

Abstract
The genesis of designing bivalent or bitopic molecules that engender unique pharmacological properties began with Portoghese’s work directed toward opioid receptors, in the early 1980’s. This strategy has evolved as an attractive way to engineer highly selective compounds for targeted G-protein coupled receptors (GPCRs) with optimized efficacies and/or signaling bias. The emergence of X-ray crystal structures of many GPCRs and the identification of both orthosteric and allosteric binding sites have provided further guidance to ligand drug design that includes a primary pharmacophore (PP), a secondary pharmacophore (SP) and a linker between them. It is critical to note the synergistic relationship among all three of these components as they contribute to the overall interaction of these molecules with their receptor proteins, and that strategically designed combinations have and will continue to provide the GPCR molecular tools of the future.
Graphical Abstract
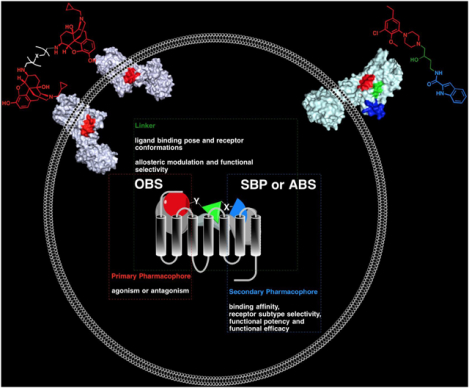
INTRODUCTION: BIVALENT LIGANDS
Bivalent ligands directed toward opioid receptors were first introduced into the literature by none other than Phil Portoghese, in 1982.1 These molecules were defined as having two pharmacophores linked by a “connecting chain” or “spanner” which in this Perspective will be referred to as a “linker”.2 Specifically, these pharmacophores are what we now designate as primary pharmacophores (PP), with each terminus targeting the orthosteric binding site (OBS) of the targeted receptor – in this early case, opioid receptors. Portoghese posited that these bivalent molecules would exhibit increased potency over the monovalent analogue due to a gain in entropic energy provided the linker was sufficiently long to reach the OBS of two adjacent opioid receptors (Figure 1). Further, it was envisioned that the linker length could serve as a molecular yardstick to determine the distance between these two sites when bound by the bivalent ligand.
Figure 1.
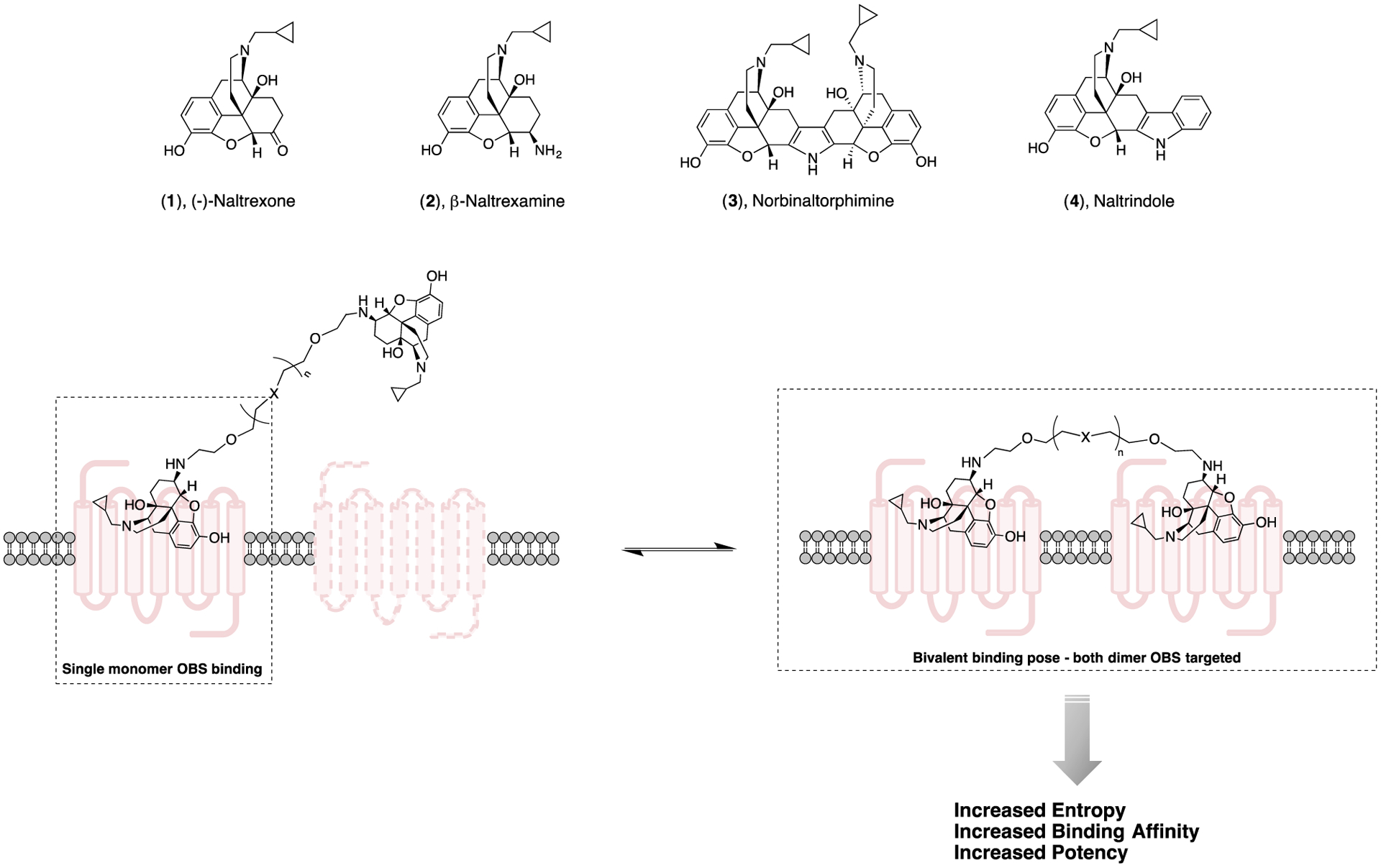
Bivalent ligand cartoon with naltrexone (1), β-naltrexamine (2), norbinaltorphimine (3) and naltrindole (4). The bivalent ligand is represented while engaging in binding with GPCR monomer and homo/hetero-dimer.
In this Perspective we will not attempt to review every bivalent or bitopic ligand designed and investigated, but rather start with a historical view of how the pioneering efforts of Portoghese have influenced an enormous and exciting field of drug design. We will provide the rationale for designing bivalent and/or bitopic molecules and hone into how these early discoveries have influenced our own drug design on dopamine D2-like compounds. Specifically, we will discuss the importance and influence of the PP, the secondary pharmacophore (SP) and the linker between them. Ultimately, we will propose that it is the fine-tuned design of the entire molecule, which has ultimately lead to remarkable tools that have helped shape our thinking regarding G-protein coupled receptor (GPCR) structure and function and will undoubtedly inform the next generation of therapeutic agents.
Of note, for the purpose of this perspective, the term “bivalent” molecule will refer to a molecule that has a PP on both ends of a linker that will vary in size/length and may be rigid or flexible. It is worth noting that the PPs maybe be identical (homobivalent) or distinct (heterobivalent). In contrast, “bitopic” molecules consist of a PP and a SP that is structurally different and is expected to interact with a secondary binding pocket (SBP) that results in a unique pharmacological profile of the molecule, either through receptor selectivity, functional selectivity and/or allostery. The PP and SP are connected by a linker of defined length and molecular composition. The role of the linker once was considered to only keep the PPs, in the case of bivalent molecules, or the PP and the SP, in the case of bitopic molecules, at a specific distance from one another. However, herein we propose that the linker not only plays a critical role in the proximity of the two pharmacophores, but its chemical composition is also fundamental in the overall pharmacological profile of the molecule.
History: the Portoghese opioid receptor bivalent ligand legacy
The initial work on bivalent molecules directed toward opioid receptors was done on a 6-amino-derivative of naltrexone (1), naltrexamine (2), and varying length polyethyleneglycol linkers.1 Using the guinea pig ileum and mouse vas deferens models as measures of “narcotic antagonistic activity” against the opioid agonists morphine (mu), ethylketocyclazocine (kappa) or DADLE (delta), the authors concluded that the potencies they measured with two of the bivalent ligands exceeded those of the monovalent ligands and were dependent on the “spanner” length.1 Subsequently, Portoghese expanded this idea to agonists with different linkers and began to see significant differences in potencies and efficacies.3–5 It must be noted that these compounds were designed before the concepts of receptor oligomers had been proposed as functional units. Clearly, over time the conceptual framework has evolved, but the practice of using bivalent (and bitopic) molecules to tweak affinities and efficacies remains relevant.
Early on, the opioid receptor subtypes (mu, kappa, delta) posed a medicinal chemistry challenge of determining structure-activity relationships (SAR) that could be delineated between receptor subtypes, so as to synthesize highly subtype selective ligands. One approach to this challenge was to design bivalent, non-peptide opioid antagonists.6 The concept that high receptor subtype selectivity could be achieved with bivalent molecules was predicated on a critical role of the linker. Even before data emerged supporting the existence of homomers (sometimes referred to as homodimers and existing as two copies of the same protein that are functionally linked) or heteromers (sometimes referred to as heterodimers and consist of one copy of two different proteins that are functionally linked), Portoghese hypothesized that close spatial arrangements of neighboring OBSs could be bridged with the binding of a bivalent ligand, and when these vicinal sites were both bound by the same ligand, affinity would be improved (Figure 1). Indeed, a second potential way of binding would be where one terminus binds to the OBS and the other terminus, despite being structurally a PP, binds to a secondary site, within the same receptor, which would also convey higher affinity binding due to additional contact points. The clever approach to testing this hypothesis was to synthesize two bivalent ligands for comparison, one with both termini having the active (−)-stereochemistry, one with an active (−)-1 terminus and one inactive (+)-1 terminus. Although the latter molecule had higher affinity for the mu opioid receptors than the monovalent compound, it was 1/30th as active as the (−)/(−) bivalent ligand, suggesting that the (+)-terminus was interacting with another site, which conveyed favorable interactions, but was not as favorable as the (−)/(−) bivalent molecule. At this time, another scenario was not contemplated and that is if one terminus binds to the OBS, by virtue of being tethered, the second terminus is in close proximity to the OBS, so that when one pharmacophore dissociates from the receptor, the other could readily bind. In this way, affinity for the receptor in the (−)/(−) bivalent molecule would also be improved entropically (Figure 1).
Interestingly, when (−)-1 was fused to another (−)-1 via a pyrrole ring to give norbinaltorphimine (3; Figure 1), a highly potent and selective kappa receptor antagonist resulted.6 Cleverly, the meso isomer was also synthesized, in which (−)-1 was fused identically with (+)-1, and discovered to be 5 times as potent as 3 (norbinaltorphimine) in the smooth muscle preparations, suggesting that both termini were not binding to vicinal receptors, but rather the whole molecule that combined both termini with the rigid linker conferred a highly selective and potent kappa antagonist.7–8 It was concluded that one end binds to what we now refer to as the OBS and the other end binds to an SBP that is unique to kappa receptors. Portoghese suggested that this model resembled the “message-address” concept first presented by Schwyzer9 wherein the “message” is the PP, binding to the OBS, whereas the “address” is the SP and confers additional binding interactions that can improve affinity and selectivity for the target kappa opioid receptor. He went on to discover naltrindole (4) (Figure 1), a highly selective and potent bivalent delta antagonist.10 Remarkably, these tools are still used today.
BIVALENT MOLECULES FOR OTHER GPCRs TO STUDY HOMOMERS AND HETEROMERS
GPCR oligomers, in the form of dimers (homomers or heteromers) have been proposed as the “actual targets” of neurotransmitters and molecules that bind to the OBS.11 Although biochemical evidence suggested the existence of receptor dimers, it was not until the 1990’s that this concept gained traction,12 well after the first bivalent ligands were described. Indeed, the first opioid receptor dimerization was described for the delta opioid receptor subtype in 199713 and later confirmed using time resolved fluorescence resonance energy transfer (FRET) and bioluminescence resonance energy transfer (BRET) techniques.14 Shortly after the Devi group described delta opioid receptor dimers, they discovered that heteromers consisting of delta and kappa opioid receptors form functional heteromers, opening a whole new vista for GPCR research,15 but also the possibility that bivalent molecules could be designed to specifically target receptor heteromers that would only exist in discrete brain regions, hence potentially affording site-directed therapeutic agents.16 The notable success of Portoghese’s pioneering bivalent approach with the opioid receptors prompted multiple efforts to find the applicability of these bivalent molecules in various different GPCR systems. GPCRs functioning in these oligomeric states produce distinct signaling responses from the monomers alone,17 consequently bivalent molecules have provided powerful molecular tools for the study of these GPCRs.18–19
Following Portoghese’s footsteps, LeBoulluec et al. reported the first bivalent ligands targeting serotonin (5-HT) receptors using the 5-carboxamidoindole moiety as the PP in a series of homobivalent ligands as early as 1995, reporting increased affinity towards the 5-HT1A and 5-HT1D receptors and increased selectivity for the 5-HT1D receptor over the PP alone.20 At this time the improved affinity was not attributed to the targeting of receptor dimers, likely due to the lack of evidence for GPCR dimerization available at the time. These efforts were quickly driven forward by Halazy et al. to expand the use of bivalent molecules in the study of the serotonergic system21 developing a vast array of high affinity selective ligands. Since then, evidence of oligomerization among the 5-HT receptors has become increasingly available22 leading to increased efforts toward discovering novel bivalent ligands with increased subtype selectivity capable of discriminating among receptor subtypes.23–29 Likewise, bivalent ligands have also played a prominent role in the study of cannabinoid receptors (CB1/2), starting with the efforts of Zhang et al.30 a large library of bivalent ligands have been developed to study homo- and heteromers formed by the cannabinoid receptors.31–33 Despite remaining unclear whether current bivalent ligands of the cannabinoid receptor system do indeed simultaneously bind two receptors within a dimer as postulated,34 bivalent ligands remain attractive tools in the study of these receptor complexes.35–38
Heteromers of neurotransmitter receptors are thought to play an important role in neurotransmission within the central nervous system (CNS),39–40 as a result, these can be potential targets for a variety of therapeutic applications. Indeed, the prevalence of heteromers within the adrenergic receptor systems41–44 make them interesting targets for the treatment of various cardiovascular ailments.45–47 For example, Karellas et al. introduced heterobivalent molecules targeting beta2-adrenergic receptor/adenosine A1 receptor (β2AR/A1AR) heteromers in an effort to understand the cross talk among these two cellular cAMP regulating receptors;48 it was proposed that careful balance between the signaling pathways generated by the two receptor monomers conforming this heteromer could aid in the prevention of certain cardiac arrhythmias.47 Due to the importance of functions regulated by this receptor system, it has become the focus of significant analysis via heterobivalent ligands.49
The study of dopamine receptor heteromers with the use of heterobivalent ligands is equally promising. Adenosine A2A receptor (A2AR) and dopamine D2 receptor heteromers (D2R/A2AR) are of particular interest due to their purported role in Parkinson’s Disease.50–51 Indeed, the potential of dopamine receptor agonists as agents to treat Parkinson’s disease is well documented.52 Moreover, it has been hypothesized that antagonism at A2AR in conjunction with agonism at the D2R has the potential to increase the therapeutic utility of the D2R agonists by counteracting some of the common side effects and helping prevent disease progression.53–55 As a result, significant focus has been directed at heterobivalent ligands that can target these heteromers. For example, Soriano et al. reported the synthesis of heterobivalent ligands utilizing XCC (2-(4-(2,6-dioxo-1,3-dipropyl-2,3,4,5,6,7-hexahydro-1H-purin-8-yl)phenoxy)acetic acid) as the A2AR antagonist pharmacophore and (±)-PPHT-NH2 (6-((4-aminophenethyl)(propyl)amino)-5,6,7,8-tetrahydronaphthalen-1-ol) as the D2R agonist.56 Jörg et al. later reported excellent affinities in a series of bivalent ligands containing the canonical ropinirole as the D2R agonist and ZM 241385 (4-(2-(7-amino-2-(furan-2-yl)-[1,2,4]triazolo[1,5-a][1,3,5]triazin-5-ylamino)ethyl)phenol) as A2AR antagonist.57 Similar efforts have been aimed at targeting Adenosine A1 receptor-dopamine D1 receptor (A1R-D1R) heteromers.58 More recently heterobivalent ligands were used to target D2R and neurotensin NTS1 heteromers, with affinities up to three orders of magnitude higher and selectivity greater than seen in cells expressing solely D2R,59 as well as the dopamine D2R-metabotropic glutamate 5 (D2R/mGluR5) heteromer,60 yet another testament to the potential of heterobivalent ligands in the study of heteromers.
Due to the extensive amount of homo- and heteromers observed among dopamine receptors,61–64 many of which are directly linked to key roles in the pathology of substance use disorders,62, 65–67 significant effort has been directed toward developing bivalent ligands that effectively target dopamine receptor oligomers selectively. Several research groups have focused on the synthesis of bivalent ligands to explore the pharmacological implications of targeting dopamine receptor dimers. The Gmeiner group has published several papers reporting the synthesis of a variety of bivalent probes, connecting two agonist or antagonist PPs, binding in the OBS with polyethylene glycol or polymethylene unit linkers. Their main goal was to study receptor dimerization and target dopamine receptor homo- and heteromers to evaluate their role in schizophrenia and psychotic behaviors.68–71 Despite the high potential of this drug design, it still presents several limitations, especially in the ability to fully understand the binding modes and kinetics of these highly flexible and high molecular weight compounds. Moreover, when designing these bivalent ligands targeting receptor dimers, the optimum length for the linkers becomes even more important, in order to be able to effectively target a dimer entity and minimize the “flip-flop” binding mechanism where only the PP or the SP binds alternatively to the OBS or allosteric binding site (ABS), within monomers. Interest in the study of the D2-D2 dimer has led to various other efforts in developing bivalent ligands,72 which can target this homomer, as also seen in Gogoi et al. with the synthesis of a homobivalent ligands using 5-OH-DPAT as the PP,73 and McRobb et al. who report a clozapine based homobivalent ligand, which results in increased affinity and functional potency over the monomer parent drug.74
BITOPIC LIGANDS: MOLECULES TO STUDY ALLOSTERISM AND FUNCTIONAL BIAS OF GPCRS
General Drug Design and Pharmacological Implications
GPCRs are one of the most targeted family of receptors for drug design and development of novel therapeutics and pharmacological tools. GPCRs can be activated, and their signaling modulated, by several endogenous neurotransmitters, as well as synthetic ligands. Their predominant role in regulation of neurological and physiological processes, centrally or peripherally mediated, make them interesting targets for studying signaling mechanisms in cellular processes. Clearly, improving our understanding of the mechanisms underlying these signaling processes can lead to the development of safer and more specifically directed therapeutics. Roth and colleagues75 recently summarized how advances in molecular pharmacology are expanding the common knowledge of GPCRs. Combination of molecular studies and receptor pharmacology are shedding light on unique mechanistic consequences of structurally, chemically and functionally specific ligand-receptor interactions from carefully designed synthetic molecules. Over the last decade, the Christopoulos research group has studied the concept of GPCRs as “dynamic proteins” and has investigated the implications of multiple receptor active-states linked to distinct functional responses and pathophysiological outcomes.76
Thanks to the recent advances in X-ray crystallographic techniques and cryo-microscopy technology, the number of GPCR structures being resolved is exponentially increasing. Advanced cryo-microscopy is pushing the boundaries of GPCR structural studies, allowing visualization of protein conformational changes upon ligand binding, and how those adjustments can impact the ability to recruit/activate second messengers. Indeed, GPCRs are known to possess multiple binding sites,76 with the OBS being the most targeted for development of agonists or antagonists alike.77 However, allosteric ligands, binding at spatially distinct and less structurally conserved sites, can also stabilize unique receptor states, and exploit key pharmacological responses modulating the functional profiles of orthosteric ligands. GPCR allostery, or more generally targeting SBPs within receptors, is becoming the key to enhance receptor-subtype specificity, study the kinetic processes behind ligand-receptor interactions, and understand receptor functional cooperativity.
Improved understanding of GPCR functional states, drug development guided by computational chemistry, structure-activity and structure-function relationship studies (SAR and SFR), combined with advances in analytical pharmacology models are “bridging the gap”77 towards the design of complex bitopic molecules.78 Bitopic orthosteric-allosteric ligands can be seen as a natural extension of the classic bivalent drug design approach. Linking PPs that bind to the OBS, with SPs that bind to an allosteric binding site (ABS), or more generally an SBP, merge and combine the pharmacological properties of otherwise independent ligands in a new “hybrid” molecule with a unique pharmacological profile (Figure 2).
Figure 2.
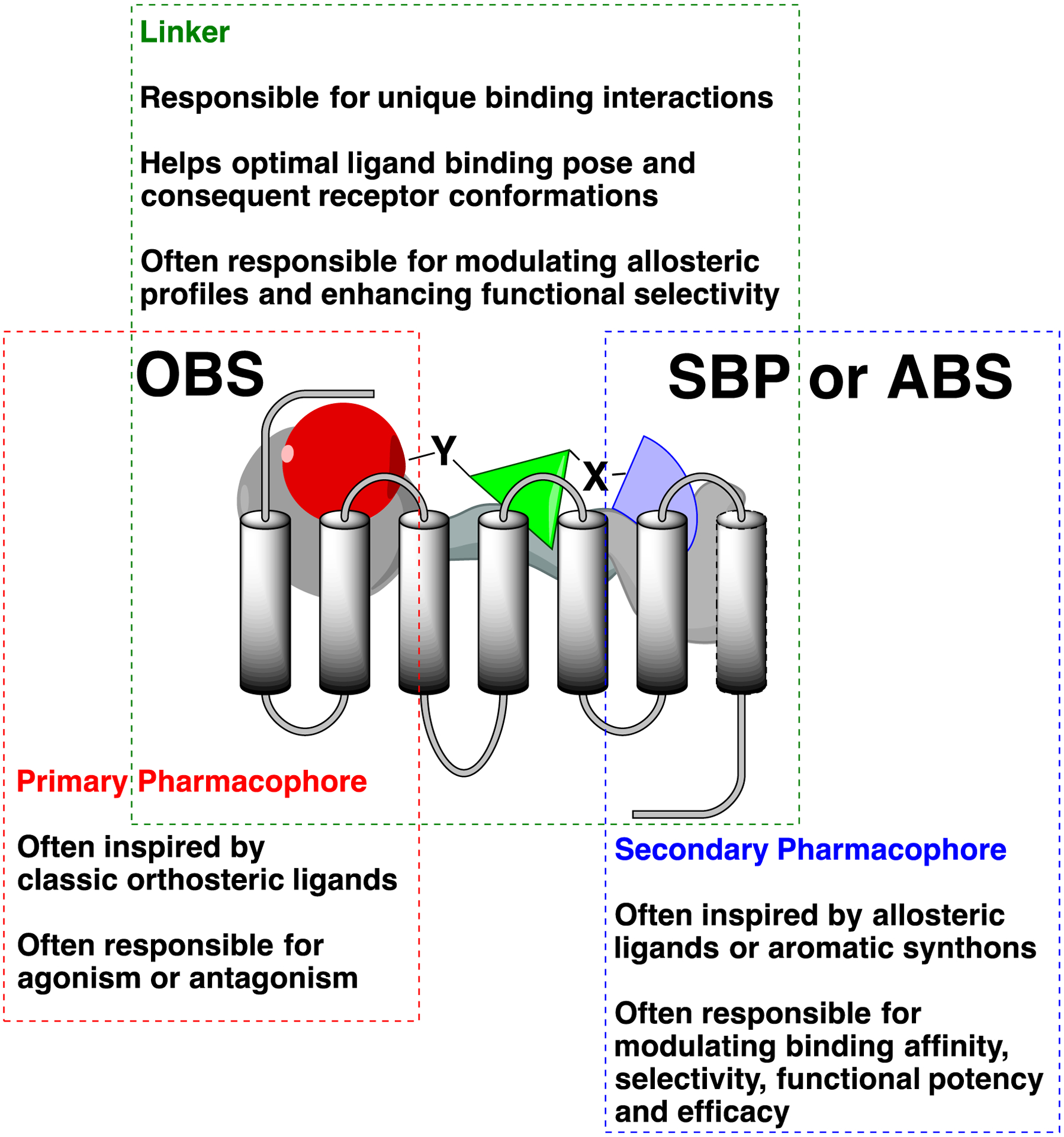
Conceptual framework of bitopic ligands and the role of each segment.
The main pharmacological consequences of bitopic ligand effects are not just an increased affinity and selectivity for the target receptor, but often their highly specific binding interactions translate into biased stimuli (functional selectivity), positive or negative allosteric modulation or cooperativity. Biased agonism or functional selectivity, is defined as the ability of a ligand to activate only discrete functional pathways associated with receptor signaling machinery, as a consequence of uniquely induced protein conformations. This can be seen as a process starting with the bitopic ligand concomitant interactions with the OBS and SBP, followed by structural changes in the protein conformation, leading to more favorable recruitment of either G-proteins or β-arrestins, thereby enacting a functionally selective signaling response. Christopoulos defines the phenomenon of biased signaling as an extension of the allosteric nature of all the GPCRs seen as “signaling machines”.78 Particularly, he describes how the binding of extracellular ligands “transfers” structural information to distinct intracellular sites, which then are responsible for propagating functional responses through specific signaling proteins.
Developing agonists that selectively activate a GPCR signaling pathway over another is essential to identifying which signaling cascade might be responsible for altered physiological responses and drug “on-target” side effects, and consequently how to develop safer treatments by minimizing these undesired responses. Due to their complex structural nature, the study of the pharmacological effects of bitopic ligands and the meaningful evaluation of their bias selectivity is still a significant challenge. In order to assess functional selectivity, drug-elicited effects need to be tested in multiple parallel functional assays for the GPCR target of interest, and the results compared with reference agonists to properly perform bias factor analyses, taking into account assay sensitivities and receptor expression levels. Lane has been at the forefront of developing mathematical models for cooperativity and operational models for functional selectivity studies.79–82 He has described several analytical considerations to be taken into account when working with bitopic ligands: in particular, he describes how bitopic ligands binding can occur via a “flip-flop” mechanism (a mechanism almost impossible to identify in normal experimental conditions). However, when the bitopic ligands engage the receptor simultaneously in OBS and ABS/SBP, we should expect cooperative binding, with the strength of equilibrium constant and functional responses different compared to a pure allosteric or orthosteric binding mode. From our work, and extensive characterization of our dopaminergic bitopic ligand library, it is evident how a bitopic interaction mode induces functional responses, binding affinities and subtype receptor selectivity significantly different from the PP, SP, or allosteric modulators, when tested alone.83–87
Though in this Perspective we will primarily focus on the development of bitopic ligands for the dopaminergic system, this strategy has played an important role in the development of ligands for a variety of GPCRs. Among the first receptor systems to be studied with bitopic molecules were the muscarinic acetylcholine receptors. The extensive body of literature detailing the allosteric pharmacology of these receptors88 make them suitable targets for rational bitopic ligand design, furthermore, they pose an attractive target for medicinal chemists due to the pivotal role they play in multiple body functions and their distribution throughout the nervous system.89–91
One of the first muscarinic bitopic molecules reported is methoctramine, a polymethylene tetraamine first reported by Melchiorre et al.92 as a selective cardiac M2 receptor antagonist. A homobivalent ligand with two 2-methoxybenzylamine moieties at either end of an extended linker, the ligand was found to be a potent M2 antagonist as a result of its bitopic binding mode, binding both to the OBS as well as an ABS in the outer loop of the receptor.93 Over the last three decades, a remarkable increase in bitopic ligands with ever improving affinities and selectivities, ranging from agonists,94–95 partial agonists96–98 and antagonists99–103 have been developed to study the family of muscarinic receptors.
Another receptor system gaining attention in recent years are the sphingosine-1-phosphate GPCRs (S1P1 through S1P5), largely due to the relevant role they play in the regulation of the cardiovascular, lymphatic, and nervous systems.104 They are considered essential to key features of vascular development, maintenance of vasculature, and lymphocyte migration.105 As a result, this receptor system has been an attractive target for the treatment of a variety of ailments ranging from autoimmune and cardiovascular pathologies to cancer and inflammatory diseases.104, 106 Achieving ligands with high degrees of subtype selectivity among these receptors has been a challenge due to the significant homogeneity shared among them.107 However, in an effort to develop subtype selective ligands capable of elucidating the different functions of the S1P receptors, Kohno et al.108 synthesized multiple bitopic ligands among which SPM-242 ((S)-2-amino-4-(2-chloro-4-((3-hydroxyphenyl)thio)phenyl)-1-hydroxybutan-2-yl-dihydrogen phosphate), containing a phosphate moiety to recognize the OBS and a 2-[(3-chlorophenyl)thio]-phenol secondary pharmacophore, has raised considerable interest. Biological evaluation of this lead molecule showed it functions via a bitopic binding mode and is a selective antagonist for the S1P3 subtype.107 Chemical manipulation of this scaffold, reducing the 2-hydroxymethyl moiety in the linker to an ethyl chain and ortho-trifluoromethylation of the phenol in the secondary group, engendered ligand SPM-354 ((S)-2-amino-4-[2-chloro-4-(2-hydroxy-5-trifluoromethylphenylthio)phenyl] -2-propylbutylphosphoric acid monoester) which exhibited significantly increased selectivity for S1P3 over other subtypes relative to parent molecule, improved potency and efficacy as well as improved metabolic stability.109 The results observed by these bitopic molecules were unparalleled by hitherto available S1P receptor ligands and provide an avenue for the study of the S1P3 receptor subtype and its role in in vivo cardiovascular function.109
The adenosine receptor (AR) tells another success story for bitopic ligands. As mentioned earlier, the adenosine receptors have long been an attractive target for medicinal chemists due to their active role in mediating a variety of biological processes. The A1AR receptor plays an important physiological role in the prevention of ischemia/reperfusion damage to heart tissue,110 and while adenosine receptor agonists have shown promising results as treatments111–112 their use as clinical tools has been encumbered by numerous deleterious side effects at pharmacologically active doses, the most commonly cited being bradycardia.113 It was consequently postulated that a rationally designed biased agonist may favor activation of the cellular pathway that promotes cardio-protection while avoiding stimulation of the pathways which may lead to adverse side effects. Valant et al. reported the first A1AR receptor bitopic biased agonist, VCP746 (5-amino-4-benzoyl-N-(6-((9-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-9H-purin-6-yl)amino)hexyl)-3-(3- (trifluoromethyl)phenyl)thiophene-2-carboxamide) constructed from adenosine (as primary pharmacophore) and VCP171 ((2-amino-4-(3-(trifluoromethyl)phenyl)thiophen-3-yl)(phenyl)methanone), a known AR positive allosteric modulator.114 The resulting bitopic molecule was found to induce the cardioprotective effects of AR agonism while not inducing bradycardia; agonism at the A1AR biased away from calcium mobilization relative to all other pathways contributes to prevention of the cytotoxic levels of calcium observed during ischemia/reperfusion injury events.115 This ligand effectively activates the therapeutic pathways of the A1AR receptor while remaining inactive towards the pathways responsible for the undesired side effects. Following the original report, a series of strategically designed bitopic biased ligands has provided additional tools for medicinal chemists to further investigate the effect of the A1AR downstream pathways.116
Dopamine receptor bitopic ligands: application to receptor selectivity, allostery and functional bias
Dopamine receptors, particularly the D2-like family that includes subtypes D2R, D3R and D4R, are some of the most studied and targeted GPCRs. They are implicated in many physiological processes such as reward, addiction, locomotion and movement coordination, as well as metabolism and hormonal regulation.117 They all share high homology in their OBS, making the development of selective agonists/antagonists a major challenge. The crystal structures for the D3R was resolved in 2010 in complex with the D2R/D3R antagonist/inverse agonist, eticlopride (5) and has significantly guided the effort towards the design of more selective D3R antagonists.118 More recently, in 2017 and 2018, the Roth research group crystallized and resolved the structures of D2R and D4R in complex with risperidone and nemonapride, respectively.119–120 These findings have illuminated the dopamine D2-like receptor structures and enabled structure-based campaigns for the design and synthesis of novel D2-like subtype selective ligands.121–122
In addition to developing SAR for the optimization of the PP and SP in our drug design, we also focus on the most understudied component of bitopic molecules, which we will highlight in this perspective: the linker. Often seen as a mere connecting portion of the molecule, we have pushed our drug design in the direction of evaluating linkers with different length, substituents, polarity, rigidity and chirality. We have demonstrated how properly designed linkers can create specific interactions with amino acid residues and guide the favorable (or unfavorable) binding poses of the ligands, inducing unique bias and allosteric pharmacological responses, together with unprecedented receptor-subtype affinity and selectivity. The following discussion will mainly focus on our successful drug design of highly selective bitopic dopaminergic agonists and antagonists, highlighting how the advances in molecular and structural pharmacology inspired the discovery of these new classes of ligands.
When Chien et al. published the D3R crystal structure,118 they also reported computational studies performed using our ligand, ((R)-PG648 (6) R-22; Figure 3), to show how the 2,3-dichloro-phenylpiperazine PP and the 1H-indole-2-carboxamide SP were binding in two distinct sites. As such, the PP recognized the OBS, and nicely overlapped with eticlopride (5), whereas the SP was posed in the less conserved SBP. The presence of a tractable SBP is now well established for these D2-like receptor subtypes and can be targeted to improve affinity and subtype selectivity.122–123 In addition, the crystal structure and subsequent molecular dynamics studies provided the first indication that a bitopic binding mode was important for the design of ligands with higher D3R subtype affinity and selectivity. Furthermore, our attention was directed to the key role played by the linker, the 3-OH substituent in particular, whose interactions oriented the molecule towards its optimal pose. These observations prompted us toward significant efforts in designing new bitopic D3R agonists and antagonists, exploring structural features of PP, SP and linker substitutions, leading to successful SAR campaigns in preparing highly subtype selective ligands and small molecule tools with unique pharmacological profiles.
Figure 3.
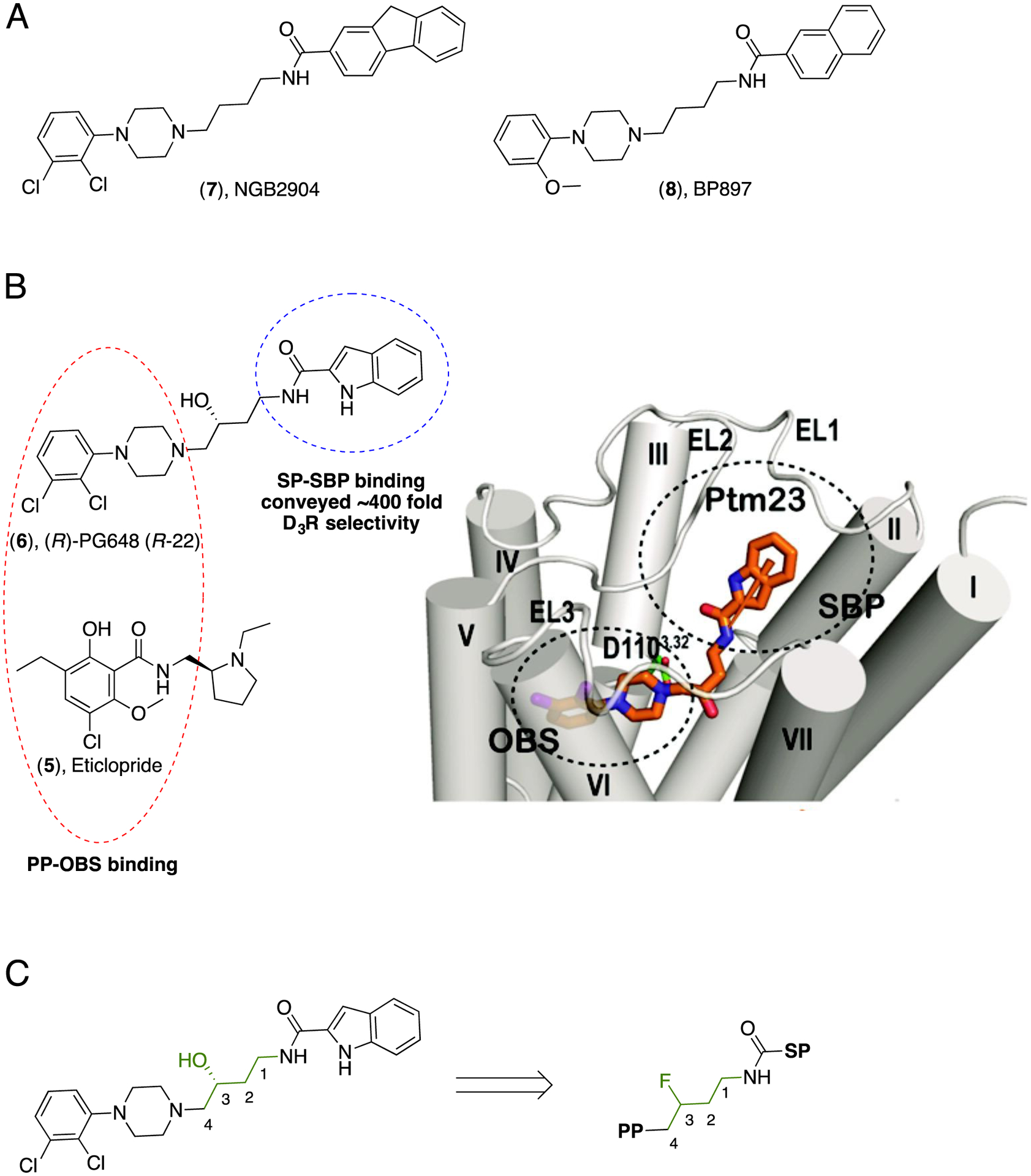
A) Structures of NGB2904 (7) and BP897 (8) representing aryl-phenylpiperazine as the PP. B) Structures of eticlopride (5) and (R)-PG648 (6) and highlighting the overlap between their PP binding in the OBS. Docking pose of 6 in D3R showing its bitopic binding mode, with simultaneous interactions with both OBS and SBP. Figure published in Newman, AH et al. J. Med. Chem. 2012, 55, 6689–6699 [Copyright © 2012 American Chemical Society]. C) Structural investigation of the linker, exploring the bioisosteric substitution of the -OH with a -F atom.
Identification of the D3R PP and SP leading to discovery of their bitopic nature
When we first embarked on the discovery of novel and D3R selective antagonists,124–127 we were frankly not thinking of these molecules as being bitopic. Based on the structures of NGB2904 (7)128 and BP897 (8),129 early D3R-selective antagonists/partial agonists (Figure 3A), we first attempted to identify novel scaffolds based on fragments of these lead molecules that appeared in other compounds in our in-house library. We settled for optimizing 6, retaining the 2,3-dichloro-phenylpiperazine motif, modifying the tricyclic terminus and investigating optimal linker length between the two. Our early efforts124 identified key SAR for high affinity at D3R and modest selectivity over the homologous D2R and we and others continued to build on this scaffold in following years.126, 130–133
As mentioned, the advance of the D3R crystal structure in the presence of the D2R/D3R antagonist/inverse agonist, eticlopride (5), confirmed what we had suspected: the 2,3-dichloro-phenylpiperzine served as the PP of our molecules,118 whereas the 1H-indole-2-carboxamide terminus of the lead compound 6,134 serving as the SP, occupied a SBP that was unique to D3R and conveyed ~400-fold D3R selectivity over D2R (Figure 3B). Subsequently, we deconstructed our D3R scaffold into its “synthons” and confirmed that the PP indeed showed essentially no D3R-selectivity over D2R until it was linked to the SP.135 In addition, the affinity and efficacy of this scaffold appeared to be primarily attributed to the PP, whereas the SP was responsible for D3R selectivity. Further elucidation of the SBP identified a single Glycine (Gly98) residue in extracellular loop 1 (EL1) to be a key contributor to D3R selectivity.136 Importantly, we found the SP had virtually no binding affinity to the D3R, as measured with radioligand binding assays, until it was linked to the PP. Moreover, as suggested in the D3R crystal structure paper, the role of the 3-OH substituent in the linker became apparent in further conveying D3R selectivity to these molecules.136
Role of the linker in D3R selectivity: partial agonists/antagonists
Early on in our SAR campaign, we took a page out of the “Portoghese play book” and began to explore functionalization of the 4C-linker between the PP and SP. The possibility that these compounds were bitopic had begun to emerge, but the role of the linker besides being key to keeping the PP and SP at an optimal distance from one another had not been investigated. We discovered, for example that a trans olefin could replace the saturated butyl linking chain and retain high affinity D3R binding (e.g., PG01037, (9) Ki= 1 nM, Figure 4) and D3R/D2R selectivity of >100125, 137–138 but that a rigid alkyne was not tolerated.137
Figure 4.
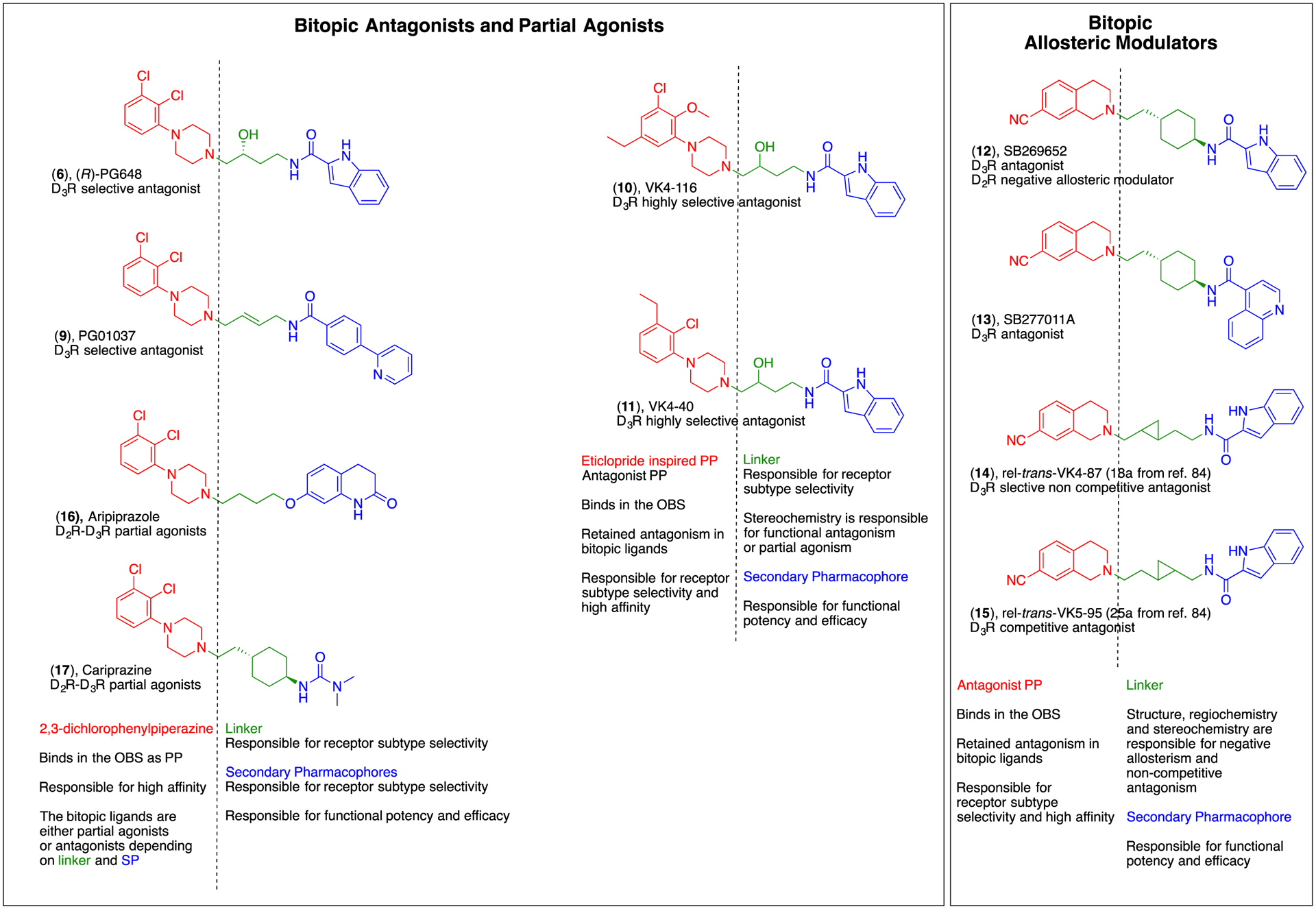
Summary of the main dopaminergic bitopic compounds, studied over the years, which inspired SAR and drug design to generate highly selective agonists, partial agonists and antagonists. The PP are highlighted in red, in linkers in green and the SP in blue. The descriptions below every structure highlight the primary role played by each fragment of the bitopic molecules toward unique pharmacological profiles.84, 87, 148, 149
In order to reduce the lipophilicity of these molecules to improve their in vivo profiles, we began to explore the addition of an -OH group in either the 2- or 3-position of the linking chain.134 Indeed, we discovered that these analogues were well tolerated at D3R but notably their D3R selectivity was further improved with the addition of the 3-OH (e.g., rac-6, D3R/D2R ~400-fold)134 and not the 2-position (Figure 3). These observations were confirmed computationally and a Tyrosine residue on trans-membrane helix 7 (TM7) was identified as a contributor to the D3R selectivity observed118 providing the first clue that not only was the SP important for D3R selectivity, but functionalization of the linker could also play a role (Figure 3, 4). Subsequent separation of the enantiomers of rac-6 identified enantioselectivity at D3R, without concomitant enantioselectivity for D2R. The R-enantiomer, 6, was identified as the eutomer in this series, further highlighting the importance of the linker composition and the subtle but tractable differences in these binding sites.134 Of note, this substitution was limited, as for example acetylation134 at the 3-position was not tolerated. Importantly, other groups subsequently showed that hydroxylation as well as other structural modifications to the linking chain were well tolerated at D3R, in this scaffold.139
More recently, capitalizing on the D3R crystal structure and incorporating it into the small molecule SAR that has been well established, we have discovered a new series of highly D3R selective antagonists/partial agonists, namely VK4-116 (10) and VK4-40 (11) (Figure 4.)87, 123 In this series, by simply modifying the substitution on the 4-phenylpiperazine, retaining the 3-OH linker and the privileged 1H-indole-2-carboxamide SP, highly D3R selective analogues with high affinity emerged.87 This investigation further highlights the importance of the PP, SP, and the length and composition of the linker, substantiating the concept that the “whole is greater than the sum of its parts.” Importantly, 10, one of the most D3R selective compounds reported to date, demonstrated remarkable metabolic stability and early in vivo profile to support further pursuit as a lead molecule for the prevention and treatment of opioid use disorder, which will be described in further detail below.
Given fluorine (-F) is often regarded as a bio-isostere to a hydroxyl (OH) group140–142 we went on to explore the linker substitution with a 3-F (Figure 3C). This SAR campaign led to some of the most D3R selective ligands reported at that time143–144 and we have further pursued these leads to discover new and improved molecules for in vivo study.145 Importantly, we determined that the amide linked heterocyclic terminus was critical for this scaffold to bind to D3R, as reducing this to the amine, significantly decreased D3R binding affinity.143 However, we subsequently showed that replacing the amide with a triazole scaffold using click chemistry was also well-tolerated.146 In this series, the lead compound, which shares the PP, SP and 3-OH linker with our previous lead compound rac-6 (PG648), demonstrated D3R Ki=6 nM with a D3R/D2R selectivity of 165. Importantly, other compounds in that series, which also possessed the 3-OH linker, demonstrated even more robust D3R/D2R selectivity, albeit at lower affinities for D3R, a trend we have also observed in the 3-F series.144–145
As previously mentioned, Lane and his research group embraced this same bitopic drug design approach and have devoted extensive efforts in developing analytical pharmacology methods to fully characterize the binding and functional profiles of small molecules simultaneously binding the OBS and SBP within the same receptor. In 2015, in back to back papers, Shonberg and Mistry reported how the well-known bitopic ligand SB269652 (12) acts as a negative allosteric modulator of the dopamine D2R receptor (Figure 5).147–148 They described, for the first time, a small molecule negative allosteric modulator for D2R engaging the receptor in a bitopic mode. They also studied how the bitopic engagement of a D2R protomer can limit dopamine (DA) binding in the potential formation of dimers. They performed extensive SAR studies to determine the role played by each portion of the 12 bitopic molecule in its biological profile. Shonberg reported that the 1,2,3,4-tetrahydroisoquinoline PP with its basic tertiary amine is important to maintain high affinity, overall allostery and functional profiles, since it is the “head” or PP of the molecule competing with DA for the OBS. He also described the 1H-indole-2-carboxamide SP, as a “tail” group whose lipophilicity is a common structural feature in numerous D2-like subtype selective bitopic ligands. Moreover, SAR based on the linker portion of the molecule provided the observation that subtle changes in the alkyl chain length and rigidity completely alter the orientation of SP causing a switch from negative allosterism to competitive antagonism. Mistry generated new SAR based on the fragmentation of the 12 structure, identifying D2R allosteric fragments based on the 1H-indole-2-carboxamide SP scaffold. In this report, an allosteric pocket in the D2R between TM2 and TM7 was discovered, which can be targeted to design novel allosteric modulator and bitopic ligands. However, the most important conclusion from these studies is the observation that despite the role played by the single pharmacophore, in order to simultaneously bind both OBS and ABS, PP and SP need to be connected in close proximity, and perfect alignment of all the fragments by the linker is essential. These studies further highlight the importance of the linker and how structural modifications generate preferable SAR. Based on these findings Capuano, Lane and Christopoulos recently published a new generation of analogues of 12, where changes in the 1H-indole-2-carboxamide SP dramatically affected functional affinity, cooperativity and overall pharmacological properties with respect to the parent molecule.149 This underscores again how bitopic ligands are composed of interconnected structural scaffolds, and how modifications in a part of the molecule directly affects the overall structural pose within the receptor, recognition of receptor binding sites, and ultimately translation to changes within the intracellular signaling machinery.79–80, 149
Figure 5.
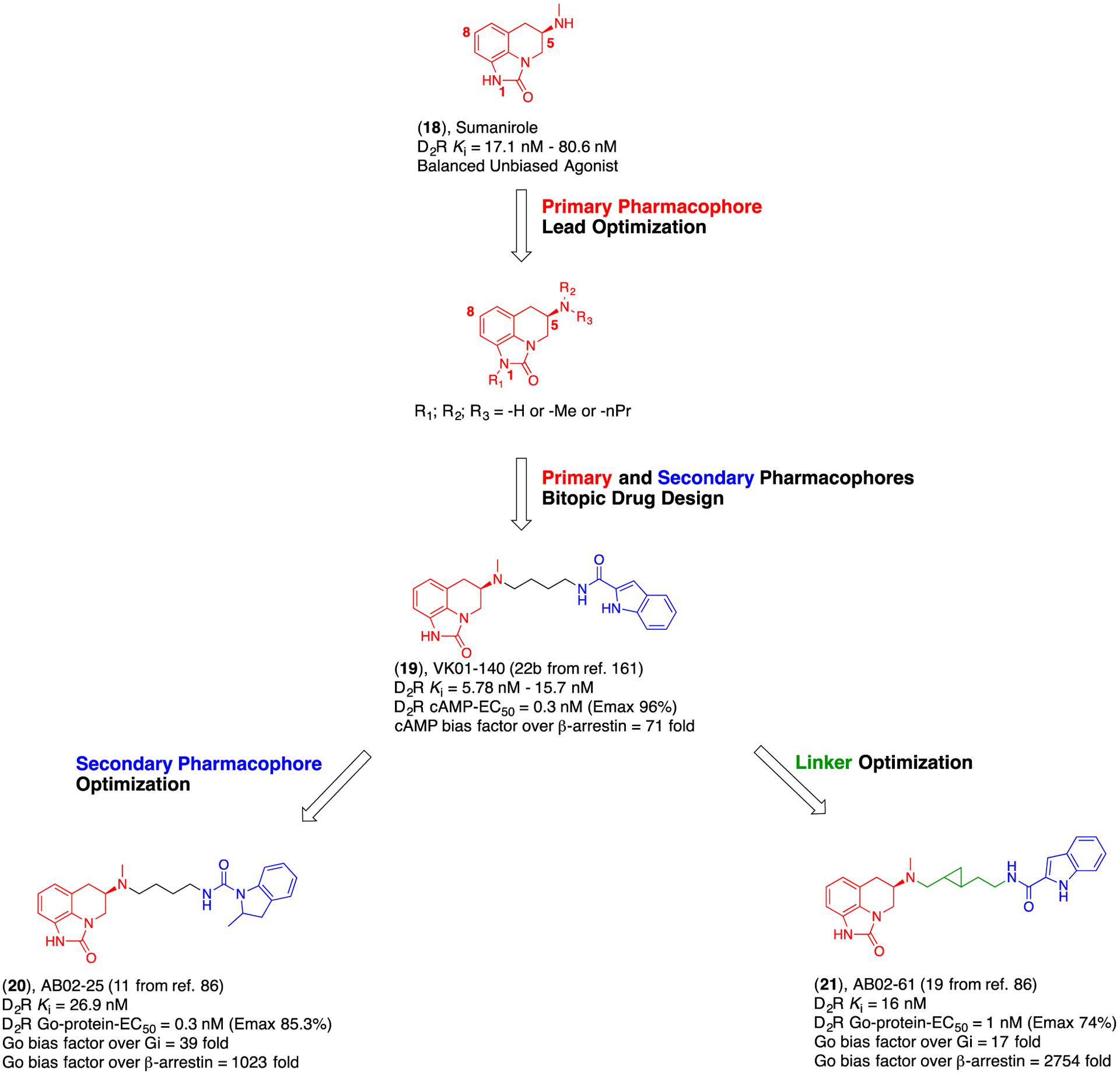
Drug design strategy, from study of the PP, to structural optimization of SP and linker to improve D2R affinity, selectivity and specific pharmacological profiles achievable only with bitopic ligands.86, 161, 162
Parallel studies from our own lab also used the 12 scaffold, but in comparison to its analogue SB277011A (13; Figure 4), a classic D3R antagonist used in many in vivo studies that has not been shown to have any allosteric properties at either D2R or D3R. We set out to better understand the structural basis of these observations at D3R, carefully deconstructing each molecule and then reconstructing novel analogues, including the addition of a cyclopropylated linker.84 Once again, we saw that the PPs had modest binding affinities and selectivities for D3R over D2R and showed competitive interactions at D3R. Likewise, the SP did not bind to the OBS at concentrations of up to 100 μM. However, depending on the length and composition of the linker, several analogues with the same PP as 12 showed allosterism at D3R. Furthermore, these data suggest that the specific orientation of the SP within the SBP can confer noncompetitive or allosteric modes of interaction but that relatively subtle changes to structure and orientation of the SP, likely due to the composition of the linker, can cause a switch between apparently allosteric and competitive antagonism (Figure 4).84 In addition, compound 14 (18a from reference 84; VK4-87, Figure 4) demonstrated ~4-fold higher D3R affinity than the parent compound (Ki=0.89 nM) and >200-fold D3R/D2R selectivity, further exemplifying the importance of its bitopic nature not only for functional allostery but for improving affinity and selectivity at the target receptor.84 In contrast, compound 15 (25a from reference 84; Figure 4) also showed negative allosterism at D3R, but had both lower affinity and selectivity for D3R than either the parent compound or 14. Of note, the only difference between these two molecules is the location of the cyclopropyl group in the linker. Additional investigation of compound 14 is under way.
Role of the linker in D3R selectivity: Agonists
As we previously described, the pharmacological effects of bitopic ligands are not limited to allosteric modulation, but growing evidence points to how specific ligand-receptor interactions, particularly regarding bitopic agonists, translate into functional selectivity or bias stimuli. Recent efforts from different research groups have converged toward the design and synthesis of D2R/D3R agonists to identify structural requirements necessary to selectively activate either the G-proteins or the β-arrestin signaling pathways. For example, characterization of the physiological responses associated with activation of independent GPCR pathways has been extremely successful in the opioid research field. Understanding the physiological importance of activation of the mu–opioid receptor G-protein pathway, in comparison to the potentially undesired effects mediated by the β-arrestin pathway, is leading to the development of promising new agonists for pain management.150–154 However, for the DA receptors, the link between signaling pathways and biological and behavioral responses remains elusive. Nevertheless, with the emerging toolbox of bitopic ligands with functionally selective profiles, promise to overcome these challenges is on the horizon.
SAR campaigns to identify highly selective D2R or D3R-subtype selective agonists have remained largely unsuccessful, likely due to these molecules occupying the highly homologous OBS. The use of the bitopic molecule strategy to identify functionally biased and full agonists has grown more recently. Indeed, in the past few years there have been several published studies on D2R bitopic biased agonists selective for G-protein or β-arrestin activation. Most of the published structures rely on common scaffolds or molecular fragments often based on the partial agonist aripiprazole (16) or cariprazine (17).155–157 Despite the availability of many new compounds, the D2-like bias agonism research field still presents several limitations. Inconsistencies in (i) data analyses and assessment of bias factor comparisons, (ii) multiple differing functional assays, and (iii) overall medicinal chemistry has made generating highly subtype selective full agonists challenging. As such, a clear translation of small molecules in vitro results to in vivo animal models of pathophysiological behaviors has thus far been impeded. Indeed, most of the studies aimed to dissect the role of DA signaling pathways in the central nervous system in vivo are still relying on genetic manipulations rather than pure pharmacological approaches or a combination thereof.158–160
Novel and bitopic D2R agonists based on sumanirole
In our first attempt to design selective D2R agonists, Zou and Keck et al.,161 reported initial SAR based on the D2R/D3R-preferential agonist, sumanirole (18; Figure 5) as the PP scaffold. This work was focused on understanding how regiochemical substitutions in positions 1-, 5-, and 8- of the sumanirole pharmacophore would affect its binding poses, and consequent affinity and agonist potency profiles. The initial SAR investigation built the foundation for follow up studies directed toward exploring the chemical space around the D2R OBS, targeting the SBP, and studying ligand-receptor interactions that are mediated by the linker portion of the molecule. Zou and Keck reached the important conclusions that i) simple sumanirole N-1 alkylation was tolerated and produced compounds with overall reduced potency at D3R, but not significant changes in D2R vs. D3R subtype selectivity, suggesting that the N-1 interactions at the OBS might translate in different functional activations for the highly homologous D2R and D3R; ii) N-5 substitution with the canonical n-butyl-arylamide linkers and SP, found in classic D3R antagonists described above, induced significant increases in affinity and full agonist potency for the D2R, despite having a relatively nonselective subtype binding profile; iii) it is important to use the agonist [3H]-7-OH-DPAT radioligand when testing binding affinities for D2R and D3R agonists. Agonist competition experiments performed in the presence of an antagonist radioligand (e.g., [3H]-N-methylspiperone) tend to underestimate affinities and dramatically shift selectivity ratios.
These initial observations on how N-1 and N-5 substitutions can be responsible for changes in either affinity or potency of new sumanirole (18) analogues inspired us to synthesize a new library of bitopic molecules and further characterize the structural requirements for both SP and linkers to optimally target the SBP. In 2017, Bonifazi and Yano et al. published the design and synthesis of complex bitopic analogues (Figure 5), as well as their functional profiles, to identify biased agonism among the possible D2R-mediated signaling pathways analyzed.162 In particular, they explored the structure-activity and structure-function relationships of the new analogues via bitopic substitution on 18’s N-5 and/or N-1 position mediated by linkers with different lengths and polarities. In agreement with the literature previously described, they confirmed how bitopic interactions often translate into highly functionally selective profiles. For example, it was observed that N-5 alkylation led to low nanomolar affinity (Ki) at D2R and enhanced sub-nanomolar potency (EC50) toward the G-protein signaling (cAMP inhibition) pathway, identifying highly G-protein biased agonists based on potency. In contrast, when studying the effect of N-1 bitopic alkylation on functional efficacy, they reported that despite a markedly reduced D2R affinity, the analogues presented lower potency, but near full activation (Emax) of the G-protein pathway, obtaining G-protein biased agonists based on efficacy. Overall, this work highlighted the flexibility of the sumanirole N-5 position to increase D2R affinities, potencies and push the boundaries of biased agonism towards new limits previously unexplored. Moreover, they identified the optimal length and composition (polarity) of the linker.
After studying the regiochemical requirement of the sumanirole PP, in 2019, Bonifazi and Yano et al. published a follow up study where they explored new allosteric aromatic SP structures, chirality in the SP, and carefully analyzed the functional effects of a bitopic ligand presenting more rigid cyclopropyl-containing linkers (Figure 5).86 The trans-cyclopropyl linkers used were inspired by the previously reported D3R non-competitive antagonist 14 (18a from reference 84; Figure 4).84 As described above, the non-competitive nature of this compound was exclusively dependent on the length and regiochemistry of the cyclopropyl moiety embedded in the linker. They subsequently showed that by introducing subtle modifications, such as a 2-methyindoline-amide allosteric SP (20) or trans-cyclopropyl indole-amide linker (21) to the N-5 sumanirole (18) bitopic analogues, they could achieve the highest D2R G-protein biased agonism observed to date (bias factor >1000 fold over β-arrestin recruitment).86 In addition, the presence of the trans-cyclopropyl linker provided privileged recruitment and activation of Go-protein over Gi-protein (bias factor >10 fold). This was the first time that biased agonism was observed within D2R coupled G-proteins belonging to the same sub-family, and it was the first time where the linker was deemed a critical mediator of such an extraordinary bias response.
Over the years, our lab has meticulously established how PPs are responsible for maintaining the agonist or antagonist profile of the bitopic molecules,85, 135 meanwhile the SPs modulate receptor subtype selectivity and functional potency of these bitopic molecules. We have also provided structural evidence showing how the linker plays a key role in inducing functional selectivity and allosteric modulation, likely due to its ability to effect specific ligand-receptor conformations. This underscores how bitopic ligands combine the best properties of single synthons into new complex structures, and ultimately affects downstream signal messengers through discrete drug-protein interactions.
Novel and bitopic D3R agonists based on PD128,907 and PF592,379
The SAR story that we have developed over the years regarding D3R antagonist selectivity and D2R biased agonism inspired us to take the challenge of designing D3R selective agonists and provide the pharmacology community with an essential counterpart tool to the highly selective antagonists already generated.84–85, 87, 123, 163 Recently, Battiti, Cemaj and Guerrero et al.,164 published a comprehensive SAR study aimed at identifying the optimal structural features for the PP and linker to optimize affinity and selectivity in D3R agonist drug design. Our chemical campaign was particularly focused on the significance of chirality and aimed to explore chiral combinations between PP and linker for optimal D3R binding (Figure 6).
Figure 6.
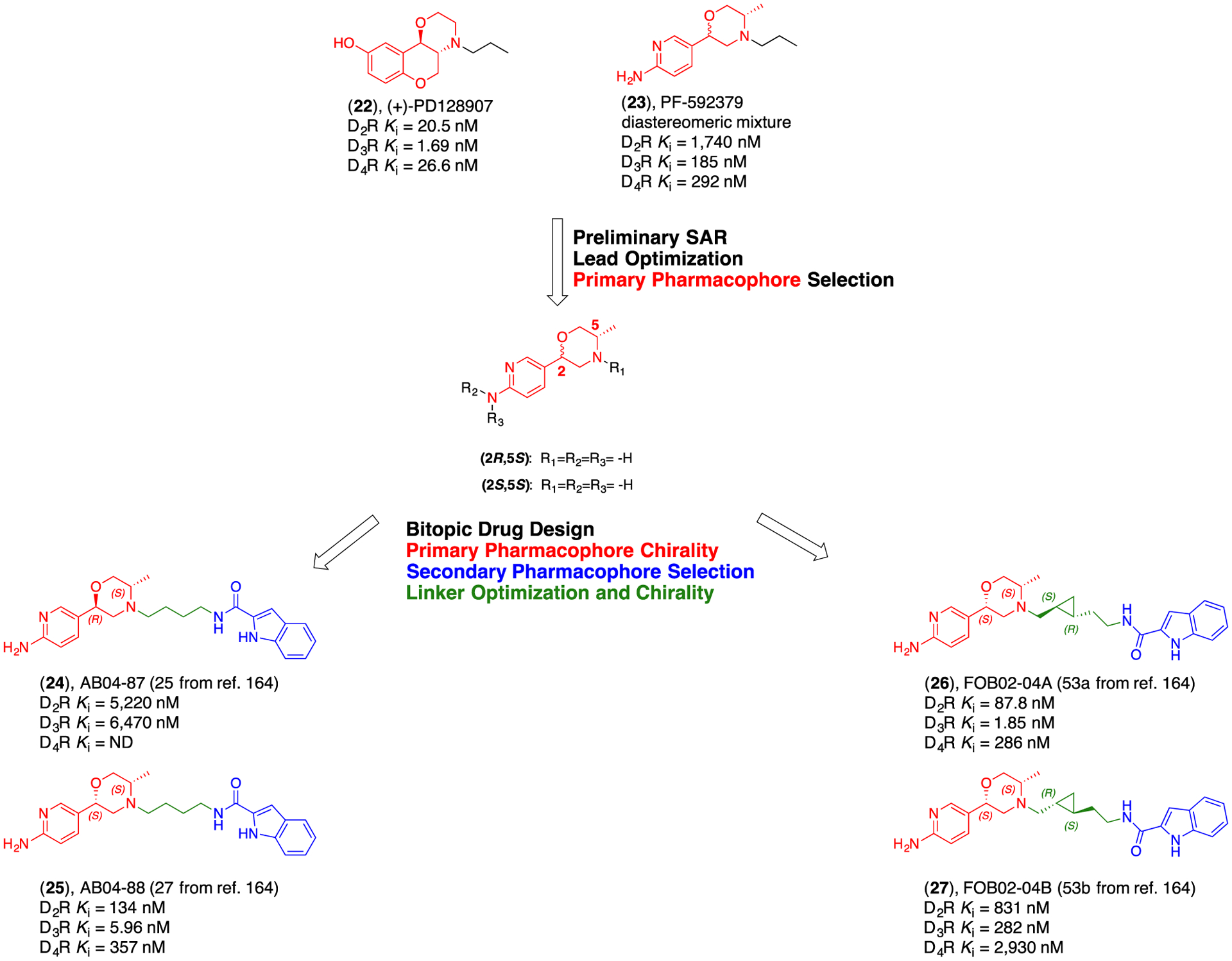
Drug design strategy, from study of the PP, to structural optimization of SP and linker to improve D3R affinity, selectivity and specific pharmacological profiles achievable only with bitopic ligands.164
We designed our new bitopic ligands based on the well-known D3R agonists, (+)-PD128907 (22) and PF592379 (23) as the PPs (Figure 6). Both compounds present two chiral centers in their core structures, and both present multiple positions for potential alkylation and chemical modification. Despite the intractable nature of the 22 scaffold (e.g., all chemical modifications resulted in lower affinities and D3R selectivities than the parent molecule), 23 proved to be an extremely interesting scaffold for chemical manipulation.
While 23 is reported as the (2R,5S)-eutomer at the morpholine ring (Figure 6), we demonstrated that, when assembled into a bitopic configuration, the (2S,5S) conformation becomes the privileged architecture, with increased affinity and selectivity for the D3R. When bitopic ligands presenting (2R,5S)- and (2S,5S)- morpholine isomers were tested in parallel, we observed >1000 fold diastereospecificity in D3R binding for the (2S,5S)-enantiomer. Moreover, the (2S,5S) bitopic conformation significantly improved the subtype selectivity, reducing the overall D2R affinity. In addition, a cyclopropyl moiety was incorporated in the linker as previously described for compound 14 (18a from reference 84; Figure 4) and we achieved full resolution of the four chiral centers. The identified lead eutomer demonstrated significantly higher D3R selectivity than PF592379 (23) and is among the highest D3R selective agonists described in the literature to date. We found that a very specific chiral combination is necessary for the trans-cyclopropyl function, since inversion in configuration toward the less active isomer decreased the affinity >150 fold (compounds 24-27; Figure 6). Moreover, this is the first time it has been observed that the PP and linker are structurally and conformationally dependent on one another in achieving the optimal ligand binding pose at D3R for an agonist. We demonstrated that the right geometry for the linker, not only changes the stereochemical requirements of the PP, but it might also direct the SP toward different SBP interactions, opening the opportunity of a completely new range of possible ligand-receptor conformations, which can translate to yet unexplored intracellular functional activities. This work is just the first step in understanding how linkers, when optimized in their length, relative regiochemical positions of the PP and SP, and relative and absolute stereochemistry can dramatically modulate pharmacological profiles. Due to the high degree of homology among the D2-like dopamine receptors, exploring less studied chemical spaces and binding sites different from OBS and SBP might be the key to achieving high subtype selectivity. Even in the presence of highly homologous and structurally conserved receptors, subtle differences in amino-acid residue spatial orientation can be targeted with carefully designed ligands presenting suitable stereochemistry. We expect that future computational studies will aid in better understanding D3R agonist-receptor interactions, as well as inspire new studies on the significance of specific ligand induced receptor conformations to elicit desirable pharmacological profiles.
IN VIVO STUDIES WITH LEAD D3R SELECTIVE LIGANDS – RELEVANCE TO DRUG DEVELOPMENT
Thus far in this perspective, we have focused on the roles and importance of the PP, SP and linker between them for affinity, receptor subtype selectivity, allostery and functional selectivity or signaling bias. However, beyond discovering how each of these pieces works in concert to bind GPCRs in specific conformations that elicit these profiles, we ultimately want to know what these compounds do in vivo and if we can relate these profiles to behavior. In this pursuit, the PP, SP and linker also play a critical role as these exquisitely designed small molecules ultimately have to possess the appropriate biophysical properties to penetrate the blood brain barrier (in the case of our D2R/D3R ligands), be metabolically stable, have appropriate pharmacokinetics, including oral bioavailability and to not have off-target effects that can pose behavioral or other toxicities. Indeed, we can develop an interesting compound that binds with high affinity and selectivity to our desired receptor subtype, with the in vitro efficacy we also desire, however it might be inactive in vivo because of structural flaws that make it a poor drug. This challenge has been particularly difficult with the bitopic molecules directed toward the D3R due to their lipophilicity, metabolic instability, short half-lives and propensity to be P-glycoprotein (PGP) transported-substrates.130, 165–166 Keseru has described highly “decorated” and potent compounds through target-based SAR have resulted in an abundance of compounds with high affinity and/or selectivity for their biological target, but that are large and lipophilic, resulting in a poor absorption, distribution, metabolism, excretion, and toxicity (ADMET) profiles and ultimate failure preclinically or in the clinic.167–168
There are many reviews in the literature123, 168–171 addressing Lipinski’s Rule of 5 and more recent permutations and computational models used to predict these physical properties to be used in drug design; these will not be reviewed here. Instead, a quick review of our own program developing high affinity and selective D3R antagonists/partial agonists toward substance use disorders will be used as an illustration of our evolution in drug design and again how the combination of SP, PP and linker played a role in developing these in vivo tools.
As described above, our starting lead molecule, NGB2904 (7; Figure 3A), came out of the lab of the late Andrew Thurkauf at Neurogen.128 We collaborated with Thurkauf briefly124 and discovered some early analogues, but none that were of particular interest, until we started to modify the linker,137 incorporating a trans olefin to give PG01037 (9; Figure 4).125, 130, 138 To date, there are nearly 40 papers cited in PubMed using 9 as a tool for both in vitro and in vivo studies and it has been commercially available for several years. Indeed, its debut in the behavioral literature as a selective D3R antagonist, was in a series of papers from the Woods lab showing that it selectively blocked the yawning effect of several D3R agonists in rats.172–174 These studies were replicated in male and female rhesus monkeys under various drug combination conditions.175–176 Subsequently, several other labs have used this compound and its close analogues in models of L-DOPA-induced dyskinesia,177–178 cocaine or methamphetamine use disorders and impulsivity.126, 179–190 The key to the success of 9 was that we were able to improve its D3R selectivity and also reduce its lipophilicity over 7, via incorporating that trans olefin linker and modifying the SP, by replacing the tricyclic fluorenyl function with a 2-pyridyl-phenyl group.125, 138 Nevertheless, its subsequent characterization as a PGP transported substrate191 dampened enthusiasm for taking this compound into further development for human use.
While 9 proved to a good in vivo tool, subsequent drug design led to compounds with subnanomolar affinity and/or >300-fold D3R/D2R selectivity. Importantly, a seminal modification was the addition of a 3-OH into the linker to give the next lead molecule rac-6 (PG648; Figure 4).134 As described above, rac-6 and its (R)-enantiomer (6) have binding affinities for D3R of ~1 nM and are ~400-selective over D2R, as well as having a clean off target profile.123, 127, 134 In this molecule, we retained the 2,3-dichlorophenylpiperazine PP, but modified the SP to the recently identified D2R/D3R privileged 2-indoleamide. We showed that rac-6 was indeed effective in rodent models of methamphetamine use disorders, but to our disappointment, it was not effective in similar models in nonhuman primates.127 There may be many reasons for this, not the least of which is that nonhuman primates, like humans, are far more diverse in their response to drugs of abuse and potential medications and the n’s are always low, making statistical significance more challenging to obtain than in rodent studies. Nevertheless, one factor that may have contributed to the lack of effectiveness in these models was that we later discovered rac-6 was metabolically unstable in rhesus monkey liver microsomes (unpublished data) providing an example of species differences that one also has to consider when developing lead molecules. Again, different functional groups are more metabolically vulnerable than others across species and the key is to find compounds that are predicted to be stable across species, before spending resources developing compounds that are only effective in rodents. This is of course not always easy in academic labs, where resources like these are not as common as in the pharmaceutical industry. Nevertheless, our own experiences have led us to be more thoughtful of metabolism across species earlier in our drug discovery process, which is not always predictable based on structure. Indeed, we originally thought that the 3-OH group may be metabolically vulnerable, which lead us to synthesize the 3-F analogues.143–144 Subsequent metabolic ID in our newer lead compounds suggest that in fact, the 3-OH does not appear to be metabolically vulnerable.87, 192
Although we have synthesized many analogues more recently, further examining all parts of these bitopic molecules and developing both SAR as well as identifying in vivo tools, our efforts have now directed us toward the overall profile of our new lead molecules, 10 (VK4-116) and 11 (VK4-40; Figure 4). These two analogues share the 3-OH linker and the privileged indole-2-carboxamide, but differ in the PP, which was inspired by the D2R/D3R antagonist/inverse agonist eticlopride (5), as described above. We posited that the SP, PP and linker of these molecules had proven to have both the pharmacological specificity and metabolic stability in other molecules from our lab and others and hoped that the combination would lead to new molecules with outstanding D3R affinity, selectivity and ADMET properties that predict in vivo efficacy.87, 145 We have synthesized the enantiomers and discovered the R-enantiomer for both to be the eutomer. Extensive behavioral testing of 10 and (R)-11 has been done and further studies are underway.192–193 The R-enantiomers of these two lead molecules have been tested in telemeterized rats alone and in the presence of either cocaine or oxycodone, with impressive results showing neither compound exacerbates the cardiovascular effects of these drugs of abuse and, in some cases, serve to reduce blood pressure and/or heart rate which is increased in their presence.194 Further development of these lead molecules is underway in hopes that at least one will be safe enough for clinical trials. This is especially important, not only for the hope of development as a non-opioid therapeutic for the treatment and possibly prevention of opioid use disorder, but to also determine if the preclinical models we are testing these compounds in (rodents and nonhuman primates) translate into humans as, although we know that many of these models have translational validity based on the success of the opioids, namely methadone and buprenorphine, we do not know if the non-opioid treatments that are being developed will show the same profile195 and this is critical to our success.
SUMMARY: THE WHOLE IS GREATER THAN THE SUM OF ITS PARTS
In summary, from the seminal work of Portoghese through the contributions of many, the discovery of bivalent and bitopic molecules has provided a fantastic toolbox of molecules with which to understand basic biology of the GPCRs we have targeted. As X-ray crystal structures of receptor targets are becoming increasingly available, identification of potential SBPs which can be exploited with bitopic molecules opens an avenue for ever more selective and potent rationally designed small molecules. Moreover, the promise that some of these molecules may make it to the clinic because of their unique ability to bind the targeted receptors in such a way that a therapeutic arises with all (or many) of the right attributes to be an effective drug exists. Importantly, while we have focused on the structures of the PPs, SPs, and the linker, it is undoubtedly the combined effect of these strategically connected pieces that contribute to the overall pharmacological profile of the drug, including its drug-like properties, which are critical to its success. Thus, it is paramount to consider the whole molecule as not just the sum of its parts, but that the right combination can result in molecules that are greater/better/more promising leads. Our adventure into bivalent and bitopic molecules, inspired by Portoghese and beyond, will continue. We hope that others will follow in these footsteps when discovering molecules for their target of choice as the fundamental principles will be the same. There is no doubt that these bitopic and bivalent molecules will provide the tools necessary to parse signaling bias and relate those signaling pathways to behaviors that can be targeted or avoided, depending on the outcome. Paying attention to the chemical properties of the PP and SP and the length and composition of the linker between them has proven fruitful for our work on the D3R and D2R and we look forward to the discoveries of others using these medicinal chemistry principles.
Acknowledgements.
AHN would like to acknowledge her admiration for Dr. Phil Portoghese - his pioneering work on bivalent and bitopic ligands has been a great inspiration to many. She would also like to acknowledge her mentor, Dr. Kenner Rice, for his continuous support throughout her career, and thank the many collaborators, post-doctoral and post-baccalaureate fellows, and graduate students that have worked with her over the years and continue to inspire her every day. The authors would like to thank Dr. Therese Ku for proofreading the manuscript and providing insightful suggestions. And a special thanks to J. Cao for >20 years of technical support to the Medicinal Chemistry Section. This work was funded by Z1A DA000424 and Z1A DA 000609. The authors declare no conflicts of interest.
Abbreviations Used:
- GPCR
G-protein coupled receptor
- D3R
dopamine D3 receptor
- D2R
dopamine D2 receptor
- PP
primary pharmacophore
- SP
secondary pharmacophore
- OBS
orthosteric binding site
- SBP
secondary binding pocket
- ABS
allosteric binding site
- TM
transmembrane
- ADMET
absorption, distribution, metabolism, excretion, toxicity
BIOGRAPHIES
Amy Hauck Newman received her doctorate in Medicinal Chemistry from the Medical College of Virginia, Virginia Commonwealth University. After postdoctoral studies at the NIH and a brief time at WRAIR, she joined the National Institute on Drug Abuse-Intramural Research Program, NIH. She is currently Acting Scientific Director, Chief of the Molecular Targets and Medications Discovery Branch and Director of the NIDA-IRP Medications Development Program. She has coauthored more than 280 original articles and reviews on the design, synthesis, and evaluation of CNS active agents, with an emphasis on selective ligands for the dopaminergic system, as potential treatment medications for substance use disorders. She is also an inventor on several licensed NIH patents. She was the first woman to receive the Philip Portoghese Lectureship Award.
Francisco O. Battiti received his BS in Chemistry and BSLA in Physics from the University of Illinois at Urbana-Champaign in 2018, conducting research on functionalization in dearomatization strategies for natural product synthesis under the mentorship of Prof. David Sarlah. Francisco joined the Medicinal Chemistry Section of the Molecular Targets and Medications Discovery Branch as a post-baccalaureate IRTA Fellow and is a recipient of the Scientific Director’s Diversity in Research Fellowship, in the laboratory of Dr. Amy H. Newman, in 2018.
Alessandro Bonifazi received his M.S., Pharm. D. and doctorate degree in Pharmaceutical Sciences at the University of Camerino, Italy (School of Advanced Studies) – under the mentorship of Prof. Wilma Quaglia, in 2014. He joined the Medicinal Chemistry Section in the Molecular Targets and Medications Discovery Branch, as a Postdoctoral Fellow in the laboratory of Dr. Amy H. Newman in 2014. In 2019 he was appointed Research Fellow in the same laboratory. In addition to performing and developing multiple cell- and tissue-based radioligand binding assays, his research interests include the structure-based rational design and synthesis of highly selective D2 and D3 receptor agonists with the aim to elucidate ligand-receptor interactions necessary to activate specific signaling pathways.
Footnotes
This Award address was presented at the 252nd National Meeting of the American Chemical Society in Philadelphia, PA, August 2016
REFERENCES
- 1.Erez M; Takemori AE; Portoghese PS Narcotic antagonistic potency of bivalent ligands which contain beta-naltrexamine. Evidence for bridging between proximal recognition sites. J. Med. Chem 1982, 25, 847–849. [DOI] [PubMed] [Google Scholar]
- 2.Portoghese PS; Ronsisvalle G; Larson DL; Yim CB; Sayre LM; Takemori AE Opioid agonist and antagonist bivalent ligands as receptor probes. Life Sci 1982, 31, 1283–1286. [DOI] [PubMed] [Google Scholar]
- 3.Portoghese PS; Ronsisvalle G; Larson DL; Takemori AE Synthesis and opioid antagonist potencies of naltrexamine bivalent ligands with conformationally restricted spacers. J. Med. Chem 1986, 29, 1650–1653. [DOI] [PubMed] [Google Scholar]
- 4.Portoghese PS; Larson DL; Sayre LM; Yim CB; Ronsisvalle G; Tam SW; Takemori AE Opioid agonist and antagonist bivalent ligands. The relationship between spacer length and selectivity at multiple opioid receptors. J. Med. Chem 1986, 29, 1855–1861. [DOI] [PubMed] [Google Scholar]
- 5.Portoghese PS; Rein MD; Takemori AE Synthesis and biological activity of analogues of beta-chlornaltrexamine and beta-funaltrexamine at opioid receptors. J. Med. Chem 1986, 29, 1861–1864. [DOI] [PubMed] [Google Scholar]
- 6.Portoghese PS Bivalent ligands and the message-address concept in the design of selective opioid receptor antagonists. Trends Pharmacol. Sci 1989, 10, 230–235. [DOI] [PubMed] [Google Scholar]
- 7.Portoghese PS; Nagase H; Lipkowski AW; Larson DL; Takemori AE Binaltorphimine-related bivalent ligands and their kappa opioid receptor antagonist selectivity. J. Med. Chem 1988, 31, 836–841. [DOI] [PubMed] [Google Scholar]
- 8.Portoghese PS; Nagase H; Takemori AE Only one pharmacophore is required for the kappa opioid antagonist selectivity of norbinaltorphimine. J. Med. Chem 1988, 31, 1344–1347. [DOI] [PubMed] [Google Scholar]
- 9.Schwyzer R ACTH: a short introductory review. Ann. N. Y. Acad. Sci 1977, 297, 3–26. [DOI] [PubMed] [Google Scholar]
- 10.Portoghese PS; Sultana M; Takemori AE Naltrindole, a highly selective and potent non-peptide delta opioid receptor antagonist. Eur. J. Pharmacol 1988, 146, 185–186. [DOI] [PubMed] [Google Scholar]
- 11.Franco R; Casado V; Cortes A; Ferrada C; Mallol J; Woods A; Lluis C; Canela EI; Ferre S Basic concepts in G-protein-coupled receptor homo- and heterodimerization. ScientificWorldJournal 2007, 7, 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bouvier M Oligomerization of G-protein-coupled transmitter receptors. Nat. Rev. Neurosci 2001, 2, 274–286. [DOI] [PubMed] [Google Scholar]
- 13.Cvejic S; Trapaidze N; Cyr C; Devi LA Thr353, located within the COOH-terminal tail of the delta opiate receptor, is involved in receptor down-regulation. J. Biol. Chem 1996, 271, 4073–4076. [DOI] [PubMed] [Google Scholar]
- 14.McVey M; Ramsay D; Kellett E; Rees S; Wilson S; Pope AJ; Milligan G Monitoring receptor oligomerization using time-resolved fluorescence resonance energy transfer and bioluminescence resonance energy transfer. The human delta -opioid receptor displays constitutive oligomerization at the cell surface, which is not regulated by receptor occupancy. J. Biol. Chem 2001, 276, 14092–14099. [DOI] [PubMed] [Google Scholar]
- 15.Jordan BA; Devi LA G-protein-coupled receptor heterodimerization modulates receptor function. Nature 1999, 399, 697–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gomes I; Ayoub MA; Fujita W; Jaeger WC; Pfleger KD; Devi LA G protein-coupled receptor heteromers. Annu. Rev. Pharmacol. Toxicol 2016, 56, 403–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonzalez-Maeso J GPCR oligomers in pharmacology and signaling. Mol. Brain 2011, 4, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gurevich VV; Gurevich EV How and why do GPCRs dimerize? Trends Pharmacol. Sci 2008, 29, 234–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terrillon S; Bouvier M Roles of G-protein-coupled receptor dimerization. EMBO Rep 2004, 5, 30–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.LeBoulluec KL; Mattson RJ; Mahle CD; McGovern RT; Nowak HP; Gentile AJ Bivalent indoles exhibiting serotoninergic binding affinity Bioorg. Med. Chem 1995, 5, 123–126. [Google Scholar]
- 21.Halazy S; Perez M; Fourrier C; Pallard I; Pauwels PJ; Palmier C; John GW; Valentin JP; Bonnafous R; Martinez J Serotonin dimers: application of the bivalent ligand approach to the design of new potent and selective 5-HT(1B/1D) agonists. J. Med. Chem 1996, 39, 4920–4927. [DOI] [PubMed] [Google Scholar]
- 22.Herrick-Davis K Functional significance of serotonin receptor dimerization. Exp. Brain Res 2013, 230, 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soulier JL; Russo O; Giner M; Rivail L; Berthouze M; Ongeri S; Maigret B; Fischmeister R; Lezoualc’h F; Sicsic S; Berque-Bestel I Design and synthesis of specific probes for human 5-HT4 receptor dimerization studies. J. Med. Chem 2005, 48, 6220–6228. [DOI] [PubMed] [Google Scholar]
- 24.Lezoualc’h F; Jockers R; Berque-Bestel I Multivalent-based drug design applied to serotonin 5-HT(4) receptor oligomers. Curr. Pharm. Des 2009, 15, 719–729. [DOI] [PubMed] [Google Scholar]
- 25.Soto CA; Shashack MJ; Fox RG; Bubar MJ; Rice KC; Watson CS; Cunningham KA; Gilbertson SR; Anastasio NC Novel bivalent 5-HT2A receptor antagonists exhibit high affinity and potency in vitro and efficacy in vivo. ACS Chem. Neurosci 2018, 9, 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Russo O; Berthouze M; Giner M; Soulier JL; Rivail L; Sicsic S; Lezoualc’h F; Jockers R; Berque-Bestel I Synthesis of specific bivalent probes that functionally interact with 5-HT(4) receptor dimers. J. Med. Chem 2007, 50, 4482–4492. [DOI] [PubMed] [Google Scholar]
- 27.Castriconi F; Paolino M; Donati A; Giuliani G; Anzini M; Mennuni L; Sabatini C; Lanza M; Caselli G; Makovec F; Sbraccia M; Molinari P; Costa T; Cappelli A Multivalent ligands for the serotonin 5-HT4 receptor. Medchemcomm 2017, 8, 647–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cappelli A; Manini M; Paolino M; Gallelli A; Anzini M; Mennuni L; Del Cadia M; De Rienzo F; Menziani MC; Vomero S Bivalent ligands for the serotonin 5-HT3 receptor. ACS Med. Chem. Lett 2011, 2, 571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perez M; Pauwels PJ; Fourrier C; Chopin P; Valentin JP; John GW; Marien M; Halazy S Dimerization of sumatriptan as an efficient way to design a potent, centrally and orally active 5-HT1B agonist. Bioorg. Med. Chem. Lett 1998, 8, 675–680. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y; Gilliam A; Maitra R; Damaj MI; Tajuba JM; Seltzman HH; Thomas BF Synthesis and biological evaluation of bivalent ligands for the cannabinoid 1 receptor. J. Med. Chem 2010, 53, 7048–7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nimczick M; Decker M New approaches in the design and development of cannabinoid receptor ligands: multifunctional and bivalent compounds. ChemMedChem 2015, 10, 773–786. [DOI] [PubMed] [Google Scholar]
- 32.Huang G; Pemp D; Stadtmuller P; Nimczick M; Heilmann J; Decker M Design, synthesis and in vitro evaluation of novel uni- and bivalent ligands for the cannabinoid receptor type 1 with variation of spacer length and structure. Bioorg. Med. Chem. Lett 2014, 24, 4209–4214. [DOI] [PubMed] [Google Scholar]
- 33.Perrey DA; Gilmour BP; Thomas BF; Zhang Y Toward the development of bivalent ligand probes of cannabinoid CB1 and orexin OX1 receptor heterodimers. ACS Med. Chem. Lett 2014, 5, 634–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glass M; Govindpani K; Furkert DP; Hurst DP; Reggio PH; Flanagan JU One for the price of two…are bivalent ligands targeting cannabinoid receptor dimers capable of simultaneously binding to both receptors? Trends Pharmacol. Sci 2016, 37, 353–363. [DOI] [PubMed] [Google Scholar]
- 35.Wager-Miller J; Westenbroek R; Mackie K Dimerization of G protein-coupled receptors: CB1 cannabinoid receptors as an example. Chem. Phys. Lipids 2002, 121, 83–89. [DOI] [PubMed] [Google Scholar]
- 36.Ellis J; Pediani JD; Canals M; Milasta S; Milligan G Orexin-1 receptor-cannabinoid CB1 receptor heterodimerization results in both ligand-dependent and -independent coordinated alterations of receptor localization and function. J. Biol. Chem 2006, 281, 38812–38824. [DOI] [PubMed] [Google Scholar]
- 37.Schoffelmeer AN; Hogenboom F; Wardeh G; De Vries TJ Interactions between CB1 cannabinoid and mu opioid receptors mediating inhibition of neurotransmitter release in rat nucleus accumbens core. Neuropharmacology 2006, 51, 773–781. [DOI] [PubMed] [Google Scholar]
- 38.Kearn CS; Blake-Palmer K; Daniel E; Mackie K; Glass M Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors enhances heterodimer formation: a mechanism for receptor cross-talk? Mol. Pharmacol 2005, 67, 1697–1704. [DOI] [PubMed] [Google Scholar]
- 39.Ferre S; Ciruela F; Woods AS; Lluis C; Franco R Functional relevance of neurotransmitter receptor heteromers in the central nervous system. Trends Neurosci 2007, 30, 440–446. [DOI] [PubMed] [Google Scholar]
- 40.Borroto-Escuela DO; Carlsson J; Ambrogini P; Narvaez M; Wydra K; Tarakanov AO; Li X; Millon C; Ferraro L; Cuppini R; Tanganelli S; Liu F; Filip M; Diaz-Cabiale Z; Fuxe K Understanding the role of GPCR heteroreceptor complexes in modulating the brain networks in health and disease. Front. Cell. Neurosci 2017, 11, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu J; He J; Castleberry AM; Balasubramanian S; Lau AG; Hall RA Heterodimerization of alpha 2A- and beta 1-adrenergic receptors. J. Biol. Chem 2003, 278, 10770–10777. [DOI] [PubMed] [Google Scholar]
- 42.Jordan BA; Trapaidze N; Gomes I; Nivarthi R; Devi LA Oligomerization of opioid receptors with beta 2-adrenergic receptors: a role in trafficking and mitogen-activated protein kinase activation. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barki-Harrington L; Luttrell LM; Rockman HA Dual inhibition of beta-adrenergic and angiotensin II receptors by a single antagonist: a functional role for receptor-receptor interaction in vivo. Circulation 2003, 108, 1611–1618. [DOI] [PubMed] [Google Scholar]
- 44.Hague C; Lee SE; Chen Z; Prinster SC; Hall RA; Minneman KP Heterodimers of alpha1B- and alpha1D-adrenergic receptors form a single functional entity. Mol. Pharmacol 2006, 69, 45–55. [DOI] [PubMed] [Google Scholar]
- 45.Lalchandani SG; Lei L; Zheng W; Suni MM; Moore BM; Liggett SB; Miller DD; Feller DR Yohimbine dimers exhibiting selectivity for the human alpha 2C-adrenoceptor subtype. J. Pharmacol. Exp. Ther 2002, 303, 979–984. [DOI] [PubMed] [Google Scholar]
- 46.Kizuka H; Hanson RN Beta-adrenoceptor antagonist activity of bivalent ligands. 1. Diamide analogues of practolol. J. Med. Chem 1987, 30, 722–726. [DOI] [PubMed] [Google Scholar]
- 47.Song Y; Shryock JC; Knot HJ; Belardinelli L Selective attenuation by adenosine of arrhythmogenic action of isoproterenol on ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol 2001, 280, H2789–2795. [DOI] [PubMed] [Google Scholar]
- 48.Karellas P; McNaughton M; Baker SP; Scammells PJ Synthesis of bivalent beta2-adrenergic and adenosine A1 receptor ligands. J. Med. Chem 2008, 51, 6128–6137. [DOI] [PubMed] [Google Scholar]
- 49.Barlow N; Baker SP; Scammells PJ Effect of linker length and composition on heterobivalent ligand-mediated receptor cross-talk between the A1 adenosine and beta2 adrenergic receptors. ChemMedChem 2013, 8, 2036–2046. [DOI] [PubMed] [Google Scholar]
- 50.Ferre S; Quiroz C; Woods AS; Cunha R; Popoli P; Ciruela F; Lluis C; Franco R; Azdad K; Schiffmann SN An update on adenosine A2A-dopamine D2 receptor interactions: implications for the function of G protein-coupled receptors. Curr. Pharm. Des 2008, 14, 1468–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hillion J; Canals M; Torvinen M; Casado V; Scott R; Terasmaa A; Hansson A; Watson S; Olah ME; Mallol J; Canela EI; Zoli M; Agnati LF; Ibanez CF; Lluis C; Franco R; Ferre S; Fuxe K Coaggregation, cointernalization, and codesensitization of adenosine A2A receptors and dopamine D2 receptors. J. Biol. Chem 2002, 277, 18091–18097. [DOI] [PubMed] [Google Scholar]
- 52.Fahn S The history of dopamine and levodopa in the treatment of Parkinson’s disease. Mov. Disord 2008, 23 Suppl 3, S497–508. [DOI] [PubMed] [Google Scholar]
- 53.Pinna A Novel investigational adenosine A2A receptor antagonists for Parkinson’s disease. Expert Opin. Investig. Drugs 2009, 18, 1619–1631. [DOI] [PubMed] [Google Scholar]
- 54.Simola N; Morelli M; Pinna A Adenosine A2A receptor antagonists and Parkinson’s disease: state of the art and future directions. Curr. Pharm. Des 2008, 14, 1475–1489. [DOI] [PubMed] [Google Scholar]
- 55.Morelli M; Wardas J Adenosine A(2a) receptor antagonists: potential therapeutic and neuroprotective effects in Parkinson’s disease. Neurotox. Res 2001, 3, 545–556. [DOI] [PubMed] [Google Scholar]
- 56.Soriano A; Ventura R; Molero A; Hoen R; Casado V; Cortes A; Fanelli F; Albericio F; Lluis C; Franco R; Royo M Adenosine A2A receptor-antagonist/dopamine D2 receptor-agonist bivalent ligands as pharmacological tools to detect A2A-D2 receptor heteromers. J. Med. Chem 2009, 52, 5590–5602. [DOI] [PubMed] [Google Scholar]
- 57.Jorg M; May LT; Mak FS; Lee KC; Miller ND; Scammells PJ; Capuano B Synthesis and pharmacological evaluation of dual acting ligands targeting the adenosine A2A and dopamine D2 receptors for the potential treatment of Parkinson’s disease. J. Med. Chem 2015, 58, 718–738. [DOI] [PubMed] [Google Scholar]
- 58.Shen J; Zhang L; Song WL; Meng T; Wang X; Chen L; Feng LY; Xu YC; Shen JK Design, synthesis and biological evaluation of bivalent ligands against A(1)-D(1) receptor heteromers. Acta Pharmacol. Sin 2013, 34, 441–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hubner H; Schellhorn T; Gienger M; Schaab C; Kaindl J; Leeb L; Clark T; Moller D; Gmeiner P Structure-guided development of heterodimer-selective GPCR ligands. Nat. Commun 2016, 7, 12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Qian M; Wouters E; Dalton JAR; Risseeuw MDP; Crans RAJ; Stove C; Giraldo J; Van Craenenbroeck K; Van Calenbergh S Synthesis toward bivalent ligands for the dopamine D2 and metabotropic glutamate 5 receptors. J. Med. Chem 2018, 61, 8212–8225. [DOI] [PubMed] [Google Scholar]
- 61.Kaczor AA; Jorg M; Capuano B The dopamine D2 receptor dimer and its interaction with homobivalent antagonists: homology modeling, docking and molecular dynamics. J. Mol. Model 2016, 22, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Navarro G; Moreno E; Bonaventura J; Brugarolas M; Farre D; Aguinaga D; Mallol J; Cortes A; Casado V; Lluis C; Ferre S; Franco R; Canela E; McCormick PJ Cocaine inhibits dopamine D2 receptor signaling via sigma-1-D2 receptor heteromers. PLoS One 2013, 8, e61245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perreault ML; Hasbi A; O’Dowd BF; George SR Heteromeric dopamine receptor signaling complexes: emerging neurobiology and disease relevance. Neuropsychopharmacology 2014, 39, 156–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Milligan G G protein-coupled receptor dimerisation: molecular basis and relevance to function. Biochim. Biophys. Acta 2007, 1768, 825–835. [DOI] [PubMed] [Google Scholar]
- 65.Filip M; Zaniewska M; Frankowska M; Wydra K; Fuxe K The importance of the adenosine A(2A) receptor-dopamine D(2) receptor interaction in drug addiction. Curr. Med. Chem 2012, 19, 317–355. [DOI] [PubMed] [Google Scholar]
- 66.Hasbi A; O’Dowd BF; George SR Dopamine D1-D2 receptor heteromer signaling pathway in the brain: emerging physiological relevance. Mol. Brain 2011, 4, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hasbi A; Perreault ML; Shen MYF; Fan T; Nguyen T; Alijaniaram M; Banasikowski TJ; Grace AA; O’Dowd BF; Fletcher PJ; George SR Activation of dopamine D1-D2 receptor complex attenuates cocaine reward and reinstatement of cocaine-seeking through inhibition of DARPP-32, ERK, and DeltaFosB. Front. Pharmacol 2017, 8, 924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huber D; Lober S; Hubner H; Gmeiner P Bivalent molecular probes for dopamine D2-like receptors. Bioorg. Med. Chem 2012, 20, 455–466. [DOI] [PubMed] [Google Scholar]
- 69.Kuhhorn J; Gotz A; Hubner H; Thompson D; Whistler J; Gmeiner P Development of a bivalent dopamine D(2) receptor agonist. J. Med. Chem 2011, 54, 7911–7919. [DOI] [PubMed] [Google Scholar]
- 70.Salama I; Lober S; Hubner H; Gmeiner P Synthesis and binding profile of haloperidol-based bivalent ligands targeting dopamine D(2)-like receptors. Bioorg. Med. Chem. Lett 2014, 24, 3753–3756. [DOI] [PubMed] [Google Scholar]
- 71.Hiller C; Kuhhorn J; Gmeiner P Class A G-protein-coupled receptor (GPCR) dimers and bivalent ligands. J. Med. Chem 2013, 56, 6542–6559. [DOI] [PubMed] [Google Scholar]
- 72.Pulido D; Casado-Anguera V; Perez-Benito L; Moreno E; Cordomi A; Lopez L; Cortes A; Ferre S; Pardo L; Casado V; Royo M Design of a true bivalent ligand with picomolar binding affinity for a G protein-coupled receptor homodimer. J. Med. Chem 2018, 61, 9335–9346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gogoi S; Biswas S; Modi G; Antonio T; Reith ME; Dutta AK Novel bivalent ligands for D2/D3 dopamine receptors: Significant co-operative gain in D2 affinity and potency. ACS Med. Chem. Lett 2012, 3, 991–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McRobb FM; Crosby IT; Yuriev E; Lane JR; Capuano B Homobivalent ligands of the atypical antipsychotic clozapine: design, synthesis, and pharmacological evaluation. J. Med. Chem 2012, 55, 1622–1634. [DOI] [PubMed] [Google Scholar]
- 75.Wacker D; Stevens RC; Roth BL How ligands illuminate GPCR molecular pharmacology. Cell 2017, 170, 414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Christopoulos A Advances in G protein-coupled receptor allostery: from function to structure. Mol. Pharmacol 2014, 86, 463–478. [DOI] [PubMed] [Google Scholar]
- 77.Lane JR; Sexton PM; Christopoulos A Bridging the gap: bitopic ligands of G-protein-coupled receptors. Trends Pharmacol. Sci 2013, 34, 59–66. [DOI] [PubMed] [Google Scholar]
- 78.Valant C; Robert Lane J; Sexton PM; Christopoulos A The best of both worlds? Bitopic orthosteric/allosteric ligands of G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol 2012, 52, 153–178. [DOI] [PubMed] [Google Scholar]
- 79.Draper-Joyce CJ; Michino M; Verma RK; Klein Herenbrink C; Shonberg J; Kopinathan A; Scammells PJ; Capuano B; Thal DM; Javitch JA; Christopoulos A; Shi L; Lane JR The structural determinants of the bitopic binding mode of a negative allosteric modulator of the dopamine D2 receptor. Biochem. Pharmacol 2018, 148, 315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lane JR; Donthamsetti P; Shonberg J; Draper-Joyce CJ; Dentry S; Michino M; Shi L; Lopez L; Scammells PJ; Capuano B; Sexton PM; Javitch JA; Christopoulos A A new mechanism of allostery in a G protein-coupled receptor dimer. Nat. Chem. Biol 2014, 10, 745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shonberg J; Lopez L; Scammells PJ; Christopoulos A; Capuano B; Lane JR Biased agonism at G protein-coupled receptors: the promise and the challenges--a medicinal chemistry perspective. Med. Res. Rev 2014, 34, 1286–1330. [DOI] [PubMed] [Google Scholar]
- 82.Klein Herenbrink C; Sykes DA; Donthamsetti P; Canals M; Coudrat T; Shonberg J; Scammells PJ; Capuano B; Sexton PM; Charlton SJ; Javitch JA; Christopoulos A; Lane JR The role of kinetic context in apparent biased agonism at GPCRs. Nat. Commun 2016, 7, 10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Taylor M; Grundt P; Griffin SA; Newman AH; Luedtke RR Dopamine D3 receptor selective ligands with varying intrinsic efficacies at adenylyl cyclase inhibition and mitogenic signaling pathways. Synapse 2010, 64, 251–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kumar V; Moritz AE; Keck TM; Bonifazi A; Ellenberger MP; Sibley CD; Free RB; Shi L; Lane JR; Sibley DR; Newman AH Synthesis and pharmacological characterization of novel trans-cyclopropylmethyl-linked bivalent ligands that exhibit selectivity and allosteric pharmacology at the dopamine D3 receptor (D3R). J. Med. Chem 2017, 60, 1478–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Michino M; Boateng CA; Donthamsetti P; Yano H; Bakare OM; Bonifazi A; Ellenberger MP; Keck TM; Kumar V; Zhu C; Verma R; Deschamps JR; Javitch JA; Newman AH; Shi L Toward understanding the structural basis of partial agonism at the dopamine D3 receptor. J. Med. Chem 2017, 60, 580–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bonifazi A; Yano H; Guerrero AM; Kumar V; Hoffman AF; Lupica CR; Shi L; Newman AH Novel and potent dopamine D2 receptor Go-protein biased agonists. ACS Pharmacol. Transl. Sci 2019, 2, 52–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kumar V; Bonifazi A; Ellenberger MP; Keck TM; Pommier E; Rais R; Slusher BS; Gardner E; You ZB; Xi ZX; Newman AH Highly selective dopamine D3 receptor (D3R) antagonists and partial agonists based on eticlopride and the D3R crystal structure: New leads for opioid dependence treatment. J. Med. Chem 2016, 59, 7634–7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gregory KJ; Sexton PM; Christopoulos A Allosteric modulation of muscarinic acetylcholine receptors. Curr. Neuropharmacol 2007, 5, 157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brown DA Muscarinic acetylcholine receptors (mAChRs) in the nervous system: some functions and mechanisms. J. Mol. Neurosci 2010, 41, 340–346. [DOI] [PubMed] [Google Scholar]
- 90.Felder CC; Bymaster FP; Ward J; DeLapp N Therapeutic opportunities for muscarinic receptors in the central nervous system. J. Med. Chem 2000, 43, 4333–4353. [DOI] [PubMed] [Google Scholar]
- 91.Abrams P; Andersson KE; Buccafusco JJ; Chapple C; de Groat WC; Fryer AD; Kay G; Laties A; Nathanson NM; Pasricha PJ; Wein AJ Muscarinic receptors: their distribution and function in body systems, and the implications for treating overactive bladder. Br. J. Pharmacol 2006, 148, 565–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Melchiorre C; Cassinelli A; Quaglia W Differential blockade of muscarinic receptor subtypes by polymethylene tetraamines. Novel class of selective antagonists of cardiac M-2 muscarinic receptors. J. Med. Chem 1987, 30, 201–204. [DOI] [PubMed] [Google Scholar]
- 93.Mohr K; Trankle C; Kostenis E; Barocelli E; De Amici M; Holzgrabe U Rational design of dualsteric GPCR ligands: quests and promise. Br. J. Pharmacol 2010, 159, 997–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Digby GJ; Utley TJ; Lamsal A; Sevel C; Sheffler DJ; Lebois EP; Bridges TM; Wood MR; Niswender CM; Lindsley CW; Conn PJ Chemical modification of the M(1) agonist VU0364572 reveals molecular switches in pharmacology and a bitopic binding mode. ACS Chem. Neurosci 2012, 3, 1025–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Keov P; Lopez L; Devine SM; Valant C; Lane JR; Scammells PJ; Sexton PM; Christopoulos A Molecular mechanisms of bitopic ligand engagement with the M1 muscarinic acetylcholine receptor. J. Biol. Chem 2014, 289, 23817–23837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Valant C; Gregory KJ; Hall NE; Scammells PJ; Lew MJ; Sexton PM; Christopoulos A A novel mechanism of G protein-coupled receptor functional selectivity. Muscarinic partial agonist McN-A-343 as a bitopic orthosteric/allosteric ligand. J. Biol. Chem 2008, 283, 29312–29321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bock A; Chirinda B; Krebs F; Messerer R; Batz J; Muth M; Dallanoce C; Klingenthal D; Trankle C; Hoffmann C; De Amici M; Holzgrabe U; Kostenis E; Mohr K Dynamic ligand binding dictates partial agonism at a G protein-coupled receptor. Nat. Chem. Biol 2014, 10, 18–20. [DOI] [PubMed] [Google Scholar]
- 98.Chen X; Klockner J; Holze J; Zimmermann C; Seemann WK; Schrage R; Bock A; Mohr K; Trankle C; Holzgrabe U; Decker M Rational design of partial agonists for the muscarinic M1 acetylcholine receptor. J. Med. Chem 2015, 58, 560–576. [DOI] [PubMed] [Google Scholar]
- 99.Daval SB; Kellenberger E; Bonnet D; Utard V; Galzi JL; Ilien B Exploration of the orthosteric/allosteric interface in human M1 muscarinic receptors by bitopic fluorescent ligands. Mol. Pharmacol 2013, 84, 71–85. [DOI] [PubMed] [Google Scholar]
- 100.Schmitz J; van der Mey D; Bermudez M; Klockner J; Schrage R; Kostenis E; Trankle C; Wolber G; Mohr K; Holzgrabe U Dualsteric muscarinic antagonists--orthosteric binding pose controls allosteric subtype selectivity. J. Med. Chem 2014, 57, 6739–6750. [DOI] [PubMed] [Google Scholar]
- 101.Bonifazi A; Yano H; Del Bello F; Farande A; Quaglia W; Petrelli R; Matucci R; Nesi M; Vistoli G; Ferre S; Piergentili A Synthesis and biological evaluation of a novel series of heterobivalent muscarinic ligands based on xanomeline and 1-[3-(4-butylpiperidin-1-yl)propyl]-1,2,3,4-tetrahydroquinolin-2-one (77-LH-28–1). J. Med. Chem 2014, 57, 9065–9077. [DOI] [PubMed] [Google Scholar]
- 102.Eberlein WG; Engel W; Mihm G; Rudolf K; Wetzel B; Entzeroth M; Mayer N; Doods HN Structure-activity relationships and pharmacological profile of selective tricyclic antimuscarinics. Trends Pharmacol. Sci 1989, Suppl, 50–54. [PubMed] [Google Scholar]
- 103.Steinfeld T; Mammen M; Smith JA; Wilson RD; Jasper JR A novel multivalent ligand that bridges the allosteric and orthosteric binding sites of the M2 muscarinic receptor. Mol. Pharmacol 2007, 72, 291–302. [DOI] [PubMed] [Google Scholar]
- 104.Blaho VA; Hla T An update on the biology of sphingosine 1-phosphate receptors. J. Lipid. Res 2014, 55, 1596–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Proia RL; Hla T Emerging biology of sphingosine-1-phosphate: its role in pathogenesis and therapy. J. Clin. Invest 2015, 125, 1379–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Park SJ; Im DS Sphingosine 1-phosphate receptor modulators and drug discovery. Biomol. Ther. (Seoul) 2017, 25, 80–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jo E; Bhhatarai B; Repetto E; Guerrero M; Riley S; Brown SJ; Kohno Y; Roberts E; Schurer SC; Rosen H Novel selective allosteric and bitopic ligands for the S1P(3) receptor. ACS Chem. Biol 2012, 7, 1975–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kohno Y; Tanaka K; Kuriyama K; Hori W Aminophosphonic Acid Derivatives, Addition Salts Thereof And S1p Receptor Modulators US 7456157 B2, 2008. [Google Scholar]
- 109.Sanna MG; Vincent KP; Repetto E; Nguyen N; Brown SJ; Abgaryan L; Riley SW; Leaf NB; Cahalan SM; Kiosses WB; Kohno Y; Brown JH; McCulloch AD; Rosen H; Gonzalez-Cabrera PJ Bitopic sphingosine 1-phosphate receptor 3 (S1P3) antagonist rescue from complete heart block: Pharmacological and genetic evidence for direct S1P3 regulation of mouse cardiac conduction. Mol. Pharmacol 2016, 89, 176–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Reichelt ME; Willems L; Molina JG; Sun CX; Noble JC; Ashton KJ; Schnermann J; Blackburn MR; Headrick JP Genetic deletion of the A1 adenosine receptor limits myocardial ischemic tolerance. Circ. Res 2005, 96, 363–367. [DOI] [PubMed] [Google Scholar]
- 111.Donato M; Gelpi RJ Adenosine and cardioprotection during reperfusion--an overview. Mol. Cell. Biochem 2003, 251, 153–159. [PubMed] [Google Scholar]
- 112.Headrick JP; Hack B; Ashton KJ Acute adenosinergic cardioprotection in ischemic-reperfused hearts. Am. J. Physiol. Heart Circ. Physiol 2003, 285, H1797–1818. [DOI] [PubMed] [Google Scholar]
- 113.Mustafa SJ; Morrison RR; Teng B; Pelleg A Adenosine receptors and the heart: role in regulation of coronary blood flow and cardiac electrophysiology. Handb. Exp. Pharmacol 2009, (193), 161–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Valant C; May LT; Aurelio L; Chuo CH; White PJ; Baltos JA; Sexton PM; Scammells PJ; Christopoulos A Separation of on-target efficacy from adverse effects through rational design of a bitopic adenosine receptor agonist. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 4614–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Baltos JA; Gregory KJ; White PJ; Sexton PM; Christopoulos A; May LT Quantification of adenosine A(1) receptor biased agonism: Implications for drug discovery. Biochem. Pharmacol 2016, 99, 101–112. [DOI] [PubMed] [Google Scholar]
- 116.Aurelio L; Baltos JA; Ford L; Nguyen ATN; Jorg M; Devine SM; Valant C; White PJ; Christopoulos A; May LT; Scammells PJ A Structure-activity relationship study of bitopic N(6)-substituted adenosine derivatives as biased adenosine A1 receptor agonists. J. Med. Chem 2018, 61, 2087–2103. [DOI] [PubMed] [Google Scholar]
- 117.Beaulieu JM; Gainetdinov RR The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev 2011, 63, 182–217 [DOI] [PubMed] [Google Scholar]
- 118.Chien EY; Liu W; Zhao Q; Katritch V; Han GW; Hanson MA; Shi L; Newman AH; Javitch JA; Cherezov V; Stevens RC Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang S; Che T; Levit A; Shoichet BK; Wacker D; Roth BL Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 2018, 555, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang S; Wacker D; Levit A; Che T; Betz RM; McCorvy JD; Venkatakrishnan AJ; Huang XP; Dror RO; Shoichet BK; Roth BL D4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science 2017, 358, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mannel B; Jaiteh M; Zeifman A; Randakova A; Moller D; Hubner H; Gmeiner P; Carlsson J Structure-guided screening for functionally selective D2 dopamine receptor ligands from a virtual chemical library. ACS Chem. Biol 2017, 12, 2652–2661. [DOI] [PubMed] [Google Scholar]
- 122.Michino M; Beuming T; Donthamsetti P; Newman AH; Javitch JA; Shi L What can crystal structures of aminergic receptors tell us about designing subtype-selective ligands? Pharmacol. Rev 2015, 67, 198–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Keck TM; Burzynski C; Shi L; Newman AH Beyond small-molecule SAR: using the dopamine D3 receptor crystal structure to guide drug design. Adv. Pharmacol 2014, 69, 267–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Robarge MJ; Husbands SM; Kieltyka A; Brodbeck R; Thurkauf A; Newman AH Design and synthesis of [(2,3-dichlorophenyl)piperazin-1-yl]alkylfluorenylcarboxamides as novel ligands selective for the dopamine D3 receptor subtype. J. Med. Chem 2001, 44, 3175–3186. [DOI] [PubMed] [Google Scholar]
- 125.Grundt P; Carlson EE; Cao J; Bennett CJ; McElveen E; Taylor M; Luedtke RR; Newman AH Novel heterocyclic trans olefin analogues of N-{4-[4-(2,3-dichlorophenyl)piperazin-1-yl]butyl}arylcarboxamides as selective probes with high affinity for the dopamine D3 receptor. J. Med. Chem 2005, 48, 839–848. [DOI] [PubMed] [Google Scholar]
- 126.Heidbreder CA; Newman AH Current perspectives on selective dopamine D(3) receptor antagonists as pharmacotherapeutics for addictions and related disorders. Ann. N. Y. Acad. Sci 2010, 1187, 4–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Keck TM; John WS; Czoty PW; Nader MA; Newman AH Identifying medication targets for psychostimulant addiction: Unraveling the dopamine D3 receptor hypothesis. J. Med. Chem 2015, 58, 5361–5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yuan J; Chen X; Brodbeck R; Primus R; Braun J; Wasley JW; Thurkauf A NGB 2904 and NGB 2849: two highly selective dopamine D3 receptor antagonists. Bioorg. Med. Chem. Lett 1998, 8, 2715–2718. [DOI] [PubMed] [Google Scholar]
- 129.Pilla M; Perachon S; Sautel F; Garrido F; Mann A; Wermuth CG; Schwartz JC; Everitt BJ; Sokoloff P Selective inhibition of cocaine-seeking behaviour by a partial dopamine D3 receptor agonist. Nature 1999, 400, 371–375. [DOI] [PubMed] [Google Scholar]
- 130.Newman AH; Grundt P; Nader MA Dopamine D3 receptor partial agonists and antagonists as potential drug abuse therapeutic agents. J. Med. Chem 2005, 48, 3663–3679. [DOI] [PubMed] [Google Scholar]
- 131.Micheli F Recent advances in the development of dopamine D3 receptor antagonists: a medicinal chemistry perspective. ChemMedChem 2011, 6, 1152–1162. [DOI] [PubMed] [Google Scholar]
- 132.Micheli F; Heidbreder C Dopamine D3 receptor antagonists: a patent review (2007 – 2012). Expert Opin. Ther. Pat 2013, 23, 363–381. [DOI] [PubMed] [Google Scholar]
- 133.Micheli F Novel, selective, and developable dopamine D3 antagonists with a modified “amino” region. ChemMedChem 2017, 12, 1254–1260. [DOI] [PubMed] [Google Scholar]
- 134.Newman AH; Grundt P; Cyriac G; Deschamps JR; Taylor M; Kumar R; Ho D; Luedtke RR N-(4-(4-(2,3-dichloro- or 2-methoxyphenyl)piperazin-1-yl)butyl)heterobiarylcarboxamides with functionalized linking chains as high affinity and enantioselective D3 receptor antagonists. J. Med. Chem 2009, 52, 2559–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Newman AH; Beuming T; Banala AK; Donthamsetti P; Pongetti K; LaBounty A; Levy B; Cao J; Michino M; Luedtke RR; Javitch JA; Shi L Molecular determinants of selectivity and efficacy at the dopamine D3 receptor. J. Med. Chem 2012, 55, 6689–6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Michino M; Donthamsetti P; Beuming T; Banala A; Duan L; Roux T; Han Y; Trinquet E; Newman AH; Javitch JA; Shi L A single glycine in extracellular loop 1 is the critical determinant for pharmacological specificity of dopamine D2 and D3 receptors. Mol. Pharmacol 2013, 84, 854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Newman AH; Cao J; Bennett CJ; Robarge MJ; Freeman RA; Luedtke RR N-(4-[4-(2,3-dichlorophenyl)piperazin-1-yl]butyl, butenyl and butynyl)arylcarboxamides as novel dopamine D(3) receptor antagonists. Bioorg. Med. Chem. Lett 2003, 13, 2179–2183. [DOI] [PubMed] [Google Scholar]
- 138.Grundt P; Prevatt KM; Cao J; Taylor M; Floresca CZ; Choi JK; Jenkins BG; Luedtke RR; Newman AH Heterocyclic analogues of N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)arylcarboxamides with functionalized linking chains as novel dopamine D3 receptor ligands: potential substance abuse therapeutic agents. J. Med. Chem 2007, 50, 4135–4146. [DOI] [PubMed] [Google Scholar]
- 139.Chen J; Levant B; Jiang C; Keck TM; Newman AH; Wang S Tranylcypromine substituted cis-hydroxycyclobutylnaphthamides as potent and selective dopamine D(3) receptor antagonists. J. Med. Chem 2014, 57, 4962–4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Shah P; Westwell AD The role of fluorine in medicinal chemistry. J. Enzyme Inhib. Med. Chem 2007, 22, 527–540. [DOI] [PubMed] [Google Scholar]
- 141.Meanwell NA Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem 2018, 61, 5822–5880. [DOI] [PubMed] [Google Scholar]
- 142.Zafrani Y; Yeffet D; Sod-Moriah G; Berliner A; Amir D; Marciano D; Gershonov E; Saphier S Difluoromethyl bioisostere: Examining the “lipophilic hydrogen bond donor” concept. J. Med. Chem 2017, 60, 797–804. [DOI] [PubMed] [Google Scholar]
- 143.Banala AK; Levy BA; Khatri SS; Furman CA; Roof RA; Mishra Y; Griffin SA; Sibley DR; Luedtke RR; Newman AH N-(3-fluoro-4-(4-(2-methoxy or 2,3-dichlorophenyl)piperazine-1-yl)butyl)arylcarboxamides as selective dopamine D3 receptor ligands: critical role of the carboxamide linker for D3 receptor selectivity. J. Med. Chem 2011, 54, 3581–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kumar V; Banala AK; Garcia EG; Cao J; Keck TM; Bonifazi A; Deschamps JR; Newman AH Chiral resolution and serendipitous fluorination reaction for the selective dopamine D3 receptor antagonist BAK2–66. ACS Med. Chem. Lett 2014, 5, 647–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Shaik AB; Kumar V; Bonifazi A; Guerrero AM; Cemaj SL; Gadiano A; Lam J; Xi ZX; Rais R; Slusher B; Newman AH Unpublished Results [DOI] [PMC free article] [PubMed]
- 146.Keck TM; Banala AK; Slack RD; Burzynski C; Bonifazi A; Okunola-Bakare OM; Moore M; Deschamps JR; Rais R; Slusher BS; Newman AH Using click chemistry toward novel 1,2,3-triazole-linked dopamine D3 receptor ligands. Bioorg. Med. Chem 2015, 23, 4000–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Shonberg J; Herenbrink CK; Lopez L; Christopoulos A; Scammells PJ; Capuano B; Lane JR A structure-activity analysis of biased agonism at the dopamine D2 receptor. J. Med. Chem 2013, 56, 9199–9221. [DOI] [PubMed] [Google Scholar]
- 148.Mistry SN; Shonberg J; Draper-Joyce CJ; Klein Herenbrink C; Michino M; Shi L; Christopoulos A; Capuano B; Scammells PJ; Lane JR Discovery of a novel class of negative allosteric modulator of the dopamine D2 receptor through fragmentation of a bitopic ligand. J. Med. Chem 2015, 58, 6819–6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kopinathan A; Draper-Joyce C; Szabo M; Christopoulos A; Scammells PJ; Lane JR; Capuano B Subtle modifications to the indole-2-carboxamide motif of the negative allosteric modulator N-((trans)-4-(2-(7-Cyano-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)cyclohexyl)-1H-indole-2-carboxamide (SB269652) yield dramatic changes in pharmacological activity at the dopamine D2 receptor. J. Med. Chem 2019, 62, 371–377. [DOI] [PubMed] [Google Scholar]
- 150.Schmid CL; Kennedy NM; Ross NC; Lovell KM; Yue Z; Morgenweck J; Cameron MD; Bannister TD; Bohn LM Bias factor and therapeutic window correlate to predict safer opioid analgesics. Cell 2017, 171, 1165–1175 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Fossler MJ; Sadler BM; Farrell C; Burt DA; Pitsiu M; Skobieranda F; Soergel DG Oliceridine (TRV130), a novel G protein-biased ligand at the mu-opioid receptor, demonstrates a predictable relationship between plasma concentrations and pain relief. I: Development of a pharmacokinetic/pharmacodynamic model. J. Clin. Pharmacol 2018, 58, 750–761. [DOI] [PubMed] [Google Scholar]
- 152.Janecka A; Piekielna-Ciesielska J; Wtorek K Biased agonism as an emerging strategy in the search for better opioid analgesics. Curr. Med. Chem 2019, Epub ahead of print DOI: 10.2174/0929867326666190506103124 [DOI] [PubMed] [Google Scholar]
- 153.Ehrlich AT; Kieffer BL; Darcq E Current strategies toward safer mu opioid receptor drugs for pain management. Expert Opin. Ther. Targets 2019, 23, 315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Stanczyk MA; Kandasamy R Biased agonism: the quest for the analgesic holy grail. Pain. Rep 2018, 3, e650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Chen X; McCorvy JD; Fischer MG; Butler KV; Shen Y; Roth BL; Jin J Discovery of G protein-biased D2 dopamine receptor partial agonists. J. Med. Chem 2016, 59, 10601–10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Moller D; Banerjee A; Uzuneser TC; Skultety M; Huth T; Plouffe B; Hubner H; Alzheimer C; Friedland K; Muller CP; Bouvier M; Gmeiner P Discovery of G protein-biased dopaminergics with a pyrazolo[1,5-a]pyridine substructure. J. Med. Chem 2017, 60, 2908–2929. [DOI] [PubMed] [Google Scholar]
- 157.Shen Y; McCorvy JD; Martini ML; Rodriguiz RM; Pogorelov VM; Ward KM; Wetsel WC; Liu J; Roth BL; Jin J D2 dopamine receptor G protein-biased partial agonists based on cariprazine. J. Med. Chem 2019, 62, 4755–4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Donthamsetti P; Gallo EF; Buck DC; Stahl EL; Zhu Y; Lane JR; Bohn LM; Neve KA; Kellendonk C; Javitch JA Arrestin recruitment to dopamine D2 receptor mediates locomotion but not incentive motivation. Mol. Psychiatry 2018, Epub ahead of print DOI: 10.1038/s41380-018-0212-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rose SJ; Pack TF; Peterson SM; Payne K; Borrelli E; Caron MG,Engineered D2R variants reveal the balanced and biased contributions of G-protein and beta-arrestin to dopamine-dependent functions. Neuropsychopharmacology 2018, 43, 1164–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Urs NM; Gee SM; Pack TF; McCorvy JD; Evron T; Snyder JC; Yang X; Rodriguiz RM; Borrelli E; Wetsel WC; Jin J; Roth BL; O’Donnell P; Caron MG Distinct cortical and striatal actions of a beta-arrestin-biased dopamine D2 receptor ligand reveal unique antipsychotic-like properties. Proc. Natl. Acad. Sci. U. S. A 2016, 113, E8178–E8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zou MF; Keck TM; Kumar V; Donthamsetti P; Michino M; Burzynski C; Schweppe C; Bonifazi A; Free RB; Sibley DR; Janowsky A; Shi L; Javitch JA; Newman AH Novel analogues of (R)-5-(Methylamino)-5,6-dihydro-4H-imidazo[4,5,1-ij]quinolin-2(1H)-one (Sumanirole) provide clues to dopamine D2/D3 receptor agonist selectivity. J. Med. Chem 2016, 59, 2973–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Bonifazi A; Yano H; Ellenberger MP; Muller L; Kumar V; Zou MF; Cai NS; Guerrero AM; Woods AS; Shi L; Newman AH Novel bivalent ligands based on the sumanirole pharmacophore reveal dopamine D2 receptor (D2R) biased agonism. J. Med. Chem 2017, 60, 2890–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Boateng CA; Bakare OM; Zhan J; Banala AK; Burzynski C; Pommier E; Keck TM; Donthamsetti P; Javitch JA; Rais R; Slusher BS; Xi ZX; Newman AH High affinity dopamine D3 receptor (D3R)-selective antagonists attenuate heroin self-administration in wild-type but not D3R knockout mice. J. Med. Chem 2015, 58, 6195–6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Battiti FO; Cemaj SL; Guerrero AM; Shaik AB; Lam J; Rais R; Slusher B; Deschamps JR; Imler GH; Newman AH; Bonifazi A The significance of chirality in drug design and synthesis of bitopic ligands as D3 receptor (D3R) selective agonists. J. Med. Chem 2019, 62, 6287–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Newman AH; Blaylock BL; Nader MA; Bergman J; Sibley DR; Skolnick P Medication discovery for addiction: translating the dopamine D3 receptor hypothesis. Biochem. Pharmacol 2012, 84, 882–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Liow JS; Morse CL; Lu S; Frankland M; Tye GL; Zoghbi SS; Gladding RL; Shaik AB; Innis RB; Newman AH; Pike VW [O-methyl-(11)C]N-(4-(4-(3-Chloro-2-methoxyphenyl)-piperazin-1-yl)butyl)-1H-indol e-2-carboxamide ([(11)C]BAK4–51) is an efflux transporter substrate and ineffective for PET imaging of brain D(3) receptors in rodents and monkey. Molecules 2018, 23, 2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tarcsay A; Nyiri K; Keseru GM Impact of lipophilic efficiency on compound quality. J. Med. Chem 2012, 55, 1252–1260. [DOI] [PubMed] [Google Scholar]
- 168.Hann MM; Keseru GM Finding the sweet spot: the role of nature and nurture in medicinal chemistry. Nat. Rev. Drug Discov 2012, 11, 355–365. [DOI] [PubMed] [Google Scholar]
- 169.Lipinski CA Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [DOI] [PubMed] [Google Scholar]
- 170.Lipinski CA; Lombardo F; Dominy BW; Feeney PJ Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev 2001, 46, 3–26. [DOI] [PubMed] [Google Scholar]
- 171.Wager TT; Hou X; Verhoest PR; Villalobos A Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci 2010, 1, 435–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Collins GT; Witkin JM; Newman AH; Svensson KA; Grundt P; Cao J; Woods JH Dopamine agonist-induced yawning in rats: a dopamine D3 receptor-mediated behavior. J. Pharmacol. Exp. Ther 2005, 314, 310–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Collins GT; Newman AH; Grundt P; Rice KC; Husbands SM; Chauvignac C; Chen J; Wang S; Woods JH Yawning and hypothermia in rats: effects of dopamine D3 and D2 agonists and antagonists. Psychopharmacology (Berl) 2007, 193, 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Collins GT; Truccone A; Haji-Abdi F; Newman AH; Grundt P; Rice KC; Husbands SM; Greedy BM; Enguehard-Gueiffier C; Gueiffier A; Chen J; Wang S; Katz JL; Grandy DK; Sunahara RK; Woods JH Proerectile effects of dopamine D2-like agonists are mediated by the D3 receptor in rats and mice. J. Pharmacol. Exp. Ther 2009, 329, 210–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Martelle SE; Nader SH; Czoty PW; John WS; Duke AN; Garg PK; Garg S; Newman AH; Nader MA Further characterization of quinpirole-elicited yawning as a model of dopamine D3 receptor activation in male and female monkeys. J. Pharmacol. Exp. Ther 2014, 350, 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Czoty PW; John WS; Newman AH; Nader MA Yawning elicited by intravenous ethanol in rhesus monkeys with experience self-administering cocaine and ethanol: Involvement of dopamine D3 receptors. Alcohol 2018, 69, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kumar R; Riddle LR; Griffin SA; Chu W; Vangveravong S; Neisewander J; Mach RH; Luedtke RR Evaluation of D2 and D3 dopamine receptor selective compounds on L-dopa-dependent abnormal involuntary movements in rats. Neuropharmacology 2009, 56, 956–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Riddle LR; Kumar R; Griffin SA; Grundt P; Newman AH; Luedtke RR Evaluation of the D3 dopamine receptor selective agonist/partial agonist PG01042 on L-dopa dependent animal involuntary movements in rats. Neuropharmacology 2011, 60, 284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Higley MJ; Sabatini BL Competitive regulation of synaptic Ca2+ influx by D2 dopamine and A2A adenosine receptors. Nat. Neurosci 2010, 13, 958–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Orio L; Wee S; Newman AH; Pulvirenti L; Koob GF The dopamine D3 receptor partial agonist CJB090 and antagonist PG01037 decrease progressive ratio responding for methamphetamine in rats with extended-access. Addict. Biol 2010, 15, 312–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Blaylock BL; Gould RW; Banala A; Grundt P; Luedtke RR; Newman AH; Nader MA Influence of cocaine history on the behavioral effects of Dopamine D(3) receptor-selective compounds in monkeys. Neuropsychopharmacology 2011, 36, 1104–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Caine SB; Thomsen M; Barrett AC; Collins GT; Grundt P; Newman AH; Butler P; Xu M Cocaine self-administration in dopamine D(3) receptor knockout mice. Exp. Clin. Psychopharmacol 2012, 20, 352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Yan Y; Kong H; Wu EJ; Newman AH; Xu M Dopamine D3 receptors regulate reconsolidation of cocaine memory. Neuroscience 2013, 241, 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Yan Y; Newman AH; Xu M Dopamine D1 and D3 receptors mediate reconsolidation of cocaine memories in mouse models of drug self-administration. Neuroscience 2014, 278, 154–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.John WS; Banala AK; Newman AH; Nader MA Effects of buspirone and the dopamine D3 receptor compound PG619 on cocaine and methamphetamine self-administration in rhesus monkeys using a food-drug choice paradigm. Psychopharmacology (Berl) 2015, 232, 1279–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Martelle JL; Claytor R; Ross JT; Reboussin BA; Newman AH; Nader MA Effects of two novel D3-selective compounds, NGB 2904 [N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)-9H-fluorene-2-carboxamide] and CJB 090 [N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)-4-(pyridin-2-yl)benzamide], on the reinforcing and discriminative stimulus effects of cocaine in rhesus monkeys. J. Pharmacol. Exp. Ther 2007, 321, 573–582. [DOI] [PubMed] [Google Scholar]
- 187.Martelle JL; Nader MA A within-subject assessment of the discriminative stimulus and reinforcing effects of self-administered cocaine in rhesus monkeys. Psychopharmacology (Berl) 2009, 203, 343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Achat-Mendes C; Platt DM; Newman AH; Spealman RD The dopamine D3 receptor partial agonist CJB 090 inhibits the discriminative stimulus but not the reinforcing or priming effects of cocaine in squirrel monkeys. Psychopharmacology (Berl) 2009, 206, 73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Besson M; Belin D; McNamara R; Theobald DE; Castel A; Beckett VL; Crittenden BM; Newman AH; Everitt BJ; Robbins TW; Dalley JW Dissociable control of impulsivity in rats by dopamine D2/3 receptors in the core and shell subregions of the nucleus accumbens. Neuropsychopharmacology 2010, 35, 560–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Moreno M; Economidou D; Mar AC; Lopez-Granero C; Caprioli D; Theobald DE; Fernando A; Newman AH; Robbins TW; Dalley JW Divergent effects of D(2)/(3) receptor activation in the nucleus accumbens core and shell on impulsivity and locomotor activity in high and low impulsive rats. Psychopharmacology (Berl) 2013, 228, 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Mason CW; Hassan HE; Kim KP; Cao J; Eddington ND; Newman AH; Voulalas PJ Characterization of the transport, metabolism, and pharmacokinetics of the dopamine D3 receptor-selective fluorenyl- and 2-pyridylphenyl amides developed for treatment of psychostimulant abuse. J. Pharmacol. Exp. Ther 2010, 333, 854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.You ZB; Bi GH; Galaj E; Kumar V; Cao J; Gadiano A; Rais R; Slusher BS; Gardner EL; Xi ZX; Newman AH Dopamine D3R antagonist VK4–116 attenuates oxycodone self-administration and reinstatement without compromising its antinociceptive effects. Neuropsychopharmacology 2019, 44, 1415–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Jordan CJ; Humburg B; Rice M; Bi GH; You ZB; Shaik AB; Cao J; Bonifazi A; Gadiano A; Rais R; Slusher B; Newman AH; Xi ZX The highly selective dopamine D3R antagonist, R-VK4–40 attenuates oxycodone reward and augments analgesia in rodents. Neuropharmacology 2019, Epub ahead of print DOI: 10.1016/j.neuropharm.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Jordan CJ; Humburg BA; Thorndike EB; Shaik AB; Xi ZX; Baumann MH; Newman AH; Schindler CW Unpublished Results [DOI] [PMC free article] [PubMed]
- 195.Banks ML; Townsend EA; Negus SS Testing the 10 most wanted: a preclinical algorithm to screen candidate opioid use disorder medications. Neuropsychopharmacology 2019, 44, 1011–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]