

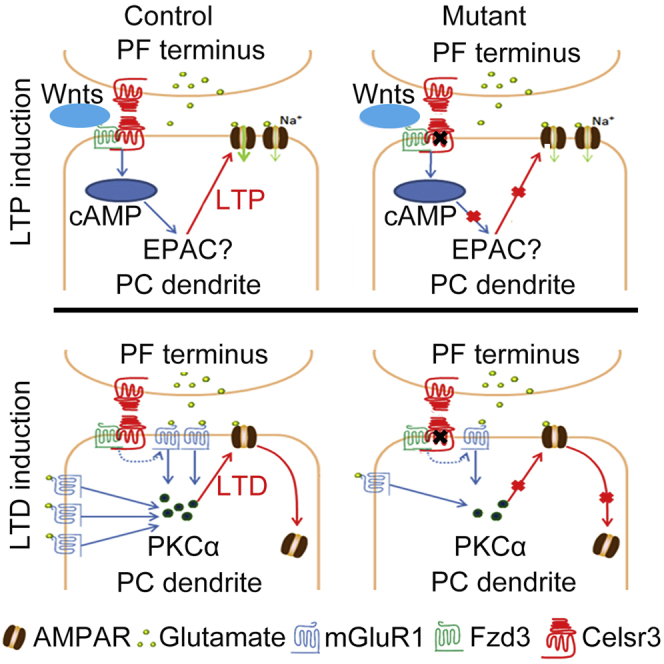
Summary
Atypical cadherin Celsr3 is critical for brain embryonic development, and its role in the postnatal cerebellum remains unknown. Using Celsr3-GFP mice, Celsr3 shows high expression in postnatal Purkinje cells (PCs). Mice with conditional knockout (cKO) of Celsr3 in postnatal PCs exhibit deficit in motor coordination and learning, atrophic PC dendrites, and decreased synapses. Whole-PC recording in cerebellar slices discloses a reduction frequency of mEPSC and defective postsynaptic plasticity (LTP and LTD) in Celsr3 cKO mutants. Wnt5a perfusion enhances LTP formation, which could be occluded by cAMP agonist and diminished by cAMP antagonist in control, but not in Celsr3 cKO or Fzd3 cKO cerebellar slices. Celsr3 cKO resulted in the failure of mGluR1 agonist-induced LTD and paired stimulation-induced PKCα overexpression in PC dendrites, and downregulation of mGluR1 expression compvared to controls. In conclusion, Celsr3 is required for PCs maturation and regulates postsynaptic LTP and LTD through Wnt5a/cAMP and mGluR1/PKCα signaling respectively.
Subject areas: Developmental neuroscience, Cognitive neuroscience
Graphical abstract
Highlights
-
•
Celsr3 cKO in postnatal PCs impairs mouse motor coordination and learning
-
•
Celsr3 inactivation affects the maturation of PC dendrites and synapses
-
•
Celsr3 is required for the cerebellar LTP induction via the Wnt5a/cAMP signaling
-
•
Celsr3 regulates the cerebellar LTD induction through the mGluR1/PKCα pathway
Developmental neuroscience; Cognitive neuroscience
Introduction
The formation of cerebellar circuitry is important for locomotor plasticity and adaptive learning. Cerebellar development is protracted and thereby at risk of genetic or environmental disruption. The cerebellum maintains wide connections with the cerebral cortex, brain stem, and spinal cord, and integrates various modalities of sensory and motor information to refine and modulate motor coordination, balance, posture, and skilled learning (Cerminara, et al., 2015; Ito, 2006). GABAergic Purkinje cells (PCs) are a key component of the cerebellar circuitry. Their somas are organized into a monolayer, their dendrites ramify sagittally in the molecular layer, and their axons innervate deep cerebellar nuclei that project to extracerebellar targets. Two excitatory glutamatergic inputs synapse on PCs: Climbing fibers (CFs) from the inferior olivary complex and parallel fibers (PFs) from granule cells (Jorntell and Hansel, 2006). Each PC receives monosynaptic inputs from one CF and each CF projects to about 10 PCs. PFs are bifurcations of granule cell axons, and contribute more than 100,000 synapses on PC dendrites, making it the strongest excitatory projection in the brain (Eccles, et al., 1966).
Both postsynaptic long-term potentiation (LTP) and long-term depression (LTD) occur at PF-PC synapses, and are induced by a low-frequency 1 Hz stimulation of PFs (LTP) and both PFs-CFs (LTD) (Hoxha, et al., 2016; Hirano, 2013; Sakurai, 1987; Ito, et al., 1982). LTP and LTD implicate α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid glutamate receptors (AMPARs), which are recruited (LTP) or internalized (LTD) in PC dendritic spines, generating different intracellular signals (Martin, et al., 2020; Gutierrez-Castellanos, et al., 2017). LTP and LTD can occur in vivo and may play different roles in motor learning: LTP is responsible for the gain-decrease vestibulo-ocular reflex (VOR) adaptation, whereas LTD mediates gain-increase VOR adaptation (Boyden and Raymond, 2003).
Celsr3 encodes an atypical cadherin receptor that plays critical roles in neural development (Goffinet and Tissir, 2017; Boutin, et al., 2012). Frizzled3 (Fzd3) protein acts as a receptor of Wnt ligands to regulate multiple processes of brain development (Wang, et al., 2016b). Studies in mutant mice showed that Celsr3 and Fzd3 cooperatively regulate axon wiring, probably by forming a receptor complex (Chai, et al., 2015). Celsr3 expression is downregulated after birth in most brain regions, but not in the cerebellum (Tissir and Goffinet, 2006). As the development of dendrites and acquisition of electrophysiological properties of PCs occur postnatally, we investigated the potential implication of Celsr3 in PC maturation. We studied Celsr3 expression in the postnatal cerebellum, generated conditional mutant mice with PC-specific Celsr3 inactivation, and studied cerebellar morphology, synaptic plasticity, and behavior. Our results indicate that Ceslr3 is required for optimal PC maturation and regulates the induction of PF-PC postsynaptic LTP and LTD, respectively through Wnt5a/cAMP, and mGluR1/PKCɑ signaling.
Results
Celsr3 is highly expressed in postnatal PCs
The maturation of PCs occurs during the first three postnatal weeks. Embryonic PC clusters transform into stripes around birth, and progressively form the adult array of parasagittal stripes around postnatal day (P) 20 (Hawkes, 2018); the refinement of PC dendrites lasts even longer (McKay and Turner, 2005). We studied Celsr3 expression in Celsr3-GFP postnatal mice, using double immunofluorescent staining against GFP (Figures 1A, 1E, and 1I) and the PC marker Calbindin (Maskey, et al., 2010). At P3, cerebellar folding became apparent. Calbindin-positive PCs were arrayed parallel to the cerebellar surface, and their efferent axons collected in the depth of the cerebellum (Figure 1B). When the GFP and Calbindin signals were compared (Figures 1A and 1B), merged images indicated that all Calbindin-positive cells were positive for Celsr3-GFP, whereas a few GFP-positive cells located more deeply were Calbindin negative (Figure 1D). At P10, somas of Calbindin-positive cells formed a monolayer and their dendrites ramified profusely in the molecular layer (Figure 1F). The GFP-positive signal was evident in PCs bodies and dense neurites (Figure 1E). A few GFP-positive, Calbindin-negative cells were scattered below the PC layer (Figure 1H); they were positive for Calretinin and negative for Parvalbumin (Figure S1), indicating that a small minority of Calretinin-positive granule cells also express Celsr3 (Rogers, 1989). At P21, Calbindin-positive PC dendrites filled the molecular layer, and individual neurites were readily identified (Figure 1J). The GFP-signal was restricted to PCs and the molecular layer (Figure 1I), and overlapped with Calbindin-positive labeling (Figure 1L). These results indicate that Celsr3 remains highly expressed in PCs cell bodies and neurites in postnatal cerebellum.
Figure 1.

Celsr3 is highly expressed in postnatal PCs
Parasagittal cerebellar sections from Celsr3-GFP mice were processed by double immunofluorescent staining with anti-Calbindin (red) and anti-GFP antibodies (green)
A-D: At postnatal day (P) 3, GFP-positive signal was identified in PCs and some fibers in deeper regions (A), with distribution similar to that of Calbindin-immunoreactivity (B), as confirmed in the merged figure (D).A few GFP-positive cells below the PC layer were negative for Calbindin (arrowheads in A, D).
E-H: At P10, GFP-immunoreactivity was visualized in cells and dendrites (E), and anti-Calbindin immunostaining showed that PC somas formed a monolayer and their dendrites invaded the molecular layer (F). Co-expression of Calbindin and Celsr3-GFP was confirmed in the merged image (H). A few GFP-positive cells were negative for Calbindin (indicated by arrowheads in E, H). I-L: At P21, co-expression of Celsr3-GFP(I) and Calbindin (J) shown in PCs (L). DAPI staining disclosed the nuclei in blue (C, G, K). Insets in D, H and L show enlarged images indicated by boxed regions respectively.
Celsr3cKO in postnatal PCs impairs motor coordination and learning
High expression of Celsr3 in postnatal PCs prompted us to assess its role in cerebellum-associated behaviors. To explore this, we produced Celsr3 cKO mice (see STAR Methods) in which Cre recombinase expression, driven by PCP2, is initiated in PCs around P10 (Zhang, et al., 2004). Celsr3 cKO animals survive and grow similarly to control animals. Behaviors were studied using young adult animals (2–3 months old). We first assessed locomotor performances in the rotarod test. In control animals, the time spent in the rotarod increased from the second day, with a significant difference between the first day and second day: 194.60 ± 18.88 s versus 248.00 ± 18.82 s (Figure 2A; P< 0.05, ANOVA post-hoc test; n = 8), indicating that animals learn to adapt to acceleration. In contrast, in Celsr3 cKO animals, the stay time on the accelerated rotarod increased only on the fifth (Figure 2A; P< 0.05, Student’s t-test; n = 8). At each time point, the latency before falls was significantly shorter in Celsr3 cKO than in control mice (Figure 2A, P< 0.01 at day1–3 and P< 0.05 at day 4, 5; Student’s t-test). This indicates slower adaptive learning and impaired motor coordination in the mutant.
Figure 2.

Celsr3 cKO mice show impaired motor coordination and adaptive motor learning
(A) In rotarod tests, the falling latency was significantly shorter at each time point in Celsr3 cKO than in control mice. Compared to day 1, stay duration time increased at day 2 in controls, but not until day 5 in mutants. ∗, P< 0.05; ∗∗, P< 0.01, ANOVA post-hoc test; n = 8 mice in each group.
(B) In Catwalk tests, the swing speed and stand index of hindlimbs, and the maximal contact areas of hindpaws were significantly decreased in Celsr3 cKO compared to controls. ∗, P< 0.05; Student’s t-test; n = 8 mice in each group.
(C) In grid tests which evaluate walking coordination, the error ratio was significantly increased in Celsr3 cKO versus control mice. ∗∗, P< 0.01; Student’s t-test; n = 6 mice in each group.
(D and E) There was no significant difference between Celsr3 cKO and control mice in the open-field test (D) and the grid strength test (E). P> 0.05; Student’s t-test; n = 8 mice in each group.
Walking coordination was assessed by Catwalk and grid tests. In the Catwalk, the swing speed and footprint area of hind paws were significantly decreased and the stand index of hind paws was increased in Celsr3 cKO animals compared to controls (Figure 2B; P< 0.05, Student’s t-test, n = 8 in each group). In grid tests, the error ratio was significantly increased in Celsr3 cKO mice (Figure 2C, 17 ± 2% (Celsr3 cKO) versus 9 ± 2% (control) in total 50 steps (P< 0.01, Student’s t-test; n = 6 in each group).
Spontaneous locomotor behavior, measured in the open field, showed no significant differences in the walking distance (30-min recording) in Celsr3 cKO and control mice: 12,712.86 ± 2797.05 cm versus 11,255.00 ± 2097.53 cm (Figure 2D; P> 0.05, Student’s t-test; n = 8). Similarly, the forelimb grip strength showed no significant differences between both groups: 88.89 ± 4.21 g (Celsr3 cKO) versus 88.22 ± 8.67 g (control) (Figure 2E; P> 0.05, Student’s t-test; n = 8 in each group).
Celsr3cKO mice have reduced synapse density on PC dendrites
Cerebellar foliation and cytoarchitecture appeared similar in Celsr3 cKO and control animals (Figures 3A and 3B), and the mean cerebellar volume was comparable in both groups (Figure 3K). Calbindin-positive PC somas were organized into monolayers and their dendrites filled the molecular layer similarly in control and mutant samples (Figures 3C and 3D). In addition, DiI placement in the cerebellar cortex labeled PC axons projecting to the deep nuclei in both groups (Figure S2). To assess the synapse density on PCs, we first carried out double immunofluorescent staining for Calbindin and the presynaptic marker synaptophysin 38 (SY38), or the postsynaptic density protein 95 (PSD95). In high magnification confocal images, SY38-positive and PSD95-positive profiles were widely distributed in somas and dendrites of Calbindin-positive PCs in both genotypes (Figures 3E–3H), but the density of SY38-positive and PSD95-positive profiles was significantly lower in Celsr3 cKO than in control mice (Figures 3I and 3J; P< 0.05, Student’s t-test, n = 4 animals in each group). We analyzed cerebellar tissues by transmission electron microscopy. As reported previously (Fan, et al., 2018), asymmetrical synapses on PC dendrites were readily identified in the two groups (Figures S3A, S3B, S3A′, and S3B′), with a significantly lower density of synapses in Celsr3 cKO versus control samples (Figure S3C; 13.7 ± 0.96/100 μm2 in control versus 7.9 ± 0.85/100 μm2 in mutant, n = 3, P< 0.05; Student’s t-test), but no differences of the thickness of postsynaptic densities (Figure S3D).
Figure 3.

The density of synaptic profiles is decreased in Celsr3 cKO mice
(A and B) Parasagittal cerebellar sections from control (A) and Celsr3 cKO mice (B) stained for Calbindin (red). Overall cerebellar architecture is similar in both genotypes.
(C and D) At higher magnification, Calbindin-positive PC somas and dendritic field are comparable in control (C) and mutant (D) mice. C and D correspond to boxed squares in A and B.
(E and F) Double immunostaining for Calbindin (red) and synaptophysin 38 (SY38, green) in control (E) and mutant (F). SY38-positive dots are abundant in the molecular and the PC layer, and many of them are Calbindin-positive, appearing in yellow.
(G and H) Double immunostaining was for Calbindin (red) and postsynaptic density protein 95 (PSD95, green) in the control (G) and the mutant (H).
(I and J) Decreased densities of SY38-posititive (I) and PSD95-positive (J) profiles in Celsr3 cKO mice compared to control mice. ∗, P< 0.05, Student’s t-test; n = 4 animals in each group. Gross morphology of the cerebellum is comparable in both genotypes groups (arrows).
Celsr3cKO mice show decreased mEPSC frequency and PC dendritic atrophy
Given the impaired locomotor performances and decreased synapse density in Celsr3 cKO mice, we asked whether Celsr3 inactivation in PCs may impact synaptic transmission. We recorded mEPSCs of individual PCs in acute cerebellar slices of young adult mice (8–10 weeks old). Compared to normal samples, there was a significant reduction of mEPSC frequency (Figures 4A and 4C; P< 0.05, Student’s t-test), but no differences of mEPSC amplitude (Figures 4B and 4C; P> 0.05, Student’s t-test) in mutant PCs (17 cells from 6 mutant mice and 15 cells from 7 control mice), indicating a deficiency of synaptic transmission. The morphology of individual PCs was examined using biocytin-injection followed with streptavidin-FITC immunofluorescent staining (Figure 4F). Analysis of 3D-reconstructed PCs showed a significant decrease of dendritic area, volume, and spine density in mutant versus control cells (Figures 4D–4F; P< 0.05 in dendritic area and volume and P< 0.01 in spine density; Student’s t-test, n = 6 and 5 PCs in the mutant and control respectively).
Figure 4.

Celsr3 cKO mice show decreased mEPSC frequency and PC dendritic atrophy
(A-C)Whole-cell recording of PCs in acute cerebellar slices. There is significant reduction of mEPSC frequency (A), but not amplitude (B) in Celsr3 cKO compared to control slices. ∗P< 0.05, Student’s t-test; 15 cells from 7 control mice and 17 cells from 6 mutant mice.
(C) Examples of recordings.
(D-F)Analysis of biocytin-injected PCs. There is a significant reduction of dendritic area (D), dendritic volume (E) and spine density (F) in Celsr3 cKO compared to control mice. Examples of PCs from control and Celsr3 cKO mice (F). ∗, P< 0.05; ∗∗, P< 0.01; Student’s t-test; n = 5 PCs in the control and n = 6 PCs in Celsr3 cKO.
Inactivation of Celsr3 in PCs prevents induction of PF-PC LTP and LTD
To test the role of Celsr3 in cerebellar plasticity, in acute cerebellar slices, stimulating PFs at 1 Hz for 300 s induced LTP in control but not in Celsr3 cKO mice (Figure 5A; P< 0.05, n = 15 PCs from 6 control mice; n = 9 PCs from 4 mutant mice, Student’s t-test), whereas the paired plus ratio (PPR) was comparable in both groups (Figure 5C; P> 0.05, n = 8 PCs from 3 mice in each group, Student’s t-test), indicating that presynaptic neurotransmitter release is unaffected in Celsr3 cKO mice (Fioravante and Regehr, 2011). Similarly, paired stimulation of PFs and CFs at 1 Hz for 300 s induced LTD in control but not in Celsr3 cKO mice (Figure 5B; P< 0.05, n = 15 PCs from 6 control mice; n = 9 PCs from 4 mutant mice, Student’s t-test), with no difference of the PPR (Figure 5D; P> 0.05, n = 8 PCs from 3 mice, Student’s t-test). Furthermore, the amplitude of CF-PC EPSC and the density of VGlut2-positive particles (CF terminals) on Calbindin-positive PC dendrites were comparable in control and Celsr3 cKO mice (Figure S4). Altogether, these results demonstrate that Celsr3 in PCs is required for induction of PF-PC postsynaptic plasticity, and acts in a cell-autonomous manner.
Figure 5.

Celsr3 mutant PCs show impaired postsynaptic plasticity
LTP or LTD induced in acute cerebellar slices
(A) Left panel: percentage of baseline EPSC amplitudes in control (black) and Celsr3 cKO (red) plotted before (first 10 min) and after (starting at 15 min) PF stimulation. PF stimulation induced an increase of EPSC amplitudes (LTP) in the control but not in the mutant. Right panel: representative traces of EPSCs before (black) and after (red) stimulation. The time of LTP induction was indicated by the arrow. P< 0.05, n = 15 PCs from 6 mice in the control; n = 9 PCs from 4 mice in the mutant, Student’s t-test.
(B) Left panel: percentage of baseline EPSC amplitudes in the control (black) and the Celsr3 cKO (red) plotted before (first 15 min) and after (starting at 20 min) combined stimulation of PFs and CFs. Stimulation induced a decrease of EPSC amplitudes (LTD) in the control but not in the mutant. Right panel, representative traces of EPSCs before (black) and after (red) stimulation. The time of LTD induction was indicated by the arrow. P< 0.05, n = 15 PCs from 6 mice in the control; n = 9 PCs from 4 mice in the mutant, Student’s t-test.
(C and D) The paired pulse ratios (PPRs) were comparable before (Pre-LTP, Pre-LTD) and after (Post-LTP, Post-LTD) the induction of LTP and LTD in control (C) and mutants (D). Representative traces of EPSCs before (black) and after (red) LTP or LTD induction.P> 0.05, n = 8 PCs from 3 mice in each group, Student’s t-test.
Facilitation of LTP induction by Celsr3 requires Wnt5a/cAMP signaling
The second messenger cyclic adenosine monophosphate (cAMP) is a key modulator of the PF-PC postsynaptic LTP, which is triggered by G-protein-coupled postsynaptic receptors such as adrenergic receptors (Martin, et al., 2020). To test whether cAMP activation induces LTP in PCs, the cAMP agonist forskolin was perfused in cerebellar slices during baseline recording of PCs. LTP was similarly induced in both control and mutant slices (Figure 6A; P> 0.05, n = 6 PCs from 3 mice in each group, Student’s t-test), indicating that cAMP-dependent LTP induction can proceed independently of Celsr3. But could cAMP activation itself be regulated by Celsr3?
Figure 6.

Wnt5a/Celsr3/cAMP signaling is required for LTP induction
(A) Perfusion of the cAMP agonist forskolin induces EPSP typical features of LTP in both control and Celsr3 cKO groups (P> 0.05, n = 6 PCs from 3 mice in each group, Student’s t-test). Washing out of forskolin does not diminish LTP immediately.
(B) LTP cannot be induced in Fzd3 cKO PCs compared to the control (P< 0.05, n = 6 PCs from 3 mice in each group, Student’s t-test). The time of LTP induction is indicated by the arrow.
(C) Perfusion of Wnt5a induces LTP in controls, but not in Fzd3 or Celsr3 mutants (P< 0.05, n = 6 PCs from 3 mice in each group, Student’s t-test). In the control, washing out of Wnt5a does not diminish LTP immediately.
(D) Wnt5a and forskolin perfusion do not enhance the LTP induction upon single drug perfusion, and LTP induction by forskolin and Wnt5a is prevented by pre-perfusion of the cAMP antagonist-SQ22536. P< 0.05, n = 6 PCs from 3 mice in each group, Student’s t-test.
(E) The PPR is unaffected by administration of forskolin, Wnt5a or SQ22536. P> 0.05, n = 6 PCs from 3 mice in each group, Student’s t-test.
Wnt5a activates cAMP signaling through Fzd3 receptor (Hansen, et al., 2009) and induces LTP in hippocampal neurons (Zhang, et al., 2015; Cerpa, et al., 2010). We tested whether Wnt5a perfusion could induce LTP of PCs. Interestingly, Wnt5a induced LTP efficiently in control but not in Celsr3 cKO slices (Figure 6C; P< 0.05, n = 6 PCs from 3 mice in each group, Student’s t-test), suggesting that Celsr3 inactivation in PCs prevents Wnt5a-induced cAMP-dependent LTP induction.
Previous studies indicated that Celsr3 and Fzd3 form receptor complexes that regulate developmental events, including some driven by non-canonical Wnt ligands (Wang, et al., 2016a; Chai, et al., 2015; Chai, et al., 2014). We wondered whether Fzd3 inactivation in PCs might also impair Wnt5a-induced LTP. To test this, we generated Fzd3 cKO mice using PCP2-Cre (see STAR Methods). Similar to the finding in Celsr3 cKO animals (Figure 5A), the PF-PC postsynaptic LTP could not be induced by stimulating PFs (Figure 6B; P< 0.05, n = 6 PCs from 3 mice in each group, Student’s t-test) or by Wnt5a perfusion in Fzd3 cKO compared to control slices (Figure 6C; P< 0.05, n = 6 PCs from 3 mice in each group, Student’s t-test).
To test whether cAMP is a downstream component of Wnt5a-Celsr3/Fzd3 signaling during LTP induction, cerebellar slices were subjected to Wnt5a and forskolin perfusion with or without pre-perfusion of the cAMP inhibitor SQ22536. The effect of forskolin and Wnt5a could be occluded by each other (Figure 6D). In addition, we found that pre-perfusion of SQ22356 impaired Wnt5a and forskolin-induced LTP in PCs of normal slices (Figure 6D; P< 0.05, n = 6 PCs from 3 mice in each group, Student’s t-test), indicating that LTP induction by Wnt5a requires cAMP activity. Of note, perfusion of forskolin, Wnt5a or SQ22536 did not affect the PPR in PFs-PCs (Figure 6E; P> 0.05, n = 6 PCs from 3 mice in each group, Student’s t-test), confirming that Wnt5a-Celsr3/Fzd3-cAMP signaling regulates LTP induction postsynaptically.
Celsr3 regulates the PF-PC postsynaptic LTD through the mGluR1/PKCα pathway
PF-PC LTD induction requires mGluR1-dependent synaptic signaling and enhanced ionotropic glutamate receptor internalization via PKCα phosphorylation (Hoxha, et al., 2016). To explore whether Celsr3 regulates mGluR1 signaling during LTD induction, we perfused acute cerebellar slices with the mGluR1 selective agonist DHPG. As predicted, in control slices, DHPG efficiently induced PF-PC LTD (Figure 7A), while the PPR was unaffected (Figure 7A; P> 0.05, n = 5 PCs from 3 mice in each group, Student’s t-test). By contrast, in Celsr3 cKO slices, DHPG perfusion failed to induce LTD in PCs (Figure 7A; P< 0.05, n = 9 PCs from 4 mice in the control; n = 6 PCs from 3 mice in the mutant, Student’s t-test).
Figure 7.

Celsr3 inactivation impairs mGluR1-dependent LTD induction
(A) Left panel, plots of EPSC baseline percentages show mGluR1 agonist DHPG-induced LTP in the control (black) but not in the Celsr3 cKO (red) (P< 0.05, n = 9 PCs from 4 mice in the control; n = 6 PCs from 3 mice in the mutant, Student’s t-test). Right panel: Representative traces of EPSCs before (black) and after (red) LTD induction. The PPR was comparable with and without DHPG perfusion (P> 0.05, n = 5 PCs from 3 mice in each group, Student’s t-test).
(B) In control slices, LTD induction is accompanied by a significant increase of PKCα (red) overexpression in PC dendrites (green, biocytin-labelling) as compared to sham stimulation in the control (left panel). But no PKCα overexpression is observed after PF or sham stimulation in Celsr3 cKO slices (right panel). Quantification in the histogram (∗∗∗, P< 0.001; n = 4 PCs from 3 mice in each group; Student’s t-test).
(C) PCs were immunostained for Calbindin (green) and mGluR1 (red). A significant decrease of mGluR1-postitive particle density in spines is seen in the Celsr3 cKO compared to the control (∗, P< 0.05; Student’s t-test; n = 5).
(D) Western blots of cerebellar samples show decreased mGluR1 protein in the mutant compared to the control.(∗, P< 0.05; Student’s t-test; at least triplicate).
By which mechanism could Celsr3 inactivation impair mGluR1 activation? A possibility could be that Celsr3 regulates mGluR1 distribution in PCs. To test this, we studied mGluR1 expression in Calbindin-labeled PCs (Figure 7C). In PC dendrites, the density of total and spine-associated mGluR1-positive profiles was significantly decreased in Celsr3 mutant versus control cells (Figure 7C; P< 0.05, n = 3 mice in each group, Student’s t-test). In line with this, Western blot analysis of cerebellar samples showed that mGluR1 protein levels were lower in mutant than control tissues (Figure 7D; P< 0.05, n = 3 mice in each group, Student’s t-test).
LTD can be induced upon combined PC depolarization and glutamate application, and PC depolarization induces PKCα translocation to plasma membrane (Tsuruno and Hirano, 2007). To compare PKCα translocation during LTD induction in control and mutant cells, we selected well separated biocytin-labeled PCs in the same slice, and submitted one of them to combined stimulation to induce LTD, while the other was given sham stimulation. We fixed slices immediately after induction and carried out PKCα immunostaining. Biocytin labeled dendrites of individual PCs in all samples (Figure 7B). In control animals, there was a significant increase of PKCα immunoreactivity in the dendritic field of PCs with LTD induction compared to those with sham stimulation (Figure 7B; P< 0.001, n = 4 PCs from 3 mice in each group, Student’s t-test). In contrast, in Celsr3 mutant samples, PKCα immunoreactivity was similarly weak upon PF/CF paired and sham stimulation (Figure 7B; P> 0.05, n = 4 PCs from 3 mice in each group, Student’s t-test), indicating a failure of PKCα overexpression. In addition, the basal expression of PKCα was comparable in control and Celsr3 cKO mutant cerebellar slices (Figure S5). Thus, paired stimulation of PFs and CFs triggers PKCα overexpression in PC dendritic field in a Celsr3-dependent manner.
Discussion
Atypical cadherin Celsr3 plays critical roles during brain development, in collaboration with Fzd3 (Feng, et al., 2016; Chai, et al., 2014; Qu, et al., 2014; Feng, et al., 2012; Qu, et al., 2010; Ying, et al., 2009; Zhou, et al., 2008; Tissir, et al., 2005). Its putative role during postnatal brain maturation has not been addressed in detail, because Celsr3 expression is sharply downregulated after birth, except in a few regions such as the cerebellum. Here, we show that Celsr3 is required for postnatal cerebellar Purkinje cell maturation and synaptic plasticity, which has potential implications in neural disorders implicating the cerebellum.
To avoid issues related to abnormal prenatal development, we studied mutant mice with PC-specific inactivation (Celsr3 cKO), driven by PCP2-Cre which is activated from P10 (Zhang, et al., 2004; Tomomura, et al., 2001). In those mice, Celsr3 expression during neurogenesis and migration is unaffected. Accordingly, mutant PCs are normally organized into a monolayer and the architecture of the cerebellum is preserved. In contrast, there is impaired PC maturation at later stages, leading to decreased dendritic volume, surface, and synapse density. In whole cell recordings, mutant PC’s mEPSC frequency is decreased. Both decreased release of presynaptic neurotransmitter and reduced synapses on PCs can result in the reduction of mEPSC frequency (Choy, et al., 2018; Chen, et al., 2010). In Celsr3 cKO mice, the PPR of PF-PC is not affected, and Celsr3 is mainly inactivated in PCs according to Celsr3 expression pattern. These suggest that Celsr3 might mainly modulate excitatory synaptic transmission to PCs through influencing synapse maturation. In line with this, it is reported that Celsr3 knockout results in a dramatic loss of glutamatergic synapse formation in hippocampal pyramidal neurons (Thakar, et al., 2017).
In rotarod, Catwalk, and grid tests, Celsr3 cKO mice display abnormalities of motor coordination, adaptive learning, and walking balance. However, PC axons project normally to the deep cerebellar nuclei, so that those deficits are likely due to impairment of synaptic function. This is supported by morphological data, spontaneous mEPSC recording, and the defective induction of the PF-PC postsynaptic LTP and LTD in Celsr3 cKO mice. The unaffected PPR in Celsr3 cKO slices suggests that Celsr3 modulates synaptic plasticity through the postsynaptic component.
Neither LTP nor LTD can be induced in Celsr3 cKO mutants. In the cerebellum, PF-PC LTP is induced by AMPARs insertion in dendritic spine membranes. Multiple intracellular molecules are involved in the process, such as protein phosphatases (PP1, PP2A, and PP2B), T-type Ca2+ channels, and nitric oxide (Gutierrez-Castellanos, et al., 2017; Belmeguenai and Hansel, 2005; Kakegawa and Yuzaki, 2005; Lev-Ram, et al., 2002). Recent studies show that cAMP signaling activates the GluA2/GluA3-dependent AMPARs by enhancing their conductance, which induces LTP (Martin, et al., 2020; Gutierrez-Castellanos, et al., 2017). In our study, the cAMP agonist forskolin was equally efficient in inducing LTP in control and Celsr3 cKO slices. This implies that Celsr3 acts upstream of cAMP, or that Celsr3-dependent LTP induction does not rely on cAMP signaling. Wnt5a acts as an activator of cAMP signaling through Fzd receptors (Ding, et al., 2017; Hansen, et al., 2009) and induces LTP in the hippocampus (Zhang, et al., 2015; Cerpa, et al., 2010). In our setting, a short perfusion of a low concentration of Wnt5a was sufficient to induce LTP in control PCs, and this was inhibited by forskolin, indicating that Wnt5a acts upstream of cAMP signaling. However, Wnt5a failed to induce LTP in mutant PCs. Taken together these results indicate that Wnt5a/Celsr3/cAMP is a candidate signaling cascade for LTP induction in PCs. Previous studies showed that Wnt5a/Celsr3 regulated glutamatergic synapse formation in hippocampal slices, albeit at a high Wnt5a concentration (100 ng/mL), and that long Wnt5a administration (36 h) is detrimental to synapse structure (Thakar, et al., 2017). In contrast, our data indicate that a low Wnt5a concentration (20 ng/mL) and a short-time perfusion (LTP appears after 5 min) contribute to promote synaptic plasticity. In addition, the Wnt5a-induced LTP in PCs is different from that in hippocampus. The induction of Wnt5a-dependent LTP in hippocampus is slow, taking approximately 20 min (Cerpa, et al., 2011; Cerpa, et al., 2010). This might be related to the fact that the LTP induction of PCs needs a moderate increase of intracellular calcium in the postsynaptic component (Zang and De Schutter, 2019), whereas the LTP in hippocampus depends on a large increase of intracellular calcium (Graupner and Brunel, 2012).
Studies in knockout mice showed that Celsr3 and Fzd3 share similar roles in brain development, and it was proposed that both proteins form a receptor complex to steer axon pathfinding driven by Wnt ligands (Wang, et al., 2016a; Chai, et al., 2015; Chai, et al., 2014). Our finding that LTP could not be induced in Fzd3 cKO mutant PCs byPF stimulation or Wnt5a perfusion supports that mechanism.
PF-PC LTD induction requires a postsynaptic signaling cascade implicating mGluR1 and PKCɑ, and can be blocked by inhibiting mGluR1 function using antibodies, or by inactivating PKCɑ (Leitges, et al., 2004; Shigemoto, et al., 1994). Celsr3 inactivation in PCs results in a decrease of mGluR1 distribution in dendritic spines and reduced protein expression. This may attribute to PC dendrite atrophy in Celsr3 cKO mutants since mGluR1 is enriched in PC dendritic spines (Mansouri, et al., 2015). Alternatively, Celsr3 inactivation may directly disturb mGluR1 expression and distribution, which is supported by the decreased intensity of mGluR1-positive particles on PC spines in the mutant (shown in Figure 7C). The impaired aggregation of PKCɑ in dendrites upon the paired stimulation of PFs and CFs is also consistent with the previous report that activating mGluR1 could induce PKCɑ overexpression in dendrites of rod bipolar cells (Hellmer, et al., 2018). During LTD induction, the stimulation could induce a quick translocation of PKCɑ in a short time window using PKCɑ-GFP transgenic mice (Tsuruno and Hirano, 2007). In Celsr3 cKO mice, no change of PKCɑ immunofluorescent intensity after the simulation may indirectly reflect the impaired translocation from somas to dendrites as the previous report (Hirono, et al., 2001). Our Celsr3 mutant mice not only had defective LTD, but disclosed behavioral anomalies similar to those observed in mGluR1 mutants (Aiba, et al., 1994). This suggests strongly that Celsr3 is required for normal mGluR1 distribution in PCs, and regulates LTD induction via mGluR1/PKCɑ signaling.
In conclusion, expression of Celsr3 in postnatal PCs is required for the maturation of their dendrites and for PF-PC postsynaptic plasticity. Celsr3 regulates postsynaptic LTP via Wnt5a/cAMP signaling, probably in complex with Fzd3, and LTD through mGluR1/PKCα signaling.
Limitations of study
We acknowledge some limitations in our study. First, we did not observe the translocation of PKCα in the recording cerebellar slices. Second, the present study does not clarify how Celsr3 regulates the expression and distribution of mGluR1in PCs.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-mGluR1 | CST | Cat#12551s; RRID:AB_2797953 |
| Rabbit polyclonal anti-PKCα | Sigma | Cat#P4334; RRID:AB_477345 |
| Rabbit polyclonal anti-βIII-Tubulin | Abcam | Cat#ab18207; RRID:AB_444319 |
| Rabbit polyclonal anti-Calretinin | Merk Millipore | Cat#AB5054; RRID:AB_2068506 |
| Mouse monoclonal anti-Parvalbumin | Merk Millipore | Cat#MAB1572; RRID:AB_2174013 |
| Mouse polyclonal anti-Calbindin | Sigma | Cat#C9848; RRID:AB_476894 |
| Chicken polyclonal anti-green fluorescent protein | Abcam | Cat#ab13970; RRID:AB_300798 |
| Mouse monoclonal anti-postsynaptic density protein 95 | Merk Millipore | Cat# MAB1596; RRID:AB_2092365 |
| Mouse monoclonal anti-synaptophysin 38 | Abcam | Cat#ab8049; RRID:AB_2198854 |
| Rabbit polyclonal anti-mGluR1 | Abcam | Cat#ab27199; RRID:AB_470927 |
| Guinea pig polyclonal anti-Vglut2 | Merk Millipore | Cat#ab2251; RRID:AB_1587626 |
| Chemicals, peptides, and recombinant proteins | ||
| Forskolin | MCE | Cat# HY-15371 |
| SQ22536 | MCE | Cat# HY-100396 |
| NBQX | Sigma | Cat# N183 |
| DHPG | MCE | Cat# HY-12598A |
| Biocytin | Sigma | Cat# B4261 |
| Wnt5a | R&D | Cat#645-WN |
| TTX | Tocris | Cat#1078 |
| Streptavidin-FITC | BD Biosciences | Cat#434311 |
| Picrotoxin | Abcam | Cat#ab120315 |
| Experimental models: Organisms/strains | ||
| Mouse: PCP2-Cre | Jackson Laboratory | JAX: 010536 |
| Mouse: Celsr3-GFP | (Ying, et al., 2009) | NA |
| Mouse: Fzd3f/f and Fzd3KI/+ | (Qu, et al., 2014) | NA |
| Mouse: Celsr3+/− | (Tissir, et al., 2005) | NA |
| Mouse: Celsr3f/f | (Zhou, et al., 2008) | NA |
| Software and algorithms | ||
| pClamp | Molecular devices | https://www.moleculardevices.com/applications/patch-clamp-electrophysiology#relatedproducts |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Imaris | Bitplane | https://imaris.oxinst.cn/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed and will be fulfilled by the Lead Contact, Libing Zhou (tlibingzh@jnu.edu.cn)
Materials availability
This study did not generate new unique reagents.
Data and code availability
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Animals
All animal experimental procedures were approved by the Laboratory Animal Ethics Committee at Jinan University. Animals were housed under 12-h light/dark cycle (light on from 8:00 A.M. to 8:00 P.M.) with free access to water and food. PCP2-Cre transgenic mice, in which Cre driven upon PCP2 expression is restricted in cerebellar PCs (Zhang, et al., 2004), were purchased from the Jackson Laboratory (Stock number: 010536). PCP2-Cre mice were crossed with Celsr3+/− or Fzd3KI/+ mice to yield double heterozygous males PCP2-Cre;Celsr3+/− or PCP2-Cre;Fzd3KI/+, which were then crossed with Celsr3f/f or Fzd3f/f females to obtain PCP2-Cre;Celsr3f/- (Celsr3 cKO) and PCP2-Cre;Fzd3 f/KI (Fzd3 cKO) mice, in which only one Celsr3 or Fzd3 “floxed” allele is inactivated by Cre recombinase in order to increase inactivation efficiency (Zhou, et al., 2008). Littermate Celsr3f/+ or Fzd3f/+ mice were used as controls. Celsr3-GFP transgenic mice (Ying, et al., 2009) (provided by Qiang Wu, Shanghai Jiaotong University) were used to detect Celsr3 protein expression with anti-GFP immunostaining. Genotypes were determined by PCR. Males and females (2–3 months, 25–30 g) were used indiscriminately.
Method details
Behavioral studies of adult mice (2–3 months, 25–30 g) were performed by experimenters blind to animal genotypes.
Open-field test
Spontaneous motor activity was measured in an open field made of transparent Plexiglas panels (50 × 50 × 35 cm) with an open top, under bright illumination (500 lux). Each mouse was placed in a corner of a compartment and its ambulation was recorded for 30 min. Mice were studied for 3 consecutive days. Data were analyzed using EnthoVision XT 7.0 (Noldus, Netherlands).
Rotarod test
Motor coordination was evaluated using the rotarod test (Ugo Basile, Italy). After a 3-min adaption on the stationary rod, animals were subjected to the rotating rod with increasing speed from 4 to 40 rpm, and the falling latency was monitored for a maximum of 5 min. Animals were tested for 5 consecutive days, three trials per day with a 30-min interval. The average of three trials in each animal was taken as one experiment.
Grid test
Ataxic behavior was measured in the grid test apparatus, a 25 × 25 cm wire mesh with 1 cm2 grids positioned 1 cm above a glass floor. Full (score: 2) and partial foot faults (score: 1) were scored if the mouse slipped on the wire mesh floor (partial) and stepped on the glass plate below (full). The foot fault rate was calculated with 50 steps (scores/50∗100%).
Grip strength test
Grip force of mouse forelimb was assessed by a 47200 Grip Strength Meter (Ugo Basile, Italy). Mice were tested 3 times with a 3-min interval. The results were averaged as one experiment.
Catwalk
Gait was analyzed using the Catwalk system (Noldus Information Technology, Leesburg, VA). Briefly, mice traversed a glass walkway (109 × 15 × 0.6 cm) with dark plastic walls spaced 15 cm apart in a dark room. Light from an encased fluorescent bulb was internally reflected within the glass walkway and scattered when the plantar surface of the paw contacted the floor, producing paw prints. Paw prints were recorded by a high-speed CCD camera mounted below the walkway. Trials in which the animal stopped or changed direction were excluded from analysis, and three uninterrupted trials were analyzed and averaged to obtain the final gait analysis values. Paw print data were analyzed using the Catwalk software (v 7.1).
Immunostaining
After anesthesia, mice were perfused with 4% paraformaldehyde (PFA) and cerebellar tissues were dissected out and fixed in the same fixative overnight at 4°C. Sagittal sections (25-μm thickness) were prepared using a cryostat (Leica), washed in 0.01M phosphate buffered saline (PBS) 3 times, blocked in 3% BSA, 1% goat serum and 0.01% X-triton in 0.01M PBS for 1 h, and incubated with primary antibodies overnight at 4°C. The primary antibodies were: Rabbit anti-Calretinin (1:400, AB5054, Invitrogen), mouse anti-Parvalbumin (1:1000, MAB1572, Millipore), mouse anti-Calbindin (1:1000, C9848, Sigma), rabbit anti-Calbindin (1:1000; ab11426, Abcam), chicken anti-green fluorescent protein (GFP; 1:800; ab13970, Abcam), mouse anti-postsynaptic density protein 95 (PSD95; 1:500; MAB1596, Abcam), guinea pig anti-vGlut2 (1:2000; ab2251, Millipore) and mouse anti-synaptophysin 38 (SY38; 1:500; ab8049, Abcam). Signal was detected with Alexa Fluor 546 or 488 fluorescent secondary antibodies (1:1000, Invitrogen). After double immunofluorescent staining for Calbindin and PSD95 or SY38, images in the molecular layer were collected with a 63× oil objective and confocal microscopy (LSM700, Zeiss, Germany), and PSD95-positive or SY38-positive profiles were counted using ImageJ. In each animal, the mean density calculated using 5-10 sections was taken as one sample, and 4 animals were used in mutant and control groups respectively.
For imaging of thick slices after recording, biocytin-injected neurons were immunostained with streptavidin-FITC (1:200; BD Biosciences Pharmingen) and images of labeled individual PCs were acquired using a confocal microscope (LSM 700, Zeiss, Germany) with a 20× objective. Sequential optical sections of 1024 × 1024 pixels were taken at 1.0-μm intervals along the z-axis, and three-dimensional (3D) reconstruction of whole cells was made using Imaris 8.0 (Bitplane, USA) to calculate the entire dendritic surface area, the dendritic filament, volume and the spine density. To study the expression of mGluR1and PKCα, thick slices (300 μm) were incubated with rabbit anti-mGluR1 (1:200; Abcam) and anti-PKCα (1:5000; Sigma) for 96 h at 4°C, and the images were scanned using a confocal microscope (LSM 700, Zeiss, Germany).
Transmission electron microscopy
Animals were perfused with 2% PFA and 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4), followed by dissection of cerebellar tissues and overnight post-fixation. One-mm thick vibratome sections were prepared and areas containing PC dendrite were selected for trimming into 1-mm3 samples. After further fixation in 2.5% glutaraldehyde for 6 h, the samples were washed 3 times in distilled water, incubated in 1% uranium acetate for 1 h, followed with washing in distilled water, dehydration in graded alcohol and propylene oxide and infiltration overnight in a mixture of propylene oxide and Epon (1:1; TAAB, United Kingdom). Samples were then embedded in Epon, with polymerization for 48 h at 60°C, and 60–80 nm ultrathin sections were prepared using an ultramicrotome. Transferred to a copper mesh, the sections were stained with lead citrate and images were captured with a transmission electron microscope (Hitachi, H-7650) equipped with an AMT 2k CCD camera. In the superficial molecular layer, 20 fields were selected under 17,500х magnification and synapses were counted blindly in each field (6∗9 μm2) by two different observers. To measure the thickness of the postsynaptic density, 5 fields were selected and photographed under 37,000х magnification.
Immunoblotting
Cerebellar samples were homogenized in RIPA lysis buffer (containing PMSF, 1:50) and protein concentration was measured using the BCA assay (Bio-Rad). Samples corresponding to 30 μg proteins were analyzed by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to PVDF membrane (Immobilon P; Millipore) by electroblotting. Membranes were blocked with 5% low-fat milk and 0.01% Tween 20 in PBS and incubated with primary antibodies overnight at 4°C. Signal was disclosed using enhanced chemiluminescence (Pierce Biotechnology). Primary antibodies were: rabbit anti-mGluR1 (1:1000, CST), rabbit anti βIII-Tubulin (1:10,000, Abcam).
Whole-cell patch-clamp recording and single cell tracing
Acute parasagittal cerebellar slices (300-μm thickness) were prepared from adult animals (8–10 weeks, 25–30 g), in cold extracellular solution. The solution for slice preparation and perfusion contained (in mM): 125 NaCl, 2.5 KCl, 2 MgCl2, 2 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, 10 glucose, 300–310 mOsm/L, and was bubbled with 95% O2 and 5% CO2. After a recovery period of at least 30 min at 35°C, slices were moved to a submersion-type recording chamber and perfused with the extracellular solution at 27 ± 1°C (flow rate: 1–2 mL/min). PCs in lobule IV-V were visualized under an upright microscope (Eclipse FN1, Nikon, Japan). Whole-cell patch-clamp recordings were made using a MutiClamp 700B amplifier (pClamp, Molecular devices, USA). Patch pipettes (resistance 4–6 MΩ) were filled with the intracellular solution containing (in mM): 135 CsMeSO3, 8 CsCl, 4 Na2-phosphocreatine, 2 Mg-ATP, 0.4 Na-GTP, 10 HEPES, 0.25 EGTA, 0.2% biocytin (sigma), pH 7.2–7.3 with KOH, 280–300 mOsm/L. The PCs were held at −70 mV, and the miniature excitatory postsynaptic currents (mEPSCs) were sampled at 10 kHz and filtered with Bessel-filter at 4kHz, using clamp 10.0 (pClamp, Molecular devices, USA). Picrotoxin (0.1 mM) and tetrodotoxin (TTX, 1 μM, Tocris) were included in the extracellular solution during mEPSC recording. Experiments were not considered further if total series of resistance changed more than 20%. The mEPSC detection was made using a sliding template algorithm with Clamp Fit 10 (pClamp, Molecular devices, USA). Noise in recorded data was minimized with a Gaussian low pass filter. The criteria for inclusion were a 6-pA amplitude threshold and a rise time (10–90%) longer than 1 ms. Overlapping events were rejected. The traces of mEPSCs from control and mutant mice were averaged for comparison.
LTP and LTD induction
For the induction of LTP and LTD, the intracellular solution contained (in mM): 133 KmeSO3, 7.4 KCl, 0.3 MgCl2, 10 HEPES, 0.1 EGTA, 3 MgATP, 0.3 Na2GTP, pH 7.3 with KOH, 270–290 mOsm/L, and 0.1 mM picrotoxin was added to block GABA effects. PCs were held in the voltage clamp at −70 mV and signals were acquired at 10 kHz using a Multiclamp 700B amplifier with low-pass filter at 4 kHz. Cells with more than 25 mΩ of access resistance were discarded, and input and access resistances were monitored for stability throughout the recording. Stimulating electrodes were placed in the upper tier of the molecular layer for PF stimulation, and in the granule layer for CF stimulation. A 0.05-Hz pulse was delivered to PFs for baseline recording. After the baseline was stable, LTP was induced by stimulating PF at 1 Hz for 300 s, and LTD was induced by a paired stimulation of PFs and CFs at 1 Hz for 300 s. Two test responses to a PF pulse (with 50 ms interval) were evoked every 20 s in voltage-clamp mode for paired-pulse ratio (PPR). The PPR was calculated as the ratio of the amplitude of the second evoked excitatory postsynaptic current (eEPSC) to that of the first. PPRs were averaged per 10 min and normalized for presentation. For the CF-PC EPSC recording, the stimulating electrode was placed in the granule cell layer, 50–100 μm away from PC somas, and PCs were held at −70 mV in the presence of 0.5 μM NBQX (N183, Sigma) to maintain voltage clamp.
Drug application
During LTP recording, the following membrane permeable drugs were added to the extracellular solution: 50 μM forskolin (cAMP activator, MCE), 100 μM DHPG (mGluR1 selective agonist, MCE), 20 ng/mL Wnt5a (R&D Systems) and/or 50 μM SQ22536 (cAMP antagonist, MCE).
Quantification and statistical analysis
Results are expressed as mean ± SEM. ANOVA post-hoc test was used for analysis of rotarod tests and Student’s t-test was used for the rest analysis. Statistical significance is noted as ∗, ∗∗ or ∗∗∗, P< 0.05, P< 0.01 or P< 0.001, respectively.
Acknowledgments
We wish to thank Meizhi Wang for technical support and Dongdong Yu for mouse breeding. This work was supported by the following grants:National Natural Science Foundation of China (81971148, 31771133 and 31811530148, L.Z.), Key project of Natural Science Foundation of Guangdong province (2016A030311053, L.Z.), Outstanding Scholar Program of Bioland Laboratory (Guangzhou Regenerative Medicine and Health Guangdong Laboratory; 2018GZR110102002), Program of Introducing Talents of Discipline to Universities (B14036).
Author contributions
Conceptualization, Q.Z. and L.Z.; Methodology, Q.Z., J.Q., and W.Z.; Investigation, Q.Z., J.Q., Y.L., W.Z., and S.H.; Writing – Original Draft, Q.Z.; Writing –Review & Editing, L.Z. and F.T.; Funding Acquisition, L.Z.; Resources, L.Z and Y.B.; Supervision, L.Z.
Declaration of interests
The authors declare no competing financial interests.
Published: July 23, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102812.
Supplemental information
References
- Aiba A., Kano M., Chen C., Stanton M.E., Fox G.D., Herrup K., Zwingman T.A., Tonegawa S. Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell. 1994;79:377–388. [PubMed] [Google Scholar]
- Belmeguenai A., Hansel C. A role for protein phosphatases 1, 2A, and 2B in cerebellar long-term potentiation. J.Neurosci. 2005;25:10768–10772. doi: 10.1523/JNEUROSCI.2876-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutin C., Goffinet A.M., Tissir F. Celsr1-3 cadherins in PCP and brain development. Curr.Top. Dev. Biol. 2012;101:161–183. doi: 10.1016/B978-0-12-394592-1.00010-7. [DOI] [PubMed] [Google Scholar]
- Boyden E.S., Raymond J.L. Active reversal of motor memories reveals rules governing memory encoding. Neuron. 2003;39:1031–1042. doi: 10.1016/s0896-6273(03)00562-2. [DOI] [PubMed] [Google Scholar]
- Cerminara N.L., Lang E.J., Sillitoe R.V., Apps R. Redefining the cerebellar cortex as an assembly of non-uniform Purkinje cell microcircuits. Nat. Rev. Neurosci. 2015;16:79–93. doi: 10.1038/nrn3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerpa W., Farias G.G., Godoy J.A., Fuenzalida M., Bonansco C., Inestrosa N.C. Wnt-5a occludes Abeta oligomer-induced depression of glutamatergic transmission in hippocampal neurons. Mol. Neurodegener. 2010;5:3. doi: 10.1186/1750-1326-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerpa W., Gambrill A., Inestrosa N.C., Barria A. Regulation of NMDA-receptor synaptic transmission by Wnt signaling. J. Neurosci. 2011;31:9466–9471. doi: 10.1523/JNEUROSCI.6311-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai G., Goffinet A.M., Tissir F. Celsr3 and Fzd3 in axon guidance. Int. J. Biochem.Cell Biol. 2015;64:11–14. doi: 10.1016/j.biocel.2015.03.013. [DOI] [PubMed] [Google Scholar]
- Chai G., Zhou L., Manto M., Helmbacher F., Clotman F., Goffinet A.M., Tissir F. Celsr3 is required in motor neurons to steer their axons in the hindlimb. Nat. Neurosci. 2014;17:1171–1179. doi: 10.1038/nn.3784. [DOI] [PubMed] [Google Scholar]
- Chen S.X., Tari P.K., She K., Haas K. Neurexin-neuroligin cell adhesion complexes contribute to synaptotropic dendritogenesis via growth stabilization mechanisms in vivo. Neuron. 2010;67:967–983. doi: 10.1016/j.neuron.2010.08.016. [DOI] [PubMed] [Google Scholar]
- Choy J.M.C., Agahari F.A., Li L., Stricker C. Noradrenaline increases mEPSC frequency in pyramidal cells in layer II of rat barrel cortex via calcium release from presynaptic stores. Front.Cell Neurosci. 2018;12:213. doi: 10.3389/fncel.2018.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S., Xu Z., Yang J., Liu L., Huang X., Wang X., Zhuge Q. The involvement of the decrease of astrocytic Wnt5a in the cognitive decline in minimal hepatic encephalopathy. Mol. Neurobiol. 2017;54:7949–7963. doi: 10.1007/s12035-016-0216-5. [DOI] [PubMed] [Google Scholar]
- Eccles J.C., Llinas R., Sasaki K. The mossy fibre-granule cell relay of the cerebellum and its inhibitory control by Golgi cells. Exp. Brain Res. 1966;1:82–101. doi: 10.1007/BF00235211. [DOI] [PubMed] [Google Scholar]
- Fan W.J., Yan M.C., Wang L., Sun Y.Z., Deng J.B., Deng J.X. Synaptic aging disrupts synaptic morphology and function in cerebellar Purkinje cells. Neural Regen. Res. 2018;13:1019–1025. doi: 10.4103/1673-5374.233445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J., Xian Q., Guan T., Hu J., Wang M., Huang Y., So K.F., Evans S.M., Chai G., Goffinet A.M. Celsr3 and Fzd3 organize a pioneer neuron scaffold to steer growing thalamocortical axons. Cereb.Cortex. 2016;26:3323–3334. doi: 10.1093/cercor/bhw132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J., Xu Y., Wang M., Ruan Y., So K.F., Tissir F., Goffinet A., Zhou L. A role for atypical cadherin Celsr3 in hippocampal maturation and connectivity. J. Neurosci. 2012;32:13729–13743. doi: 10.1523/JNEUROSCI.1965-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioravante D., Regehr W.G. Short-term forms of presynaptic plasticity. Curr.Opin.Neurobiol. 2011;21:269–274. doi: 10.1016/j.conb.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffinet A.M., Tissir F. Seven pass cadherins CELSR1-3. Semin.Cell Dev. Biol. 2017;69:102–110. doi: 10.1016/j.semcdb.2017.07.014. [DOI] [PubMed] [Google Scholar]
- Graupner M., Brunel N. Calcium-based plasticity model explains sensitivity of synaptic changes to spike pattern, rate, and dendritic location. Proc. Natl. Acad. Sci. U. S. A. 2012;109:3991–3996. doi: 10.1073/pnas.1109359109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Castellanos N., Da Silva-Matos C.M., Zhou K., Canto C.B., Renner M.C., Koene L.M.C., Ozyildirim O., Sprengel R., Kessels H.W., De Zeeuw C.I. Motor learning requires purkinje cell synaptic potentiation through activation of AMPA-receptor subunit GluA3. Neuron. 2017;93:409–424. doi: 10.1016/j.neuron.2016.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen C., Howlin J., Tengholm A., Dyachok O., Vogel W.F., Nairn A.C., Greengard P., Andersson T. Wnt-5a-induced phosphorylation of DARPP-32 inhibits breast cancer cell migration in a CREB-dependent manner. J. Biol. Chem. 2009;284:27533–27543. doi: 10.1074/jbc.M109.048884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes R. The Ferdinando Rossi memorial lecture: zones and stripes-pattern formation in the cerebellum. Cerebellum. 2018;17:12–16. doi: 10.1007/s12311-017-0887-0. [DOI] [PubMed] [Google Scholar]
- Hellmer C.B., Clemons M.R., Nawy S., Ichinose T. A group I metabotropic glutamate receptor controls synaptic gain between rods and rod bipolar cells in the mouse retina. Physiol. Rep. 2018;6 doi: 10.14814/phy2.13885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano T. Long-term depression and other synaptic plasticity in the cerebellum. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2013;89:183–195. doi: 10.2183/pjab.89.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirono M., Kishimoto Y., Sakai I., Miyazawa T., Kishio M., Inoue H., Nakao K., Ikeda M., Kawahara S., Kirino Y. Phospholipase Cbeta4 and protein kinase Calpha and/or protein kinase CbetaI are involved in the induction of long term depression in cerebellar Purkinje cells. J. Biol. Chem. 2001;276:45236–45242. doi: 10.1074/jbc.M105413200. [DOI] [PubMed] [Google Scholar]
- Hoxha E., Tempia F., Lippiello P., Miniaci M.C. Modulation, plasticity and pathophysiology of the parallel fiber-Purkinje cell synapse. Front.Synaptic Neurosci. 2016;8:35. doi: 10.3389/fnsyn.2016.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M. Cerebellar circuitry as a neuronal machine. Prog.Neurobiol. 2006;78:272–303. doi: 10.1016/j.pneurobio.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Ito M., Sakurai M., Tongroach P. Climbing fibre induced depression of both mossy fibre responsiveness and glutamate sensitivity of cerebellar Purkinje cells. J. Physiol. 1982;324:113–134. doi: 10.1113/jphysiol.1982.sp014103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorntell H., Hansel C. Synaptic memories upside down: bidirectional plasticity at cerebellar parallel fiber-Purkinje cell synapses. Neuron. 2006;52:227–238. doi: 10.1016/j.neuron.2006.09.032. [DOI] [PubMed] [Google Scholar]
- Kakegawa W., Yuzaki M. A mechanism underlying AMPA receptor trafficking during cerebellar long-term potentiation. Proc. Natl. Acad. Sci. U. S. A. 2005;102:17846–17851. doi: 10.1073/pnas.0508910102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitges M., Kovac J., Plomann M., Linden D.J. A unique PDZ ligand in PKCalpha confers induction of cerebellar long-term synaptic depression. Neuron. 2004;44:585–594. doi: 10.1016/j.neuron.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Lev-Ram V., Wong S.T., Storm D.R., Tsien R.Y. A new form of cerebellar long-term potentiation is postsynaptic and depends on nitric oxide but not cAMP. Proc.Natl. Acad.Sci.U. S. A. 2002;99:8389–8393. doi: 10.1073/pnas.122206399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri M., Kasugai Y., Fukazawa Y., Bertaso F., Raynaud F., Perroy J., Fagni L., Fagni L., Kaufmann W.A., Watanabe M., Shigemoto R., Ferraguti F. Distinct subsynaptic localization of type 1 metabotropic glutamate receptors at glutamatergic and GABAergic synapses in the rodent cerebellar cortex. Eur. J. Neurosci. 2015;41(2):157–167. doi: 10.1111/ejn.12779. [DOI] [PubMed] [Google Scholar]
- Martin R., Garcia-Font N., Suarez-Pinilla A.S., Bartolome-Martin D., Ferrero J.J., Lujan R., Torres M., Sanchez-Prieto J. Beta-adrenergic receptors/Epac signaling increases the size of the readily releasable pool of synaptic vesicles required for parallel fiber LTP. J.Neurosci. 2020;40:8604–8617. doi: 10.1523/JNEUROSCI.0716-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskey D., Pradhan J., Kim H.J., Park K.S., Ahn S.C., Kim M.J. Immunohistochemical localization of calbindin D28-k, parvalbumin, and calretinin in the cerebellar cortex of the circling mouse. Neurosci.Lett. 2010;483:132–136. doi: 10.1016/j.neulet.2010.07.077. [DOI] [PubMed] [Google Scholar]
- McKay B.E., Turner R.W. Physiological and morphological development of the rat cerebellar Purkinje cell. J. Physiol. 2005;567:829–850. doi: 10.1113/jphysiol.2005.089383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y., Glasco D.M., Zhou L., Sawant A., Ravni A., Fritzsch B., Damrau C., Murdoch J.N., Evans S., Pfaff S.L. Atypical cadherins Celsr1-3 differentially regulate migration of facial branchiomotor neurons in mice. J. Neurosci. 2010;30:9392–9401. doi: 10.1523/JNEUROSCI.0124-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y., Huang Y., Feng J., Alvarez-Bolado G., Grove E.A., Yang Y., Tissir F., Zhou L., Goffinet A.M. Genetic evidence that Celsr3 and Celsr2, together with Fzd3, regulate forebrain wiring in a Vangl-independent manner. Proc. Natl. Acad. Sci. U. S. A. 2014;111:E2996–E3004. doi: 10.1073/pnas.1402105111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J.H. Immunoreactivity for calretinin and other calcium-binding proteins in cerebellum. Neuroscience. 1989;31:711–721. doi: 10.1016/0306-4522(89)90435-1. [DOI] [PubMed] [Google Scholar]
- Sakurai M. Synaptic modification of parallel fibre-Purkinje cell transmission in in vitro Guinea-pig cerebellar slices. J. Physiol. 1987;394:463–480. doi: 10.1113/jphysiol.1987.sp016881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemoto R., Abe T., Nomura S., Nakanishi S., Hirano T. Antibodies inactivating mGluR1 metabotropic glutamate receptor block long-term depression in cultured Purkinje cells. Neuron. 1994;12:1245–1255. doi: 10.1016/0896-6273(94)90441-3. [DOI] [PubMed] [Google Scholar]
- Thakar S., Wang L., Yu T., Ye M., Onishi K., Scott J., Qi J., Fernandes C., Han X., Yates J.R., 3rd Evidence for opposing roles of Celsr3 and Vangl2 in glutamatergic synapse formation. Proc. Natl. Acad. Sci. U. S. A. 2017;114:E610–E618. doi: 10.1073/pnas.1612062114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissir F., Bar I., Jossin Y., De Backer O., Goffinet A.M. Protocadherin Celsr3 is crucial in axonal tract development. Nat. Neurosci. 2005;8:451–457. doi: 10.1038/nn1428. [DOI] [PubMed] [Google Scholar]
- Tissir F., Goffinet A.M. Expression of planar cell polarity genes during development of the mouse CNS. Eur. J. Neurosci. 2006;23:597–607. doi: 10.1111/j.1460-9568.2006.04596.x. [DOI] [PubMed] [Google Scholar]
- Tomomura M., Rice D.S., Morgan J.I., Yuzaki M. Purification of Purkinje cells by fluorescence-activated cell sorting from transgenic mice that express green fluorescent protein. Eur. J. Neurosci. 2001;14:57–63. doi: 10.1046/j.0953-816x.2001.01624.x. [DOI] [PubMed] [Google Scholar]
- Tsuruno S., Hirano T. Persistent activation of protein kinase Calpha is not necessary for expression of cerebellar long-term depression. Mol.Cell Neurosci. 2007;35:38–48. doi: 10.1016/j.mcn.2007.01.016. [DOI] [PubMed] [Google Scholar]
- Wang W., Jossin Y., Chai G., Lien W.H., Tissir F., Goffinet A.M. Feedback regulation of apical progenitor fate by immature neurons through Wnt7-Celsr3-Fzd3 signalling. Nat. Commun. 2016;7:10936. doi: 10.1038/ncomms10936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Chang H., Rattner A., Nathans J. Frizzled receptors in development and disease. Curr.Top. Dev. Biol. 2016;117:113–139. doi: 10.1016/bs.ctdb.2015.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying G., Wu S., Hou R., Huang W., Capecchi M.R., Wu Q. The protocadherin gene Celsr3 is required for interneuron migration in the mouse forebrain. Mol.Cell Biol. 2009;29:3045–3061. doi: 10.1128/MCB.00011-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang Y., De Schutter E. Climbing fibers provide graded error signals in cerebellar learning. Front. Syst. Neurosci. 2019;13:46. doi: 10.3389/fnsys.2019.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.L., Zhang J., Li S.F., Lei L., Xie H.Y., Deng F., Feng J.C., Qi J.S. Wnt-5a prevents Abeta-induced deficits in long-term potentiation and spatial memory in rats. Physiol. Behav. 2015;149:95–100. doi: 10.1016/j.physbeh.2015.05.030. [DOI] [PubMed] [Google Scholar]
- Zhang X.M., Ng A.H., Tanner J.A., Wu W.T., Copeland N.G., Jenkins N.A., Huang J.D. Highly restricted expression of Cre recombinase in cerebellar Purkinje cells. Genesis. 2004;40:45–51. doi: 10.1002/gene.20062. [DOI] [PubMed] [Google Scholar]
- Zhou L., Bar I., Achouri Y., Campbell K., De Backer O., Hebert J.M., Jones K., Kessaris N., de Rouvroit C.L., O'Leary D. Early forebrain wiring: genetic dissection using conditional Celsr3 mutant mice. Science. 2008;320:946–949. doi: 10.1126/science.1155244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.