

ABSTRACT
The generation of oxidative stress is a host strategy used to control Staphylococcus aureus infections. Sulfur-containing amino acids, cysteine and methionine, are particularly susceptible to oxidation because of the inherent reactivity of sulfur. Due to the constant threat of protein oxidation, many systems evolved to protect S. aureus from protein oxidation or to repair protein oxidation after it occurs. The S. aureus peptide methionine sulfoxide reductase (Msr) system reduces methionine sulfoxide to methionine. Staphylococci have four Msr enzymes, which all perform this reaction. Deleting all four msr genes in USA300 LAC (Δmsr) sensitizes S. aureus to hypochlorous acid (HOCl) killing; however, the Δmsr strain does not exhibit increased sensitivity to H2O2 stress or superoxide anion stress generated by paraquat or pyocyanin. Consistent with increased susceptibility to HOCl killing, the Δmsr strain is slower to recover following coculture with both murine and human neutrophils than USA300 wild type. The Δmsr strain is attenuated for dissemination to the spleen following murine intraperitoneal infection and exhibits reduced bacterial burdens in a murine skin infection model. Notably, no differences in bacterial burdens were observed in any organ following murine intravenous infection. Consistent with these observations, USA300 wild-type and Δmsr strains have similar survival phenotypes when incubated with murine whole blood. However, the Δmsr strain is killed more efficiently by human whole blood. These findings indicate that species-specific immune cell composition of the blood may influence the importance of Msr enzymes during S. aureus infection of the human host.
KEYWORDS: MRSA, methionine sulfoxide, methionine sulfoxide reductases, oxidative stress, pathogenesis, Staphylococcus aureus
INTRODUCTION
Staphylococcus aureus is a leading cause of bacterial infections in humans (1). Nearly two-thirds of the population will be colonized by S. aureus during their lives, and S. aureus is capable of infecting every tissue of the human host (2). Common S. aureus infections include bacteremia, skin and soft tissue infections, infective endocarditis, biofilms on indwelling medical devices, and osteomyelitis (1). Further complicating the treatment of S. aureus infections, community-acquired methicillin-resistant S. aureus (CA-MRSA) strains, which are resistant to β-lactam antibiotics, are major disease-causing strains in recent decades (3, 4). Both the Centers for Disease Control and Prevention (5) and the World Health Organization (6) have identified S. aureus as a pathogen that necessitates the development of more effective treatments. Therefore, expanding our understanding of how the vertebrate host protects against S. aureus and how S. aureus avoids killing by the host will identify and validate new targets for novel antistaphylococcal therapies.
The human immune system has many strategies to generate reactive oxygen intermediates; thus, S. aureus regularly encounters oxidative stress in the host (7). During infection, among the first immune cells that S. aureus encounters are neutrophils. One mechanism neutrophils use to clear S. aureus infection is the generation of reactive oxidants, including hypochlorous acid (HOCl) through the enzymatic activity of myeloperoxidase (8). Cellular respiration produces superoxide anion as a by-product, which is a precursor to most relevant cellular oxidants, including peroxyl radical, hydrogen peroxide, and hydroxyl radical (9). Other bacterial species also generate oxidative stress that can harm S. aureus. Pseudomonas aeruginosa, a common pathogen found to coinfect the lungs of cystic fibrosis patients with S. aureus, generates phenazines, redox-active molecules that reduce molecular oxygen to superoxide anion (10). Because it is under constant threat by oxidants, S. aureus has multiple systems to sense oxidative stress, scavenge reactive oxidants, and repair the damage to cellular macromolecules caused by oxidants.
One system that S. aureus uses to repair oxidative damage to the proteome are the peptide methionine sulfoxide reductases (Msr). These enzymes reduce oxidized methionine, known as methionine sulfoxide, to methionine (11). Methionine and cysteine are the only amino acids that contain a reactive sulfur atom. Unlike the thiol of cysteine, the thioether of methionine does not appear to directly have a role in catalytic reactions (12). Methionine residues are hydrophobic and thus have a stabilizing role in the core of many protein structures through interactions with hydrophobic amino acids and sulfur-π interactions with aromatic amino acids (13, 14). One study replaced all methionine residues of cytochrome P450 with norleucine (15). The result was no loss of basal enzymatic activity, but without sulfur-π interactions, the norleucine-containing enzyme was less resistant to heat inactivation, further indicating a stabilizing role for methionine in protein structure (15). Another study demonstrated that oxidation strengthens and promotes aberrant methionine-aromatic interactions, altering protein structure and function (14). Despite its unclear role in protein structure, methionine is essential, and Msr enzymes appear to be present in all living organisms, emphasizing the importance of reduced methionine (11). The ability to repair damaged proteins conserves cellular resources that are needed for survival during infection. Without this repair mechanism, S. aureus would need to continually resynthesize each protein that is oxidized at a critical methionine residue.
Methionine sulfoxide is a chiral molecule; therefore, most organisms have two Msr enzymes. MsrA repairs the S-epimer of methionine sulfoxide, while MsrB repairs the R-epimer (16). Staphylococci are unique in having four Msr enzymes, three MsrAs (MsrA1, MsrA2, and MsrA3) and one MsrB, and it is unclear why a small subset of bacterial species have maintained four genes encoding these enzymes (17, 18). When no stressors are present, msrA1 and msrB, which are in an operon together, are the most highly expressed msr genes in S. aureus SH1000 (17). During stationary phase, msrA1 and msrB are maximally expressed, while msrA2 and msrA3 are maximally expressed during mid-exponential phase (17). Antibiotics induce msrA1 and msrB expression (17, 19), and msrA1 expression is under the control of VraRS in S. aureus USA300 LAC during neutrophil infection (19). Osmotic stress induces the expression of msrA3, while no stimulus has been identified that induces the expression of msrA2 (17). Therefore, most studies consider MsrA1 and MsrB to be the most important Msr enzymes in S. aureus. In an intraperitoneal murine coinfection model comparing SH1000 and various isogenic msr deletion strains, dissemination defects were identified in the liver and spleen, but only for strains lacking msrA1 (18).
Here, we present data indicating that HOCl is the primary oxidant from which Msr enzymes protect S. aureus USA300, the dominant CA-MRSA strain in the United States (20). Protection was not observed by Msr enzymes for any of the other relevant biological oxidants tested. Additionally, strains lacking all msr genes did not exhibit differential phenotypes in vivo during a murine intravenous infection model, while modest protection by Msr activity was observed in both intraperitoneal and skin infection models. Consistent with the differential killing by HOCl, both murine and human neutrophils induced a similar differential killing phenotype in strains with and without Msr enzymes. Finally, the ability to reduce oxidized methionine protects S. aureus from killing by human whole blood but not murine whole blood. This emphasizes that the Msr enzymes are important for S. aureus infection of the human host and may be targeted to help the immune response abate S. aureus infections in human patients.
RESULTS
Staphylococci have four methionine sulfoxide reductases.
The loci harboring adjacent msrA1 and msrB genes are conserved across staphylococcal species and include genes for a total of four methionine sulfoxide reductases: one B reductase and three A reductases (see Table S1 in the supplemental material). Despite this conserved architecture, the importance of three separate A reductases remains unclear. In contrast to the presence of four msr genes across the genus Staphylococcus, most members of the Bacillales order, other than isolated exceptions such as Macrococcus caseolyticus, carry fewer than four msr genes. A sequence-based comparison of Msr proteins from different bacterial species across the Bacillales order confirmed the existence of several subgroups, including MsrA1, MsrA2, MsrA3, and MsrB clusters (Table S1). These findings suggest the possibility of unique niches during the staphylococcal life cycle where oxidative stress is experienced, selecting for the maintenance of additional msr genes that are absent in related bacterial genera.
Δmsr strain cannot repair methionine oxidation.
All four methionine sulfoxide reductase genes, msrA1 (SAUSA300_1317), msrA2 (SAUSA300_1256), msrA3 (SAUSA300_2594), and msrB (SAUSA300_1316), were deleted from USA300 wild type to generate the Δmsr strain. Figure 1A demonstrates that the Δmsr strain does not have any Msr activity, indicating that all Msr-encoding genes were deleted. To assess the extent of methionine oxidation in whole cells, USA300 wild-type and Δmsr strains were treated with HOCl, the oxidant generated through the enzymatic activity of myeloperoxidase (8). Following treatment, the cell lysates were incubated with Ox4 (21), which reacts with methionine but not methionine sulfoxide. A biotin tag attached through click chemistry followed by SDS-PAGE allowed for the visualization of methionine oxidation with a streptavidin-conjugated fluorophore, where increased labeling indicates decreased methionine sulfoxide across the proteome. When both strains were treated with HOCl, the Δmsr strain contained more oxidized methionine residues than the USA300 wild type, presumably due to the inability of the Δmsr strain to repair methionine sulfoxide (Fig. 1B). These data confirm that HOCl oxidizes methionine residues in S. aureus and that accumulation of this damage is prevented by Msr enzymes.
FIG 1.
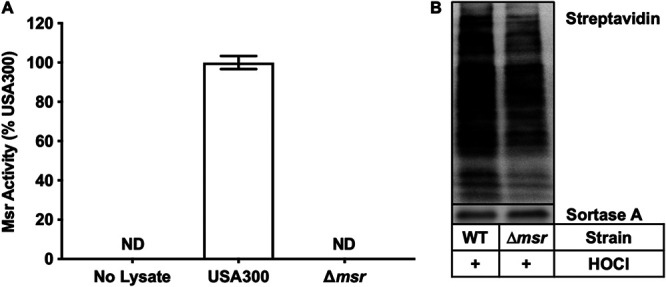
Methionine sulfoxide reductases repair oxidized methionine residues. (A) USA300 wild-type and Δmsr strains were lysed, and Msr activity was quantified by HPLC. The Δmsr strain has no measurable Msr activity. Data are means ± standard deviations from biological triplicate measurements. (B) USA300 wild-type and Δmsr strains were treated with HOCl; then, using a methionine reactive probe, Ox4, the extent of methionine oxidation was assessed. A darker signal indicates less methionine oxidation, and as expected, the Δmsr strain, which is unable to repair methionine sulfoxide, has decreased levels of free methionine.
Methionine sulfoxide reductases protect S. aureus from HOCl killing.
USA300 wild type and Δmsr strains were exposed to oxidative stress to determine the specific Msr enzymes that are most effective at protecting S. aureus. HOCl was more effective at killing the Δmsr strain than at killing the USA300 wild type (Fig. 2A and Fig. S1A), indicating that Msr activity is critical for protecting S. aureus from HOCl-mediated killing. Individual tagged Msr enzymes were constitutively expressed in the Δmsr strain (Fig. S1B) to determine if a specific Msr enzyme has a greater protective effect for S. aureus against HOCl. The delay in observable growth following HOCl treatment in the Δmsr strain was complemented in trans by expressing either MsrA1 (Fig. S1C) or MsrB (Fig. S1F) in the Δmsr background but not by expressing MsrA2 (Fig. S1D) or MsrA3 (Fig. S1E), indicating that certain Msr enzymes protect S. aureus from HOCl. To define the specific Msr enzymes that are involved in protecting S. aureus from HOCl, individual deletions of msrA1, msrA2, msrA3, and msrB, a complete msrA deletion (msrA1, msrA2, and msrA3), and a strain with both msrA1 and msrB deleted were created and tested. The ΔmsrA1B strain was the only partial msr deletion strain that was completely inhibited for growth when treated with HOCl (see Fig. S2C). The ΔmsrA (Fig. S2A), ΔmsrB (Fig. S2B), and ΔmsrA1 (Fig. S2D) strains were all inhibited differentially compared to the wild type (WT). The protection afforded by the Msr enzymes correlates with the known expression of these enzymes in another S. aureus strain, SH1000. MsrA1 and MsrB were highly expressed under basal conditions and thus protective, while MsrA2 (Fig. S2E) and MsrA3 (Fig. S2F) were expressed at very low levels (17) and were not protective under these treatment conditions. However, expression alone does not explain the differences in protection by Msr enzymes, because only MsrA1 and MsrB protect S. aureus from HOCl when each individual Msr is constitutively expressed (Fig. S1B to F).
FIG 2.

Methionine sulfoxide reductases protect S. aureus from HOCl killing. (A) USA300 wild-type and Δmsr strains were treated with 0 or 100 μM HOCl. Growth was monitored by optical density at 600 nm for 24 h. Data are means ± standard deviations for measurements acquired in biological triplicates. (B) USA300 wild-type and Δmsr strains were treated with 0 or 3.2 mM paraquat (PQ). Growth was monitored by optical density at 600 nm for 24 h. Data are means ± standard deviations for measurements acquired in six biological replicates. (C) USA300 wild-type and Δmsr strains were treated with 0 or 16 μg/ml pyocyanin (PCN). Growth was monitored by optical density at 600 nm for 24 h. Data are means ± standard deviations for measurements acquired in biological quadruplicates. (D) USA300 wild-type and Δmsr strains were treated with 0 or 500 μM H2O2. Growth was monitored by optical density at 600 nm for 24 h. Data are means ± standard deviations for measurements acquired in biological triplicates.
USA300 wild-type and Δmsr strains were equally inhibited for growth by the superoxide anion generators paraquat (Fig. 2B) (22) and pyocyanin (Fig. 2C) (10), indicating that Msr activity does not play a major role in protecting S. aureus from superoxide anion stress. The Δmsr strain was slightly protected compared to USA300 wild type when H2O2 was added to the cultures (Fig. 2D), which was not due to the Δmsr strain having a compensatory activation of catalase (see Fig. S3). Due to the protection afforded to the Δmsr strain against H2O2 killing, each of the S. aureus msr mutants was also tested to determine their susceptibility to H2O2 killing. There was no differential killing between the USA300 wild type and each respective msr deletion strain (see Fig. S4A to F), indicating that the protection observed may be a cumulative effect of having all four msr genes inactivated. In total, these data show that of the relevant oxidants tested here, HOCl is the major contributor to S. aureus killing through methionine oxidation.
Methionine sulfoxide reductases are not required for full S. aureus virulence in an intravenous murine infection model.
Eight-week-old female BALB/cJ mice were infected retro-orbitally with USA300 wild-type or Δmsr strains. Infections were allowed to proceed for 4 or 10 days. These two infection time points were chosen to test the hypothesis that the Msr enzymes protect S. aureus from host-derived oxidative stress. Mice lacking a functional NADPH oxidase system and infected intravenously with S. aureus do not have a differential bacterial burden phenotype until 6 days postinfection compared to mice with a fully functional NADPH oxidase system (23). There were no differences in weight loss between mice infected with the USA300 wild type and mice infected with the Δmsr strain during both 4- (see Fig. S5A) and 10-day infections, a common indicator of disease severity (Fig. S5B). Consistent with the lack of weight loss differences, bacterial burdens were similar between the strains in the heart (Fig. 3A), lungs (Fig. 3B), liver (Fig. 3C), kidneys (Fig. 3D), and spleen (Fig. 3E) at both 4 and 10 days postinfection. These data indicate that methionine sulfoxide reductases are not necessary for full S. aureus virulence during murine intravenous infection.
FIG 3.

S. aureus lacking all four Msr enzymes is fully virulent in a murine model of bacteremia. Female 8-week-old BALB/cJ mice were infected retro-orbitally with USA300 wild-type or Δmsr strains. After 4 or 10 days of monitoring, the heart (A), kidneys (B), liver (C), lungs (D), and spleen (E) were harvested, homogenized, and dilution plated on solid medium to enumerate viable bacteria. Gray symbols indicate bacterial burdens below the limit of detection, and so the limit of detection was used for statistical analyses. Data are means ± standard deviations, and P values were calculated by Mann-Whitney test.
Methionine sulfoxide reductases enhance spleen colonization by S. aureus in an intraperitoneal murine infection model.
Given that methionine sulfoxide reductases are necessary for survival during oxidative stress in vitro but dispensable for full virulence during murine intravenous infection, we tested the hypothesis that Msr activity is important for S. aureus pathogenesis through alternative routes of infection. Eight-week-old female BALB/cJ mice were infected intraperitoneally with USA300 wild-type or Δmsr strains. No bacteria were detected in the heart or kidneys, and similar USA300 wild-type and Δmsr bacterial burdens were observed in the liver (Fig. 4A) and lungs (Fig. 4B). However, 3-fold more bacteria were present in the spleens of USA300 wild-type infected mice than in Δmsr strain-infected mice (Fig. 4C). These data indicate that the contribution of Msr activity to S. aureus pathogenesis is potentially impacted by the route of infection.
FIG 4.

S. aureus lacking all four Msr enzymes is attenuated for dissemination to the spleen in a murine model of intraperitoneal infection. Female 8-week-old BALB/cJ mice were infected intraperitoneally with USA300 wild-type or Δmsr strains. After 4 days of monitoring, the heart, kidneys, liver (A), lungs (B), and spleen (C) were harvested, homogenized, and dilution plated on solid medium to enumerate viable bacteria. No viable bacteria were detected in the heart or kidneys. Gray symbols indicate bacterial burdens below the limit of detection, and so the limit of detection was used for statistical analyses. Data are means ± standard deviations, and P values were calculated by Mann-Whitney test.
Methionine sulfoxide reductases enhance S. aureus pathogenesis in a murine skin infection model.
S. aureus is a common cause of skin infections (20), and S. aureus skin infections are mediated by neutrophils (24). Therefore, we tested the role of Msr activity in S. aureus skin infections. Wounds were created on the backs of 8-week-old female BALB/cJ mice, inoculated with the USA300 wild-type or Δmsr strain, and monitored for 48 h. USA300 wild-type-infected mice had 2-fold higher bacterial burdens in the skin than Δmsr strain-infected mice (Fig. 5). These data reveal that the Msr enzymes protect S. aureus during murine skin infections and further demonstrate that the role of Msr enzymes during S. aureus pathogenesis in mice is modest. Future infection experiments testing longer time points may reveal a stronger role for the Msr enzymes in S. aureus cutaneous pathogenesis.
FIG 5.
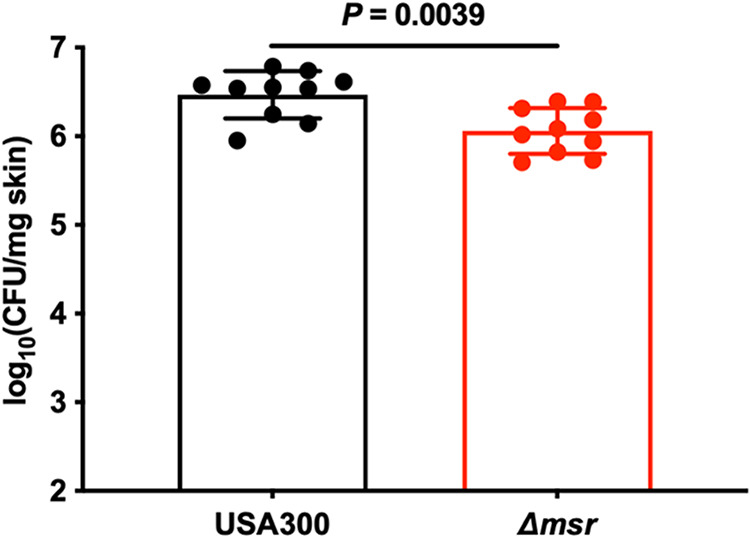
S. aureus lacking all four Msr enzymes is attenuated for pathogenesis in a murine skin infection model. Skin wounds were created on the backs of female 8-week-old BALB/cJ mice and then infected with USA300 wild-type or Δmsr strains. After 2 days of monitoring, the skin lesions were harvested, homogenized, and dilution plated on solid medium to enumerate viable bacteria. Data are means ± standard deviations, and the P value was calculated by Mann-Whitney test.
Methionine sulfoxide reductases allow S. aureus to resume growth following neutrophil challenge.
Msr enzymes protect S. aureus from HOCl killing, which is primarily generated in neutrophils through the enzymatic activity of myeloperoxidase (25). Therefore, we tested the hypothesis that Msr activity is important for S. aureus to survive neutrophil killing. Both human (Fig. 6A) and murine (Fig. 6B) neutrophils were tested for their ability to kill USA300 wild-type and Δmsr strains. Human neutrophils killed both S. aureus strains faster and to a greater extent, decreasing the viable cell population by 85% in 1 h (Fig. 6A). At 1 h, murine neutrophils decreased the viable population of both strains by only 25%, and this phenotype was most pronounced at 6 h, when the viable population was decreased by 75% (Fig. 6B). At the 6-h time point for human neutrophils and 8 h time point for murine neutrophils, S. aureus had overcome the stress induced by neutrophils and began to multiply. USA300 wild type grew better than the Δmsr strain following incubation with neutrophils from both species, which is consistent with the role of the Msr enzymes in repairing oxidized methionine residues. A strain that cannot repair critical methionine sulfoxide residues will experience a delay in growth compared to a strain with a fully functional set of Msr enzymes due to those proteins needing to be synthesized de novo. Consistent with the in vitro data, neutrophils killed S. aureus, and the repair of oxidized proteins by Msr activity allowed USA300 wild type to grow better than the Δmsr strain following neutrophil coculture.
FIG 6.

Msr enzymes protect S. aureus against neutrophil killing and human whole-blood killing. (A) USA300 wild-type and Δmsr strains were incubated with PMNs (MOI = 1) isolated from human blood, and killing was assessed by plating for CFU. (B) USA300 wild-type and Δmsr strains were incubated with PMNs (MOI = 1) isolated from murine bone marrow, and killing was assessed by plating for CFU. (C) USA300 wild-type and Δmsr strains were incubated with human whole blood (MOI = 1), and killing was assessed by plating for CFU. (D) USA300 wild-type and Δmsr strains were incubated with murine whole blood (MOI = 1), and killing was assessed by plating for CFU. All data are means ± standard deviations, and P values were calculated by t test. ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Methionine sulfoxide reductases protect S. aureus during infection of human but not murine blood.
HOCl, an abundant neutrophil oxidant, appears to be the main mechanism of killing S. aureus that is reversed by Msr activity. Human blood contains 10-fold more neutrophils than murine blood (26–28), indicating that Msr activity, while not necessary for full virulence in murine systemic infections, may be important for full S. aureus virulence in the human host. Therefore, we hypothesize that immune cell differences between human and murine blood contribute to a species-specific role for these enzymes during S. aureus pathogenesis. To test this hypothesis, whole-blood killing of USA300 wild-type and Δmsr strains using both human (Fig. 6C) and murine (Fig. 6D) blood was performed. Indeed, human whole blood exhibited enhanced killing of the Δmsr strain compared to the killing of USA300 wild type at each time point tested (Fig. 6C), and no differential killing was observed at any time point tested for murine blood (Fig. 6D). These data indicate that Msr enzymes, while not required for virulence in murine septicemia, may contribute to S. aureus virulence in the human host.
DISCUSSION
Oxidative stress is a ubiquitous stressor encountered by pathogens, resulting in the oxidation of cellular macromolecules, alteration of function for those molecules, and decreased growth or cell death for the organism (29, 30). Due to the common mechanism of macromolecule oxidation, organisms evolved systems to prevent oxidation of these molecules or repair oxidation once it occurs. The system studied in the manuscript, the S. aureus Msr system, reduces methionine sulfoxide to methionine (11). Msr activity was not detected in the Δmsr strain, a strain lacking all four msr genes; therefore, we do not expect that any additional Msr enzymes are expressed in the USA300 wild-type strain. Here, we report that this Msr-deficient S. aureus strain is more susceptible to killing by HOCl, a neutrophil-derived oxidant, than the wild-type strain. Finally, Msr activity protects S. aureus in a human blood infection but not a murine blood infection, indicating a potential species-specific role for Msr enzymes in S. aureus pathogenesis.
Immune cells recruited to the site of infection to clear S. aureus generate antibacterial oxidative stress (23, 31). Other bacterial species can also generate oxidative stress that is toxic to S. aureus. For example, P. aeruginosa, a common bacterial species found with S. aureus infecting the lungs of cystic fibrosis patients, produces large amounts of pyocyanin, a redox-active molecule that reduces oxygen to superoxide anion (10). Deleting all four msr genes in the USA300 wild-type background of S. aureus revealed which oxidants are most important for controlling S. aureus infections through the oxidation of methionine residues in proteins. HOCl, the enzymatic product of myeloperoxidase and the primary oxidant generated by neutrophils was the only oxidant that killed the Δmsr strain more effectively than USA300 wild type. These data are consistent with a previous study that demonstrated differential killing by HOCl between a wild type and the ΔmsrA1B strain in a USA300 wild-type background (19). In immune cell killing assays, both human and murine polymorphonuclear leukocytes (PMNs) are effective at killing S. aureus, and the Δmsr strain demonstrates a decreased ability to grow when incubated with neutrophils compared to the growth of USA300 wild type. This is potentially due to the inability of the Δmsr strain to repair oxidized methionine residues, and thus needing to synthesize oxidized proteins de novo, delaying growth following neutrophil challenge. Superoxide anion stress generated by the redox-active molecules paraquat or pyocyanin caused delays in visible growth in both strains equally, while the Δmsr strain was slightly protected from H2O2. These results are consistent with what was previously reported for S. aureus SH1000, where the Δmsr strain is slightly more protected from H2O2-induced delays in observable growth than the wild type (18). While the mechanism of protection is not understood, a compensatory change in catalase activity in the Δmsr strain is not responsible for the observed protection. These data are consistent with Msr having a role in S. aureus pathogenesis by protecting S. aureus from neutrophil killing with HOCl.
With strong differential phenotypes both in vitro and in cell culture killing models using PMNs, the role of Msr activity in vivo was tested. The first infections performed were intravenous monoinfections of USA300 wild-type or Δmsr strains through retro-orbital injection followed by 4 or 10 days of monitoring and bacterial burden enumeration in the organs. The HOCl produced by myeloperoxidase uses oxidants generated by NADPH oxidase activity (8). Differential bacterial burdens observed during intravenous infection of mice deficient for NADPH oxidase were not detected until 6 days postinfection compared to that in wild-type mice (23), justifying the different time points here. There was no difference between the strains in terms of weight loss, a common indicator of disease severity, or organ bacterial burdens at either time point tested. One potential explanation for this phenotype is that mice have >10-fold lower levels of neutrophils in their blood than humans (28). Therefore, circulating neutrophils may be used more extensively by the human host than by the murine host to control S. aureus infections.
Other routes of infection were tested because each route of infection has a unique set of immune cells that first encounter S. aureus. Both strains were injected intraperitoneally, and mice were observed for 4 days. Intraperitoneal infections of S. aureus had decreased dissemination to other organs compared to that with intravenous infections. Therefore, no bacteria were detected in the heart and kidneys. Also, no difference in bacterial burdens was detected in the liver and lungs. In the spleen, the Δmsr strain was slightly attenuated for dissemination compared USA300 wild type. In the murine skin infection model, the Δmsr strain was also attenuated compared to USA300 wild type. These data suggest that Msr enzymes protect S. aureus during intraperitoneal and skin infection models but not during intravenous infection. This is potentially due to different cell types that S. aureus encounters in the different host niches, and future cell culture killing experiments with different immune cell subsets will determine which cells are responsible for the observed phenotypes.
Differences in humans and mice are well known for many aspects of S. aureus pathogenesis (32, 33). Therefore, we tested the ability of murine and human whole blood to kill USA300 wild-type and Δmsr strains to determine if species differences are a plausible explanation for why there is no differential infection phenotype during a murine intravenous infection. In murine blood, both USA300 wild-type and Δmsr strains were killed similarly. However, in human blood, Msr enzymes provided protection to S. aureus at all time points tested. Human blood has >10-fold more neutrophils than murine blood (28), and these results may indicate that oxidative stress has a less important role in clearing S. aureus infections in murine blood than in human blood. Additionally, other leukocytes are present at different levels in murine blood than in human blood, including B cells, basophils, monocytes, and several subsets of T cells (28). These differences potentially could also explain why there is differential killing in human blood compared to that in mouse blood, but future studies are needed to fully elucidate this mechanism.
Collectively, these data demonstrate that Msr enzymes protect S. aureus from certain types of oxidative stress, notably, HOCl, and that the reduction of oxidized methionine has a minor role in pathogenesis in a mouse. However, the differences observed in killing between human and murine blood hint at the possibility that methionine oxidation may have a greater role in controlling S. aureus infections in the human host and that future studies of S. aureus Msr enzymes may need to be performed in humanized infection models. The distinct differences observed in terms of the oxidants that Msr enzymes are protective against indicate that humans and mice may use different oxidants at the host-pathogen interface to control S. aureus infections. Future studies in human whole blood and using different types of isolated immune cells will give a better understanding of the cells used by the host to control S. aureus infection and the cell types that Msr enzymes are most protective against.
MATERIALS AND METHODS
Ethics statement.
Human blood samples for neutrophil isolation were obtained as buffy coats from healthy, anonymous consenting adult donors (New York Blood Center). Freshly isolated human whole blood was collected in accordance with a protocol approved by the NYU Grossman School of Medicine Institutional Review Board for Human Subjects (Torres Lab IRB number i14-02129). All donors provided written consent to participate in the study.
All mouse experiments were approved by the Vanderbilt University Institutional Animal Care and Use Committee and performed according to institutional policies, the Animal Welfare Act, the National Institutes of Health guidelines, and the American Veterinary Association guidelines on euthanasia.
Materials.
Staphylococcus aureus strains (Table 1), plasmids (Table 2), and primers (Table 3) used in this study are listed in their respective tables. All lab plasticware was from USA Scientific Ocala, FL, or Corning, Corning, NY. All S. aureus growth was performed on tryptic soy agar (TSA) or in tryptic soy broth (TSB) (Becton, Dickinson, Franklin Lakes, NJ). All other chemicals were obtained from Sigma, St. Louis, MO, unless otherwise noted. PCRs were performed using 2× Phusion HF master mix (Thermo Fisher, Waltham, MA). All Sanger sequencing was performed by Genewiz, South Plainfield, NJ.
TABLE 1.
Staphylococcus aureus bacterial strains used in this study
| Strain | Genotype | Description | Reference |
|---|---|---|---|
| RN4220 | WT | Restriction enzyme-deficient cloning intermediate strain | 35 |
| USA300 | WT | WT community-acquired methicillin-resistant isolate (CA-MRSA) | 20 |
| USA300 | ΔmsrA1 | In-frame unmarked deletion of msrA1 (SAUSA300_1317) generated by allelic exchange | This work |
| USA300 | ΔmsrA2 | In-frame unmarked deletion of msrA2 (SAUSA300_1256) generated by allelic exchange | This work |
| USA300 | ΔmsrA3 | In-frame unmarked deletion of msrA3 (SAUSA300_2594) generated by allelic exchange | This work |
| USA300 | ΔmsrB | In-frame unmarked deletion of msrB (SAUSA300_1316) generated by allelic exchange | This work |
| USA300 | ΔmsrA1B | In-frame unmarked deletion of msrA1 (SAUSA300_1317) and msrB (SAUSA300_1316) generated by allelic exchange | This work |
| USA300 | ΔmsrA | In-frame unmarked deletion of msrA1 (SAUSA300_1317), msrA2 (SAUSA300_1256), and msrA3 (SAUSA300_2594) generated by allelic exchange | This work |
| USA300 | Δmsr | In-frame unmarked deletion of msrA1 (SAUSA300_1317), msrA2 (SAUSA300_1256), msrA3 (SAUSA300_2594), and msrB (SAUSA300_1316) generated by allelic exchange | This work |
| JE2 | WT | Wild-type, USA300 community-acquired methicillin-resistant isolate (CA-MRSA) | 41 |
| JE2 | katA | katA::Tn (NE1366; SAUSA300_1232::Tn) | 41 |
TABLE 2.
Plasmids used in this study
| Plasmid | Description | Reference |
|---|---|---|
| pKOR1 | Temp-sensitive vector for allelic exchange | 34 |
| pKOR1.ΔmsrA1 | Allelic exchange vector for the deletion of msrA1 | This work |
| pKOR1.ΔmsrA2 | Allelic exchange vector for the deletion of msrA2 | This work |
| pKOR1.ΔmsrA3 | Allelic exchange vector for the deletion of msrA3 | This work |
| pKOR1.ΔmsrB | Allelic exchange vector for the deletion of msrB | This work |
| pKOR1.ΔmsrA1B | Allelic exchange vector for the deletion of msrA1B | This work |
| pOS1.Plgt | Expression vector with lgt (constitutive) promoter | 38 |
| pOS1.Plgt.msrA1.Etag | Expression vector with msrA1 containing a C-terminal Etag for blotting, driven by the lgt (constitutive) promoter | This work |
| pOS1.Plgt.msrA2.Etag | Expression vector with msrA2 containing a C-terminal Etag for blotting, driven by the lgt (constitutive) promoter | This work |
| pOS1.Plgt.msrA3.Etag | Expression vector with msrA3 containing a C-terminal Etag for blotting, driven by the lgt (constitutive) promoter | This work |
| pOS1.Plgt.msrB.Etag | Expression vector with msrB containing a C-terminal Etag for blotting, driven by the lgt (constitutive) promoter | This work |
TABLE 3.
Primers used in this study
| Name | Sequence |
|---|---|
| WNB00047 | ATAGTGAGTCGTATTACATGGTC |
| WNB00048 | GGGCCCGAGCTTAAGACTG |
| WNB00058 | CACTAACCTGCCCCGTTAGTTG |
| WNB00059 | ACACTTTATGCTTCCGGCTCG |
| WNB00123 | CATGTAATACGACTCACTATAAACTAGATGAGAAAAATACAATTCC |
| WNB00186 | TCTTGATGGTATGAATTACCTCCTCTATCTATCTAATTATAAATTTAG |
| WNB00187 | GGTAATTCATACCATCAAGATTATTACAAAAAGAAC |
| WNB00188 | CCAGTCTTAAGCTCGGGCCCTCACAGCATTTTCATCATAAATATTAATTG |
| WNB00161 | CATGTAATACGACTCACTATAAACTAGATGAGAAAAATACAATTCC |
| WNB00162 | CTTTCCACTCATGAATTACCTCCTCTATCTATCTAATTATAAATTTAG |
| WNB00163 | GGTAATTCATGAGTGGAAAGTATGTTTAAAAAATTATTC |
| WNB00164 | CCAGTCTTAAGCTCGGGCCCTTCTTGGTCTTGATTGCTTG |
| WNB00189 | CATGTAATACGACTCACTATAATTATTTGTAGTTATAGGGCAATTG |
| WNB00190 | CTTTCCACTCTTAAGCATTTTGATTCCC |
| WNB00191 | AAATGCTTAAGAGTGGAAAGTATGTTTAAAAAATTATTC |
| WNB00168 | CAACGTAGAGGGCGTTGATG |
| WNB00169 | CCTGAAACACCTTCATTAAAGGATTTCG |
| WNB00170 | GTGCACAATGCGAGAACTC |
| WNB00202 | TTGCTAATGGTGCGGCG |
| WNB00119 | CATGTAATACGACTCACTATGACTTAACATCTTTTTCACCTC |
| WNB00120 | AGTATAAATGTTTTGACATCCTTTCATTAG |
| WNB00121 | GATGTCAAAACATTTATACTTGATGAAACTGATTG |
| WNB00122 | CCAGTCTTAAGCTCGGGCCCGTGATGAAAATGCCTTTG |
| WNB00165 | CGAACAAGGCGACTTCTATCTG |
| WNB00166 | GGAGATTATTAATCAGTACGGCGG |
| WNB00167 | GCTTAGGCGCCGAAG |
| WNB00127 | CATGTAATACGACTCACTATTGGATTTGGTAAAAACATTCAAAAG |
| WNB00128 | CCTCTGAAAAATTTACACCTCATCATCCTTTTTTATATTTTTAAC |
| WNB00129 | AGGTGTAAATTTTTCAGAGGAGAATAGATGTTTAAATTAAC |
| WNB00130 | CCAGTCTTAAGCTCGGGCCCTACGTCGGCATTGCAGAAG |
| WNB00171 | GCTTGTGAAATTGGCGGAG |
| WNB00172 | AAAGGCGACGCTGACATAC |
| WNB00173 | GTGTGTTAGGTCTTGTTGTAGC |
| WNB00137 | ATGTTCACCTCAATTGTATTTATC |
| WNB00205 | GGCGCGCCGGTGCCGTAT |
| WNB00218 | AATACAATTGAGGTGAACATATGACAAAAGAATATGCAACATTAGCAGG |
| WNB00220 | CGCGCCAGCATTTTGATTCCC |
| WNB00222 | AATACAATTGAGGTGAACATATGAATATTAATACAGCTTATTTTG |
| WNB00223 | GGATACGGCACCGGCGCGCCTTGCTTATTTTTGTATTCTTGG |
| WNB00224 | AATACAATTGAGGTGAACATATGGCAGTTGTTTATGTAGC |
| WNB00225 | GGATACGGCACCGGCGCGCCCTTGTATTTATTTAGCAAATCTTTTG |
| WNB00226 | AATACAATTGAGGTGAACATATGCTTAAAAAAGATAAAAGTGAAC |
| WNB00227 | GGATACGGCACCGGCGCGCCCTTATCAAAATGTGATATTAAATCG |
| WNB00080 | CCAAAGCGCTAACCTTTTAGC |
| WNB00081 | GTTAGCTCACTCATTAGGCACC |
S. aureus bacterial cultures.
Unless otherwise noted, all S. aureus strains were streaked on TSA and incubated 18 h at 37°C. Single colonies were picked, and 5 ml of TSB in 15-ml polypropylene tubes with aeration lids was inoculated. Tubes were incubated with shaking for 16 h at a 45° angle and 180 rpm in an Innova44 shaking incubator (Eppendorf, Hauppauge, NY) set to 37°C.
Allelic exchange.
Allelic exchange was performed using the pKOR1 plasmid as described with slight modifications to construct design and assembly (34). The pKOR1 vector was linearized by PCR using primers WNB00047 and WNB00048. The 1-kb upstream flanking region of msrA1 was PCR amplified using primers WNB00123 and WNB00186, while the 1-kb downstream flanking region was amplified with primers WNB00187 and WNB00188 for the construction of pKOR1.ΔmsrA1. The 1-kb upstream flanking region of msrA2 was PCR amplified using primers WNB00119 and WNB00120, while the 1-kb downstream flanking region was amplified with primers WNB00121 and WNB00122 for the construction of pKOR1.ΔmsrA2. The 1-kb upstream flanking region of msrA3 was PCR amplified using primers WNB00127 and WNB00128, while the 1-kb downstream flanking region was amplified with primers WNB00129 and WNB00130 for the construction of pKOR1.ΔmsrA3. The 1-kb upstream flanking region of msrA1 was PCR amplified using primers WNB00161 and WNB00162, while the 1-kb downstream flanking region of msrB was amplified with primers WNB00163 and WNB00164 for the construction of pKOR1.ΔmsrA1B. The 1-kb upstream flanking region of msrB was PCR amplified using primers WNB00189 and WNB00190, while the 1-kb downstream flanking region was amplified with primers WNB00191 and WNB00164 for the construction of pKOR1.ΔmsrB. All primers were designed using the NEBuilder tool and assembled using HiFi master mix as described by the manufacturer (New England BioLabs, Ipswich, MA).
Each pKOR1 plasmid was chemically transformed into Escherichia coli DH5α, checked by colony PCR using WNB00058 and WNB00059, electroporated into the cloning intermediate S. aureus RN4220 (35), followed by electroporation into S. aureus USA300 (20). Plasmids were isolated at each step using a Qiagen (Hilden, Germany) miniprep kit as described by the manufacturer. The plasmid isolated from E. coli DH5α was sequenced using primers WNB00058 and WNB00059 to confirm that the construct was correct. Following allelic exchange, genomic DNA was amplified across each deleted gene and validated by Sanger sequencing. Primers WNB00168 and WNB00169 were used to amplify across msrA1B, and primer WNB00170 was used in sequencing to confirm ΔmsrA1 and ΔmsrA1B, while WNB00202 was used in sequencing to confirm ΔmsrB. Primers WNB00165 and WNB00166 were used to amplify across msrA2, and primer WNB00167 was used in sequencing to confirm ΔmsrA2. Primers WNB00171 and WNB00172 were used to amplify across msrA3, and primer WNB00173 was used in sequencing to confirm ΔmsrA3. ΔmsrA was created by first making ΔmsrA2 with pKOR1.ΔmsrA2, then ΔmsrA2 ΔmsrA3 using pKOR1.ΔmsrA3, and then the final strain using pKOR1.ΔmsrA1. Δmsr was created by first making ΔmsrA2 with pKOR1.ΔmsrA2, then ΔmsrA2 ΔmsrA3 using pKOR1.ΔmsrA3, and then the final strain using pKOR1.ΔmsrA1B.
Msr activity assay.
Methionine sulfoxide reductase activity was quantified as previously described with some minor modifications (16, 19). USA300 wild type and Δmsr cell pellets were suspended in 500 μl phosphate-buffered saline (PBS) plus 10 mM MgCl2 plus 40 μg lysostaphin and incubated 30 min at 37°C. To lyse the cells, 500 μl 2% IGEPAL plus 800 μM phenylmethylsulfonyl fluoride (PMSF) was added to each sample and incubated 10 min on ice. Each sample was lysed by sonication with an ultrasonic Dismembrator 150E (Fisher Scientific, Waltham, MA) three times, resting on ice for 10 min between each round of sonication. Insoluble debris was pelleted in a microcentrifuge (Eppendorf) at maximum speed and 4°C for 10 min. Protein was quantified in the supernatant by using bicinchoninic acid (BCA) (Thermo Fisher). Each lysate was diluted to 1 mg/ml in 200 μl PBS prewarmed to 37°C. Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) was added to 100 mM and 9-fluorenylmethoxy carbonyl (Fmoc)-methionine sulfoxide was added to 1 mM to initiate the reaction. Samples were incubated with mixing for 30 min at 37°C and the reaction was quenched by adding 200 μl ice-cold methanol and placing the samples at −20°C for 2 h. Precipitated methionine sulfoxide reductase was pelleted in a microcentrifuge at maximum speed and 4°C for 15 min, and 10 μl of each sample and standard was analyzed on an Agilent 1260 Infinity II system. Analytes were separated by gradient high-pressure liquid chromatography (HPLC) on a Supelco Ascentis Express C18 column (50 by 2.1 mm, 5 μm) with a Phenomenex SecurityGuard C18 cartridge (3.2 by 8 mm) at a flow rate of 0.4 ml/min using 0.1% formic acid in water and 0.1% formic acid in acetonitrile as the A and B mobile phases, respectively. The gradient was held at 15% B for 0.5 min and then ramped to 100% B over the next 6.5 min. The column was washed at 100% B for 3 min and then equilibrated to 15% B for 45 min. Fmoc-methionine and Fmoc-methionine sulfoxide were detected using absorbance at 265 nm, and retention times were confirmed using each standard. Relative activity was determined by comparing the Fmoc-methionine area under the curve (AUC) for each sample lysate to the Fmoc-methionine AUC for the USA300 wild-type lysate.
Methionine sulfoxide blotting.
USA300 wild-type and Δmsr cell pellets were suspended in 500 μl PBS plus 10 mM MgCl2 plus 40 μg lysostaphin and incubated 30 min at 37°C. To lyse the cells, 500 μl 2% IGEPAL plus 800 μM PMSF was added to each sample and incubated 10 min on ice. Each sample was lysed by sonication with an ultrasonic Dismembrator 150E (Fisher Scientific, Waltham, MA) three times, resting on ice for 10 min between each round of sonication. Insoluble debris was pelleted in a microcentrifuge (Eppendorf) at maximum speed and 4°C for 10 min. Protein was quantified in the supernatant by using BCA (Thermo Fisher). Each lysate was diluted to 1 mg/ml in 200 μl PBS. NaOCl (Clorox) was added to 20 μM, and samples were incubated with mixing at 25°C for 30 min. SDS, iodoacetamide, and TCEP were added to each sample at 1%, 10 mM, and 2.5 mM, respectively, and incubated 5 min at 70°C. Ox4, a methionine-reactive clickable probe (21), was added to a final concentration of 500 μM and incubated with mixing at 25°C for 30 min. CuSO4, sodium ascorbate, azido-biotin, and Tris[(1-benzyl-4-triazolyl)methyl]amine (TBTA) were added to 1 mM, 1 mM, 1 mM, and 500 μM, respectively, and incubated with mixing at 25°C for 2 h to facilitate the attachment of azido-biotin to Ox4-modified proteins by copper-catalyzed azide-alkyne cycloaddition (click chemistry) (36). Proteins were separated from excess click chemistry reagents by adding 1 ml ice-cold acetone to each sample and incubating 15 min on ice followed by microcentrifugation at maximum speed and 4°C for 10 min. The protein pellet was washed once more with 1 ml ice-cold acetone. Proteins were suspended in 250 μl PBS plus 1% SDS and 50 μl 6× Laemmli buffer followed by sonication with an ultrasonic Dismembrator and incubation at 90°C for 10 min. Fifteen microliters of each sample was separated on a 4% to 20% polyacrylamide gel (Bio-Rad, Hercules, CA) at 120 V. The gel was transferred to 0.45-μm nitrocellulose (Bio-Rad) in a Trans-Blot Turbo (Bio-Rad) at 25 V for 18 min. Ponceau S staining was used to ensure consistent loading and complete transfer, and then the blot was blocked with PBS Odyssey blocking buffer (LI-COR, Lincoln, NE). Anti-sortase A (rabbit) primary (1°) antibody (37) and streptavidin IR-Dye 800CW (LI-COR) were diluted 20,000-fold and 10,000-fold, respectively, in PBS Odyssey blocking buffer, added to the blot, and rocked overnight at 4°C, shielded from light. After washing with PBS plus 0.1% Tween 20, anti-rabbit Alexa Fluor 680 (goat) secondary (2°) antibody (Invitrogen) and streptavidin IR-Dye 800CW were both diluted 10,000-fold in PBS Odyssey blocking buffer, added to the blot, and rocked 1 h at 25°C, shielded from light. After washing with PBS plus 0.1% Tween 20, the blot was imaged on a ChemiDoc MP (Bio-Rad). The streptavidin signal quantifies the amount of methionine (unoxidized) across the proteome, while the sortase A signal ensures equal loading of the samples.
S. aureus kinetic growth curves.
S. aureus overnight cultures were grown as described above and diluted 100-fold. Into each well of a 96-well plate, 100 μl of TSB containing the stressor to be tested and a 10-fold dilution of each bacterial strain were added giving a final bacterial dilution of 1,000-fold. All growth curves were performed on an EPOCH 2 plate reader (BioTek, Winooski, VT) set to 37°C and using linear shaking at 567 cpm (3 mm) for 24 h, taking the optical density at 600 nm every 30 min. All data were plotted and statistical analyses performed in Prism 7 (GraphPad, La Jolla, CA).
S. aureus kinetic growth curves treated with HOCl or H2O2.
S. aureus overnight cultures were grown in biological triplicates as described above and diluted 100-fold into PBS. Into each well of a 96-well plate, 50 μl of PBS containing vehicle, 100 μM NaOCl (Clorox, Oakland, CA), or 500 μM H2O2 and 10 μl of each bacterial strain were added. A range of HOCl and H2O2 concentrations were tested, but only the stated concentrations are displayed in the manuscript. After incubating 15 min at 25°C, 60 μl of TSB was added to each well, and the kinetic growth curve was started. All growth curves were performed on an EPOCH 2 plate reader (BioTek, Winooski, VT) set to 37°C and using linear shaking at 567 cpm (3 mm) for 24 h, taking the optical density at 600 nm every 30 min.
Constitutive expression of individual tagged Msr enzymes.
Complementation constructs expressing individual, tagged msr genes were created using the pOS1 vector (38). The pOS1 vector containing the lipoprotein diacylglyceryl transferase (lgt) promoter (39) and a sequence encoding a C-terminal Etag peptide tag (40) was linearized by PCR using primers WNB00137 and WNB00205. Each msr was amplified and the appropriate overhangs for HiFi assembly were added by PCR: msrA1 using primers WNB00218 and WNB00220, msrA2 using primers WNB00222 and WNB00223, msrA3 using primers WNB00224 and WNB00225, and msrB using WNB00226 and WNB00227. All primers were designed using the NEBuilder tool (New England BioLabs) and assembled into the final vector using HiFi master mix as described by the manufacturer. Each complementation vector was chemically transformed into E. coli DH5α, checked by colony PCR using WNB00080 and WNB00081, and electroporated into S. aureus RN4220 (35) followed by electroporation into S. aureus USA300 Δmsr. Plasmids were isolated at each step using a Qiagen miniprep kit as described by the manufacturer. Each plasmid isolated from DH5α was sequenced using primers WNB00080 and WNB00081 to make sure the construct was correct.
Western blotting for Etag-conjugated Msr enzymes.
USA300 pOS1.Plgt, Δmsr pOS1.Plgt, Δmsr pOS1.Plgt.msrA1.Etag, Δmsr pOS1.Plgt.msrA2.Etag, Δmsr pOS1.Plgt.msrA3.Etag, and Δmsr pOS1.Plgt.msrB.Etag cell pellets were suspended in 500 μl PBS plus 10 mM MgCl2 plus 40 μg lysostaphin and incubated 30 min at 37°C. To lyse the cells, 500 μl of 2% IGEPAL plus 800 μM PMSF was added to each sample and incubated 10 min on ice. Each sample was lysed by sonication with an ultrasonic Dismembrator 150E three times, resting on ice for 10 min between each round of sonication. Insoluble debris was pelleted in a microcentrifuge at maximum speed and 4°C for 10 min. Protein was quantified in the supernatant by using BCA. Lysates were diluted to 1 mg/ml with PBS and 6× Laemmli buffer followed by incubation at 90°C for 10 min. Ten microliters of each sample was separated on a 4% to 20% polyacrylamide gel at 120 V. The gel was transferred to 0.45-μm nitrocellulose (Bio-Rad) in a Trans-Blot Turbo at 25 V for 18 min. Ponceau S staining was used to ensure consistent loading and complete transfer, and then the blot was blocked with PBS Odyssey blocking buffer. Anti-sortase A (rabbit) 1° antibody (37) and anti-Etag (goat) 1° antibody (Abcam, Cambridge, United Kingdom) were diluted 20,000-fold and 1,000-fold, respectively, in PBS Odyssey blocking buffer, added to the blot, and rocked overnight at 4°C, shielded from light. After washing with PBS plus 0.1% Tween 20, anti-rabbit Alexa Fluor 680 2° antibody (Invitrogen) and anti-goat IR-Dye 800CW (LI-COR) were both diluted 10,000-fold in PBS Odyssey blocking buffer, added to the blot, and rocked 1 h at 25°C, shielded from light. After washing with PBS plus 0.1% Tween 20, the blot was imaged on a ChemiDoc MP (Bio-Rad). The Etag signal quantifies the amount of each tagged Msr, while the sortase A signal ensures equal loading of the samples.
S. aureus kinetic growth curves for strains constitutively expressing tagged Msr enzymes and treated with HOCl.
USA300 pOS1.Plgt, Δmsr pOS1.Plgt, Δmsr pOS1.Plgt.msrA1.Etag, Δmsr pOS1.Plgt.msrA2.Etag, Δmsr pOS1.Plgt.msrA3.Etag, and Δmsr pOS1.Plgt.msrB.Etag overnight cultures were grown in biological triplicates in TSB plus 10 μg/ml chloramphenicol and diluted 100-fold into PBS. Into each well of a 96-well plate, 100 μl of PBS containing vehicle, 100 μM NaOCl (Clorox, Oakland, CA), and 10 μl of each bacterial strain were added. After incubating 30 min at 25°C, 50 μl of TSB plus 0 μg/ml chloramphenicol was added to each well, and the kinetic growth curve was started. All growth curves were performed on an EPOCH 2 plate reader (Bio Tek, Winooski, VT) set to 37°C and using linear shaking at 567 cpm (3 mm) for 24 h, taking the optical density at 600 nm every 30 min.
S. aureus kinetic growth curves treated with superoxide anion generating compounds.
S. aureus overnight cultures were grown in six biological replicates for paraquat and four biological replicates for pyocyanin as described above and diluted 100-fold into TSB. Into each well of a 96-well plate, 100 μl of TSB containing vehicle, 3.2 mM paraquat, or 16 μg/ml pyocyanin and 10 μl of each bacterial strain were added, and the kinetic growth curve was started. A range of paraquat and pyocyanin concentrations were tested, but only the stated concentrations are displayed in the manuscript. All growth curves were performed on an EPOCH 2 plate reader (Bio Tek, Winooski, VT) set to 37°C and using linear shaking at 567 cpm (3 mm) for 24 h, taking the optical density at 600 nm every 30 min.
HOCl CFU plating.
S. aureus overnight cultures were grown in biological triplicates as described above and diluted 100-fold into PBS. Into each well of a 96-well plate, 100 μl of PBS containing 0, 12.5, 25, or 50 μM NaOCl and 10 μl of each bacterial strain were added. After incubating 30 min at 25°C, each well was diluted, 100 through 10−7 in PBS in 10-fold intervals. Each dilution was spot plated (10 μl) on TSA and incubated overnight at 37°C followed by CFU enumeration.
Catalase activity assay.
USA300 wild-type, Δmsr, JE2 wild-type (41), and JE2 with katA disrupted by transposon insertion (41) overnight cultures were grown in biological triplicates as described above. Each overnight culture (1 ml) was pelleted by microcentrifugation and suspended in 200 μl PBS plus 10 mM MgCl2 plus 10 μg lysostaphin and incubated 15 min at 37°C. Two hundred microliters PBS plus 2% IGEPAL plus 500 μM PMSF was added to each sample and incubated 10 min on ice. Each sample was lysed by sonication with an ultrasonic Dismembrator 150E two times, resting on ice for 10 min between each round of sonication. Insoluble debris was pelleted in a microcentrifuge at maximum speed and 4°C for 10 min. Protein was quantified in the supernatant by using BCA. Each lysate was diluted to 0.25 mg/ml in 200 μl PBS. Catalase activity was assayed in technical triplicates as described in the Cayman catalase activity assay (707002).
Murine retro-orbital infection with USA300 wild-type and Δmsr strains.
USA300 wild-type and Δmsr overnight cultures grown as described above were diluted 100-fold in TSB and grown for 3 h at 37°C on a roller drum at setting 6 to reach mid-exponential phase. Cells were pelleted by centrifugation at 4°C and washed with 10 ml ice-cold PBS. The 600-nm optical density of each sample was adjusted to 0.4 with ice-cold PBS, and CFU for each sample was quantified by dilution plating to ensure a correct inoculum. Eight-week-old female BALB/cJ mice (Jackson Laboratory, Bar Harbor, ME) were anesthetized intraperitoneally with 2,2,2-tribromoethanol diluted in PBS. Following anesthetization, the mice were injected retro-orbitally with 4 × 107 CFU of each culture in 100 μl PBS. Mice were monitored twice per day and weighed once per day for 4 or 10 days. On the fourth or tenth day, the mice were euthanized by CO2 asphyxiation, and the heart, kidneys, liver, lungs, and spleen were removed. Livers were added individually to Whirl-Pak bags (Nasco, Fort Atkinson, WI) containing 1 ml PBS and homogenized with 100 strokes of a rolling pin at 4°C. Heart, kidneys, and spleen were added individually to 1.5-ml NAVY Bullet Blender tubes containing 550 μl PBS and homogenized using a Bullet Blender (Next Advance, Troy, NY) at 4°C on setting 8 for 5 min and then on setting 12 for 5 min. Lungs were added individually to 1.5-ml NAVY Bullet Blender tubes containing 800 μl PBS and homogenized on a Bullet Blender at 4°C on setting 8 for 5 min and then on setting 12 for 5 min. Each sample was serially diluted in 10-fold intervals in PBS. Each dilution was spot plated on TSA and incubated overnight at 37°C followed by CFU enumeration.
Murine intraperitoneal infection with USA300 wild-type and Δmsr strains.
USA300 wild-type and Δmsr overnight cultures grown as described above were diluted 100-fold in TSB and grown for 3 h at 37°C with orbital shaking to reach mid-exponential phase. Cells were pelleted by centrifugation at 4°C and washed with 10 ml ice-cold PBS. The 600-nm optical density of each sample was adjusted to 0.4 with ice-cold PBS, and CFU for each sample was quantified by dilution plating to ensure a correct inoculum. Eight-week-old female BALB/cJ mice were injected intraperitoneally with 4 × 107 CFU of each culture in 100 μl PBS. Mice were monitored twice per day and weighed once per day for 4 days. On the fourth day, the mice were euthanized by CO2 asphyxiation, and the heart, kidneys, liver, lungs, and spleen were removed. Livers were added individually to 5-ml NAVY Bullet Blender tubes containing 1 ml PBS and homogenized using a Bullet Blender at 4°C twice on setting 8 for 5 min. Heart, kidneys, and spleen were added individually to 1.5-ml NAVY Bullet Blender tubes containing 550 μl PBS and homogenized using a Bullet Blender at 4°C on setting 8 for 5 min and then on setting 12 for 5 min. Lungs were added individually to 1.5-ml NAVY Bullet Blender tubes containing 800 μl PBS and homogenized using a Bullet Blender at 4°C on setting 8 for 5 min and then on setting 12 for 5 min. Each sample was serially diluted in 10-fold intervals in PBS. Each dilution was spot plated on TSA and incubated overnight at 37°C followed by CFU enumeration.
Murine skin infection with USA300 wild-type and Δmsr strains.
USA300 wild-type and Δmsr overnight cultures grown as described above were diluted 100-fold in TSB and grown for 3 h at 37°C with orbital shaking to reach mid-exponential phase. Cells were pelleted by centrifugation at 4°C and washed with 10 ml ice-cold PBS. The 600-nm optical density of each sample was adjusted to 1.8 with ice-cold PBS, and CFU for each sample was quantified by dilution plating to ensure a correct inoculum. Eight-week-old female BALB/cJ mice were anesthetized by isoflurane inhalation. Fur was removed and a skin wound was created on the back of each mouse with 15 pulls of Tensoplast medical tape (BSN Medical, Hamburg, Germany). Each wound was inoculated with 1 × 107 CFU in 5 μl PBS. Mice were monitored twice per day and weighed once per day for 2 days. On the second day, the mice were euthanized by CO2 asphyxiation. The skin wounds were excised, weighed, and added individually to 1.5-ml NAVY Bullet Blender tubes containing 500 μl PBS and homogenized on a Bullet Blender at 4°C 4× on setting 12 for 5 min each. Each sample was serially diluted in 10-fold intervals in PBS. Each dilution (10 μl) was spot plated on TSA and mannitol salt agar (MSA) and incubated overnight at 37°C followed by CFU enumeration and normalization to the weight of excised skin. CFU on both TSA and MSA were the same, indicating that the wound was not contaminated with other bacteria during the infection, and so the CFU numbers reported are from TSA.
Human neutrophil killing.
USA300 wild-type and Δmsr overnight cultures grown as described above were diluted 100-fold in TSB and grown for 3 h at 37°C with orbital shaking to reach mid-exponential phase. Cells were pelleted by centrifugation at 4°C and washed with 10 ml ice-cold PBS. The 600-nm optical density of each sample was adjusted to 1 with ice-cold RPMI 1640 medium (Corning) plus 10 mM HEPES (Corning) plus 0.1% human serum albumin (HSA). Twenty percent pooled normal human serum (NHS) (Seracare Lifesciences Inc., Milford, MA) was added to each sample, and the bacteria were opsonized for 30 min at 37°C with orbital shaking. Following opsonization, the bacteria were pelleted by centrifugation and washed twice with RPMI medium plus 10 mM HEPES plus 0.1% HSA. Each sample was suspended in 800 μl RPMI medium plus 10 mM HEPES plus 0.1% HSA to reach a cell density of 2.5 × 108 CFU/ml.
Each well of a 96-well plate was coated with 100 μl of 20% pooled NHS in RPMI medium plus 10 mM HEPES plus 0.1% HSA and incubated 30 min at 37°C in 5% CO2. Each well was then washed twice with RPMI medium plus 10 mM HEPES plus 0.1% HSA. After isolation from human leukopacks (42), the polymorphonuclear neutrophils (PMNs) were plated at 2.5 × 105 cells per well in 90 μl of RPMI medium plus 10 mM HEPES plus 0.1% HSA and incubated 30 min at 37°C in 5% CO2 to allow the PMNs to adhere. To reach a multiplicity of infection (MOI) of 1, 10 μl of a 10-fold dilution of each bacterial strain was added to each well. The plates were synchronized at 1,500 rpm for 5 min and then incubated at 37°C in 5% CO2 for 1, 2, 3, 4, or 6 h. At each time point, 11 μl of 1% saponin was added to each well, and plates were incubated on ice for 20 min to lyse the PMNs. Each well was pipetted extensively and dilution plated on TSA. Plates were incubated overnight for CFU enumeration.
Murine neutrophil killing.
USA300 wild-type and Δmsr overnight cultures grown as described above were diluted 100-fold in non-heat-inactivated fetal bovine serum (FBS) and placed on ice for 1 h to allow for opsonization. Bone marrow was isolated from the femurs and tibias of six 8-week-old female BALB/cJ mice. Neutrophils were isolated from the bone marrow using density centrifugation (43), and 104 neutrophils were transferred to a low-attachment, round-bottom 96-well plate to rest for 1 h at 37°C and 5% CO2 in 250 μl D10 medium (Dulbecco’s modified Eagle medium [DMEM] plus 10% FBS). Opsonized bacteria were transferred to the plates containing neutrophils (MOI = 1) or lacking neutrophils and incubated at 37°C and 5% CO2. At 0, 1, 2, 4, 6, and 8 h, the wells containing bacteria were serially diluted, and the dilutions were spot plated to TSA and incubated overnight at 37°C followed by CFU enumeration.
Human whole-blood killing.
USA300 wild-type and Δmsr overnight cultures grown as described above were diluted 100-fold in TSB and grown for 3 h at 37°C with orbital shaking to reach mid-exponential phase. Cells were pelleted by centrifugation at 4°C and washed with 10 ml ice-cold PBS. The 600-nm optical density of each sample was adjusted to 1 with PBS (1 × 109 CFU/ml). Each bacterial strain was further diluted 50-fold in PBS. Venous blood was collected from eight healthy donors (3 ml per donor) in Hirudin-coated tubes (Roche, Basel, Switzerland). Five microliters of bacteria was combined with 95 μl of blood to give an MOI of 1 based on the calculation that human blood has 1 × 106 PMN/ml. Tubes were held at a 90° angle on a tube revolver (Thermo Fisher) and mixed by circular rotation at speed 10 at 37°C for 1, 2, 4, or 6 h. At each time point, the samples were combined 1:1 with 2% saponin plus 200 U/ml streptokinase plus 1 mg/ml trypsin plus 0.02 mg/ml DNase 1 plus 0.1 mg/ml RNase on ice. Samples were incubated with orbital shaking at 37°C for 10 min and then rocked at 4°C for 10 min. Each sample was dilution plated on TSA and incubated overnight at 37°C for CFU enumeration.
Murine whole-blood killing.
USA300 wild-type and Δmsr overnight cultures were grown as described above and then put on ice. Cells were pelleted by centrifugation at 4°C and washed with 10 ml ice-cold PBS. The 600-nm optical density of each sample was adjusted to 1 with PBS (1 × 109 CFU/ml). Each bacterial strain was further diluted 50-fold in PBS. Eight 8-week-old female BALB/cJ mice were anesthetized intraperitoneally with 150 μl of 7.5 mg/ml ketamine plus 1.5 mg/ml xylazine in saline. Blood was drawn from each mouse by cardiac puncture into K3EDTA collection tubes. The blood was rested on ice for 1 h. Human and murine neutrophils have different immune cell compositions (28), and so to be able to compare the two whole-blood experiments, the ratio of blood to bacteria was kept the same as in the human whole-blood killing experiments. Three microliters of bacteria was combined with 27 μl of murine whole blood, and samples were mixed by orbital shaking at 37°C for 1, 2, 4, or 6 h. At each time point, the samples were dilution plated on TSA and incubated overnight at 37°C for CFU enumeration.
ACKNOWLEDGMENTS
We thank the Skaar laboratory for critical reading and feedback on the manuscript.
This work was funded by R01 AI069233 (E.P.S.), R01 Al073843 (E.P.S.), R01 AI099394 (V.J.T.), R01 AI105129 (V.J.T.), R01 AI137336 (V.J.T.), T32 HL069765 (W.N.B.), T32 AI007474 (W.N.B.), 18POST34030426 (W.N.B.), F32 HL144081 (A.J.M.), F32 AI157215 (A.W.), R01 GM139245 (C.J.C.), NSF-GRFP DGE 1106400 (A.H.C.), and R35 GM118190 (F.D.T.). V.J.T. is a Burroughs Wellcome Fund Investigator in the pathogenesis of infectious diseases.
Footnotes
Supplemental material is available online only.
Contributor Information
Eric P. Skaar, Email: eric.skaar@vumc.org.
Nancy E. Freitag, University of Illinois at Chicago
REFERENCES
- 1.Tong SY, Davis JS, Eichenberger E, Holland TL, Fowler VG, Jr.. 2015. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev 28:603–661. 10.1128/CMR.00134-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Graham PL, III, Lin SX, Larson EL. 2006. A U.S. population-based survey of Staphylococcus aureus colonization. Ann Intern Med 144:318–325. 10.7326/0003-4819-144-5-200603070-00006. [DOI] [PubMed] [Google Scholar]
- 3.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK, Active Bacterial Core Surveillance MRSA Investigators . 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771. 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 4.Zimlichman E, Levin-Scherz J. 2013. The coming golden age of disruptive innovation in health care. J Gen Intern Med 28:865–867. 10.1007/s11606-013-2335-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.CDC. 2019. Antibiotic resistance threats in the United States. https://www.cdc.gov/DrugResistance/Biggest-Threats.html. Accessed 1 March 2021.
- 6.WHO. 2017. Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. https://www.who.int/medicines/publications/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf.
- 7.Hampton MB, Kettle AJ, Winterbourn CC. 1996. Involvement of superoxide and myeloperoxidase in oxygen-dependent killing of Staphylococcus aureus by neutrophils. Infect Immun 64:3512–3517. 10.1128/IAI.64.9.3512-3517.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson MM, Requena JR, Crowley JR, Thorpe SR, Heinecke JW. 1999. The myeloperoxidase system of human phagocytes generates N-epsilon-(carboxymethyl)lysine on proteins: a mechanism for producing advanced glycation end products at sites of inflammation. J Clin Invest 104:103–113. 10.1172/JCI3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sawyer DT, Valentine JS. 1981. How super is superoxide. Acc Chem Res 14:393–400. 10.1021/ar00072a005. [DOI] [Google Scholar]
- 10.Noto MJ, Burns WJ, Beavers WN, Skaar EP. 2017. Mechanisms of pyocyanin toxicity and genetic determinants of resistance in Staphylococcus aureus. J Bacteriol 199:e00221-17. 10.1128/JB.00221-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ezraty B, Aussel L, Barras F. 2005. Methionine sulfoxide reductases in prokaryotes. Biochim Biophys Acta 1703:221–229. 10.1016/j.bbapap.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 12.Holliday GL, Mitchell JB, Thornton JM. 2009. Understanding the functional roles of amino acid residues in enzyme catalysis. J Mol Biol 390:560–577. 10.1016/j.jmb.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 13.Valley CC, Cembran A, Perlmutter JD, Lewis AK, Labello NP, Gao J, Sachs JN. 2012. The methionine-aromatic motif plays a unique role in stabilizing protein structure. J Biol Chem 287:34979–34991. 10.1074/jbc.M112.374504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewis AK, Dunleavy KM, Senkow TL, Her C, Horn BT, Jersett MA, Mahling R, McCarthy MR, Perell GT, Valley CC, Karim CB, Gao J, Pomerantz WC, Thomas DD, Cembran A, Hinderliter A, Sachs JN. 2016. Oxidation increases the strength of the methionine-aromatic interaction. Nat Chem Biol 12:860–866. 10.1038/nchembio.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cirino PC, Tang Y, Takahashi K, Tirrell DA, Arnold FH. 2003. Global incorporation of norleucine in place of methionine in cytochrome P450 BM-3 heme domain increases peroxygenase activity. Biotechnol Bioeng 83:729–734. 10.1002/bit.10718. [DOI] [PubMed] [Google Scholar]
- 16.Moskovitz J, Singh VK, Requena J, Wilkinson BJ, Jayaswal RK, Stadtman ER. 2002. Purification and characterization of methionine sulfoxide reductases from mouse and Staphylococcus aureus and their substrate stereospecificity. Biochem Biophys Res Commun 290:62–65. 10.1006/bbrc.2001.6171. [DOI] [PubMed] [Google Scholar]
- 17.Singh K, Singh VK. 2012. Expression of four methionine sulfoxide reductases in Staphylococcus aureus. Int J Microbiol 2012:719594. 10.1155/2012/719594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh VK, Vaish M, Johansson TR, Baum KR, Ring RP, Singh S, Shukla SK, Moskovitz J. 2015. Significance of four methionine sulfoxide reductases in Staphylococcus aureus. PLoS One 10:e0117594. 10.1371/journal.pone.0117594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pang YY, Schwartz J, Bloomberg S, Boyd JM, Horswill AR, Nauseef WM. 2014. Methionine sulfoxide reductases protect against oxidative stress in Staphylococcus aureus encountering exogenous oxidants and human neutrophils. J Innate Immun 6:353–364. 10.1159/000355915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tenover FC, McDougal LK, Goering RV, Killgore G, Projan SJ, Patel JB, Dunman PM. 2006. Characterization of a strain of community-associated methicillin-resistant Staphylococcus aureus widely disseminated in the United States. J Clin Microbiol 44:108–118. 10.1128/JCM.44.1.108-118.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin S, Yang X, Jia S, Weeks AM, Hornsby M, Lee PS, Nichiporuk RV, Iavarone AT, Wells JA, Toste FD, Chang CJ. 2017. Redox-based reagents for chemoselective methionine bioconjugation. Science 355:597–602. 10.1126/science.aal3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Day BJ, Shawen S, Liochev SI, Crapo JD. 1995. A metalloporphyrin superoxide dismutase mimetic protects against paraquat-induced endothelial cell injury, in vitro. J Pharmacol Exp Ther 275:1227–1232. [PubMed] [Google Scholar]
- 23.Pizzolla A, Hultqvist M, Nilson B, Grimm MJ, Eneljung T, Jonsson IM, Verdrengh M, Kelkka T, Gjertsson I, Segal BH, Holmdahl R. 2012. Reactive oxygen species produced by the NADPH oxidase 2 complex in monocytes protect mice from bacterial infections. J Immunol 188:5003–5011. 10.4049/jimmunol.1103430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cho JS, Pietras EM, Garcia NC, Ramos RI, Farzam DM, Monroe HR, Magorien JE, Blauvelt A, Kolls JK, Cheung AL, Cheng G, Modlin RL, Miller LS. 2010. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J Clin Invest 120:1762–1773. 10.1172/JCI40891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosen H, Klebanoff SJ, Wang Y, Brot N, Heinecke JW, Fu X. 2009. Methionine oxidation contributes to bacterial killing by the myeloperoxidase system of neutrophils. Proc Natl Acad Sci U S A 106:18686–18691. 10.1073/pnas.0909464106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doeing DC, Borowicz JL, Crockett ET. 2003. Gender dimorphism in differential peripheral blood leukocyte counts in mice using cardiac, tail, foot, and saphenous vein puncture methods. BMC Clin Pathol 3:3. 10.1186/1472-6890-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mestas J, Hughes CC. 2004. Of mice and not men: differences between mouse and human immunology. J Immunol 172:2731–2738. 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 28.Bjornson-Hooper ZB, Fragiadakis GK, Spitzer MH, Madhireddy D, McIlwain D, Nolan GP. 11 March 2019. A comprehensive atlas of immunological differences between humans, mice, and non-human primates bioRxiv 10.1101/574160. [DOI] [PMC free article] [PubMed]
- 29.Beavers WN, Skaar EP. 2016. Neutrophil-generated oxidative stress and protein damage in Staphylococcus aureus. Pathog Dis 74:ftw060. 10.1093/femspd/ftw060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaupp R, Ledala N, Somerville GA. 2012. Staphylococcal response to oxidative stress. Front Cell Infect Microbiol 2:33. 10.3389/fcimb.2012.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hampton MB, Kettle AJ, Winterbourn CC. 1998. Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood 92:3007–3017. 10.1182/blood.V92.9.3007. [DOI] [PubMed] [Google Scholar]
- 32.Parker D. 2017. Humanized mouse models of Staphylococcus aureus infection. Front Immunol 8:512. 10.3389/fimmu.2017.00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim HK, Missiakas D, Schneewind O. 2014. Mouse models for infectious diseases caused by Staphylococcus aureus. J Immunol Methods 410:88–99. 10.1016/j.jim.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bae T, Schneewind O. 2006. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55:58–63. 10.1016/j.plasmid.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 35.Kreiswirth BN, Lofdahl S, Betley MJ, O'Reilly M, Schlievert PM, Bergdoll MS, Novick RP. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:709–712. 10.1038/305709a0. [DOI] [PubMed] [Google Scholar]
- 36.Beavers WN, Rose KL, Galligan JJ, Mitchener MM, Rouzer CA, Tallman KA, Lamberson CR, Wang X, Hill S, Ivanova PT, Brown HA, Zhang B, Porter NA, Marnett LJ. 2017. Protein modification by endogenously generated lipid electrophiles: mitochondria as the source and target. ACS Chem Biol 12:2062–2069. 10.1021/acschembio.7b00480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mazmanian SK, Ton-That H, Su K, Schneewind O. 2002. An iron-regulated sortase anchors a class of surface protein during Staphylococcus aureus pathogenesis. Proc Natl Acad Sci U S A 99:2293–2298. 10.1073/pnas.032523999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schneewind O, Model P, Fischetti VA. 1992. Sorting of protein A to the staphylococcal cell wall. Cell 70:267–281. 10.1016/0092-8674(92)90101-h. [DOI] [PubMed] [Google Scholar]
- 39.Beavers WN, Monteith AJ, Amarnath V, Mernaugh RL, Roberts LJ, II, Chazin WJ, Davies SS, Skaar EP. 2019. Arachidonic acid kills Staphylococcus aureus through a lipid peroxidation mechanism. mBio 10:e01333-19. 10.1128/mBio.01333-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davies SS, Talati M, Wang X, Mernaugh RL, Amarnath V, Fessel J, Meyrick BO, Sheller J, Roberts LJ, II.. 2004. Localization of isoketal adducts in vivo using a single-chain antibody. Free Radic Biol Med 36:1163–1174. 10.1016/j.freeradbiomed.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 41.Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW. 2013. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 4:e00537-12. 10.1128/mBio.00537-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reyes-Robles T, Lubkin A, Alonzo F, III, Lacy DB, Torres VJ. 2016. Exploiting dominant-negative toxins to combat Staphylococcus aureus pathogenesis. EMBO Rep 17:428–440. 10.15252/embr.201540994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferrante A, Thong YH. 1980. Optimal conditions for simultaneous purification of mononuclear and polymorphonuclear leucocytes from human blood by the Hypaque-Ficoll method. J Immunol Methods 36:109–117. 10.1016/0022-1759(80)90036-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download IAI00146-21_Supp_1_seq14.pdf, PDF file, 1.2 MB (1.2MB, pdf)