

Abstract
Fibrodysplasia ossificans progressiva (FOP) and diffuse intrinsic pontine glioma (DIPG) are debilitating diseases that share causal mutations in ACVR1, a TGF‐β family type I receptor. ACVR1R206H is a frequent mutation in both diseases. Pathogenic signaling via the SMAD1/5 pathway is mediated by Activin A, but how the mutation triggers aberrant signaling is not known. We show that ACVR1 is essential for Activin A‐mediated SMAD1/5 phosphorylation and is activated by two distinct mechanisms. Wild‐type ACVR1 is activated by the Activin type I receptors, ACVR1B/C. In contrast, ACVR1R206H activation does not require upstream kinases, but is predominantly activated via Activin A‐dependent receptor clustering, which induces its auto‐activation. We use optogenetics and live‐imaging approaches to demonstrate Activin A‐induced receptor clustering and show it requires the type II receptors ACVR2A/B. Our data provide molecular mechanistic insight into the pathogenesis of FOP and DIPG by linking the causal activating genetic mutation to disrupted signaling.
Keywords: Activin A, ACVR1R206H , DIPG, FOP, receptor clustering
Subject Categories: Cancer, Signal Transduction
Activation of mutated TGF‐β family type I receptors independent of upstream type II receptor kinase activity promotes SMAD signaling in human glioma cells.
Introduction
Fibrodysplasia ossificans progressiva (FOP) and diffuse intrinsic pontine glioma (DIPG) are devastating human diseases, both associated with activating mutations in ACVR1, a type I TGF‐β family receptor (Taylor et al, 2014b). FOP is a monogenic, autosomal dominant disease caused by germline mutations in ACVR1 and characterized by progressive, episodic heterotopic ossification of soft tissue (Shore et al, 2006; Furuya et al, 2008; Kaplan et al, 2009). DIPG, in contrast, is an invariably fatal childhood glioma arising in the ventral pons of the hindbrain, with a median survival of 9–12 months. Approximately 20% of DIPG tumors harbor somatic ACVR1 mutations (Buczkowicz et al, 2014; Fontebasso et al, 2014; Taylor et al, 2014a; Wu et al, 2014; Mackay et al, 2017).
TGF‐β family ligands require two different receptors for signaling, type I and type II receptors, which are thought to function as a heterotetramer, comprising two type I and two type II receptors. Ligand binding brings the receptors together, whereupon the constitutively active type II receptor phosphorylates and activates the type I receptor (Wrana et al, 1994). The activated type I receptors then phosphorylate the intracellular effectors of the pathway, the receptor‐activated SMADs (R‐SMADs). Phosphorylated R‐SMADs form complexes with SMAD4 and SMAD heterotrimers accumulate in the nucleus to regulate gene expression (Miller & Hill, 2016). There are five type II receptors and seven type I receptors, and different ligands use different combinations of receptors to signal. The identity of the SMADs phosphorylated by a particular ligand–receptor complex is dictated by the type I receptor engaged (Massague, 2012). Thus, ACVR1B and ACVR1C, which bind Activins and Nodal, and TGFBR1, which binds TGF‐βs, induce phosphorylation of SMAD2 and SMAD3. The other type I receptors, which bind BMPs and GDFs, induce phosphorylation of SMAD1, SMAD5, and SMAD9 (Massague, 2012). ACVR1 is in this latter group and has a preference for BMPs (Miller & Hill, 2016).
An exception to these rules emerged with the discovery that TGF‐β signaling through ACVRL1 and ACVR1 led to SMAD1/5 phosphorylation (Goumans et al, 2002; Lebrin et al, 2004; Daly et al, 2008; Ramachandran et al, 2018). TGF‐β‐induced SMAD1/5 phosphorylation is required for cell proliferation in endothelial cells (Goumans et al, 2002; Lebrin et al, 2004) and for anchorage‐independent growth, cell migration, and epithelial‐to‐mesenchymal transition (EMT) in other cell types (Miettinen et al, 1994; Daly et al, 2008; Liu et al, 2009; Ramachandran et al, 2018). The underlying mechanism of receptor activation in this case represents a departure from the classical mechanism and involves a type I receptor activating another type I receptor. We recently demonstrated that TGFBR1, activated as a result of TGF‐β stimulation, can phosphorylate and activate ACVR1, which subsequently phosphorylates SMAD1/5 (Ramachandran et al, 2018). It is not yet known whether this mechanism of activating ACVR1 is unique to TGF‐β or whether other receptor complexes that normally signal through SMAD2/3 can also fulfill this function.
The mutations in ACVR1 that lead to FOP occur in the cytoplasmic portion of the receptor and are concentrated around both the GS domain, a glycine‐serine rich juxtamembrane region, and the kinase domain (Han et al, 2018). Although approximately 13 mutations have been identified, almost 97% of cases of FOP have a single amino acid change, Arg206His (hereafter ACVR1R206H) (Shore et al, 2006; Han et al, 2018). This same mutation accounts for about a fifth of DIPG mutations (Han et al, 2018).
ACVR1 mutations in FOP and DIPG, and in particular ACVR1R206H, lead to enhanced SMAD1/5‐induced signaling (Fukuda et al, 2009; Shen et al, 2009; van Dinther et al, 2010; Mucha et al, 2018). However, the underlying mechanism has proven controversial. One possible explanation has centered around the immunophilin FKBP1A (also known as FKBP12), which binds the GS domain of the type I receptors, maintaining them in an inactive conformation (Wang et al, 1994; Okadome et al, 1996). Disease mutations in ACVR1 were thought to disrupt binding of FKBP1A, thereby removing pathway inhibition. However, the vast majority of ACVR1 mutants retain the ability to bind FKBP1A and can be inhibited by it, suggesting that this mechanism does not explain the enhanced activity of ACVR1R206H (Groppe et al, 2011; Chaikuad et al, 2012; Machiya et al, 2018).
The roles of type II receptors and activating ligands in the activity of ACVR1R206H are also not fully understood. In Drosophila, the hyperactivity of transgenic ACVR1R206H is dependent on the presence of a type II receptor (Le & Wharton, 2012). Similarly, transgenic mice overexpressing ACVR1Q207D, another activating mutation, only developed heterotopic ossification in the presence of the type II receptors ACVR2A and BMPR2, demonstrating a requirement of the type II receptors for the pathology of mutant ACVR1 in FOP (Bagarova et al, 2013). Whether the type II receptors perform a catalytic or structural role has remained unclear. As ACVR1 is best characterized as a BMP receptor (Luo et al, 2010), initial work focused on the BMPs as the activating ligands (Fukuda et al, 2009; Shen et al, 2009; Song et al, 2010; van Dinther et al, 2010; Chaikuad et al, 2012). However, two recent studies using murine models and patient‐derived induced pluripotent stem cells concluded that Activin A was the relevant ligand signaling through ACVR1R206H in FOP (Hatsell et al, 2015; Hino et al, 2015). In both cases, Activin A exhibited strong differential signaling through the mutant receptor, compared to the wild‐type receptor, leading to robust SMAD1/5 phosphorylation, induction of downstream target genes, and the initiation of heterotopic ossification in mice. Notably, this pathological signaling could be blocked using the soluble type II receptor extracellular domains, acting as ligand traps or Activin A neutralizing antibodies (Hatsell et al, 2015; Lees‐Shepard et al, 2018).
Here, we unravel the mechanism whereby Activin A signals through ACVR1R206H. We use extensive genome editing in HEK293T cells, as well as glioma cells from DIPG patients to both model the disease mutation, ACVR1R206H, and resolve the role of additional receptors in Activin A‐induced ACVR1R206H activity. We provide strong evidence that type I receptor activation of other type I receptors, as we previously established for TGF‐β, is a general mechanism and not restricted to TGF‐β receptors. We demonstrate that wild‐type ACVR1 (hereafter ACVR1WT) is essential for Activin A‐induced SMAD1/5 phosphorylation, and that similar to TGFBR1, ACVR1B/C activate ACVR1WT downstream of Activin A to mediate this phosphorylation. In contrast, ACVR1R206H is predominantly auto‐activated by clustering, independent of the kinase activity of type II receptors or ACVR1B/C. Finally, we use live‐imaging approaches to demonstrate Activin A‐induced receptor clustering and show that this requires the type II receptors, ACVR2A/B. These results elucidate the signaling mechanism of ACVR1R206H, which is crucial for understanding the pathogenesis of FOP and DIPG.
Results
ACVR1R206H exhibits increased responsiveness to Activin A, relative to wild‐type ACVR1
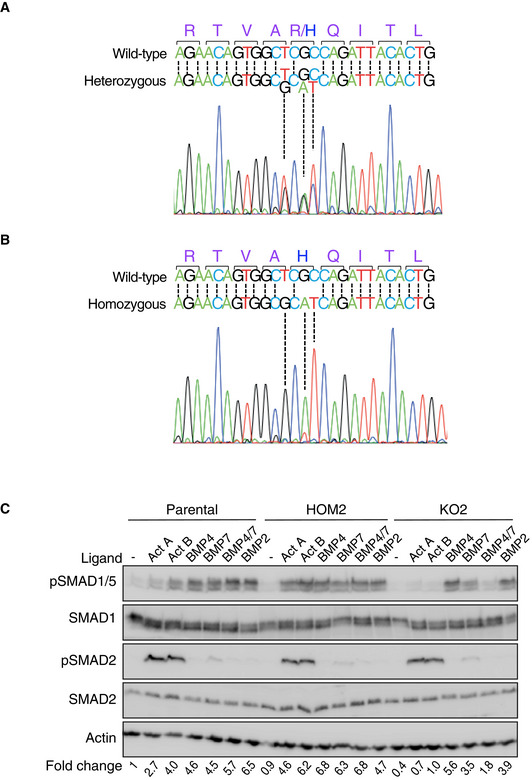
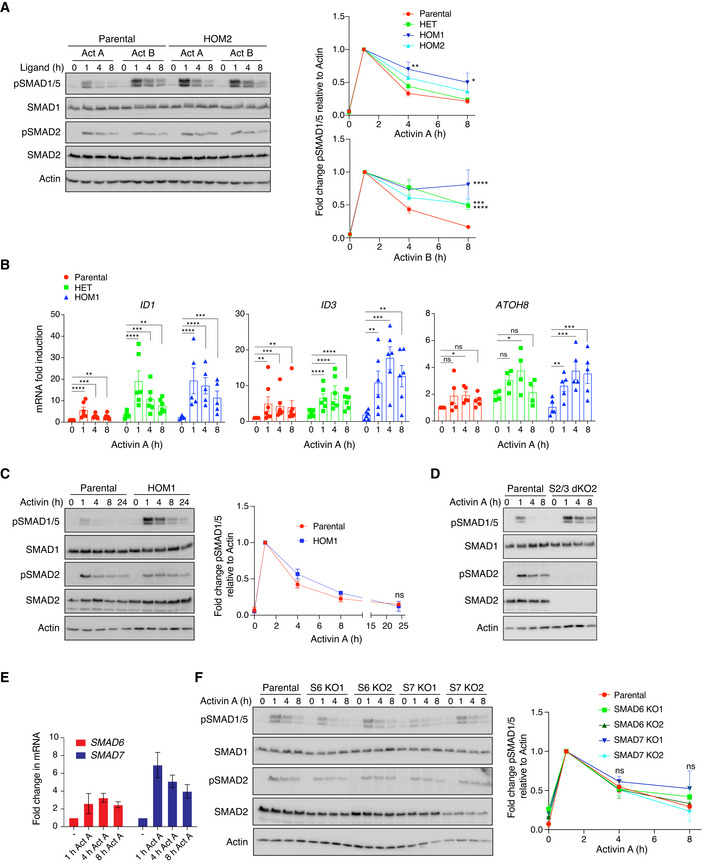
To dissect the mechanism whereby the mutant ACVR1R206H receptor signals, we introduced this point mutation into the endogenous ACVR1 locus in a clone of HEK293T cells (hereafter referred to as parental) using CRISPR/Cas9 genome editing. A heterozygous clone (HET) and several homozygous clones (HOM1 and HOM2) were obtained (Fig EV1A and B). We first characterized the responsiveness of these clones to different TGF‐β family ligands (Activin A, Activin B, BMP4, BMP7, BMP4/7 heterodimer, and BMP2), using SMAD1/5 phosphorylation (pSMAD1/5) as a readout. Treatment of parental cells for 1 h with all these ligands resulted in induction of pSMAD1/5, with Activin A being the least efficient (Figs 1A and C, and EV1C). Strikingly, cells that contained the ACVR1R206H allele (HET, HOM1, and HOM2) all showed substantially increased sensitivity to Activin A (Figs 1A and EV1C), whereas induction of pSMAD1/5 in response to the other ligands in HET, HOM1, and HOM2 cells was comparable to that in the parental cells (Figs 1A and C, and EV1C). We also generated several ACVR1 knockout clones (KO1 and KO2) (Appendix Supplementary Methods). These knockout cells were markedly inhibited in their response to Activin A, Activin B, BMP4/7, and to a lesser extent BMP7, but not to BMP4 and BMP2 (Figs 1A and C, and EV1C). As expected, Activin A and Activin B also led to SMAD2 phosphorylation, that was unaffected by the presence of AVCR1R206H or loss of ACVR1 (Figs 1A and EV1C). Thus, ACVR1WT is essential for the induction of pSMAD1/5 in response to Activin A, Activin B, and BMP4/7, and ACVR1R206H selectively confers increased responsiveness to Activin A.
Figure EV1. Knock‐in of ACVR1R206H and its responsiveness to TGF‐β family ligands.
-
A, BSanger sequencing traces of the ACVR1 locus at nucleotides 604‐630 in HET (A) or HOM1 (B). In each case, the wild‐type sequence is shown for reference.
-
CParental HEK293T, HOM2, and KO2 cells were treated with the indicated ligands for 1 h. Fold changes indicated are the levels of pSMAD1/5 relative to actin, expressed as fold change relative to the untreated parental HEK293T sample. A representative experiment is shown. In all panels, Western blots of whole cell lysates were probed with the antibodies indicated. Act, Activin and KO2, ACVR1 knockout clone 2.
Source data are available online for this figure.
Figure 1. Characterization of ACVR1 responsiveness to TGF‐β family ligands.
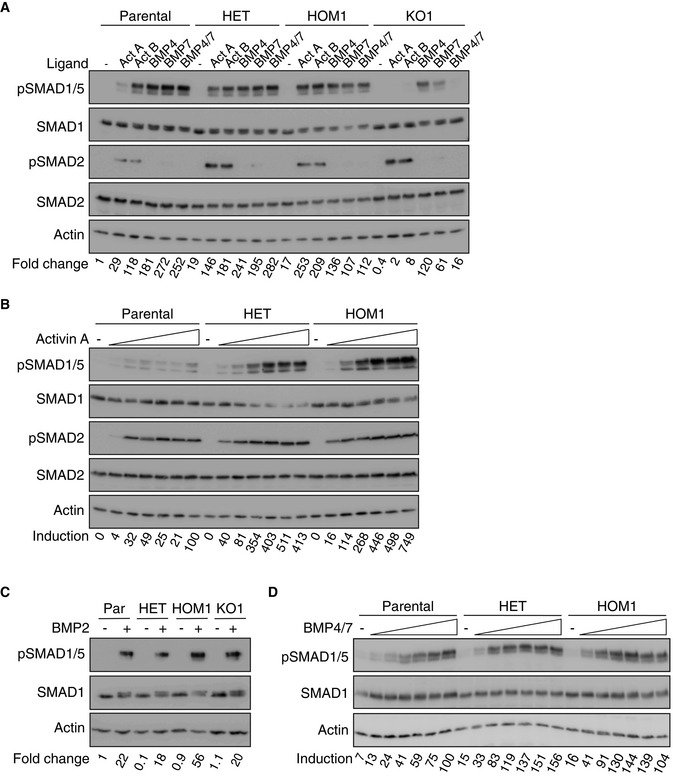
- Parental HEK293T, HET, HOM1, and KO1 cells were stimulated with the indicated ligands for 1 h.
- Cells were stimulated with increasing (doubling) doses of Activin A (0.625–20 ng/ml) for 1 h.
- Cells were stimulated with BMP2 for 1 h.
- Cells were stimulated with increasing (doubling) doses of BMP4/7 (0.625–20 ng/ml) for 1 h.
Data information: In all cases, whole cell extracts were Western‐blotted using the indicated antibodies. Representative experiments are shown. The levels of pSMAD1/5 relative to actin were determined and are expressed as a fold change relative to the parental HEK293T untreated sample in (A and C), or normalized to the 20 ng/ml ligand dose in parental HEK293T cells in (B and D). Act, Activin A; Par, parental HEK293T cells; and KO1, ACVR1 KO clone 1.
Source data are available online for this figure.
To understand how ligand dose affected signaling, we compared the responses of parental, HET, and HOM1 cells to increasing ligand doses, focusing on Activin A and BMP4/7 (as a control ligand that also signals through ACVR1). In parental cells, a low level of pSMAD1/5 was achieved with 1.25 ng/ml Activin A and this was essentially unchanged at higher ligand concentrations (Fig 1B). However, in both HET and HOM1 cells, Activin A led to a dose‐dependent quantitative increase in pSMAD1/5 that plateaued at around 5 ng/ml Activin A (Fig 1B). Activin A led to similar SMAD2 phosphorylation in parental, HET, and HOM1 cells, plateauing in the range of 0.625–1.25 ng/ml (Fig 1B). Parental, HET, and HOM1 cells all displayed a similar dose response to BMP4/7, with SMAD1/5 phosphorylation plateauing at around 2.5–5 ng/ml BMP4/7 (Fig 1D), indicating that signaling via this ligand is not sensitive to the mutation status of ACVR1. Thus, in parental cells, the similarity of pSMAD1/5 and pSMAD2 induction with increasing Activin A concentrations suggested the involvement of shared activated receptors. In contrast, the graded phosphorylation of SMAD1/5 with increasing Activin A dose in cells containing ACVR1R206H, not seen with pSMAD2, suggested that Activin A triggers an alternative receptor activation mechanism in cells bearing this mutation.
ACVR1R206H confers an extended duration of SMAD1/5 phosphorylation relative to ACVR1WT
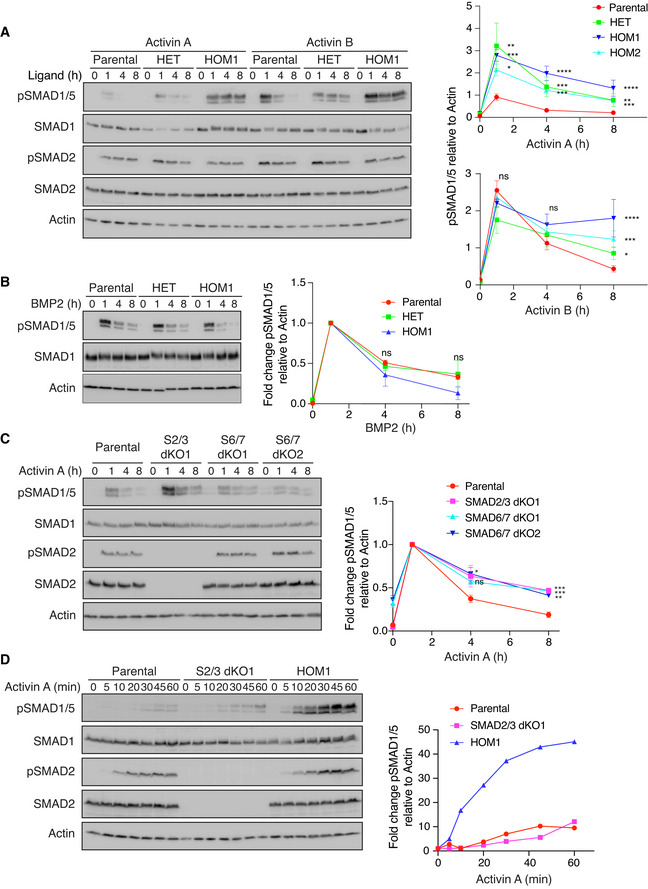
To investigate the effect of the R206H mutation in ACVR1 on signaling dynamics, we performed time courses of Activin A and Activin B stimulation in the different cell lines, with BMP2 treatment time courses as a control. Parental cells exhibited a transient pSMAD1/5 response to Activin A and B, whereas in HET, HOM1, and HOM2 cells, the duration of SMAD1/5 phosphorylation was extended. This was most prominent in the HOM1 cells, whereas HET cells displayed kinetics intermediate between HOM1 and parental cells (Figs 2A and EV2A). These phosphorylation kinetics are also mirrored in the expression of pSMAD1/5 target genes, ID1, ID3, and ATOH8 (Grönroos et al, 2012; Ramachandran et al, 2018), where the induction of target gene expression seen in parental cells is more robust and sustained in HET and HOM1 cells (Fig EV2B). The differential pSMAD1/5 observed between the parental and HOM1 cells was temporary; however, as by 24 h of Activin A treatment, SMAD1/5 phosphorylation in HOM1 cells had returned to basal levels seen in parental cells (Fig EV2C). As expected, the presence of the point mutation in ACVR1 did not alter responsiveness to BMP2, which signals through other TGF‐β family type I receptors, over the time course (Fig 2B).
Figure 2. ACVR1R206H and SMAD2/3 signaling influence the kinetics of SMAD1/5 phosphorylation.
-
A, BParental HEK293T, HET, and HOM1 cells were treated with Activin A or Activin B (A) or BMP2 (B) for the indicated times. Quantifications are shown on the right.
-
CParental HEK293T, S2/3 dKO1, S6/7 dKO1, and S6/7 dKO2 cells were treated with Activin A for the indicated times.
-
DParental HEK293T, S2/3 dKO1, and HOM1 cells were treated with Activin A for the indicated times.
Data information: In (A–C), quantifications are the average normalized intensities ± SEM of between 3 and 10 (A and C) or 2 (B) independent experiments, and in the case of Activin A and Activin B, include a second ACVR1R206H homozygous clone (HOM2, see Fig EV2A). In (A), quantifications are the levels of pSMAD1/5 normalized to actin, and in (B and C), the quantifications are expressed as fold changes relative to 1 h of ligand stimulation. ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001. The P‐values are from two‐way ANOVA with Dunnett’s post hoc test. In (D), quantifications are from a representative experiment and are the intensities of pSMAD1/5 normalized to actin, expressed as fold change relative to untreated cells for each clone. In all panels, Western blots of whole cell lysates were probed with the antibodies indicated. S2/3 dKO1, SMAD2/3 double knockout clone 1 and S6/7 dKO1/2, SMAD6/7 double knockout clones 1 and 2.
Source data are available online for this figure.
Figure EV2. Kinetics of SMAD1/5 phosphorylation.
- Parental HEK293T and HOM2 cells were treated with Activin A or Activin B for the indicated times. Quantifications are presented on the right. These are the same quantifications as shown in Fig 2A, but here they are shown expressed as fold changes relative to 1 h of ligand stimulation to see more clearly how signaling attenuated over time.
- Parental HEK293T, HET, and HOM1 cells were treated with Activin A for the indicated times, and gene expression was measured by qPCR. Expression of ID1, ID3, and ATOH8 was normalized to GAPDH and is presented as fold change relative to parental untreated. Data are the average ± SEM of 4–7 experiments.
- Parental HEK293T and HOM1 cells were treated with Activin A for the indicated times, up to 24 h. Quantifications are presented on the right and are the averaged normalized intensities ± SEM of 3 independent experiments. Note that the 0‐h, 1‐h, 4‐h, and 8‐h data from these 3 experiments are also included in the data shown in (A).
- Parental HEK293T and S2/3 dKO2 cells were treated with Activin A for the indicated times.
- Parental HEK293T cells were treated with Activin A for the indicated times, and gene expression was measured by qPCR. Expression of SMAD6 and SMAD7 was normalized to GAPDH and is presented as fold change relative to untreated. Data are the average ± SEM of 4 experiments.
- Parental HEK293T, S6 KO1, S6 KO2, S7 KO1, and S7 KO2 cells were treated with Activin A for the indicated times.
Data information: In (A, C, D, and F), Western blots of whole cell lysates were probed with the antibodies indicated. Quantifications are the average normalized intensities ± SEM of 3 independent experiments. In all cases, quantifications are the levels of pSMAD1/5 normalized to actin, additionally expressed as fold changes relative to 1 h of ligand stimulation. The P‐values are from two‐way ANOVA with Dunnett’s post hoc test. ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001. In (B), the P‐values are from one‐way ANOVA with Dunnett’s post hoc test. ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001. Act, Activin; S2/3, SMAD2/3; S6, SMAD6; S7, SMAD7; KO, knockout; and dKO, double knockout.
Source data are available online for this figure.
To shed light on the mechanism governing pSMAD1/5 signaling duration, we investigated whether the SMAD2/3 arm of signaling was involved. Strikingly, combined knockout of SMAD2 and SMAD3 in parental cells extended the duration of Activin A‐mediated SMAD1/5 phosphorylation (Figs 2C and EV2D, Appendix Supplementary Methods), suggesting that downstream targets of this signaling arm could be responsible. The most likely targets were the I‐SMADs SMAD6 and SMAD7, as these are induced in response to Activin A in parental cells (Fig EV2E), and known to be involved in mediating receptor turnover (reviewed in Miller and Hill (2016)). We therefore knocked out SMAD6 and SMAD7, individually and simultaneously using CRISPR/Cas9 genome editing (Appendix Supplementary Methods). Combined knockout of both SMAD6 and SMAD7 also extended the duration of Activin A‐induced pSMAD1/5 (Fig 2C), while knocking out each alone had little effect (Fig EV2F). Thus, in parental cells, SMAD2/3 signaling, at least in part via the induction of SMAD6/7, acts to specifically attenuate SMAD1/5 phosphorylation downstream of Activin A.
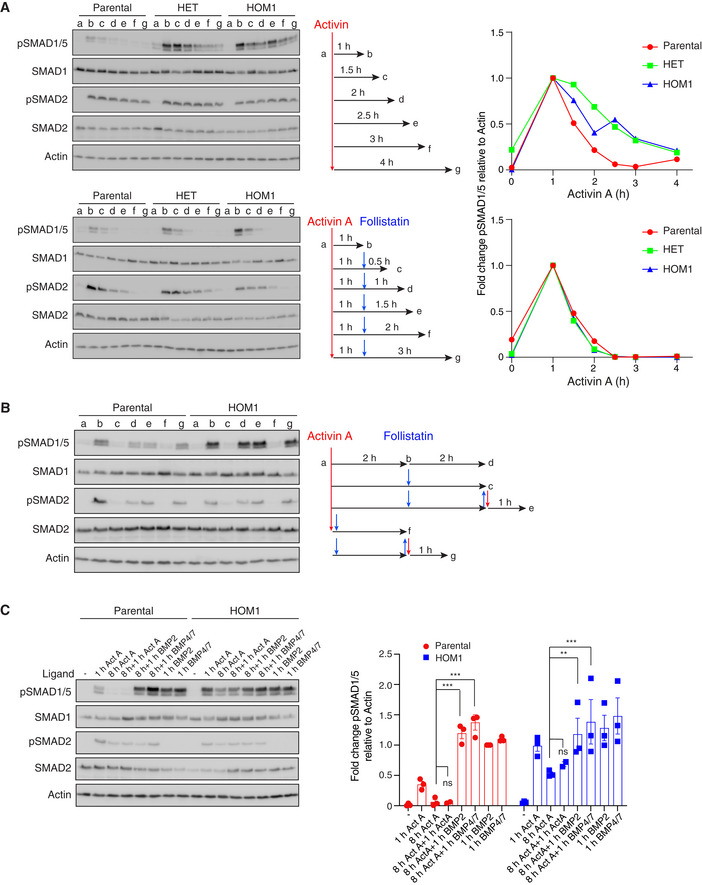
We next addressed why ACVR1R206H mediated an extended signaling duration compared to ACVR1WT. While TGF‐β family signaling is initiated at the cell surface, the majority of signaling occurs in the internalized endosomal compartment (Vizán et al, 2013; Miller et al, 2018; Miller et al, 2019). We therefore investigated whether the longer duration of ACVR1R206H signaling might result from extended signaling from these intracellular compartments. Cells were pulsed with Activin A for 1 h and then chased with its specific antagonist follistatin (Nakamura et al, 1990) for different durations, up to 4 h. As the addition of follistatin prevents further binding of Activin A to receptors at the cell surface, any SMAD phosphorylation detected in these conditions would be the result of ongoing signaling from endosomes. Parental, HET, and HOM1 cells all demonstrated similar half‐lives for phosphorylated SMAD2 and SMAD1/5 (Fig EV3A). Thus, ACVR1R206H does not obviously alter signaling dynamics from endosomes. This left open the possibility that the underlying mechanism of altered signaling dynamics could be at the level of surface receptors.
Figure EV3. ACVR1 signaling from endosomes and recycling to the cell surface.
- Parental HEK293T, HET, and HOM1 cells were untreated (a), stimulated with Activin A for 1 h (b), and then left (upper panel) or chased with follistatin (lower panel) for a further 0.5 h (c), 1 h (d), 1.5 h (e), 2 h (f), or 3 h (g) as indicated in the experimental scheme. Quantifications are the levels of pSMAD1/5 normalized to actin levels expressed as fold changes relative to the level after 1 h of Activin treatment for one representative experiment.
- Parental HEK293T and HOM1 cells were untreated (a), treated with Activin A for 2 h (b), and then left untreated (d) or treated with follistatin (c) for a further 2 h. Subsequently, follistatin was washed out and the cells re‐stimulated with Activin A for a further 1 h (e). As controls, cells were simultaneously treated with Activin A and follistatin (f) to confirm the efficacy of follistatin or pre‐treated with follistatin for 2 h followed by follistatin wash out and re‐stimulation with Activin A for 1 h (g) to assay for efficient wash out.
- Parental HEK293T and HOM1 cells were untreated or treated with Activin A for 1 or 8 h. After 8 h, cells were re‐stimulated with Activin A, BMP2, or BMP4/7 for a further 1 h. Naive cells were also treated with BMP2 and BMP4/7 for 1 h as controls. Quantifications are the levels of pSMAD1/5 normalized to actin, expressed as fold change relative to 1‐h BMP2 stimulation of the parental HEK293T cells, and are the average ± SEM of 3 independent experiments.
Data information: In all panels, Western blots of whole cell lysates were probed with the antibodies indicated. The P‐values in (C) are from one‐way ANOVA with Sidak’s post hoc test. ns, not significant; **P < 0.01; and ***P < 0.001.
Source data are available online for this figure.
To address this, we investigated whether ACVR1 was more rapidly depleted from the cell surface after Activin A treatment in parental cells, compared with HOM1 cells. To this end, we treated parental and HOM1 cells with Activin A for 2 h and then added the antagonist follistatin for a further 2 h to reduce phosphorylated R‐SMAD levels back to basal levels (Fig EV3B). The follistatin was then washed out and the cells re‐stimulated with Activin A for 1 h. In both parental and HOM1 cells, Activin A led to re‐phosphorylation of both SMAD1/5 and SMAD2, demonstrating that ACVR1WT and ACVR1R206H (and the classic Activin type I receptors ACVR1B and ACVR1C) are still present at the cell surface following an acute Activin A treatment (Fig EV3B). Moreover, we showed that ACVR1WT and ACVR1R206H at the cell surface even after 8‐h Activin treatment can be engaged by other ligands, including BMP4/7, to induce SMAD1/5 phosphorylation (Fig EV3C). Thus, the intracellular signaling dynamics and recycling of receptors to the cell surface are broadly similar for ACVR1WT and ACVR1R206H.
ACVR1R206H alters the catalytic efficiency of ACVR1 in response to Activin A
We have shown that the R206H mutation in ACVR1 increases both the magnitude and the duration of SMAD1/5 phosphorylation in response to Activin A. To begin to determine the underlying mechanism, we performed a high‐resolution time course of Activin A induction in parental, HET, and HOM1 cells, sampling every 5–15 min for the first hour of signaling. A higher initial rate of SMAD1/5 phosphorylation was measured in the HET and HOM1 cells, relative to parental cells, suggesting that the R206H mutation endowed an increase in the catalytic efficiency of ACVR1 (Fig 2D, Appendix Fig S1A). As a control, we found that the initial rates of BMP2‐mediated pSMAD1/5 were unaffected by the presence of the mutation in ACVR1 (Appendix Fig S1B). Furthermore, the extended pSMAD1/5 duration in response to Activin A in the SMAD2/3 dKO cells was not due to an increased catalytic activity of ACVR1WT, as the time course of Activin A‐induced pSMAD1/5 in these cells was similar to that seen in parental cells (Fig 2D). Thus, although both ACVR1R206H and loss of SMAD2/3 lead to an increase in levels and duration of Activin A‐induced pSMAD1/5, the underlying mechanisms are distinct.
ACVR1B/1C are required for the activation of ACVR1WT, but not ACVR1R206H
We next investigated how Activin A induces signaling through ACVR1WT and ACVR1R206H. We hypothesized that Activin A might induce the activation of ACVR1 by a mechanism analogous to the one we previously described for TGF‐β (Ramachandran et al, 2018). In that case, TGF‐β binding to TGFBR2 leads to the canonical activation of TGFBR1, which subsequently phosphorylates and activates ACVR1. We thus investigated whether the Activin type I receptors, ACVR1B and ACVR1C, were involved in Activin A‐induced signaling via wild‐type and mutant ACVR1. Additionally, we noted above that the wild‐type and mutant ACVR1 behaved differently to escalating doses of Activin A (Fig 1B), with ACVR1WT being relatively insensitive to the dose of Activin A, while the mutant receptor responded in a dose‐dependent manner. This suggested that higher doses of Activin A might activate the mutant receptor through a distinct mechanism to low doses of Activin A.
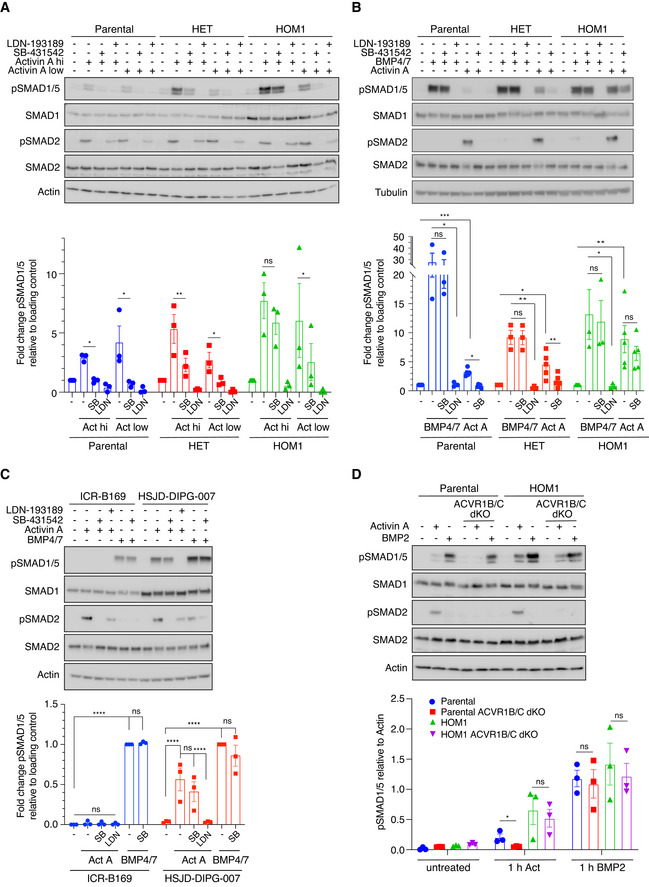
To test these ideas, we treated parental, HET, and HOM1 cells with either a high (20 ng/ml) or a low (1.25 ng/ml) dose of Activin A, in the absence or presence of SB‐431542, that inhibits the kinase activity of ACVR1B/C (Inman et al, 2002), or LDN‐193189, that inhibits the kinase activity of ACVR1/BMPR1A/BMPR1B (Cuny et al, 2008). In all cells and at both Activin A doses, SB‐431542 completely prevented Activin A‐induced pSMAD2, demonstrating that it is sufficient to block all ACVR1B/C activity. In parental cells, SB‐431542 also completely inhibited the phosphorylation of SMAD1/5 at both concentrations of Activin A (Fig 3A and B). Similarly, in HET and HOM1 cells, pSMAD1/5 levels induced with a low dose of Activin A were substantially reduced by treatment with SB‐431542 (Fig 3A). However, strikingly, in HET, and in particular in HOM1 cells treated with a high dose of Activin A, significant pSMAD1/5 persisted, even after treatment with SB‐431542 (Fig 3A and B). In all cell lines and at both Activin A doses, treatment with LDN‐193189 completely blocked SMAD1/5 phosphorylation downstream of Activin A, without affecting SMAD2 phosphorylation. Similarly, LDN‐193189, but not SB‐431542, completely blocked BMP4/7‐induced pSMAD1/5 (Fig 3B). Thus, we conclude that in all cases, Activin A‐ and BMP4/7‐induced pSMAD1/5 requires the kinase activity of ACVR1. For ACVR1WT, Activin A‐induced pSMAD1/5 additionally requires the kinase activity of ACVR1B/C. For the mutant receptor, a similar mechanism operates at low doses of Activin A, but at higher Activin A concentrations, activation of ACVR1R206H occurs via a second mechanism that is largely independent of ACVR1B/C.
Figure 3. Distinct type I receptor requirements for Activin A‐mediated SMAD1/5 phosphorylation by ACVR1WT and ACVR1R206H .
- Parental HEK293T, HET, and HOM1 cells were treated with high (20 ng/ml) or low (1.25 ng/ml) concentrations of Activin A for 1 h, in the presence or absence of either 1 µM LDN‐193189 or 10 µM SB‐431542. Quantifications are the levels of pSMAD1/5 normalized to the loading control (actin), expressed as fold change relative to untreated for each clone. Quantifications are the average normalized intensities ± SEM of 3 independent experiments.
- Parental HEK293T, HET, and HOM1 cells were treated with either Activin A or BMP4/7 (both at 20 ng/ml) for 1 h, in the presence or absence of either 1 µM LDN‐193189 or 10 µM SB‐431542. Quantifications are the levels of pSMAD1/5 normalized to the loading control (actin or tubulin), expressed as fold change relative to untreated for each clone. Quantifications are the average normalized intensities ± SEM of 3‐5 independent experiments. Note that the Activin A quantifications also includes data generated with high dose Activin A in Fig 3A as they are independent replicates.
- ICR‐B169 and HSJD‐DIPG‐007 cells were treated with either Activin A or BMP4/7 (both at 20 ng/ml) for 1 h, in the presence or absence of either 1 µM LDN‐193189 or 10 µM SB‐431542. Quantifications are the levels of pSMAD1/5 normalized to the loading control (actin) expressed as fold change relative to BMP4/7 induction in each clone and are the average normalized intensities ± SEM of 3 independent experiments.
- Parental HEK293T, ACVR1B/C dKO, HOM1, and HOM1 ACVR1B/C dKO cells were treated with 20 ng/ml Activin or BMP2 for 1 h. Quantifications are the average intensities of pSMAD1/5 normalized to actin ± SEM of 3 independent experiments.
Data information: In all panels, Western blots of whole cell lysates were probed with the antibodies indicated. hi, high; Act, Activin A; SB, SB‐431542; and LDN, LDN‐193189. The P‐values (for A, B, and D) are from a t‐test with Holm–Sidak correction, for comparison between indicated pairs. ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001. The P‐values in (C) are from two‐way ANOVA with Tukey’s post hoc correction.
Source data are available online for this figure.
To investigate whether these mechanisms are relevant in DIPG, we treated cell lines derived from DIPG patients that are either ACVR1 +/+ (ICR‐B169) or ACVR1 +/ R206H (HSJD‐DIPG‐007) (Carvalho et al, 2019; C. Jones, personal communication) with Activin A. ICR‐B169 cells did not detectably induce pSMAD1/5 in response to Activin A (Fig 3C). However, in agreement with observations in HET and HOM1 cells, Activin A induced pSMAD1/5 in HSJD‐DIPG‐007 cells that was inhibited by LDN‐193189, but unaffected by SB‐431542, although the latter completely inhibited Activin A‐induced pSMAD2 (Fig 3C). In addition, BMP4/7‐induced pSMAD1/5 was inhibited by LDN‐193189 in both ICR‐B169 and HSJD‐DIPG‐007 cells (Fig 3C). Unexpectedly, BMP4/7 also induced low levels of pSMAD2 in HSJD‐DIPG‐007 cells. We have previously observed that recombinant ACVR1 is capable of efficiently phosphorylating SMAD2 in vitro (Ramachandran et al, 2018) raising the possibility that ACVR1R206H may confer a neofunction to BMP4/7.
To further confirm these results obtained with small molecule inhibitors, we took a genetic approach and simultaneously knocked out ACVR1B and ACVR1C in both parental and HOM1 cells using CRISPR/Cas9 genome editing (Appendix Supplementary Methods). As expected, in parental cells lacking both ACVR1B/C, Activin A‐induced pSMAD1/5 and pSMAD2 were lost, while SMAD1/5 phosphorylation in response to BMP2 was unaffected (Fig 3D). Strikingly, in HOM1 cells, knockout of both ACVR1B/C led to a complete loss of pSMAD2 but did not inhibit pSMAD1/5 phosphorylation in response to Activin A (Fig 3D). Again, BMP2‐mediated SMAD1/5 phosphorylation was unaffected by the absence of ACVR1B/C.
The data described above demonstrate that higher doses of Activin A induce pSMAD1/5 via two different mechanisms, depending on the mutation status of ACVR1; ACVR1WT signaling requires ACVR1B/C, while ACVR1R206H does not. We therefore considered the possibility that ACVR1R206H could similarly be activated by TGF‐β via a mechanism independent of TGFBR1. HEK293T cells respond very poorly to TGF‐β stimulation (Levy et al, 2007), so we used mouse embryonic fibroblasts (MEFs) from ACVR1+/R206H mice and MEFs derived from their matched wild‐type littermates (ACVR1+/+) (Culbert et al, 2014) for this experiment. TGF‐β induced pSMAD1/5 and pSMAD2 in both lines (Appendix Fig S2). In ACVR1+/+ MEFs, treatment with the TGFBR1 kinase inhibitor, SB‐431542, inhibited both pSMAD1/5 and pSMAD2 downstream of TGF‐β to basal levels, as observed previously for other cell lines (Ramachandran et al, 2018). Strikingly, in ACVR1+/R206H MEFs, SMAD1/5 phosphorylation downstream of TGF‐β was unaffected by SB‐431542, but only blocked by LDN‐193189 (Appendix Fig S2). Thus, multiple non‐canonical ligands can trigger the additional mechanism of receptor activation conferred by ACVR1R206H.
The kinase activity of type II receptors is not required for ACVR1R206H function
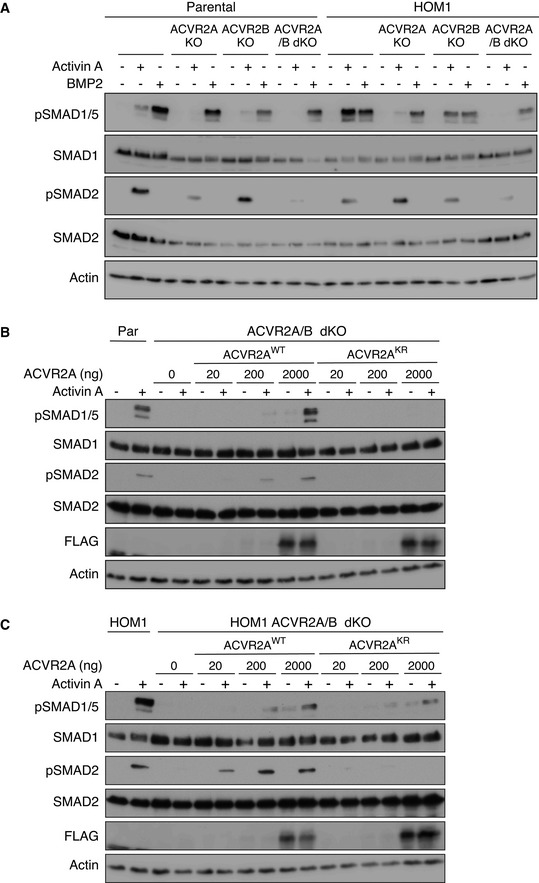
To address the role of the type II receptors in Activin A‐induced signaling via ACVR1R206H, we deleted ACVR2A and/or ACVR2B in both parental HEK293T and HOM1 cells using CRISPR/Cas9 (Appendix Supplementary Methods). In both cell lines, ACVR2A contributed more to Activin A‐induced SMAD2 and SMAD1/5 phosphorylation than ACVR2B. Combined loss of ACVR2A/B led to a loss of pSMAD2 and pSMAD1/5 in both lines (Fig 4A). Importantly, phosphorylation of SMAD2 was rescued in ACVR2A/B dKO lines (both parental and HOM1) by transfecting in wild‐type ACVR2A (ACVR2AWT) (Fig 4B and C).
Figure 4. The structural, but not enzymatic, function of ACVR2A/B is essential for ACVR1R206H signaling.
- Parental HEK293T, ACVR2A KO, ACVR2B KO, ACVR2A/B dKO, HOM1, HOM1 ACVR2A KO, HOM1 ACVR2B KO, and HOM1 ACVR2A/B dKO cells were treated with Activin A or BMP2 for 1 h.
- ACVR2A/B dKO cells were transfected with increasing concentrations of either wild‐type or kinase‐dead ACVR2A (ACVR2AWT or ACVR2AKR, respectively) and treated with Activin A for 1 h. Parental HEK293T cells served as a control.
- HOM1 ACVR2A/B dKO cells were transfected with increasing concentrations of either wild‐type or kinase‐dead ACVR2A (ACVR2AWT or ACVR2AKR) and treated with Activin A for 1 h. HOM1 cells served as a control.
Data information: In all panels, Western blots of whole cell lysates were probed with the antibodies indicated. Representative experiments are shown. Par, parental HEK293T cells.
Source data are available online for this figure.
To disentangle the ligand binding function of ACVR2A from its catalytic activity, we used a kinase‐dead ACVR2A, in which the invariant lysine in the ATP binding pocket had been mutated to arginine (ACVR2AK219R, hereafter called ACVR2AKR) (Bagarova et al, 2013). In ACVR2A/B dKO parental cells, only ACVR2AWT, but not ACVR2AKR, could rescue both pSMAD2 and pSMAD1/5 (Fig 4B). Similarly, in ACVR2A/B dKO HOM1 cells, ACVR2AWT could restore both pSMAD2 and pSMAD1/5. In marked contrast, in ACVR2A/B dKO HOM1 cells, ACVR2AKR restored SMAD1/5 phosphorylation, but not SMAD2 phosphorylation (Fig 4C). Thus, in parental cells, phosphorylation of SMAD2 and SMAD1/5 are linked and share aspects of a common receptor activation mechanism for signaling. In contrast, in ACVR1R206H‐containing cells, SMAD2 and SMAD1/5 phosphorylation are uncoupled from each other, and SMAD1/5 phosphorylation is achieved by a mechanism distinct from the classic type II–type I receptor activation cascade.
Clustering of ACVR1R206H is sufficient to initiate signaling
The ability of ACVR2AKR to restore pSMAD1/5 in Activin A‐induced ACVR2A/B dKO HOM1 cells and the observation that pSMAD1/5 persisted in Activin A‐induced ACVR1B/C dKO HOM1 cells suggested that clustering of ACVR1R206H by the Activin A–ACVR2A–ACVR1R206H complex is sufficient to lead to the activation of ACVR1R206H, without the requirement for any upstream kinases. To test this hypothesis, we employed optogenetics to achieve light‐dependent clustering of either ACVR1WT or ACVR1R206H. The constructs were analogous to those published previously and comprise the light–oxygen–voltage (LOV) domain of aureochrome1 from Vaucheria frigida, fused to the intracellular domains of ACVR1WT or ACVR1R206H, along with an N‐terminal myristoylation motif to anchor them to the plasma membrane (Ramachandran et al, 2018). FLAG‐SMAD1 was also transfected in these experiments to increase the dynamic range of the assay. Light‐dependent clustering of Opto‐ACVR1WT was insufficient to induce SMAD1/5 phosphorylation, as previously observed (Fig 5A, lanes 3–8) (Ramachandran et al, 2018). However, robust light‐dependent phosphorylation of FLAG‐SMAD1 was observed with Opto‐ACVR1R206H, particularly when lower amounts of receptor were transfected (Fig 5A, lanes 9–14). Transfection of higher amounts of Opto‐ACVR1R206H led to significant light‐independent phosphorylation of FLAG‐SMAD1, most likely by achieving supraphysiological levels of expression leading to artificial receptor clustering. Opto‐ACVR1R206H‐mediated phosphorylation of FLAG‐SMAD1 was completely inhibited by LDN‐193189, demonstrating that the kinase activity of ACVR1R206H was necessary and sufficient for SMAD1/5 phosphorylation (Fig 5B, lanes 9 and 10).
Figure 5. Receptor clustering is sufficient for ACVR1R206H signaling.
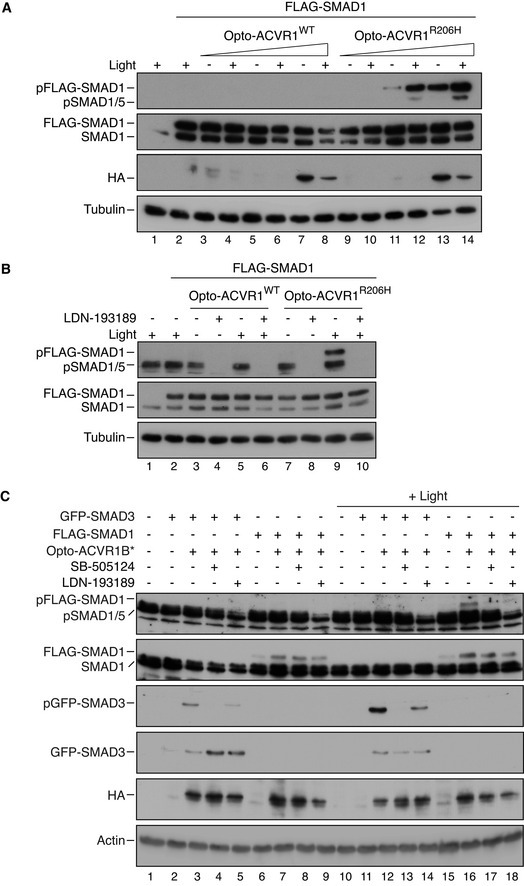
-
A, BNIH‐3T3 cells were either untransfected or transfected with FLAG‐SMAD1 together with increasing (doubling) amounts of Opto‐ACVR1WT or Opto‐ACVR1R206H (5–20 ng). Post‐transfection, cells were either kept in the dark or exposed to blue light for 1 h. In (B), cells were treated in the presence or absence of 1 µM LDN‐193189.
-
CNIH‐3T3 cells were either untransfected or transfected with either FLAG‐SMAD1 or GFP‐SMAD3 together with Opto‐ACVR1B*. Post‐transfection, cells were either kept in the dark or exposed to blue light for 1 h, in the presence or absence of 50 µM SB‐505124 or 1 µM LDN‐193189.
Data information: In all panels, Western blots of whole cell lysates were probed with antibodies against pSMAD1/5 (that detected both endogenous and pFLAG‐SMAD1), SMAD1 (which detects both endogenous and FLAG‐SMAD1), HA (which detects the Opto‐receptors), pSMAD3 (which detects pGFP‐SMAD3), SMAD3 (which detects GFP‐SMAD3), and either tubulin or actin as the loading controls. Lane numbers are given below each blot for clarity.
Source data are available online for this figure.
The inability of Opto‐ACVR1WT to mediate FLAG‐SMAD1 phosphorylation is consistent with the requirement for an upstream kinase for activation of ACVR1WT. Given the biochemical and genetic data placing ACVR1B/C upstream of ACVR1WT upon Activin A induction (Fig 3), we investigated whether constitutively active ACVR1B (Opto‐ACVR1B*) could induce SMAD1/5 phosphorylation via wild‐type ACVR1. As a control, we demonstrated the ability of Opto‐ACVR1B* to induce phosphorylation of GFP‐SMAD3 upon stimulation with blue light (Fig 5C, compare lanes 3 and 12). Strikingly, Opto‐ACVR1B* also induced phosphorylation of FLAG‐SMAD1 in a light‐dependent manner (Fig 5C, compare lanes 7 and 16). We concluded that this was mediated via endogenous ACVR1, as it was inhibited by LDN‐193189, as well as by the ACVR1B/C inhibitor SB‐505124 (Fig 5C, lanes 17 and 18).
Activin A induces ACVR2A/ACVR2B‐dependent clustering in parental and HOM1 cells
From the optogenetic experiments, we concluded that receptor clustering is sufficient to induce the auto‐activation of ACVR1R206H. We hypothesized that in vivo, Activin A binding to ACVR2A/B provides the mechanism by which receptor clustering into higher‐order structures is achieved and, therefore, developed methods to directly visualize it for endogenous receptors. First, we performed confocal microscopy and live cell imaging, applying Number and Brightness (N&B) analysis (see Methods), which has been extensively used to detect endogenous protein clustering (Digman et al, 2008; Nagy et al, 2010; Ross et al, 2011; Zamai et al, 2019). To this end, we generated recombinant dimeric mature Activin A with an N‐terminal His tag, labeled at a single amino group with the far‐red fluorophore Atto 647N (His‐Activin A Atto647N).
The brightness of the His‐Activin A Atto647N dimer was first calibrated in solution, in the absence of cells (Fig 6A, red box in the brightness versus intensity plot (i) and color‐coded (i.e., red) pixels in the map below (ii)). The B histograms (panel iii) show the frequency and distribution of the apparent brightness values (B) in the image stack; dimeric Activin A in solution has a clearly defined, bell‐shaped peak (Fig 6Aiii). In the presence of either parental (Fig 6Bi,ii) or HOM1 cells (Fig 6Ei,ii), labeled Activin A was detected as dimeric Activin A (red box and color‐coded pixels), but strikingly, also as tetramers (green box and color‐coded pixels) and higher‐order oligomers (blue and pink boxes and color‐coded pixels). Furthermore, in the presence of either parental (Fig 6Biii) or HOM1 cells (Fig 6Eiii), the B histogram distributions were right‐shifted, demonstrating an increase in the frequency of pixels with higher brightness values, corresponding to higher‐order oligomers (Fig 6Biii and Eiii). These differences in Bav were reproducibly detected in multiple images (Fig 6Biv and Eiv) and were also significant across several biological replicates (Fig 6H).
Figure 6. ACVR2A/B are required for Activin clustering.
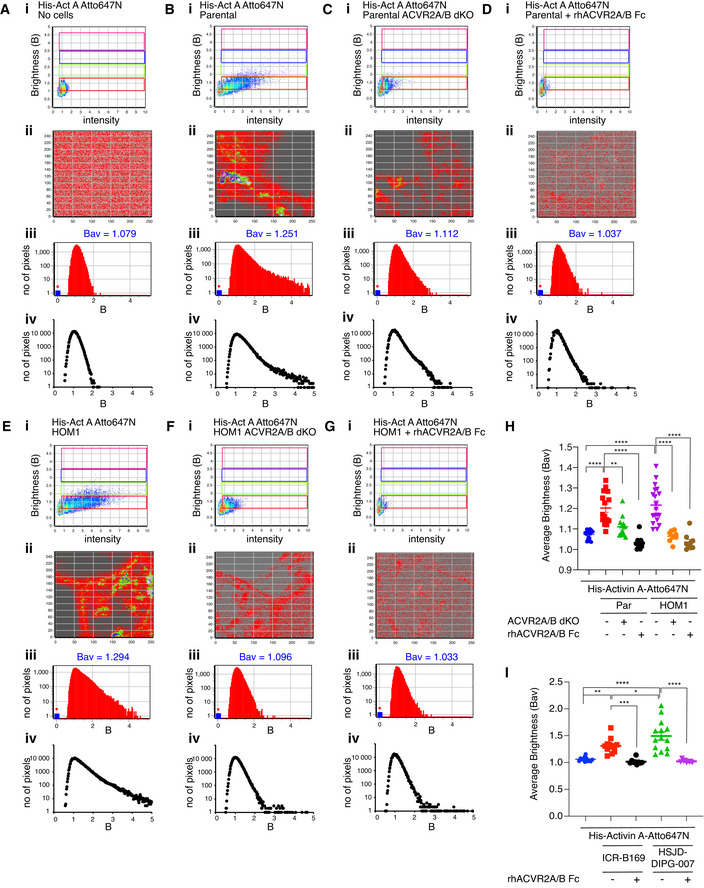
-
A–GHis‐Activin A‐Atto647N was imaged free in solution with no cells (A) or with parental HEK293T cells (B), ACVR2A/B dKO cells (C), parental HEK293T cells pre‐incubated with 1.5 μg/ml of rhACVR2A Fc and 15 μg/ml of rhACVR2B Fc (D), HOM1 cells (E), HOM1 ACVR2A/B dKO cells (F), and HOM1 cells pre‐incubated with 1.5 μg/ml of rhACVR2A Fc and 15 μg/ml of rhACVR2B Fc (G). Number and Brightness (N&B) analysis was performed on raster‐scanned images, and a representative experiment is shown. Panel i, Brightness (B) vs intensity 2D plots. Red rectangular box, pixels that contribute to brightness of free, dimeric His‐Activin A‐Atto647N in solution. Green rectangular box, pixels that contribute to His‐Activin A‐Atto647N clustered into putative tetramers. Blue and pink rectangular boxes, pixels that contribute to His‐Activin A‐Atto647N in higher‐order clusters. Panel ii, Selection map of the image captured reconstructed with the pixels in red, green, blue, and pink rectangular boxes shown in the upper panel (i). Panel iii, B histograms, frequency distribution of the number of pixels across various brightness of His‐Activin A‐Atto647N of the selected image. Bav indicates the average brightness for each image. Panel iv, cumulative B histograms from a representative experiment with 4 independent regions of interest.
-
HAverage brightness of His‐Activin A‐Atto647N in conditions (A–G). Mean ± SEM are plotted. Each point represents one analyzed image, collected from 2–3 biological replicates.
-
IAverage brightness of His‐Activin A‐Atto647N in ICR‐B169 and HSJD‐DIPG‐007 cells alone or with pre‐incubation with 1.5 µg/ml of rhACVR2A Fc and 15 µg/ml of rhACVR2B Fc. Source data are in Fig EV4, EV5. Mean ± SEM are plotted. Each point represents one analyzed image, collected from 2–4 biological replicates.
Data information: In (H and I), the P‐values are from one‐way ANOVA with Tukey’s post hoc correction. *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001.
Source data are available online for this figure.
To test the dependency of this clustering on the type II receptors, we used ACVR2A/B dKO parental or HOM1 cells or pre‐incubated parental and HOM1 cells with ligand traps comprising the extracellular domain of human ACVR2A and ACVR2B fused to the Fc portion of human IgG1 (rhACVR2A/B Fc) to inhibit Activin A binding to endogenous ACVR2A/B (Fig EV4A). When labeled Activin A was added to ACVR2A/B dKO cells, or cells were pre‐incubated with rhACVR2A/B Fc, the occurrence of tetramers and higher‐order oligomers was significantly reduced in both parental and HOM1 cells (Fig 6C, D, F and G, respectively). This was evident in the B histograms, which showed a left‐shifted distribution, demonstrating a decrease in the number of pixels with higher brightness values. In the case of the ligand traps, the Bav values were reduced to the value of dimeric Activin A in solution (Fig 6D, G and H), while in ACVR2A/B dKO cells, the average brightness was slightly higher than that of dimeric Activin A, although the difference was not significant (Fig 6C, F and H). Thus, in both parental and HOM1 cells, Activin A‐induced receptor clustering requires ACVR2A/B.
Figure EV4. Neutralization of Activin A signaling and Activin A clustering in DIPG cells.
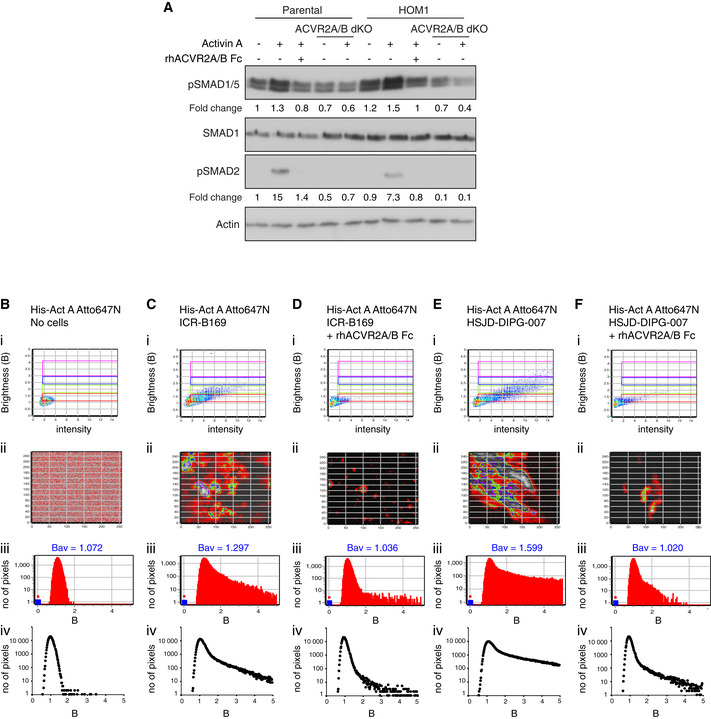
-
AParental HEK293T and HOM1 cells were treated with Activin A either with or without pre‐incubation with both 0.7 μg/ml of rhACVR2A Fc and 9 μg/ml of rhACVR2B Fc for 15 min. ACVR2A/B dKO and HOM1 ACVR2A/B dKO cells were also treated with Activin A for 1 h. Fold changes indicated are the levels of pSMAD1/5 (upper panel) or pSMAD2 (lower panel) relative to actin, expressed as fold change relative to the untreated parental HEK293T sample. A representative experiment is shown. In all panels, Western blots of whole cell lysates were probed with the antibodies indicated.
-
B–FHis‐Activin A‐Atto647N was imaged free in solution with no cells (B) or with ICR‐B169 cells (C), ICR‐B169 cells pre‐incubated with 1.5 μg/ml of rhACVR2A Fc and 15 μg/ml of rhACVR2B Fc (D), HSJD‐DIPG‐007 cells (E), DIPG‐007 cells pre‐incubated with 1.5 μg/ml of rhACVR2A Fc and 15 μg/ml of rhACVR2B Fc (F). Number and Brightness (N&B) analysis was performed on raster‐scanned images, and a representative experiment is shown. Panel i, Brightness (B) vs intensity 2D plots. Red rectangular box, pixels that contribute to brightness of free, dimeric His‐Activin A‐Atto647N in solution. Green rectangular box, pixels that contribute to His‐Activin A‐Atto647N clustered into putative tetramers. Blue and pink rectangular boxes, pixels that contribute to His‐Activin A‐Atto647N in higher‐order clusters. Panel ii, Selection map of the image captured reconstructed with the pixels in red, green, blue, and pink rectangular boxes shown in the upper panel (i). Panel iii, B histograms, frequency distribution of the number of pixels across various brightness of His‐Activin A‐Atto647N of the selected image. Bav indicates the average brightness for each image. Panel iv, cumulative B histograms from a representative experiment with 4–5 independent regions of interest.
Source data are available online for this figure.
We next investigated whether Activin A also triggers receptor clustering in disease relevant models using the DIPG cell lines ICR‐B169 and HSJD‐DIPG‐007. In both cell lines, labeled Activin A clustered into tetramers and higher‐order oligomers, compared with Activin A in the absence of cells (Fig EV4B, Ci,ii and Ei,ii, green, blue and pink boxes, respectively) that was reflected in right‐shifted histograms and Bav values in multiple images (Fig EV4Ciii,iv and Eiii,iv) across several biological replicates (Fig 6I). Interestingly, HSJD‐DIPG‐007 cells demonstrated higher Activin A‐induced clustering compared to ICR‐B169 cells (Figs EV4E and C, and 6I). In both cell lines, pre‐incubation with ligand traps significantly reduced higher‐order receptor oligomerization with a concomitant decrease in Bav values (Figs EV4D and F, and 6I). Thus, in ACVR1WT and ACVR1R206H glioma cells, Activin A induces receptor clustering in a manner dependent on the presence of the type II receptors.
Finally, to demonstrate receptor clustering more visually, we turned to total internal reflection fluorescence (TIRF) microscopy of artificial planar lipid bilayers and specifically focused on the HOM1 cells. This method has been used extensively to study T‐ and B‐cell receptor clustering in the immunological synapse, as well as tyrosine kinase receptor clustering (Grakoui et al, 1999; Tolar et al, 2009; Salaita et al, 2010). We attached His‐Activin A Atto647N to nickel‐containing phospholipids in an artificial mobile lipid bilayer on a glass coverslip. The His‐Activin A‐Atto647N was anchored, but could diffuse freely within the bilayer in the XY plane. Upon plating of cells on the bilayer, Activin A‐induced receptor clustering is visualized as a localized reduction in mobility of the fluorescent Activin A that is evident as a focused increase in fluorescence intensity.
As a technical control to demonstrate the mobility of the bilayer, we used 4‐Hydroxy‐3‐iodo‐5‐nitrophenyl)acetyl (NIP)1‐H12‐Hylight647 ((NIP)1‐Hylight647), a fluorescently labeled His‐tagged small molecule B‐cell antigen, previously used on lipid bilayers (Tolar et al, 2009), that does not bind Activin receptors (Movie EV1). Plating HOM1 cells on this bilayer revealed that the cells spread, but (NIP)1‐Hylight647 remained dispersed and freely diffusible, with only occasional small clusters (Fig 7A and D; Movie EV2). Time‐lapse TIRF imaging of His‐Activin A‐Atto647N on the bilayer for 20 s before the addition of cells demonstrated that it too was mobile and dispersed (Movie EV3; Fig 7C). Importantly, upon plating HOM1 cells on the lipid bilayer containing His‐Activin A‐Atto647N, time‐lapse TIRF imaging for 20 min showed that shortly after cell spreading, prominent fluorescent clusters appeared (Fig 7B; Movie EV4). The first clusters were already apparent at the 0 min time point, taken approximately 5 min after cell plating, but became more intense and numerous over time (Fig 7B). Quantification of cluster intensities showed that areas of the Activin A‐containing bilayer in contact with cells (as defined by the brightfield images) had significantly brighter clusters, and the brightness increased over time, compared to areas of the bilayer without cells (Fig 7D).
Figure 7. TIRF imaging of Activin A‐induced clustering on artificial lipid bilayers.
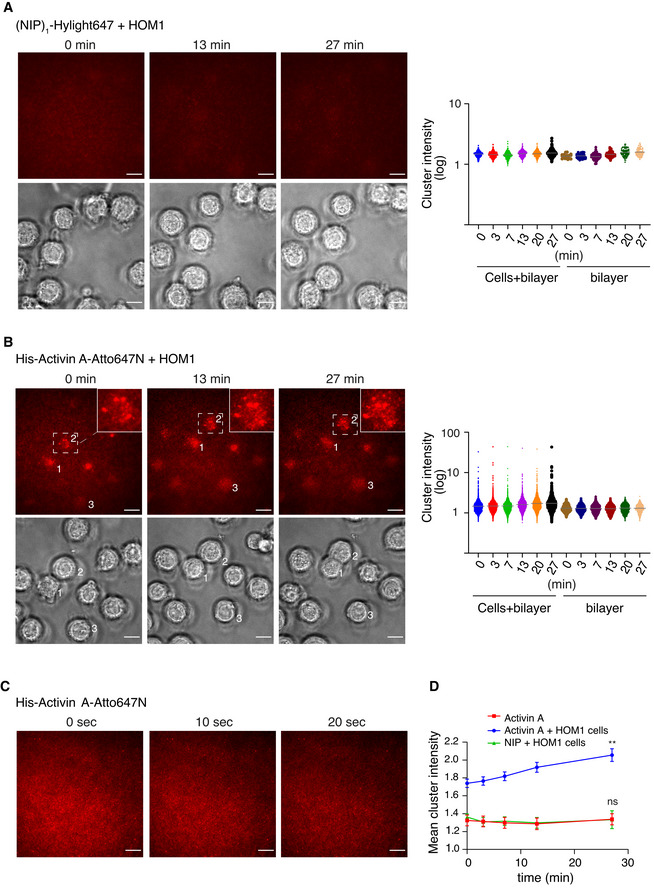
-
A, BHEK293T HOM1 cells were plated on a planar lipid bilayer containing (NIP)1‐H12‐Hylight647 (A) or His‐Activin A‐Atto647N (B) and an automated time‐lapse was started, where 2–4 positions within a well were imaged every 40 s during a 27‐min time period; 3 time points from one position are shown. TIRF, upper panels and brightfield, lower panels. The cluster intensities from all positions were quantified at 6 time points indicated (right panel) and plotted as individual cluster values. The horizontal gray bar denotes the mean. Cells + bilayer denotes bilayer areas under the cells, as identified from the brightfield images, and bilayer denotes areas where no cells were present. A representative experiment from 2 biological replicates quantified is shown. Scale bar 10 μm. In (B), the region under cell 2 is shown enlarged in the top right‐hand corner.
-
CMobile His‐Activin A‐Atto647N on a planar lipid bilayer was imaged for a 20 s period; 3 time points are shown. Scale bar 10 μm.
-
DMean cluster intensity per cell over time. Quantifications are the mean cluster intensities at 5 time points of 35 cells (NIP + cells), 99 cells (Activin A + cells), and 28 bilayer areas ± SEM, from a representative experiment imaged as in (A and B). The P‐values are from F‐test. ns, not significant and **P < 0.01.
Source data are available online for this figure.
As a further control to demonstrate that the clustering was not caused by nonspecific aggregation of the Atto647N fluorophore which is known to be hydrophobic, we conjugated His‐tagged Activin to another far‐red fluorophore (CF640R), that is hydrophilic and less prone to aggregation (Zanetti‐Domingues et al, 2013). The results were consistent with what we had shown for Activin A‐Atto647N. Time‐lapse TIRF imaging of His‐Activin‐A‐CF640R on the bilayer for 20 s (Movie EV5) or 7.5 min (Fig EV5A; Movie EV6) demonstrated its mobility, and upon plating the HOM1 cells on the His‐Activin‐A‐CF640R‐containing lipid bilayers, clusters of labeled Activin A appeared and increased in intensity over time (Fig EV5B, C and D; Movie EV7). Thus, we conclude that Activin A binding to receptors in HOM1 cells induces substantial receptor clustering.
Figure EV5. TIRF imaging of Activin A‐induced clustering on artificial lipid bilayers.
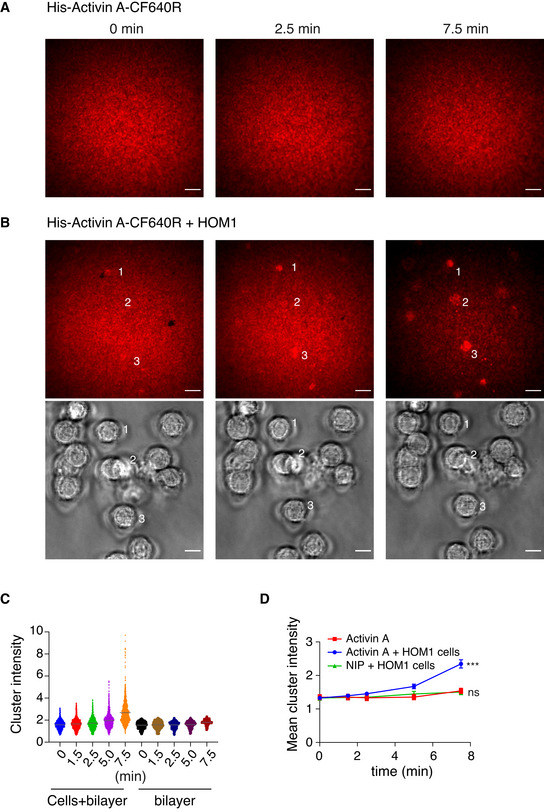
- Mobile His‐Activin A‐CF640R on a planar lipid bilayer was imaged for 7.5 min; 3 time points are shown. Scale bar 10 μm.
- HEK293T HOM1 cells were plated on a planar lipid bilayer containing His‐Activin A‐CF640R and an automated time‐lapse was started, where 3 positions within a well were imaged during a 7.5‐min time period; 3 time points for 1 position are shown. TIRF, upper panels and brightfield, lower panels. Scale bar 10 μm.
- The cluster intensity from all positions was quantified at 5 time points indicated and plotted as individual cluster values. Cells + bilayer denotes bilayer areas under the cells, as identified from the brightfield images, and bilayer denotes areas where no cells are present. The horizontal gray bar denotes the mean.
- Mean cluster intensity per cell over time. Quantifications are the mean cluster intensities at 5 time points of 13 cells (NIP + cells), 61 cells (Activin A + cells), and 16 bilayer areas ± SEM, from a representative experiment imaged as in (A). The P‐values are from F‐test. ns, not significant and ***P < 0.001.
Source data are available online for this figure.
Discussion
Here, we demonstrate the mechanism whereby ACVR1WT and ACVR1R206H are activated by Activin A to induce the SMAD1/5 signaling pathway. We have shown that for ACVR1WT, this occurs by formation of a canonical Activin A–ACVR2A/B–ACVR1B/C complex, whereupon the activated ACVR1B/C phosphorylates and activates ACVR1. This is analogous to the mechanism we previously showed underpins the activation of SMAD1/5 by TGF‐β (Ramachandran et al, 2018). For both ligands, the fundamental new concept is the ability of one activated type I receptor to phosphorylate and activate another type I receptor. For ACVR1R206H, we have shown that this mechanism also operates, particularly at low Activin A levels. However, for the mutant receptor the predominant mechanism involves direct clustering of ACVR2A/B–ACVR1R206H complexes by Activin A. In this case, the role of the type II receptor is structural, as its kinase activity is not required. This view is supported by optogenetics experiments, showing that light‐induced dimerization of opto‐ACVR1R206H is sufficient to induce a robust phosphorylation of SMAD1/5. For cells containing either ACVR1WT or ACVR1R206H, including glioma cells from DIPG patients, we demonstrate that Activin A induces formation of higher‐order clusters, as determined by N&B analysis of live confocal images and as directly visualized by the clustering of fluorescent Activin A on synthetic lipid bilayers. Thus, ACVR1R206H establishes a new paradigm in TGF‐β family signaling, where only its kinase activity is required for signaling.
Ligand selectivity of ACVR1WT versus ACVR1R206H
Here, we have demonstrated that Activin A induces pSMAD1/5 via ACVR1WT and ACVR1R206H, and a recent study in a multiple myeloma cell line also demonstrated this activity in ACVR1WT cells (Olsen et al, 2020). We note that previous studies have readily detected Activin A‐induced pSMAD1/5 in ACVR1WT cells, but this has gone unacknowledged (Hatsell et al, 2015; Hino et al, 2015; Aykul et al, 2020), leading to the conclusion that the R206H mutation conferred a neofunction with respect to Activin induction. Activin A was suggested to form a non‐signaling complex with ACVR2A/B and ACVR1, but a functional signaling complex when ACVR1 was mutated (Hatsell et al, 2015; Aykul et al, 2020). One possible reason for this interpretation could be the assays used. The BRE‐Luciferase reporter has been widely used in other studies as a readout of Activin A‐induced SMAD1/5 signaling. However, the assay requires sufficient time for luciferase levels to accumulate and while we have shown previously that it is an excellent readout of BMP activity via pSMAD1/5, it does not respond to TGF‐β‐induced SMAD1/5 signaling, as it is potently inhibited by the SMAD3 arm of the TGF‐β pathway (Daly et al, 2008; Grönroos et al, 2012). As Activin A also stimulates the SMAD3 pathway, we have avoided this reporter assay as a readout for Activin‐induced pSMAD1/5 signaling and instead have predominantly used phosphorylation of SMAD1/5 as a direct pathway readout. We have also shown by qPCR that the extent of pSMAD1/5 induced by both ACVR1WT and ACVR1R206H is mirrored in the induction of SMAD1/5 target genes, demonstrating that in all cases it is functional. From our data, we conclude that the R206H mutation does not confer a neofunction on ACVR1. Instead, pSMAD1/5 in response to Activin A is a widespread phenomenon in cells that respond to Activin A, reflected at both the protein level and by target gene expression. ACVR1R206H enables the phosphorylation of SMAD1/5 in response to Activin A via a distinct mechanism from wild type, requiring only receptor clustering via the type II receptor.
Interestingly, using MEFs from an FOP mouse model, we have also shown that TGF‐β can activate ACVR1R206H, but not ACVR1WT, via this novel mechanism, although presumably TGFBR2 is the relevant type II receptor in this case. This result suggests that TGF‐β might also be a relevant ligand in vivo for activating the pSMAD1/5 pathway via ACVR1R206H. Of note, we do not see a clear differential between the ability of ACVR1WT and ACVR1R206H to induce pSMAD1/5 when cells are treated with Activin B. We have not delved into the underlying mechanism in detail, but it seems to reflect a higher efficiency of signaling via ACVR1WT in response to Activin B compared with Activin A. It is possible that the Activin type I receptor plays a role in this, as Activin A has a preference for ACVR1B, while Activin B can signal through ACVR1B or ACVR1C (Goebel et al, 2019).
ACVR1R206H has higher catalytic activity than the wild‐type receptor, and can be activated without the requirement for upstream kinases
The tight time course of Activin A induction performed for wild‐type versus mutant ACVR1 revealed that the kinetics of SMAD1/5 phosphorylation and thus receptor activation efficiency is different, with the mutant receptor responding much more rapidly than the wild type. This is consistent with previous structural work showing that the R206H mutation destabilizes the inactive state of the receptor (Chaikuad et al, 2012). In wild‐type ACVR1, R206, which is part of the GS domain, is important for the interaction of the GS domain with the N‐lobe of the kinase domain, hence maintaining the inactive state. The His mutation destabilizes these interactions and also reduces the binding of FKBP1A. Thus, the mutant receptor is in a conformation poised for activation (Shen et al, 2009; Chaikuad et al, 2012).
We hypothesize that the partially active conformation of ACVR1R206H explains the lack of requirement for an upstream kinase for activation. Our optogenetic experiments with this mutant receptor alone demonstrate that dimerization is sufficient for robust activation. We propose that in these homomeric complexes, the kinase domains phosphorylate and activate each other, allowing SMAD1/5 binding and phosphorylation. A similar process occurs in the type II receptor rescue experiments, when kinase‐dead ACVR2A is used. In this case, we propose that Activin A binding to ACVR2AKR and ACVR1R206H brings the mutated type I receptors in close proximity and hence triggers their auto‐activation. In the case of ACVR1WT, Activin‐induced SMAD1/5 phosphorylation is completely inhibited by the ACVR1B/C inhibitor, SB‐431542, demonstrating that ACVR1 activation by Activin A is indirect and mediated via activated ACVR1B/C. We have additionally confirmed this with optogenetic and knockout experiments.
Our data and the work of others raise the interesting question of why for ACVR1WT, an Activin A–ACVR2A/B–ACVR1 complex does not lead to signaling, whereas ACVR1WT can be directly activated in a conventional ligand–type II–ACVR1 complex by BMPs (this work; Macías‐Silva et al, 1998; Hatsell et al, 2015; Aykul et al, 2020). This observation suggests that type II receptors cannot phosphorylate and activate ACVR1 when brought into close proximity by Activin A, but can when the ligand is a BMP. This question has not been answered, but we can speculate that the reason may lie in the different affinity of Activin for the receptor complex, compared with the BMPs, or possibly it is due to the specific type II receptor in the complex (Hinck et al, 2016). Notably, ACVR2A and ACVR2B bind Activin 100–1,000 times more avidly than they do BMPs, and BMPR2 binds BMPs, but not Activins (Hinck et al, 2016). Alternatively, the orientation of the kinase domains in the different complexes may be different for Activin–receptor complexes versus BMP–receptor complexes. Indeed, a recent structure of a GDF11–ACVR2B–TGFBR1 complex, which is thought to mirror an Activin‐containing complex, has a distinct architecture and mode of receptor binding compared with either BMP‐ or TGF‐β‐containing receptor complexes (Goebel et al, 2019).
Activin A‐induced phosphorylation of SMAD1/5 is transient
Our data demonstrate that Activin A‐induced phosphorylation of SMAD1/5 is attenuated over time in both wild‐type and mutant cells, with the duration of signaling slightly prolonged in the HOM1 cells compared to parental. We have shown that in both cases, prolonged signaling requires the constant presence of extracellular ligand, as a follistatin chase after a 1‐h Activin A stimulation curtails signaling in both parental and HOM1 cells. This indicates that signaling requires constant activation of new receptors at the cell surface. TGF‐β family signal attenuation is driven by receptor degradation mediated via the ESCRT machinery (Miller et al, 2018; Miller et al, 2019). We show here that pathway attenuation requires the Activin‐induced SMAD2/3 pathway, at least partly via its ability to induce expression of SMAD6 and SMAD7. We do not yet understand the molecular mechanism underlying the slightly prolonged Activin A‐induced pSMAD1/5 signaling in HOM1 cells, compared with parental cells, but it could reflect a decreased degradation rate for ACVR1R206H versus ACVR1WT. We speculate that this could result from a difference in binding affinity of the receptors for SMAD6/7 and/or a difference in efficiency of receptor ubiquitination that is required for receptor trafficking to the lysosomes (Miller et al, 2018; Miller et al, 2019).
Activin A stimulation leads to receptor clustering and formation of higher‐order receptor complexes
For receptor tyrosine kinases, it has been well established that dimerization and higher‐order hetero‐oligomerization are required for efficient receptor activation (Schlessinger, 2000), but the formation of higher‐order ligand–receptor complexes has not previously been demonstrated for TGF‐β family receptors. Here, we utilize two live‐imaging methods for studying Activin A‐induced receptor clustering and demonstrate the rapid formation of higher‐order complexes in both parental and HOM1 cells. The formation of these complexes does not depend on the mutational status of ACVR1, but is entirely dependent on the presence of ACVR2A/B. We propose that formation of higher‐order receptor complexes enables the type I receptors to activate each other, either in parental cells when activated ACVR1B/C phosphorylate and activate ACVR1 or in the mutant cells where ACVR1R206H receptors activate each other. We note that this might allow the mutant ACVR1 to initiate signaling in additional cell types lacking ACVR1B/C, and when Activin A levels are high, such as during inflammation in the context of FOP (Alessi Wolken et al, 2018). We concluded previously that TGF‐β also induces formation of higher‐order receptor complexes (Ramachandran et al, 2018). It will be important in the future to demonstrate this directly using the approaches pioneered here.
Materials and Methods
Cell lines
All cell lines were maintained at 37°C and 10% CO2, except the DIPG cell lines which were kept at 5% CO2, and have been certified mycoplasma negative by the Francis Crick Institute Cell Services. HEK293T cells were obtained from the Francis Crick Institute Cell Services, and NIH‐3T3 cells were obtained from Dr Richard Treisman (Francis Crick Institute, London). HEK293T cells were authenticated using short tandem repeat (STR) profiling, while NIH‐3T3 cells had species confirmation at the Francis Crick Institute Cell Services. Mouse embryonic fibroblasts (MEFs) from Acvr1 +/+ and Acvr1 +/R206H mice were isolated as previously described (Xu, 2005; Culbert et al, 2014). The ICR‐B169 and HSJD‐DIPG‐007 cell lines derived from DIPG patient gliomas (Carvalho et al, 2019) were obtained from Professor Chris Jones (ICR, London) in whose laboratory STR profiling had been performed. Patients consented to inclusion in a local IRB/Research Ethics Committee‐approved project at the respective institutions (Hospital San Joan de Deu, Barcelona for HSJD‐DIPG‐007; ICR, London for ICR‐B169). These ethical approvals cover the use of the cell lines in collaborations beyond the institute the cell lines were established in. For further details about these lines, see https://www.crukchildrensbraintumourcentre.org/research/resources/cell‐line‐repository/. They were cultured as described in Taylor et al (2014a). In brief, cells were cultured under adherent stem cell conditions in flasks coated with Laminin (R&D systems) in neurobasal‐A medium (Invitrogen), supplemented with B‐27 (Invitrogen) and the growth factors epidermal growth factor (EGF), basal fibroblast growth factor (bFGF), platelet‐derived growth factor (PDGF)‐AA, and PDGF‐BB (all from Shenandoah Biotech).
HEK293T, human embryonic kidney cells, transformed with SV40 T‐antigen, maintained in Dulbecco’s modified Eagle’s medium (DMEM) + 10% fetal calf serum (FCS) + 1% penicillin/streptomycin (Pen/Strep).
NIH‐3T3 cells, mouse embryonic fibroblasts, maintained in DMEM + 10% FCS + 1% Pen/Strep.
MEFs (Acvr1 +/+ and Acvr1 +/R206H), mouse embryonic fibroblasts from Acvr1 +/+ and Acvr1 +/R206H mice, maintained in DMEM + 10% FCS + 1% Pen/Strep.
The following were all derived from HEK293T cells and were all maintained in DMEM + 10% FCS + 1% Pen/Strep. Sequences of the genome‐edited alleles are given in Appendix Supplementary Methods.
HEK293T parental clone, single cell clone isolated and expanded from the original HEK293T pool that responded to Activin A and BMP2 like the original pool and in which all genome editing was performed.
HET, HEK293T clone in which one allele was ACVR1WT and the other ACVR1R206H.
HOM1, HEK293T clone in which only ACVR1R206H was detected.
HOM2, HEK293T clone in which only ACVR1R206H was detected.
ACVR1 KO1, HEK293T clone in which ACVR1 had frameshift mutations leading to predicted protein truncations and loss of function.
ACVR1 KO2, HEK293T clone in which ACVR1 had frameshift mutations leading to predicted protein truncations and loss of function.
S2/3 dKO1, HEK293T clone in which both SMAD2 and SMAD3 had frameshift mutations leading to predicted protein truncations and loss of function.
S2/3 dKO2, HEK293T clone in which both SMAD2 and SMAD3 had frameshift mutations leading to predicted protein truncations and loss of function.
S6 KO1, HEK293T clone in which SMAD6 had frameshift mutations leading to predicted protein truncations and loss of function.
S6 KO2, HEK293T clone in which SMAD6 had frameshift mutations leading to predicted protein truncations and loss of function.
S7 KO1, HEK293T clone in which SMAD7 had frameshift mutations leading to predicted protein truncations and loss of function.
S7 KO2, HEK293T clone in which SMAD7 had frameshift mutations leading to predicted protein truncations and loss of function.
S6/7 dKO1, HEK293T clone in which both SMAD6 and SMAD7 had frameshift mutations leading to predicted protein truncations and loss of function.
S6/7 dKO2, HEK293T clone in which both SMAD6 and SMAD7 had frameshift mutations leading to predicted protein truncations and loss of function.
ACVR1B/C dKO, HEK293T clone in which both ACVR1B and ACVR1C had frameshift mutations leading to predicted protein truncations and loss of function.
HOM1 ACVR1B/C dKO, HEK293T HOM1 in which both ACVR1B and ACVR1C had frameshift mutations leading to predicted protein truncations and loss of function.
ACVR2A KO, HEK293T clone in which ACVR2A had frameshift mutations leading to predicted protein truncations and loss of function.
ACVR2B KO, HEK293T clone in which ACVR2B had frameshift mutations leading to predicted protein truncations and loss of function.
ACVR2A/B dKO, HEK293T clone in which both ACVR2A and ACVR2B had frameshift mutations leading to predicted protein truncations and loss of function.
HOM1 ACVR2A KO, HEK293T HOM1 in which ACVR2A had frameshift mutations leading to predicted protein truncations and loss of function.
HOM1 ACVR2B KO, HEK293T HOM1 in which ACVR2B had frameshift mutations leading to predicted protein truncations and loss of function.
HOM1 ACVR2A/B dKO, HEK293T HOM1 in which both ACVR2A and ACVR2B had frameshift mutations leading to predicted protein truncations and loss of function.
HSJD‐DIPG‐007, primary culture derived from DIPG patient with ACVR1 R206H and K27M H3F3A mutations.
ICR‐B169, primary culture derived from DIPG patient with wild‐type ACVR1 and K27M H3F3A mutation.
Ligands, chemicals and cell culture
Activin A (PeproTech) and Activin B (PeproTech) were reconstituted in phosphate buffered saline (PBS) supplemented with 0.1% bovine serum albumin (BSA). TGF‐β (PeproTech), BMP4 (PeproTech), BMP7 (PeproTech), the BMP4‐BMP7 heterodimer (BMP4/7) (R&D Systems), and BMP2 (R&D Systems) were reconstituted in 4.4 mM HCl supplemented with 0.1% BSA. Unless otherwise noted, all ligands were used at 20 ng/ml except TGF‐β, which was used at 2 ng/ml. Growth factors (bFGF, EGF, PDGF‐AA, PDGF‐BB) used in the tumor stem cell medium were reconstituted in stem cell medium just before use, and used at 20 ng/ml (bFGF, EGF) or 10 ng/ml (PDGF‐AA, PDGF‐BB) final concentration. Cells were treated with the indicated ligands for the times indicated in the figures. SB‐431542 (Tocris, UK) was used at a final concentration of 10 µM, SB‐505124 (Tocris) was used at a final concentration of 50 μM, and LDN‐193189 (a gift from Paul Yu) was used at a final concentration of 1 µM. For Activin A blocking experiments, follistatin 288 (Sigma) was reconstituted in PBS and used at a final concentration of 500 ng/ml. Recombinant human (rh) ACVR2A Fc chimera protein (R&D Systems) and rhACVR2B Fc chimera (R&D Systems) were reconstituted in PBS and used according to the manufacturer’s instructions. Prior to ligand stimulation, HEK293T cells and NIH‐3T3 cells were starved overnight in DMEM containing 0.5% FCS. For the MEFs, cells were starved in OptiMEM with 10 µM SB‐431542 overnight and then the SB‐431542 was washed out prior to ligand stimulation.
Preparation of fluorescently tagged recombinant Activin
Mature Activin A (amino acids 311‐426, UniProt: P08476) with N‐terminal hexa‐His and StrepTags (tag sequence: MNTIHHHHHHNTSSATMNSWSHPQFEKSM) was expressed in E. coli, refolded, and purified to homogeneity. Washed inclusion bodies were first solubilized in 20 mM Tris(2‐carboxyethyl)phosphine (TCEP), 6 M Guanidinium HCl, 50 mM Tris–HCl pH 8.0, and 2 mM EDTA. Solubilized protein was buffer‐exchanged to 6 M urea and 20 mM HCl and then diluted 10× into refolding solution of 1 M pyridinyl propyl sulfonate, 50 mM Tris–HCl, 50 mM ethanolamine, 1 mM EDTA, 2 mM cysteine, and 0.2 mM cystine, pH 8.0. Refolded protein was purified by a combination of cation exchange and reversed phase chromatography. Pure, dimeric Activin A was dried under vacuum and stored in aliquots. For labeling, Activin A was solubilized in 10 mM HCl and diluted to 1 mg/ml concentration in a solution containing 50 mM Hepes pH 7.4, 30% acetonitrile. NHS‐activated fluorophores (Atto 647N (Sigma‐Aldrich) or CF640R (Insight Biotechnology)), both 10 mM stock solutions in DMSO, were added to the protein in 2–8× molar excess and incubated for 2–3 h at room temperature. The labeling reaction was terminated by addition of 0.1% trifluoroacetic acid. The sample was loaded onto an ACE C4 reversed phase column and eluted by acetonitrile gradient, following absorbance at maximum absorption wavelength of the fluorophore. Labeled protein fractions were analyzed by SDS–PAGE and degree of labeling determined spectrophotometrically as the ratio of fluorophore concentration and protein concentration. Fractions with lowest degree of labeling (less than two labels per dimeric Activin A) were used for the experiments. Labeled protein was dried under vacuum and stored in single use aliquots in black tubes at −80°C.
Knock‐in of ACVR1R206H at the endogenous locus
From wild‐type HEK293T cells, a parental clone was selected whose response to Activin A and BMP2 phenocopied the wild‐type pool. Guide RNAs (gRNAs) for CRISPR/Cas9‐mediated genome editing were expressed from the pSpCas9(BB)‐2A‐GFP (PX458) plasmid (Ran et al, 2013). All gRNA sequences and cloning oligonucleotides are listed in Table EV1. A gRNA immediately downstream of codon 206 was chosen for the knock‐in of the ACVR1R206H point mutation (homology‐directed repair gRNA) and cloned into PX458. A 100 nucleotide single‐stranded DNA (ssDNA) of HPLC purity was provided as the repair template (Table EV1). The ssDNA was modified by phosphorothioate bonds between the first two and last two nucleotides at the 5’ and 3’ ends, respectively. The repair template alters codon 205 (nucleotides 613‐615) from GCT → GCG (Ala → Ala) and codon 206 (nucleotide 616‐618) from CGC → CAT (Arg → His). The silent mutation of codon 205 was introduced to increase the specificity of the downstream screening primer. The mutation of codon 206 additionally destroys the PAM sequence for the gRNA. Four million HEK293T cells were plated in a 10‐cm dish in a total volume of 10 ml media. The following day, a transfection mix was made with 7.5 µg of pX458‐gRNA plasmid, 1 µl of 100 µM ssDNA repair template, and 30 µl of FuGENE HD in a total of 500 µl OptiMEM. Complexes were allowed to form at room temperature for 15 min and then added to HEK293T cells. Forty‐eight hour post‐transfection, HEK293T cells were single cell sorted for GFP expression into 96‐well plates. Ten days post‐sorting, clones were consolidated and replica plated into two 96‐well plates—one plate was used as the cell stock, while the other was used for screening. Once confluent, cells were lysed with the QuickExtract DNA Extraction Solution (Lucigen) according to the manufacturer’s instructions. The same universal reverse primer (Table EV1) was used to screen all clones. Two different forward primers were used: either the ACVR1WT forward primer to detect the ACVR1WT allele or the ACVR1R206H forward primer to detect the ACVR1R206H knock‐in allele. Clones that were positive for the ACVR1R206H knock‐in by PCR were selected for further analysis and verified by Sanger sequencing. Of note, during the CRISPR/Cas9‐mediated genome editing, homology‐directed repair did not always occur leading to the generation of ACVR1 knockout clones that were also used in this study.
CRISPR/Cas9‐mediated Gene Knockouts
Guide RNAs targeting ACVR1B, ACVR1C, ACVR2A, ACVR2B, SMAD2, SMAD3, SMAD6, and SMAD7 (Table EV1) were cloned into PX458. gRNAs were designed to target the intracellular domain of the receptors just upstream of the GS domain (type I receptors) or kinase domain (type II receptors). SMAD knockout clones were generated in the HEK293T parental clone only. All receptor knockouts were generated in both HEK293T parental and HEK293T HOM1 cells. Transfections and cell sorting were performed as described above, with the omission of the ssDNA repair template. Clones were verified by Sanger sequencing.
Transient transfection of ACVR2A
Wild‐type ACVR2A (ACVR2AWT) and kinase‐dead ACVR2A in which codon 219 was mutated from lysine to arginine (ACVR2AK219R, referred to as ACVR2AKR) (Table EV1) were cloned into pcDNA3.1 Hygro (+) (Thermo Fisher), both with a C‐terminal FLAG tag. Plasmids were transiently transfected using FuGENE HD into both ACVR2A/B dKO and HOM1 ACVR2A/B dKO cells. Cells were transfected with 2 µg, 200 ng, or 20 ng of the appropriate plasmids in a 6‐well format; where less than 2 µg of ACVR2AWT/KR plasmid was transfected, the quantity was made up to 2 µg with empty pcDNA3.1 Hygro (+). Twenty‐four hours post‐transfection, cells were starved overnight. The following days, cells were stimulated with 20 ng/ml Activin A.
Generation of Opto‐Receptors and LED light photoactivation
Opto‐ACVR1WT was previously described (Ramachandran et al, 2018), and site‐directed mutagenesis was performed to generate Opto‐ACVR1R206H by mutating codon 206 from CGC to CAC (Table EV1). Both receptors have a C‐terminal HA tag. Opto‐ACVR1B* was generated in pCS2+ as previously described (Ramachandran et al, 2018) and included an N‐terminal myristoylation domain, the intracellular portion (residues 150–505) of the constitutively active mouse ACVR1B (ACVR1BT206D), a light‐oxygen‐voltage (LOV) domain, and a C‐terminal HA tag (Table EV1). Blue light‐dependent dimerization of Opto‐receptors was performed as previously described (Ramachandran et al, 2018). In brief, NIH‐3T3 cells were transfected with a total of 2 µg of plasmid DNA that included 25 ng of FLAG‐SMAD1 (Lechleider et al, 2001), alone or in combination with varying amounts of either Opto‐ACVR1WT or Opto‐ACVR1R206H. For experiments with Opto‐ACVR1B*, cells were transfected as above, but with either 15 ng FLAG‐SMAD1 or 5 ng GFP‐SMAD3 (Nicolas et al, 2004) alone, or in combination with 10 ng Opto‐ACVR1B*. Twenty‐four hours post‐transfection, cells were starved overnight in DMEM with 0.5% FCS. Cells were then left untreated or pre‐treated with 1 μM LDN‐193189 or 50 μM SB‐505124 for 5 min and then exposed to blue light from an LED array for 1 h at 37°C in a humidified incubator. Control cells were kept in the dark by wrapping in aluminum foil and placed in the same incubator
Antibodies, immunoblotting, and quantification
All primary and secondary antibodies are listed in Table EV2. Western blots using whole cell extracts were performed as previously described (Miller et al, 2018). For quantification analysis, see below.
qPCR
Oligonucleotides used are listed in Table EV1. Total RNA extractions and reverse transcriptions were performed as previously described (Grönroos et al, 2012). The cDNA was diluted 10‐fold and then used for qPCR. All qPCRs were performed with the PowerUp SYBR Green Master Mix (Thermo Fisher) with 300 nM of each primer and 2 µl of diluted cDNA. Fluorescence acquisition was performed on a 7500 FAST machine (Thermo Fisher). Quantification of relative gene expression was performed using the comparative Ct method with target gene expression normalized to GAPDH.
Confocal microscopy
For live cell confocal imaging, parental, ACVR2A/B dKO, HOM1, and HOM1 ACVR2A/B dKO cells were grown in phenol‐free DMEM (Thermo Fisher Scientific) supplemented with 10% FCS, 1 mM sodium pyruvate, and 1% Pen/Strep. Cells were plated on 35 mm High Precision 1.5 MatTek glass‐bottom dishes (MatTek, Ashland, MA, USA) coated with Poly‐L‐Lysine, or laminin for DIPG cells, and starved overnight in phenol‐free DMEM with 0.5% FCS or 3–4 h in tumor stem cell medium without growth factors for DIPG cells. On the day of the experiment, cells were stained for 30 min with 5 μl/ml of CellBrite Green cytoplasmic membrane dye (Biotium) in phenol‐free DMEM and then washed according to the manufacturers’ instructions.
Imaging was performed on a Zeiss Invert 880 NLO confocal microscope and ZEN platform, with a C‐Apochromat 63×/1.2 water objective. His‐Activin A Atto647N at 40 ng/ml in phenol‐free DMEM was imaged at the start of every Number & Brightness experiment, for calibration of the brightness value for dimeric His‐Activin A. For cell imaging, 40 ng/ml of His‐Activin A‐Atto647N was added to the cells 15 min before the start of imaging. During imaging, cells were kept at 37°C. The green membrane dye was used to select a region of the apical or lateral cell membrane. Images were acquired in raster scan mode, with a frame size of 256×256, pixel size of 0.05 μm and pixel dwell time of 33 μs (scan time/frame of 5.03 s). The 633 nm laser was set at 3%, and a time series of 100 images per ROI were collected in photon‐counting mode (Digman et al, 2008; Digman et al, 2013). The image stacks were transferred to the simFCS software (Laboratory for Fluorescence Dynamics, Irvine, CA) for N&B analysis (Digman et al, 2008; Digman et al, 2013).
Preparation of planar lipid bilayers containing His‐Activin A and NIP‐based antigens
Planar lipid bilayers (PLBs) were prepared by fusing small unilamellar lipid vesicles (SUVs) with clean glass coverslips essentially as described (Tolar et al, 2009). Briefly, 1,2‐dioleoyl‐sn‐glycero‐3‐phosphocholine (DOPC) and 1,2‐dioleoyl‐sn‐Glycero‐3‐{[N(5‐amino‐1‐carboxypentyl) imino diacetic acid]succinyl} (nickel salt) (DOGS‐NiNTA, both from Avanti) were combined in a 10:1 molar ratio in chloroform and dried to a film with a gentle stream of argon and then further under vacuum for 1 h. The lipid film was then resuspended in PBS to 5 mM, vortexed until clear, and then bath sonicated for 30 min to produce SUVs, which were clarified by ultracentrifugation. Glass coverslips (Menzel‐Gläser) were cleaned with fresh piranha solution (98% H2SO4 and 30% H2O2 in a 2:1 ratio) for 30 min, rinsed thoroughly with Milli‐Q H2O, dried with argon, and glued to the bottom of Labtek 8‐well imaging chambers (Fisher Scientific). PLBs were prepared from a 0.5 mM solution of SUVs in PBS, and excess lipids were washed away with PBS. PLBs were blocked for 30 min with 0.05% BSA in PBS (w/v), incubated with His‐tagged Activin A‐Atto647N, His‐tagged Activin A‐CF640R, or His‐tagged 4‐hydroxy‐5‐iodo‐3‐nitrophenyl acetyl (NIP)1‐H12‐Hylight647 for 15 min, and excess was washed away with PBS. For imaging, PBS was replaced by phenol‐red free DMEM and approximately 250,000 HOM1 cells in phenol‐red free DMEM were added to the bilayer and allowed to attach for a few minutes. Time‐lapse TIRF imaging was performed to visualize clustering over time.
TIRF microscopy
TIRF imaging was performed on a Nikon Eclipse Ti microscope with an ORCA‐Flash 4.0 V3 digital complementary metal‐oxide semiconductor (CMOS) camera (Hamamatsu Photonics) and 100x/1.49 Apo TIRF objective (Nikon), controlled through Metamorph software (Molecular Devices). Illumination was provided by a 637 nm laser (Cairn) through an iLas2 Targeted Laser Illuminator (Gataca Systems) which produces a 360° spinning beam with adjustable TIRF illumination angle. Automated time‐lapse imaging of 3–4 areas of the bilayers containing His‐Activin A‐Atto647N, or 1–2 areas containing (NIP)1‐H12‐Hylight647, with HOM1 clones was performed every 40 s, with 100 ms exposure, for 20 min. For Activin A‐CF640R with HOM1 clones, time‐lapse imaging was performed every 30 s, with 100 ms exposure, for 7.5 min. Acquired time‐lapse images were processed into movies at 10 frames per second using FIJI, which was also used for image processing.
Quantification and statistical analysis
All experiments have been performed with a minimum of two independent biological replicates. Where indicated, data from a representative experiment are presented.
Western blot quantification was performed using the FIJI/ImageJ software; background‐subtracted densitometry measurements for pSMAD1/5 were normalized to the loading control (actin or tubulin) and are shown as fold change relative to the indicated condition, unless otherwise specified in the figure legends. Where appropriate, normalized pSMAD1/5 measurements for the different cell lines were used as input into ICEKAT (Olp et al, 2020), and initial rates were calculated using default parameters.
Number and Brightness (N&B) analysis provides a single‐pixel resolution map of the number (N) and Brightness (B, i.e., oligomerization state) of diffusing particles within the focal spot, by calculating the average (k) and variance (σ) of the intensity distribution at each pixel according to the following formulas (for a detector in photon‐counting mode (Digman et al, 2008)):
The value B is related to the molecular brightness ε, defined as counts per second per molecule, and reflects the clustering of fluorophore containing molecules. N is directly proportional to the number of particles n. A map of the parameters B and N is used for identifying the regions of an image, i.e., regions of a cell, where protein clustering is occurring. When a given pixel contains a mixture of species with different levels of brightness, the N&B method averages the brightness. Thus, the N&B method cannot accurately measure the size of aggregates but reports on their presence. Photobleaching leads to a decrease in mean intensity over time and can, thus, confound the final results (Trullo et al, 2013). To overcome this, we have used a very photostable dye, Atto‐647N, which is known to have minimal photobleaching. Images with obvious acquisition artefacts were removed. A high pass filter algorithm (detrend filter) was applied to the time sequence at each pixel of the stack, when mean intensity fluctuations were > 10%, to correct for sample movement and photobleaching during image acquisition (Ossato et al, 2010; Trullo et al, 2013). The N&B analysis was performed in simFCS (Laboratory for Fluorescence Dynamics).
Fluorescent clusters were detected in areas of the bilayer underneath or outside of cells by a spot detection algorithm using MATLAB similarly as previously described (Roper et al, 2019). Briefly, images were flatfield‐corrected for illumination and background‐subtracted. Spots were then enhanced by convolution with a 325‐nm‐sized Gaussian filter followed by subtraction of an averaging filter of the same size. The enhanced images were thresholded using an empirically determined intensity value that was kept constant for each experiment. Thresholded areas were watershed‐split and used as masks to identify individual clusters and quantify their number and fluorescence intensity in the original corrected images.
Statistical analysis was performed in Prism 8 (GraphPad). At least three independent experiments were performed for statistical analysis. Normalized densitometry measurements from Western blots were log‐transformed for analysis by parametric tests. For qPCR data, the ΔΔCT value was used in the statistical analysis, and the normalized gene expression data were plotted as individual data points for each biological replicate, with height of the bar representing the mean, and error bars the standard error of the mean (SEM). For pairwise comparison between two groups, paired Student’s t‐test was used, with correction for multiple testing. For comparisons between more than two groups, analysis of variance (ANOVA) was performed, followed by Dunnett’s multiple comparison test (for comparison of a number of treatments with a single control) or Tukey’s multiple comparison test (for pairwise comparison between all samples). Quantifications of Western blots with 2–3 replicates per condition are presented as individual data points with height of the bar representing the mean, and error bars the standard error of the mean (SEM). For comparison of clustering over time, simple linear regression was performed on mean cluster intensity/cell on Activin and NIP bilayers, and mean cluster intensity on Activin bilayers without cells (corresponding to areas of approximate cell size). Slopes were tested for a non‐zero slope, and significant differences between slopes were determined using F‐test. Outliers were included in statistical analysis. A P < 0.05 was considered statistically significant.
Conflict of interest
The authors declare that they have no conflict of interest.
Author contributions
AR and CSH conceived and designed the study. AR and MM planned and performed the experiments, and analyzed the data with technical help from LW, DM, PT, and IG. BKB, DMC, EMS, CJ, and MH produced and provided resources. MM performed all the statistical analyses. AR and CSH wrote the manuscript with help from MM. CSH provided supervision and CSH, PT and CJ provided funding for the study.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Source Data for Expanded View/Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank Paul Yu for LDN‐193189 and Bob Lechleider for the FLAG‐SMAD1 expression plasmid. We thank Alexandra Stanley for help with the MEFs from Acvr1 +/+ and Acvr1 +/ R206H mice, Michelle Digman and Enrico Gratton for advice concerning the Number and Brightness analysis, Gavin Kelly and Stuart Horswell for statistics advice, and Louise Richardson for the synopsis image. We are very grateful to the Francis Crick Institute Light Microscopy, particularly Rocco D’Antuono, to the Flow Cytometry Facility, and to the Genomics Equipment Park. We thank all the members of the Hill laboratory and Davide Coda for discussion and very useful comments on the manuscript. For the purpose of Open Access, the corresponding author has applied a CC BY public copyright license to any Author Accepted Manuscript version arising from this submission. This work was supported by the Francis Crick Institute which receives its core funding from Cancer Research UK (FC001095 and FC001185), the UK Medical Research Council (FC001095 and FC001185) and the Wellcome Trust (FC001095 and FC001185), and by Brain Research UK.
The EMBO Journal (2021) 40: e106317.
Contributor Information
Anassuya Ramachandran, Email: anassuya.ramachandran@auckland.ac.nz.
Caroline S Hill, Email: caroline.hill@crick.ac.uk.
Data availability
This study does not include data amenable for deposition in external repositories.
References
- Alessi Wolken DM, Idone V, Hatsell SJ, Yu PB, Economides AN (2018) The obligatory role of Activin A in the formation of heterotopic bone in Fibrodysplasia Ossificans Progressiva. Bone 109: 210–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aykul S, Corpina RA, Goebel EJ, Cunanan CJ, Dimitriou A, Kim H, Zhang Q, Rafique A, Leidich R, Wang X et al (2020) Activin A forms a non‐signaling complex with ACVR1 and type II Activin/BMP receptors via its finger 2 tip loop. eLife 9: e54582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagarova J, Vonner AJ, Armstrong KA, Borgermann J, Lai CS, Deng DY, Beppu H, Alfano I, Filippakopoulos P, Morrell NW et al (2013) Constitutively active ALK2 receptor mutants require type II receptor cooperation. Mol Cell Biol 33: 2413–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, Morrison A, Lewis P, Bouffet E, Bartels U et al (2014) Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet 46: 451–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho D, Taylor KR, Olaciregui NG, Molinari V, Clarke M, Mackay A, Ruddle R, Henley A, Valenti M, Hayes A et al (2019) ALK2 inhibitors display beneficial effects in preclinical models of ACVR1 mutant diffuse intrinsic pontine glioma. Commun Biol 2: 156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaikuad A, Alfano I, Kerr G, Sanvitale CE, Boergermann JH, Triffitt JT, von Delft F, Knapp S, Knaus P, Bullock AN (2012) Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva. J Biol Chem 287: 36990–36998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culbert AL, Chakkalakal SA, Theosmy EG, Brennan TA, Kaplan FS, Shore EM (2014) Alk2 regulates early chondrogenic fate in fibrodysplasia ossificans progressiva heterotopic endochondral ossification. Stem Cells 32: 1289–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuny GD, Yu PB, Laha JK, Xing X, Liu JF, Lai CS, Deng DY, Sachidanandan C, Bloch KD, Peterson RT (2008) Structure‐activity relationship study of bone morphogenetic protein (BMP) signaling inhibitors. Bioorg Med Chem Lett 18: 4388–4392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly AC, Randall RA, Hill CS (2008) Transforming growth factor β‐induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage‐independent growth. Mol Cell Biol 28: 6889–6902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digman MA, Dalal R, Horwitz AF, Gratton E (2008) Mapping the number of molecules and brightness in the laser scanning microscope. Biophys J 94: 2320–2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digman MA, Stakic M, Gratton E (2013) Raster image correlation spectroscopy and number and brightness analysis. Methods Enzymol 518: 121–144 [DOI] [PubMed] [Google Scholar]
- van Dinther M, Visser N, de Gorter DJ, Doorn J, Goumans MJ, de Boer J, ten Dijke P (2010) ALK2 R206H mutation linked to fibrodysplasia ossificans progressiva confers constitutive activity to the BMP type I receptor and sensitizes mesenchymal cells to BMP‐induced osteoblast differentiation and bone formation. J Bone Miner Res 25: 1208–1215 [DOI] [PubMed] [Google Scholar]
- Fontebasso AM, Papillon‐Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset P‐O, Bechet D, Faury D, De Jay N, Ramkissoon LA et al (2014) Recurrent somatic mutations in ACVR1 in pediatric midline high‐grade astrocytoma. Nat Genet 46: 462–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda T, Kohda M, Kanomata K, Nojima J, Nakamura A, Kamizono J, Noguchi Y, Iwakiri K, Kondo T, Kurose J et al (2009) Constitutively activated ALK2 and increased SMAD1/5 cooperatively induce bone morphogenetic protein signaling in fibrodysplasia ossificans progressiva. J Biol Chem 284: 7149–7156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya H, Ikezoe K, Wang L, Ohyagi Y, Motomura K, Fujii N, Kira J, Fukumaki Y (2008) A unique case of fibrodysplasia ossificans progressiva with an ACVR1 mutation, G356D, other than the common mutation (R206H). Am J Med Genet A 146A: 459–463 [DOI] [PubMed] [Google Scholar]
- Goebel EJ, Corpina RA, Hinck CS, Czepnik M, Castonguay R, Grenha R, Boisvert A, Miklossy G, Fullerton PT, Matzuk MM et al (2019) Structural characterization of an activin class ternary receptor complex reveals a third paradigm for receptor specificity. Proc Natl Acad Sci U S A 116: 15505–15513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P (2002) Balancing the activation state of the endothelium via two distinct TGF‐β type I receptors. EMBO J 21: 1743–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML (1999) The immunological synapse: a molecular machine controlling T cell activation. Science 285: 221–227 [DOI] [PubMed] [Google Scholar]
- Grönroos E, Kingston IJ, Ramachandran A, Randall RA, Vizán P, Hill CS (2012) Transforming growth factor β inhibits bone morphogenetic protein‐induced transcription through novel phosphorylated Smad1/5‐Smad3 complexes. Mol Cell Biol 32: 2904–2916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groppe JC, Wu J, Shore EM, Kaplan FS (2011) In vitro analyses of the dysregulated R206H ALK2 kinase‐FKBP12 interaction associated with heterotopic ossification in FOP. Cells Tissues Organs 194: 291–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han HJ, Jain P, Resnick AC (2018) Shared ACVR1 mutations in FOP and DIPG: opportunities and challenges in extending biological and clinical implications across rare diseases. Bone 109: 91–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatsell SJ, Idone V, Wolken DM, Huang L, Kim HJ, Wang L, Wen X, Nannuru KC, Jimenez J, Xie L et al (2015) ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci Transl Med 7: 303ra137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinck AP, Mueller TD, Springer TA (2016) Structural biology and evolution of the TGF‐β family. Cold Spring Harb Perspect Biol 8: a022103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hino K, Ikeya M, Horigome K, Matsumoto Y, Ebise H, Nishio M, Sekiguchi K, Shibata M, Nagata S, Matsuda S et al (2015) Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc Natl Acad Sci U S A 112: 15438–15443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, Laping NJ, Hill CS (2002) SB‐431542 is a potent and specific inhibitor of transforming growth factor‐β superfamily type I activin receptor‐like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol 62: 65–74 [DOI] [PubMed] [Google Scholar]
- Kaplan FS, Xu M, Seemann P, Connor JM, Glaser DL, Carroll L, Delai P, Fastnacht‐Urban E, Forman SJ, Gillessen‐Kaesbach G et al (2009) Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum Mutat 30: 379–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le VQ, Wharton KA (2012) Hyperactive BMP signaling induced by ALK2(R206H) requires type II receptor function in a Drosophila model for classic fibrodysplasia ossificans progressiva. Dev Dyn 241: 200–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrin F, Goumans MJ, Jonker L, Carvalho RL, Valdimarsdottir G, Thorikay M, Mummery C, Arthur HM, ten Dijke P (2004) Endoglin promotes endothelial cell proliferation and TGF‐β/ALK1 signal transduction. EMBO J 23: 4018–4028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechleider RJ, Ryan JL, Garrett L, Eng C, Deng C, Wynshaw‐Boris A, Roberts AB (2001) Targeted mutagenesis of Smad1 reveals an essential role in chorioallantoic fusion. Dev Biol 240: 157–167 [DOI] [PubMed] [Google Scholar]
- Lees‐Shepard JB, Yamamoto M, Biswas AA, Stoessel SJ, Nicholas SE, Cogswell CA, Devarakonda PM, Schneider MJ Jr, Cummins SM, Legendre NP et al (2018) Activin‐dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat Commun 9: 471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy L, Howell M, Das D, Harkin S, Episkopou V, Hill CS (2007) Arkadia activates Smad3/Smad4‐dependent transcription by triggering signal‐induced SnoN degradation. Mol Biol Cell 27: 6068–6083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu IM, Schilling SH, Knouse KA, Choy L, Derynck R, Wang XF (2009) TGFβ‐stimulated Smad1/5 phosphorylation requires the ALK5 L45 loop and mediates the pro‐migratory TGFβ switch. EMBO J 28: 88–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Tang M, Huang J, He B‐C, Gao J‐L, Chen L, Zuo G‐W, Zhang W, Luo Q, Shi Q et al (2010) TGFβ/BMP type I receptors ALK1 and ALK2 are essential for BMP9‐induced osteogenic signaling in mesenchymal stem cells. J Biol Chem 285: 29588–29598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machiya A, Tsukamoto S, Ohte S, Kuratani M, Fujimoto M, Kumagai K, Osawa K, Suda N, Bullock AN, Katagiri T (2018) Effects of FKBP12 and type II BMP receptors on signal transduction by ALK2 activating mutations associated with genetic disorders. Bone 111: 101–108 [DOI] [PubMed] [Google Scholar]
- Macías‐Silva M, Hoodless PA, Tang SJ, Buchwald M, Wrana JL (1998) Specific activation of Smad1 signaling pathways by the BMP7 type I receptor, ALK2. J Biol Chem 273: 25628–25636 [DOI] [PubMed] [Google Scholar]
- Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal‐Salom J, Taylor KR, Bjerke L, Clarke M, Vinci M, Nandhabalan M et al (2017) Integrated molecular meta‐analysis of 1,000 pediatric high‐grade and diffuse intrinsic pontine glioma. Cancer Cell 32: 520–537.e525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J (2012) TGFβ signalling in context. Nat Rev Mol Cell Biol 13: 616–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinen PJ, Ebner R, Lopez AR, Derynck R (1994) TGF‐β induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol 127: 2021–2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DSJ, Bloxham RD, Jiang M, Gori I, Saunders RE, Das D, Chakravarty P, Howell M, Hill CS (2018) The dynamics of TGF‐β signaling are dictated by receptor trafficking via the ESCRT machinery. Cell Rep 25: 1841–1855.e1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DSJ, Hill CS (2016) TGF‐β superfamily signalling. In Encyclopedia of Cell Biology, Bradshaw RA, Stahl PD (eds.), pp 37–50. Amsterdam: Elsevier Inc; [Google Scholar]
- Miller DSJ, Schmierer B, Hill CS (2019) TGF‐β family ligands exhibit distinct signalling dynamics that are driven by receptor localisation. J Cell Sci 132: jcs234039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucha BE, Hashiguchi M, Zinski J, Shore EM, Mullins MC (2018) Variant BMP receptor mutations causing fibrodysplasia ossificans progressiva (FOP) in humans show BMP ligand‐independent receptor activation in zebrafish. Bone 109: 225–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy P, Claus J, Jovin TM, Arndt‐Jovin DJ (2010) Distribution of resting and ligand‐bound ErbB1 and ErbB2 receptor tyrosine kinases in living cells using number and brightness analysis. Proc Natl Acad Sci U S A 107: 16524–16529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Takio K, Eto Y, Shibai H, Titani K, Sugino H (1990) Activin‐binding protein from rat ovary is follistatin. Science 247: 836–838 [DOI] [PubMed] [Google Scholar]
- Nicolas FJ, De Bosscher K, Schmierer B, Hill CS (2004) Analysis of Smad nucleocytoplasmic shuttling in living cells. J Cell Sci 117: 4113–4125 [DOI] [PubMed] [Google Scholar]
- Okadome T, Oeda E, Saitoh M, Ichijo H, Moses HL, Miyazono K, Kawabata M (1996) Characterization of the interaction of FKBP12 with the transforming growth factor‐β type I receptor in vivo . J Biol Chem 271: 21687–21690 [DOI] [PubMed] [Google Scholar]
- Olp MD, Kalous KS, Smith BC (2020) ICEKAT: an interactive online tool for calculating initial rates from continuous enzyme kinetic traces. BMC Bioinformatics 21: 186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen OE, Hella H, Elsaadi S, Jacobi C, Martinez‐Hackert E, Holien T (2020) Activins as dual specificity TGF‐β family molecules: SMAD‐activation via Activin‐ and BMP‐type 1 receptors. Biomolecules 10: 519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossato G, Digman MA, Aiken C, Lukacsovich T, Marsh JL, Gratton E (2010) A two‐step path to inclusion formation of huntingtin peptides revealed by number and brightness analysis. Biophys J 98: 3078–3085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran A, Vizán P, Das D, Chakravarty P, Vogt J, Rogers KW, Muller P, Hinck AP, Sapkota GP, Hill CS (2018) TGF‐β uses a novel mode of receptor activation to phosphorylate SMAD1/5 and induce epithelial‐to‐mesenchymal transition. eLife 7: e31756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper SI, Wasim L, Malinova D, Way M, Cox S, Tolar P (2019) B cells extract antigens at Arp2/3‐generated actin foci interspersed with linear filaments. eLife 8: e48093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross JA, Digman MA, Wang L, Gratton E, Albanesi JP, Jameson DM (2011) Oligomerization state of dynamin 2 in cell membranes using TIRF and number and brightness analysis. Biophys J 100: L15–L17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salaita K, Nair PM, Petit RS, Neve RM, Das D, Gray JW, Groves JT (2010) Restriction of receptor movement alters cellular response: physical force sensing by EphA2. Science 327: 1380–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger J (2000) Cell signaling by receptor tyrosine kinases. Cell 103: 211–225 [DOI] [PubMed] [Google Scholar]
- Shen Q, Little SC, Xu M, Haupt J, Ast C, Katagiri T, Mundlos S, Seemann P, Kaplan FS, Mullins MC et al (2009) The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP‐independent chondrogenesis and zebrafish embryo ventralization. J Clin Invest 119: 3462–3472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M et al (2006) A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 38: 525–527 [DOI] [PubMed] [Google Scholar]
- Song GA, Kim HJ, Woo KM, Baek JH, Kim GS, Choi JY, Ryoo HM (2010) Molecular consequences of the ACVR1(R206H) mutation of fibrodysplasia ossificans progressiva. J Biol Chem 285: 22542–22553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor KR, Mackay A, Truffaux N, Butterfield YS, Morozova O, Philippe C, Castel D, Grasso CS, Vinci M, Carvalho D et al (2014a) Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet 46: 457–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor KR, Vinci M, Bullock AN, Jones C (2014b) ACVR1 mutations in DIPG: lessons learned from FOP. Cancer Res 74: 4565–4570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolar P, Hanna J, Krueger PD, Pierce SK (2009) The constant region of the membrane immunoglobulin mediates B cell‐receptor clustering and signaling in response to membrane antigens. Immunity 30: 44–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trullo A, Corti V, Arza E, Caiolfa VR, Zamai M (2013) Application limits and data correction in number of molecules and brightness analysis. Microsc Res Tech 76: 1135–1146 [DOI] [PubMed] [Google Scholar]
- Vizán P, Miller DS, Gori I, Das D, Schmierer B, Hill CS (2013) Controlling long‐term signaling: receptor dynamics determine attenuation and refractory behavior of the TGF‐β pathway. Sci Signal 6: ra106 [DOI] [PubMed] [Google Scholar]
- Wang T, Donahoe PK, Zervos AS (1994) Specific interaction of type I receptors of the TGF‐β family with the immunophilin FKBP‐12. Science 265: 674–676 [DOI] [PubMed] [Google Scholar]
- Wrana JL, Attisano L, Wieser R, Ventura F, Massague J (1994) Mechanism of activation of the TGF‐β receptor. Nature 370: 341–347 [DOI] [PubMed] [Google Scholar]
- Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J et al (2014) The genomic landscape of diffuse intrinsic pontine glioma and pediatric non‐brainstem high‐grade glioma. Nat Genet 46: 444–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J (2005) Preparation, culture, and immortalization of mouse embryonic fibroblasts. Curr Protoc Mol Biol 70: 28.1.1–28.1.8 [DOI] [PubMed] [Google Scholar]
- Zamai M, Trullo A, Giordano M, Corti V, Arza Cuesta E, Francavilla C, Cavallaro U, Caiolfa VR (2019) Number and brightness analysis reveals that NCAM and FGF2 elicit different assembly and dynamics of FGFR1 in live cells. J Cell Sci 132: jcs220624 [DOI] [PubMed] [Google Scholar]
- Zanetti‐Domingues LC, Tynan CJ, Rolfe DJ, Clarke DT, Martin‐Fernandez M (2013) Hydrophobic fluorescent probes introduce artifacts into single molecule tracking experiments due to non‐specific binding. PLoS One 8: e74200 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Source Data for Expanded View/Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
This study does not include data amenable for deposition in external repositories.