number of reasons. These include a shortfall of accepted and validated standards for care, lack of practitioners trained to provide support and evaluate people with DS-AD, inadequate data on the natural history of persons with DS-AD, lack of DS-specific diagnostic and treatment approaches, lack of evidence of efficacy of existing treatments, and inadequate resources for caregivers.
Fig. 1.
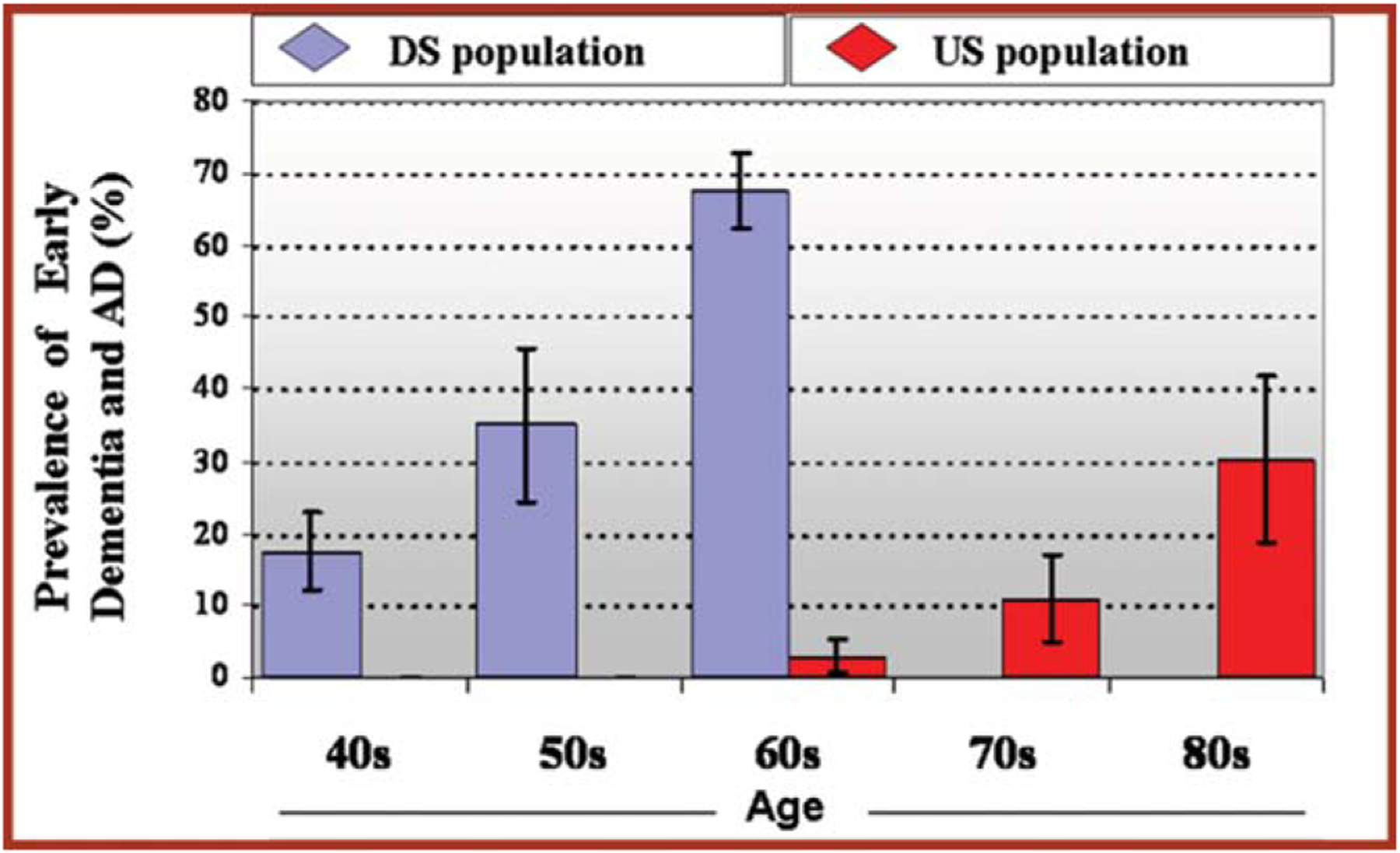
Prevalence of Early Dementia and AD by Age [218].
The reasons for the increased incidence of DS-AD are multifactorial but a primary driver is likely related to genes on HSA21. Among the genes expressed on HSA21 is the gene for amyloid precursor protein (APP). Overexpression of this protein due to trisomy leads to excess production of the toxic, AD associated protein, β-amyloid. β-amyloid accumulates within senile plaques in the brain but also can affect the blood vessels of the brain (cerebral amyloid angiopathy or CAA) [222]. Although most people with DS are rarely affected by atherosclerosis and arteriosclerosis, which are two risk factors for cerebrovascular pathology, there is significantly more CAA in DS than in people without DS or when compared to older people with AD [223, 224]. Neuroinflammation may also show unique signatures in the brains of people with DS, with features both common to and unique from sporadic AD [225]. Thus, there are multiple pathological events that occur either serially or in tandem to accelerate the development of AD in people with DS.
Genetic, cell biology, cellular / animal models, and natural history studies have provided insight into the aging process and the increased risk of dementia in individuals with DS. It is well known that the brain undergoes accelerated aging changes in DS, but biological mechanisms for this have not been fully explored. These studies suggest that increased expression of HSA21 genes induces many cellular changes, including dysfunction of endosomal and lysosomal systems, composition of neurotrophic signaling, increase in amyloid aggregation and cognitive defects, and alterations of microglia and astrocyte function [219, 226–232]. Genome-wide association studies (GWAS) have identified 25 candidate DS-AD risk alleles. In small DS participant datasets, longitudinal telomere length changes correlate with cognitive decline and accelerated epigenetic aging in DS is under investigation. Investigators probing the gut microbiome in people with DS have identified bacterial species that correlate with Aberrant Behavior Checklist (ABC) scores [233]. In addition, novel biomarker methods including neuron-derived exosomes and more sensitive plasma-based assays allow early diagnostics and a potential to better evaluate efficacy of interventions [234, 235].
Clinical phenotyping, biomarker development and validation in DS, and neuropathological diagnostic research have also progressed in recent years, enabling several clinical trials of potential AD therapeutics in individuals with DS. In addition, animal and cellular models in development have enabled exploration of therapeutic approaches, with early evidence that immune approaches may be feasible [236]. Many collaborations and partnerships have been established in recent years to accelerate research on DS-AD, including:
Horizon 21, a multisite study in Europe recruiting a trial-ready cohort [237]
The Alzheimer’s Biomarker Consortium-Down Syndrome (ABC-DS) – defining conversion to dementia based on biomarkers [238]
Longitudinal Investigation For the Enhancement of Down Syndrome Research (LIFE-DSR) – an observational cohort study [239]
Alzheimer’s Clinical Trials Consortium - Down Syndrome (ACTC-DS) is preparing to recruit a trial-ready cohort [240]
3-Star study of the candidate vaccine, ACI-24 sponsored by AC Immune is nearing completion; AC Immune will also soon launch a Phase 2 trial of β-amyloid vaccine in adults with DS [241]
20. Alzheimer’s disease and aging research gaps
More research support is needed to better understand the genetics and epigenetics of DS-AD. The underlying mechanisms that lead to high APP expression in DS-AD pathogenesis is not well understood. For example, genetic and epigenetic regulation of APP expression remains largely unexplored in people with DS. Epigenetic aging studies have been virtually unexplored in DS and could lead to novel genetic pathways that are amenable to intervention. Another yet unexplored area is the influence of specific microRNAs in the AD pathology. Chr. 21 contains several microRNAs, of which some have been shown to affect AD pathology. There is a need to perform sampling at multiple time points to evaluate trajectories across the lifespan. To explore the role of additional genetic AD risk factors, it will also be valuable to compare GWAS hits in DS-AD to those from late onset AD (LOAD) including their association with age of onset, cognitive decline, and variation in risk by sex or ethnicity. Other risk factors also need research attention such as sex, race, ethnicity, the role of inflammation, the immune system, and potential links to cerebrovascular pathology in DS-AD across the lifespan. There is a need to further refine the amyloid/tau/ neurodegeneration framework in DS for comparison to LOAD and dominantly inherited AD and to identify where and how neuroinflammation and cerebrovascular pathology contribute. The efficacy of treatments and care models in DS-AD are knowledge gaps. The use of approved AD drugs in people with DS needs study to determine if these widely used drugs are safe and/or appropriate for DS-AD. It is unknown if the therapeutic window of approved AD drugs is different for DS-AD as compared with LOAD. In addition, new care models need to be developed that enhance the quality of life and longevity in individuals with DS-AD.
21. Community engagement
Community engagement is a continuum depending upon the level of collaboration between researchers and the community yet is essential since members of the DS community can give meaningful insight into issues that are important to them across the lifespan. Community members should thus be included in all aspects of research studies, starting with the design of the study, the determination of inclusion and exclusion criteria, the selection of outcome measures, the recruitment of participants, and the development of informed consent procedures. As a research study progresses, members of the DS community should continue to be included in discussions of the progress of the study, interim results, and communication protocols with research participants.
Community engagement research focuses on ensuring that the inclusion of community members in studies achieves the goals of improving their understanding of the purpose of research, optimizes recruitment for clinical studies, improves the reliability and validity of measurement tools, and improves the translation of research into practice. The Community Engagement working group recommended the need to communicate the message better so that people with DS understand the reason for the research. They also called for the development of strategies to increase participation of people with DS in clinical trials and to engage individuals with DS in the design of such trials.
22. Community engagement gaps
A recent survey of 256 individuals with DS found that 86% want new treatments and interventions, but 64% never participated in research studies. In addition, for those that have participated in research studies, direct interviews with individuals with DS and their caregivers found a lack of meaningful engagement with them at each step of the research process. Increased efforts are needed to better understand the factors that lead to individuals with DS being significantly under-represented and oftentimes excluded from research studies, including potential discrimination, rigid inclusion and exclusion criteria, low prioritization and limited community outreach and awareness. More infrastructure is needed for outreach and information sharing and dissemination. In particular, new outreach approaches are needed for the older adult community who tend to live outside the family unit in independent living, groups homes and long-term care facilities.
23. General research needs
Each workgroup identified research needs for their specific focus area and many of these research gaps have been highlighted above. However, the workgroups also identified areas that represent clear DS research needs that cross multiple disciplines. The table below summarizes eight areas of overall research need that, if funded, would significantly improve the understanding of DS and lead to improvements in treatments and quality of life. The eight overall topic areas are clinical and genetic phenotyping, cellular and animal models of DS, longitudinal studies, randomized controlled trials in the DS population, centralized biorepository, open access centralized DS data, research training for clinicians and scientists, and research inclusion. The workgroups also developed a comprehensive list of recommendations in response to the DS RFI. The full list of recommendations can be found at: https://www.nih.gov/sites/default/files/research-training/initiatives/include/down-syndrome-RFI-responses-combined.pdf.
24. Conclusion
While it is true that the life expectancy for people with DS has increased significantly in recent decades thanks to advances in medical care, this review clearly demonstrates that many scientific gaps remain that prevent further advances in the discovery of new treatments and improvements in quality of life. These research recommendations were developed specifically in response to the NIH RFI and it is the hope of the coauthors that the NIH will not only include the recommendations submitted and published here in their Research Plan on Down Syndrome, but also provide sufficient funding for implementation. The coauthors also recognize that, for these recommendations to achieve their desired result, a coordinated approach with other funders of research, including other U.S. government agencies, and non-government organizations is needed. In addition, it is hoped that this paper will serve as a call to action for policymakers and the DS community around a focused and comprehensive national Down syndrome research strategy.
Table 1.
Overall Priority Recommendations for DS Research
| Recommendations | Milestones |
|---|---|
| Clinical and Genetic Phenotyping | |
| Establish a panel of DS experts to define clinical and genetic phenotypes | - Define DS phenotypes for clinical trials from existing data - Develop clinical guidelines and personalized medicine approaches unique to DS - Identify changes related to stage of life and aging including inflammation and metabolism |
| Conduct an “All of US” DS sub-study of 5000 participants | - Use GWAS and clinical data to help define DS phenotypes |
| Expand genetic and epigenetic profiling beyond Chr21 | - Elucidate complex gene-network effects and incorporate knowledge from non-DS populations |
| Gather more unbiased -Omics data | - Global and tissue specific metabolomics data to help establish metabolic phenotypes and to discover new biomarkers of metabolic disease. - Lipidomics data to better understand the risk of diabetes and obesity in DS - Comprehensive ‘omics’ in brain samples to define genome, epigenome, metabolome, transcriptome and proteome - Microbiome (i.e. gut, oral) to better understand the potential associations of the microbiome to diseases common in DS |
| Expand the DS Connect portal and collect DS to compare with data from: | - the general population - people with intellectual disabilities but without DS - siblings of people with DS who do not have DS themselves - people with autosomal dominant AD |
| Cellular and animal models of DS | |
| Current understanding of DS neurobiology is derived largely from mouse studies (Ts65Dn, Tc1) including anatomical and behavioral analyses of organismal brain development. Develop and validate mouse models that better translate to characteristics in individuals with DS | - Support for new in vivo models that will better represent HSA21 genetic changes are needed. - Determine the extent of correspondence between findings in models and human biomarkers. - Perform comparative phenotyping including aging and lifespan of all DS mouse models. - Comprehensive ‘omics’ including the metabolome in all DS mouse models (including aging studies) are needed to better characterize these models. - Define and compare genetics, mechanisms and significance of dysregulated endosomes, exosomes, autophagosome, and proteostasis in all DS mouse models. - Map pathogenesis pathways in the DS mouse models, testing for the contribution of individual dysregulated genes. - Design new treatment paradigms and pathways for testing in DS mouse models (dose-response/toxicity studies). |
| Support new models (including in mouse, rat, or non-human best model DSprimate) that | - Develop animal models that can reflect the structural changes in human DS brain and other differences such as the immune system. |
| Develop novel cellular models of DS | - Support development of human DS cellular models (e.g. induced pluripotent stem cell (iPSC) neuronal cultures), exploring variation by both sex and genetic ancestry. - Develop the use of human iPSC models for more organ systems and pathways affected in DS. |
| Increase collaboration between bench to bedside researchers to share results and enhance translational research in DS. | - Ensure that clinical scientists have the tools to implement advances from bench research, and that bench discoveries are important to clinical researchers. - Identify gaps and discrepancies between basic research and clinical observations and address them. - Facilitate collaborations between neuropathologists and DS clinicians to assess translational relevance of model systems and biomarkers to DS neurobiology. - Explore links between cellular phenotypes/mechanisms in DS mouse models with clinical findings including fluid biomarkers. - Translate insights from mouse models to clinic to inform possible treatments and novel trial designs. |
| Longitudinal Studies | |
| Support studies in the DS population over the life course and help to define clinical phenotypes to inform future research including clinical trials. | - Expand and extend on-going longitudinal studies to keep these important cohorts generating valuable data from more people for more years. - Conduct longitudinal studies across all age groups including adolescents and young adults. - Facilitate international communication and data sharing of between research studies – e.g. ABC-DS/Horizon 21. |
| Within existing and new longitudinal cohorts, support efforts to more clearly define and chart trajectory of: | - Cognitive function. - Behavioral assessments. - Include participant and caregiver reported outcomes. - Include biomarkers as available (imaging, fluid and genetic) - Sex effects. - Contribution of comorbid conditions such as thyroid function, heart disease, obesity, metabolic disorders, autoimmune disorders, OSA to health. - Environmental influences such as education, home setting, work, medications, and use of supplements. - Incorporate new technology, data, and/or samples as appropriate. For example, as new low-cost genome sequencing technology becomes available, studies could add genomics to existing cohorts. |
| Establish new DS cohorts with specific goals | Develop partial trisomy and mosaic DS cohorts, including bio-samples, iPSC lines and brain banks. |
| Randomized Controlled Trials (RCT) in the DS Population | |
| Expand support for drug and devise RCT’s across the DS lifespan | - Build and sustain trial-ready DS cohorts across the lifespan. - Assess benefits across the lifespan to improve cognitive outcomes in childhood and delay declines in adulthood - Determine the age when an intervention will have the most benefit for people with DS. - Support for more drug repurposing trials in DS where target and mechanistic rationales exist. |
| Support for RCT’s on the efficacy of life-style interventions in the DS population across the lifespan. Explore outcomes of physical fitness, health, behavior, cognition, and development. | - Test interventions that effectively increase physical activity and reduce sedentary behavior. - Develop effective lifestyle interventions that foster healthy behaviors. - Test interventions that reduce obesity and improve overall health outcomes. - Test the efficacy of technologies (i.e. animal-assisted therapies, digital, wearable technology) to promote healthy behaviors. |
| Support research to enable RCT’s in DS. | - Find an appropriate control population with which to compare DS participants (related to BMI, BP, activity levels, intellectual disability, etc.). - Conduct studies with large enough sample sizes to produce statistical power and significance to generalize the results to the larger DS population. - Explore differences in drug metabolism (pharmacokinetics / pharmacodynamics) and drug safety in both children and adults with DS compared with the general population for experimental drug candidates and with FDA-approved drugs. |
| Establish the safety and efficacy of FDA approved drugs in both children and adults with DS via RCT’s. | - Conduct RTC’s in drugs commonly used to treat mood disorders, cognitive deficits, autoimmune disorders, and other manifestations. - Document side effect, safety and efficacy data including behavioral and cognitive effects of approved drugs. - Improve assessment and management of side effects during treatment. (i.e. pain/nausea during cancer therapy). |
| Expand support for clinical trial recruitment. | - Support efforts to inform participants and caregivers of the value of research activities and encourage trial participation. - Build infrastructure to facilitate enrollment in clinical trials with disease-specific or condition-specific sub-groups. - Expand expertise in recruiting specific age ranges particularly for adults and underrepresented groups. - Expand DS Connect or establish other approaches to support clinical trial recruitment. |
| Expand support and training in the conduct of DS RCT’s | - Provide training to sites that may not have clinical research experience or DS clinical experience. |
| Develop and disseminate methodology for studying cognitive/behavior outcome measures in the context of large, multi-site trials. | - Develop or employ outcome measures that have demonstrable clinical utility (i.e. predict real-world changes in behavior, cognition, and/or adaptive skill independence). - Identify the participants that may be appropriate for a clinical trial given the selected outcome measures. |
| Expand clinical trial data sharing | - Harmonize DS clinical protocols with between networks in the US and internationally to enable more meaningful data sharing. |
| Centralized Biorepository | |
| Expand support for centralized (or virtual) biorepository of DS samples collected across the life span. | - Expand support for brain banks and fluid biobanks from well-characterized participants with clinical, behavioral, and functional data and with REDCap accessibility. - Establish a robust plan for banking of cells, plasma, serum, CSF, and brains either centrally or tracked via a virtual repository. - Produce and store specific cells types such as peripheral blood mononuclear cells (PBMC’s), iPSC’s and exosomes (brain derived and peripheral). - Establish a fair and equitable process for reviewing and approving request for access to samples. - The collection and storage of the samples should be standardized using established “best practices”. - Integrate biobanking efforts with existing “best practices” for genomic data-sharing, including file formats, storage/hosting solutions, and versioning protocols. - Prioritize (epi)genome-wide profiling over candidate-gene profiling to address diminishing costs of throughput while preserving scarce and highly valuable tissue samples. - Integrate the biorepository data with DS Connect to increase the value of both resources. |
| Open Access Centralized DS Data | |
| Establish a single network for DS data. | - Continue efforts to establish data standards and data sharing in the DS research community. - Establish a centralized data repository or federated network where researcher can go as a “one-stop-shop” for DS data. - The centralized data repository mentioned above could include information on access to biorepositories and access to well-characterized sample. - Support long standing DS clinics to digitize their clinical data into an open access, searchable format - Support the creation of curated data sets that included assessment, survey, and transcription DS data leading to large data sets that support the use of computational modeling. |
| Research Training for Clinicians and Scientists | |
| Support training clinical trials and clinical neuroscience in DS. | - Provide training for clinicians and researchers in DS who are both established and early in their career to attract them to the field. - Support the development of Master Clinics for Adults with DS (MCADS) that operate on a hub and spoke model to provide adults access to expertise across the US, train physicians in DS medical care and that enable clinical trial readiness activities. - Encourage interdisciplinary research collaboration and support cross disciplinary training with a focus on DS. |
| Research Inclusion | |
| Expand the inclusion of individuals with DS who have been significantly under-represented and oftentimes excluded from all sorts of research. | - Develop strategies to increase the participation of people with DS in non-DS focused research. - Develop strategies to increase participation in research focused on DS-specific priorities of people with DS. - Increase the participation of people with DS in the design of studies for both DS and non-DS specific research. |
Footnotes
Dedication
This paper is dedicated to the memory of Dr. Angelika Amon who was a leader in Down syndrome research and who made significant contributions to this review.
References
- [1].Mai CT CT, Isenburg JL, Canfield MA, Meyer RE, Correa A, Alverson CJ, et al. , National population-based estimates for major birth defects, 2010–2014, Birth Defects Res 111(18) (2019), 1420–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sullivan KD, Evans D, Pandey A, Hraha TH, Smith KP, Markham N, et al. , Trisomy 21 causes changes in the circulating proteome indicative of chronic autoinflammation, Sci Rep 7(1) (2017), 14818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bull MJ, Down Syndrome, N Engl J Med 382(24) (2020), 2344–2352. [DOI] [PubMed] [Google Scholar]
- [4].Versacci P, Di Carlo D, Digilio MC and Marino B, Cardiovascular disease in Down syndrome, Curr Opin Pediatr 30(5) (2018), 616–622. [DOI] [PubMed] [Google Scholar]
- [5].Hasle H, Friedman JM, Olsen JH and Rasmussen SA, Low risk of solid tumors in persons with Down syndrome, Genet Med 18(11) (2016), 1151–1157. [DOI] [PubMed] [Google Scholar]
- [6].de Graaf G, Buckley F and Skotko B, People living with Down syndrome in the USA, https://dsuri.net/us-population-factsheet.
- [7].Baburamani AA, Vontell RT, Uus A, Pietsch M, Patkee PA, Wyatt-Ashmead J, et al. , Assessment of radial glia in the frontal lobe of fetuses with Down syndrome, Acta Neuropathol Commun 8(1) (2020), 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Karlsen AS and Pakkenberg B, Total numbers of neurons and glial cells in cortex and basal ganglia of aged brains with Down syndrome–a stereological study, Cereb Cortex 21(11) (2011), 2519–2524. [DOI] [PubMed] [Google Scholar]
- [9].Kanaumi T, Milenkovic I, Adle-Biassette H, Aronica E and Kovacs GG, Non-neuronal cell responses differ between normal and Down syndrome developing brains, Int J Dev Neurosci 31(8) (2013), 796–803. [DOI] [PubMed] [Google Scholar]
- [10].Marin-Padilla M, Structural abnormalities of the cerebral cortex in human chromosomal aberrations: a Golgi study, Brain Res 44(2) (1972), 625–629. [DOI] [PubMed] [Google Scholar]
- [11].Kaufmann WE and Moser HW, Dendritic anomalies in disorders associated with mental retardation, Cereb Cortex 10(10) (2000), 981–991. [DOI] [PubMed] [Google Scholar]
- [12].Esbensen AJ, Hooper SR, Fidler D, Hartley SL, Edgin J, d’Ardhuy XL, et al. Outcome Measures for Clinical Trials in Down Syndrome, Am J Intellect Dev Disabil 122(3) (2017) 247–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].M Channell M, Thurman AJ, Kover ST and Abbeduto L, Patterns of change in nonverbal cognition in adolescents with Down syndrome, Res Dev Disabil 35(11) (2014), 2933–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gilmore L and Cuskelly M, Associations of Child and Adolescent Mastery Motivation and Self-Regulation With Adult Outcomes: A Longitudinal Study of Individuals With Down Syndrome, Am J Intellect Dev Disabil 122(3) (2017), 235–46. [DOI] [PubMed] [Google Scholar]
- [15].Keeling LA, Spiridigliozzi GA, Hart SJ, Baker JA, Jones HN and Kishnani PS, Challenges in measuring the effects of pharmacological interventions on cognitive and adaptive functioning in individuals with Down syndrome: A systematic review, Am J Med Genet A 173(11) (2017), 3058–3066. [DOI] [PubMed] [Google Scholar]
- [16].Roberts LV and Richmond JL, Preschoolers with Down syndrome do not yet show the learning and memory impairments seen in adults with Down syndrome, Dev Sci 18(3) (2015), 404–419. [DOI] [PubMed] [Google Scholar]
- [17].Edgin JO, Anand P, Rosser T, Pierpont EI, Figueroa C, Hamilton D, et al. , The Arizona Cognitive Test Battery for Down Syndrome: Test-Retest Reliability and Practice Effects, Am J Intellect Dev Disabil 122(3), (2017) 215–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hessl D, Sansone SM, Berry-Kravis E, Riley K, Widaman KF, Abbeduto L, et al. , The NIH Toolbox Cognitive Battery for intellectual disabilities: three preliminary studies and future directions, J Neurodev Disord 8(1) (2016), 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Liogier d’Ardhuy X, Edgin JO, Bouis C, de Sola S, Goeldner C, Kishnani P, et al. , Assessment of Cognitive Scales to Examine Memory, Executive Function and Language in Individuals with Down Syndrome: Implications of a 6-month Observational Study, Front Behav Neurosci 9 (2015), 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Phillips BA, Loveall SJ, Channell MM and Conners FA, Matching variables for research involving youth with Down syndrome: Leiter-R versus PPVT-4, Res Dev Disabil 35(2) (2014), 429–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].de Sola S, de la Torre R, Sánchez-Benavides G, Benejam B, Cuenca-Royo A, Del Hoyo L, et al. , A new cognitive evaluation battery for Down syndrome and its relevance for clinical trials, Front Psychol 6 (2015), 708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Startin CM, Rodger E, Fodor-Wynne L, Hamburg S and Strydom A, Developing an Informant Questionnaire for Cognitive Abilities in Down Syndrome: The Cognitive Scale for Down Syndrome (CS-DS), PloS One 11(5) (2016), e0154596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Amadó A, Serrat E and Vallès-Majoral E, The Role of Executive Functions in Social Cognition among Children with Down Syndrome: Relationship Patterns, Front Psychol 7 (2016), 1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Barisnikov K and Lejeune F, Social knowledge and social reasoning abilities in a neurotypical population and in children with Down syndrome, PloS One 13(7) (2018), e0200932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Malmir M, Seifenaraghi M, Farhud DD, Afrooz GA and Khanahmadi M, Mother’s Happiness with Cognitive - Executive Functions and Facial Emotional Recognition in School Children with Down Syndrome, Iran J Public Health 44(5) (2015), 646–653. [PMC free article] [PubMed] [Google Scholar]
- [26].Morales GE, Lopez EO, Castro-Campos C, Charles DJ and Mezquita-Hoyos YN, Contributions to the Cognitive Study of Facial Recognition on Down Syndrome: A New Approximation to Exploring Facial Emotion Processing Style, J Intellect Disabil - Diagn Treat 2(2) (2014), 124–132. [Google Scholar]
- [27].Næss K-AB, Nygaard E, Ostad J, Dolva A-S and Lyster S-AH , The profile of social functioning in children with Down syndrome, Disabil Rehabil 39(13) (2017), 1320–1331. [DOI] [PubMed] [Google Scholar]
- [28].de Santana CCVP, et al. , Recognition of facial emotional expressions and its correlation with cognitive abilities in children with Down syndrome, Psychology & Neuroscience, 7(2) (2014), 73–81. [Google Scholar]
- [29].Bello A, Onofrio D and Caselli MC, Nouns and predicates comprehension and production in children with Down syndrome, Res Dev Disabil 35(4) (2014), 761–775. [DOI] [PubMed] [Google Scholar]
- [30].Daunhauer LA, Fidler DJ and Will E, School function in students with Down syndrome, Am J Occup Ther 68(2) (2014), 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jacola LM, Hickey F, Howe SR, Esbensen A and Shear PK, Behavior and adaptive functioning in adolescents with Down syndrome: specifying targets for intervention, J Ment Health Res Intellect Disabil 7(4) (2014), 287–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Tomaszewski B, Fidler D, Talapatra D and Riley K, Adaptive behaviour, executive function and employment in adults with Down syndrome, J Intellect Disabil Res 62(1) (2018), 41–52. [DOI] [PubMed] [Google Scholar]
- [33].Trezise KL, M Gray K, Taffe J and Sheppard DM, Working memory in adolescent males with Down syndrome and males with autism and intellectual disability: Implications for the classroom, J Intellect Dev Disabil 39(1) (2014), 24–34. [Google Scholar]
- [34].Valencia-Naranjo N and Robles-Bello MA, Learning potential and cognitive abilities in preschool boys with fragile X and Down syndrome, Res Dev Disabil 60 (2017), 153–161. [DOI] [PubMed] [Google Scholar]
- [35].Will E, Fidler DJ, Daunhauer L and Gerlach-McDonald B, Executive function and academic achievement in primary-grade students with Down syndrome, J Intellect Disabil Res 61(2) (2017), 181–195. [DOI] [PubMed] [Google Scholar]
- [36].Chen C-C(JJ), Ringenbach SDR, Albert A and Semken K, Fine Motor Control is Related to Cognitive Control in Adolescents with Down Syndrome, Int J Disabil Dev Educ 61(1) (2014), 6–15. [Google Scholar]
- [37].Abd El-Hady SS, Abd El-Azim FH and El-Talawy HAE-AM, Correlation between cognitive function, gross motor skills and health – Related quality of life in children with Down syndrome, Egypt J Med Hum Genet 19(2) (2018), 97–101. [Google Scholar]
- [38].Ferreira-Vasques AT and Lamônica DAC, Motor, linguistic, personal and social aspects of children with Down syndrome, J Appl Oral Sci 23(4) (2015), 424–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hocking DR, Menant JC, E Kirk H, Lord S and Porter MA, Gait profiles as indicators of domain-specific impairments in executive control across neurodevelopmental disorders, Res Dev Disabil 35(1) (2014), 203–214. [DOI] [PubMed] [Google Scholar]
- [40].Horvat M, Croce R and Fallaize A, Information Processing and Motor Control in Down Syndrome, J Down Syndr Chr Abnorm 2(1) (2016). Available from: https://www.omicsonline.org/open-access/information-processing-and-motor-control-in-down-syndrome-jdsca-1000107.php?aid=70786 [Google Scholar]
- [41].Schott N and Holfelder B, Relationship between motor skill competency and executive function in children with Down’s syndrome, J Intellect Disabil Res 59(9) (2015), 860–872. [DOI] [PubMed] [Google Scholar]
- [42].Alesi M, Battaglia G, Roccella M, Testa D, Palma A and Pepi A, Improvement of gross motor and cognitive abilities by an exercise training program: three case reports, Neuropsychiatr Dis Treat 10 (2014), 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chen C-CJJ and Ringenbach SDR, Dose-response relationship between intensity of exercise and cognitive performance in individuals with Down syndrome: a preliminary study, J Intellect Disabil Res 60(6) (2016), 606–614. [DOI] [PubMed] [Google Scholar]
- [44].Holzapfel SD, Ringenbach SDR, Mulvey GM, Sandoval-Menendez AM, Cook MR, Ganger RO, et al. , Improvements in manual dexterity relate to improvements in cognitive planning after assisted cycling therapy (ACT) in adolescents with down syndrome, Res Dev Disabil 45–46 (2015), 261–270. [DOI] [PubMed] [Google Scholar]
- [45].Cooper S-A, Caslake M, Evans J, Hassiotis A, Jahoda A, McConnachie A, et al. , Toward onset prevention of cognitive decline in adults with Down syndrome (the TOP-COG study): study protocol for a randomized controlled trial, Trials 15 (2014), 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Das D, Phillips C, Hsieh W, Sumanth K, Dang V and Salehi A, Neurotransmitter-based strategies for the treatment of cognitive dysfunction in Down syndrome, Prog Neuropsychopharmacol Biol Psychiatry 54 (2014), 140–148. [DOI] [PubMed] [Google Scholar]
- [47].Hart SJ, Visootsak J, Tamburri P, Phuong P, Baumer N, Hernandez M-C, et al. , Pharmacological interventions to improve cognition and adaptive functioning in Down syndrome: Strides to date, Am J Med Genet A 173(11) (2017), 3029–3041. [DOI] [PubMed] [Google Scholar]
- [48].Kirk HE, Gray K, Riby DM and Cornish KM, Cognitive training as a resolution for early executive function difficulties in children with intellectual disabilities, Res Dev Disabil 38 (2015), 145–160. [DOI] [PubMed] [Google Scholar]
- [49].Kirk H, Gray K, Ellis K, Taffe J and Cornish K, Impact of Attention Training on Academic Achievement, Executive Functioning, and Behavior: A Randomized Controlled Trial, Am J Intellect Dev Disabil 122(2) (2017), 97–117. [DOI] [PubMed] [Google Scholar]
- [50].Mancini E, Beglinger C, Drewe J, Zanchi D, Lang UE and Borgwardt S, Green tea effects on cognition, mood and human brain function: A systematic review, Phytomedicine Int J Phytother Phytopharm 34 (2017), 26–37. [DOI] [PubMed] [Google Scholar]
- [51].Martínez-Cué C, Delatour B and Potier M-C, Treating enhanced GABAergic inhibition in Down syndrome: use of GABA α5-selective inverse agonists, Neurosci Biobehav Rev 46(Pt 2) (2014), 218–227. [DOI] [PubMed] [Google Scholar]
- [52].Camp JS, Karmiloff-Smith A, Thomas MSC and Farran EK, Cross-syndrome comparison of real-world executive functioning and problem solving using a new problem-solving questionnaire, Res Dev Disabil 59 (2016), 80–92. [DOI] [PubMed] [Google Scholar]
- [53].Lee NR, Anand P, Will E, Adeyemi EI, Clasen LS, Blumenthal JD, et al. , Everyday executive functions in Down syndrome from early childhood to young adulthood: evidence for both unique and shared characteristics compared to youth with sex chromosome trisomy (XXX and XXY), Front Behav Neurosci 9 (2015), 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Purser HRM, Farran EK, Courbois Y, Lemahieu A, Sockeel P, Mellier D, et al. , The development of route learning in Down syndrome, Williams syndrome and typical development: investigations with virtual environments, Dev Sci 18(4) (2015), 599–613. [DOI] [PubMed] [Google Scholar]
- [55].D’Souza D, Booth R, Connolly M, Happé F and Karmiloff-Smith A, Rethinking the concepts of “local or global processors”: evidence from Williams syndrome, Down syndrome, and Autism Spectrum Disorders, Dev Sci 19(3) (2016), 452–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Udhnani M, Perez M, Clasen LS, Adeyemi E and Lee NR, Relations between Everyday Executive Functioning and Language in Youth with Down Syndrome and Youth with Autism Spectrum Disorder, Dev Neuropsychol 45(2) (2020), 79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wilde L and Oliver C, Brief Report: Contrasting Profiles of Everyday Executive Functioning in Smith-Magenis Syndrome and Down Syndrome, J Autism Dev Disord 47(8) (2017), 2602–2609. [DOI] [PubMed] [Google Scholar]
- [58].Alsaied T, Marino BS, Esbensen AJ, Anixt JS, Epstein JN and Cnota JF, Does Congenital Heart Disease Affect Neurodevelopmental Outcomes in Children with Down Syndrome? Congenit Heart Dis 11(1) (2016), 26–33. [DOI] [PubMed] [Google Scholar]
- [59].Ashworth A, Hill CM, Karmiloff-Smith A and Dimitriou D, The Importance of Sleep: Attentional Problems in School-Aged Children With Down Syndrome and Williams Syndrome, Behav Sleep Med 13(6) (2015), 455–471. [DOI] [PubMed] [Google Scholar]
- [60].Breslin J, Spanò G, Bootzin R, Anand P, Nadel L and Edgin J, Obstructive sleep apnea syndrome and cognition in Down syndrome, Dev Med Child Neurol 56(7) (2014), 657–664. [DOI] [PubMed] [Google Scholar]
- [61].Brooks LJ, Olsen MN, Bacevice AM, Beebe A, Konstantinopoulou S and Taylor HG, Relationship between sleep, sleep apnea, and neuropsychological function in children with Down syndrome, Sleep Breath Schlaf Atm 19(1) (2015) 197–204. [DOI] [PubMed] [Google Scholar]
- [62].Channell MM, Phillips BA, Loveall SJ, Conners FA, Bussanich PM and Klinger LG, Patterns of autism spectrum symptomatology in individuals with Down syndrome without comorbid autism spectrum disorder, J Neurodev Disord 7(1) (2015), 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Esbensen AJ and Hoffman EK, Impact of sleep on executive functioning in school-age children with Down syndrome, J Intellect Disabil Res 62(6) (2018), 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Horne RS, Wijayaratne P, Nixon GM and Walter LM, Sleep and sleep disordered breathing in children with down syndrome: Effects on behaviour, neurocognition and the cardiovascular system, Sleep Med Rev 44 (2019), 1–11. [DOI] [PubMed] [Google Scholar]
- [65].Joyce A and Dimitriou D, Sleep-disordered breathing and cognitive functioning in preschool children with and without Down syndrome, J Intellect Disabil Res 61(8) (2017), 778–791. [DOI] [PubMed] [Google Scholar]
- [66].Joyce A, Elphick H, Farquhar M, Gringras P, Evans H, Bucks RS, et al. , Obstructive Sleep Apnoea Contributes to Executive Function Impairment in Young Children with Down Syndrome, Behav Sleep Med 16 (2019), 1–11. [DOI] [PubMed] [Google Scholar]
- [67].Mason GM, Spanó G and Edgin J, Symptoms of attention-deficit/hyperactivity disorder in Down syndrome: effects of the dopamine receptor D4 gene, Am J Intellect Dev Disabil 120(1) (2015), 58–71. [DOI] [PubMed] [Google Scholar]
- [68].Muchová J, Žitňanová I and Ďuračková Z, Oxidative stress and Down syndrome. Do antioxidants play a role in therapy? Physiol Res 63(5) (2014), 535–542. [DOI] [PubMed] [Google Scholar]
- [69].Nixon GM, Biggs SN, Jitpiriyaroj S and Horne RSC, The Relationship Between Sleep-Disordered Breathing Severity and Daytime Adaptive Functioning in Children with Down Syndrome, CNS Neurosci Ther 22(11) (2016), 936–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Pritchard AE, Kalback S, McCurdy M and Capone GT, Executive functions among youth with Down Syndrome and co-existing neurobehavioural disorders, J Intellect Disabil Res 59(12) (2015), 1130–1141. [DOI] [PubMed] [Google Scholar]
- [71].Rachubinski AL, Hepburn S, Elias ER, Gardiner K and Shaikh TH, The co-occurrence of Down syndrome and autism spectrum disorder: is it because of additional genetic variations? Prenat Diagn 37(1) (2017), 31–6. [DOI] [PubMed] [Google Scholar]
- [72].Rosser TC, Edgin JO, Capone GT, Hamilton DR, Allen EG, Dooley KJ, et al. , Associations Between Medical History, Cognition, and Behavior in Youth With Down Syndrome: A Report From the Down Syndrome Cognition Project, Am J Intellect Dev Disabil 123(6) (2018), 514–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Kasari C and Sigman M, Expression and understanding of emotion in atypical development: Autism and Down syndrome. In: Emotional development in atypical children. Hillsdale, NJ, US: Lawrence Erlbaum Associates, Inc; 1996, pp. 109–30. [Google Scholar]
- [74].Vetrano DL, Carfì A, Brandi V, L’Angiocola PD, Di Tella S, Cipriani MC, et al. , Left ventricle diastolic function and cognitive performance in adults with Down syndrome, Int J Cardiol 203 (2016), 816–818. [DOI] [PubMed] [Google Scholar]
- [75].Zis P, Strydom A, Buckley D, Adekitan D and McHugh PC, Cognitive ability in Down syndrome and its relationship to urinary neopterin, a marker of activated cellular immunity, Neurosci Lett 636 (2017), 254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Nugent J, Gorman G and Erdie-Lalena CR, Disparities in Access to Healthcare Transition Services for Adolescents with Down Syndrome, J Pediatr 197 (2018), 214–220. [DOI] [PubMed] [Google Scholar]
- [77].Barbosa TMMF, Lima ILB, Alves GÂDS and Delgado IC, Contributions of speech-language therapy to the integration of individuals with Down syndrome in the workplace, CoDAS 30(1) (2018), e20160144. [DOI] [PubMed] [Google Scholar]
- [78].Matthews TJ, Allain DC, Matthews AL, Mitchell A, Santoro SL and Cohen L, An assessment of health, social, communication, and daily living skills of adults with Down syndrome, Am J Med Genet A 176(6) (2018), 1389–1397. [DOI] [PubMed] [Google Scholar]
- [79].Makary AT, Testa R, Tonge BJ, Einfeld SL, Mohr C and Gray KM, Association between adaptive behaviour and age in adults with Down syndrome without dementia: examining the range and severity of adaptive behaviour problems, J Intellect Disabil Res 59(8) (2015), 689–702. [DOI] [PubMed] [Google Scholar]
- [80].Alonso-Sardón M, Iglesias-de-Sena H, Fernández-Martín LC and Mirón-Canelo JA, Do health and social support and personal autonomy have an influence on the health-related quality of life of individuals with intellectual disability? BMC Health Serv Res 19(1) (2019), 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Sandjojo J, Zedlitz AMEE, Gebhardt WA, Hoekman J, Dusseldorp E, den Haan JA, et al. , Training staff to promote self-management in people with intellectual disabilities, J Appl Res Intellect Disabil 31(5) (2018), 840–850. [DOI] [PubMed] [Google Scholar]
- [82].Oppewal A, Hilgenkamp TIM, van Wijck R, Schoufour JD and Evenhuis HM, Physical fitness is predictive for a decline in the ability to perform instrumental activities of daily living in older adults with intellectual disabilities: Results of the HA-ID study, Res Dev Disabil 41–42 (2015), 76–85. [DOI] [PubMed] [Google Scholar]
- [83].Bouck EC, Satsangi R and Bartlett W, Supporting grocery shopping for students with intellectual disability:a preliminary study, Disabil Rehabil Assist Technol 12(6) (2017), 605–613. [DOI] [PubMed] [Google Scholar]
- [84].Allen KD, Vatland C, Bowen SL and Burke RV, An evaluation of parent-produced video self-modeling to improve independence in an adolescent with intellectual developmental disorder and an autism spectrum disorder: a controlled case study, Behav Modif 39(4) (2015), 542–556. [DOI] [PubMed] [Google Scholar]
- [85].Tassé MJ, Wagner JB and Kim M, Using technology and remote support services to promote independent living of adults with intellectual disability and related developmental disabilities, J Appl Res Intellect Disabil 33(3) (2020), 640–647. [DOI] [PubMed] [Google Scholar]
- [86].Brigida AL and Siniscalco D, Induced pluripotent stem cells as a cellular model for studying Down Syndrome, J Stem Cells Regen Med 12(2) (2016), 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Schill EM, Wright CM, Jamil A, LaCombe JM, Roper RJ and Heuckeroth RO, Down syndrome mouse models have an abnormal enteric nervous system, JCI Insight 18 (2019), 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Ma D, Cardoso MJ, Zuluaga MA, Modat M, Powell NM, Wiseman FK, et al. , Substantially thinner internal granular layer and reduced molecular layer surface in the cerebellar cortex of the Tc1 mouse model of down syndrome - a comprehensive morphometric analysis with active staining contrast-enhanced MRI, NeuroImage 223 (2020), 117271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Gupta M, Dhanasekaran AR and Gardiner KJ, Mouse models of Down syndrome: gene content and consequences, Mamm Genome 27(11–12) (2016), 538–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Zhao X and Bhattacharyya A, Human Models Are Needed for Studying Human Neurodevelopmental Disorders, Am J Hum Genet 103(6) (2018), 829–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Florio M, Heide M, Pinson A, Brandl H, Albert M, Winkler S, et al. , Evolution and cell-type specificity of human-specific genes preferentially expressed in progenitors of fetal neocortex, eLife 21 (2018), 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Sousa AMM, Meyer KA, Santpere G, Gulden FO, and Sestan N, Evolution of the Human Nervous System Function, Structure, and Development, Cell 170(2) (2017), 226–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Capone G, Goyal P, Ares W and Lannigan E, Neurobehavioral disorders in children, adolescents, and young adults with Down syndrome, Am J Med Genet C Semin Med Genet 142C(3) (2006), 158–172. [DOI] [PubMed] [Google Scholar]
- [94].Oxelgren UW, Myrelid Å, Annerén G, Ekstam B, Göransson C, Holmbom A, et al. , Prevalence of autism and attention-deficit-hyperactivity disorder in Down syndrome: a population-based study, Dev Med Child Neurol 59(3) (2017), 276–283. [DOI] [PubMed] [Google Scholar]
- [95].O’Brien G and Pearson J, Autism and learning disability, Autism Int J Res Pract 8(2) (2004), 125–140. [DOI] [PubMed] [Google Scholar]
- [96].Warner G, Moss J, Smith P and Howlin P, Autism characteristics and behavioural disturbances in 500 children with Down’s syndrome in England and Wales, Autism Res 7(4) (2014), 433–41. [DOI] [PubMed] [Google Scholar]
- [97].Mircher C, Cieuta-Walti C, Marey I, Rebillat A-S, Cretu L, Milenko E, et al. , Acute Regression in Young People with Down Syndrome, Brain Sci 7(6) (2017), 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Chicoine B and Capone G, Regression in adolescents and adults with Down syndrome. In: Physical health of adults with intellectual and developmental disabilities, Springer, Cham; 2019. [Google Scholar]
- [99].Worley G, Crissman BG, Cadogan E, Milleson C, Adkins DW and Kishnani PS, Down Syndrome Disintegrative Disorder: New-Onset Autistic Regression, Dementia, and Insomnia in Older Children and Adolescents With Down Syndrome, J Child Neurol 30(9) (2015), 1147–1152. [DOI] [PubMed] [Google Scholar]
- [100].Rosso M, Fremion E, Santoro SL, Oreskovic NM, Chitnis T, Skotko BG, et al. , Down Syndrome Disintegrative Disorder: A Clinical Regression Syndrome of Increasing Importance, Pediatrics 145(6) (2020), e20192939. [DOI] [PubMed] [Google Scholar]
- [101].Cardinale KM, Bocharnikov A, Hart SJ, Baker JA, Eckstein C, Jasien JM, et al. , Immunotherapy in selected patients with Down syndrome disintegrative disorder, Dev Med Child Neurol 61(7) (2019), 847–851. [DOI] [PubMed] [Google Scholar]
- [102].Smith M, Manduchi B, Burke É, Carroll R, McCallion P, McCarron M, Communication difficulties in adults with Intellectual Disability: Results from a national cross-sectional study, Res Dev Disabil 97 (2020), 103557. [DOI] [PubMed] [Google Scholar]
- [103].Abbeduto L, Arias-Trejo N, Thurman AJ, Ramos-Sanchez J, del Hoyo Soriano L, Language Development in Down Syndrome. In: Burack JA, Edgin JO, Abbeduto L, Busciglio J editors. The Oxford Handbook of Down Syndrome and Development [Internet]. Oxford University Press; 2020. [cited 2020 Dec 7]. Available from: https://oxfordhandbooks.com/view/10.1093/oxfordhb/9780190645441.001.0001/oxfordhb-9780190645441-e-18 [Google Scholar]
- [104].Abbeduto L, Warren SF and A Conners F, Language development in Down syndrome: from the prelinguistic period to the acquisition of literacy, Ment Retard Dev Disabil Res Rev 13(3) (2007), 247–261. [DOI] [PubMed] [Google Scholar]
- [105].Kent RD and Vorperian HK, Speech impairment in Down syndrome: a review, J Speech Lang Hear Res 56(1) (2013), 178–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Kumin L, Intelligibility of speech in children with Down syndrome in natural settings: parents’ perspective, Percept Mot Skills 78(1) (1994), 307–313. [DOI] [PubMed] [Google Scholar]
- [107].Lemons CJ and Fuchs D, Phonological awareness of children with Down syndrome: its role in learning to read and the effectiveness of related interventions, Res Dev Disabil 31(2) (2010), 316–330. [DOI] [PubMed] [Google Scholar]
- [108].Coppens-Hofman MC, Maassen BAM, van Schrojenstein Lantman-de Valk HMJ and Snik AFM, Speech difficulties and poor speech intelligibility in adults with Down syndrome: A review of the literature, J Hear Sci 2(1) (2012), 9–16.24319590 [Google Scholar]
- [109].Kover ST, McDuffie A, Abbeduto L and Brown WT, Effects of sampling context on spontaneous expressive language in males with fragile X syndrome or Down syndrome, J Speech Lang Hear Res 55(4) (2012), 1022–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Loveall SJ, Channell MM, Abbeduto L and Conners FA, Verb production by individuals with Down syndrome during narration, Res Dev Disabil 85 (2019), 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Conners FA, Tungate AS, Abbeduto L, Merrill EC and Faught GG, Growth and Decline in Language and Phonological Memory Over Two Years Among Adolescents With Down Syndrome, Am J Intellect Dev Disabil 123(2) (2018), 103–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Warren SF, Bredin-Oja SL, Hahn LJ and Brady N, Communication and Language Interventions for Children With Down Syndrome, In: Burack JA, Edgin JO, Abbeduto L, Busciglio J editors. The Oxford Handbook of Down Syndrome and Development [Internet]. Oxford University Press; 2020. [cited 2020 Dec 7]. Available from: https://oxfordhandbooks.com/view/10.1093/oxfordhb/9780190645441.001.0001/oxfordhb-9780190645441-e-26 [Google Scholar]
- [113].Watt T, Robertson K and Jacobs RJ, Refractive error, binocular vision and accommodation of children with Down syndrome, Clin Exp Optom 98(1) (2015) 3–11. [DOI] [PubMed] [Google Scholar]
- [114].Alio JL JL, Vega-Estrada A, Sanz P, Osman AA, Kamal AM, Mamoon A, et al. , Corneal Morphologic Characteristics in Patients With Down Syndrome, JAMA Ophthalmol 136(9) (2018), 971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Mangalesh S, Vinekar A, Jayadev C, Kemmanu V, Bhat M, Sivakumar M, et al. , Spectral Domain Optical Coherence Tomography in Detecting Sub-Clinical Retinal Findings in Asian Indian Children with Down Syndrome, Curr Eye Res 44(8) (2019), 901–907. [DOI] [PubMed] [Google Scholar]
- [116].O’Brien S, Wang J, Smith HA, Donaldson DL, Haider KM, Roberts GJ, et al. , Macular structural characteristics in children with Down syndrome, Graefes Arch Clin Exp Ophthalmol 253(12) (2015), 2317–2323. [DOI] [PubMed] [Google Scholar]
- [117].Cregg M, Woodhouse JM, Stewart RE, Pakeman VH, Bromham NR, Gunter HL, et al. , Development of refractive error and strabismus in children with Down syndrome, Invest Ophthalmol Vis Sci 44(3) (2003), 1023–1030. [DOI] [PubMed] [Google Scholar]
- [118].Raut P, Sriram B, Yeoh A, Hee KYM, Lim SB, Daniel ML, High prevalence of hearing loss in Down syndrome at first year of life, Ann Acad Med Singap 40(11) (2011), 493–498. [PubMed] [Google Scholar]
- [119].Tedeschi AS, Roizen NJ, Taylor HG, Murray G, Curtis CA and Parikh AS, The prevalence of congenital hearing loss in neonates with Down syndrome, J Pediatr 166(1) (2015), 168–171. [DOI] [PubMed] [Google Scholar]
- [120].Austeng ME, Akre H, Falkenberg E-S, Øverland B, Abdelnoor M, Kværner KJ, Hearing level in children with Down syndrome at the age of eight, Res Dev Disabil 34(7) (2013), 2251–2256. [DOI] [PubMed] [Google Scholar]
- [121].Hildmann A, Hildmann H and Kessler A, Hearing disorders in children with Down’s syndrome, Laryngorhinootologie, 81(1) (2002), 3–7. [DOI] [PubMed] [Google Scholar]
- [122].Keiser H, Montague J, Wold D, Maune S and Pattison D, Hearing loss of Down syndrome adults, Am J Ment Defic 85(5) (1981), 467–472. [PubMed] [Google Scholar]
- [123].Picciotti PM, Carfì A, Anzivino R, Paludetti G, Conti G, Brandi V, et al. , Audiologic Assessment in Adults With Down Syndrome, Am J Intellect Dev Disabil 122(4) (2017), 333–341. [DOI] [PubMed] [Google Scholar]
- [124].Saliba I, Sbeity S, El-Zir E, Yammine FG, Noun CT and Haddad A, Down syndrome: an electrophysiological and radiological profile, The Laryngoscope 124(4) (2014), E141–147. [DOI] [PubMed] [Google Scholar]
- [125].Mazzoni DS, Ackley RS and Nash DJ, Abnormal pinna type and hearing loss correlations in Down’s syndrome, J Intellect Disabil Res 38(Pt 6) (1994), 549–560. [DOI] [PubMed] [Google Scholar]
- [126].Satwant S, Subramaniam KN, Prepageran N, Raman R and Jalaludin MA, Otological disorders in Down’s Syndrome, Med J Malaysia 57(3) (2002), 278–282. [PubMed] [Google Scholar]
- [127].Balkany TJ, Mischke RE, Downs MP and Jafek BW, Ossicular abnormalities in Down’s syndrome, Otolaryngol Head Neck Surg 87(3) (1979), 372–384. [DOI] [PubMed] [Google Scholar]
- [128].Fausch C, Röösli C, The incudomalleolar articulation in Down syndrome (trisomy 21): a temporal bone study, Otol Neurotol 36(2) (2015) 348–353. [DOI] [PubMed] [Google Scholar]
- [129].Blaser S, Propst EJ, Martin D, Feigenbaum A, James AL, Shannon P, et al. , Inner ear dysplasia is common in children with Down syndrome (trisomy 21), The Laryngoscope 116(12) (2006), 2113–2119. [DOI] [PubMed] [Google Scholar]
- [130].Clark CM, Patel HH, Kanekar SG and Isildak H, Enlarged vestibular aqueducts and other inner-ear abnormalities in patients with Down syndrome, J Laryngol Otol 131(4) (2017), 298–302. [DOI] [PubMed] [Google Scholar]
- [131].Intrapiromkul J, Aygun N, Tunkel DE, Carone M and Yousem DM, Inner ear anomalies seen on CT images in people with Down syndrome, Pediatr Radiol 42(12) (2012), 1449–1455. [DOI] [PubMed] [Google Scholar]
- [132].Evenhuis HM, van Zanten GA, Brocaar MP and Roerdinkholder WH, Hearing loss in middle-age persons with Down syndrome, Am J Ment Retard 97(1) (1992), 47–56. [PubMed] [Google Scholar]
- [133].McCarron M, Gill M, McCallion P and Begley C, Health co-morbidities in ageing persons with Down syndrome and Alzheimer’s dementia, J Intellect Disabil Res 49(Pt 7) (2005), 560–566. [DOI] [PubMed] [Google Scholar]
- [134].Asim A, Agarwal S, Panigrahi I, Sarangi AN, Muthuswamy S and Kapoor A, CRELD1 gene variants and atrioventricular septal defects in Down syndrome, Gene 641 (2018), 180–185. [DOI] [PubMed] [Google Scholar]
- [135].Irving K, Hogervorst E, Oliveira D and Kivipelto M, New Developments in Dementia Prevention Research: State of the Art and Future Possibilities, 1st Edition (Hardback) – Routledge [Internet]. Routledge.com. Routledge; [cited 2020 Feb 26]. Available from: https://www.routledge.com/New-Developments-in-Dementia-Prevention-Research-State-of-the-Art-and/Irving-Hogervorst-Oliveira-Kivipelto/p/book/9780815358336 [Google Scholar]
- [136].Morales-Demori R, Congenital heart disease and cardiac procedural outcomes in patients with trisomy 21 and Turner syndrome, Congenit Heart Dis 12(6) (2017), 820–827. [DOI] [PubMed] [Google Scholar]
- [137].Pierpont ME, Brueckner M, Chung WK, Garg V, Lacro RV, McGuire AL, et al. , Genetic Basis for Congenital Heart Disease: Revisited: A Scientific Statement From the American Heart Association, Circulation 138(21) (2018), e653–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Stoll C, Dott B, Alembik Y and M-P. Roth, Associated congenital anomalies among cases with Down syndrome, Eur J Med Genet 58(12) (2015), 674–680. [DOI] [PubMed] [Google Scholar]
- [139].Pfitzer C, Helm PC, Rosenthal L-M, Berger F, Bauer UMM and Schmitt KR, Dynamics in prevalence of Down syndrome in children with congenital heart disease, Eur J Pediatr 177(1) (2018), 107–115. [DOI] [PubMed] [Google Scholar]
- [140].Ramachandran D, Mulle JG, Locke AE, Bean LJH, Rosser TC, Bose P, et al. , Contribution of copy-number variation to Down syndrome-associated atrioventricular septal defects, Genet Med 17(7) (2015), 554–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Ramachandran D, Zeng Z, Locke AE, Mulle JG, Bean LJH, Rosser TC, et al. , Genome-Wide Association Study of Down Syndrome-Associated Atrioventricular Septal Defects, G3 Bethesda 5(10) (2015), 1961–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Sailani MR, Makrythanasis P, Valsesia A, Santoni FA, Deutsch S, Popadin K, et al. , The complex SNP and CNV genetic architecture of the increased risk of congenital heart defects in Down syndrome, Genome Res 23(9) (2013), 1410–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Rambo-Martin BL, Mulle JG, Cutler DJ, Bean LJH, Rosser TC, Dooley KJ, et al. , Analysis of Copy Number Variants on Chromosome 21 in Down Syndrome-Associated Congenital Heart Defects, G3 Bethesda 8(1) (2018), 105–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Ackerman C, Locke AE, Feingold E, Reshey B, Espana K, Thusberg J, et al. , An excess of deleterious variants in VEGF-A pathway genes in Down-syndrome-associated atrioventricular septal defects, Am J Hum Genet 91(4) (2012), 646–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Trevino CE, Holleman AM, Corbitt H, Maslen CL, Rosser TC, Cutler DJ, et al. , Identifying genetic factors that contribute to the increased risk of congenital heart defects in infants with Down syndrome, Sci Rep 10(1) (2020), 18051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Li H, Cherry S, Klinedinst D, DeLeon V, Redig J, Reshey B, et al. , Genetic modifiers predisposing to congenital heart disease in the sensitized Down syndrome population, Circ Cardiovasc Genet 5(3) (2012), 301–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Li H, Edie S, Klinedinst D, Jeong JS, Blackshaw S, Maslen CL, et al. , Penetrance of Congenital Heart Disease in a Mouse Model of Down Syndrome Depends on a Trisomic Potentiator of a Disomic Modifier, Genetics 203(2) (2016), 763–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Ripoll C, Rivals I, Ait Yahya-Graison E, Dauphinot L, Paly E, Mircher C, et al. , Molecular signatures of cardiac defects in Down syndrome lymphoblastoid cell lines suggest altered ciliome and Hedgehog pathways, PloS One 7(8) (2012), e41616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Sobey CG, Judkins CP, Sundararajan V, Phan TG, Drummond GR and Srikanth VK, Risk of Major Cardiovascular Events in People with Down Syndrome, PloS One 10(9) (2015), e0137093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Hilgenkamp TIM, Wee SO, Schroeder EC, Baynard T and Fernhall B, Peripheral blood flow regulation in response to sympathetic stimulation in individuals with Down syndrome, Artery Res 24 (2018), 16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].DeRuisseau LR, Receno CN, Heffernan KS and Cunningham CM, Heart rate and blood pressure in male Ts65Dn mice: a model to investigate cardiovascular responses in Down syndrome, Physiol Rep 7(17) (2019), e14205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Kumar P, Panigrahi I, Sankhyan N, Ahuja C and Goyadi PK, Down Syndrome with Moyamoya Disease: A Case Series, J Pediatr Neurosci 13(2) (2018), 201–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Santoro JD, Lee S, Mlynash M, Nguyen T, Lazzareschi DV, Kraler LD, et al. , Blood Pressure Elevation and Risk of Moyamoya Syndrome in Patients With Trisomy 21, Pediatrics 142(4) (2018), e20180840. [DOI] [PubMed] [Google Scholar]
- [154].Ptomey LT, Szabo AN, Willis EA, Gorczyca AM, Greene JL, Danon JC, et al. , Changes in cognitive function after a 12-week exercise intervention in adults with Down syndrome, Disabil Health J 11(3) (2018), 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Lee C-F, Lee C-H, Hsueh W-Y, Lin M-T, Kang K-T, Prevalence of Obstructive Sleep Apnea in Children With Down Syndrome: A Meta-Analysis, J Clin Sleep Med 14(5) (2018), 867–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Maris M, Verhulst S, Wojciechowski M, Van de Heyning P and Boudewyns A, Prevalence of Obstructive Sleep Apnea in Children with Down Syndrome, Sleep 39(3) (2016), 699–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Watts R and Vyas H, An overview of respiratory problems in children with Down’s syndrome, Arch Dis Child 98(10) (2013), 812–7. [DOI] [PubMed] [Google Scholar]
- [158].Hamilton J, Yaneza MMC, Clement WA and Kubba H, The prevalence of airway problems in children with Down’s syndrome, Int J Pediatr Otorhinolaryngol 81 (2016), 1–4. [DOI] [PubMed] [Google Scholar]
- [159].Beckhaus AA and Castro-Rodriguez JA, Down Syndrome and the Risk of Severe RSV Infection: A Meta-analysis, Pediatrics 142(3) (2018), e20180225. [DOI] [PubMed] [Google Scholar]
- [160].Jackson A, Maybee J, Moran MK, Wolter-Warmerdam K and Hickey F, Clinical Characteristics of Dysphagia in Children with Down Syndrome, Dysphagia 31(5) (2016), 663–671. [DOI] [PubMed] [Google Scholar]
- [161].Bush D, Galambos C, Ivy DD, Abman SH, Wolter-Warmerdam K and Hickey F, Clinical Characteristics and Risk Factors for Developing Pulmonary Hypertension in Children with Down Syndrome, J Pediatr 202 (2018), 212–219.e2. [DOI] [PubMed] [Google Scholar]
- [162].King P and Tulloh R, Management of pulmonary hypertension and Down syndrome, Int J Clin Pract 65(s174) (2011), 8–13. [DOI] [PubMed] [Google Scholar]
- [163].Cooney TP and Thurlbeck WM, Pulmonary hypoplasia in Down’s syndrome, N Engl J Med 307(19) (1982), 1170–1173. [DOI] [PubMed] [Google Scholar]
- [164].Foley C and Killeen OG, Musculoskeletal anomalies in children with Down syndrome: an observational study, Arch Dis Child 104(5) (2019), 482–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Dierssen M, Fructuoso M, Martínez de Lagrán M, Perluigi M and Barone E, Down Syndrome Is a Metabolic Disease: Altered Insulin Signaling Mediates Peripheral and Brain Dysfunctions, Front Neurosci 14 (2020), 670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Peiris H, Duffield MD, Fadista J, Jessup CF, Kashmir V, Genders AJ, et al. , A Syntenic Cross Species Aneuploidy Genetic Screen Links RCAN1 Expression to β-Cell Mitochondrial Dysfunction in Type 2 Diabetes, PLoS Genet 12(5) (2016), e1006033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Melville CA, Cooper S-A, McGrother CW, Thorp CF and Collacott R, Obesity in adults with Down syndrome: a case-control study, J Intellect Disabil Res 49(2) (2005), 125–133. [DOI] [PubMed] [Google Scholar]
- [168].Bertapelli F, Pitetti K, Agiovlasitis S and Guerra-Junior G, Overweight and obesity in children and adolescents with Down syndrome-prevalence, determinants, consequences, and interventions: A literature review, Res Dev Disabil 57 (2016), 181–192. [DOI] [PubMed] [Google Scholar]
- [169].Stancliffe RJ, Lakin KC, Larson S, Engler J, Bershadsky J, Taub S, et al. , Overweight and Obesity Among Adults With Intellectual Disabilities Who Use Intellectual Disability/Developmental Disability Services in 20 U.S. States, Am J Intellect Dev Disabil 116(6) (2011), 401–418. [DOI] [PubMed] [Google Scholar]
- [170].Rubin SS, Rimmer JH, Chicoine B, Braddock D and McGuire DE, Overweight prevalence in persons with Down syndrome, Ment Retard 36(3) (1998), 175–181. [DOI] [PubMed] [Google Scholar]
- [171].Ptomey LT, Walpitage DL, Mohseni M, Dreyer Gillette ML, Davis AM, Forseth B, et al. , Weight status and associated comorbidities in children and adults with Down syndrome, autism spectrum disorder and intellectual and developmental disabilities, J Intellect Disabil Res 64(9) (2020), 725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Stefanowicz-Bielska A, Wierzba J, Stefanowicz J and Chamienia A, Factors affecting the prevalence of overweight and obesity in children with Down syndrome, Minerva Pediatr (2020. May 15). [DOI] [PubMed] [Google Scholar]
- [173].Foerste T, Sabin M, Reid S and Reddihough D, Understanding the causes of obesity in children with trisomy 21: hyperphagia vs physical inactivity, J Intellect Disabil Res 60(9) (2016), 856–64. [DOI] [PubMed] [Google Scholar]
- [174].Magge SN, Zemel BS, Pipan ME, Gidding SS and Kelly A, Cardiometabolic Risk and Body Composition in Youth With Down Syndrome, Pediatrics 144(2) (2019), e20190137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Buonuomo PS, Bartuli A, Mastrogiorgio G, Vittucci A, Di Camillo C, Bianchi S, et al. , Lipid profiles in a large cohort of Italian children with Down syndrome, Eur J Med Genet 59(8) (2016), 392–395. [DOI] [PubMed] [Google Scholar]
- [176].Adelekan T, Magge S, Shults J, Stallings V and Stettler N, Lipid profiles of children with Down syndrome compared with their siblings, Pediatrics 129(6) (2012), e1382–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Valentini D, Alisi A, di Camillo C, Sartorelli MR, Crudele A, Bartuli A, et al. , Nonalcoholic Fatty Liver Disease in Italian Children with Down Syndrome: Prevalence and Correlation with Obesity-Related Features, J Pediatr 189 (2017), 92–97.e1. [DOI] [PubMed] [Google Scholar]
- [178].Tenneti N, Dayal D, Sharda S, Panigrahi I, Didi M, Attri SV, et al. , Concentrations of leptin, adiponectin and other metabolic parameters in non-obese children with Down syndrome, J Pediatr Endocrinol Metab 30(8) (2017), 831–837. [DOI] [PubMed] [Google Scholar]
- [179].Fox B, Moffett GE, Kinnison C, Brooks G and Case LE, Physical Activity Levels of Children With Down Syndrome, Pediatr Phys Ther 31(1) (2019), 33–41. [DOI] [PubMed] [Google Scholar]
- [180].Oreskovic NM, Cottrell C, Torres A, Patsiogiannis V, Santoro S, Nichols D, et al. , Physical activity patterns in adults with Down Syndrome, J Appl Res Intellect Disabil 33(6) (2020), 1457–1464. [DOI] [PubMed] [Google Scholar]
- [181].Baynard T, Pitetti KH, Guerra M, Unnithan VB and Fernhall B, Age-related changes in aerobic capacity in individuals with mental retardation: a 20-yr review, Med Sci Sports Exerc 40(11) (2008), 1984–1989. [DOI] [PubMed] [Google Scholar]
- [182].Croce RV, Pitetti KH, Horvat M and Miller J, Peak torque, average power, and hamstrings/quadriceps ratios in nondisabled adults and adults with mental retardation, Arch Phys Med Rehabil 77(4) (1996), 369–372. [DOI] [PubMed] [Google Scholar]
- [183].Phillips AC and Holland AJ, Assessment of objectively measured physical activity levels in individuals with intellectual disabilities with and without Down’s syndrome, PloS One 6(12) (2011), e28618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Pitetti K, Baynard T and Agiovlasitis S, Children and adolescents with Down syndrome, physical fitness and physical activity, J Sport Health Sci 2(1) (2013), 47–57. [Google Scholar]
- [185].Pitchford EA, Adkins C, Hasson RE, Hornyak JE and Ulrich DA, Association between Physical Activity and Adiposity in Adolescents with Down Syndrome, Med Sci Sports Exerc 50(4) (2018), 667–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Silva V, Campos C, Sá A, Cavadas M, Pinto J, Simões P, et al. , Wii-based exercise program to improve physical fitness, motor proficiency and functional mobility in adults with Down syndrome, J Intellect Disabil Res 61(8) (2017), 755–765. [DOI] [PubMed] [Google Scholar]
- [187].Shields N, Hussey J, Murphy J, Gormley J and Hoey H, An exploratory study of the association between physical activity, cardiovascular fitness and body size in children with Down syndrome, Dev Neurorehabilitation 20(2) (2017), 92–98. [DOI] [PubMed] [Google Scholar]
- [188].Hasle H, Clemmensen IH and Mikkelsen M, Risks of leukaemia and solid tumours in individuals with Down’s syndrome, Lancet 355(9199) (2000), 165–169. [DOI] [PubMed] [Google Scholar]
- [189].Taub JW, Berman JN, Hitzler JK, Sorrell AD, Lacayo NJ, Mast K, et al. , Improved outcomes for myeloid leukemia of Down syndrome: a report from the Children’s Oncology Group AAML0431 trial, Blood 129(25) (2017), 3304–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Uffmann M, Rasche M, Zimmermann M, von Neuhoff C, Creutzig U, Dworzak M, et al. , Therapy reduction in patients with Down syndrome and myeloid leukemia: the international ML-DS 2006 trial, Blood 129(25) (2017), 3314–3321. [DOI] [PubMed] [Google Scholar]
- [191].Buitenkamp TD, Izraeli S, Zimmermann M, Forestier E, Heerema NA, van den Heuvel-Eibrink MM, et al. , Acute lymphoblastic leukemia in children with Down syndrome: a retrospective analysis from the Ponte di Legno study group, Blood 123(1) (2014), 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Matloub Y, Rabin KR, Ji L, Devidas M, Hitzler J, Xu X, et al. , Excellent long-term survival of children with Down syndrome and standard-risk ALL: a report from the Children’s Oncology Group, Blood Adv 3(11) (2019), 1647–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Cheung YT and Krull KR, Neurocognitive outcomes in long-term survivors of childhood acute lymphoblastic leukemia treated on contemporary treatment protocols: A systematic review, Neurosci Biobehav Rev 53 (2015), 108–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Iyer NS, Balsamo LM, Bracken MB and Kadan-Lottick NS, Chemotherapy-only treatment effects on long-term neurocognitive functioning in childhood ALL survivors: a review and meta-analysis, Blood 126(3) (2015), 346–353. [DOI] [PubMed] [Google Scholar]
- [195].Goldsby RE, Stratton KL, Raber S, Ablin A, Strong LC, Oeffinger K, et al. , Long-term sequelae in survivors of childhood leukemia with Down syndrome: A childhood cancer survivor study report: Survivors of Leukemia With Down Syndrome, Cancer 124(3) (2018), 617–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Salazar EG, Li Y, Fisher BT, Rheingold SR, Fitzgerald J, Seif AE, et al. , Supportive care utilization and treatment toxicity in children with Down syndrome and acute lymphoid leukaemia at free-standing paediatric hospitals in the United States, Br J Haematol 174(4) (2016), 591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Roncadin C, Hitzler J, Downie A, Montour-Proulx I, Alyman C, Cairney E, et al. , Neuropsychological late effects of treatment for acute leukemia in children with Down syndrome: Leukemia and Down Syndrome: Late Effects, Pediatr Blood Cancer 62(5) (2015), 854–858. [DOI] [PubMed] [Google Scholar]
- [198].Schoch J, Rohrer TR, Kaestner M, Abdul-Khaliq H, Gortner L, Sester U, et al. , Quantitative, Phenotypical, and Functional Characterization of Cellular Immunity in Children and Adolescents With Down Syndrome, J Infect Dis 215(10) (2017), 1619–1628. [DOI] [PubMed] [Google Scholar]
- [199].Espinosa JM and Sullivan KD, A signature for success, eLife 16 (2015), 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Espinosa JM, Down Syndrome and COVID-19: A Perfect Storm? Cell Rep Med 1(2) (2020), 100019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Huggard D, Doherty DG and Molloy EJ, Immune Dysregulation in Children With Down Syndrome, Front Pediatr 8 (2020), 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Löwensteyn YN, Phijffer EWEM, Simons JVL, Scheltema NM, Mazur NI, Nair H, et al. , Respiratory Syncytial Virus-related Death in Children With Down Syndrome: The RSV GOLD Study, Pediatr Infect Dis J 39(8) (2020), 665–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Clift AK, Coupland CAC, Keogh RH, Hemingway H and Hippisley-Cox J, COVID-19 Mortality Risk in Down Syndrome: Results From a Cohort Study Of 8 Million Adults, Ann Intern Med (2020. October 21) M20–4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Malle L, Gao C, Hur C, Truong HQ, Bouvier NM, Percha B, et al. , Individuals with Down syndrome hospitalized with COVID-19 have more severe disease, Genet Med (2020. October 16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Hennequin M, Faulks D, Veyrune JL and Bourdiol P, Significance of oral health in persons with Down syndrome: a literature review, Dev Med Child Neurol 41(4) (1999), 275–283. [DOI] [PubMed] [Google Scholar]
- [206].Cuoghi OA, Topolski F, Perciliano de Faria L, Occhiena CM, Ferreira NDSP, Ferlin CR, et al. , Prevalence of Dental Anomalies in Permanent Dentition of Brazilian Individuals with Down Syndrome, Open Dent J 10 (2016), 469–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Sekerci AE, Cantekin K, Aydinbelge M, Ucar Fİ, Prevalence of dental anomalies in the permanent dentition of children with Down syndrome, J Dent Child 81(2) (2014), 78–83. [PubMed] [Google Scholar]
- [208].Moreira MJS, Schwertner C, Jardim JJ and Hashizume LN, Dental caries in individuals with Down syndrome: a systematic review, Int J Paediatr Dent 26(1) (2016), 3–12. [DOI] [PubMed] [Google Scholar]
- [209].Deps TD, Angelo GL, Martins CC, Paiva SM, A Pordeus I and Borges-Oliveira AC, Association between Dental Caries and Down Syndrome: A Systematic Review and Meta-Analysis, PLOS ONE 10(6) (2015), e0127484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Nuernberg MAA, Ivanaga CA, Haas AN, Aranega AM, Casarin RCV, Caminaga RMS, et al. , Periodontal status of individuals with Down syndrome: sociodemographic, behavioural and family perception influence, J Intellect Disabil Res 63(10) (2019), 1181–1192. [DOI] [PubMed] [Google Scholar]
- [211].Reuland-Bosma W, Periodontal disease in Down’s syndrome, Ned Tijdschr Tandheelkd 97(11) (1990), 468–471. [PubMed] [Google Scholar]
- [212].van de Wiel B, van Loon M, Reuland W and Bruers J, Periodontal disease in Down’s syndrome patients. A retrospective study, Spec Care Dent Off Publ 38(5) (2018), 299–306. [DOI] [PubMed] [Google Scholar]
- [213].Hsieh K, Murthy S, Heller T, Rimmer JH and Yen G, Reported gum disease as a cardiovascular risk factor in adults with intellectual disabilities, J Intellect Disabil Res 62(3) (2018), 187–198. [DOI] [PubMed] [Google Scholar]
- [214].Jones D and Morrison J, Preventative therapies and periodontal interventions for Down syndrome patients, Evid Based Dent 17(4) (2016), 101–102. [DOI] [PubMed] [Google Scholar]
- [215].Kamer AR, Pirraglia E, Tsui W, Rusinek H, Vallabhajosula S, Mosconi L, et al. , Periodontal disease associates with higher brain amyloid load in normal elderly, Neurobiol Aging 36(2) (2015), 627–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Kamer AR, Fortea JO, Videla S, Mayoral A, Janal M, Carmona-Iragui M, et al. , Periodontal disease’s contribution to Alzheimer’s disease progression in Down syndrome, Alzheimers Dement 2 (2016), 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Kamer AR, Craig RG, Niederman R, Fortea J and de Leon MJ, Periodontal disease as a possible cause for Alzheimer’s disease, Periodontol 2000 83(1) (2020), 242–271. [DOI] [PubMed] [Google Scholar]
- [218].Zigman WB, Schupf N, Devenny DA, Miezejeski C, Ryan R, Urv TK, et al. , Incidence and prevalence of dementia in elderly adults with mental retardation without down syndrome, Am J Ment Retard 109(2) (2004), 126–141. [DOI] [PubMed] [Google Scholar]
- [219].Head E, Lott IT, Wilcock DM and Lemere CA, Aging in Down Syndrome and the Development of Alzheimer’s Disease Neuropathology, Curr Alzheimer Res 13(1) (2016), 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].McCarron M, McCallion P, Reilly E, Dunne P, Carroll R and Mulryan N, A prospective 20-year longitudinal follow-up of dementia in persons with Down syndrome, J Intellect Disabil Res 61(9) (2017), 843–852. [DOI] [PubMed] [Google Scholar]
- [221].Hithersay R, Startin CM, Hamburg S, Mok KY, Hardy J, Fisher EMC, et al. , Association of Dementia With Mortality Among Adults With Down Syndrome Older Than 35 Years, JAMA Neurol 76(2) (2019), 152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Carmona-Iragui M, Videla L, Lleó A and Fortea J, Down syndrome, Alzheimer disease, and cerebral amyloid angiopathy: The complex triangle of brain amyloidosis, Dev Neurobiol 79(7) (2019), 716–37. [DOI] [PubMed] [Google Scholar]
- [223].Head E, Phelan MJ, Doran E, Kim RC, Poon WW, Schmitt FA, et al. , Cerebrovascular pathology in Down syndrome and Alzheimer disease, Acta Neuropathol Commun 5(1) (2017), 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Carmona-Iragui M, Balasa M, Benejam B, Alcolea D, Fernández S, Videla L, et al. , Cerebral amyloid angiopathy in Down syndrome and sporadic and autosomal-dominant Alzheimer’s disease, Alzheimers Dement 13(11) (2017), 1251–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Wilcock DM and Griffin WST, Down’s syndrome, neuroinflammation, and Alzheimer neuropathogenesis, J Neuroinflammation 10 (2013), 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Colacurcio DJ, Pensalfini A, Jiang Y and Nixon RA, Dysfunction of autophagy and endosomal-lysosomal pathways: Roles in pathogenesis of Down syndrome and Alzheimer’s Disease, Free Radic Biol Med 114 (2018), 40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Gauthier SA, Pérez-González R, Sharma A, Huang F-K, Alldred MJ, Pawlik M, et al. , Enhanced exosome secretion in Down syndrome brain - a protective mechanism to alleviate neuronal endosomal abnormalities, Acta Neuropathol Commun 5(1) (2017), 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Kim S, Sato Y, Mohan PS, Peterhoff C, Pensalfini A, Rigoglioso A, et al. , Evidence that the rab5 effector APPL1 mediates APP-βCTF-induced dysfunction of endosomes in Down syndrome and Alzheimer’s disease, Mol Psychiatry 21(5) (2016), 707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Jiang Y, Sato Y, Im E, Berg M, Bordi M, Darji S, et al. , Lysosomal Dysfunction in Down Syndrome Is APP-Dependent and Mediated by APP-βCTF (C99), J Neurosci 39(27) (2019), 5255–5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Nixon RA, Mathews PM, Cataldo AM, The neuronal endosomal-lysosomal system in Alzheimer’s disease, J Alzheimers Dis 3(1) (2001), 97–107. [DOI] [PubMed] [Google Scholar]
- [231].Millan Sanchez M, Heyn SN, Das D, Moghadam S, Martin KJ, Salehi A Neurobiological elements of cognitive dysfunction in down syndrome: exploring the role of APP, Biol Psychiatry 71(5) (2012), 403–409. [DOI] [PubMed] [Google Scholar]
- [232].Iulita MF, Do Carmo S, Ower AK, Fortress AM, Flores Aguilar L, Hanna M, et al. , Nerve growth factor metabolic dysfunction in Down’s syndrome brains, Brain J Neurol 137(Pt 3) (2014), 860–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Biagi E, Candela M, Centanni M, Consolandi C, Rampelli S, Turroni S, et al. , Gut microbiome in Down syndrome, PloS One 9(11) (2014), e112023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Hamlett ED, Goetzl EJ, Ledreux A, Vasilevko V, Boger HA, LaRosa A, et al. , Neuronal exosomes reveal Alzheimer’s disease biomarkers in Down syndrome, Alzheimers Dement 13(5) (2017), 541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Fortea J, Vilaplana E, Carmona-Iragui M, Benejam B, Videla L, Barroeta I, et al. , Clinical and biomarker changes of Alzheimer’s disease in adults with Down syndrome: a cross-sectional study, Lancet 395(10242) (2020), 1988–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Hamlett ED, Boger HA, Ledreux A, Kelley CM, Mufson EJ, Falangola MF, et al. , Cognitive Impairment, Neuroimaging, and Alzheimer Neuropathology in Mouse Models of Down Syndrome, Curr Alzheimer Res 13(1) (2016), 35–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Strydom A, Coppus A, Blesa R, Danek A, Fortea J, Hardy J, et al. , Alzheimer’s disease in Down syndrome: An overlooked population for prevention trials, Alzheimers Dement Transl Res Clin Interv 4(1) (2018), 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Handen BL, Lott IT, Christian BT, Schupf N, OBryant S, Mapstone M, et al. , The Alzheimer’s Biomarker Consortium-Down Syndrome: Rationale and methodology, Alzheimers Dement 12(1) (2020), e12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].About LIFE-DSR - For the Enhancement of Down Syndrome Research [Internet]. [cited 2020 Dec 8]. Available from: http://ds-ctn.org/LiFE-DSR.html
- [240].ACTC [Internet]. Alzheimer’s Clinical Trials Consortium Down Syndrome. [cited 2020 Dec 8]. Available from: https://www.actc-ds.org/
- [241].Safety, Tolerability and Immunogenicity of ACI-24 Vaccine in Adults With Down Syndrome - Full Text View - ClinicalTrials.gov [Internet]. [cited 2020 Dec 8]. Available from: https://clinicaltrials.gov/ct2/show/NCT02738450