

Abstract
Heterotrimeric G proteins were originally discovered through efforts to understand the effects of hormones, such as glucagon and epinephrine, on glucose metabolism. On the other hand, many cellular metabolites, including glucose, serve as ligands for G protein-coupled receptors. Here we investigate the consequences of glucose-mediated receptor signaling, and in particular the role of a Gα subunit Gpa2 and a non-canonical Gβ subunit, known as Asc1 in yeast and RACK1 in animals. Asc1/RACK1 is of particular interest because it has multiple, seemingly unrelated, functions in the cell. The existence of such “moonlighting” operations has complicated the determination of phenotype from genotype. Through a comparative analysis of individual gene deletion mutants, and by integrating transcriptomics and metabolomics measurements, we have determined the relative contributions of the Gα and Gβ protein subunits to glucose-initiated processes in yeast. We determined that Gpa2 is primarily involved in regulating carbohydrate metabolism while Asc1 is primarily involved in amino acid metabolism. Both proteins are involved in regulating purine metabolism. Of the two subunits, Gpa2 regulates a greater number of gene transcripts and was particularly important in determining the amplitude of response to glucose addition. We conclude that the two G protein subunits regulate distinct but complementary processes downstream of the glucose-sensing receptor, as well as processes that lead ultimately to changes in cell growth and metabolism.
Author summary
Despite the societal importance of glucose fermentation in yeast, the mechanisms by which these cells detect and respond to glucose have remained obscure. Glucose detection requires a cell surface receptor coupled to a G protein that is comprised of two subunits, rather than the more typical heterotrimer: an α subunit Gpa2 and the β subunit Asc1 (or RACK1 in humans). Asc1/RACK1 also serves as a subunit of the ribosome, where it regulates the synthesis of proteins involved in glucose fermentation. This manuscript uses global metabolomics and transcriptomics to demonstrate the distinct roles of each G protein subunit in transmitting the glucose signal. Whereas Gpa2 is primarily involved in the metabolism of carbohydrates, Asc1/RACK1 contributes to production of amino acids necessary for protein synthesis and cell division. These findings reveal the initial steps of glucose signaling and several unique and complementary functions of the G protein subunits. More broadly, the integrated approach used here is likely to guide efforts to determine the topology of complex G protein and metabolic signaling networks in humans.
Introduction
All cells respond to changes in extracellular and environmental conditions, and many of these inputs are detected by receptors coupled to guanine nucleotide-binding proteins (G proteins). While G protein-coupled receptors (GPCRs) have established roles in detecting odors, light, hormones, and neurotransmitters, more recent investigations have uncovered an important role for GPCRs in responding to nutrients and metabolites such as glucose, amino acids, purine nucleotides, and carboxylic acids including fatty acids [1]. GPCR activation leads to the synthesis of chemical second messengers, changes in cell metabolism and transcriptional reprograming. Thus, G proteins act as signal transducers, transmitting a specific extracellular signal to a variety of intracellular second messengers and chemical metabolites. In some cases, the initiating and ensuing signals are one and the same.
The yeast Saccharomyces cerevisiae has two G protein signaling systems, one that responds to mating pheromone and another that responds to glucose. These systems do not share components but appear to act in a coordinated fashion; when glucose is limiting, the mating response is delayed until the cells have undergone two complete rounds of cell division [2]. Of these GPCR systems, the pheromone pathway is the best characterized and is typical of those found in humans. A peptide ligand binds to a cell surface receptor, which then activates a heterotrimeric G protein, comprised of an α subunit and a tightly associated βγ subunit dimer. Gα then exchanges GDP for GTP and dissociates from Gβγ. The Gα subunit activates a phosphatidylinositol 3-kinase while Gβγ initiates a mitogen-activated protein kinase (MAPK) cascade [3,4].
The second GPCR pathway responds to glucose [5]. The presumptive glucose receptor (Gpr1) is coupled to a typical Gα (Gpa2) [6–8], but there is no corresponding Gβγ. Rather, Gpa2 appears to assemble with a multifunctional protein known as RACK1 in animals [9] Gib2 in Cryptococcus neoformans [10,11], and Asc1 in Saccharomyces cerevisiae [12]. Whereas Gpa2 activates adenylyl cyclase, leading to a transient increase in cellular cAMP [13], Asc1 has the opposite effect on cAMP production [12]. It is also common in animal cells that Gα and Gβγ bind to and regulate the same effector enzymes, including adenylyl cyclase, in opposition to one another [14].
While Asc1/RACK1 has characteristics of a Gβ subunit, it also has other important functions in the cell. RACK1 was originally identified as an adaptor for protein kinase C in animals, and was proposed to have a role in kinase-mediated signal transduction [15]. RACK1 has also been demonstrated to interact with several GPCRs and G protein βγ subunits [16–19]. Most prominently, Asc1/RACK1 is part of the 40S subunit of the ribosome [20–25]. In that capacity, Asc1 plays an important role in recruiting quality control systems that diminish frameshifting errors when translation is stalled [26,27]. Moreover, Asc1 regulates a subset of transcripts primarily related to glycolysis, respiration, oxidative stress and fermentation [28]. Thus Asc1 is part of two distinct molecular complexes, one involved in glucose sensing and the other in glucose utilization. While the function of Asc1 in the ribosome has been well characterized, its role as a G protein is largely unexplored.
Here, we sought to determine the role of Asc1, in comparison with Gpa2, to glucose signaling. To gain a better understanding of their relative contributions to cell physiology, we undertook an integrated metabolomics and transcriptomics analysis, comparing the glucose response in mutants that lack the Gα or Gβ subunit. By this approach, one that is largely unprecedented in the GPCR field, we have identified the earliest events leading to glucose fermentation. Our analysis is likely to guide similar efforts to determine the topology of complex G protein and metabolic signaling networks in humans.
Results
We determined previously that Asc1 binds to Gpa2, that these proteins have opposing effects on adenylyl cyclase activity, and that they act in response to the glucose receptor Gpr1 [12]. Here we sought a deeper understanding of the molecular and cellular consequences of G protein activation. To that end, we undertook a multi-platform investigation, performing untargeted metabolomics and transcriptomics analysis, in cells lacking each of these proteins, in response to glucose. By measuring changes in gene expression and perturbations in host metabolism we sought to gain an understanding of Asc1, apart from its role in translation, and how it complements the functions of Gpa2.
Wildtype, gpr1, gpa2 and asc1 cells (all without nutritional auxotrophic markers) were grown for 1 h in low (L, 0.05%) glucose, then divided and either left untreated or treated with high (H, 2%) glucose for 2 minutes (metabolomics) or 10 minutes (transcriptomics). These time points were selected based on prior data, showing an early and transient spike of cAMP and a subsequent induction of genes within 10 minutes of glucose treatment (see Methods) [29]. We then analyzed our data using Principal Component Analysis (PCA). This unsupervised multivariate analysis method is particularly useful for the visualization of the relationship between observations and variables. When applied to our transcriptomics data, PCA indicated good differentiation of groups based on the proximity of data points for a given treatment and genotype (S1A Fig). This analysis revealed that PC1, which aligns primarily with treatment, accounts for 90% of variance while PC2, which aligns primarily with genotype, represents 5% of variance. Thus the first 2 components explained 95% of the variance. For metabolomics, the first 2 components explained 63% of the variance (S1B Fig). For both measurements, and as expected, gpr1 aligned closely with gpa2 [6–8,12,13]. Both measures are consistent with previously established opposing effects of Gα and Gβ on processes downstream of the G protein.
Our next objective was to identify the specific pathways and processes regulated by each G protein subunit. To that end, we analyzed the transcriptomics and metabolomics data, with and without glucose addition, for wildtype, gpr1, gpa2 and asc1 cells. Herein we use the term “concentration analysis” when comparing the data for mutant and wildtype cells after the addition of glucose (Figs 1 and S2A), and we use the term “sensitivity analysis” when comparing the difference in response at high and low glucose for the mutants (mutantH-mutantL) and the wildtype (wtH-wtL) cells (Figs 1 and S2B). Thus, concentration analysis reveals cell status after glucose addition, while sensitivity analysis reveals response amplitude (H-L) differences for the different strains. Such amplitude differences are important when a negative regulator restores the system to baseline in the face of sustained (or step-wise) activation by a positive regulator.
Fig 1. Analysis pipeline.

Workflow of concentration analysis and sensitivity analysis for transcriptomics and metabolomics data.
In summary, we present integrated transcription data and untargeted metabolomics data obtained for the cell extracts, as follows:
Concentration Analysis
gpa2 mutant vs. wildtype at high (2%) glucose
asc1 mutant vs. wildtype at high (2%) glucose
We then compare the results of A and B, calculated as detailed in Methods.
Sensitivity Analysis
difference between gpa2 mutant at high and low glucose (gpa2H-gpa2L) vs. difference between wildtype at high and low glucose (wtH-wtL)
difference between asc1 mutant at high and low glucose (asc1H-asc1L) vs. difference between wildtype at high and low glucose (wtH-wtL)
We then compare the results of A and B, calculated as detailed in Methods.
Concentration analysis
We began by establishing the transcriptional and metabolic profile of wildtype cells. We defined the differentially-expressed genes (DEGs) as having an adjusted p-value <0.05, absolute log2 fold-change value >1 and baseMean >100. Then, using the ClusterProfiler package in R [30] we performed gene set enrichment analysis (GSEA) in the Kyoto Encyclopedia of Genes and Genomes (KEGG) [31–33]. This database provides an overview of biological pathways in the cell, as determined by genome sequencing and other high-throughput methods. GSEA determines whether a defined set of genes shows statistically significant and concordant differences between two phenotypes. Since it is a rank-based analysis no cutoff is needed (see Tables 1–5, ‘Transcriptomics’).
Table 1. Single-omics analysis results for wildtype comparing 2% (H) and 0.05% (L) glucose.
First block shows GSEA for transcriptomics with adjusted p-value <0.05, arranged in ascending order; second block shows MetaboAnalystR pathway enrichment analysis for metabolomics with combined p-value <0.05 arranged in ascending order, as detailed in Methods. Enriched pathways refer to classifications provided in KEGG.
| Transcriptomics | Metabolomics | ||||
|---|---|---|---|---|---|
| enriched pathways | adjusted p-value | enriched pathways | adjusted p-value | enriched pathways | combined p-value |
| Ribosome | 0.0023 | Lysine biosynthesis | 0.0051 | Starch and sucrose metabolism | 0.0010 |
| Cell cycle—yeast | 0.0023 | Fructose and mannose metabolism | 0.0075 | Tyrosine metabolism | 0.0065 |
| Biosynthesis of amino acids | 0.0023 | Aminoacyl-tRNA biosynthesis | 0.0080 | Galactose metabolism | 0.0073 |
| RNA transport | 0.0023 | Sulfur metabolism | 0.0094 | Cysteine and methionine metabolism | 0.0184 |
| Ribosome biogenesis in eukaryotes | 0.0023 | One carbon pool by folate | 0.0098 | Glycolysis / Gluconeogenesis | 0.0198 |
| Cysteine and methionine metabolism | 0.0023 | Valine, leucine and isoleucine biosynthesis | 0.0101 | Inositol phosphate metabolism | 0.0207 |
| RNA polymerase | 0.0023 | Proteasome | 0.0101 | Amino sugar and nucleotide sugar metabolism | 0.0233 |
| Purine metabolism | 0.0028 | Autophagy—yeast | 0.0184 | Purine metabolism | 0.0343 |
| DNA replication | 0.0028 | Pyrimidine metabolism | 0.0184 | N-Glycan biosynthesis | 0.0361 |
| Galactose metabolism | 0.0028 | Amino sugar and nucleotide sugar metabolism | 0.0188 | Pentose phosphate pathway | 0.0364 |
| Glyoxylate and dicarboxylate metabolism | 0.0028 | Selenocompound metabolism | 0.0226 | Butanoate metabolism | 0.0443 |
| Citrate cycle (TCA cycle) | 0.0028 | Mismatch repair | 0.0231 | ||
| Peroxisome | 0.0028 | Glycine, serine and threonine metabolism | 0.0322 | ||
| Starch and sucrose metabolism | 0.0028 | Glycolysis / Gluconeogenesis | 0.0387 | ||
| Oxidative phosphorylation | 0.0033 | Autophagy—other | 0.0416 | ||
| Carbon metabolism | 0.0041 | Meiosis—yeast | 0.0474 | ||
Table 5. Single- and Multi-omics integration results for asc1 by sensitivity analysis.
First block shows GSEA for transcriptomics with adjusted p-value <0.05, arranged in ascending order; second block shows MetaboAnalystR pathway enrichment analysis for metabolomics with combined p-value <0.05, arranged in ascending order; third block shows MetaboAnalystR joint pathway analysis with adjusted p-value <0.05, arranged in ascending order, as detailed in Methods.
| Transcriptomics | Metabolomics | Integration | |||
|---|---|---|---|---|---|
| enriched pathways | adjusted p-value | enriched pathways | combined p-value | enriched pathways | adjusted p-value |
| Ribosome | 0.0071 | Lysine degradation | 0.0082 | Ribosome | 4.76E-98 |
| Ribosome biogenesis in eukaryotes | 0.0071 | Cysteine and methionine metabolism | 0.0473 | Ribosome biogenesis in eukaryotes | 1.87E-21 |
| Glyoxylate and dicarboxylate metabolism | 0.0071 | RNA polymerase | 0.0015 | ||
| RNA polymerase | 0.0071 | Galactose metabolism | 0.0015 | ||
| Peroxisome | 0.0071 | Pentose phosphate pathway | 0.0422 | ||
| Galactose metabolism | 0.0104 | Purine metabolism | 0.0422 | ||
| Citrate cycle (TCA cycle) | 0.0141 | Arginine biosynthesis | 0.0422 | ||
| RNA transport | 0.0196 | ||||
| Starch and sucrose metabolism | 0.0241 | ||||
| Carbon metabolism | 0.0479 | ||||
| Propanoate metabolism | 0.0479 | ||||
| Aminoacyl-tRNA biosynthesis | 0.0498 | ||||
When comparing wildtype before and after glucose addition (0.05% vs. 2%), we observed transcriptomic changes in 32 pathways, including ribosome, DNA replication, transcription, cell cycle as well as carbon, amino acids, lipids and nucleotide metabolism (Table 1). These differences were expected, and reflect processes needed to transition from a low glucose phase where metabolism supports cellular homeostasis (e.g. autophagy, addressing reactive oxygen species, maintaining osmotic balance) to a high glucose phase where metabolism supports cell growth and division (structural rearrangements as well as anabolic processes to make building blocks that support cell proliferation) [34].
We next conducted untargeted metabolomics by mass spectrometry. Pathway enrichment analysis was performed in MetaboAnalystR [31,32], using the Fisher’s method to integrate Mummichog [33] and GSEA results to produce the combined p-values reported here (Table 1). The pathway enrichment analysis module Mummichog is optimized for detecting prominent changes while GSEA excels at detecting concordant small changes in peak intensity. Because of the uncertainty associated with peak annotation for LC-MS data, the reliability of pathway enrichment is improved when combining the results from two different statistical methods [31,32]. When comparing 0.05% to 2% glucose in wildtype, 11 pathways were perturbed with a combined p-value <0.05 (Table 1). These include perturbations in the metabolism of carbohydrates, amino acids, nucleotides and lipids, and are concordant with changes in gene transcription. Again, these differences reflect processes needed to prepare the cell for growth and division. In addition, they are likely related to the role of Asc1 and Gpa2 in haploid invasive growth [12,35,36], a process where cells form long branching filaments and exhibit increased adherence and invasion of the substratum during periods of glucose limitation [37].
We next considered the effects of glucose in each of the mutant strains and how each mutant compares with wildtype. Based on GSEA, the gpa2 mutant exhibited changes in transcripts linked to oxidative phosphorylation and ribosome biogenesis (Tables 2 and S1). The asc1 mutant was enriched for twelve pathways including ribosome biogenesis and metabolism of amino acids (Tables 3 and S2).
Table 2. Single- and multi-omics integration results for gpa2 by concentration analysis.
First block shows GSEA for transcriptomics with adjusted p-value <0.05, arranged in ascending order; second block shows MetaboAnalystR pathway enrichment analysis for metabolomics with combined p-value <0.05, arranged in ascending order; third block shows MetaboAnalystR joint pathway analysis with adjusted p-value <0.05, arranged in ascending order, as detailed in Methods.
| Transcriptomics | Metabolomics | Integration | |||
|---|---|---|---|---|---|
| enriched pathways | adjusted p-value | enriched pathways | combined p-value | enriched pathways | adjusted p-value |
| Ribosome biogenesis in eukaryotes | 0.0084 | Purine metabolism | 0.0021 | Oxidative phosphorylation | 3.13E-14 |
| Oxidative phosphorylation | 0.0084 | Fructose and mannose metabolism | 0.0047 | Galactose metabolism | 1.60E-11 |
| Amino sugar and nucleotide sugar metabolism | 0.0079 | ABC transporters | 1.31E-08 | ||
| Galactose metabolism | 0.0079 | Glycolysis or Gluconeogenesis | 8.02E-05 | ||
| Glutathione metabolism | 0.0155 | Fructose and mannose metabolism | 8.18E-05 | ||
| Tyrosine metabolism | 0.0224 | Starch and sucrose metabolism | 8.18E-05 | ||
| Arginine biosynthesis | 0.0234 | Arginine biosynthesis | 0.0012 | ||
| Biotin metabolism | 0.0252 | Pentose phosphate pathway | 0.0012 | ||
| Aminoacyl-tRNA biosynthesis | 0.0360 | Purine metabolism | 0.0017 | ||
| Starch and sucrose metabolism | 0.0396 | Amino sugar and nucleotide sugar metabolism | 0.0073 | ||
| Phosphatidylinositol signaling system | 0.0424 | beta-Alanine metabolism | 0.0119 | ||
| Alanine, aspartate and glutamate metabolism | 0.0139 | ||||
| Cysteine and methionine metabolism | 0.0149 | ||||
| Citrate cycle (TCA cycle) | 0.0221 | ||||
Table 3. Single- and multi-omics integration results for asc1 by concentration analysis.
First block shows GSEA for transcriptomics with adjusted p-value <0.05, arranged in ascending order; second block shows MetaboAnalystR pathway enrichment analysis for metabolomics with combined p-value <0.05, arranged in ascending order; third block shows MetaboAnalystR joint pathway analysis with adjusted p-value <0.05, arranged in ascending order, as detailed in Methods.
| Transcriptomics | Metabolomics | Integration | |||
|---|---|---|---|---|---|
| enriched pathways | adjusted p-value | enriched pathways | combined p-value | enriched pathways | adjusted p-value |
| Starch and sucrose metabolism | 0.0155 | Arginine biosynthesis | 0.0029 | Arginine biosynthesis | 7.47E-10 |
| Biosynthesis of amino acids | 0.0156 | Glutathione metabolism | 0.0054 | ABC transporters | 7.47E-10 |
| Sulfur metabolism | 0.0156 | Aminoacyl-tRNA biosynthesis | 0.0214 | Purine metabolism | 1.41E-09 |
| Ribosome | 0.0156 | Purine metabolism | 0.0481 | Tryptophan metabolism | 0.0074 |
| Galactose metabolism | 0.0156 | Galactose metabolism | 0.0074 | ||
| Meiosis—yeast | 0.0156 | Cysteine and methionine metabolism | 0.0087 | ||
| Glycine, serine and threonine metabolism | 0.0243 | Alanine, aspartate and glutamate metabolism | 0.0087 | ||
| One carbon pool by folate | 0.0243 | Histidine metabolism | 0.0087 | ||
| Aminoacyl-tRNA biosynthesis | 0.0243 | Arginine and proline metabolism | 0.0136 | ||
| 2-Oxocarboxylic acid metabolism | 0.0387 | Phenylalanine metabolism | 0.0191 | ||
| Lysine biosynthesis | 0.0477 | Lysine biosynthesis | 0.0348 | ||
| Ribosome biogenesis in eukaryotes | 0.0477 | Amino sugar and nucleotide sugar metabolism | 0.0465 | ||
According to criteria outlined above, we found 197 and 94 DEGs for gpa2 vs. wildtype and for asc1 vs. wildtype, respectively. Fig 2A depicts a Venn diagram, comparing the DEGs for gpa2 vs. wildtype and for asc1 vs. wildtype (S3 Table). We then conducted over-representation analysis (ORA) for the unique as well as shared intersects of the diagram. In contrast to GSEA, ORA requires a threshold, defined above for DEGs. In this way, we were able to compare DEGs in the two mutants and gain a detailed understanding of how they are similar and how they differ from one another. As shown in Fig 2B, the shared DEGs were over-represented for arginine biosynthesis. Arginine contributes to nitrogen balance, through urea production, and also regulates protein translation [38]. In comparison the unique DEGs were over-represented for five pathways related to amino acid metabolism (asc1, Fig 2C) and eight pathways linked to carbohydrate, lipid and energy metabolism (gpa2, Fig 2D). Most of the opposing effects were related to retrotransposon elements and are unlikely to be related to glucose metabolism or signaling. Thus, pathway analysis revealed that Asc1 mainly affects transcripts related to amino acids while Gpa2 mainly affects transcripts related to carbohydrate metabolism.
Fig 2. Concentration analysis of differentially expressed genes (DEGs) after glucose addition.

A) Venn diagram of subsets of DEGs, for asc1 and gpa2 vs. wildtype, after glucose addition to 2%. Upper semicircle shows up-regulated DEGs and lower semicircle shows down-regulated DEGs. Numbers in parenthesis are shared DEGs regulated in the opposite direction, placed in the area corresponding to the direction of regulation. DEGs used for ORA analysis that are B) shared and change in the same direction; C) unique to asc1; D) unique to gpa2. Listed are all pathways and their functional categories with adjusted p-value <0.05.
We next conducted untargeted metabolomics, followed by metabolite identification/annotations and pathway enrichment analysis, for gpa2 and asc1. When compared to wildtype, gpa2 cells were enriched in ten pathways including arginine biosynthesis as well as purine and carbohydrate metabolism (Tables 2 and S1), while asc1 cells were enriched in four pathways including arginine biosynthesis and purine metabolism (Tables 3 and S2). Peak annotations derived from MetaboAnalystR are hereafter referred to as “metabolites”. The Venn diagram shows shared and unique significantly perturbed metabolites (SPMs, defined as those with adjusted p-value <0.05) for each mutant vs. wildtype comparison (Fig 3A and S4 Table). When comparing the two mutants, ORA revealed that those SPMs that changed in the same direction were enriched in pathways related to carbohydrate metabolism: amino sugar and nucleotide sugar metabolism, as well as fructose and mannose metabolism (Fig 3B). Because nucleotide sugars (e.g. UDP-glucose) are substrates for protein and lipid glycosylation, they may reflect preparation for new cell wall synthesis [39]. Glycosylation of the mucin Msb2 is needed to ensure signal fidelity downstream of the filamentous/invasive growth pathway [40]. SPMs perturbed in the opposite direction were enriched in purine metabolism (Fig 3C). The perturbations unique to individual mutants were over-represented for two pathways related to amino acid metabolism (asc1, Fig 3D) and two pathways linked to non-glucose carbohydrate metabolism (gpa2, Fig 3E). Contrary to what we observed for transcriptomics, pathway enrichment analysis for metabolomics revealed that more pathways were affected by the loss of GPA2 than by the loss of ASC1 (Tables 2 and 3). ORA analysis with subsets of SPMs corroborated the trends observed in transcriptomics. Based on these data we conclude that Asc1 mainly affects metabolites related to amino acids, Gpa2 mainly affects carbohydrate metabolism, and the two proteins have opposing effects on purine metabolism. Thus, the metabolomics data presented here mirror the transcriptomics data presented above.
Fig 3. Concentration analysis of significantly perturbed metabolites (SPMs) after glucose addition.

A) Venn diagram of subsets of SPMs, for asc1 and gpa2 vs. wildtype, after glucose addition. Upper semicircle shows up-regulated SPMs and lower semicircle shows down-regulated SPMs. Numbers in parenthesis are shared SPMs regulated in the opposite direction, placed in the area corresponding to the direction of regulation. SPMs used for ORA analysis that are B) shared and change in the same direction; C) shared and change in the opposite direction; D) unique to asc1; E) unique to gpa2. Listed are all pathways and their functional categories with adjusted p-value <0.05.
To gain a deeper understanding of the functional relationship between changes in gene transcription and changes in the levels of metabolites, we employed the joint pathway analysis module in MetaboAnalystR. In this application, we input all significantly perturbed genes (DEGs) and significantly perturbed metabolites (SPMs), and queried for those over-represented in KEGG. By integrating the data in this manner, we sought to obtain more information than could be gleaned from transcriptomics and metabolomics separately. Moreover, a protein can impact a single (rate limiting) part of a metabolic pathway and have the same effect as another protein that impacts multiple components. MetaboAnalystR also provides the ‘impact score’ (S1 and S2 Tables), which weighs topological importance of components within a pathway. Because of space limitations, we only focused on the adjusted p-value for each pathway in the integration analysis.
We found that, when compared to wildtype cells, both mutants affected genes or metabolites involved in the synthesis of amino acids (cysteine, methionine, arginine, alanine, aspartic acid and glutamic acid), the metabolism of purines, galactose, amino sugars and nucleotide sugars, as well as genes/metabolites involved in ABC transporters (Tables 2 and 3 and S1 and S2). These results reveal a shared role of Asc1 and Gpa2 in regulating the metabolism of carbohydrates, as expected for any component of the glucose-sensing pathway. In addition, both subunits affected the metabolism of amino acids, particularly branches of that pathway most closely linked to the TCA cycle. Amino acids can be used for energy production in the TCA cycle, which is common when glucose is being syphoned off for anabolic processes such as making nucleotides via the pentose phosphate pathway (S3–S9 Figs). The gpa2 strain was unique in regulating glycolysis or gluconeogenesis, oxidative phosphorylation, TCA cycle, and the pentose phosphate pathway as well as the metabolism of β-alanine, starch, sucrose, fructose and mannose (Table 2). Thus, it appears that the Gα subunit Gpa2 mainly regulates the conversion of glucose to ATP as well as general carbohydrate metabolism. In contrast, the Gβ subunit Asc1 was unique in regulating a variety of amino acids (Table 3). Thus, it appears that the Gβ subunit specifically regulates the utilization of nitrogen in part through the metabolism of arginine and other amino acids. Notably, most of the changes observed for gpa2 and asc1 were also observed in cells lacking their shared activator, the GPCR Gpr1 (S5 Table). More broadly, the two G protein subunits regulate distinct but complementary processes downstream of the glucose sensing receptor.
To visualize the functional relationship of Gpa2 and Asc1, we projected the inputs of our integration analysis onto the pertinent yeast metabolic pathways in KEGG. It was evident that changes in the gpa2 strain compared to wildtype were concentrated in regions related to glucose and ATP as well as non-glucose carbohydrates (Figs 4A and S10), while the changes in asc1 were concentrated in various types of amino acid metabolism (Figs 4B and S11). Both mutants also impacted purine metabolism, but they did so in opposition to one another as detailed above (Fig 3C). These changes mirror the opposing effects of Gpa2 and Asc1 on cAMP. More broadly, these findings highlight the differences between the two G protein subunits and their effects downstream of the glucose-sensing receptor. Whereas Gα regulates carbohydrate utilization, Gβ regulates amino acids.
Fig 4. KEGG pathways regulated by GPA2 or ASC1 as determined by concentration analysis.

The relevant part of a specific KEGG pathway is shown with genes displayed as rectangles and metabolites displayed as circles. KEGG compound name for each metabolite is labeled beside the circle. Standard gene names are labeled inside the rectangle. For enzyme complexes, the gene name for the major component is shown followed with an ellipsis. The directions of irreversible enzymatic reactions are shown by the arrows. Reversible reactions are connected by straight lines. DEGs and SPMs are highlighted in blue (gpa2) and red (asc1). Shared DEGs and SPMs are colored half blue and half red. A) as compared with asc1, gpa2 affected more components in pentose phosphate pathway (functional category: carbohydrate); B) as compared with gpa2, asc1 affected more components in tryptophan metabolism (functional category: amino acid).
MetaboAnalystR is well suited for annotating a large number of signals. A complementary approach is to use our in-house library annotation, which includes retention time (RT), exact mass, and MS/MS library (OL) developed with data acquired for standards run under the same conditions as the study samples, as well as matching to public databases (PD), as described in Supporting Information. Metabolites are reported (S6 Table) based on the confidence in the assignment; for example OL1 is a match to the in-house library by retention time, exact mass, and MS/MS fragmentation, whereas PDa is matched to a public database by mass and experimental MS/MS. The signals identified and annotated by this method yielded pathways that mirrored those obtained using MetaboAnalystR.
In summary, our concentration analysis provides new and complementary information about glucose signaling. In particular, using an integrated transcriptomics and metabolomics approach [41,42], we were able to confirm and consolidate changes seen at the metabolic or transcriptional level alone. For example, the integration analysis confirmed results obtained at the single -omics level; that Gpa2 affects pathways related to carbohydrate utilization while Asc1 affects pathways related to amino acid metabolism. Second, integration analysis revealed information that might have been hidden using single -omics analysis methods. Only when the two datasets were combined did several important pathways meet the threshold of significance. For example, when comparing gpa2 to wildtype, integration analysis established a substantial and statistically significant role for the Gα protein Gpa2 in glycolysis and gluconeogenesis. Of the fourteen features that emerged from our integrated analysis of this pathway, seven came from metabolomics and seven came from transcriptomics, neither of which met the threshold of significance on its own (Tables 2 and S1). Likewise, when comparing asc1 to wildtype, integration analysis helped us to establish a specific, substantial and statistically significant role for the Gβ protein Asc1 in cysteine and methionine metabolism (Tables 3 and S2). Since the metabolic pathway is comprised of enzymes (gene products) and metabolites (enzyme products) it is noteworthy that both are regulated in a similar manner, even if the numbers obtained from each analytical method are small. Finally, our integration analysis allowed us to narrow the role for Gpa2 from very general effects on carbohydrate metabolism to a more specific role in regulating glucose and ATP. Conversely, our integration analysis allowed us to show a broader role for Asc1 in amino acid metabolism, one not limited to a specific subset of amino acids.
Sensitivity analysis
The previous section compares the role of each signaling component in establishing transcript and metabolite concentration after glucose addition. Another way to explore the consequences of glucose sensing is to instead measure changes in response amplitude (sensitivity analysis), defined here as the difference between mutant (high minus low glucose, mutantH-mutantL) and wildtype (high minus low glucose, wtH-wtL). To put this in a biological context, a change in response amplitude reflects the role of a given component (Asc1 or Gpa2) in regulating the relative level of metabolites and/or genes, in response to a perturbation. A case in point is the relative change in cAMP after glucose addition, where the fold-change in its abundance is detected by the cell, regardless of the starting concentration. Differences detected using this approach are illustrated in S2B Fig.
We first compared transcriptional changes in wildtype and mutant cells, using the interaction term in DESeq2 [43]. In this analysis we found altered sensitivity for 877 and 587 genes (s-DEGs) when comparing gpa2 with wildtype and comparing asc1 with wildtype, respectively. In sensitivity analysis we defined s-DEGs as having altered sensitivity changes with an adjusted p-value <0.05, absolute log2 fold-change value >1 and baseMean >100. Using GSEA we determined that Gpa2 regulates the sensitivity of transcripts linked to ten pathways including N-glycan biosynthesis, carbohydrate metabolism and steroid biosynthesis (Tables 4 and S1). These changes are likely related to cell membrane and cell wall biosynthesis, leading to cell division. Asc1 regulates twelve pathways, including ribosome biogenesis and carbohydrate metabolism (Tables 5 and S2). ORA on the Venn diagram revealed that shared s-DEGs were enriched for arginine and purine metabolism (Fig 5A and 5B and S7 Table). For purine metabolism, the mutants had opposing effects for the concentration analysis (as discussed earlier) but correspondent effects for the sensitivity analysis. This suggests that purine metabolism is an important target of this glucose-sensing pathway and is congruent with the role of glucose as a precursor in purine biosynthesis. s-DEGs unique to asc1 were over-represented for three pathways related to ribosome, ribosome biogenesis and monobactam biosynthesis (Fig 5C); s-DEGs unique to gpa2 were over-represented for five pathways linked to amino acid and lipid metabolism (Fig 5D). These results reveal that Gpa2 uniquely affects the sensitivity of genes related to lipid metabolism (processes related to cell membrane synthesis), as well as some amino acids, while Asc1 mainly affects the sensitivity of genes related to ribosome biogenesis (processes related to new protein synthesis).
Table 4. Single- and Multi-omics integration results for gpa2 by sensitivity analysis.
First block shows GSEA for transcriptomics with adjusted p-value <0.05, arranged in ascending order; second block shows MetaboAnalystR pathway enrichment analysis for metabolomics with combined p-value <0.05, arranged in ascending order; third block shows MetaboAnalystR joint pathway analysis with adjusted p-value <0.05, arranged in ascending order, as detailed in Methods.
| Transcriptomics | Metabolomics | Integration | |||
|---|---|---|---|---|---|
| enriched pathways | adjusted p-value | enriched pathways | combined p-value | enriched pathways | adjusted p-value |
| N-Glycan biosynthesis | 0.0050 | Fructose and mannose metabolism | 0.0044 | Glycolysis or Gluconeogenesis | 2.32E-06 |
| Various types of N-glycan biosynthesis | 0.0050 | Glycolysis / Gluconeogenesis | 0.0183 | Galactose metabolism | 2.32E-06 |
| DNA replication | 0.0050 | Methane metabolism | 0.0211 | Cell cycle | 1.61E-05 |
| Aminoacyl-tRNA biosynthesis | 0.0050 | Porphyrin and chlorophyll metabolism | 0.0473 | Pentose phosphate pathway | 2.03E-05 |
| Cysteine and methionine metabolism | 0.0050 | Starch and sucrose metabolism | 3.41E-05 | ||
| Starch and sucrose metabolism | 0.0050 | MAPK signaling pathway | 0.0002 | ||
| Galactose metabolism | 0.0050 | Meiosis | 0.0010 | ||
| Cell cycle | 0.0056 | Fructose and mannose metabolism | 0.0010 | ||
| Mismatch repair | 0.0063 | Purine metabolism | 0.0025 | ||
| Steroid biosynthesis | 0.0308 | Cysteine and methionine metabolism | 0.0025 | ||
| Protein processing in endoplasmic reticulum | 0.0096 | ||||
| Peroxisome | 0.0124 | ||||
| Spliceosome | 0.0230 | ||||
| Pyruvate metabolism | 0.0238 | ||||
| DNA replication | 0.0238 | ||||
| Longevity regulating pathway | 0.0238 | ||||
| Glycerolipid metabolism | 0.0238 | ||||
| beta-Alanine metabolism | 0.0238 | ||||
| Steroid biosynthesis | 0.0238 | ||||
| Amino sugar and nucleotide sugar metabolism | 0.0238 | ||||
| Alanine, aspartate and glutamate metabolism | 0.0238 | ||||
| Inositol phosphate metabolism | 0.0241 | ||||
Fig 5. Sensitivity analysis of differentially expressed genes (s-DEGs) after glucose addition.

A) Venn diagram of subsets of s-DEGs, before and after glucose addition for each mutant (mutantH-mutantL) and wildtype (wtH-wtL). Upper semicircle shows up-regulated s-DEGs and lower semicircle shows down-regulated s-DEGs. Numbers in parenthesis are shared s-DEGs regulated in the opposite direction, placed in the area corresponding to the direction of regulation. Subset of s-DEGs used for ORA analysis that are B) shared and change in the same direction; C) unique to asc1; D) unique to gpa2; E) shared and change in the opposite direction. Listed are all pathways and their functional categories with adjusted p-value <0.05.
While each of the mutants had unique effects, there were an additional 99 s-DEGs regulated by both mutants but in opposite directions (S7 Table). Whereas ORA in concentration analysis revealed opposing effects for a small group of DEGs related to transposon elements, ORA in sensitivity analysis revealed opposing effects for s-DEGs related to carbohydrate metabolism (Fig 5E). For the 62 s-DEGs that were up-regulated in asc1 and down-regulated in gpa2, most were due to a change in basal expression (at low glucose) (S12 Fig). For the remaining 37 s-DEGs the pattern was more complex, with mutants affecting basal expression or induced expression or both (S13 Fig).
We then performed generalized linear modeling to detect significant differences in metabolite/peak levels, comparing (wtH-wtL) and (mutantH-mutantL), followed by pathway enrichment analysis in MetaboAnalystR, as detailed above. By this approach we determined that the gpa2 strain was primarily enriched for carbohydrate-related pathways, while asc1 was enriched for pathways involved in lysine, cysteine, and methionine metabolism (Tables 4 and 5 and S1 and S2). Here we define s-SPMs as those with an adjusted p-value <0.05. ORA indicates that the s-SPMs unique to gpa2 were enriched for steroid biosynthesis (Fig 6A and 6B and S8 Table). These results confirm the role of Gpa2 in lipid metabolism, as shown above. Most notably, the mutants had opposing effects on the sensitivity of an additional 42 compounds primarily related to carbohydrate metabolism (Fig 6C), consistent with the opposing effects of gpa2 and asc1 revealed through transcriptomics (Fig 5E).
Fig 6. Sensitivity analysis of significantly perturbed metabolites (s-SPMs) after glucose addition.

A) Venn diagram of subsets of s-SPMs, before and after glucose addition for each mutant (mutantH-mutantL) and wildtype (wtH-wtL). Upper semicircle shows up-regulated s-SPMs and lower semicircle shows down-regulated s-SPMs. Numbers in parenthesis are shared s-SPMs regulated in the opposite direction, placed in the area corresponding to the direction of regulation. Subset of s-SPMs used for ORA analysis that are B) unique to gpa2; C) shared and changed in the opposite direction. Shared s-SPMs changed in the same direction and s-SPMs unique to asc1 are not over-represented in any pathway and are therefore not shown. Listed are all pathways and their functional categories with adjusted p-value <0.05.
The results above reveal important differences between the two G protein subunits, as determined by concentration and sensitivity analysis. In general, the two mutants responded in opposing ways to glucose addition, and did so at both the metabolic and transcriptional levels. Across all gene level comparisons, only arginine metabolism was affected similarly by the two mutants (Figs 2 and 5). Given the opposing roles of Asc1 and Gpa2 in other processes it is noteworthy that aspects of arginine metabolism were also affected, and in the same direction, by a mutant lacking the shared activator Gpr1 (S5 Table). More specifically, all three mutants led to significant induction of four genes (AGP1, MEP1, DAL2, and GDH1) ten minutes after glucose addition.
Quantifiable changes in these gene transcripts may prove useful as reporters of glucose-mediated GPCR signaling. Accordingly, we performed qPCR to quantify the expression level of these four genes before and ten minutes after glucose addition in wildtype and all three mutants: asc1, gpa2 and gpr1. As shown in Fig 7, all four genes responded as expected based on our RNA-seq data. Of these, the best performing transcript was AGP1, which was significantly induced (adjusted p<0.05) after only ten minutes, in all three mutants compared to wildtype (S9 Table), as determined by both concentration (Fig 7A and 7B) and sensitivity analysis (Fig 7C and 7D). AGP1 encodes a broad-specificity, low-affinity amino acid permease, located at the plasma membrane [44]. Longer time courses should lead to even larger cumulative changes indicative of GPCR signaling.
Fig 7. qPCR and RNA-seq analysis.

A, B) Bar plots of concentration analysis results of A) RNA-seq data and B) qPCR data for AGP1 (green), MEP1 (yellow), DAL2 (brown) and GDH1 (orange). X-axes show mutants; Y-axes show fold induction over wildtype. C, D) Bar plots of sensitivity analysis results of C) RNA-seq data and D) qPCR data for AGP1 (green), MEP1 (yellow), DAL2 (brown) and GDH1 (orange). X-axes show mutants; Y-axes show change in induction between low and high glucose treatment. Error bars, standard error of the mean of biological replicates. Significance marks for qPCR analysis are as follows: p <0.05(*), p <0.15(+) by Mann-Whitney U test and adjusted with the Benjamini-Hochberg procedure.
We then applied integration analysis and found that both mutants affected genes or metabolites involved in the pentose phosphate pathway, as well as purine and galactose metabolism (Tables 4 and 5 and S1 and S2). The gpa2 strain uniquely regulated a variety of cellular functions (Table 4), while asc1 uniquely regulated ribosome function as well as arginine metabolism (Table 5). Thus, the relationship of Asc1 to amino acids and ribosome function is reflected in both concentration and sensitivity analysis. By concentration analysis, the effects of gpa2 were primarily related to carbohydrate metabolism, while sensitivity analysis revealed a greater diversity of processes including amino acids and lipids, in addition to carbohydrates. This indicates that while Gpa2 is crucial in maintaining carbohydrate metabolism, it also interacts with genes and metabolites related to other non-carbohydrate species to ensure their proper response amplitude upon sugar addition. Taken together, it is evident that concentration and sensitivity analysis provide information that is complementary and biologically meaningful.
Finally, in order to visualize the functional relationship of Gpa2 and Asc1, we projected the inputs of our integration analysis onto pertinent yeast metabolic pathways in KEGG. It is evident that Gpa2 affects the sensitivity of a larger number of genes and metabolites, as compared with Asc1 (S14 Fig). The effects of gpa2 were pervasive and included glucose, other carbohydrates, lipids, amino acids and purine nucleotides (Figs 8A and S14). In contrast, the effects of asc1 centered on pentose phosphate, purine and arginine metabolism (S15 Fig). In addition, and as anticipated given its role in ribosome assembly, asc1 uniquely affected processes related to RNA polymerase and ribosome biogenesis (Fig 8B and Table 5). These findings reveal that, although the two G protein subunits each had substantial effects by concentration analysis, the Gα subunit had by far the largest effect on sensitivity analysis. Collectively, and as summarized in Fig 9, these data indicate that Gpa2 affects the amplitude of the response to glucose, and does so for an especially large group of genes and metabolites.
Fig 8. KEGG pathways regulated by GPA2 or ASC1 as determined by sensitivity analysis.

The relevant part of a specific KEGG pathway is shown with genes displayed as rectangles and metabolites displayed as circles. KEGG compound name for each metabolite is labeled beside the circle. Standard gene names are labeled inside the rectangle. For enzyme complexes, the gene name for the major component is shown followed with an ellipsis. The directions of irreversible enzymatic reactions are shown by the arrows. Reversible reactions are connected by straight lines. s-DEGs and s-SPMs are highlighted in blue (gpa2) and red (asc1). Shared s-DEGs and s-SPMs are colored half blue and half red. A) as compared with asc1, gpa2 affected more components in steroid biosynthesis (functional category: lipid); B) as compared with gpa2, asc1 affected more components in ribosome biogenesis (showing 90S pre-ribosome components, functional category: genetic information processing).
Fig 9. Summary of interactions.

Gpa2 is primarily involved in regulating carbohydrate metabolism while Asc1 is primarily involved in amino acid metabolism, as indicated with thick lines (concentration analysis). Gpa2 and Asc1 have shared effects on arginine metabolism and opposing effects on purine metabolism, as indicated with solid and dashed lines (concentration analysis). Gpa2 regulates a greater number of gene transcripts and was particularly important in determining the amplitude of response to glucose addition, as indicated with the larger sphere (sensitivity analysis).
Discussion
The yeast S. cerevisiae has two functionally distinct GPCRs and two Gα proteins, but only a single canonical Gβγ. Here we considered the role of a second, “atypical”, Gβ protein Asc1 and compared its function with that of the cognate Gα protein Gpa2. Through a comprehensive analysis of glucose-dependent metabolic and transcriptional changes, we have uncovered new and common biochemical processes mediated by these two G protein subunits.
Like other Gβ proteins, Asc1 binds preferentially to Gα-GDP, slows GDP-GTP exchange [12] and has a seven-bladed propeller domain structure [24,45,46]. Like other Gβ proteins, Asc1 binds directly to the effector adenylyl cyclase; whereas Gpa2 stimulates the production of cAMP however, Asc1 has the opposite effect [12,13]. Despite their opposing effects on second messenger production, Asc1 and Gpa2 have similar effects on haploid invasive growth [12,35,36], a process by which the cells form long branching filaments and exhibit increased adherence and invasion of the substratum. This growth phenotype occurs during periods of glucose limitation [37], possibly in an attempt to direct colony expansion to sites of greater nutrient availability. The fact that the Gα and Gβ have opposing effects on cAMP but similar effects on invasive growth suggested to us that these proteins regulate processes other than adenylyl cyclase activation.
To gain a better understanding of their relative contributions to cell physiology, we undertook an integrated metabolomics and transcriptomics analysis, comparing mutants that lack one or the other of the G protein subunits. Our analysis focused on short term changes that occur in response to glucose addition, as opposed to the chronic effects resulting from deletion of gpr1, gpa2 or asc1 (while disruption of the pheromone-responsive Gα protein Gpa1 leads to permanent signaling, this behavior is not typical of other G protein systems). Correspondingly, we did not consider the effects of permanently activating Gpa2, for example by mutationally disrupting GTPase activity.
We then used several analytical methods to determine the direction and magnitude of changes in transcription and metabolism. For transcriptomics, GSEA provides an enrichment score that reflects whether a pathway is up- or down-regulated. For metabolomics, GSEA and Mummichog use different algorithms to assign directionality within a pathway, accounting for the fact that abundance of some components within that pathway might decrease while others increase. Whereas GSEA emphasizes concerted small changes Mummichog emphasizes prominent changes. Because these methods weigh the data differently, they can be difficult to reconcile with regard to pathway direction. Finally, our integration analysis (MetaboAnalyst) provides adjusted p-values, but not directionality, as an output. While our conclusions do not depend on a determination of directionality, the data provided can be used by other investigators for that purpose. Building on these findings, we are currently developing targeted assays of key metabolites, to be measured in conjunction with transcriptional reporters, in various genetic backgrounds and in response to different carbohydrate ligands, ligand concentrations, and time scales. In this way we are laying the foundation for a more comprehensive and mechanistic understanding of glucose receptor pharmacology.
As noted above, Gpa2 and Asc1 have opposing effects on cAMP production [14], as well as other metabolic processes in the cell (this work). Such competing interactions mirror those that occur in animal cells, where Gα and Gβγ proteins often have antagonistic effects on adenylyl cyclase activity [14]. While seemingly paradoxical, this design has several useful properties [47]. First, such “antagonistic bifunctionality” results in an input-output relationship that is inherently insensitive to naturally occurring fluctuations in protein abundance (“robustness”). Assuming that every Gα is partnered with a Gβ, any increase or decrease in one subunit would lead to a concomitant change in the other. A potential advantage of this arrangement is to buffer against fluctuations in the abundance of either the positive or negative regulator, which might in turn lead to substantial cell-to-cell differences in pathway activity (“noise”). Second, if the positive and negative regulators operate on different time scales they can produce outputs that are transient. This might occur if there is a delay between the inactivation of Gα (following GTP hydrolysis) and the inactivation of Gβγ (following reassociation with Gα). Third, a built-in delay can confer a property known as fold-change detection. In this scenario, the amplitude of the output is proportional to the relative difference in, rather than the absolute concentration of, the input. This can occur when the negative regulator restores the output to the pre-stimulus baseline despite sustained activation by the positive regulator. Indeed, our fold-change analysis, or sensitivity analysis, allowed us to detect phenotypes that were not evident by concentration analysis alone. All of these features are characteristic of other G protein signaling systems. An especially well-documented example is light detection by the GPCR rhodopsin, which exhibits exceptionally low noise, a transient neural output, and a wide-dynamic range of light detection [48].
Asc1 could also affect glucose signaling in other, more indirect ways. For example, others reported copurification of Asc1 with Ras1 (in C. neoformans) and a Ras GTPase-activating protein (S. cerevisiae) [49]. Together with Gpa2 [50–61], Ras1 and Ras2 activate adenylyl cyclase [52,62–64] leading to an increase in cellular cAMP [8,54,65]. Currently we are investigating novel roles of Ras1 and Ras2, using similar multi-omics approaches as those detailed above.
While many of the effects of Asc1 overlap with those of Gpa2, others appear to be unrelated to glucose sensing. Most prominently, Asc1 serves as an accessory component of the ribosome, where it is required for some protein-protein interactions within the complex and for proper protein translation [20–25,28]. It is also possible that the effects of glucose on cell metabolism are coordinated with changes in protein translation. That is to say, the protein translation machinery is made aware of changes in nutrient abundance, through the coordinated action of a shared protein Asc1. Indeed, Asc1 appears to associate with the ribosome only under glucose-rich conditions [25], and regulates a subset of transcripts related to glycolysis, respiration, oxidative stress and glucose fermentation [28].
Thus, Asc1 functions as part of two distinct multi-protein complexes, one that responds to a cell surface receptor and the other that controls protein synthesis. While seemingly incongruous, this is a phenomenon for which there is ample precedent. The recruitment of a protein that originally evolved with one function to serve a second unrelated function is an example of Darwinian exaptation, more colloquially known as “protein moonlighting” because it is analogous to a single person with two different jobs [66]. The first description of a moonlighting protein was argininosuccinate lyase, which has a well-established enzymatic function but which also serves as a structural protein responsible for the transparency of the lens and cornea [67,68]. Like Asc1, a large fraction of other proteins involved in glucose metabolism exhibit moonlighting behaviors; this includes the majority of enzymes involved in glycolysis and the tricarboxylic acid cycle [69,70]. There is also evidence for moonlighting by typical G proteins. The mammalian Gβ2 binds to Gα and Gγ subunits but also assembles with the ubiquitin ligase DDB1-CUL4A-ROC1, where it helps to recruit the receptor kinase GRK2 for targeted degradation [71]. Our identification of Asc1 as the Gβ for Gpa2 is like that of many other moonlighting proteins, which have been identified because of their presence in unexpected multiprotein complexes or locations.
Finally, while it has most of the functions of a typical Gβ, Asc1 has a structure that makes it unique among Gβ protein subtypes [24,45,46]. This too has precedent. Whereas Gpa1 binds to a typical Gβ (Ste4), which is necessary for pheromone-induced mating, it also associates with an atypical Gβ called Vps15, which promotes autophagy through activation of phosphatidylinositol 3-kinase [4,72]. Our x-ray structure determination revealed that Vps15 has a 7-bladed propeller domain structure analogous to that of other Gβ proteins [73], as well as a protein kinase domain of unknown function. The discovery of atypical and multifunctional Gβ proteins, including Asc1 and Vps15, suggests that the superfamily of Gβ subunits may be far larger and more complex than previously recognized [74].
It has long been appreciated that the glucose-sensing pathway in yeast employs a G protein-coupled receptor. Despite the nutritional and societal importance of glucose fermentation in yeast however, the mechanisms and molecular consequences of glucose sensing have remained obscure. By integrating transcriptomic and metabolomic measurements, we have taken a major step towards elucidating the contributions of the Gα and Gβ protein subunits in the glucose sensing pathway. We anticipate that the approaches implemented here will be useful for investigating other GPCR pathways and other moonlighting proteins, including G proteins, that participate in multiple, seemingly unrelated, biological processes.
Methods
Yeast strains
The prototrophic (wildtype) strain used throughout was constructed from BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0). HIS3, LEU2, MET15 and URA3 were integrated at the endogenous loci with sequence amplified by PCR from S288C strain DNA. All single mutants (gpr1, gpa2, asc1) were constructed by transforming the wildtype strain with corresponding sequence from the Yeast Knock-Out collection that replaces the target gene with KanMX4 [75].
Cell preparation
All strains were cultivated in the same way and maintained at 30 oC unless otherwise indicated. Cells were inoculated into Synthetic Complete (SC) (2% glucose) overnight and grown to saturation, then back diluted and kept in log phase overnight. The next morning, the culture was harvested when OD reached 1.0. For each genotype, 90 ml of culture (OD = 1.0) was split into 2 tubes (45 ml each) for later high or low glucose treatment. For each tube, cells were centrifuged and resuspended twice with SC (0.05% glucose). Cells were then resuspended into 10 ml SC (0.05% glucose) and cultivated for 1 h.
For high glucose treatment, 245 μL 65.5% glucose was added to 10 mL cell culture and for low glucose treatment, 245 μL 0.05% glucose was added to 10 mL cell culture, each for exactly 2 minutes (metabolomics) or 10 minutes (transcriptomics). The 10 minute time point was selected based on a pilot time course experiment (2, 5, 10, 15, 20, 30 and 45 minutes), showing that this is the earliest time point that captures most of the transcripts affected by the glucose treatment.
For metabolomics, each replicate consisting of 3 mL of cell culture was mixed with 45 mL cold pure methanol on dry ice. After 5 minutes, cells were centrifuged in a precooled rotor (-80°C). After discarding the supernatant, cell pellets were immediately stored at -80°C. A small aliquot of each sample was saved to manually determine cell density with a hemocytometer.
For RNA-seq, 500 μL of cell culture was aliquoted into a 1.7 mL microfuge tube and centrifuged at 1000 x g for 1 minute at 4°C. After discarding supernatant, the cell pellet was flash frozen by liquid nitrogen and stored at -80°C.
Sample preparation for RNA-seq
Cell pellets stored at -80°C were resuspended with 600 μL buffer RLT 1% (v/v) 2-mercaptoethanol from the QIAGEN RNeasy Mini Kit (Cat No.: 74106) and then added into 2 mL OMNI prefilled ceramic bead tubes (SKU: 19–632). Tubes were loaded onto an OMNI Bead Mill Homogenizer (SKU:19-040E) for 3 beating cycles. For each cycle, samples were agitated at 5 m/s for 1 minute at 4 oC and then cooled on ice for 3 minutes between each cycle. The resulting lysate was clarified by centrifugation at 11,000 xg and then used for total RNA extraction with QIAGEN RNeasy Mini Kit (Cat No.: 74106) with on-column DNase digestion according to manufacturer’s instructions. Extracted total RNA for each sample was checked for purity and quantified with Invitrogen Qubit 2.0 Fluorometer (Cat No.: Q32866) and Qubit RNA HS Assay kit (Cat No.: Q32855) according to manufacturer’s instructions.
Sample preparation for metabolomics
Frozen cell pellets were resuspended with extraction reagent (8:2 methanol-water solution) to 3x108 cells/mL and then transferred into 2 mL ceramic bead MagNalyser tubes. Blank samples were prepared by adding 1300 μL of extraction reagent with no cells to a MagNalyser tube with ceramic beads. Tubes were subjected to homogenization, with Bead Ruptor Elite Bead Mill Homogenizer (OMNI International) at 6.0 m/s for 40 seconds in 2 cycles at room temperature. This step was repeated twice. All samples were then centrifuged at 16,000 xg for 10 minutes at 4°C. 500 μL of the supernatant was transferred into low-bind 1.7 mL microfuge tubes. Total pools were made by combining an additional 65 μL of the supernatant from each sample and then aliquoting this mixture into low-bind 1.7 mL tubes at a volume of 500 μL. The remaining supernatant was stored at -80°C for repeat experiments if necessary. For all experimental samples, pooled samples and blanks were dried using a speedvac vacuum concentrator overnight. Dried samples were stored at -80°C.
Before LC-MS analysis, 100 μL of reconstitution buffer (95:5 water:methanol with 500 ng/mL tryptophan d-5) was added to each dried sample. All tubes were vortexed at 5000 rpm for 10 minutes and then centrifuged at room temperature at 16,000 xg for 4 minutes. Supernatant was transferred into autosampler vials for LC-MS.
RNA library preparation
RNA libraries were prepared with Kapa stranded mRNA-seq kits, with KAPA mRNA Capture Beads (KAPA code: KK8421; Roche Cat No.: 07962207001) through the UNC High Throughput Sequencing Facility. All procedures were according to manufacturer’s instructions.
RNA sequence analysis
Quality of raw sequence was checked with the FASTQC algorithm (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Sequence alignment to genome indices, generated based on Saccharomyces cerevisiae data downloaded from Ensembl.org, was performed with the STAR algorithm [76]. Quantification on a transcriptome level was performed with the SALMON algorithm [77]. The quantified data were then analyzed with the DESeq2 package in R [78], which provides a means to determine differences in transcript abundance using a negative binomial generalized linear model [43]. Differentially Expressed Genes (DEGs for concentration analysis, s-DEGs for sensitivity analysis) are defined as having adjusted p-value <0.05, absolute log2 fold-change >1 and baseMean >100. A series of baseMean thresholds were tested, including 0, 50 and 100. The conclusion remains unchanged. Therefore, the most stringent threshold (baseMean >100, which filters out >20% of genes) was chosen for the data analysis.
PCA analysis was performed using internal PCA function of DESeq2 package with variance stabilizing transformation (vst) normalized data.
For concentration analysis, sequencing results for wildtype and all mutants after glucose addition were used as input and were analyzed with the design formula = ~batch+genotype. Here ‘batch’ is incorporated to account for batch effects of sample preparation. For sensitivity analysis, sequencing results for wildtype and all mutants before and after glucose addition were used as input and were analyzed with the design formula = ~batch+genotype+treatment+genotype:treatment. Here ‘treatment’ is equivalent to glucose addition. The interaction term ‘genotype:treatment’ is included to estimate how the response amplitude of each mutant is different from wildtype, that is (mutantH-mutantL)-(wtH-wtL).
UHPLC high-resolution Orbitrap MS metabolomics data acquisition
Metabolomics data were acquired on a Vanquish UHPLC system coupled to a QExactive HF-X Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Fisher Scientific, San Jose, CA), as described previously [79]. Our UHPLC-HRMS reversed phase platform was established based on published methods [80,81]. Metabolites were separated using an HSS T3 C18 column (2.1 × 100 mm, 1.7 μm, Waters Corporation) at 50°C with binary mobile phase of water (A) and methanol (B), each containing 0.1% formic acid (v/v). The UHPLC linear gradient started from 2% B, and increased to 100% B in 16 minutes, then held for 4 minutes, with the flow rate at 400 μL/minute. The untargeted data were acquired in positive mode from 70 to 1050 m/z using the data-dependent acquisition mode.
Metabolomics data normalization and filtration
Progenesis QI (version 2.1, Waters Corporation) was used for peak picking, alignment, and normalization as described previously [79]. Samples were randomized and run within two batches with blanks and pools interspersed at a rate of 10%. Starting from the un-normalized data for each of the batch runs, the data were filtered so as to only include signals with an average intensity fold change of 3.0 or greater in the total pools compared to the blanks. Individual samples (including pools, blanks, and study samples) were then normalized to a reference sample that was selected by Progenesis from the total pools via a function named “normalize to all”. Signals were then excluded that were significantly different between pools of batch 1 and pools of batch 2 based on an ANOVA comparison calculated in Progenesis (q <0.05). After normalization and filtration, 2397 signals passed the QC procedures and were used for further analysis.
The filtered and normalized data were mean-centered and Pareto scaled prior to conducting the unsupervised principal components analysis using the ropls R package
In-house compound identification and annotation
Peaks were identified or annotated by Progenesis QI through matching to an in-house experimental standards library generated by acquiring data for approximately 1000 compounds under conditions identical to study samples, as well as to public databases (including HMDB, METLIN and NIST), as described previously [79]. Identifications and annotations were assigned using available data for retention time (RT), exact mass (MS), MS/MS fragmentation pattern, and isotopic ion pattern. The identification or annotation of each signal is provided in Supporting Information. Signals/metabolites that matched to the in-house experimental standards library by (a) RT, MS, and MS/MS are labeled as OL1, or (b) by RT and MS are labeled OL2a. An OL2b label was provided for signals that match by MS and MS/MS to the in-house library that were outside the retention time tolerance (± 0.5 minutes) for the standards run under identical conditions. Signals matched to public databases are labeled as PDa (MS and experimental MS/MS), PDb (MS and theoretical MS/MS), PDc (MS and isotopic similarity or adducts), and PDd (MS only) are also provided (Supporting Information).
Transcriptomics pathway enrichment analysis and over-representation analysis
Pathway enrichment analysis for transcriptomics data was performed with ClusterProfiler package in R [30]; Log2 fold-change for each comparison (mutantH vs. wtH for concentration analysis or mutantH-mutantL vs. wtH-wtL for sensitivity analysis) was extracted from corresponding DESeq2 analysis. GSEA analysis was then performed with gseKEGG function, with organism set to ‘sce’ (Saccharomyces cerevisiae), permutation number set to 1000, minimal and maximal size for each analyzed geneset as 3 and 200, p-value cutoff set to 0.05, p-value adjustment method set to ‘BH’ (Benjamini-Hochberg). The KEGG sce database was used throughout, for both metabolomics and transcriptomics.
Over-representation analysis for the corresponding subsection of the Venn diagram was performed with the enrichKEGG function in ClusterProfiler package, with organism set to ‘sce’ (Saccharomyces cerevisiae), minimal and maximal size for each analyzed geneset as 3 and 200, p-value cutoff set to 0.05, p-value adjustment method set to ‘BH’ (Benjamini-Hochberg).
Compound annotation, metabolic pathway enrichment analysis and over-representation analysis
Compound annotation and pathway enrichment analysis for metabolomics was performed with the MetaboAnalystR 3.0 package in R [41,42] (https://www.metaboanalyst.ca/docs/RTutorial.xhtml). For compound annotations, molecular weight tolerance (ppm) was set to 3.0, analytical mode was set to positive and retention time was included. Pathway enrichment analysis was performed with ‘integ’ module (using both Mummichog V2.0 and GSEA) with the yeast KEGG database. The p-value threshold for Mummichog was set at 0.05.
For concentration analysis, normalized peak data, from Progenesis QI for wildtype and mutants after glucose addition, were used as input for MetaboAnalystR. For sensitivity analysis, normalized peak data for wildtype and mutants before and after glucose addition were used as inputs for generalized linear model: AUC~genotype+treatment+genotype:treatment. AUC is the normalized area under curve for each peak. Treatment means before or after glucose addition. The interaction term estimated how the response amplitude of each mutant is different from wildtype, that is (mutantH-mutantL)-(wtH-wtL). The modeled p-value and t score for the interaction term associated with each peak were then used as inputs for pathway enrichment analysis. Significantly perturbed metabolites (SPMs for concentration analysis, s-SPMs for sensitivity analysis) were defined as annotations that have adjusted p-value <0.05 (FDR) from the output of MetaboAnalystR. Significantly perturbed pathways were defined as having combined p-value <0.05 (Mummichog and GSEA).
Over-representation analysis for the corresponding subsection of the Venn diagram was performed with the Enrichment Analysis module in MetaboAnalystR, with KEGG ID for each metabolites as the input. FDR adjusted p-value <0.05 was the threshold for over-represented pathways.
Integration of transcriptomics and metabolomics data
Integration analysis was performed with the ‘joint pathway analysis’ module of MetaboAnalystR (https://www.metaboanalyst.ca/docs/RTutorial.xhtml). For both concentration and sensitivity analysis, gene input together with log2 fold-change was generated based on the corresponding DESeq2 analysis, with the threshold set as adjusted p-value <0.05, absolute log2 fold-change >1 and baseMean >100 (DEGs or s-DEGs); metabolite input together with log2 fold-change was generated based on MetaboAnalystR analysis, with the threshold set as adjusted p-value <0.05 (SPMs or s-SPMs). Integration analysis was performed on ‘all pathways’, which includes both metabolic pathways as well as gene-only pathways. Enrichment analysis was performed using ‘Hypergeometric test’. Topology measure was set to ‘Degree Centrality’. Integration method was set to ‘combine queries’, which is a tight integration method with genes and metabolites pooled into a single query and used to perform enrichment analysis within their "pooled universe". Significantly enriched pathways were defined as having FDR adjusted p-value <0.05.
Yeast RNA extraction, DNase treatment, and reverse transcription
RNA was extracted from cells using RNeasy mini kit (Qiagen) directly as described in “Sample preparation for RNA-seq” or using hot acid phenol. Briefly, cells were resuspended in TES solution (10 mM Tris-HCl, pH 7.5; 10 mM EDTA; 0.5% SDS) and incubated with acid phenol at 65°C for one hour. Following, phenol-chloroform precipitation was used to separate the RNA, RQ1 DNase (Promega) was used to degrade any residual DNA and the samples were further purified using RNeasy mini kit. Final concentration was determined using a NanoDrop One (Thermo Scientific). To produce cDNA, 250 ng RNA was reverse transcribed with a High-Capacity cDNA Reverse Transcription Kit (Thermo Scientific) following manufacturer’s protocol.
qPCR
qPCR primers were designed to select for short, unique regions of YCL025C (AGP1), YGR121C (MEP1), YIR029W (DAL2), YOR275C (GDH1) and reference gene YER100W (UBC6) and ordered from Integrated DNA Technologies, as follows:
YER100W_FWD primer: 5’ GAAGCCACGACAGGATCAAT 3’
YER100W_REV primer: 5’ ATCCCCCTCATCCAATTTTC 3’
YCL025C_FWD primer: 5’ TGGATGATGGTTTTGGGTTT 3’
YCL025C_REV primer: 5’ CTTCTGCATAACCACGAGCA 3’
YGR121C_FWD primer: 5’ GTTGGTCTGTGCTCCGGTAT 3’
YGR121C_REV primer: 5’ CTCCAAAAAGGGCATTGAAA 3’
YIR029W_FWD primer: 5’ GCTGGGAAACACGAAGACAT 3’
YIR029W_REV primer: 5’ CTTCTTCGCCCTCGTCATAG 3’
YOR375C_FWD primer: 5’ TGCCAATTGTTTCTGTTCCA 3’
YOR375C_REV primer: 5’ ACAAGTTCACGGAAGGATGG 3’
cDNA was diluted 50-fold following reverse transcription and amplified by qPCR in technical triplicate with SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) according to manufacturer’s instructions modified with 45 cycles and 45 seconds anneal/extension time. Expected sequence length was verified by agarose gel electrophoresis. The threshold cycle (Ct) was determined for each using CFX Maestro Software (BIO-RAD) and ΔΔCt values were calculated in reference to wild type cells and YER100W expression levels. All values were normalized to wild type ΔΔCt values. Concentration analysis was performed on cells after high glucose treatment (see “Cell preparation”) following ΔΔCt analysis as described above. Sensitivity analysis was performed by comparison of ΔΔCt values of high glucose treatment minus low glucose treatment. p-values were calculated using independent, non-parametric, one-tailed (with direction as determined by RNA-seq data) Mann-Whitney U tests and adjusted for multiple comparisons following the Benjamini-Hochberg Procedure.
Supporting information
For A) transcriptomics and B) metabolomics, X-axis shows PC1 with the percentage of explained variance and Y-axis shows PC2 with the percentage of explained variance. Data are scaled as detailed in Methods. Wildtype (black), asc1 (red), gpa2 (blue), gpr1 (purple). Low glucose (L, 0.05% glucose)-triangles, high glucose (H, 2% glucose)-circles.
(TIF)
Illustration of different modes of changes as captured by concentration analysis (A) and sensitivity analysis (B). The Y-axis represents either normalized gene counts or normalized peak area for metabolites. The X-axis represents different genotypes. A hypothetical wildtype is shown in black. The triangle represents measurement at low glucose and the square represents measurement at high glucose. The connecting grey line represents the response amplitude, detected by sensitivity analysis (wtH-wtL). Hypothetical mutants with increased response amplitude are colored green, while mutants with decreased response amplitude are colored red.
(TIF)
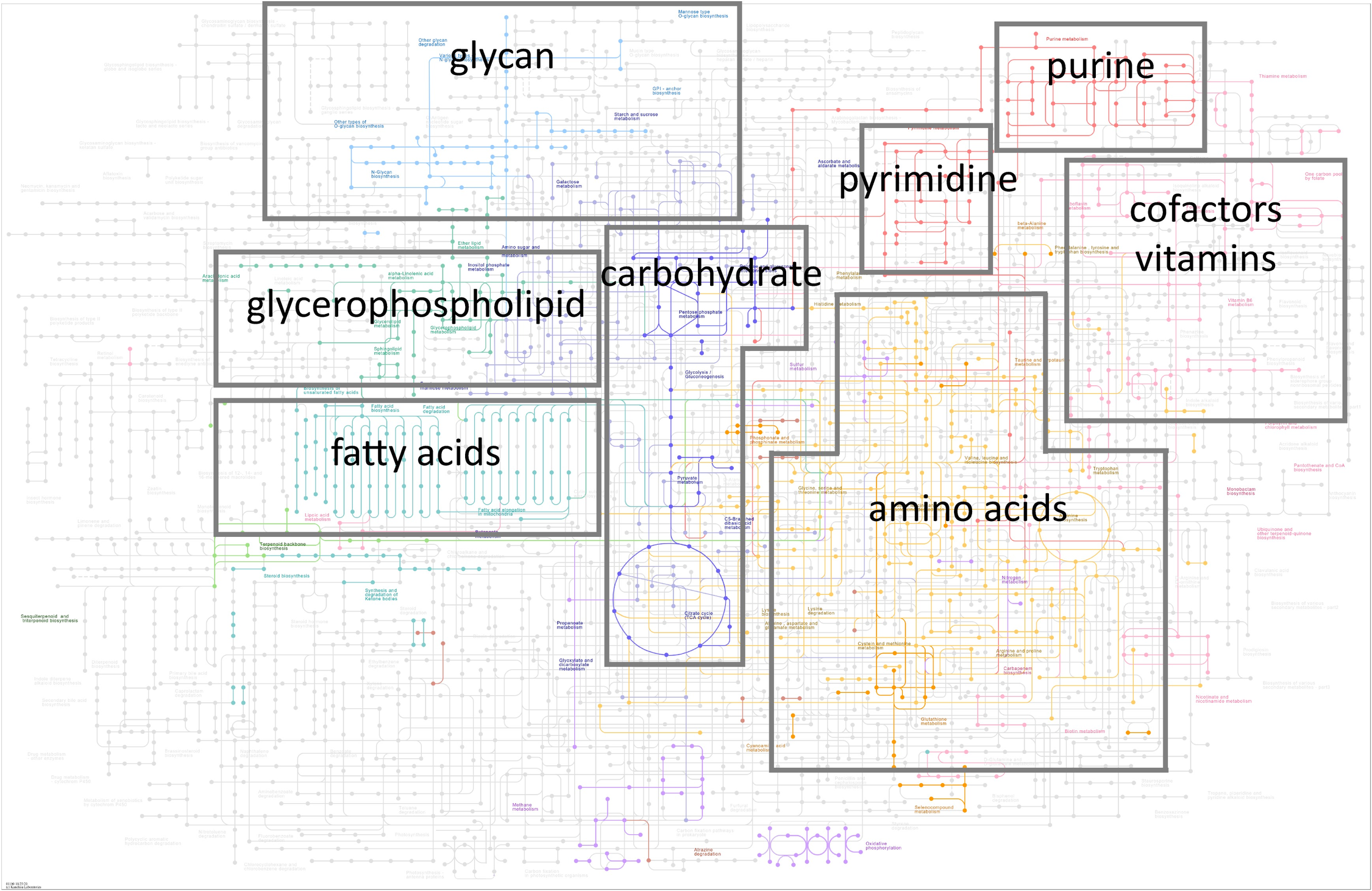
Map is color coded to delineate carbohydrate metabolism (blue), glycan biosynthesis and metabolism (cyan), amino acid metabolism (yellow), nucleotide metabolism (red), lipid metabolism (teal), metabolism of cofactors and vitamins (pink). Major species are highlighted with grey bounding box.
(PNG)
{kind=link}
(PNG)
{kind=link}
(PNG)
{kind=link}
(PNG)
{kind=link}
(PNG)
{kind=link}
(PNG)
{kind=link}
(PNG)
{kind=link}
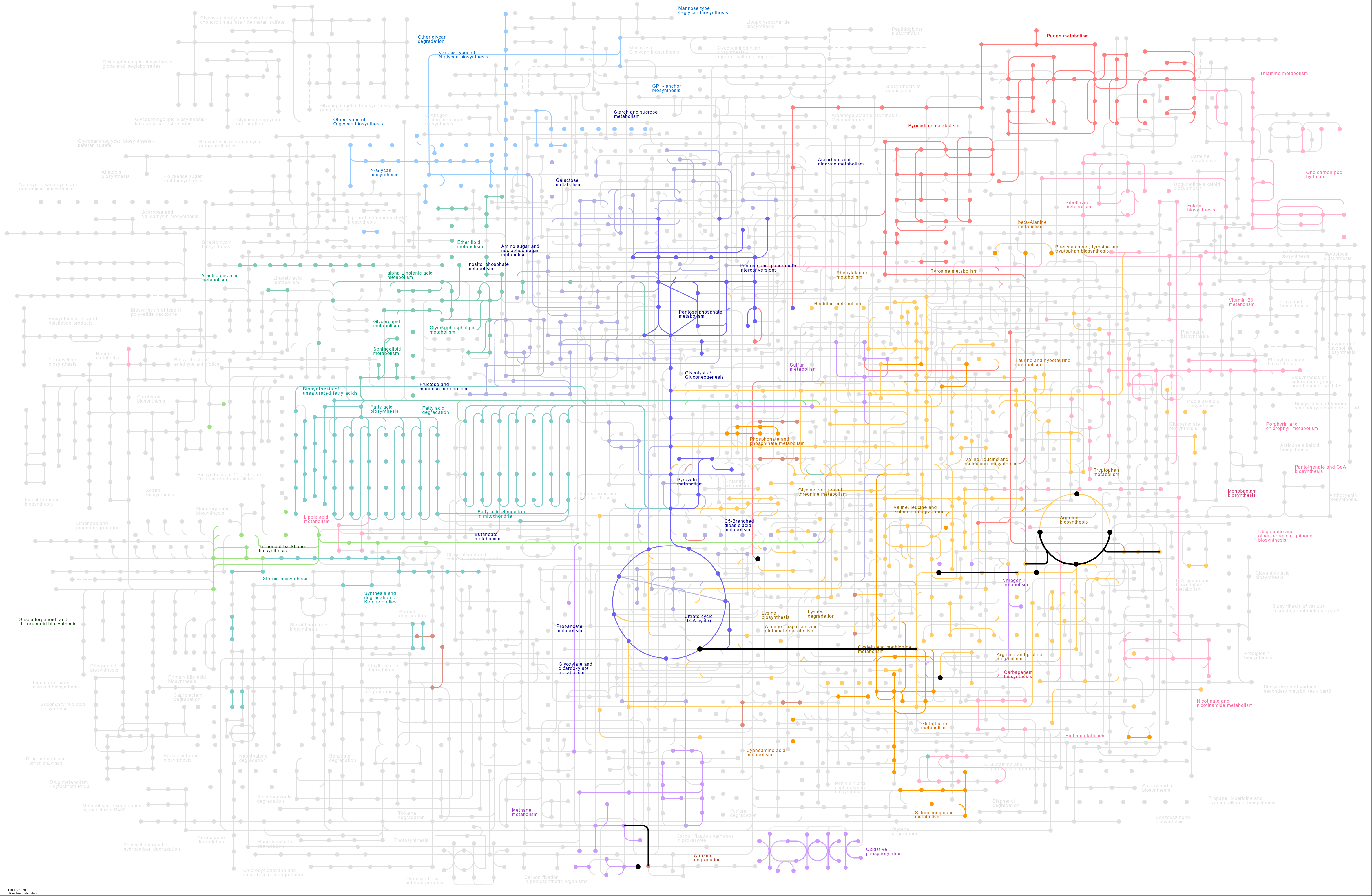
The map is color coded as in S3 Fig. Highlighted are the DEGs (black lines) and SPMs (black dots) for gpa2 integration analysis and gray boxes are used to delineate clusters associated with a specific pathway.
(PNG)
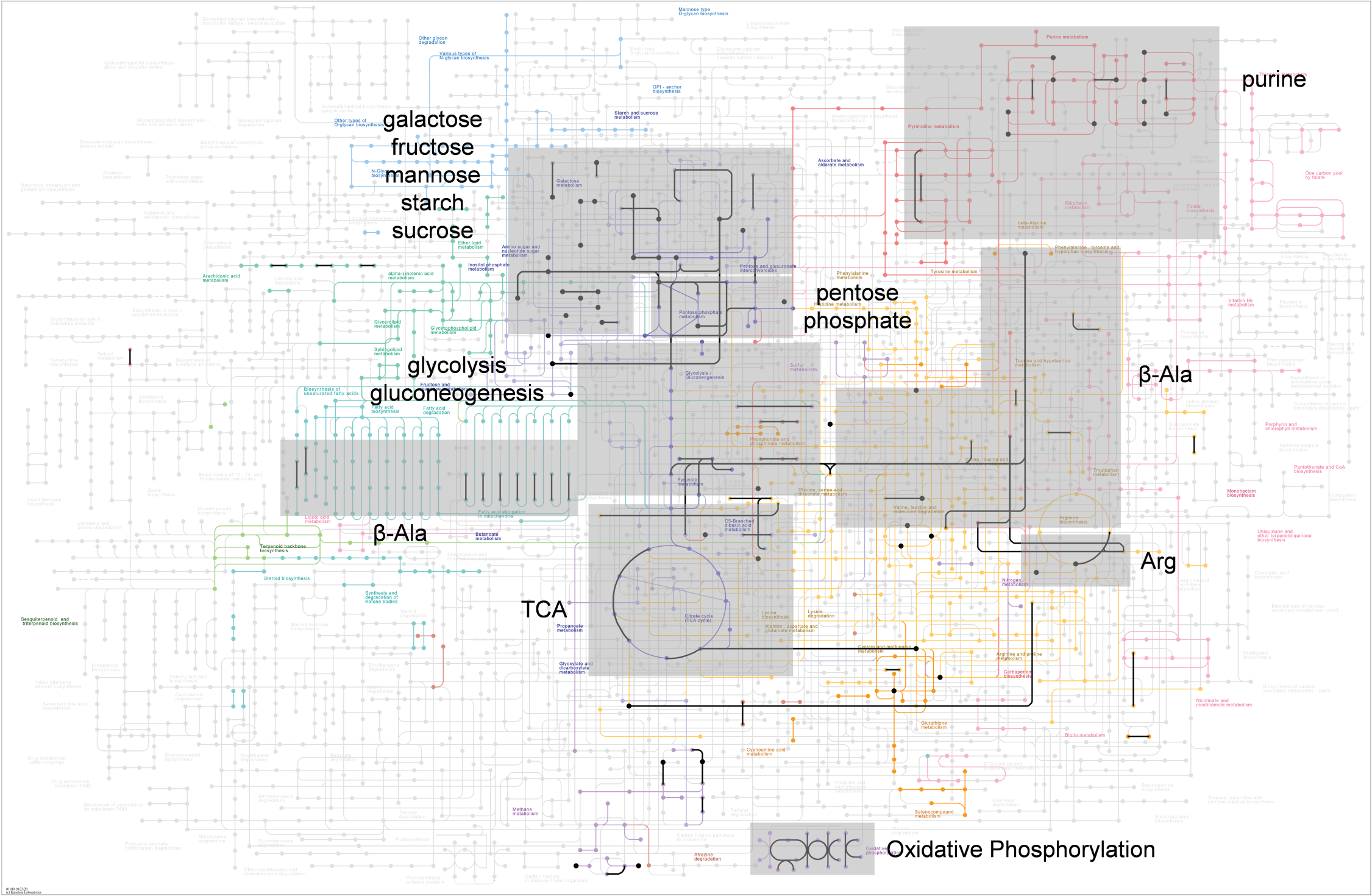{kind=link}
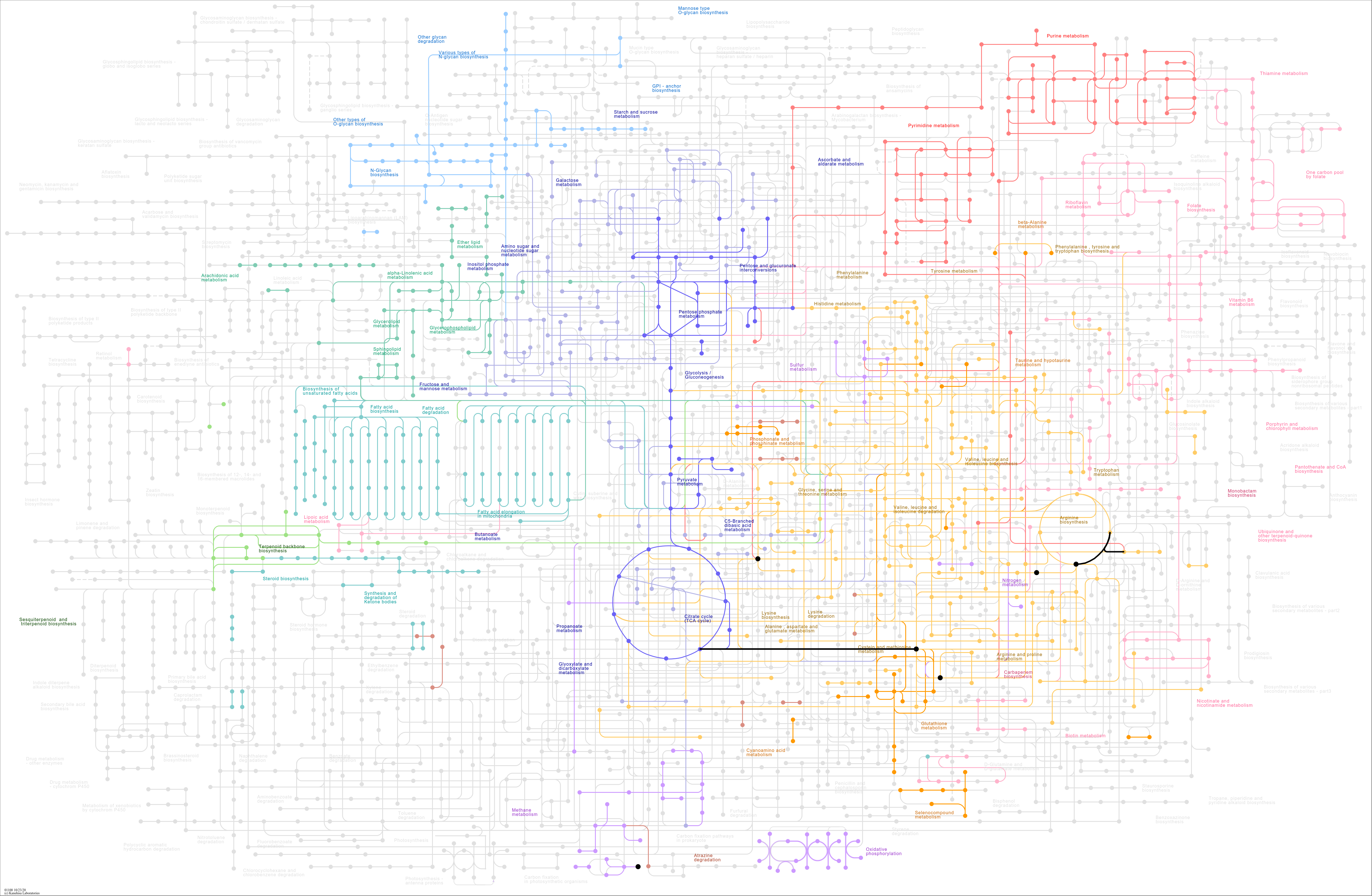
The map is color coded as in S3 Fig. Highlighted are the DEGs (black lines) and SPMs (black dots) for asc1 integration analysis and gray boxes are used to delineate clusters associated with a specific pathway.
(PNG)
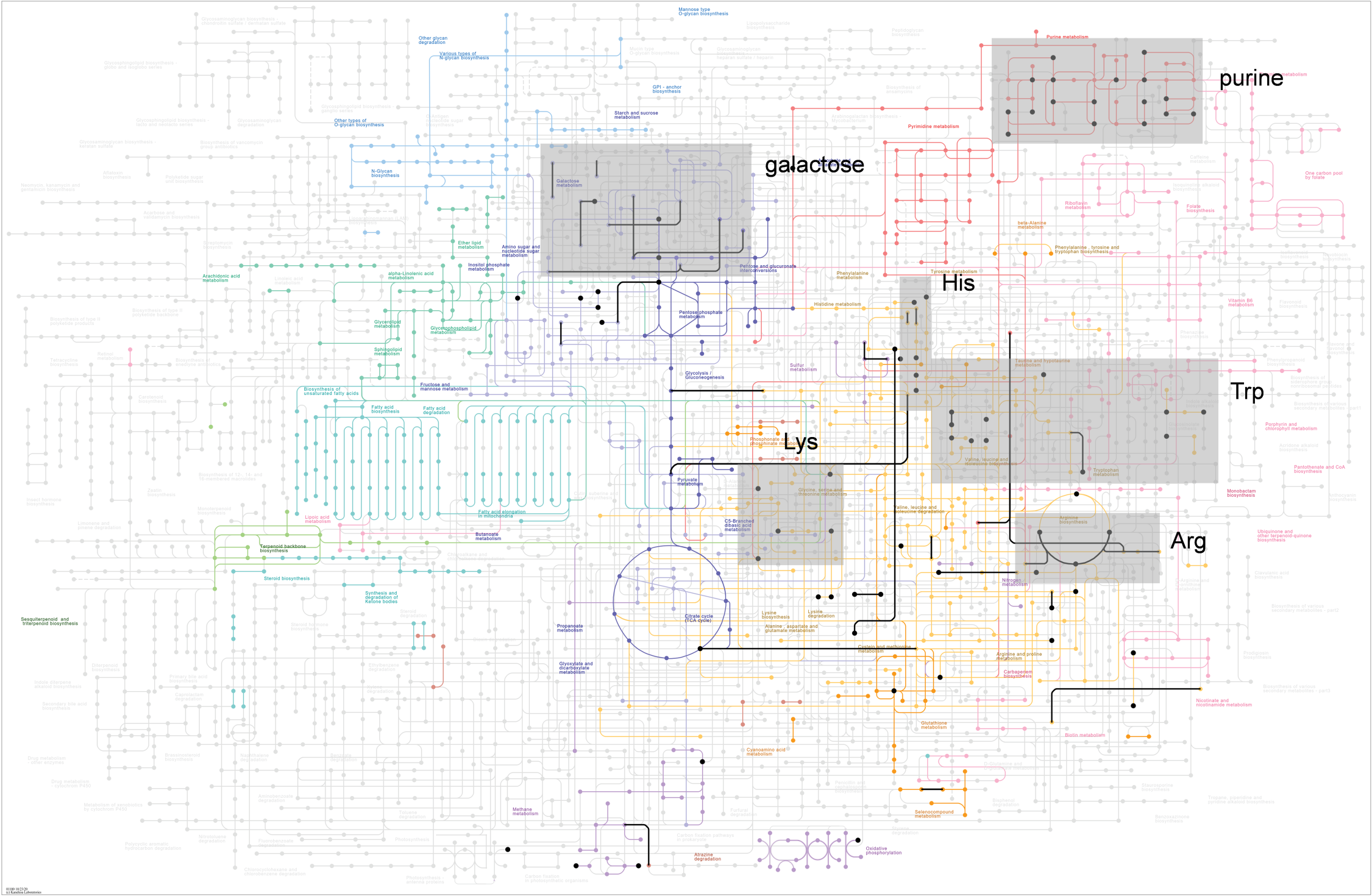{kind=link}
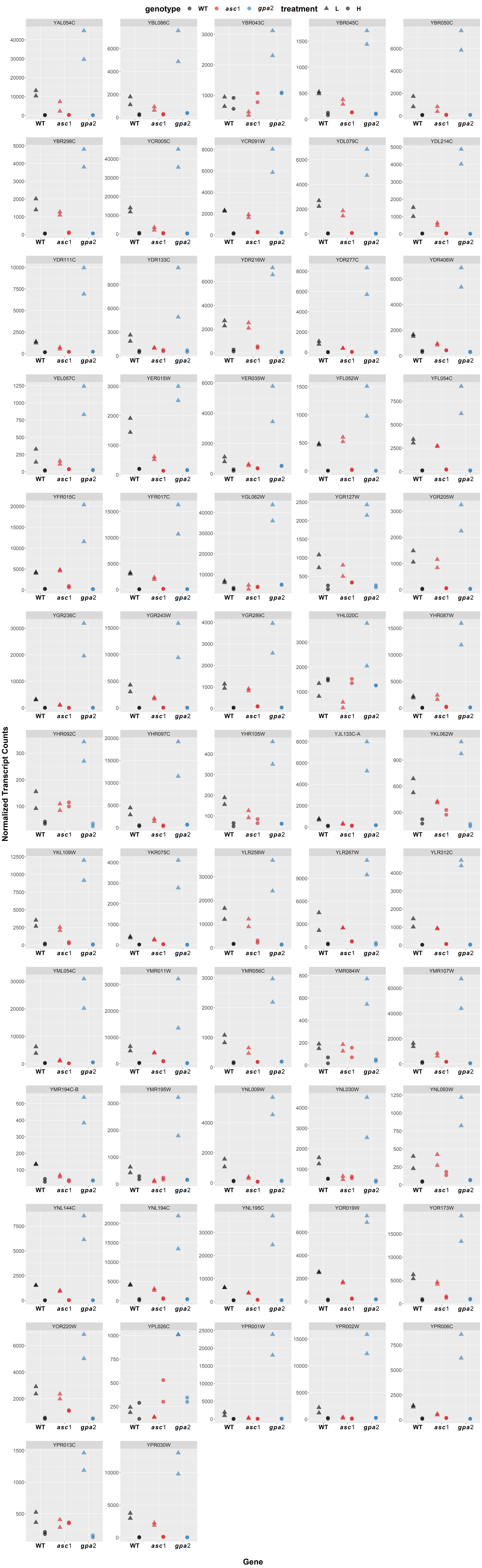
Each gene has its own grid. Y-axis shows normalized transcript counts for low glucose (triangle), high glucose (square) in wildtype (black), asc1 (red) and gpa2 (blue).
(PNG)
{kind=link}
Each gene has its own grid. Y-axis shows normalized transcript counts for low glucose (triangle), high glucose (square) in wildtype (black), asc1 (red) and gpa2 (blue).
(PNG)
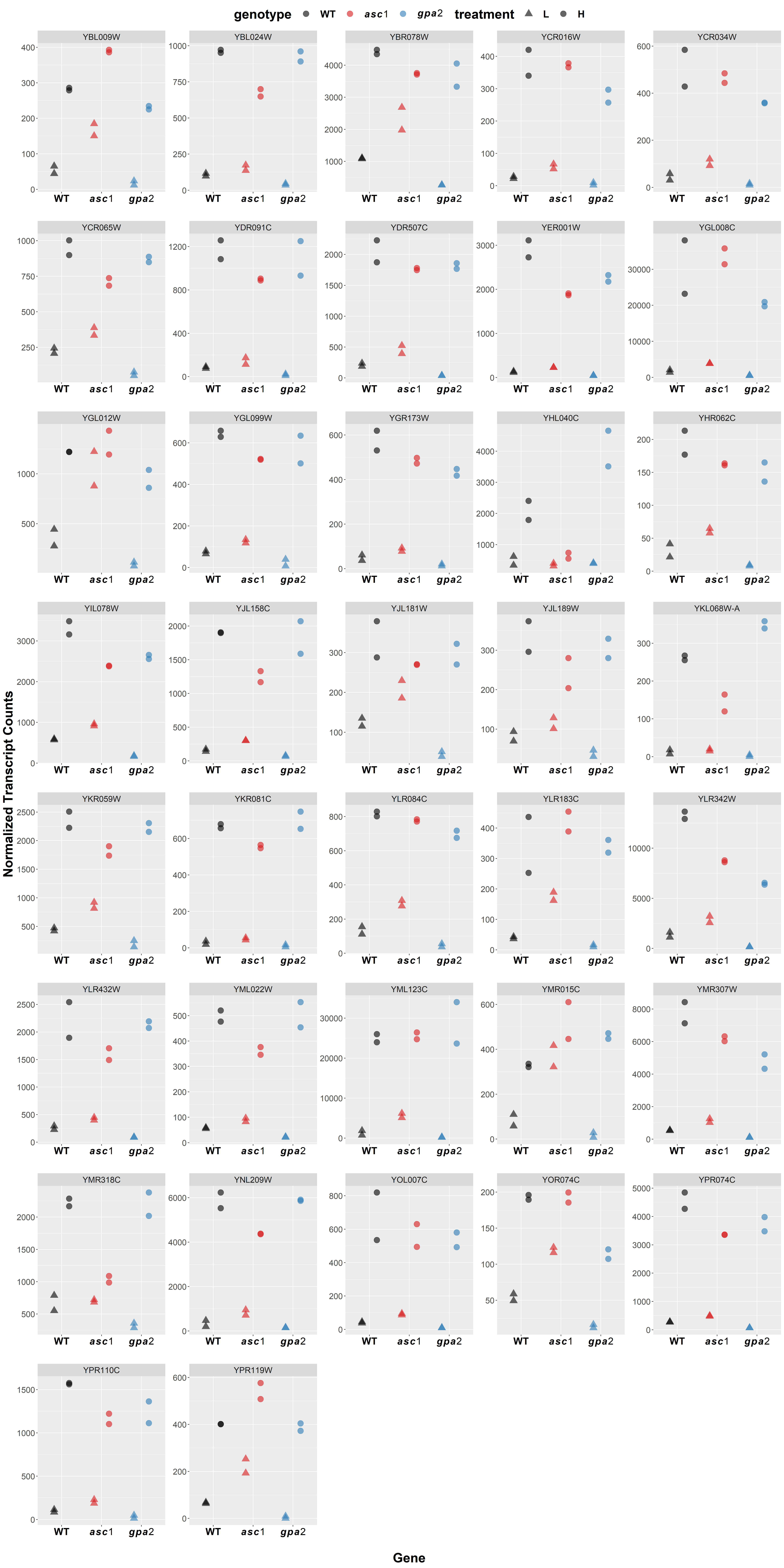{kind=link}
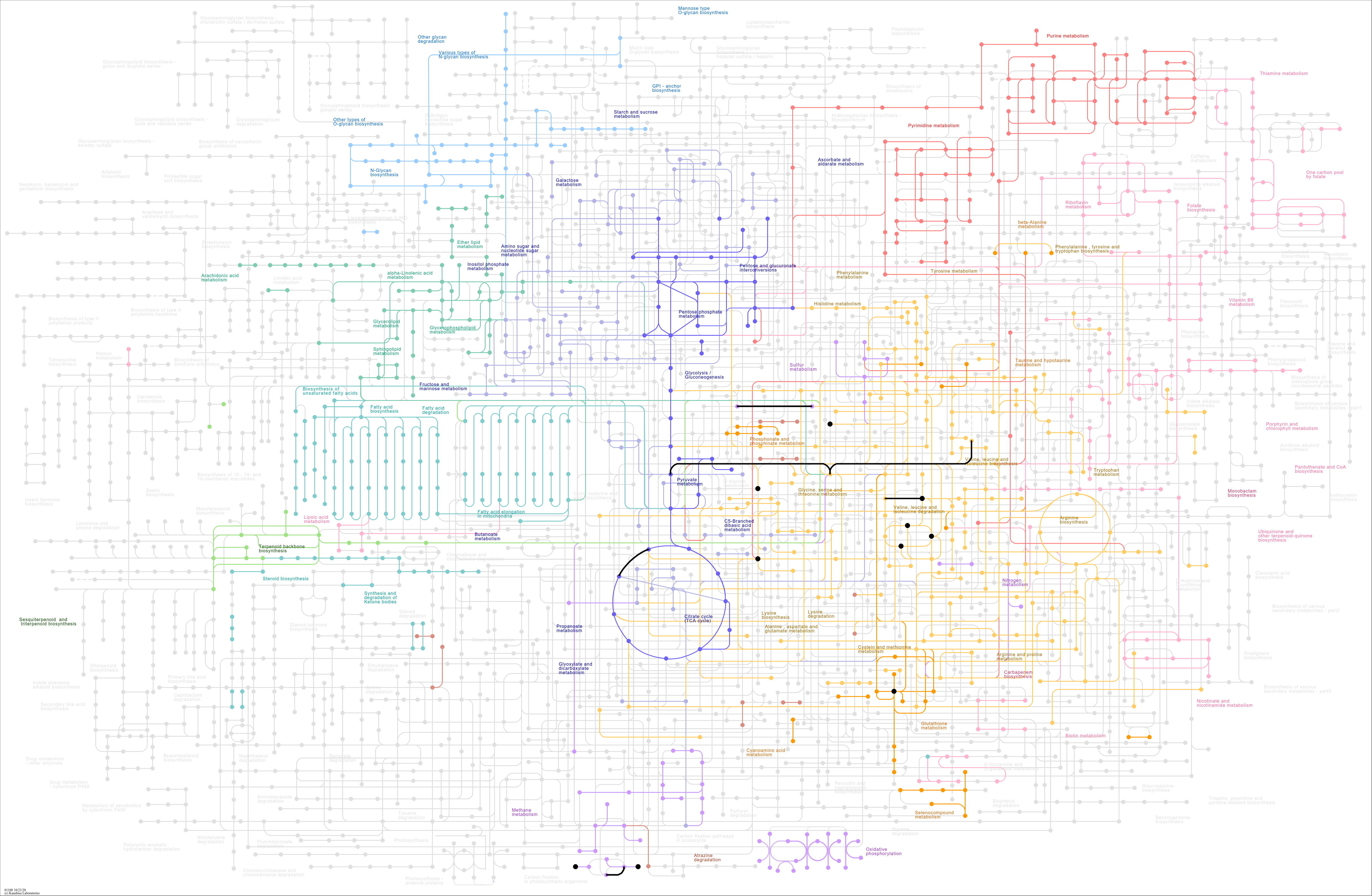
The map is color coded as in S3 Fig. Highlighted are the s-DEGs (black lines) and s-SPMs (black dots) for gpa2 integration analysis and gray boxes are used to delineate clusters associated with a specific pathway.
(PNG)
{kind=link}
The map is color coded as in S3 Fig. Highlighted are the s-DEGs (black lines) and s-SPMs (black dots) for asc1 integration analysis and gray boxes are used to delineate clusters associated with a specific pathway.
(PNG)
{kind=link}
(XLSX)
(XLSX)
(CSV)
(CSV)
First block shows GSEA for transcriptomics with adjusted p-value <0.05, arranged in ascending order; second block shows MetaboAnalystR pathway enrichment analysis for metabolomics with combined p-value <0.05, arranged in ascending order; third block shows MetaboAnalystR joint pathway analysis with adjusted p-value <0.05, arranged in ascending order.
(XLSX)
(XLSX)
(CSV)
(CSV)
Sheet 1: Fold induction data for concentration analysis. Sheet 2: Induction difference data for sensitivity analysis. Sheet 3: Benjamini-Hochberg corrected Mann–Whitney U test results for concentration analysis. Sheet 4: Benjamini-Hochberg corrected Mann–Whitney U test results for sensitivity analysis.
(XLSX)
Acknowledgments
We thank Patrick Maxwell for helpful discussion. We thank Amy Perou and Piotr Mieczkowski at the High Throughput Sequencing Facility at UNC Chapel Hill for collecting RNAseq data. We thank members of the Susan Sumner Laboratory at the Nutrition Research Institute at UNC Chapel Hill, where all metabolomics data capture, as well as in-house analysis, identification and annotation was performed.
Data Availability
RNA-seq data: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE171846 Metabolomics data: http://dx.doi.org/10.21228/M80D82 For review the GEO data can be accessed as follows: Go to https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE171846 Enter token qfedswaahrslxqf into the box For review the Metabolomics workbench can be accessed as follows: http://dev.metabolomicsworkbench.org:22222/data/DRCCMetadata.php?Mode=Study&StudyID=ST001786&Access=EsfC4623.
Funding Statement
Supported by National Institutes of Health grant R35GM118105 (https://www.nigms.nih.gov) to HGD, T32 GM008570 (https://www.nigms.nih.gov) to SEH, and institutional funding (https://uncnri.org/staff-members/susan-sumner-ph-d/) to SJS. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Husted AS, Trauelsen M, Rudenko O, Hjorth SA, Schwartz TW. GPCR-Mediated Signaling of Metabolites. Cell Metab. 2017;25(4):777–96. doi: 10.1016/j.cmet.2017.03.008 [DOI] [PubMed] [Google Scholar]
- 2.Clement ST, Dixit G, Dohlman HG. Regulation of yeast G protein signaling by the kinases that activate the AMPK homolog Snf1. Sci Signal. 2013;6(291):ra78. doi: 10.1126/scisignal.2004143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bardwell L. A walk-through of the yeast mating pheromone response pathway. Peptides. 2005;26(2):339–50. doi: 10.1016/j.peptides.2004.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Slessareva JE, Routt SM, Temple B, Bankaitis VA, Dohlman HG. Activation of the phosphatidylinositol 3-kinase Vps34 by a G protein alpha subunit at the endosome. Cell. 2006;126(1):191–203. doi: 10.1016/j.cell.2006.04.045 [DOI] [PubMed] [Google Scholar]
- 5.Broach JR. Nutritional control of growth and development in yeast. Genetics. 2012;192(1):73–105. doi: 10.1534/genetics.111.135731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xue Y, Batlle M, Hirsch JP. GPR1 encodes a putative G protein-coupled receptor that associates with the Gpa2p Galpha subunit and functions in a Ras-independent pathway. Embo J. 1998;17(7):1996–2007. doi: 10.1093/emboj/17.7.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yun CW, Tamaki H, Nakayama R, Yamamoto K, Kumagai H. Gpr1p, a putative G-protein coupled receptor, regulates glucose- dependent cellular cAMP level in yeast Saccharomyces cerevisiae. Biochem Biophys Res Commun. 1998;252(1):29–33. doi: 10.1006/bbrc.1998.9600 [DOI] [PubMed] [Google Scholar]
- 8.Kraakman L, Lemaire K, Ma P, Teunissen AW, Donaton MC, Van Dijck P, et al. A Saccharomyces cerevisiae G-protein coupled receptor, Gpr1, is specifically required for glucose activation of the cAMP pathway during the transition to growth on glucose. Mol Microbiol. 1999;32(5):1002–12. doi: 10.1046/j.1365-2958.1999.01413.x [DOI] [PubMed] [Google Scholar]
- 9.Ron D, Adams DR, Baillie GS, Long A, O’Connor R, Kiely PA. RACK1 to the future—a historical perspective. Cell Commun Signal. 2013;11:53. doi: 10.1186/1478-811X-11-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palmer DA, Thompson JK, Li L, Prat A, Wang P. Gib2, a novel Gbeta-like/RACK1 homolog, functions as a Gbeta subunit in cAMP signaling and is essential in Cryptococcus neoformans. J Biol Chem. 2006;281(43):32596–605. doi: 10.1074/jbc.M602768200 [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Shen G, Gong J, Shen D, Whittington A, Qing J, et al. Noncanonical Gbeta Gib2 is a scaffolding protein promoting cAMP signaling through functions of Ras1 and Cac1 proteins in Cryptococcus neoformans. J Biol Chem. 2014;289(18):12202–16. doi: 10.1074/jbc.M113.537183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeller CE, Parnell SC, Dohlman HG. The RACK1 ortholog Asc1 functions as a G-protein beta subunit coupled to glucose responsiveness in yeast. J Biol Chem. 2007;282(34):25168–76. doi: 10.1074/jbc.M702569200 [DOI] [PubMed] [Google Scholar]
- 13.Colombo S, Ma P, Cauwenberg L, Winderickx J, Crauwels M, Teunissen A, et al. Involvement of distinct G-proteins, Gpa2 and Ras, in glucose- and intracellular acidification-induced cAMP signalling in the yeast Saccharomyces cerevisiae. Embo J. 1998;17(12):3326–41. doi: 10.1093/emboj/17.12.3326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dessauer CW, Watts VJ, Ostrom RS, Conti M, Dove S, Seifert R. International Union of Basic and Clinical Pharmacology. CI. Structures and Small Molecule Modulators of Mammalian Adenylyl Cyclases. Pharmacol Rev. 2017;69(2):93–139. doi: 10.1124/pr.116.013078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ron D, Chen CH, Caldwell J, Jamieson L, Orr E, Mochly-Rosen D. Cloning of an intracellular receptor for protein kinase C: a homolog of the beta subunit of G proteins. Proc Natl Acad Sci U S A. 1994;91(3):839–43. doi: 10.1073/pnas.91.3.839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dell EJ, Connor J, Chen S, Stebbins EG, Skiba NP, Mochly-Rosen D, et al. The betagamma subunit of heterotrimeric G proteins interacts with RACK1 and two other WD repeat proteins. J Biol Chem. 2002;277(51):49888–95. doi: 10.1074/jbc.M202755200 [DOI] [PubMed] [Google Scholar]
- 17.Chen S, Dell EJ, Lin F, Sai J, Hamm HE. RACK1 regulates specific functions of Gbetagamma. J Biol Chem. 2004;279(17):17861–8. doi: 10.1074/jbc.M313727200 [DOI] [PubMed] [Google Scholar]
- 18.Parent A, Laroche G, Hamelin E, Parent JL. RACK1 regulates the cell surface expression of the G protein-coupled receptor for thromboxane A(2). Traffic. 2008;9(3):394–407. doi: 10.1111/j.1600-0854.2007.00692.x [DOI] [PubMed] [Google Scholar]
- 19.Reiner CL, McCullar JS, Kow RL, Le JH, Goodlett DR, Nathanson NM. RACK1 associates with muscarinic receptors and regulates M(2) receptor trafficking. PLoS One. 2010;5(10):e13517. doi: 10.1371/journal.pone.0013517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chantrel Y, Gaisne M, Lions C, Verdiere J. The transcriptional regulator Hap1p (Cyp1p) is essential for anaerobic or heme-deficient growth of Saccharomyces cerevisiae: Genetic and molecular characterization of an extragenic suppressor that encodes a WD repeat protein. Genetics. 1998;148(2):559–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Link AJ, Eng J, Schieltz DM, Carmack E, Mize GJ, Morris DR, et al. Direct analysis of protein complexes using mass spectrometry. Nat Biotechnol. 1999;17(7):676–82. doi: 10.1038/10890 [DOI] [PubMed] [Google Scholar]
- 22.Baum S, Bittins M, Frey S, Seedorf M. Asc1p, a WD40-domain containing adaptor protein, is required for the interaction of the RNA-binding protein Scp160p with polysomes. Biochem J. 2004;380(Pt 3):823–30. doi: 10.1042/BJ20031962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerbasi VR, Weaver CM, Hill S, Friedman DB, Link AJ. Yeast Asc1p and mammalian RACK1 are functionally orthologous core 40S ribosomal proteins that repress gene expression. Mol Cell Biol. 2004;24(18):8276–87. doi: 10.1128/MCB.24.18.8276-8287.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sengupta J, Nilsson J, Gursky R, Spahn CM, Nissen P, Frank J. Identification of the versatile scaffold protein RACK1 on the eukaryotic ribosome by cryo-EM. Nat Struct Mol Biol. 2004;11(10):957–62. doi: 10.1038/nsmb822 [DOI] [PubMed] [Google Scholar]
- 25.Opitz N, Schmitt K, Hofer-Pretz V, Neumann B, Krebber H, Braus GH, et al. Capturing the Asc1p/Receptor for Activated C Kinase 1 (RACK1) Microenvironment at the Head Region of the 40S Ribosome with Quantitative BioID in Yeast. Mol Cell Proteomics. 2017;16(12):2199–218. doi: 10.1074/mcp.M116.066654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolf AS, Grayhack EJ. Asc1, homolog of human RACK1, prevents frameshifting in yeast by ribosomes stalled at CGA codon repeats. RNA. 2015;21(5):935–45. doi: 10.1261/rna.049080.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sitron CS, Park JH, Brandman O. Asc1, Hel2, and Slh1 couple translation arrest to nascent chain degradation. RNA. 2017;23(5):798–810. doi: 10.1261/rna.060897.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rachfall N, Schmitt K, Bandau S, Smolinski N, Ehrenreich A, Valerius O, et al. RACK1/Asc1p, a ribosomal node in cellular signaling. Mol Cell Proteomics. 2013;12(1):87–105. doi: 10.1074/mcp.M112.017277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kresnowati MT, van Winden WA, Almering MJ, ten Pierick A, Ras C, Knijnenburg TA, et al. When transcriptome meets metabolome: fast cellular responses of yeast to sudden relief of glucose limitation. Mol Syst Biol. 2006;2:49. doi: 10.1038/msb4100083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7. doi: 10.1089/omi.2011.0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019;28(11):1947–51. doi: 10.1002/pro.3715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanehisa M, Furumichi M, Sato Y, Ishiguro-Watanabe M, Tanabe M. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang N, Cao L. Starvation signals in yeast are integrated to coordinate metabolic reprogramming and stress response to ensure longevity. Curr Genet. 2017;63(5):839–43. doi: 10.1007/s00294-017-0697-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lorenz MC, Pan X, Harashima T, Cardenas ME, Xue Y, Hirsch JP, et al. The G protein-coupled receptor gpr1 is a nutrient sensor that regulates pseudohyphal differentiation in Saccharomyces cerevisiae. Genetics. 2000;154(2):609–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valerius O, Kleinschmidt M, Rachfall N, Schulze F, Lopez Marin S, Hoppert M, et al. The Saccharomyces homolog of mammalian RACK1, Cpc2/Asc1p, is required for FLO11-dependent adhesive growth and dimorphism. Mol Cell Proteomics. 2007;6(11):1968–79. doi: 10.1074/mcp.M700184-MCP200 [DOI] [PubMed] [Google Scholar]
- 37.Cullen PJ, Sprague GF, Jr. Glucose depletion causes haploid invasive growth in yeast [In Process Citation]. Proc Natl Acad Sci U S A. 2000;97(25):13619–24. doi: 10.1073/pnas.240345197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Darnell AM, Subramaniam AR, O’Shea EK. Translational Control through Differential Ribosome Pausing during Amino Acid Limitation in Mammalian Cells. Mol Cell. 2018;71(2):229–43 e11. doi: 10.1016/j.molcel.2018.06.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lesage G, Bussey H. Cell wall assembly in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 2006;70(2):317–43. doi: 10.1128/MMBR.00038-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lien EC, Nagiec MJ, Dohlman HG. Proper protein glycosylation promotes mitogen-activated protein kinase signal fidelity. Biochemistry. 2013;52(1):115–24. doi: 10.1021/bi3009483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chong J, Wishart DS, Xia J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr Protoc Bioinformatics. 2019;68(1):e86. doi: 10.1002/cpbi.86 [DOI] [PubMed] [Google Scholar]
- 42.Pang Z, Chong J, Li S, Xia J. MetaboAnalystR 3.0: Toward an Optimized Workflow for Global Metabolomics. Metabolites. 2020;10(5). doi: 10.3390/metabo10050186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Love MI, Anders S, Kim V, Huber W. RNA-Seq workflow: gene-level exploratory analysis and differential expression. F1000Res. 2015;4:1070. doi: 10.12688/f1000research.7035.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schreve JL, Sin JK, Garrett JM. The Saccharomyces cerevisiae YCC5 (YCL025c) gene encodes an amino acid permease, Agp1, which transports asparagine and glutamine. J Bacteriol. 1998;180(9):2556–9. doi: 10.1128/JB.180.9.2556-2559.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coyle SM, Gilbert WV, Doudna JA. Direct link between RACK1 function and localization at the ribosome in vivo. Mol Cell Biol. 2009;29(6):1626–34. doi: 10.1128/MCB.01718-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Svidritskiy E, Brilot AF, Koh CS, Grigorieff N, Korostelev AA. Structures of yeast 80S ribosome-tRNA complexes in the rotated and nonrotated conformations. Structure. 2014;22(8):1210–8. doi: 10.1016/j.str.2014.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hart Y, Alon U. The utility of paradoxical components in biological circuits. Mol Cell. 2013;49(2):213–21. doi: 10.1016/j.molcel.2013.01.004 [DOI] [PubMed] [Google Scholar]
- 48.Arshavsky VY, Lamb TD, Pugh EN, Jr. G proteins and phototransduction. Annu Rev Physiol. 2002;64:153–87. doi: 10.1146/annurev.physiol.64.082701.102229 [DOI] [PubMed] [Google Scholar]
- 49.Phan VT, Ding VW, Li F, Chalkley RJ, Burlingame A, McCormick F. The RasGAP proteins Ira2 and neurofibromin are negatively regulated by Gpb1 in yeast and ETEA in humans. Mol Cell Biol. 2010;30(9):2264–79. doi: 10.1128/MCB.01450-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Powers S, Kataoka T, Fasano O, Goldfarb M, Strathern J, Broach J, et al. Genes in S. cerevisiae encoding proteins with domains homologous to the mammalian ras proteins. Cell. 1984;36(3):607–12. doi: 10.1016/0092-8674(84)90340-4 [DOI] [PubMed] [Google Scholar]
- 51.Toda T, Uno I, Ishikawa T, Powers S, Kataoka T, Broek D, et al. In yeast, RAS proteins are controlling elements of adenylate cyclase. Cell. 1985;40(1):27–36. doi: 10.1016/0092-8674(85)90305-8 [DOI] [PubMed] [Google Scholar]
- 52.Uno I, Mitsuzawa H, Matsumoto K, Tanaka K, Oshima T, Ishikawa T. Reconstitution of the GTP-dependent adenylate cyclase from products of the yeast CYR1 and RAS2 genes in Escherichia coli. Proc Natl Acad Sci U S A. 1985;82(23):7855–9. doi: 10.1073/pnas.82.23.7855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Field J, Nikawa J, Broek D, MacDonald B, Rodgers L, Wilson IA, et al. Purification of a RAS-responsive adenylyl cyclase complex from Saccharomyces cerevisiae by use of an epitope addition method. Mol Cell Biol. 1988;8(5):2159–65. doi: 10.1128/mcb.8.5.2159-2165.1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakafuku M, Obara T, Kaibuchi K, Miyajima I, Miyajima A, Itoh H, et al. Isolation of a second yeast Saccharomyces cerevisiae gene (GPA2) coding for guanine nucleotide-binding regulatory protein: studies on its structure and possible functions. Proc Natl Acad Sci U S A. 1988;85(5):1374–8. doi: 10.1073/pnas.85.5.1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Field J, Xu HP, Michaeli T, Ballester R, Sass P, Wigler M, et al. Mutations of the adenylyl cyclase gene that block RAS function in Saccharomyces cerevisiae. Science. 1990;247(4941):464–7. doi: 10.1126/science.2405488 [DOI] [PubMed] [Google Scholar]
- 56.Suzuki N, Choe HR, Nishida Y, Yamawaki-Kataoka Y, Ohnishi S, Tamaoki T, et al. Leucine-rich repeats and carboxyl terminus are required for interaction of yeast adenylate cyclase with RAS proteins. Proc Natl Acad Sci U S A. 1990;87(22):8711–5. doi: 10.1073/pnas.87.22.8711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mintzer KA, Field J. Interactions between adenylyl cyclase, CAP and RAS from Saccharomyces cerevisiae. Cell Signal. 1994;6(6):681–94. doi: 10.1016/0898-6568(94)90050-7 [DOI] [PubMed] [Google Scholar]
- 58.Bhattacharya S, Chen L, Broach JR, Powers S. Ras membrane targeting is essential for glucose signaling but not for viability in yeast. Proc Natl Acad Sci U S A. 1995;92(7):2984–8. doi: 10.1073/pnas.92.7.2984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kubler E, Mosch HU, Rupp S, Lisanti MP. Gpa2p, a G-protein alpha-subunit, regulates growth and pseudohyphal development in Saccharomyces cerevisiae via a cAMP-dependent mechanism. J Biol Chem. 1997;272(33):20321–3. doi: 10.1074/jbc.272.33.20321 [DOI] [PubMed] [Google Scholar]
- 60.Rolland F, De Winde JH, Lemaire K, Boles E, Thevelein JM, Winderickx J. Glucose-induced cAMP signalling in yeast requires both a G-protein coupled receptor system for extracellular glucose detection and a separable hexose kinase-dependent sensing process. Mol Microbiol. 2000;38(2):348–58. doi: 10.1046/j.1365-2958.2000.02125.x [DOI] [PubMed] [Google Scholar]
- 61.Wang Y, Pierce M, Schneper L, Guldal CG, Zhang X, Tavazoie S, et al. Ras and Gpa2 mediate one branch of a redundant glucose signaling pathway in yeast. PLoS Biol. 2004;2(5):E128. doi: 10.1371/journal.pbio.0020128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Matsumoto K, Uno I, Oshima Y, Ishikawa T. Isolation and characterization of yeast mutants deficient in adenylate cyclase and cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1982;79(7):2355–9. doi: 10.1073/pnas.79.7.2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kataoka T, Broek D, Wigler M. DNA sequence and characterization of the S. cerevisiae gene encoding adenylate cyclase. Cell. 1985;43(2 Pt 1):493–505. doi: 10.1016/0092-8674(85)90179-5 [DOI] [PubMed] [Google Scholar]
- 64.Casperson GF, Walker N, Bourne HR. Isolation of the gene encoding adenylate cyclase in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1985;82(15):5060–3. doi: 10.1073/pnas.82.15.5060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Harashima T, Heitman J. The Galpha protein Gpa2 controls yeast differentiation by interacting with kelch repeat proteins that mimic Gbeta subunits. Mol Cell. 2002;10(1):163–73. doi: 10.1016/s1097-2765(02)00569-5 [DOI] [PubMed] [Google Scholar]
- 66.Jeffery CJ. Moonlighting proteins. Trends Biochem Sci. 1999;24(1):8–11. doi: 10.1016/s0968-0004(98)01335-8 [DOI] [PubMed] [Google Scholar]
- 67.Piatigorsky J, O’Brien WE, Norman BL, Kalumuck K, Wistow GJ, Borras T, et al. Gene sharing by delta-crystallin and argininosuccinate lyase. Proc Natl Acad Sci U S A. 1988;85(10):3479–83. doi: 10.1073/pnas.85.10.3479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Piatigorsky J, Wistow GJ. Enzyme/crystallins: gene sharing as an evolutionary strategy. Cell. 1989;57(2):197–9. doi: 10.1016/0092-8674(89)90956-2 [DOI] [PubMed] [Google Scholar]
- 69.Sriram G, Martinez JA, McCabe ER, Liao JC, Dipple KM. Single-gene disorders: what role could moonlighting enzymes play? Am J Hum Genet. 2005;76(6):911–24. doi: 10.1086/430799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gancedo C, Flores CL, Gancedo JM. The Expanding Landscape of Moonlighting Proteins in Yeasts. Microbiol Mol Biol Rev. 2016;80(3):765–77. doi: 10.1128/MMBR.00012-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zha Z, Han X, Smith MD, Liu Y, Giguere PM, Kopanja D, et al. A Non-Canonical Function of Gbeta as a Subunit of E3 Ligase in Targeting GRK2 Ubiquitylation. Mol Cell. 2015;58(5):794–803. doi: 10.1016/j.molcel.2015.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rangarajan N, Gordy CL, Askew L, Bevill SM, Elston TC, Errede B, et al. Systematic analysis of F-box proteins reveals a new branch of the yeast mating pathway. J Biol Chem. 2019;294(40):14717–31. doi: 10.1074/jbc.RA119.010063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Heenan EJ, Vanhooke JL, Temple BR, Betts L, Sondek JE, Dohlman HG. Structure and function of Vps15 in the endosomal G protein signaling pathway. Biochemistry. 2009;48(27):6390–401. doi: 10.1021/bi900621w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hoffman CS. Propping up our knowledge of G protein signaling pathways: diverse functions of putative noncanonical Gbeta subunits in fungi. Sci STKE. 2007;2007(370):pe3. doi: 10.1126/stke.3702007pe3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14(2):115–32. doi: [DOI] [PubMed] [Google Scholar]
- 76.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14(4):417–9. doi: 10.1038/nmeth.4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li YY, Douillet C, Huang M, Beck R, Sumner SJ, Styblo M. Exposure to inorganic arsenic and its methylated metabolites alters metabolomics profiles in INS-1 832/13 insulinoma cells and isolated pancreatic islets. Arch Toxicol. 2020;94(6):1955–72. doi: 10.1007/s00204-020-02729-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zelena E, Dunn WB, Broadhurst D, Francis-McIntyre S, Carroll KM, Begley P, et al. Development of a robust and repeatable UPLC-MS method for the long-term metabolomic study of human serum. Anal Chem. 2009;81(4):1357–64. doi: 10.1021/ac8019366 [DOI] [PubMed] [Google Scholar]
- 81.Dunn WB, Broadhurst D, Begley P, Zelena E, Francis-McIntyre S, Anderson N, et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat Protoc. 2011;6(7):1060–83. doi: 10.1038/nprot.2011.335 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
For A) transcriptomics and B) metabolomics, X-axis shows PC1 with the percentage of explained variance and Y-axis shows PC2 with the percentage of explained variance. Data are scaled as detailed in Methods. Wildtype (black), asc1 (red), gpa2 (blue), gpr1 (purple). Low glucose (L, 0.05% glucose)-triangles, high glucose (H, 2% glucose)-circles.
(TIF)
Illustration of different modes of changes as captured by concentration analysis (A) and sensitivity analysis (B). The Y-axis represents either normalized gene counts or normalized peak area for metabolites. The X-axis represents different genotypes. A hypothetical wildtype is shown in black. The triangle represents measurement at low glucose and the square represents measurement at high glucose. The connecting grey line represents the response amplitude, detected by sensitivity analysis (wtH-wtL). Hypothetical mutants with increased response amplitude are colored green, while mutants with decreased response amplitude are colored red.
(TIF)
Map is color coded to delineate carbohydrate metabolism (blue), glycan biosynthesis and metabolism (cyan), amino acid metabolism (yellow), nucleotide metabolism (red), lipid metabolism (teal), metabolism of cofactors and vitamins (pink). Major species are highlighted with grey bounding box.
(PNG)
(PNG)
(PNG)
(PNG)
(PNG)
(PNG)
(PNG)
The map is color coded as in S3 Fig. Highlighted are the DEGs (black lines) and SPMs (black dots) for gpa2 integration analysis and gray boxes are used to delineate clusters associated with a specific pathway.
(PNG)
The map is color coded as in S3 Fig. Highlighted are the DEGs (black lines) and SPMs (black dots) for asc1 integration analysis and gray boxes are used to delineate clusters associated with a specific pathway.
(PNG)
Each gene has its own grid. Y-axis shows normalized transcript counts for low glucose (triangle), high glucose (square) in wildtype (black), asc1 (red) and gpa2 (blue).
(PNG)
Each gene has its own grid. Y-axis shows normalized transcript counts for low glucose (triangle), high glucose (square) in wildtype (black), asc1 (red) and gpa2 (blue).
(PNG)
The map is color coded as in S3 Fig. Highlighted are the s-DEGs (black lines) and s-SPMs (black dots) for gpa2 integration analysis and gray boxes are used to delineate clusters associated with a specific pathway.
(PNG)
The map is color coded as in S3 Fig. Highlighted are the s-DEGs (black lines) and s-SPMs (black dots) for asc1 integration analysis and gray boxes are used to delineate clusters associated with a specific pathway.
(PNG)
(XLSX)
(XLSX)
(CSV)
(CSV)
First block shows GSEA for transcriptomics with adjusted p-value <0.05, arranged in ascending order; second block shows MetaboAnalystR pathway enrichment analysis for metabolomics with combined p-value <0.05, arranged in ascending order; third block shows MetaboAnalystR joint pathway analysis with adjusted p-value <0.05, arranged in ascending order.
(XLSX)
(XLSX)
(CSV)
(CSV)
Sheet 1: Fold induction data for concentration analysis. Sheet 2: Induction difference data for sensitivity analysis. Sheet 3: Benjamini-Hochberg corrected Mann–Whitney U test results for concentration analysis. Sheet 4: Benjamini-Hochberg corrected Mann–Whitney U test results for sensitivity analysis.
(XLSX)
Data Availability Statement
RNA-seq data: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE171846 Metabolomics data: http://dx.doi.org/10.21228/M80D82 For review the GEO data can be accessed as follows: Go to https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE171846 Enter token qfedswaahrslxqf into the box For review the Metabolomics workbench can be accessed as follows: http://dev.metabolomicsworkbench.org:22222/data/DRCCMetadata.php?Mode=Study&StudyID=ST001786&Access=EsfC4623.