

Abstract
Alternative splicing of pre‐mRNAs can regulate gene expression levels by coupling with nonsense‐mediated mRNA decay (NMD). In order to elucidate a repertoire of mRNAs regulated by alternative splicing coupled with NMD (AS‐NMD) in an organism, we performed long‐read RNA sequencing of poly(A)+ RNAs from an NMD‐deficient mutant strain of Caenorhabditis elegans, and obtained full‐length sequences for mRNA isoforms from 259 high‐confidence AS‐NMD genes. Among them are the S‐adenosyl‐L‐methionine (SAM) synthetase (sams) genes sams‐3 and sams‐4. SAM synthetase activity autoregulates sams gene expression through AS‐NMD in a negative feedback loop. We furthermore find that METT‐10, the orthologue of human U6 snRNA methyltransferase METTL16, is required for the splicing regulation in␣vivo, and specifically methylates the invariant AG dinucleotide at the distal 3′ splice site (3′SS) in␣vitro. Direct RNA sequencing coupled with machine learning confirms m6A modification of endogenous sams mRNAs. Overall, these results indicate that homeostasis of SAM synthetase in C. elegans is maintained by alternative splicing regulation through m6A modification at the 3′SS of the sams genes.
Keywords: Caenorhabditis elegans, machine learning, N 6‐methyladenosine, nanopore direct RNA sequencing, S‐adenosyl‐L‐methionine synthetase
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; RNA Biology
Full‐length sequences of natural NMD isoforms in C. elegans are elucidated by direct RNA sequencing. SAM synthetase homeostasis is regulated via pre‐mRNA splicing of the sams genes through m6A modification at the invariant AG of the distal 3′SS.
Introduction
Alternative splicing of precursor messenger RNAs (pre‐mRNAs) contributes not only to proteome diversity (Nilsen & Graveley, 2010; Ule & Blencowe, 2019) but also to regulation of gene expression levels by generating mRNA isoforms with a premature termination codon (PTC) (Hamid & Makeyev, 2014; Sibley, 2014). Such unproductively spliced mRNAs are unstable and almost undetectable due to an mRNA surveillance system termed nonsense‐mediated mRNA decay (NMD) (Lareau & Brenner, 2015; Kishor et␣al, 2019; Kurosaki et␣al, 2019). Genetic studies in yeast and nematode identified evolutionarily conserved NMD factors including UPF1, UPF2, and UPF3 (Pulak & Anderson, 1993; He & Jacobson, 1995). Searches for mRNAs stabilized under NMD‐deficient conditions in a variety of organisms identified many splicing variants as natural NMD substrates in addition to mRNAs from snoRNA host genes, pseudogenes, long noncoding genes, and viral genes (Chapin et␣al, 2014; Kawashima et␣al, 2014; Celik et␣al, 2017; Colombo et␣al, 2017; Muir et␣al, 2018). Among the genes whose expression is regulated through alternative splicing coupled with NMD (AS‐NMD), many splicing factors and regulators have been shown to negatively autoregulate their own expression at the level of pre‐mRNA splicing (Jumaa & Nielsen, 1997; Lejeune et␣al, 2001; Sureau et␣al, 2001; Lareau et␣al, 2007; Ni et␣al, 2007; McGlincy et␣al, 2010; Turunen et␣al, 2013; Jangi et␣al, 2014; Sun et␣al, 2017).
An advantage of utilizing C. elegans as a model organism for studying AS‐NMD is in that its NMD factors are not essential for the development or fertility, allowing for genetic studies and genome‐wide survey of natural NMD substrates in␣vivo. The C. elegans genome is rich in introns like those of higher organisms (Consortium CeS, 1998), and > 25% of its protein‐coding genes undergo alternative pre‐mRNA splicing (Ramani et␣al, 2009; Ramani et␣al, 2011; Tourasse et␣al, 2017). Early studies with splicing‐sensitive microarrays identified 30 genes regulated by AS‐NMD, including those for splicing regulators (Barberan‐Soler & Zahler, 2008; Barberan‐Soler et␣al, 2009). We previously reported RNA‐seq analysis of mRNAs in an NMD‐deficient mutant smg‐2 and demonstrated that eight of ribosomal protein genes undergo negative autoregulation through AS‐NMD (Takei et␣al, 2016). Recently, AS‐NMD has been demonstrated to play crucial roles in longevity of long‐lived mutants or by dietary restriction and RNA‐seq analyses identified alternative splicing events relevant to longevity (Son et␣al, 2017; Tabrez et␣al, 2017). A caveat of the RNA‐seq analyses, however, is that an entire sequence of each mRNA molecule cannot be accurately reconstructed from the short‐read RNA‐seq data.
Post‐transcriptional base modification of mRNAs has emerged as a new layer of regulatory mechanisms controlling gene expression (Frye et␣al, 2016). Among such modifications, N 6‐methyladenosine (m6A) is the most prevalent internal modification in mRNAs in higher eukaryotic species (Wei et␣al, 1975). The m6A modification is reversible; methyltransferases (“writers”) and demethylases (“erasers”) methylate and demethylate mRNAs, respectively (Duan et␣al, 2019; Shi et␣al, 2019; Zaccara et␣al, 2019; Huang et␣al, 2020a; Huang et␣al, 2020b). Almost all of m6A modifications in mRNAs are deposited by a multicomponent m6A methyltransferase complex composed of a core component, the methyltransferase‐like 3 (METTL3)/METTL14 heterodimer, and other regulatory factors (Liu et␣al, 2014; Ping et␣al, 2014; Schwartz et␣al, 2014). The m6A modifications may alter local RNA structures to switch interaction with RNA‐binding proteins (Liu et␣al, 2015) or may affect splicing, nuclear export, translation initiation, and stability of mRNAs through specific recognition by m6A‐binding proteins (“readers”) such as YTHDC1, YTHDF1, and YTHDF2 (Wang et␣al, 2015; Xiao et␣al, 2016; Roundtree et␣al, 2017). The m6A modifications in RNAs can be removed by erasers FTO and ALKBH5 (Jia et␣al, 2011; Zheng et␣al, 2013). In C. elegans, however, orthologous genes for the m6A writers, erasers, and readers mentioned above are absent from the genome (Cunningham et␣al, 2019; Arribere et␣al, 2020) and recent studies demonstrated that only a limited fraction of mRNAs would have m6A modification (van Delft et␣al, 2017; Liberman et␣al, 2020). It is therefore still unclear whether the m6A modification also plays regulatory roles in mRNA metabolism in C. elegans.
In this study, we performed high‐throughput long‐read sequencing of mRNAs to reveal their full‐length sequences in an NMD‐deficient mutant strain with a mutation in the smg‐2 gene, encoding the crucial NMD factor UPF1. We summarize features of genes subjected to AS‐NMD. To search for a novel regulatory mechanism of AS‐NMD other than autoregulation by RNA‐binding proteins, we focused on S‐adenosyl‐L‐methionine (SAM) synthetase genes and demonstrate that m6A modification at the invariant AG dinucleotide of a 3′ splice site (SS) by a methyltransferase METT‐10 switches the choice of competing 3′SSs, leading to AS‐NMD for homeostasis of the enzyme.
Results
Full‐length sequences of NMD isoforms in C. elegans
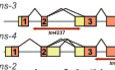
In order to reveal full‐length sequences of natural NMD substrates in C. elegans, we performed long‐read sequencing of poly(A)+ RNAs from the smg‐2 (yb979) mutant (Kuroyanagi et␣al, 2007) with a direct RNA sequencing protocol on a Nanopore MinION platform (Garalde et␣al, 2018). To minimize over representation of abundant mRNAs from housekeeping genes, we purified newly synthesized RNAs after 6‐h metabolic labeling with 4‐thiouracil (4TU) in synchronized L1 larvae and directly sequenced the nascent poly(A)+ RNAs (see Methods). After base‐calling, 1,402,229 reads were mapped to the C. elegans genome and we identified 12,517 mRNA isoforms from 8,028 genes (Dataset EV1) supported by at least 3 reads whose exon‐exon junctions were also supported by short‐read RNA‐seq data from smg‐2 mutants in previous studies (Kuroyanagi et␣al, 2013; Takei et␣al, 2016; Son et␣al, 2017). For comparison, we collected and pooled as many direct RNA sequencing data in a public database as possible for mRNAs from various stages of a wild‐type strain N2 (Roach et␣al, 2020). With the same procedure, 3,511,592 reads were mapped to the genome and 18,376 isoforms from 11,331 genes (Dataset EV1) were supported by short‐read RNA‐seq data from N2 in previous studies (Takei et␣al, 2016; Son et␣al, 2017). Figure 1A summarizes numbers of exon‐exon junctions identified in the smg‐2 mutant and/or N2 as well as those deduced from gene models in WormBase (WS271) in 6,642 genes commonly detected in the two strains. Even though the number of reads and the stage of the worms are limited in our analysis, we detected more than 700 novel junctions that were not detected in N2 or predicted in WormBase (Fig 1A), suggesting that mRNAs with unusual splicing patterns are stabilized in the smg‐2 mutant.
Figure 1. Long‐read sequencing reveals putative NMD isoforms enriched in the smg‐2 mutant.
-
AVenn diagram of exon‐exon junctions in 6,642 common genes identified among the Nanopore reads from the smg‐2 (yb979) mutant and wild‐type N2 as well as those deduced from gene models in WormBase (WS271).
-
BVolcano plot of 8,701 mRNA isoforms from 2,931 genes with multiple isoforms. X‐axis indicates a difference in percentage of each mRNA isoform within the gene (Δ% smg‐2 ‐N2). Y‐axis indicates ‐log10 of P‐value in Fisher's exact tests. Blue symbols indicate 219 mRNA isoforms significantly depleted from the smg‐2 mutant (P < 0.05, Δ% smg‐2 ‐N2< −10). Red symbols indicate 420 isoforms significantly enriched in the smg‐2 mutant (P < 0.05, Δ% smg‐2 ‐N2> 10).
-
C, DLength distribution of 3′UTRs (C) and proportions of putative PTC‐containing isoforms (D) in 10,136 mRNA isoforms detected in N2 and/or smg‐2 (All), the 219 isoforms depleted from and the 420 isoforms enriched in the smg‐2 mutant.
-
EGO terms significantly enriched in 259 genes with smg‐2‐enriched PTC isoforms (top) and in 30 genes with smg‐2‐depleted PTC isoforms (bottom). Modified Fisher's exact P‐values in the DAVID Bioinformatics system are indicated. Note that 14 of the 30 genes with the smg‐2‐depleted mRNA PTC isoforms are common with those having smg‐2‐enriched PTC isoforms. This is why the same GO terms are enriched in these two groups. The mRNAs from such genes likely contain upstream open reading frames (uORFs), which may not be directly involved in or differentially affect NMD.
In order to identify mRNA isoforms whose proportions to the total mRNAs within the genes are significantly different between the smg‐2 mutant and N2, we performed Fisher’s exact tests with read numbers in the Nanopore sequencing data for 2,931 genes with two or more isoforms and with at least 10 total reads in each strain (Dataset EV1). Here, we assumed that the proportions of the NMD isoforms within the genes remain constant throughout development and in nascent RNAs like those of ribosomal protein genes (Takei et␣al, 2016) and used the pooled data for N2. Figure 1B summarizes a difference in percentages of an mRNA isoform within the gene (Δ% smg‐2 ‐N2, percentage in smg‐2 subtracted by percentage in N2) and a P‐value for each of 8,701 isoforms. We identified 219 of smg‐2‐depleted isoforms (Δ% smg‐2 ‐N2 < −10, P < 0.05) from 208 genes and 420 of smg‐2‐enriched isoforms (Δ% smg‐2 ‐N2 > 10, P < 0.05) from 375 genes (Fig 1B and Dataset EV1). Assuming that an mRNA is translated from the first AUG codon, the smg‐2‐enriched isoforms (median = 613 nt) had significantly longer 3′ untranslated regions (3′UTRs) compared to the smg‐2‐depleted isoforms (median = 194 nt) as well as to total of 10,136 detected isoforms from 4,366 genes with at least 10 total reads in both strains (median = 218 nt) (P < 2e‐16, Wilcoxon rank sum test) (Fig 1C). Assuming that a premature termination codon (PTC) resides > 50 nucleotides (nt) upstream from the downstream‐most exon‐exon junction in an mRNA, 68.8% (289/420) of the smg‐2‐enriched mRNA isoforms had PTCs, whereas only 13.7% (30/219) of the smg‐2‐depleted isoforms and 28.1% (2,851/10,136) of all detected isoforms had PTCs (Fig 1D and Dataset EV1). We performed reverse transcription (RT)‐semi‐quantitative polymerase chain reaction (PCR) analysis of 12 genes with smg‐2‐enriched PTC isoforms and confirmed stabilization of all the PTC isoforms in the smg‐2 mutant except for extra isoforms with retained introns (Appendix␣Fig S1). These results indicate that 259 genes that produces the 289 PTC‐containing and smg‐2‐enriched mRNA isoforms are high‐confidence genes regulated by AS‐NMD in C. elegans.
Gene ontology (GO) analysis of the 259 genes that undergo AS‐NMD revealed enrichment of genes related to RNA translation and processing (Fig 1E). This is consistent with previous findings that many of RNA‐binding proteins (RBPs) including ribosomal proteins can negatively autoregulate their own expression by modulating alternative splicing patterns in both mammals and nematodes (Jangi & Sharp, 2014; Wani & Kuroyanagi, 2017).
Dynamic alternative splicing regulation of SAM synthetase genes upon feeding
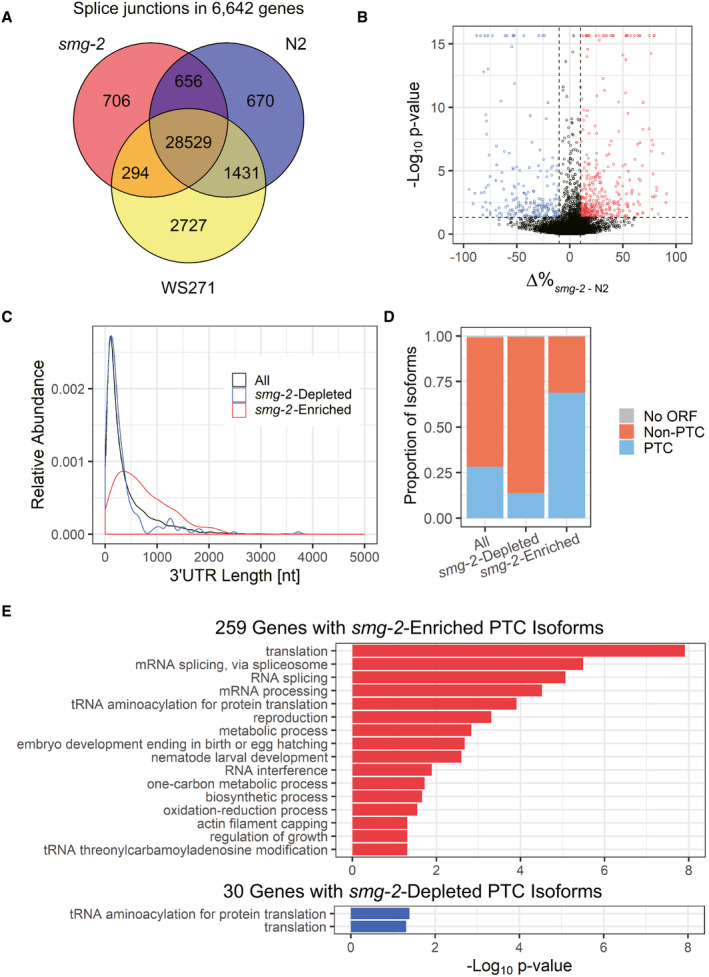
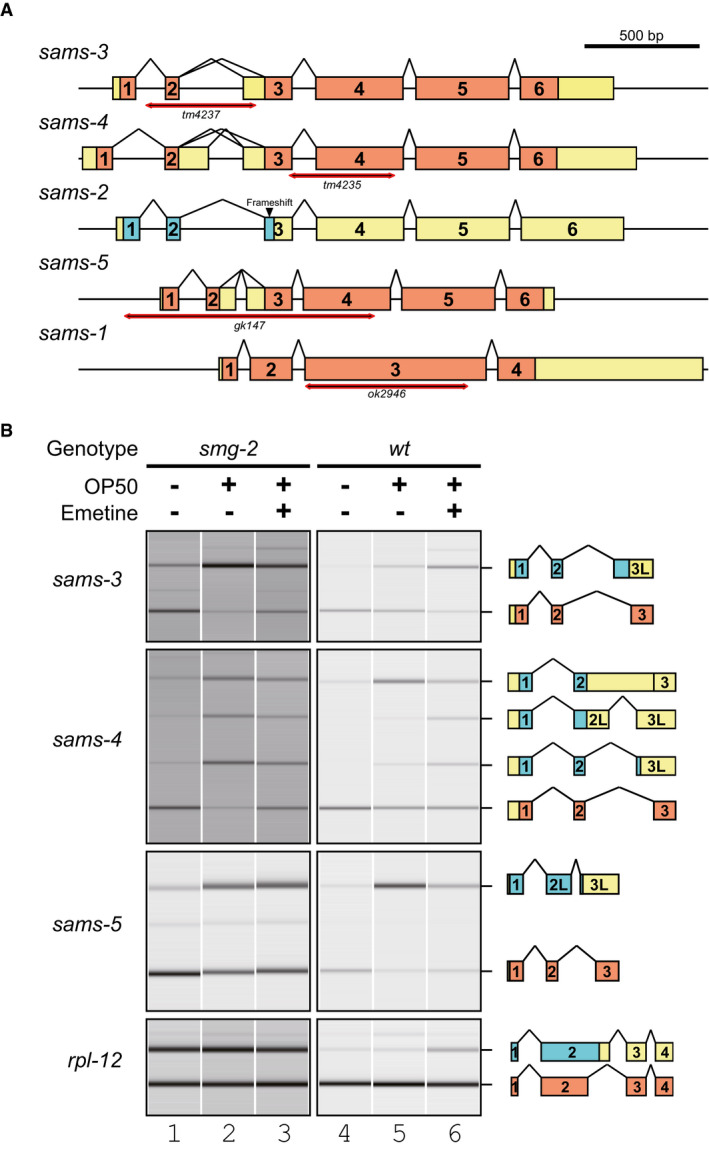
Genes related to metabolism are also significantly enriched among those that undergo AS‐NMD (Fig 1E). During the course of analysis of such genes, we found that alternative splicing of the sams‐3 and sams‐4 genes (Fig 2A) drastically changed upon feeding and fasting in the smg‐2 mutant (Fig 2B lanes 1–2 and Appendix␣Fig S2). RT‐quantitative PCR (qPCR) analysis revealed 5.6‐fold and 2.0‐fold increase in the total amounts of sams‐3 and sams‐4 mRNAs, respectively, upon feeding and confirmed divergent changes in the amounts the productive and PTC‐containing mRNA isoforms (Fig EV1A). We confirmed that the PTC‐containing sams‐3 and sams‐4 isoforms are also induced upon feeding and actually degraded by NMD in the wild‐type background by feeding L1 larvae of N2 in the absence and presence of a translation inhibitor emetine that eventually inhibits the translation‐dependent NMD process (Fig 2B lanes 4–6 and Fig EV1B). Alternative splicing of their paralogue sams‐5 was also dynamically regulated upon feeding and fasting (Figs 2 and EV1, Appendix␣Fig S2). We reasoned that expression of these genes is regulated by AS‐NMD under certain environmental conditions and therefore focused on these genes in the following sections.
Figure 2. Alternative splicing of sams‐3, sams‐4, and sams‐5 is dynamically regulated upon feeding.
-
ASchematic representation of the sams genes in C. elegans. Boxes indicate exons. Coding regions of productive mRNAs are in orange. A truncated coding region with a premature termination codon in a pseudogene sams‐2 is colored cyan. Double arrows illustrate regions deleted in mutant alleles indicated.
-
BAlternative splicing of sams‐3, sams‐4, and sams‐5 in unfed and fed worms without or with a protein synthesis inhibitor emetine. Synchronized L1 larvae of an NMD‐deficient strain KH1668: smg‐2 (yb979) (lanes 1‐3) and a wild‐type strain N2 (lanes 4‐6) were incubated in S‐complete medium alone (lanes 1, 4), with a standard E. coli strain OP50 (OD600 = 10.0) (lanes 2, 5) or OP50 supplemented with 10 mg/ml emetine (lanes 3, 6) for 3 h at 20°C. Total RNAs were extracted from whole animals and subjected to semi‐quantitative RT–PCR, whose products were analyzed by capillary electrophoresis. Representative gel‐like presentation is indicated (n = 3). Schematic structure of each PCR product is indicated on the right. Open reading frames (ORFs) for full‐length and truncated proteins are in orange and cyan, respectively. rpl‐12 was used as an unaffected control.
Figure EV1. RT–qPCR analysis of sams mRNA isoforms in fed and unfed animals.
-
A, BRelative amounts of sams‐3 (column 1), sams‐4 (column 2), and sams‐5 (column 3) mRNA isoforms as well as sams‐1, sams‐2, fat‐7, and acs‐2 mRNAs (column 4) compared to eef‐1A.1 (n = 3, mean ± standard error of mean) in the smg‐2 (yb979) mutant (A) and in N2 (B). The same RT products were analyzed in Fig 2B. None, S‐complete medium alone; OP50, OP50 in S‐complete (OD600 = 10.0); +Eme, OP50 in S‐complete supplemented with 10 mg/ml emetine. Sequences of the primers are available in Appendix␣Table S3.
The sams genes are members of a gene family encoding SAM synthetase that produces SAM from L‐methionine (L‐Met) and adenosine tri‐phosphate (ATP). The C. elegans genome contains five members of the SAM synthetase genes (Fig 2A) (Tamiya et␣al, 2013). sams‐3 and sams‐4 are highly homologous to each other (91.9% nucleotide sequence identity in the coding region), share exon organization (Fig 2A), and are divergent neighbors in the C. elegans genome. sams‐5 also shares the gene structure and the alternative splicing patterns (Fig 2). We noticed that nucleotide sequences of intron 2, the site of the alternative splicing regulation, in these genes are extraordinarily conserved for introns (87.5% identity between sams‐3 and sams‐4) (Fig EV2). In contrast, sams‐1 lacks an intron at this position (Figs 2A and EV2) and is therefore constitutively expressed. sams‐2 is the most homologous to sams‐3 (94.2% overall nucleotide sequence identity), yet is a pseudogene because of a frame‐shifting insertion in exon 3 (Fig EV2) and is constitutively spliced due to a substitution at the proximal 3′ splice site (SS) for intron 2 (Fig EV2).
Figure EV2. Nucleotide sequence alignment of genomic regions spanning from exon 2 through exon 3 of the sams genes in C. elegans .
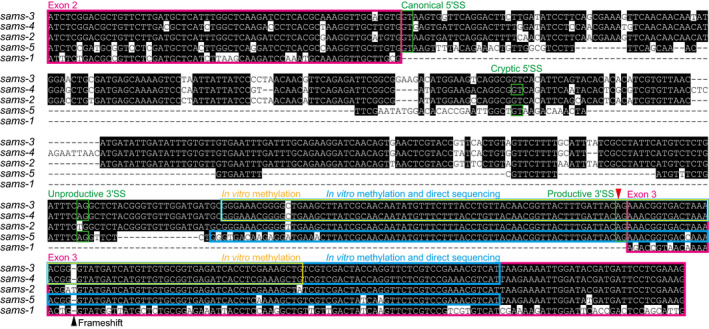
The sequences were aligned with Clustal V algorithm followed by manual adjustment. Conserved nucleotides are highlighted in black. Exons are boxed in magenta. Canonical and cryptic 5′SSs as well as proximal/unproductive and distal/productive 3′SSs are boxed in green. A yellow box indicates a sams‐3/sams‐4 RNA fragment used in in␣vitro methylation analysis with recombinant METT‐10 (Fig 5). Cyan boxes indicate sams‐3/sams‐4 and sams‐5 RNA fragments used in Nanopore direct sequencing after in␣vitro methylation (Fig 6). A red arrowhead indicates the m6A modification site. A black arrowhead indicates a frame‐shifting insertion in sams‐2. Note that sams‐1 lacks intron 2.
To ask whether the feeding‐induced change in the alternative splicing patterns of the sams genes requires newly synthesized proteins, we fed the smg‐2 worms in the presence of emetine. The ratios and the amounts of the mRNA isoforms changed to some extent even in the presence of emetine (Figs 2B and EV1). This is in good contrast to the amounts of fat‐7 and acs‐2 mRNAs whose induction and reduction, respectively, were completely suppressed by the emetine treatment (Fig EV1A). These results indicated that induction and dynamic splicing regulation of the sams genes do not depend on synthesis of new proteins upon feeding.
Negative feedback regulation of alternative splicing of the SAM synthetase genes
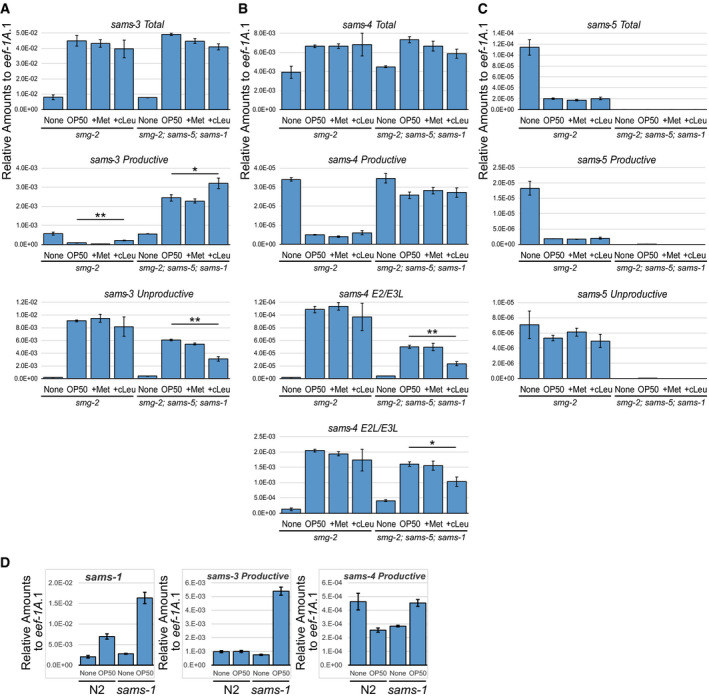
To address biological roles for the dynamic AS‐NMD of the sams genes in␣vivo, we tested whether or not SAM synthetase activity autoregulates SAMS expression. Because sams‐1 is the only sams gene that is constitutively expressed and because sams‐3 and sams‐4 are neighboring genes, we tested effects of deleting sams‐1 and sams‐5 on splicing patterns of sams‐3 and sams‐4 in the smg‐2 mutant background (Fig 3A). In unfed worms, the splicing patterns of sams‐3 and sams‐4 were not so much different between the smg‐2 (lane 1) and the smg‐2; sams‐5; sams‐1 (lane 5) mutants. Upon feeding, however, proportions of the enzyme‐coding isoforms were apparently higher in the smg‐2; sams‐5; sams‐1 mutant (lane 6) compared to the smg‐2 mutant (lane 2). RT–qPCR analysis revealed comparable induction of total sams‐3 (5.6‐fold in smg‐2 vs 6.4‐fold in smg‐2; sams‐5; sams‐1) and sams‐4 (1.7‐fold vs 1.6‐fold) mRNAs (Fig EV3, EV4, EV5), indicating that disruption of sams‐1 and sams‐5 did not affect transcriptional regulation of sams‐3 or sams‐4. Isoform‐specific qPCR analysis confirmed divergence of the feeding‐induced splicing change between the two strains (Fig EV3A and B). We also fed these worms in the presence of a substrate or a competitive inhibitor of SAM synthetase. The substrate L‐Met had little effect (lanes 3 and 7) while the inhibitor cycloleucine (Lombardini & Talalay, 1970) slightly but significantly increased the proportions (lanes 4 and 8) and the amounts (Fig EV3A) of the productive isoform of sams‐3 in either strains. These results indicated that SAM synthase activity regulates AS‐NMD of the sams‐3 and sams‐4 genes.
Figure 3. Expression of sams‐3, sams‐4, and sams‐5 is negatively regulated by SAM synthetase activity through alternative splicing.

-
AAlternative splicing of sams‐3, sams‐4, and sams‐5 in smg‐2 (yb979) and smg‐2 (yb979); sams‐5 (gk147); sams‐1 (ok2946) mutants. Synchronized L1 larvae of each strain were incubated in S‐complete medium alone (lanes 1 and 5), with OP50 (OD600 = 10.0) (lanes 2 and 6) and with OP50 supplemented with 25 mM L‐Met (lanes 3 and 7) or 25 mM cycloleucine (cLeu) (lanes 4 and 8) for 3 h at 20°C. The splicing patterns were analyzed and presented as in Fig 2B (n = 3).
-
B, CWestern blot analysis of SAMS‐1, SAMS‐3, and SAMS‐4 during larval development in the smg‐2 (yb979) (B) and wild‐type (C) backgrounds. Genotypes of the worms are smg‐2 (w) and smg‐2; sams‐5; sams‐1 (s) in (B) and wild‐type (w) and sams‐1 (s) in (C). Synchronized L1 larvae of each strain were incubated with OP50 at 20°C and subjected to Western blot analysis at indicated time points. Anti‐β‐tubulin (B) or Coomassie Brilliant Blue (CBB) staining (C) was used as a loading control. Specificity of the antibodies is confirmed in Appendix␣Fig S12. Note that upregulation of SAMS‐1 protein in the wild type during larval development in (C) is consistent with feeding‐induced upregulation of sams‐1 mRNA in Fig EV3D.
Figure EV3. RT–qPCR analysis of sams mRNA isoforms in smg‐2 and smg‐2; sams‐5; sams‐1 mutants fed in the presence of L‐Met or cycloleucine.
-
A–CRelative amounts of sams‐3 (A), sams‐4 (B), and sams‐5 (C) mRNA isoforms compared to eef‐1A.1 (n = 3, mean ± standard error of mean) in smg‐2 (yb979) and smg‐2 (yb979); sams‐5 (gk147); sams‐1 (ok2946) mutants. The same RT products were analyzed in Fig 3A. None, S‐complete medium alone; OP50, OP50 in S‐complete (OD600=10.0); +Met, OP50 in S‐complete supplemented with 25 mM L‐Met; +cLeu, OP50 in S‐complete supplemented with 25 mM cycloleucine. *P < 0.05; **P < 0.01 (Student's t‐test). Sequences of the primers are available in Appendix␣Table S3.
-
DRelative amounts of sams‐1 mRNA and sams‐3 and sams‐4 productive mRNA isoforms compared to eef‐1A.1 (n = 3, mean ± standard error of mean) in L1 larvae of N2 and the sams‐1 (ok2946) mutant incubated without (None) or with OP50 in S‐complete (OD600 = 10.0) for 3 h at 20°C.
Figure EV4. Expression of SAMS‐3 and SAMS‐4 is upregulated in a mett‐10 single mutant.
-
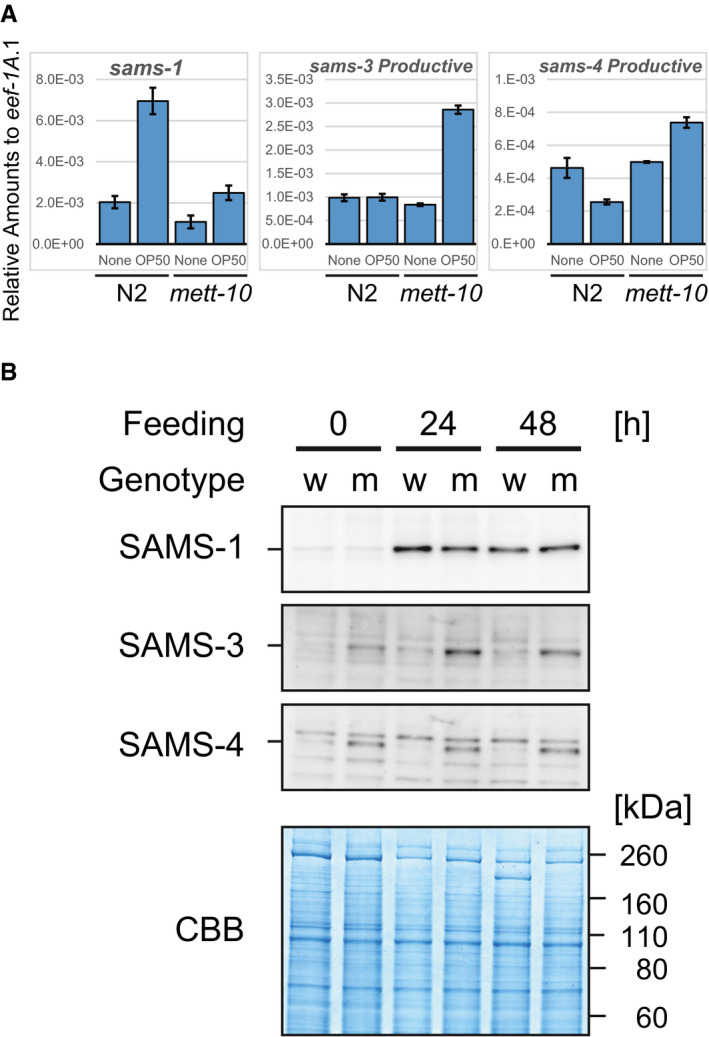
ARelative amounts of sams‐1, sams‐3, and sams‐4 productive mRNAs compared to eef‐1A.1 (n = 3, mean ± standard error of mean) in N2 and the mett‐10 (ok2204) mutant. Synchronized L1 larvae of each strain were incubated in S‐complete medium alone (None) or with OP50 (OD600 = 10.0) for 3 h at 20°C.
-
BWestern blot analysis of SAMS‐1, SAMS‐3, and SAMS‐4 during larval development of N2 (w) and the mett‐10 (ok2204) mutant (m). Synchronized L1 larvae of each strain were incubated with OP50 at 20°C and subjected to Western blot analysis at indicated time points. Coomassie Brilliant Blue (CBB) staining was used as a loading control. Representative images of three replicated experiments are shown. Note that upregulation of SAMS‐1 in both strains during larval development is consistent with feeding‐induced upregulation of sams‐1 mRNA shown in (A).
Figure EV5. Direct RNA sequencing with Nanopore technologies coupled with machine learning‐based classification reveals m6A modification of endogenous sams mRNAs.
-
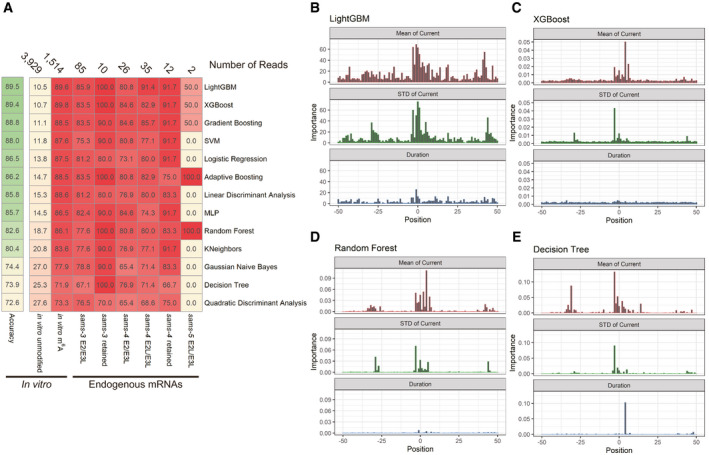
ASummary of machine learning‐based classification of sams mRNA reads in Nanopore direct RNA sequencing data with thirteen algorithms. Machine learning was conducted, and the results are shown as in Fig 6E. Numbers of reads used in the tests are indicated at the top; percentages of reads classified as “m6A‐modified” are shown with color code.
-
B–ERelative importance of the mean (top), standard deviation (middle), and duration time (bottom) of normalized Nanopore currents at nucleotide positions −50 through +50 relevant to the m6A site for LightGBM (B), XGBoost (C), Random Forest (D), and Decision Tree (E).
To test whether increase in the amounts of the productive mRNA isoforms leads to increase in the amounts of SAMS proteins, we performed Western blot analysis with antibodies specific to each of the four SAMS proteins. The amounts of SAMS‐3 and SAMS‐4 remain constant throughout larval development in the smg‐2 mutant, whereas they gradually increased in the smg‐2; sams‐5; sams‐1 mutant (Fig 3B). Consistent upregulation of SAMS‐3 and SAMS‐4 mRNAs and proteins in the sams‐1 mutant was observed in the wild‐type background (Figs 3C and EV3D). These results indicated that homeostasis of SAM synthetase activity in C. elegans is maintained by negative feedback through AS‐NMD of the sams genes.
mett‐10 regulates alternative splicing of the sams genes in␣vivo
Because SAM is the major donor of a methyl group for methyltransferases that methylate proteins, DNAs, RNAs, or lipids (Fontecave et␣al, 2004), we speculated that altered SAM synthetase activity affects alternative splicing of the sams genes through altered methylation levels of specific target molecule(s) for certain methyltransferase(s). We first analyzed histone methylation levels because some of histone marks are known to affect alternative splicing by recruiting splicing factors or by affecting the transcription elongation rate in mammals (Allo et␣al, 2009; Schor et␣al, 2009; Luco et␣al, 2010; Saint‐Andre et␣al, 2011). Chromatin immunoprecipitation coupled with high‐throughput sequencing (ChIP‐seq) analyses of histone H3 with dimethylation on lysine 4 (H3K4me2), trimethylation on lysine 27 (H3K27me3) or trimethylation on lysine 36 (H3K36me3), however, did not reveal a significant difference at the alternatively spliced exon in the sams‐3 or sams‐4 locus between the smg‐2 and smg‐2; sams‐5; sams‐1 mutants (Appendix␣Fig S3), precluding the scenario.
We next speculated methylation of the sams pre‐mRNAs themselves. Although the most abundant internal chemical modification in higher eukaryotic mRNAs is m6A (Wei et␣al, 1975), the C. elegans genome lacks orthologous genes for METTL3, METTL14, and other components of mammalian m6A methyltransferase complex that are responsible for the vast majority of the m6A modifications (Cunningham et␣al, 2019; Arribere et␣al, 2020). Recently, it has been shown that another human methyltransferase METTL16 specifically catalyzes m6A modification in a consensus sequence UACAGARAA within a loop of six hairpin structures on 3′UTR of human MAT2A mRNA that is translated into SAM synthetase (Pendleton et␣al, 2017; Shima et␣al, 2017; Doxtader et␣al, 2018). We noticed that each of sams‐3, sams‐4, and sams‐5 pre‐mRNAs potentially forms a hairpin structure with a loop sequence similar to the METTL16 consensus and that the potential m6A site is exactly at the invariant AG dinucleotide of the distal 3′SS that is specifically used for the productive mRNA isoform (Figs 4A and B and EV2). We therefore tested whether the METTL16 orthologue of C. elegans, METT‐10 (Appendix␣Fig S4), is involved in the splicing regulation of sams‐3, sams‐4, and sams‐5 in␣vivo (Fig 4C). The proportions of the NMD isoforms upon feeding drastically decreased in a smg‐2; mett‐10 double mutant (lane 4) compared to those in the smg‐2 single mutant (lane 2), indicating that mett‐10 is required for shifting the splice site choice from the distal/productive to the proximal/unproductive 3′SSs upon feeding. We also found that the productive isoforms of sams‐3 and sams‐4 mRNAs are upregulated in a mett‐10 single mutant (Fig EV4A) and SAMS‐3 and SAMS‐4 protein levels are higher in the mett‐10 mutant (Fig EV4B) compared to the wild‐type strain, confirming that mett‐10 is required for negative regulation of sams‐3 and sams‐4 in the wild‐type background.
Figure 4. mett‐10 is required for effective production of NMD isoforms from the sams genes in␣vivo .
-
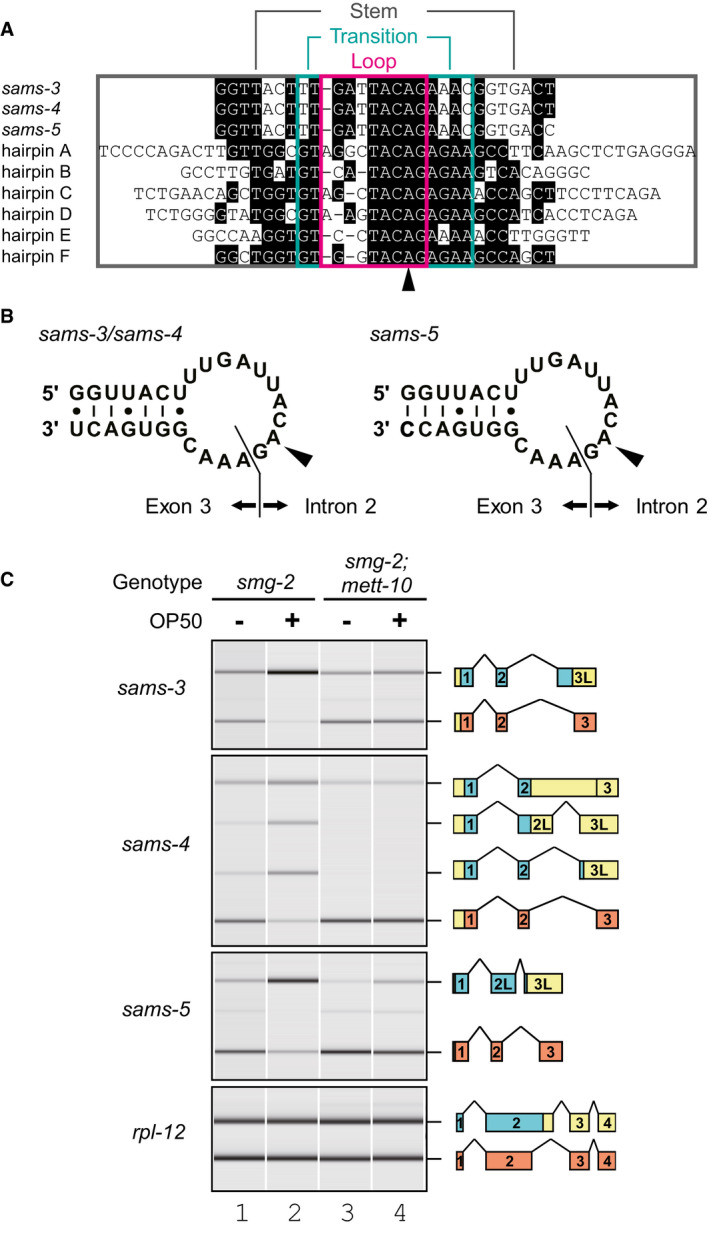
ANucleotide sequence alignment of putative stem‐loop structures in the C. elegans sams‐3, sams‐4, and sams‐5 genes together with six hairpin structures in 3′UTR of the human MAT2A gene. Conserved residues are shaded in black. Loop, transition, and stem regions of the MAT2A hairpins (Doxtader et␣al, 2018) are boxed in magenta, green, and gray, respectively. An arrowhead indicates adenine bases in the MAT2A hairpins that are specifically modified into m6A by METTL16 in␣vitro (Shima et␣al, 2017).
-
BSchematic representation of the predicted hairpin structures in sams‐3/sams‐4 (left) and sams‐5 (right) pre‐mRNAs. The boundaries between intron 2 and exon 3 are indicated. Arrowheads indicate the m6A modification sites.
-
CAlternative splicing of sams‐3, sams‐4, and sams‐5 in smg‐2 (yb979) and smg‐2 (yb979); mett‐10 (ok2204) mutants. Synchronized L1 larvae of each strain were incubated in S‐complete medium alone (lanes 1 and 3) or with OP50 (lanes 2 and 4) for 3 h at 20°C, and the splicing patterns were analyzed and presented as in Fig 2B.
METT‐10 specifically catalyzes the m6A modification at the distal 3′SS in␣vitro
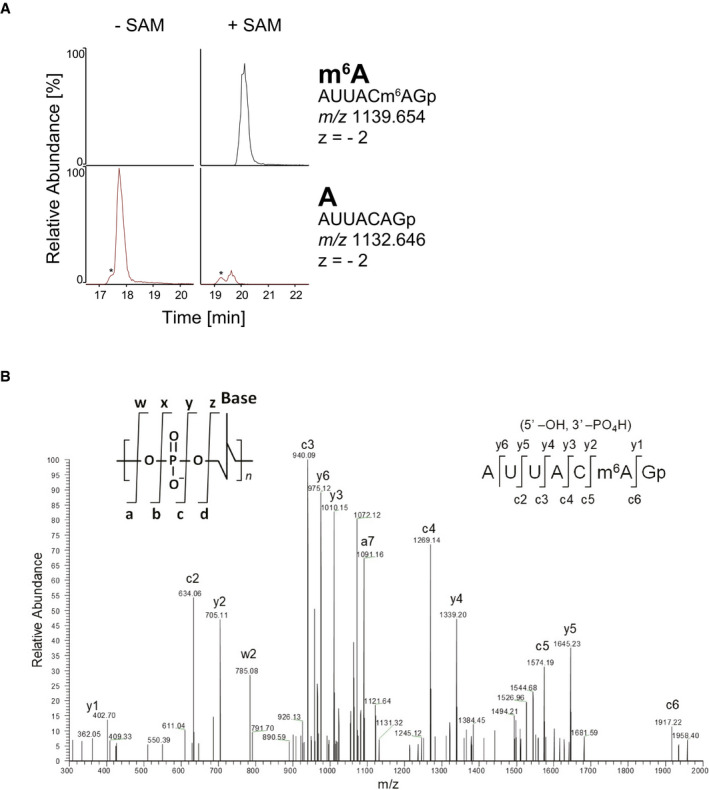
We next asked whether C. elegans METT‐10 can directly methylate the sams pre‐mRNAs in␣vitro. We prepared a 127‐nucleotide (nt) RNA spanning from intron 2 to exon 3 of the sams‐3/sams‐4 genes (Fig EV2) and performed in␣vitro methylation reactions with recombinant full‐length METT‐10 protein as well as methyltransferase domain of human METTL16. As expected, liquid chromatography (LC) coupled with tandem mass spectrometry (MS/MS) revealed that these recombinant proteins specifically and efficiently methylated the adenine base within the AG dinucleotide at the distal 3′SS in a SAM‐dependent manner (Fig 5A and B, Appendix␣Figs S5 and S6).
Figure 5. METT‐10 catalyzes m6A modification at the invariant AG dinucleotide of the distal 3′SS in␣vitro .
-
AMass chromatograms of RNA fragments to detect m6A (top) or unmodified A (bottom) after in␣vitro incubation of 127‐nt sams‐3/sams‐4 pre‐mRNA (sequence available in Fig EV2) with recombinant full‐length METT‐10 protein in the presence (left) or absence (right) of 1 mM SAM. The sequence, m/z value, and charge state for each fragment are indicated on the right. Asterisks indicate non‐specific signals.
-
BThe negatively charged ions of RNase T1 fragment is decomposed in the instrument by collision‐induced dissociation (CID) using helium gas. The product ions produced by CID are assigned on the sequence illustrated on the top right inset panel. Nomenclature of the product ions are described in the literature (McLuckey et␣al, 1992). Product ions of c and y series derive from the 5′ and 3′ termini of the fragment, respectively.
Endogenous sams mRNAs have the m6A modification at the distal 3′SS
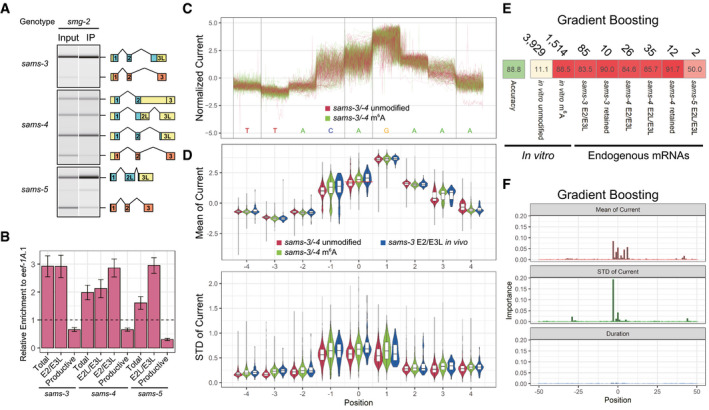
We finally investigated m6A modification of endogenous sams mRNAs. The potential m6A modification site is at the end of intron 2 and is excised from the productive mRNA, whereas it is retained in all the NMD isoforms. RNA immunoprecipitation with an antibody specific to m6A (m6A‐IP) followed by semi‐quantitative RT–PCR revealed depletion of the productive mRNA and enrichment of all the NMD isoforms for sams‐3, sams‐4, and sams‐5 (Fig 6A). RT–qPCR analysis confirmed specific enrichment of the NMD isoforms (Fig 6B), consistent with the idea that the NMD isoforms have the m6A modification in the NMD‐isoform‐specific regions.
Figure 6. Endogenous unproductive sams mRNAs have m6A modification at the AG dinucleotide of the distal 3'SS.
-
A, Bm6A‐IP specifically enriched sams mRNA isoforms that retain the AG dinucleotide at the distal/productive 3′SS. (A) Representative gel‐like images (n = 3) of semi‐quantitative RT–PCR analysis of sams‐3, sams‐4, and sams‐5 in input and immunoprecipitated (IP) RNAs from the smg‐2 (yb979) mutant. The splicing patterns were analyzed and presented as in Fig 2B. (B) Relative enrichment of total and each of mRNA isoforms compared to eef‐1A.1 mRNA by RT–qPCR. Error bars indicate standard error of mean (n = 3).
-
CExamples of normalized Nanopore currents for unmodified and m6A‐modified in‐vitro transcribed sams‐3/sams‐4 RNAs at nucleotide positions −4 through +4 relevant to the m6A site. One hundred reads are plotted for each. Color codes are indicated.
-
DDistribution of mean (top) and standard deviation (bottom) of normalized Nanopore currents at nucleotide positions −4 through +4 relevant to the m6A site for 5,000 of the unmodified and 2,680 of the m6A‐modified sams‐3/sams‐4 RNAs and 85 of an endogenous sams‐3 mRNA isoform E2/E3L. Color codes are indicated. Central bands represent the median, boxes represent the 25th and 75th percentiles, and whiskers represent the lowest and highest values. A red line at position 0 in the top panel indicates a cutoff line (mean current = 1.7557) that discriminates between the unmodified and m6A‐modified sams‐3/sams‐4 RNAs with accuracies of 64.58% for both.
-
EMachine learning‐based classification of sams RNA reads in Nanopore direct RNA sequencing data. The statistics in (D) as well as duration time of the Nanopore current at nucleotide positions −50 through +50 of up to 80% of mapped reads from the unmodified and methylated in␣vitro transcribed sams‐3/sams‐4 and sams‐5 RNAs were pooled and used for training with a machine learning algorithm Gradient Boosting and accuracy of the classifier was tested with 20% of the mapped reads. The classifier was also applied to reads identified as unproductive sams‐3, sams‐4, or sams‐5 mRNA isoforms in the analysis of endogenous mRNAs shown in Fig 1. Numbers of reads used in the tests are indicated at the top; percentages of reads classified as “m6A‐modified” are shown with color code.
-
FRelative importance of the statistics at each position for Gradient Boosting.
We further investigated m6A modification of the endogenous sams mRNAs by utilizing our direct RNA sequencing data. For comparison, we prepared barcoded, unmodified and m6A‐modified 161‐nt sams‐3/sams‐4 and 163‐nt sams‐5 pre‐mRNAs in␣vitro (see Methods for details; sequences are available in Fig EV2). Specific and efficient (> 97.5%) m6A modification at the AG dinucleotide of the distal 3′SS of the sams‐3/sams‐4 and sams‐5 RNAs methylated with the METTL16 methyltransferase domain was confirmed by LC‐MS/MS (Appendix␣Fig S7). These four RNAs were pooled and subjected to Nanopore direct RNA sequencing, which records ion current signals as an RNA strand passes through a nanopore (Garalde et␣al, 2018). The sams‐3/sams‐4 and sams‐5 RNAs were successfully distinguished by standard base‐calling and mapping. There was not a crucial difference, however, between unmodified and m6A‐modified RNAs in mean and standard deviation of the fluctuating current and duration time at each nucleotide position (Fig 6C and D, Appendix␣Fig S8); the unmodified and m6A‐modified sams‐3/sams‐4 RNAs were classified with only 64.58% of accuracy based only on the mean of the current at the m6A site (Fig 6D). We therefore conducted machine learning with a variety of algorithms to classify modification status of each of the sams RNA reads. Most of the thirteen algorithms tested successfully classified methylated test reads as “m6A‐modified” with 86–90% sensitivity and unmethylated test reads as “unmodified” with 11–19% false positives (Figs 6E and EV5A). As expected, the means of the current and the standard deviations at and near the m6A modification site contributed to the successful classification as demonstrated by some of the algorithms (Figs 6F and EV5B–E). With these classifiers, 73–100% of the endogenous sams‐3 or sams‐4 mRNA isoforms carrying the putative m6A modification site were classified as “m6A‐modified” (Figs 6E and EV5A). These results indicated that most, if not all, of the NMD isoforms of the endogenous sams mRNAs have the m6A modification at the distal/productive 3′SS.
Discussion
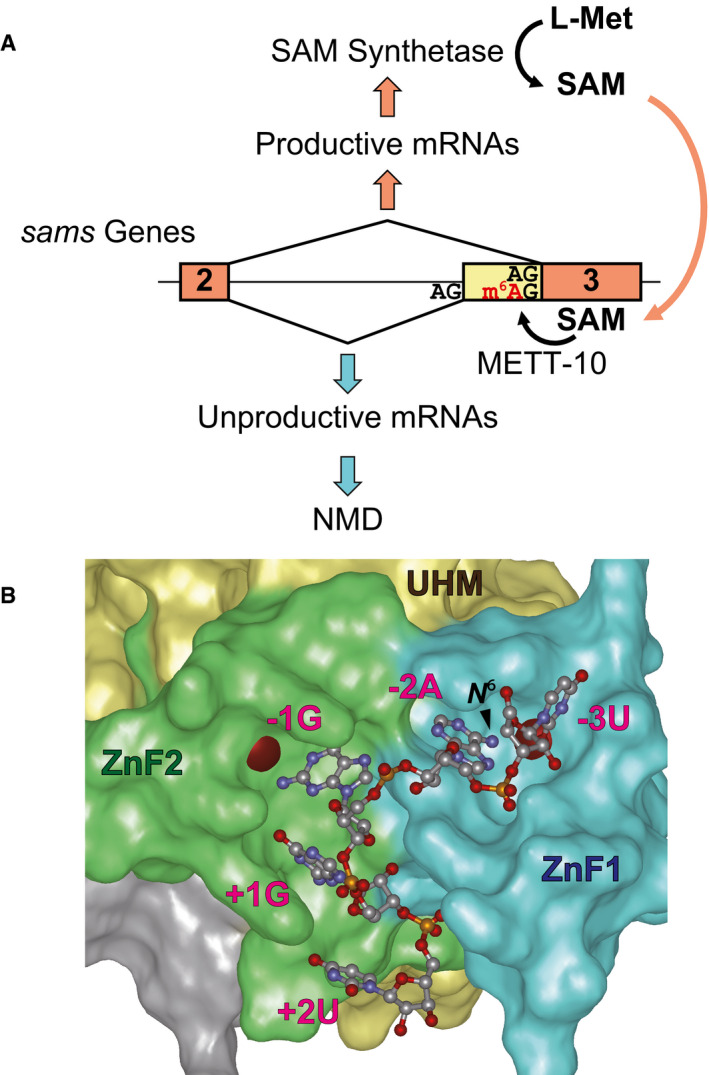
In the present study, we demonstrated that homeostasis of SAM synthetase in C. elegans is regulated at the pre‐mRNA splicing level of the sams‐3, sams‐4, and sams‐5 genes through m6A modification at the invariant AG dinucleotide of the distal 3′SS of intron 2 (Fig 7A). At low levels of SAM, the distal 3′SS remains unmodified and is preferentially used as a splice acceptor for the productive mRNA, which is translated into active SAM synthetase proteins that eventually increase the SAM level. At excess concentrations of SAM, the methyltransferase METT‐10 specifically catalyzes the m6A modification at the distal 3′SS, which leads to selection of the proximal 3′SS and production of the NMD isoforms.
Figure 7. A model for negative feedback regulation for homeostasis of SAM synthetase genes in C. elegans .
-
AA model for AS‐NMD regulation of the sams genes in C. elegans. See text for details.
-
BModeled structure of C. elegans UAF‐2 binding to 5′‐UAGGU‐3′. The structure was modeled based on sequence homology to U2AF23 from S. pombe (Appendix␣Fig S9) and crystal structure of S. pombe U2AF23/U2AF59 complex bound to the RNA (Yoshida et␣al, 2020). N‐terminal Zn finger 1 (ZnF1), U2AF homology motif (UHM), and C‐terminal Zn finger 2 (ZnF2) domains are colored in blue, yellow and green, respectively. Red spheres indicate zinc ions. The position of the amino group methylated upon m6A modification (N 6) is indicated with an arrowhead.
This is the first demonstration that the m6A modification at the 3′SS interferes with splicing in any organisms. The invariant AG dinucleotide at the 3′SS is recognized by a small subunit of an evolutionarily conserved heterodimeric protein complex U2 auxiliary factor (U2AF) (Merendino et␣al, 1999; Wu et␣al, 1999). In C. elegans, the consensus 3′SS sequence UUUUCAG is recognized by U2AF composed of the large subunit UAF‐1 and the small subunit UAF‐2 (Zorio & Blumenthal, 1999; Hollins et␣al, 2005). Crystal structure of the small subunit of U2AF binding to a 3′SS sequence has been solved only for the orthologue in fission yeast Schizosaccharomyces pombe, U2AF23, in complex with a part the large subunit U2AF59 (Yoshida et␣al, 2015; Yoshida et␣al, 2020). It has also been demonstrated that m6A modification of a 3′SS sequence 5′‐UUAGGU‐3′ at the position −2 (UUm6AGGU) dramatically decreased affinity to the U2AF23 complex in␣vitro (Yoshida et␣al, 2020). To ask whether m6A modification at the AG dinucleotide would also affect 3′SS recognition by C. elegans UAF‐2, we modeled three‐dimensional structure of UAF‐2 binding to 5′‐UAGGU‐3′ after its homology to S. pombe U2AF23 (Appendix␣Fig S9). The amino group of the adenine base at position −2 that is methylated upon m6A modification is embedded in a pocket on the surface of Zn finger domain 1 (Fig 7B) and is intimately surrounded by the identical residues as S. pombe U2AF23 forming the pocket (Yoshida et␣al, 2020), consistent with our finding that the m6A modification at the invariant AG dinucleotide interferes with its use as the 3′SS (Fig 7A) even in the absence of a reader protein in C. elegans. As for the function of the m6A modification in alternative splicing regulation of mammalian pre‐mRNAs, conflicting results have been reported (Xiao et␣al, 2016; Ke et␣al, 2017b; Louloupi et␣al, 2018; Zhou et␣al, 2019), yet there is not a reported case where the invariant AG dinucleotide at the 3′SS has the m6A modification.
This study demonstrated that homeostasis of SAM in C. elegans is maintained by an indirect negative feedback loop relying on METT‐10 that utilizes SAM to catalyze m6A modification for AS‐NMD of SAM synthetase pre‐mRNAs (Fig 7A). In human cells, the mRNA level of the SAM synthetase gene MAT2A is also indirectly regulated by the SAM level and dual roles have been reported for METTL16 that methylates MAT2A mRNA on six hairpin structures in the 3′UTR; prolonged occupancy of METTL16 on the upstream‐most hairpin due to lack of SAM promotes splicing of a retained intron to maturate the MAT2A mRNA (Pendleton et␣al, 2017), whereas m6A modification of the six hairpins by METTL16 leads to nuclear degradation of the mRNA via a nuclear reader protein YTHDC1 (Bresson et␣al, 2015; Shima et␣al, 2017). SAM is the major methyl‐donor reagent in all living organisms and is also used as a source of methylene groups, amino groups, ribosyl groups, and aminopropyl groups (Fontecave et␣al, 2004). Altered SAM levels affect a variety of biological processes including longevity (Hansen et␣al, 2005), lipid homeostasis (Li et␣al, 2011; Walker et␣al, 2011), and immune response (Ding et␣al, 2015) in C. elegans as well as longevity in Drosophila (Obata & Miura, 2015), maintenance and differentiation of human‐induced pluripotent stem cells (iPSCs) (Shiraki et␣al, 2014), and osteoclast differentiation in the mouse (Nishikawa et␣al, 2015). Autoregulation of SAM synthetase gene expression by alternative pre‐mRNA splicing in C. elegans demonstrated in this study emphasizes the importance of maintaining the SAM level in eukaryotes.
We demonstrated that METT‐10 as well as human METTL16 methyltransferase domain specifically methylated the adenine base at the AG dinucleotide of the distal 3′SS of sams‐3/sams‐4 and sams‐5 pre‐mRNAs in␣vitro. A recent study on crystal structures of the METTL16 catalytic domain bound to the hairpins of the MAT2A mRNA revealed structural basis for recognition of the target RNAs (Doxtader et␣al, 2018). Most of the amino acid residues directly involved in recognition of the RNAs are conserved in METT‐10 (Appendix␣Fig S4A). Each nucleotide in a UACAG stretch in the loop is directly recognized by METTL16 (Doxtader et␣al, 2018) and is perfectly conserved in the sams genes (Fig 4A). There is not a direct contact between METTL16 and the stem structures (Doxtader et␣al, 2018), consistent with less conserved sequences in the stem region (Fig 4A). The transition region between the loop and the stem is highly conserved among the human MAT2A hairpins (Fig 4A) and forms three unusual base pairs between GU and AGAA motifs (Doxtader et␣al, 2018). However, the sequence of the transition region is not well conserved in the sams genes (Fig 4A) and a key residue Arg200 that recognizes the transition region is not conserved in METT‐10 (Appendix␣Fig S4A). Nevertheless, METTL16 as well as METT‐10 can specifically target the AG dinucleotide of the sams pre‐mRNAs (Fig 5 and Appendix␣Figs S5–S7). This is consistent with an idea that the transition region of the stem‐loop is a key region to tune the methylation efficiency and not the target recognition based on previous findings that a G‐to‐A substitution in the latter motif or an R200Q substitution rather increased the in␣vitro methylation activity of METTL16 (Doxtader et␣al, 2018). Considering that regulation of sams gene expression is based on balanced competition between splicing and m6A modification (Fig 7A), it is reasonable to suggest that highly conserved nucleotide sequences flanking the distal as well as proximal 3′SSs of intron 2 (Appendix␣Fig S10) play critical roles in the genus Caenorhabditis. MAT2A and sams mRNAs are so far the only known mRNA targets for human METTL16 and C. elegans METT‐10, respectively. Although the m6A modification is very rare (0.0008%) in C. elegans mRNAs (Liberman et␣al, 2020), this study revealed the critical and specific role for the m6A modification in the mRNA metabolism. Quite recently, Mendel et␣al (2021) have reached essentially the same conclusion with distinct approaches.
We performed long‐read direct RNA sequencing of poly(A)+ RNAs from L1 larvae of the smg‐2 mutant of C. elegans and revealed full‐length sequences of 3,056 splice variants that are not registered as gene models in WormBase including 1,532 variants with novel junctions and 1,963 putative NMD isoforms with PTCs. This result indicates that the long‐read sequencing is a powerful tool to elucidate the full‐length sequences of mRNAs and hence the repertoire of the isoforms.
We demonstrated that a metabolite SAM can negatively regulate expression of the SAM‐synthesizing enzyme genes through AS‐NMD for homeostasis. It is common in bacteria that metabolites directly and specifically bind to mRNAs to regulate their transcription and/or translation for feedback regulation (Pavlova et␣al, 2019), including SAM (McDaniel et␣al, 2003; Winkler et␣al, 2003; Montange & Batey, 2006). This study identified many non‐RBP genes whose expression may be regulated through AS‐NMD in C. elegans (Fig 1E). mett‐10 is among the genes whose novel isoforms were discovered in this study and have a putative PTC. We found that the PTC‐containing isoform of mett‐10 is stabilized in the smg‐2 mutant (Appendix␣Fig S4B and C), confirming that mett‐10 is also regulated by AS‐NMD. Further studies on splicing regulation of such genes will elucidate further examples of gene expression control by environmental conditions at the pre‐mRNA splicing level.
We conducted machine learning to classify m6A modification status at a single specific position in individual reads for the sams mRNA isoforms from the direct Nanopore RNA sequencing data (Fig 6D) because the properties of the electric current were not so much different between unmodified and m6A‐modified RNAs prepared in␣vitro (Fig 6C). Sequencing with the Nanopore technology deconvolutes changes in the electric current as a single stranded RNA transverses a pore protein (Garalde et␣al, 2018). Since the measured current fluctuated even while the RNA strand stays in a given position, we considered not only the mean but also the standard deviation of the recorded electric current as well as the duration time at each position in the machine learning. Unbiased parameter tuning for higher accuracy revealed major contribution of the mean and the standard deviation of the electric current at the relevant positions (Figs 6E and EV5B–E), indicating that these features are useful for classifying the base modifications. The direct Nanopore RNA sequencing technology has recently been applied by other groups to detecting the m6A modifications. Liu et␣al (2019) reported that systematic errors and decreased base‐calling qualities can predict m6A RNA modifications. Parker et␣al (2020) also reported higher base‐calling error rates for an m6A‐modified RNA than an unmodified RNA and identified > 17,000 possible m6A modification sites with a motif DRAYH in mRNAs from a plant Arabidopsis thaliana by comparing the error rates for an m6A‐writer mutant line to those for a complemented line. The differential error rate and base quality, however, were not evident in our direct RNA sequencing data for in␣vitro‐modified sams‐3/sams‐4 or sams‐5 RNAs (Appendix␣Fig S11), suggesting that the differential error rate analysis approach may be context‐dependent. Lorenz et␣al developed a software MINES to detect m6A modifications in mammalian mRNAs de novo (Lorenz et␣al, 2020), yet it relies on rich data of verified m6A modification sites from cross‐linking and immunoprecipitation with high‐throughput sequencing (CLIP‐seq) and a well‐established motif DRACH for the METTL3/METTL14 complex (Linder et␣al, 2015; Ke et␣al, 2017b). Thus, classification of the base modification status at a given or new site in individual direct sequencing reads is still challenging and requires a high‐quality training data set or a large number of reads with a writer mutant control.
Materials and Methods
Worm strains
Genotypes of the strains used are summarized in Appendix␣Table S1. Some of the strains were obtained from Caenorhabditis Genetics Center (CGC) and National BioResource Project (NBRP), Japan. All worms were cultured at 20°C unless otherwise mentioned. KH2418: smg‐2 (yb979); sams‐1 (ok2946), KH2421: smg‐2 (yb979); sams‐5 (gk147); sams‐1 (ok2946); and VC2428: sams‐1 (ok2946) X were maintained in the presence of 20 mM choline chloride (Nacalai Tesque) (Walker et␣al, 2011).
Synchronized worm preparation
Gravid worms raised in S‐complete medium (Lewis & Fleming, 1995) supplemented with an E. coli strain OP50 were bleached with standard bleaching solution (Lewis & Fleming, 1995). Embryos were harvested and washed three times with M9 buffer. The embryos were incubated in M9 buffer for 18 h (except for the smg‐2; mett‐10 mutant that was incubated for 24 h) with gentle agitation for hatching and the L1 larvae were harvested and washed with S‐complete.
Direct RNA sequencing
Synchronized L1 larvae of KH1668: smg‐2 (yb979) were fed with OP50 in S‐complete medium supplemented with 4 mM 4‐thiouracil (4TU) (Tokyo Chemical Industry) for 6 h with gentle agitation. The worms were harvested and washed with M9, and total RNAs were extracted from the whole animals by using RNeasy Plus Mini (QIAGEN) as described previously (Kuroyanagi et␣al, 2010; Kuroyanagi et␣al, 2013). Thio‐labeled RNAs were purified essentially as described previously (Duffy et␣al, 2015) with minor modification; 4TU‐labeled RNAs were biotinylated with MTSEA biotin‐XX (Biotium) in 90% dimethylformamide at 10°C for 30 min and purified by using Streptavidin Mag Sepharose (GE Healthcare). Ribosomal RNAs were then removed with Ribo‐Zero Gold rRNA Removal Kit (Epidemiology) (Illumina), and the remaining RNAs were subjected to direct RNA sequencing. Nanopore libraries were prepared from 500 ng RNAs and sequenced using SQK‐RNA002 kit and FLO_MIN107 flow cells on MinION of Oxford Nanopore Technologies (ONT) according to manual instructions. Nanopore reads of Poly(A)+ RNA from the smg‐2 (yb979) mutant and in␣vitro transcribed sams RNAs were base‐called using Guppy version 2.3.4 and 3.0.3 (ONT), respectively, with default RNA parameters. Demultiplexing reads of the in␣vitro methylated RNAs and unmodified RNAs was performed by using fast5_subset (ONT) and in‐house R scripts by checking 5′‐GUCAUCCC‐3′ (unmodified) and 5′‐GUCAUGGG‐3′ (m6A) sequences within 40 nt region from the 3′‐end of the reads with 1 nt mismatch/indel allowance.
Transcript annotations from Nanopore data
Nanopore direct RNA sequencing reads for the smg‐2 (yb979) mutant and those for N2 (Roach et␣al, 2020) were mapped to ce11 reference genome in WormBase (https://parasite.wormbase.org) by using minimap2 version 2.17 (Li, 2018) with “‐x splice”. For the N2 data, reads of all developmental stages were pooled before mapping. Full‐length transcript annotations were generated from minimap2 bam outputs using spliced_bam2gff and cluster_gff in Pinfish version 0.1.0 (ONT) with settings “‐c 3 ‐d 2 ‐e 30”. The transcripts were collapsed using collapse_partials in Pinfish with “‐d 2 ‐e 30 ‐f 1000”. The generated annotations were labeled with gene names in WormBase (WS271) using bedtools intersectBed version 2.27.1 (Quinlan & Hall, 2010). To filter truncated transcripts, annotations with 5′‐ends not within +/−100 nt of annotated 5′‐ends in WormBase (WS271) were discarded. Furthermore, artificial transcripts with splice junctions caused by base‐calling error were filtered using supporting junction reads of Illumina RNA‐seq data and annotated junctions in WormBase (WS271). The supporting junctions were generated from published Illumina RNA‐seq data of N2 and the smg‐2 mutants with DDBJ accession numbers DRX002713 (Kuroyanagi et␣al, 2013), DRX021821 and DRX021822 (Takei et␣al, 2016), SRX2516749, SRX2516750, SRX2516753, and SRX2516754 (Son et␣al, 2017). The Illumina reads were mapped to ce11 reference genome using STAR version 2.5.3a (Dobin et␣al, 2013) with options “‐‐alignIntronMin 20 ‐‐alignIntronMax 10000 ‐‐alignMatesGapMax 10000”. The STAR mapping outputs include a␣list of splice junctions. Transcripts containing not‐supported, not‐annotated junctions were discarded by in‐house R and Python␣scripts.
Analysis of putative NMD isoforms using Nanopore data
Differential isoform analysis was performed on isoforms generated from pooled Nanopore data for N2 (Roach et␣al, 2020) and the smg‐2 (yb979) mutant by using Pinfish in the same way as described above. The read number of each isoform was extracted by using Pinfish and compared between N2 and the smg‐2 mutant by Fisher’s exact tests with BH adjustment (Benjamini & Hochberg, 1995) to calculate FDR. Open reading frames (ORFs) in the isoforms were predicted by using predORF in systemPipeR version 3.10 (Backman & Girke, 2016) and the upstream‐most ORFs were used. ORFs in isoforms deposited in WormBase (WS271) were re‐defined in the same way. A premature termination codon (PTC) was annotated when the stop codon locates > 50 nt upstream from the last exon‐exon junction. Gene ontology (GO) analysis was performed on smg‐2‐enriched PTC isoforms (Δ% smg‐2 ‐N2 > 10, P < 0.05), and smg‐2‐depleted PTC isoforms (Δ% smg‐2 ‐N2 < −10, P < 0.05) by using DAVID version 6.8 (Huang et␣al, 2009) with the genome background “Caenorhabditis elegans”.
Total RNA extraction and RT–PCR
Synchronized L1 larvae were incubated in 400 µl of S‐complete medium (Lewis & Fleming, 1995) in 1.5‐ml non‐stick tubes (Ambion) with continual agitation at 1,400 rpm on Thermomixer (Eppendorf). Worms were washed three times with M9 buffer, and total RNAs were extracted as described previously (Kuroyanagi et␣al, 2010; Kuroyanagi et␣al, 2013). RT–PCR was performed essentially as described previously (Kuroyanagi et␣al, 2010; Kuroyanagi et␣al, 2013). The PCR products were analyzed by using 2100 BioAnalyzer (Agilent Technologies). qPCR was performed with LightCycler 480 (Roche) and SYBR Premix DimerEraser (TaKaRa). Sequences of the primers used in the semi‐quantitative RT–PCR and RT–qPCR experiments are available in Appendix␣Tables S2 and S3, respectively. Sequences of the PCR products were confirmed by direct sequencing or by cloning and sequencing.
Western blotting
Synchronized L1 larvae were incubated in 400 µl of S‐complete medium (Lewis & Fleming, 1995) in 1.5‐ml non‐stick tubes (Ambion) with continual agitation at 1,400 rpm on Thermomixer (Eppendorf). Worm lysates were extracted from the synchronized worms, separated by neutral polyacrylamide gel electrophoresis (NuPAGE, Invitrogen), and transferred to nitrocellulose membrane (Protran NC, Amersham). Western blotting was performed with 1∶1,000‐diluted rabbit anti‐SAMS antisera and 1∶1,000‐diluted HRP‐conjugated anti‐rabbit IgG antibody (Pierce). Chemiluminescence signals (West Dura, Thermo or Western BLoT Ultra Sensitive HRP Substrate, Takara) were detected by using LAS4000 (GE Healthcare). The rabbit anti‐SAMS‐1, anti‐SAMS‐3, and anti‐SAMS‐4 antisera were raised with synthetic peptides KALKISPALLEKAKGNPI, DVELLKKIGGKTISNGN, and SADMLAKSQGPAQPDV, respectively, by Sigma‐Aldrich Japan.
ChIP‐seq analysis of histone methylations
Synchronized L1 larvae of the smg‐2 and smg‐2; sams‐5; sams‐1 mutants were fed with OP50 in S‐complete medium for 30 min. The worms were then washed with M9, frozen in liquid nitrogen, and ground to powder with mortar and pestle. The worm powder was treated with 1% formaldehyde at 20°C for 10 min, and cell nuclei were collected by centrifugation at 1,000 g. The chromatin was solubilized with Covaris M220 (Peak Incident Power = 75 W; Duty Factor = 20%; Cycles Per Burst = 200; time = 1,800 s). Nucleosomes with modified histones were immunoprecipitated with mouse monoclonal antibodies (MABI0303, MABI0323, or MABI0333, MBL, Nagoya, Japan) and DNAs were purified with NucleoSpin Gel and PCR Clean‐up (MACHEREY‐NAGEL) after proteinase K digestion of proteins at 55°C and reverse crosslinking at 65°C overnight. Sequencing libraries were prepared with TruSeq ChIP Library Prep Kit (Illumina), and sequencing (Single End 50 bp) was performed with Hiseq3000 (Illumina). Low‐quality reads were trimmed by using Trimmomatic version 0.36 (Bolger et␣al, 2014) with options “‐phred33 LEADING:30 TRAILING:30 MINLEN:50”. Mapping to the ce11 reference genome was done by using Bowtie2 version 2.4.1 (Langmead & Salzberg, 2012) with default settings. Differentially bound regions were detected by using csaw (Lun & Smyth, 2016). In the csaw analysis, the ce11 genome was binned into 200‐bp windows with a step size of 100 bp. Normalized read counts of each window were compared between the smg‐2 and smg‐2; sams‐5; sams‐1 mutants. Significantly changed regions were selected as those windows with P < 0.01. Genome browser snapshots were generated by using Integrative Genomics Viewer (Robinson et␣al, 2011).
In vitro methylation
Recombinant His‐tagged full‐length METT‐10 and human METTL16 catalytic domain were expressed in E. coli and purified with HisTrap and Capto SP cation exchange columns. A template for the 127‐nt sams‐3/sams‐4 RNA was amplified from a synthetic oligonucleotide 5′‐AACAATATGTTTCTTTACCTGTTACAACGGTTACTTTGATTACAGAAACGGTGACTAAAACGGGTATGAT‐3′ with 5′‐GCTAATACGACTCACTATAGGGAAACGGGGCTGAAGCTTATCGCAACAATATGTTTCTTTACCTGTTAC‐3′, and 5′‐CAGCTTTCGAGGTGATCTCACCGCACAACATGATCATACCCGTTTTAGTCACC‐3′. The template DNA was transcribed in␣vitro with recombinant T7 RNA polymerase. In vitro methylation of the sams‐3/sams‐4 RNA was performed with 0.2 µM recombinant protein and 2 µM RNA in methylation buffer (20 mM HEPES‐KOH pH 7.5, 150 mM NaCl, and 1 m DTT) with or without 1 mM SAM at 37°C for 3 h in a 50 μl reaction mixture.
Mass spectrometry
Identification of modified bases was performed essentially as described previously (Shima et␣al, 2017). Collision‐induced dissociation (CID) spectrum analysis was performed as described previously (Shima et␣al, 2017).
Preparation of in‐vitro methylated RNAs for direct RNA sequencing
sams‐3/sams‐4 and sams‐5 genomic DNA fragments were amplified from N2 genomic DNA by using PrimeStar GXL (Takara). The sams‐3/sams‐4 and sams‐5 DNA fragments were further amplified with a forward primer 5′‐TAATACGACTCACT ATAGGGAAACGGGGCTGA AGC‐3′ (sams‐3/sams‐4) or 5′‐TAATACGACTCACTATAGGGTGACAAGA GGAT GAAAC‐3′ (sams‐5) and a barcoded reverse primer 5′‐TTTTTTTTTTTTTTTTTTTTGGGGGGGGGATGACGTTTCGGACGAG AAC‐3′ (C9A20) or 5′‐TTTTTTTTTTTTTTTTT TTTCCCCCCCCCATGACGTTTCGGACGAGAAC‐3′ (G9A20) to attach a T7 promoter and a C9A20 or G9A20 tail. The four DNA fragments were utilized as templates for in␣vitro transcription with T7 RNA polymerase, and the RNAs were purified with PAGE and gel extraction. The G9A20‐tagged sams‐3/sams‐4 and sams‐5 RNAs were subjected to methylation reaction with 30 µM of recombinant human METTL16 catalytic domain protein in the methylation buffer described above with 1 mM SAM at 37°C for 3 h in a 50 μl reaction mixture and were purified again with PAGE and gel extraction. The four RNA samples were then pooled and subjected to direct RNA sequencing.
m6A RNA immunoprecipitation
Total RNAs were extracted from synchronized L1 larvae of the smg‐2 mutant after feeding with OP50 in S‐complete medium for 3 h. Immunoprecipitation of m6A RNA was performed by using Rabbit anti‐m6A antibody (ab151230, Abcam) and Dynabeads Protein A (Invitrogen). Antibody‐bound magnetic beads were prepared by following manual instructions and washed with Binding buffer (10 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.5% NP‐40) three times. Total RNAs in 30 μl of TE buffer were denatured for 5 min at 65°C, and then, the RNAs were added to the antibody‐bound beads in 270 μl of Binding buffer and incubated on a mixer for 2 h at 4°C (40 μg of total RNAs and 3 μg of anti‐m6A antibody per sample). After incubation, the beads were washed with Binding buffer twice and Wash buffer (10 mM Tris–HCl pH 7.5, 1 M NaCl, 1% NP‐40) three times. Washed beads were suspended in 50 μl of RLT buffer (QIAGEN) and incubated on a mixer for 5 min at r.t. to elute the beads‐bound RNAs. Finally, the eluted RNAs were purified by using RNA Clean & Concentrator‐5 kit (Zymoresearch) and were subjected to semi‐quantitative RT–PCR and RT–qPCR analysis.
Classification of m6A‐modified sams RNAs
Raw Nanopore current data for in␣vitro prepared RNAs and the endogenous mRNAs from the smg‐2 mutant were mapped to sams transcript annotations generated by Pinfish using Tombo version 1.5 (ONT). Mean, standard deviation (STD), and duration time of the normalized current at each genomic position were extracted by using Tombo Python API. For machine learning, the data of the normalized current at positions from −50 through +50 relevant to the m6A site were used. Among 15,706 reads for the unmodified RNAs and 6,063 reads for the m6A‐modified RNAs, up to 80% were used to train classifiers, and 20% were used for tests. The reads for the unmodified RNAs in the training data set were downsampled to the same number as those of the m6A‐modified RNAs in order to prevent biased learning. To select the best performing models, we compared classifiers xgboost version 1.0.0 (https://github.com/dmlc/xgboost) (Chen & Guestrin, 2016) and LightGBM version 2.3.2 (https://github.com/microsoft/LightGBM) (Ke et␣al, 2017a) and those in scikit‐learn version 0.22 (https://github.com/scikit‐learn/scikit‐learn) (Pedregosa et␣al, 2011). Scikit‐learn includes a variety of algorithms including Decision Tree, Random Forest, Logistic Regression, K‐Nearest Neighbor, SVM, Linear Discriminant Analysis, Quadratic Discriminant Analysis, Multilayer Perceptron, Gaussian Naive Bayes, and Adaptive Boosting. Each classifier was tuned by grid‐search of hyperparameters by using hyperopt version 0.2.2 (https://github.com/hyperopt/hyperopt) (Bergstra et␣al, 2013).
Protein sequence analysis
Multiple sequence alignment with the Clustal V or W algorithm was performed by using a module MegAlign in Lasergene 15 (DNASTAR). Modeling of the three‐dimensional structure of UAF‐2 was performed by using Modeller 9.24 (Webb & Sali, 2016) after the structure of S. pombe U2AF23 in complex with U2AF59 and 5′‐UAGGU‐3′ (PDB Acc. No. 7C06). The 3D structures were analyzed and visualized with a module Protean 3D in Lasergene 17 (DNASTAR).
Author contributions
EW contributed to sequencing library preparation, bioinformatics analysis, RNA‐IP, and Western blotting. MT‐O and HK contributed to RT–PCR and RT–qPCR analysis. YI and TS contributed to in␣vitro methylation, and LC‐MS/MS. SW, MK‐A, SH, and ST contributed to preliminary data acquisition. KH and AF contributed to antibody preparation and validation. YS contributed to RNA and ChIP sequencing. HK organized the study and prepared worm strains. EW and HK wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Review Process File
Acknowledgements
We thank Monima Alam, Hiroshi Kurokawa, and Yohei Watanabe for their technical assistance in worm culture. We thank Terumi Horiuchi, Kotomi Imamura, and Kazumi Abe for their technical assistance in RNA sequencing. We thank Yutaka Muto of Musashino University and Eiji Obayashi of Shimane University for providing the atomic coordinates data of S. pombe U2AF23 bound to RNA. Some C. elegans strains were provided by the Caenorhabditis Genetics Center (CGC), which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). Some C. elegans strains were provided by National Bioresource Project for the Experimental Animal “Nematode C. elegans”, which is funded by MEXT, Japan. Work in the Kuroyanagi laboratory was supported by JSPS/MEXT KAKENHI Grant Numbers JP20H04839, JP20H03181, JP17H03633, JP17H05596, JP15KK0252, JP15H01350, JP26291003, JP15H01467, JP25118506, JP24657116, and JP20112004, by JST PRESTO Grant Number 1D102 and by Nanken‐Kyoten, TMDU. Work in the T. Suzuki laboratory was supported by JSPS/MEXT KAKENHI Grant Numbers JP26113003 and JP18H05272 and by JST ERATO Grant Number JPMJER2002. Work in the Y. Suzuki laboratory was supported JSPS KAKENHI Grant Number JP16H06279 (PAGS). Work in the Fukamizu laboratory was supported by JSPS/MEXT KAKENHI Grant Numbers JP23116004, JP17H01519, and JP17K07746.
The EMBO Journal (2021) 40: e106434.
Data availability
RNA‐Seq data: DDBJ Sequence Read Archive (DRA) DRX184564 (https://ddbj.nig.ac.jp/DRASearch/experiment?acc=DRX184564).
RNA‐Seq data: DDBJ Sequence Read Archive (DRA) DRX184565 (https://ddbj.nig.ac.jp/DRASearch/experiment?acc=DRX184565).
Chip‐Seq data: DDBJ Sequence Read Archive (DRA) DRA009050 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA009050).
Codes used in the sequence analysis: GitHub KUROYANAGI‐Lab/WATABE‐SAMS (https://github.com/KUROYANAGI‐Lab/WATABE‐SAMS).
Nucleotide sequence: DDBJ/ENA/GenBank LC603057 (http://getentry.ddbj.nig.ac.jp/getentry/na/LC603057?filetype=html).
The materials generated and/or analyzed during the current study are available from KUROYANAGI Hidehito (hidehito@med.u-ryukyu.ac.jp).
References
- Alló M, Buggiano V, Fededa JP, Petrillo E, Schor I, de la Mata M, Agirre E, Plass M, Eyras E, Elela SA et␣al (2009) Control of alternative splicing through siRNA‐mediated transcriptional gene silencing. Nat Struct Mol Biol 16: 717–724 [DOI] [PubMed] [Google Scholar]
- Arribere JA, Kuroyanagi H, Hundley HA (2020) mRNA editing, processing and quality control in Caenorhabditis elegans . Genetics 215: 531–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backman TWH, Girke T (2016) systemPipeR: NGS workflow and report generation environment. BMC Bioinformatics 17: 388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberan‐Soler S, Lambert NJ, Zahler AM (2009) Global analysis of alternative splicing uncovers developmental regulation of nonsense‐mediated decay in C. elegans . RNA 15: 1652–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberan‐Soler S, Zahler AM (2008) Alternative splicing regulation during C. elegans development: splicing factors as regulated targets. PLoS Genet 4: e1000001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc: Ser B 57: 289–300 [Google Scholar]
- Bergstra J, Yamins D, Cox DD (2013) Making a science of model search: hyperparameter optimization in hundreds of dimensions for vision architectures. Proceedings of the 30th International Conference on International Conference on Machine Learning ‐ Volume 28; Atlanta, GA, USA. JMLR.org.
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresson SM, Hunter OV, Hunter AC, Conrad NK (2015) Canonical poly(A) polymerase activity promotes the decay of a wide variety of mammalian nuclear RNAs. PLoS Genet 11: e1005610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celik A, Baker R, He F, Jacobson A (2017) High‐resolution profiling of NMD targets in yeast reveals translational fidelity as a basis for substrate selection. RNA 23: 735–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapin A, Hu H, Rynearson SG, Hollien J, Yandell M, Metzstein MM (2014) In vivo determination of direct targets of the nonsense‐mediated decay pathway in Drosophila . G3 4: 485–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Guestrin C (2016) XGBoost: A Scalable Tree Boosting System. Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining; San Francisco, California, USA. Association for Computing Machinery.
- Colombo M, Karousis ED, Bourquin J, Bruggmann R, Muhlemann O (2017) Transcriptome‐wide identification of NMD‐targeted human mRNAs reveals extensive redundancy between SMG6‐ and SMG7‐mediated degradation pathways. RNA 23: 189–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium CeS (1998) Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282: 2012–2018. [DOI] [PubMed] [Google Scholar]
- Cunningham F, Achuthan P, Akanni W, Allen J, Amode M, Armean IM, Bennett R, Bhai J, Billis K, Boddu S et␣al (2019) Ensembl 2019. Nucleic Acids Res 47: D745–D751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Delft P, Akay A, Huber SM, Bueschl C, Rudolph KLM, Di Domenico T, Schuhmacher R, Miska EA, Balasubramanian S (2017) The profile and dynamics of RNA modifications in animals. ChemBioChem 18: 979–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding W, Smulan LJ, Hou NS, Taubert S, Watts JL, Walker AK (2015) s‐Adenosylmethionine levels govern innate immunity through distinct methylation‐dependent pathways. Cell Metab 22: 633–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doxtader KA, Wang P, Scarborough AM, Seo D, Conrad NK, Nam Y (2018) Structural basis for regulation of METTL16, an S‐adenosylmethionine homeostasis factor. Mol Cell 71: 1001–1011.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan HC, Wang Y, Jia G (2019) Dynamic and reversible RNA N 6‐methyladenosine methylation. Wiley Interdiscip Rev RNA 10: e1507 [DOI] [PubMed] [Google Scholar]
- Duffy EE, Rutenberg‐Schoenberg M, Stark CD, Kitchen RR, Gerstein MB, Simon MD (2015) Tracking distinct RNA populations using efficient and reversible covalent chemistry. Mol Cell 59: 858–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontecave M, Atta M, Mulliez E (2004) S‐adenosylmethionine: nothing goes to waste. Trends Biochem Sci 29: 243–249 [DOI] [PubMed] [Google Scholar]
- Frye M, Jaffrey SR, Pan T, Rechavi G, Suzuki T (2016) RNA modifications: what have we learned and where are we headed? Nat Rev Genet 17: 365–372 [DOI] [PubMed] [Google Scholar]
- Garalde DR, Snell EA, Jachimowicz D, Sipos B, Lloyd JH, Bruce M, Pantic N, Admassu T, James P, Warland A et␣al (2018) Highly parallel direct RNA sequencing on an array of nanopores. Nat Methods 15: 201–206 [DOI] [PubMed] [Google Scholar]
- Hamid FM, Makeyev EV (2014) Emerging functions of alternative splicing coupled with nonsense‐mediated decay. Biochem Soc Trans 42: 1168–1173 [DOI] [PubMed] [Google Scholar]
- Hansen M, Hsu AL, Dillin A, Kenyon C (2005) New genes tied to endocrine, metabolic, and dietary regulation of lifespan from a Caenorhabditis elegans genomic RNAi screen. PLoS Genet 1: 119–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Jacobson A (1995) Identification of a novel component of the nonsense‐mediated mRNA decay pathway by use of an interacting protein screen. Genes Dev 9: 437–454 [DOI] [PubMed] [Google Scholar]
- Hollins C, Zorio DA, MacMorris M, Blumenthal T (2005) U2AF binding selects for the high conservation of the C. elegans 3' splice site. RNA 11: 248–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Huang H, Weng H, Chen J (2020a) The biogenesis and precise control of RNA m6A methylation. Trends Genet 36: 44–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Weng H, Chen J (2020b) m6A modification in coding and non‐coding RNAs: roles and therapeutic implications in cancer. Cancer Cell 37: 270–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jangi M, Boutz PL, Paul P, Sharp PA (2014) Rbfox2 controls autoregulation in RNA‐binding protein networks. Genes Dev 28: 637–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jangi M, Sharp PA (2014) Building robust transcriptomes with master splicing factors. Cell 159: 487–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Fu YE, Zhao Xu, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang Y‐G et␣al (2011) N6‐methyladenosine in nuclear RNA is a major substrate of the obesity‐associated FTO. Nat Chem Biol 7: 885–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumaa H, Nielsen PJ (1997) The splicing factor SRp20 modifies splicing of its own mRNA and ASF/SF2 antagonizes this regulation. EMBO J 16: 5077–5085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima T, Douglass S, Gabunilas J, Pellegrini M, Chanfreau GF (2014) Widespread use of non‐productive alternative splice sites in Saccharomyces cerevisiae . PLoS Genet 10: e1004249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke G, Meng Q, Finley T, Wang T, Chen W, Ma W, Ye Q, Liu T‐Y (2017a) LightGBM: A highly efficient gradient boosting decision tree. Adv Neural Inform Process Syst 30: 3146–3154 [Google Scholar]
- Ke S, Pandya‐Jones A, Saito Y, Fak JJ, Vagbo CB, Geula S, Hanna JH, Black DL, Darnell JE Jr, Darnell RB (2017b) m(6)A mRNA modifications are deposited in nascent pre‐mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev 31: 990–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishor A, Fritz SE, Hogg JR (2019) Nonsense‐mediated mRNA decay: the challenge of telling right from wrong in a complex transcriptome. Wiley Interdiscip Rev RNA 10: e1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaki T, Popp MW, Maquat LE (2019) Quality and quantity control of gene expression by nonsense‐mediated mRNA decay. Nat Rev Mol Cell Biol 20: 406–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroyanagi H, Ohno G, Mitani S, Hagiwara M (2007) The Fox‐1 family and SUP‐12 coordinately regulate tissue‐specific alternative splicing in␣vivo . Mol Cell Biol 27: 8612–8621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroyanagi H, Ohno G, Sakane H, Maruoka H, Hagiwara M (2010) Visualization and genetic analysis of alternative splicing regulation in␣vivo using fluorescence reporters in transgenic Caenorhabditis elegans . Nat Protoc 5: 1495–1517 [DOI] [PubMed] [Google Scholar]
- Kuroyanagi H, Watanabe Y, Suzuki Y, Hagiwara M (2013) Position‐dependent and neuron‐specific splicing regulation by the CELF family RNA‐binding protein UNC‐75 in Caenorhabditis elegans . Nucleic Acids Res 41: 4015–4025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau LF, Brenner SE (2015) Regulation of splicing factors by alternative splicing and NMD is conserved between kingdoms yet evolutionarily flexible. Mol Biol Evol 32: 1072–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau LF, Inada M, Green RE, Wengrod JC, Brenner SE (2007) Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature 446: 926–929 [DOI] [PubMed] [Google Scholar]
- Lejeune F, Cavaloc Y, Stevenin J (2001) Alternative splicing of intron 3 of the serine/arginine‐rich protein 9G8 gene. Identification of flanking exonic splicing enhancers and involvement of 9G8 as a trans‐acting factor. J Biol Chem 276: 7850–7858 [DOI] [PubMed] [Google Scholar]
- Lewis JA, Fleming JT (1995) Basic culture methods. In Epstein HF, Shakes DC (eds), Caenorhabditis elegans: modern biological analysis of an organism, Vol. 48, 1, pp 3–29. San Diego, CA: Academic Press; [Google Scholar]
- Li H (2018) Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Na K, Lee HJ, Lee EY, Paik YK (2011) Contribution of sams‐1 and pmt‐1 to lipid homoeostasis in adult Caenorhabditis elegans . J Biochem 149: 529–538 [DOI] [PubMed] [Google Scholar]
- Liberman N, O'Brown ZK, Earl AS, Boulias K, Gerashchenko MV, Wang SY, Fritsche C, Fady PE, Dong A, Gladyshev VN et␣al (2020) N6‐adenosine methylation of ribosomal RNA affects lipid oxidation and stress resistance. Sci Adv 6: eaaz4370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder B, Grozhik AV, Olarerin‐George AO, Meydan C, Mason CE, Jaffrey SR (2015) Single‐nucleotide‐resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods 12: 767–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Begik O, Lucas MC, Ramirez JM, Mason CE, Wiener D, Schwartz S, Mattick JS, Smith MA, Novoa EM (2019) Accurate detection of m(6)A RNA modifications in native RNA sequences. Nat Commun 10: 4079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X et␣al (2014) A METTL3‐METTL14 complex mediates mammalian nuclear RNA N6‐adenosine methylation. Nat Chem Biol 10: 93–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T (2015) N 6‐methyladenosine‐dependent RNA structural switches regulate RNA‐protein interactions. Nature 518: 560–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardini JB, Talalay P (1970) Formation, functions and regulatory importance of S‐adenosyl‐L‐methionine. Adv Enzyme Regul 9: 349–384 [DOI] [PubMed] [Google Scholar]
- Lorenz DA, Sathe S, Einstein JM, Yeo GW (2020) Direct RNA sequencing enables m6A detection in endogenous transcript isoforms at base‐specific resolution. RNA 26: 19–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louloupi A, Ntini E, Conrad T, Orom UAV (2018) Transient N‐6‐methyladenosine transcriptome sequencing reveals a regulatory role of m6A in splicing efficiency. Cell Rep 23: 3429–3437 [DOI] [PubMed] [Google Scholar]
- Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira‐Smith OM, Misteli T (2010) Regulation of alternative splicing by histone modifications. Science 327: 996–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun AT, Smyth GK (2016) csaw: a Bioconductor package for differential binding analysis of ChIP‐seq data using sliding windows. Nucleic Acids Res 44: e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel BA, Grundy FJ, Artsimovitch I, Henkin TM (2003) Transcription termination control of the S box system: direct measurement of S‐adenosylmethionine by the leader RNA. Proc Natl Acad Sci USA 100: 3083–3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlincy NJ, Tan LY, Paul N, Zavolan M, Lilley KS, Smith CW (2010) Expression proteomics of UPF1 knockdown in HeLa cells reveals autoregulation of hnRNP A2/B1 mediated by alternative splicing resulting in nonsense‐mediated mRNA decay. BMC Genom 11: 565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLuckey SA, Van Berkel GJ, Glish GL (1992) Tandem mass spectrometry of small, multiply charged oligonucleotides. J Am Soc Mass Spectrom 3: 60–70 [DOI] [PubMed] [Google Scholar]
- Mendel M, Delaney K, Pandey RR, Chen KM, Wenda JM, Vagbo CB, Steiner FA, Homolka D, Pillai RS (2021) Splice site m(6)A methylation prevents binding of U2AF35 to inhibit RNA splicing. Cell 184: 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merendino L, Guth S, Bilbao D, Martinez C, Valcarcel J (1999) Inhibition of msl‐2 splicing by Sex‐lethal reveals interaction between U2AF35 and the 3' splice site AG. Nature 402: 838–841 [DOI] [PubMed] [Google Scholar]
- Montange RK, Batey RT (2006) Structure of the S‐adenosylmethionine riboswitch regulatory mRNA element. Nature 441: 1172–1175 [DOI] [PubMed] [Google Scholar]
- Muir VS, Gasch AP, Anderson P (2018) The substrates of nonsense‐mediated mRNA Decay in Caenorhabditis elegans . G3 8: 195–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni JZ, Grate L, Donohue JP, Preston C, Nobida N, O'Brien G, Shiue L, Clark TA, Blume JE, Ares M Jr (2007) Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense‐mediated decay. Genes Dev 21: 708–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR (2010) Expansion of the eukaryotic proteome by alternative splicing. Nature 463: 457–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa K, Iwamoto Y, Kobayashi Y, Katsuoka F, Kawaguchi S‐I, Tsujita T, Nakamura T, Kato S, Yamamoto M, Takayanagi H et␣al (2015) DNA methyltransferase 3a regulates osteoclast differentiation by coupling to an S‐adenosylmethionine‐producing metabolic pathway. Nat Med 21: 281–287 [DOI] [PubMed] [Google Scholar]
- Obata F, Miura M (2015) Enhancing S‐adenosyl‐methionine catabolism extends Drosophila lifespan. Nat Commun 6: 8332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MT, Knop K, Sherwood AV, Schurch NJ, Mackinnon K, Gould PD, Hall AJ, Barton GJ, Simpson GG (2020) Nanopore direct RNA sequencing maps the complexity of Arabidopsis mRNA processing and m6A modification. Elife 9: e49658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova N, Kaloudas D, Penchovsky R (2019) Riboswitch distribution, structure, and function in bacteria. Gene 708: 38–48 [DOI] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V et␣al (2011) Scikit‐learn: machine learning in python. J Mach Learn Res 12: 2825–2830 [Google Scholar]
- Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, Conrad NK (2017) The U6 snRNA m6A Methyltransferase METTL16 regulates SAM synthetase intron retention. Cell 169: 824–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping X‐L, Sun B‐F, Wang Lu, Xiao W, Yang X, Wang W‐J, Adhikari S, Shi Y, Lv Y, Chen Y‐S et␣al (2014) Mammalian WTAP is a regulatory subunit of the RNA N6‐methyladenosine methyltransferase. Cell Res 24: 177–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulak R, Anderson P (1993) mRNA surveillance by the Caenorhabditis elegans smg genes. Genes Dev 7: 1885–1897 [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani AK, Calarco JA, Pan Q, Mavandadi S, Wang Y, Nelson AC, Lee LJ, Morris Q, Blencowe BJ, Zhen M et␣al (2011) Genome‐wide analysis of alternative splicing in Caenorhabditis elegans . Genome Res 21: 342–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani AK, Nelson AC, Kapranov P, Bell I, Gingeras TR, Fraser AG (2009) High resolution transcriptome maps for wild‐type and nonsense‐mediated decay‐defective Caenorhabditis elegans . Genome Biol 10: R101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach NP, Sadowski N, Alessi AF, Timp W, Taylor J, Kim JK (2020) The full‐length transcriptome of C. elegans using direct RNA sequencing. Genome Res 30: 299–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP (2011) Integrative genomics viewer. Nat Biotechnol 29: 24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roundtree IA, Luo G‐Z, Zhang Z, Wang X, Zhou T, Cui Y, Sha J, Huang X, Guerrero L, Xie P et␣al (2017) YTHDC1 mediates nuclear export of N6‐methyladenosine methylated mRNAs. Elife 6: e31311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint‐Andre V, Batsche E, Rachez C, Muchardt C (2011) Histone H3 lysine 9 trimethylation and HP1γ favor inclusion of alternative exons. Nat Struct Mol Biol 18: 337–344 [DOI] [PubMed] [Google Scholar]
- Schor IE, Rascovan N, Pelisch F, Allo M, Kornblihtt AR (2009) Neuronal cell depolarization induces intragenic chromatin modifications affecting NCAM alternative splicing. Proc Natl Acad Sci USA 106: 4325–4330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Mumbach M, Jovanovic M, Wang T, Maciag K, Bushkin G, Mertins P, Ter‐Ovanesyan D, Habib N, Cacchiarelli D et␣al (2014) Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5' sites. Cell Rep 8: 284–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Wei J, He C (2019) Where, when, and how: context‐dependent functions of RNA methylation writers, readers, and erasers. Mol Cell 74: 640–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima H, Matsumoto M, Ishigami Y, Ebina M, Muto A, Sato Y, Kumagai S, Ochiai K, Suzuki T, Igarashi K (2017) S‐adenosylmethionine synthesis is regulated by selective N 6‐adenosine methylation and mRNA degradation involving METTL16 and YTHDC1. Cell Rep 21: 3354–3363 [DOI] [PubMed] [Google Scholar]
- Shiraki N, Shiraki Y, Tsuyama T, Obata F, Miura M, Nagae G, Aburatani H, Kume K, Endo F, Kume S (2014) Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab 19: 780–794 [DOI] [PubMed] [Google Scholar]
- Sibley CR (2014) Regulation of gene expression through production of unstable mRNA isoforms. Biochem Soc Trans 42: 1196–1205 [DOI] [PubMed] [Google Scholar]
- Son HG, Seo M, Ham S, Hwang W, Lee D, An SWA, Artan M, Seo K, Kaletsky R, Arey RN et␣al (2017) RNA surveillance via nonsense‐mediated mRNA decay is crucial for longevity in daf‐2/insulin/IGF‐1 mutant C. elegans . Nat Commun 8: 14749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Bao Y, Han W, Song F, Shen X, Zhao J, Zuo Ji, Saffen D, Chen W, Wang Z et␣al (2017) Autoregulation of RBM10 and cross‐regulation of RBM10/RBM5 via alternative splicing‐coupled nonsense‐mediated decay. Nucleic Acids Res 45: 8524–8540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sureau A, Gattoni R, Dooghe Y, Stevenin J, Soret J (2001) SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EMBO J 20: 1785–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabrez SS, Sharma RD, Jain V, Siddiqui AA, Mukhopadhyay A (2017) Differential alternative splicing coupled to nonsense‐mediated decay of mRNA ensures dietary restriction‐induced longevity. Nat Commun 8: 306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei S, Togo‐Ohno M, Suzuki Y, Kuroyanagi H (2016) Evolutionarily conserved autoregulation of alternative pre‐mRNA splicing by ribosomal protein L10a. Nucleic Acids Res 44: 5585–5596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamiya H, Hirota K, Takahashi Y, Daitoku H, Kaneko Y, Sakuta G, Iizuka K, Watanabe S, Ishii N, Fukamizu A (2013) Conserved SAMS function in regulating egg‐laying in C. elegans . J Recept Sig Transd 33: 56–62 [DOI] [PubMed] [Google Scholar]
- Tourasse NJ, Millet JRM, Dupuy D (2017) Quantitative RNA‐seq meta‐analysis of alternative exon usage in C. elegans . Genome Res 27: 2120–2128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turunen JJ, Verma B, Nyman TA, Frilander MJ (2013) HnRNPH1/H2, U1 snRNP, and U11 snRNP cooperate to regulate the stability of the U11–48K pre‐mRNA. RNA 19: 380–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J, Blencowe BJ (2019) Alternative splicing regulatory networks: functions, mechanisms, and evolution. Mol Cell 76: 329–345 [DOI] [PubMed] [Google Scholar]
- Walker A, Jacobs RL, Watts J, Rottiers V, Jiang K, Finnegan D, Shioda T, Hansen M, Yang F, Niebergall L et␣al (2011) A conserved SREBP‐1/phosphatidylcholine feedback circuit regulates lipogenesis in metazoans. Cell 147: 840–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, He C (2015) N 6‐methyladenosine modulates messenger RNA translation efficiency. Cell 161: 1388–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wani S, Kuroyanagi H (2017) An emerging model organism Caenorhabditis elegans for alternative pre‐mRNA processing in␣vivo . Wiley Interdiscip Rev RNA 8: e1428 [DOI] [PubMed] [Google Scholar]
- Webb B, Sali A (2016) Comparative protein structure modeling using MODELLER. Curr Protoc Bioinformatics 54: 5.6.1‐5.6.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CM, Gershowitz A, Moss B (1975) Methylated nucleotides block 5' terminus of HeLa cell messenger RNA. Cell 4: 379–386 [DOI] [PubMed] [Google Scholar]
- Winkler WC, Nahvi A, Sudarsan N, Barrick JE, Breaker RR (2003) An mRNA structure that controls gene expression by binding S‐adenosylmethionine. Nat Struct Biol 10: 701–707 [DOI] [PubMed] [Google Scholar]
- Wu S, Romfo CM, Nilsen TW, Green MR (1999) Functional recognition of the 3' splice site AG by the splicing factor U2AF35 . Nature 402: 832–835 [DOI] [PubMed] [Google Scholar]
- Xiao W, Adhikari S, Dahal U, Chen Y‐S, Hao Y‐J, Sun B‐F, Sun H‐Y, Li A, Ping X‐L, Lai W‐Y et␣al (2016) Nuclear m6A reader YTHDC1 regulates mRNA splicing. Mol Cell 61: 507–519 [DOI] [PubMed] [Google Scholar]
- Yoshida H, Park S‐Y, Oda T, Akiyoshi T, Sato M, Shirouzu M, Tsuda K, Kuwasako K, Unzai S, Muto Y et␣al (2015) A novel 3' splice site recognition by the two zinc fingers in the U2AF small subunit. Genes Dev 29: 1649–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Park SY, Sakashita G, Nariai Y, Kuwasako K, Muto Y, Urano T, Obayashi E (2020) Elucidation of the aberrant 3' splice site selection by cancer‐associated mutations on the U2AF1. Nat Commun 11: 4744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccara S, Ries RJ, Jaffrey SR (2019) Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol 20: 608–624 [DOI] [PubMed] [Google Scholar]
- Zheng G, Dahl J, Niu Y, Fedorcsak P, Huang C‐M, Li C, Vågbø C, Shi Y, Wang W‐L, Song S‐H et␣al (2013) ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 49: 18–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou KI, Shi H, Lyu R, Wylder AC, Matuszek Z, Pan JN, He C, Parisien M, Pan T (2019) Regulation of co‐transcriptional pre‐mRNA splicing by m6A through the low‐complexity protein hnRNPG. Mol Cell 76: 70–81.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorio DA, Blumenthal T (1999) Both subunits of U2AF recognize the 3' splice site in Caenorhabditis elegans . Nature 402: 835–838 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Review Process File
Data Availability Statement
RNA‐Seq data: DDBJ Sequence Read Archive (DRA) DRX184564 (https://ddbj.nig.ac.jp/DRASearch/experiment?acc=DRX184564).
RNA‐Seq data: DDBJ Sequence Read Archive (DRA) DRX184565 (https://ddbj.nig.ac.jp/DRASearch/experiment?acc=DRX184565).
Chip‐Seq data: DDBJ Sequence Read Archive (DRA) DRA009050 (https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA009050).
Codes used in the sequence analysis: GitHub KUROYANAGI‐Lab/WATABE‐SAMS (https://github.com/KUROYANAGI‐Lab/WATABE‐SAMS).
Nucleotide sequence: DDBJ/ENA/GenBank LC603057 (http://getentry.ddbj.nig.ac.jp/getentry/na/LC603057?filetype=html).
The materials generated and/or analyzed during the current study are available from KUROYANAGI Hidehito (hidehito@med.u-ryukyu.ac.jp).