

Abstract
Background:
Coinciding with other chronic comorbidities, the prevalence of periodontal disease increases with aging. Mounting evidence has established that senescent cells accumulate at sites of age-related pathologies, where they promote “non-microbial” inflammation. We hypothesized that alveolar bone osteocytes develop senescence characteristics in old age.
Methods:
Alveolar bone samples were obtained from young (6 months) and old (20 to 22 months) mice to evaluate the expression of senescence biomarkers by immunofluorescent staining. Osteocyte-enriched fractions were used to characterize the age-related senescence-associated secretory phenotype (SASP) gene expression profile. Primary alveolar bone cells were exposed to the SASP via in vitro senescent conditioned media (SCM) administration. A multiplex assay confirmed protein levels of specific cytokines. Interactions with bacterial components were evaluated by stimulating cells with lipopolysaccharide (LPS).
Results:
Increased senescence-associated distension of satellites (SADS) and p16Ink4a mRNA expression were identified in alveolar bone osteocytes with aging. These findings were associated with increased levels of DNA damage, and activated p38 MAPK, both inducers of senescence. Furthermore, interleukin-6 (IL6), IL17, IGFBP4, and MMP13 were significantly upregulated with aging in osteocyte-enriched samples. Interestingly, SCM potentiated the LPS-induced expression of IL1α, IL1β, and IL6. Cell migration and differentiation were also impeded by SCM. These in vitro effects were ameliorated by the p38 MAPK inhibitor SB202190.
Conclusions:
Accumulation of senescent osteocytes contributes to deterioration of the periodontal environment by exacerbating chronic inflammation and reducing regeneration in old age. Cellular senescence is a cell-intrinsic response to DNA damage, and a host-related mechanism associated with aging that could potentiate inflammation induced by bacterial components.
Keywords: aging, alveolar bone loss, cellular senescence, DNA damage, inflammation, periodontal diseases
1. INTRODUCTION
Aging is a time-dependent process characterized by both the progressive deterioration of multiple organs, and a chronic low-grade “sterile” inflammation in the absence of overt infection, called inflammaging.1,2 These changes have been hypothesized to drive age-related diseases as a group, and thus it is not surprising that increased severity of periodontal disease coexists with other comorbidities of aging, including osteoporosis, frailty, and neurodegenerative diseases, among others.3,4 Indeed, the prevalence of periodontal disease increases around 30 to 40 years, and is exacerbated in most of the adults over 50 years.5,6 Multiple factors have been proposed to underlie this higher prevalence and severity of periodontitis in older adults, including the accumulation of lesions with advancing age, shifts in subgingival microbiota composition, and/or declined function of the immune-inflammatory system.7 However, the exact mechanisms between aging and inflammatory periodontal tissue destruction remain poorly understood, representing an important gap of knowledge. Therefore, identifying novel therapeutic targets contributing to age-related periodontal bone loss is essential to prevent and combat its progression.
The host inflammatory reaction is the primary insult that is responsible for periodontal tissue destruction.8 There is clear evidence that bacterial infection is necessary for an early inflammatory reaction, although bacteria alone are not sufficient to promote periodontal disease development.9 A sustained production of proinflammatory mediators can induce the continued infiltration of immune cells, eventually resulting in collateral damage to healthy tissues. Of note, the host’s immunoinflammatory response, but not specific bacteria, determines whether periodontal disease develops.10 Consistent with this premise, development of periodontal alterations, including alveolar bone loss, have been reported in both old germ-free mice (12 to 30 months) and rats (18 months).11,12 Taken together, this evidence suggests that intrinsic age-related host factors might contribute to promote the increased prevalence of periodontal bone destruction with advancing aging.
Cellular senescence is a cell fate characterized by irreversible growth arrest, as a result of replicative exhaustion or various types of stress, that prevents malignant cell transformation.13 The senescent phenotype can also be induced by DNA damaging stimuli, such as ionizing radiation, oxidative stress, and LPS.14–16 Although cellular senescence was first attributed to proliferating cells, post-mitotic cells can also develop a senescent-like phenotype.17,18 One prominent physiological role of senescent cells is to coordinate a sequence of events that initiate tissue remodeling by activating and attracting immune cells, which help resolve tissue damage and eventually promote their own clearance.19 Thus, senescent cells can transiently play beneficial roles in normal physiological functions such as wound healing and during embryogenesis.20,21
In contrast, chronic senescent cell accumulation due to higher production and/or inefficient clearance with aging in humans and mice, contributes to the development of age-associated pathologies.22 Age-related accumulation of senescent cells has been attributed to impaired immune surveillance.23 Although the presence of senescent cells has mostly been associated with aging, our group has previously shown that senescent osteocytes accumulate prematurely in alveolar bone from young mice.24 Regardless of how cellular senescence is induced, a critical feature of senescent cells is the acquisition of the SASP, a dysfunctional mixture of proinflammatory cytokines and matrix degrading proteases that collectively alter the local environment.25 In addition, these secreted factors can also impact negatively the function of neighboring cells.26 Thus, age-dependent accumulation of senescent cells promotes detrimental effects in part by acting as a perpetual source of proinflammatory mediators, facilitating a sustained infiltration of immune cells.
Senescent cells are mainly found at tissues with chronic inflammation and sites of age-related pathologies, suggesting that senescence-associated inflammation contributes to the progression of those diseases.25,27 Similar to other chronic diseases, the risk of periodontal alveolar bone loss increases with aging. However, whether senescent cells accumulate in old periodontal tissues remains completely unexplored. We hypothesized that senescent osteocytes accumulate chronologically in alveolar bone, and contribute to age-related alveolar bone deterioration. Therefore, the aim of this study was to evaluate whether alveolar bone osteocytes develop a senescent phenotype with chronological aging. In addition, we also investigated the potential effect of senescent osteocytes secreted factors on different stages of bone formation.
2. MATERIALS AND METHODS
2.1. Alveolar bone osteocyte-enriched population from young and old mice
All mice experiments were approved by Institutional Animal Care and Use Committee at Mayo Clinic before study initiation. Jawbones were harvested from young (6-month-old) and old (22- to 24-month-old) C57BL/6 wild-type female mice (n = 10),* and processed independently. Soft tissues and teeth were removed, jawbones hemi-sectioned at the symphysis level, and processed as previously described.24 Briefly, hemi-jawbones were sectioned, both alveolar bone blocks pooled together, and treated as one sample. These samples were processed by serial collagenase digestion (300 active U/mL).† The resulting tissue digestion was centrifuged, and tissue immediately homogenized in lysis buffer (without in vitro cell culture). Samples were stored at −80°C for later RNA extraction.
2.2. Immunofluorescence staining (IF)
Soft tissues were removed from harvested young and old jawbones, fixed with 4% (v/v) paraformaldehyde (PFA) for 48 hours, and then processed as previously described.24 Briefly, a cryostat‡ was used to obtain 6-μm sections from optimal cutting temperature (OCT) compound embedded frozen hemi-jawbones. Samples were fixed and permeabilized and blocked with 10% goat serum for 1 hour. Tissues were incubated with γH2AX, p53 (phospho-S15), or phospho-p38 primary antibodies and goat α-rabbit Alexa 447 secondary antibodies§ at room temperature (RT) for 1 hour each. DAPI Fluoromount was used for nuclear staining. For IF staining, primary cells were treated with osteogenic differentiation media and incubated stained using an osterix antibody.** An inverted laser confocal microscope was used for staining visualization.
2.3. SADS assay
For the identification of senescent osteocytes located in alveolar bone cortices in vivo, the SADS assay was performed as previously described.17,24 Briefly, Cy3-labeled CENPB-specific peptide nucleic acid (PNA) probe†† was used for fluorescent in situ hybridization staining. DAPI was used as nuclear staining, and mounting media. SADS quantification was performed using a confocal microscope‡‡ with in-depth Z stacking (100× oil objective) at the Mayo Clinic Microscopy and Cell Analysis Core. For each sample, at least 50 osteocytes were analyzed, and those displaying ≥4 SADS were considered senescent.17 Quantification was performed three times, and the median value was used as final result for young and old samples.
2.4. Quantitative analysis of the alveolar bone osteocytes in alveolar bone
High resolution microcomputed tomography (μCT) scanning††,§§ from young and old mouse jawbones were performed ex vivo for visualization of age-related alveolar bone loss. Brightfield images were captured with an inverted laser confocal microscope with a 10× objective. The number of osteocytes was measured in alveolar bone frozen sections from young and old mice stained with DAPI. Ten random fields were used to count the number of osteocytes in each group (presented as number of osteocytes per field).
2.5. Primary cell isolation and culture
Jawbone primary cell isolation was performed as previously described.28 Briefly, jawbone fragments were digested using collagenase dissolved in α-MEM and incubated at 37°C. Cells were cultured in 6-well plates containing α-MEM supplemented with 1% penicillin/streptomycin, L-glutamine, and 10% fetal bovine serum (FBS). For osteogenic differentiation, confluent cells were treated with osteogenic media (OM; α-MEM containing 50 mg/L ascorbic acid and 10 mM β-glycerophosphate). To evaluate the effect of SCM on differentiation, cells were cultured in the presence of 25% control conditioned media (CM), or 25% SCM from irradiated osteocytes, prepared as previously described.29 In addition, cells were exposed to LPS (10 μg/mL) for 48 hours (from Porphyromonas gingivalis).*** To evaluate mineralization, cells were cultured for 2 weeks. For osteocyte differentiation, cells were cultured as previously described.30 The osteocyte cell line Ocy45431 was used to validate the results. Cells were differentiated in OM (control) and supplemented with 10 μg/mL LPS, LPS+SCM (25%, v/v), or SCM/LPS and SB202190 (10 μM)††† for 7 days.
2.6. CM preparation
To evaluate the effect of senescent osteocytes secreted factors, primary cells were cultured in 6-well plates, and induced to differentiate into osteocytes as described above. Next, cells were repeatedly exposed to LPS (10 μg/mL) to induce senescence (6 days, media changed daily), or control media alone.15,24 To eliminate potential residual LPS effect, cells were washed with 1× phosphate-buffered saline (PBS) and cultured for 1 additional week, changing media every 2 to 3 days. Cells were washed three times and cultured with serum-free media containing 1% penicillin/streptomycin at 37°C for 24 hours.32 Supernatants were collected and centrifuged at 10,000 rpm for 5 minutes to remove cellular debris. Primary osteocytes were induced to become senescent by the exposure of 10 Gray of cesium irradiation, performed as previously described.29 Stored aliquots (−80°C) were used to quantify a subset of cytokines using a multiplex bead assay.‡‡‡
2.7. Alizarin red staining (ARS)
After removing medium, differentiated cells were washed three times with 1× PBS, and fixed with PFA 4% overnight. The next day, cells were rinsed with PBS and stained with Alizarin Red solution§§§ for 10 minutes at RT. Cells were washed three times with water, air-dried overnight, and images captured using a digital camera. ARS staining absorbance was measured using a microplate reader (n = 6).
2.8. Cell migration and wound healing assay
Wound healing assay was performed as previously described.33 Briefly, osteoblastic cells were grown to confluence using 12-well plates. Cells were washed three times with PBS, and standard media replaced with media containing 1% FBS 24 hours before starting the assay. Then, the confluent cell monolayer was wounded using a pipette tip, and cells washed three times. Cells were incubated with serum-free media (control), undiluted healthy osteocyte CM (non-SCM), undiluted SCM, media containing LPS (10 μg/mL), or SCM and SB202190 combined. Cells were allowed to migrate for 48 hours, and fixed with 4% PFA (v/v). Images were taken, and the percentage of the invaded area in relation to the initial wounded area was calculated for each condition.
2.9. Senescence-associated β-galactosidase staining (SA-β-gal)
Detection of SA-β-gal positive cells was performed as previously described.34 Briefly, cells were treated for 48 hours, and fixed in 2% PFA for 15 minutes at RT. The cells were washed and stained with β-gal staining solution. Plates were protected of light, and incubated at 37°C for 24 hours. Ten random fields with at least 750 cells in total were used to determine the percentage of SA-β-gal positive cells.
2.10. RNA isolation and quantitative RT-qPCR analysis
RNA**** and gene expression†††† analysis reagents were used to perform standard RNA isolation and reverse transcriptase-quantitative polymerase chain reaction (RT-qPCR) techniques, respectively, as previously described.35 Each gene was run in triplicate and normalized to Tuba1a expression.
2.11. Statistical analyses
Data are given as mean ± SEM. Microsoft Office Excel 2003‡‡‡‡ and GraphPad software§§§§ were used to perform the analyses. Differences were calculated using Student t-test and considered significant at P <0.05,* P <0.01,** and P <0.001.*** Figures were created using GraphPad software.
3. RESULTS
3.1. Senescent osteocytes accumulate in murine alveolar bone with chronological aging
To quantify the accumulation of senescent osteocytes in alveolar bone samples, we performed a fluorescent in situ hybridization staining to identify SADS, an early and consistent marker of senescence.36 A higher percentage of osteocytes displaying elongated centromeres and “threadlike” unraveling were found in old alveolar bone samples compared with young controls (30.69% versus 73.07%) (Figs. 1A through 1C). Since DNA damage is a major causal factor of cellular senescence in aged mice,37 we next evaluated γH2AX levels, a marker of DNA damage.38 Stronger γH2AX immunofluorescent signal was observed from old alveolar sections (Fig. 1D). Furthermore, these age-dependent changes were associated to significantly higher p16Ink4a expression, a well-known senescent biomarker,39 in osteocyte-enriched populations from old samples (Fig. 1G). However, no differences were observed in phospho-p53 (p-p53) staining and mRNA expression (Figs. 1E and 1G). Because p38 MAPK is a crucial DNA damage and SASP pathway regulator,40 we evaluated its levels. Immunofluorescent staining of 6- and 22-month-old alveolar bone samples displayed an age-dependent p38 MAPK activation in vivo (Fig. 1F). To evaluate whether senescent osteocytes secrete a recognized panel of SASP factors, we directly analyzed osteocyte-enriched fractions without in vitro cell culture. Osteocytes from old alveolar bone samples displayed an upregulated mRNA expression of the SASP factors IGFBP4, IL6, IL17, and MMP13 (Fig. 1H). These findings indicate that osteocytes exhibiting senescent features accumulate in alveolar bone with aging.
FIGURE 1.
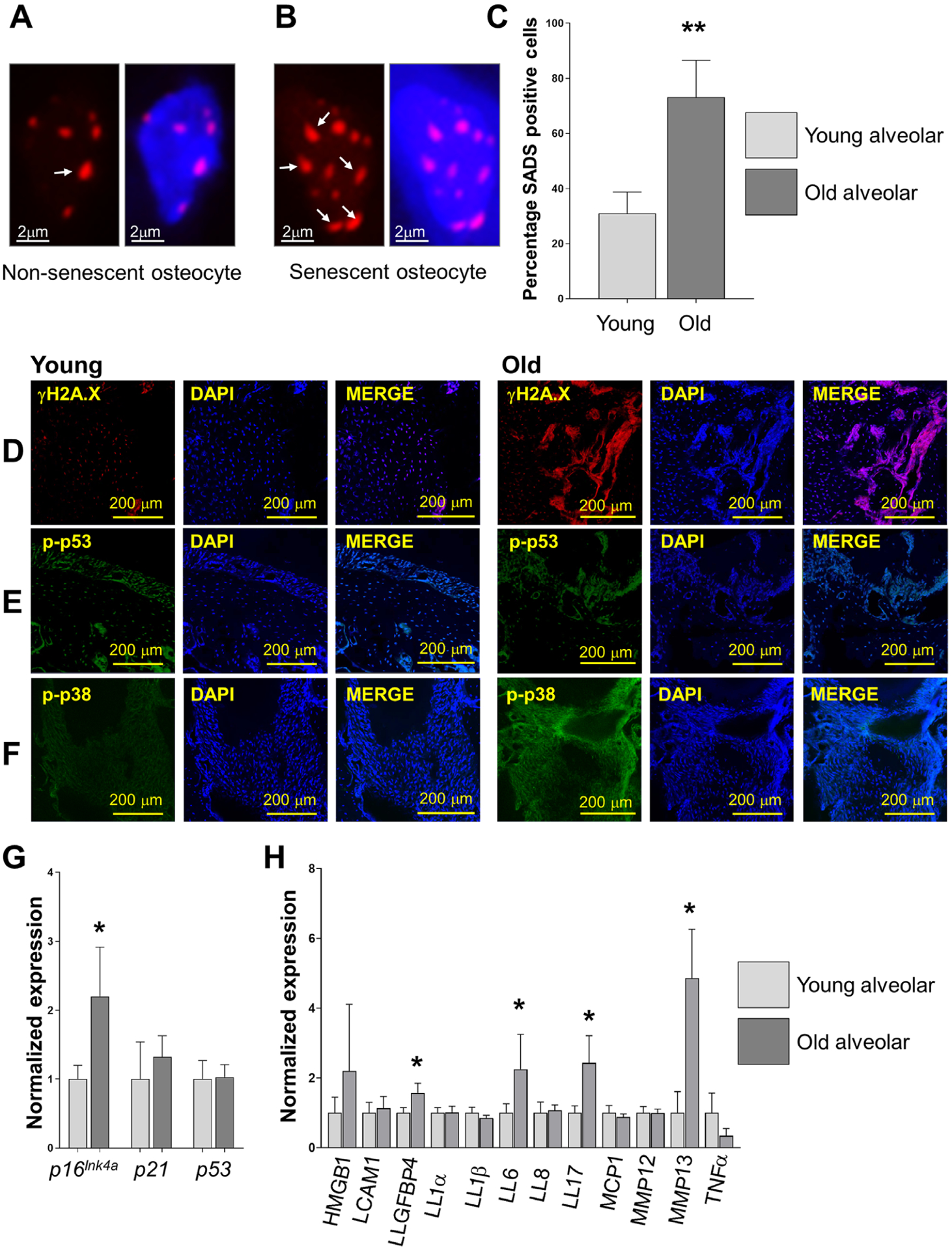
Osteocytes displaying senescent features accumulate in alveolar bone with aging. Expression of established senescence biomarkers were evaluated in alveolar bone samples from 6-month-old and 20- to 22-month-old wild-type mice. A and B) Representative images of normal and senescent alveolar bone osteocytes using the SADS assay (see white arrows for distended DNA satellites); magnification 100×. C) Percentage of alveolar bone osteocytes displaying SADS (from Panels A-B). D) γH2AX immunofluorescence signal (red) showing age-dependent DNA damage. E) Images displaying p53 signal (green). F) The DNA damage pathway p38 MAPK is displayed in green. Nuclei were stained with DAPI. G) p16Ink4a, p21, and p53 expression from osteocyte-enriched populations isolated from young and old alveolar bone without in vitro cell culture (n = 10). H) Osteocyte-enriched fraction from old alveolar bone exhibited upregulated expression of the following factors: IGFBP4, IL6, IL17, and MMP13. Data represent mean ± SEM. Significant changes are indicated by *P < 0.05; **P < 0.01; ***P < 0.001 relative to young versus old samples
3.2. Senescent osteocytes act as a sustained source of proinflammatory factors
To validate our result that senescent osteocytes are a sustained source of proinflammatory factors, we differentiated primary cells isolated from jawbone into osteocytes and induced senescence using repeated LPS exposure, as previously described.15,24 A bead-based multiplex cytokine array showed that CM from these senescent and healthy osteocytes displayed increased IL1A, IL1B, IL6, IL17, and MCP1 protein levels (Fig. 2A), as well as increased mRNA levels of IL1β and IL6 (see Figure S1 in online Journal of Periodontology). This finding was partially mirrored by a similar upregulation in IL6 and MCP1 protein levels in CM from irradiated osteocytes (see Table S1 in online Journal of Periodontology). Next, we wanted to determine the implication of senescence-associated factors and LPS in periodontal bone inflammation. A synergistic effect on IL1α, IL1β and IL6, but not IL17 and MCP1, was observed when both LPS and SCM were combined compared with LPS alone, and the p38 MAPK inhibitor SB202190 significantly reduced this effect on IL1α and IL1β, but not IL6 or MCP1 (Fig. 2B). We additionally found that MMP13 mRNA was upregulated by SCM (see Figure S2 in online Journal of Periodontology). Altogether, these results suggest that senescent osteocyte-associated factors can interact with bacterial products implicated in periodontal inflammation (e.g., LPS) and exacerbate the expression of key proinflammatory cytokines.
FIGURE 2.
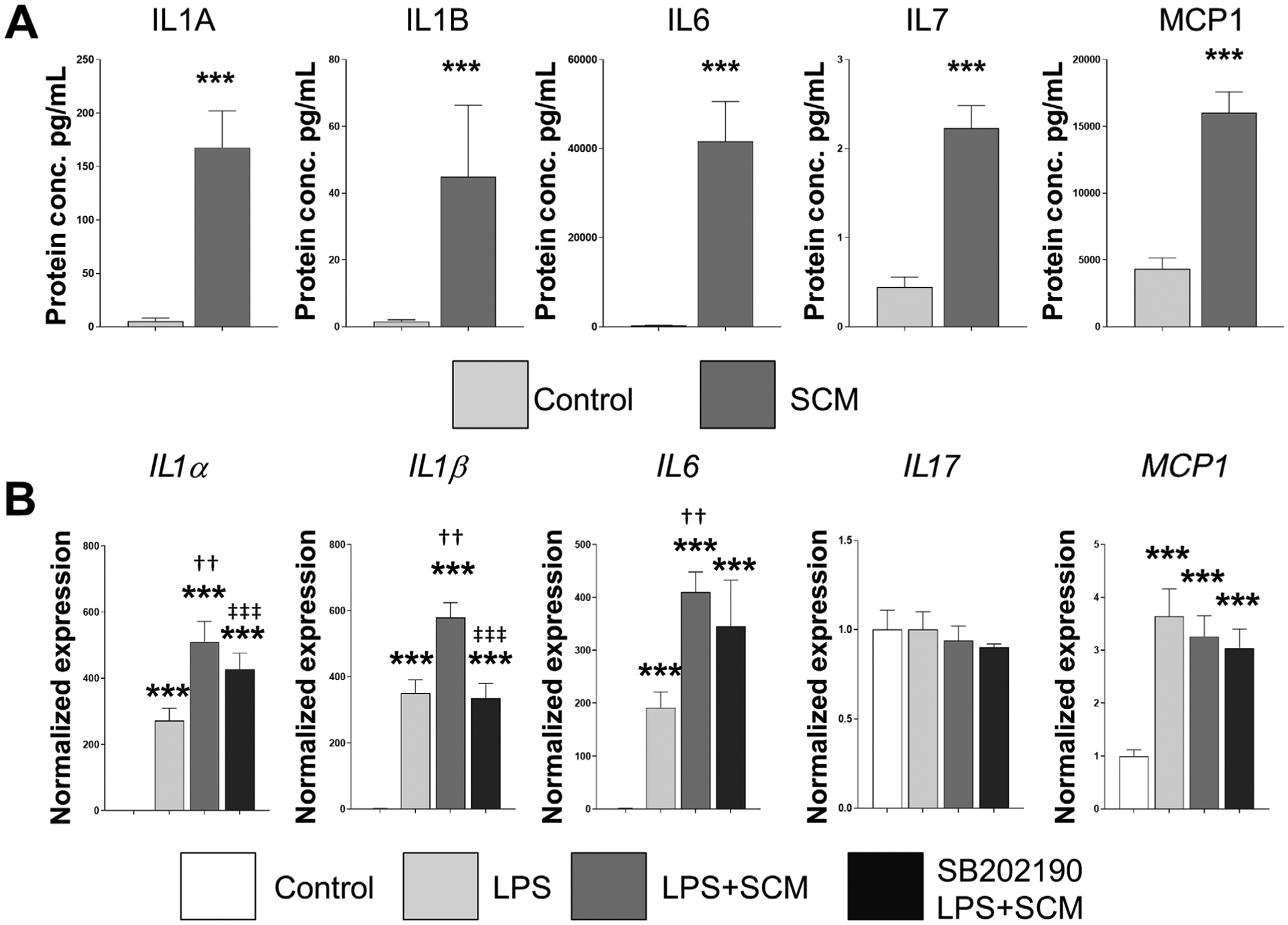
Senescence-associated factors interact with LPS. A) Jawbone cells isolated from 6-month-old mice were differentiated into osteocytes, by repeatedly exposed to LPS (10 μg/mL). CM from these senescent and healthy osteocytes was collected and analyzed by a bead-based multiplex cytokine array. Six replicates were used for each condition and protein concentration displayed in pg/mL. B) To evaluate the potential interaction between senescence-associated factors and LPS, differentiated osteocytes were exposed to media plus control CM (control), LPS (10 μg/mL) or LPS combined with SCM for 48 hours. The effect of SB202190, a selective p38 MAPK inhibitor (10 μM), was also evaluated by pretreating cells before starting the experiments. Data represent mean ± SEM. Significant changes are indicated by: * compared with control, † LPS compared with LPS+SCM, and ‡ LPS+SCM compared with LPS+SCM+SB202190. Various levels of significance are based on the number of each respective symbol (one symbol P < 0.05; two symbols P < 0.01; three symbols P < 0.001)
Previous studies have demonstrated an age-related alveolar bone loss in mice. In fact, using μCT analysis, Wu et al. found 51% less alveolar bone in aged mice compared with young controls.41 To confirm this, we found using μCT and bright-field confocal images that alveolar bone samples from 6-month-old (Fig. 3A) and 20-to 22-month old (Fig. 3B) indeed display age-related alveolar bone loss. These sections when then used to evaluate the relationship between age-related alveolar bone loss and the number of osteocytes using DAPI-stained images (Figs. 3C and 3D). Quantitative analysis of these images unveiled a decreased number of osteocytes per field in the old alveolar samples (Fig. 3E). These results taken together suggest that although osteocyte number decreases in alveolar bone with age, they exhibit a more senescence-like phenotype and are a source of proinflammatory (e.g., SASP) factors. As additional information, we also show the morphology of these osteocytes as seen in alveolar bone (see Figure S3 in online Journal of Periodontology).
FIGURE 3.
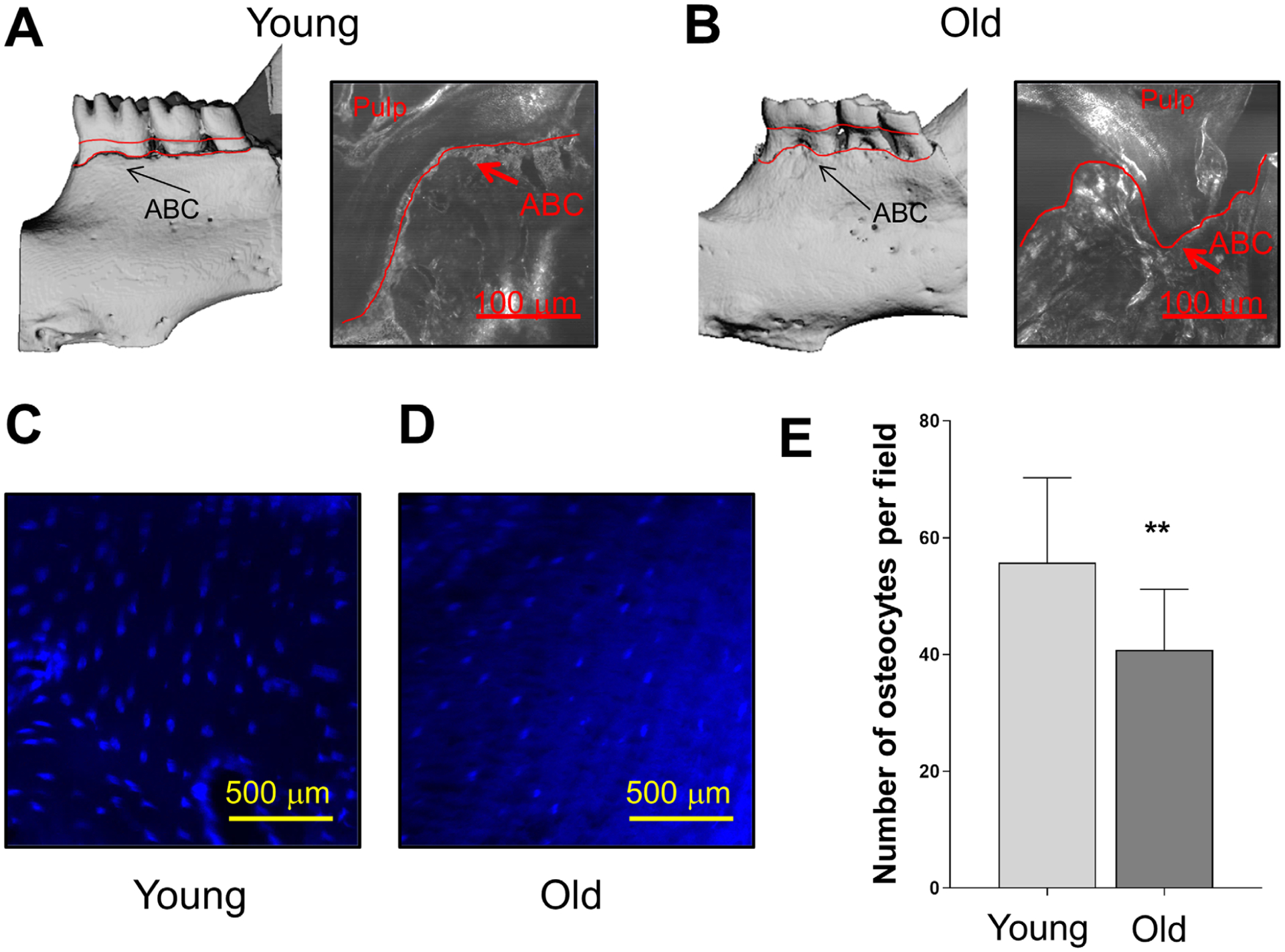
Age-related alveolar bone loss is associated with decreased osteocyte number. Representative μCT and bright-field confocal images display age-related alveolar bone loss comparing (A) 6-month-old and (B) 20- to 22-month-old mice. Increased distance from the cemento-enamel junction to alveolar bone crest (ABC) is observed in the old mice (red lines). C and D) Representative alveolar bone sections from 6- and 20- to 22-month-old wild-type mice were stained with DAPI. E) The results are presented as average number of osteocytes per field. Significant changes are indicated by **P < 0.01
3.3. Senescent osteocyte-associated factors from SCM aggravate LPS-induced inhibition of osteocyte differentiation
Since SCM is known to impair differentiation of the MC3T3-E1 osteoblastic cell line,29 and LPS is known to inhibit osteoblast differentiation, we next assessed the effect of LPS or LPS plus SCM on the mineralization capacity of the osteocyte cell model Ocy454.31 We used this model since it is a more well-established osteocytic cell model and should respond similarly to ex vivo stimuli. LPS alone decreased expression of the key osteogenic marker genes Alpl, Runx2, Osx, Isbp and Bglap and the addition of SCM (LPS+SCM) further decreased the expression, indicating that the combined stimuli aggravates osteogenic gene expression (Fig. 4A). The p38 MAPK inhibitor SB202190 reversed the effects of LPS+SCM on Bglap expression and partially reversed the effect on Runx2 expression. ARS staining of a subset of wells indicates that LPS and LPS+SCM decreased mineralization, while SB202190 partially rescued the response (Fig. 4B). The ARS experiment was repeated using differentiated primary jawbone cells under the same treatment conditions and shows comparable results with the Ocy454 cells (Fig. 4C). Quantitation of the Ocy454 mineralization experiment shows a decrease with LPS alone, an aggravated response with LPS+SCM and a rescue with the SB202190 compound (Fig. 4D). These results demonstrate that senescent osteocyte-associated factors from SCM can aggravate the negative effects of LPS on osteoblast differentiation and mineralization.
FIGURE 4.
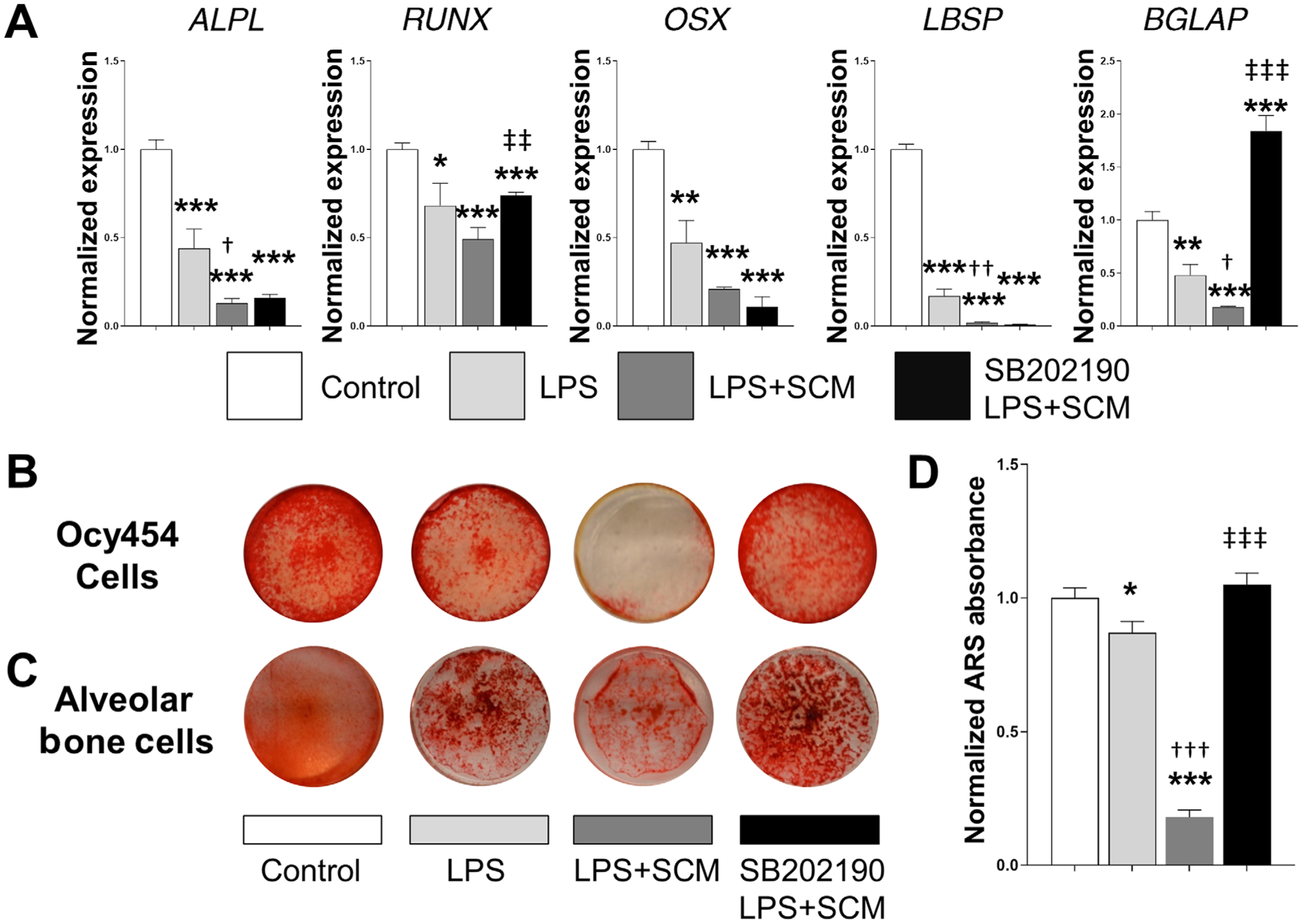
Senescent osteocyte-secreted factors aggravate osteocyte differentiation and mineralization induced by LPS. A) Differentiated Ocy454 cells were exposed to the various stimuli shown in the figure and described in the Methods, and the expression of recognized osteogenic markers was measured by RT-qPCR. Data are represented as mean ± SEM (n = 6). B) A subset of cells from panel A was used to assess bone mineralization capacity using ARS staining. C) A parallel experiment was performed using primary alveolar osteocytic cells. They were cultured under the same conditions for 15 days, and mineralized nodule formation evaluated using ARS staining. Note that in both experiments decreased mineralization produced by LPS is notably aggravated by the addition of SCM, which is reversed by SB202190. Representative images for both cell models were obtained using a digital camera. D) ARS elution was performed on the Ocy454 cells (n = 6) and normalized ARS absorbance is displayed. Significant changes are indicated by: * compared with control, †LPS compared with LPS+SCM, and ‡ for LPS+SCM compared with LPS+SCM+SB202190. Various levels of significance are based on the number of each respective symbol (one symbol P < 0.05; two symbols P < 0.01; three symbols P < 0.001)
3.4. Senescent-osteocyte secreted factors induce a deleterious paracrine effect on osteoprogenitor cell migration in vitro
Given that recruitment of osteoblast precursors is a prerequisite to rebuilt bone after resorption, and senescent cells produce strong paracrine effect on nearby cells,26 we asked whether SCM could affect osteoprogenitor cell migration. To answer this question, we used a wound healing assay. SCM significantly decreased cell migration rate compared with those cells exposed to control CM from healthy osteocytes (Figs. 5A and 5B). In addition, this effect on migration was mirrored by LPS. Next, we evaluated the effect of SCM on SA-β-gal. As shown in Figures 5C and 5D, SCM induced an increase in the percentage of β-gal-positive cells. In addition, SB202190 ameliorated the effects of SCM both on migration and β-gal-activity. No additional effects on migration and SA-β-gal activity were observed when SCM and LPS were combined (data not shown). Taken together, these findings suggest that secreted factors by senescent osteocytes can reduce osteoprogenitor cell recruitment, disrupting the subsequent process of bone formation.
FIGURE 5.
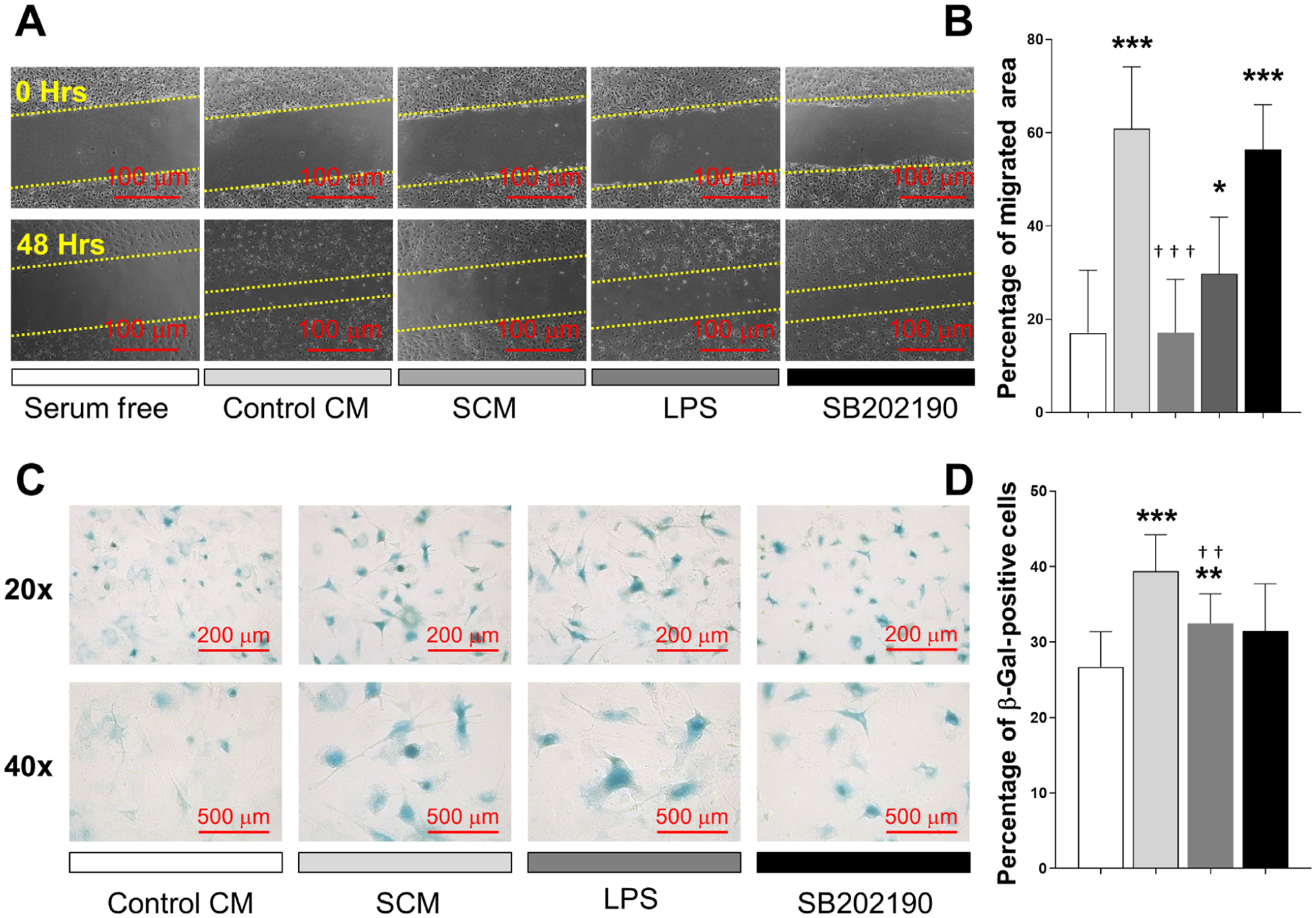
Senescent osteocyte secreted factors induce a deleterious effect on osteoprogenitor cells. A and B) A wound healing assay was used to measure the effect of SCM on chemotactic cell recruitment. Cells were exposed to serum free media as control, CM from healthy osteocytes (control CM), SCM from irradiated osteocytes, LPS (10 μg/mL), or SCM plus SB202190 (10 μM). Cells were allowed to migrate into the scratched area for 48 hours. Representative images from each condition are displayed, and results presented as percentage of migrated area. C and D) The acquisition of senescence-associated SA-β-activity was evaluated by treating primary cells with control CM, SCM, LPS (10 μg/mL) or SCM combined with SB202190. Representative images from each condition are displayed and the results are presented as percentage of β-gal positive cells. Significant changes are indicated by: * compared with serum free or control CM, and † compared with control CM versus SCM or SCM versus SB202190. Various levels of significance are based on the number of each respective symbol (one symbol P < 0.05; two symbols P < 0.01; three symbols P < 0.001)
4. DISCUSSION
Senescent cells accumulate in multiple tissues with aging, and contribute to low-grade chronic “sterile” inflammation at sites of chronic pathologies. Pro-inflammatory and matrix degrading factors secreted by senescent cells negatively alter tissue function and structure. In this study, we report for the first time that the burden of senescent osteocytes increases in alveolar bone in a time-dependent manner. Furthermore, senescent osteocytes-associated factors might interact with bacterial components exacerbating local inflammation. Likewise, these factors could also be implicated in the decreased bone regenerative capacity observed in older individuals by affecting neighboring osteoblast precursors. Therefore, our results provide novel evidence that chronological senescent osteocyte accumulation could play an important role in age-related alveolar bone loss.
Similar to other cells, periodontal cells are continuously exposed to endogenous byproducts of normal metabolism during the lifespan, which can damage DNA. Accumulation of DNA damage is considered one of the most crucial events that contributes to the aging process and development of age-related diseases.2,42 Consistent with this concept, we identified that the level of γH2AX was higher in old alveolar bone. Considering that osteocytes survive up to decades embedded in bone matrix, they are susceptible to accumulating molecular damage.43 Therefore, since cellular senescence is a response to DNA damage,14 the higher number of senescent osteocytes in old alveolar bone could be a consequence of unrepaired, or accumulated, DNA damage. On the other hand, under specific conditions the intensity of DNA damage can determine the cell fate decision. When irreparable damage is produced, cells can undergo either apoptosis or cellular senescence.44 Several studies suggest that cellular senescence is the consequence of low and persistent exposure to DNA damaging stimuli, and apoptosis the result of excessive damage.44 According to this, Gamonal et al. reported the presence of apoptotic cells in gingival tissue as well as B-cell lymphoma-2 (BCL2)-positive cells in gingival tissue from both patients with chronic periodontitis and healthy controls (aged 33 to 55 years).45 It has been suggested that inhibition of cell death, through the upregulation of the anti-apoptotic protein BCL2, might contribute to the progression of periodontal inflammation.46,47 These previous studies support our finding that senescent cells are present in periodontal tissues, because senescent cells are characterized by the anti-apoptotic pathway activation that includes increased BCL2 expression, which helps confer their resistance to apoptosis. Taken together, cellular senescence is a cell-intrinsic response to DNA damage strongly associated with the aging process, and may contribute to the development of age-related periodontal bone deterioration.
Given that cells with genomic instability represent a risk for cancer development, senescent cells communicate their damaged condition to the immune system by secreting multiple proinflammatory cytokines.19,25 How ever, it is important to mention that senescent cells accumulate with aging due to the ineffective clearance by the immune system.23 In agreement with this essential feature of senescent cells, we found that IL1α, IL1β, IL6, IL17, and MCP1 protein levels were upregulated in SCM. Furthermore, IL6, one of the most prominent SASP cytokines, was consistently upregulated in old osteocyte-enriched fractions and osteocyte SCM. In addition, old alveolar bone samples displayed higher IGFBP4 expression, a factor that similar to IL6 can induce accelerated senescence in mesenchymal stem cells.48,49 Importantly, SCM potentiated the LPS effect on IL1α, ILlβ, IL6, and MMP13 expression. MMP13 has been implicated in the degradation of collagen during periodontal disease, which can provide an increased source of nutrients in the form of collagen peptides contributing to “inflammophilic” bacteria overgrowth.50 Therefore, based on previous studies and our results, we hypothesize that senescent cells contribute to the deterioration of the periodontal environment by exacerbating local inflammation, increasing extracellular matrix remodeling, and decreasing regenerative tissue capacity.
In this study, SCM decreased the migration of osteoprogenitor cells, inhibited osteoblast differentiation, and impaired mineralization in vitro. Importantly, SCM aggravated the LPS inhibitory effect on key osteogenic genes and mineralization. We also found that age-related alveolar bone loss was associated to decreased number of osteocytes in vivo. Thus, there are at least three potential scenarios by which the accumulation of senescent cells can eventually disrupt alveolar bone formation. First, decreased recruitment and differentiation of osteoblast precursors could interrupt the normal sequence of osteocyte formation. Second, constant cell renewal to replace damaged osteocytes during the life span could induce stem cell replicative senescence, which is a major contributor for the reduced regenerative capacity observed with aging.2 Third, long-lived osteocytes and proliferating progenitor cells are particularly vulnerable to undergo age-related DNA damage, leading to cellular senescence in old age. We suggest a model to explain the increased severity of alveolar bone loss with aging based on the impact of senescent osteocytes accumulation on both inflammation and progenitor cells (Fig. 6).
FIGURE 6.
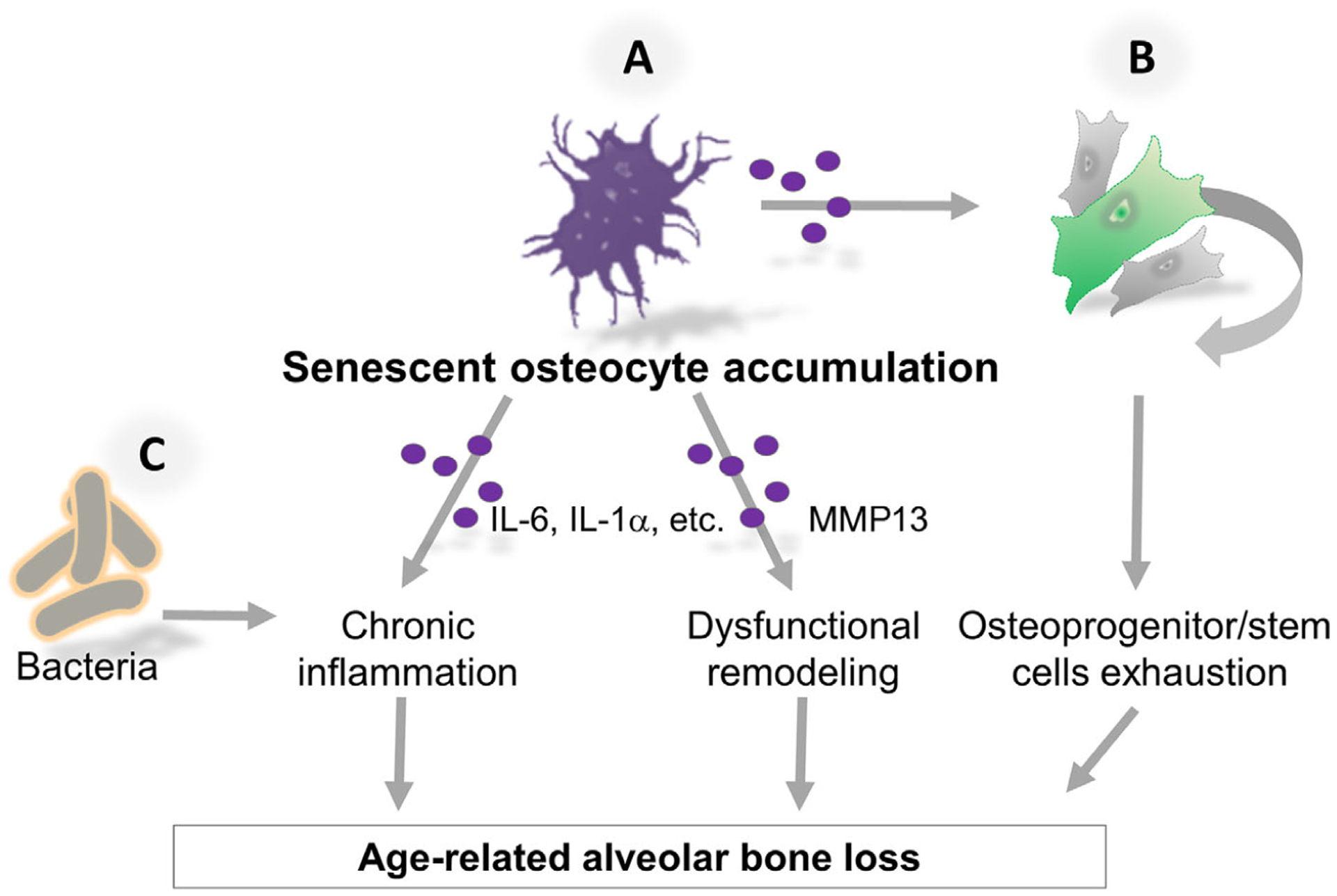
Hypothetical mechanism by which senescent cells deteriorate the old periodontal environment. A) Senescent osteocytes accumulate in alveolar bone with aging as a DNA damage response and as result of impaired clearance by the immune system. Senescent osteocytes-associated proinflammatory and proteolytic factors (purple circles) promote exacerbated local inflammation and dysfunctional bone extracellular matrix remodeling. B) The impact of both paracrine and age-related replicative senescence on stem cells contributes to the exhaustion of osteoprogenitor and stem cells. Thus, accumulation of senescent osteocytes deteriorates the periodontal environment and contributes to alveolar bone destruction with age. C) Senescent cells are an additional source of proinflammatory cytokines that could exacerbate bacterial-induced inflammation
Our study was limited to female mice. It remains to be established whether the current findings in females could also be extrapolated to males, which warrants future research. However, it’s likely that this is the case based in our knowledge and previous work, demonstrating that senescent cells, including senescent osteocytes, accumulate in bones with aging in both sexes.17 We also acknowledge that our data establish an association, and not causality, between senescent osteocyte accumulation and age-dependent alveolar bone loss. Despite these limitations, the main strength of the present study is that we established that senescent osteocytes accumulate with chronological aging in alveolar bone. This finding is not in conflict with previous mechanisms considered to explain the increased severity of periodontal disease in older individuals, including the cumulative periodontal damage over time.7 Given that some detrimental effects of SCM are ameliorated by p38 MAPK inhibitor SB202190, potential therapeutic strategies could emerge from these results. This hypothesis needs to be tested in vivo in future studies.
5. CONCLUSIONS
Cellular senescence is a host mechanism strongly associated with aging, and a cell-intrinsic response to DNA damage accumulation. Senescent osteocytes accumulate in periodontal tissues with aging and are an additional source of key proinflammatory cytokines implicated in periodontal disease pathogenesis. Senescence-associated proinflammatory factors could exacerbate local inflammation induced by bacteria. Therefore, senescent osteocytes represent a potential therapeutic target to delay the progression of alveolar bone destruction.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grants R01 AG063707 (DGM), R01 AR068275 (DGM), R21 AG065868 (JNF), K01 AR070241 (JNF), P01 AG062413 (SK). We thank Paola Divieti Pajevic (Massachusetts General Hospital and Harvard Medical School, Boston, MN) for providing the Ocy454 cells. The authors report no conflicts of interest related to this study.
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
Charles River Laboratories, Wilmington, MA.
Sigma-Aldrich, St. Louis, MO.
CM1850 UV, Leica Biosystems, Richmond, IL.
Cell Signaling Technology, MA.
ThermoFisher Scientific, Waltham, MA.
Panagene, South Korea.
LSM 780, Carl Zeiss, Oberkochen, Germany.
Viva Scan 40, Scanco USA, Southeastern, PA.
InvivoGen, San Diego, CA.
Sigma-Aldrich, St. Louis, MO.
Eve Technologies, Calgary, Alberta, Canada.
Sigma-Aldrich, St. Louis, MO.
QIAGEN, Germantown, MD.
Applied Biosystems, Foster City, CA.
Microsoft, Redmond, WA.
GraphPad, San Diego, CA.
REFERENCES
- 1.Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69Suppl 1:S4–S9. [DOI] [PubMed] [Google Scholar]
- 2.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kamer AR, Pirraglia E, Tsui W, et al. Periodontal disease associates with higher brain amyloid load in normal elderly. Neurobiol Aging. 2015;36:627–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang CJ, McCauley LK. Osteoporosis and Periodontitis. Curr Osteoporos Rep. 2016;14:284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ebersole JL, Graves CL, Gonzalez OA, et al. Aging, inflammation, immunity and periodontal disease. Periodontol 2000. 2016;72:54–75. [DOI] [PubMed] [Google Scholar]
- 6.Tonetti MS, Jepsen S, Jin L, Otomo-Corgel J. Impact of the global burden of periodontal diseases on health, nutrition and well-being of mankind: a call for global action. J Clin Periodontol. 2017;44:456–462. [DOI] [PubMed] [Google Scholar]
- 7.Feres M, Teles F, Teles R, Figueiredo LC, Faveri M. The subgingival periodontal microbiota of the aging mouth. Periodontol 2000. 2016;72:30–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Silva N, Abusleme L, Bravo D, et al. Host response mechanisms in periodontal diseases. J Appl Oral Sci. 2015;23:329–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartold PM, Van Dyke TE. Host modulation: controlling the inflammation to control the infection. Periodontol 2000. 2017;75:317–329. [DOI] [PubMed] [Google Scholar]
- 10.Bartold PM, Van Dyke TE. Periodontitis: a host-mediated disruption of microbial homeostasis. Unlearning learned concepts. Periodontol 2000. 2013;62:203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baer N, Fitzgerald RJ. Periodontal disease in the 18-month-old germfree rat. J Dent Res. 1966;45:406. [DOI] [PubMed] [Google Scholar]
- 12.Baer PN, Newton WL. Studies on peridontal disease in the mouse. 3. The germ-free mouse and its conventional control. Oral Surg Oral Med Oral Pathol. 1960;13:1134–1144. [DOI] [PubMed] [Google Scholar]
- 13.Aging Campisi J., cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.d’Adda di Fagagna F Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer. 2008;8:512–522. [DOI] [PubMed] [Google Scholar]
- 15.Feng X, Feng G, Xing J, et al. Repeated lipopolysaccharide stimulation promotes cellular senescence in human dental pulp stem cells (DPSCs). Cell Tissue Res. 2014;356:369–380. [DOI] [PubMed] [Google Scholar]
- 16.Toussaint O, Dumont P, Dierick JF, et al. Stress-induced premature senescence. Essence of life, evolution, stress, and aging. Ann N Y Acad Sci. 2000;908:85–98. [DOI] [PubMed] [Google Scholar]
- 17.Farr JN, Fraser DG, Wang H, et al. Identification of senescent cells in the bone microenvironment. J Bone Miner Res. 2016;31:1920–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jurk D, Wang C, Miwa S, et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell. 2012;11:996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482–496. [DOI] [PubMed] [Google Scholar]
- 20.Demaria M, Ohtani N, Youssef SA, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014;31:722–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Storer M, Mas A, Robert-Moreno A, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013;155:1119–1130. [DOI] [PubMed] [Google Scholar]
- 22.Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21:1424–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ovadya Y, Landsberger T, Leins H, et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat Commun. 2018;9:5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aquino-Martinez R, Rowsey JL, Fraser DG, et al. LPS-induced premature osteocyte senescence: implications in inflammatory alveolar bone loss and periodontal disease pathogenesis. Bone. 2020;132:115220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nelson G, Wordsworth J, Wang C, et al. A senescent cell bystander effect: senescence-induced senescence. Aging Cell. 2012;11:345–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ovadya Y, Krizhanovsky V. Senescent cells: sASPected drivers of age-related pathologies. Biogerontology. 2014;15:627–642. [DOI] [PubMed] [Google Scholar]
- 28.Yamaza T, Ren G, Akiyama K, Chen C, Shi Y, Shi S. Mouse mandible contains distinctive mesenchymal stem cells. J Dent Res. 2011;90:317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Farr JN, Xu M, Weivoda MM, et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat Med. 2017;23:1072–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roforth MM, Liu G, Khosla S, Monroe DG. Examination of nuclear receptor expression in osteoblasts reveals Rorbeta as an important regulator of osteogenesis. J Bone Miner Res. 2012;27:891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spatz JM, Wein MN, Gooi JH, et al. The Wnt inhibitor sclerostin is up-regulated by mechanical unloading in osteocytes in vitro. J Biol Chem. 2015;290:16744–16758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aquino-Martinez R, Monroe DG, Ventura F. Calcium mimics the chemotactic effect of conditioned media and is an effective inducer of bone regeneration. PLoS One. 2019;14:e0210301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aquino-Martinez R, Angelo AP, Pujol FV. Calcium-containing scaffolds induce bone regeneration by regulating mesenchymal stem cell differentiation and migration. Stem Cell Res Ther. 2017;8:265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Noren Hooten N, Evans MK. Techniques to induce and quantify cellular senescence. J Vis Exp. 2017(123):55533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aquino-Martinez R, Farr JN, Weivoda MM, et al. miR-219a-5p regulates rorbeta during osteoblast differentiation and in age-related bone loss. J Bone Miner Res. 2019;34:135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Swanson EC, Manning B, Zhang H, Lawrence JB. Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. J Cell Biol. 2013;203:929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell. 2009;8:311–323. [DOI] [PubMed] [Google Scholar]
- 38.Kuo LJ, Yang LX. Gamma-H2AX – a novel biomarker for DNA double-strand breaks. In Vivo. 2008;22:305–309. [PubMed] [Google Scholar]
- 39.Ressler S, Bartkova J, Niederegger H, et al. p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell. 2006;5:379–389. [DOI] [PubMed] [Google Scholar]
- 40.Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011;30:1536–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu Y, Dong G, Xiao W, et al. Effect of aging on periodontal inflammation, microbial colonization, and disease susceptibility. J Dent Res. 2016;95:460–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361:1475–1485. [DOI] [PubMed] [Google Scholar]
- 43.Jilka RL, O’Brien CA. The role of osteocytes in age-related bone loss. Curr Osteoporos Rep. 2016;14:16–25. [DOI] [PubMed] [Google Scholar]
- 44.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. [DOI] [PubMed] [Google Scholar]
- 45.Gamonal J, Bascones A, Acevedo A, Blanco E, Silva A. Apoptosis in chronic adult periodontitis analyzed by in situ DNA breaks, electron microscopy, and immunohistochemistry. J Periodontol. 2001;72:517–525. [DOI] [PubMed] [Google Scholar]
- 46.Bulut S, Uslu H, Ozdemir BH, Bulut OE. Expression of caspase-3, p53 and Bcl-2 in generalized aggressive periodontitis. Head Face Med. 2006;2:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuzenko Y, Romanyuk A, Politun A, Karpenko L. S100, bcl2 and myeloperoxid protein expirations during periodontal inflammation. BMC Oral Health. 2015;15:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kojima H, Inoue T, Kunimoto H, Nakajima K. IL-6-STAT3 signaling and premature senescence. JAKSTAT. 2013;2:e25763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Severino V, Alessio N, Farina A, et al. Insulin-like growth factor binding proteins 4 and 7 released by senescent cells promote premature senescence in mesenchymal stem cells. Cell Death Dis. 2013;4:e911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hajishengallis G The inflammophilic character of the periodontitis-associated microbiota. Mol Oral Microbiol. 2014;29:248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.