

Abstract
The field of epigenetics has exploded over the last two decades revealing an astonishing level of complexity in the way genetic information is stored and accessed in eukaryotes. This expansion of knowledge, very much ongoing, has been made possible by the availability of ever more sensitive and precise molecular tools. This review focuses on the increasingly important role that chemistry plays in this burgeoning field. In an effort to make these contributions more accessible to the non-specialist, we group available chemical approaches into those that allow the covalent structure of the protein and DNA components of chromatin to be manipulated, those that allow the activity of myriad factors that act on chromatin to be controlled, and those that allow the covalent structure and folding of chromatin to be characterized. The application of these tools is illustrated through a series of case studies which highlight how the molecular precision afforded by chemistry is being used to establish causal biochemical relationships at the heart of epigenetic regulation.
Keywords: Chemical biology, Chromatin, Epigenetics, Chemical tools
1. INTRODUCTION
Genetic information in eukaryotic cells is stored and maintained in the nucleoprotein complex called chromatin (Figure 1a). The fundamental repeating unit of chromatin is the nucleosome, a disk-shaped structure consisting of 146 base pairs of DNA spooled around an octameric protein core consisting of two copies each of histones H2A, H2B, H3 and H4 (1). At its most essential function, chromatin regulates physical access to genetic information in the cell. This occurs both through the inherent occupation of DNA within nucleosomes as well as the assembly of higher-order structures, which are promoted by a variety of inter-nucleosome interactions. The intrinsically disordered ‘tail’ domains of the four core histones mediate contacts between adjacent nucleosomes, and linker histones further compact chromatin by binding the DNA between nucleosomes. The positioning and spacing of nucleosomes is also controlled by ATP-dependent chromatin remodelers, whose activities can lead to the translocation of nucleosomes along DNA or to histone eviction and exchange (2). Additional architectural proteins promote long-range interactions that contribute to chromatin packing, looping, and folding into topologically associated domains (TADs) (3).
Figure 1.
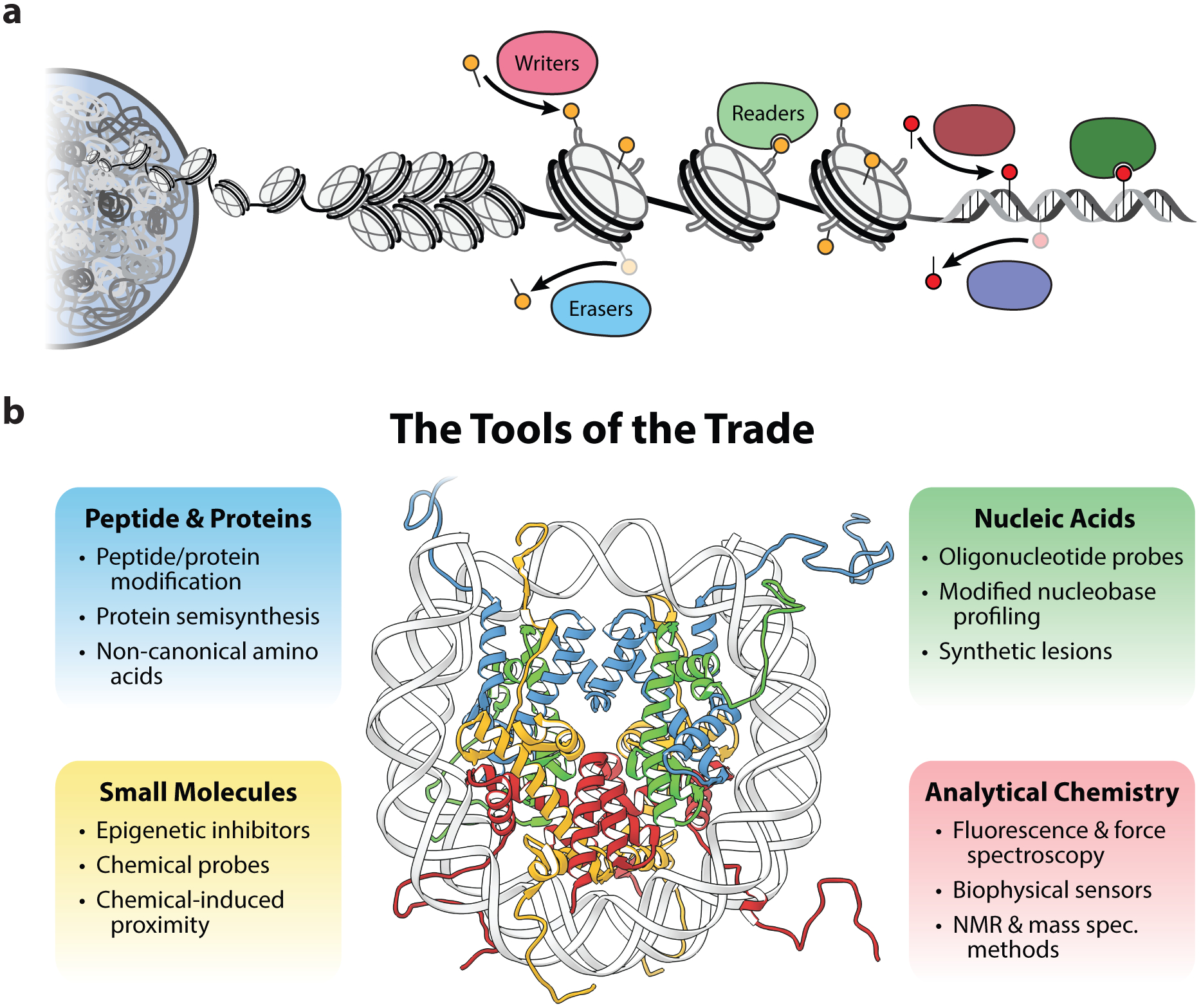
The chemical tools in chromatin biology. (a) Schematic representation of chromatin architecture in the eukaryotic genome and the proteins that install, remove, and sense epigenetic modifications on histone proteins and DNA. (b) Overview of the chemical tools for studying chromatin. The structure of the mononucleosome depicts DNA (gray) wrapped around two copies each of histones H2A (yellow), H2B (red), H3 (blue) and H4 (green). PDB, 1kx5.
To accommodate the diversity of genetic processes in the cell, including gene transcription, DNA replication, and DNA repair, chromatin structure and composition are dynamic features that must be precisely controlled. This regulation is largely achieved by the direct chemical modification of histones, DNA, and other chromatin-associated proteins. Histone post-translational modifications (PTMs or “marks”), in particular, comprise a functionally rich chemical landscape, ranging in complexity from the addition of small methyl and acetyl groups to the attachment of entire proteins, namely ubiquitin and SUMO (4). These marks act as signaling hubs that orchestrate nuclear processes by directly affecting chromatin structure or by recruiting chromatin effector proteins (5). The cellular machinery that ‘writes’, ‘erases’, or ‘reads’ histone PTMs, therefore, are crucial chromatin regulators, and mutations in these proteins are a common feature of human cancers and other disorders (6). DNA and histone marks can also be inherited during cell replication, allowing these functional states to persist via a form of epigenetic memory (7).
Due to the importance of covalent modifications to histone proteins and DNA in epigenetic regulation, chemistry has played a perhaps inevitable role in understanding these processes. Following the discoveries that histones inhibited RNA synthesis in nuclear extracts and post-translational acetylation of histones relieved this inhibition (8–10), biochemical efforts turned to the characterization of histone acetyltransferase (HAT) and histone deacetylase (HDAC) activity. The substrate scope of these enzymes was unknown, however, due to the difficulty in preparing homogenously acetylated histone proteins and peptides. Merrifield, Allfrey, and coworkers solved this challenge using burgeoning methods in solid-phase peptide synthesis (SPPS), and determined the substrate specificity of histone deacetylase (HDAC) activity on the N-terminal tail of histone H4 (11, 12). This seminal work established a template for utilizing synthetic peptides to study histone PTMs with chemical precision, an approach that remains highly productive and continues to benefit from technological advancements in peptide and protein chemistry. Despite this early progress in characterizing HDAC activity, it took over fifteen years for the first HDAC enzyme to be identified, a breakthrough that helped usher in another powerful chemical biology strategy, namely the use of a small molecule probe to fish a target protein out of a cell extract. In classic work, Schreiber and colleagues conducted the total chemical synthesis of Trapoxin A, a natural product known to inhibit HDAC activity (13), allowing them to incorporate an affinity handle into the molecule for the purposes of isolating the protein target, in this case HDAC1 (13, 14).
Since these foundational studies utilizing chemistry to understand epigenetic processes, the field of chemical biology has rapidly matured, yielding an extensive set of molecular tools and techniques. In this review we describe the chemical methods that have emerged as central to the development and understanding of chromatin biology. We group these methods into four primary categories based on their utilization of: peptide and protein synthesis, nucleic acid synthesis, small molecule probes, and analytical chemistry measurements (Figure 1b). A short discussion on emergent chemical biology tools for precise epigenomic engineering in a cellular context is also included. In an effort to illustrate the power of these tools, we also discuss a set of experimental case studies that demonstrates the implementation of these methods. Note that the works covered here are not intended to be an exhaustive summary of the various applications of these methods to chromatin biology, for which we refer the readers to recent reviews (15–18). Furthermore, while the development of genomic techniques based on DNA sequencing have been instrumental in the characterization of chromatin-related processes, these methods are outside the scope of this review (19).
2. TOOLS OF THE TRADE
2.1. Tools based on peptide and protein synthesis
Posttranslational modifications of chromatin effector proteins and histones are crucial regulatory components for the maintenance of epigenetic states in the cell. Although enzymatic approaches can be used to generate proteins with PTMs, this requires that the corresponding writer protein be purified, and such enzyme activity is often promiscuous or does not react to completion, leading to heterogeneous products. The generation of modified peptides through SPPS offers extensive control of peptide composition, and has been absolutely central for investigating the broad spectrum of PTMs found on histone proteins (15, 20). Through the course of these studies, however, the complexity of many chromatin effector proteins has been elucidated, in particular the ability to engage multiple sites on a nucleosome or multiple nucleosomes simultaneously. Fortunately, these developments have come hand in hand with advancements in protein chemistry techniques that can generate full-length modified histones. These approaches, when coupled to established biochemical methods for assembling MNs and chromatin arrays, allow for the interrogation of complex epigenetic processes on physiologically relevant substrates.
The four canonical histones have only a single conserved cysteine residue on H3, which can be innocuously mutated to alanine. Thus, histones can be modified in a site-specific manner through the introduction and labeling of cysteine residues, for which a diverse set of sulfhydryl-specific chemical probes are available (21). This relatively straightforward approach has been used extensively for biochemical and physiological applications in chromatin biology (Figure 2a). Crosslinkers installed at various positions throughout histones have elucidated intra- and internucleosomal contacts, interactions between chromatin arrays, as well as the histone-DNA interactions that contribute to overall chromatin architecture (15). The Cys-mediated introduction of fluorophores, spin labels, and paramagnetic probes has allowed for the further scrutiny of the stability and dynamics of nucleosomes and chromatin arrays (discussed further in section 2.4) (21). Cysteine alkylation provides access to a set of molecular analogs for mono-, di-, and trimethyl-lysine, differing only by a methylene to sulfide substitution (22). By analogy, the nucleophilicity of the cysteine sulfhydryl group has been exploited to generate acyl-lysine analogs such as acetylation and ubiquitylation (15). Methods have also been developed that allow cysteine to be converted into ‘desulfurized’ PTM analogs that proceed via dehydroalanine or free radical intermediates (21, 23). Finally, the temporary coupling of an asymmetrically modified pair of histone proteins via the formation of a disulfide linkage has enabled the assembly of heterotypic nucleosomes (24), a class of nucleosome substrates with distinct biochemical roles in the cell (25).
Figure 2.
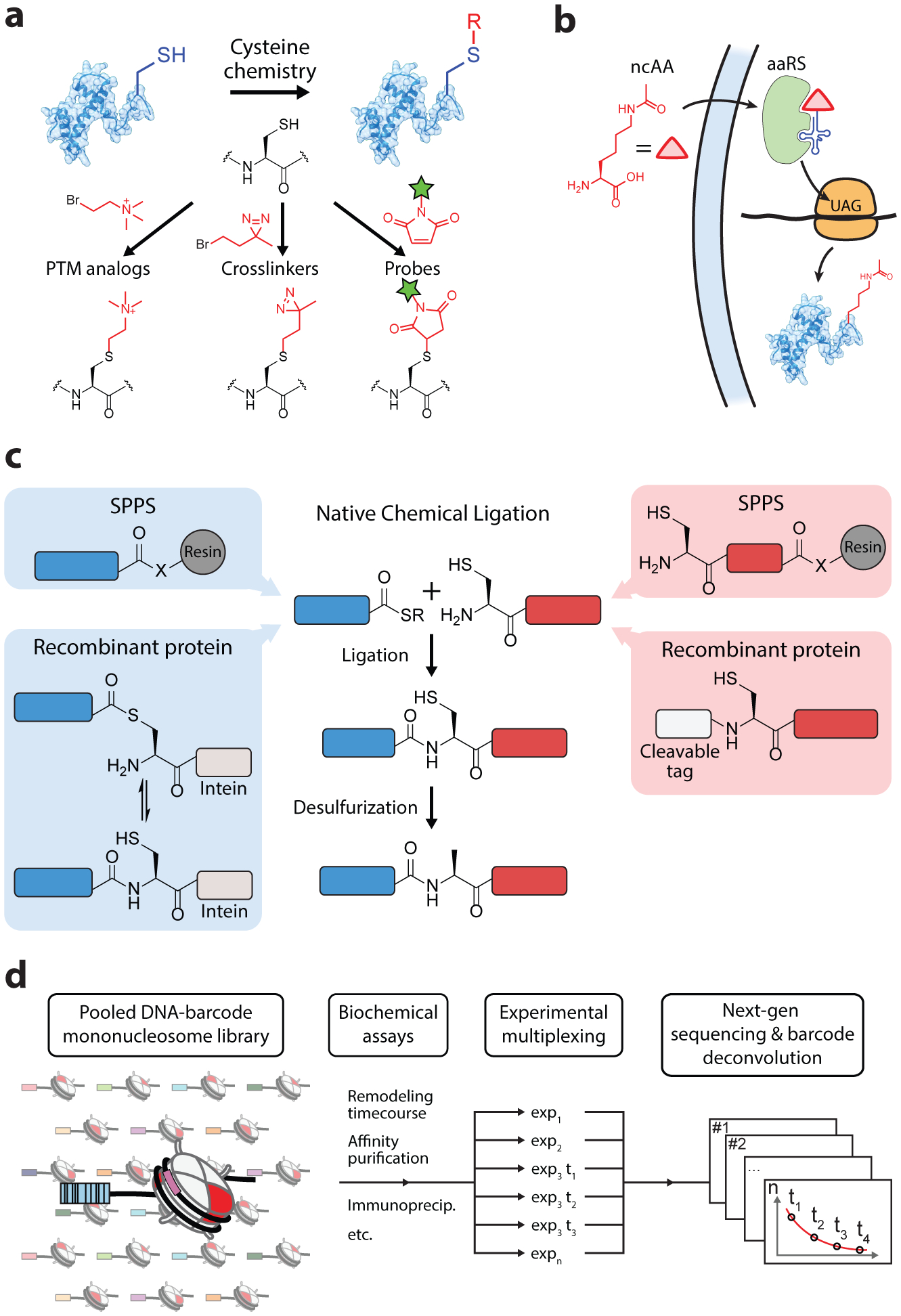
Tools based on peptide and protein synthesis. (a) Cysteine labeling for modification of proteins, which is particularly useful for histones due to their almost complete lack of Cys residues. Popular examples of reagents for cysteine alkylation and maleimide conjugation are depicted. (b) Schematic of the incorporation of a non-canonical amino acid (ncAA) into endogenous protein synthesis (acetyllysine is depicted as a prominent example). An engineered aminoacyl-tRNA synthetase (aaRS) and tRNA pair allow for the recoding of stop codons to the ncAA. (c) Protein semisynthesis by native chemical ligation. (d) Schematic of high-throughput biochemistry approaches made possible by the combination of protein semisynthesis and next-generation sequencing technologies.
Genetic code expansion with non-canonical amino acids offers an alternative approach for producing site-specifically labeled recombinant proteins. The strategy involves the decoding of an mRNA stop codon (typically the amber codon) with a complementary tRNA that is charged with a non-canonical amino acid (ncAA) (Figure 2b). During protein synthesis the ribosome reads through the amber codon, inserting the ncAA into the growing polypeptide chain. Implementation of this approach requires an engineered tRNA synthetase/tRNA pair that is orthogonal to cellular translational machinery and efficiently charges the tRNA with the ncAA of choice. Amber suppression has been used to incorporate a broad set of chemical functionalities that include fluorophores, crosslinkers, and biorthogonal handles (26). Additionally, ncAAs that contain PTMs, including acetyl-lysine, acyl-lysine derivatives, phosphoserine, and a protected monomethyl-lysine, have been co-translationally installed into histone proteins (27–30). Importantly, these applications have been extended for site-specific labeling of histones in live cells, offering a unique approach for synthetic control of the epigenome (31–35).
A significant challenge in solid-phase peptide synthesis is the exponentially increasing inefficiency that scales with peptide length, making the total synthesis of even modest-length proteins, like histones, often impractical. This limitation has been circumvented by protein semisynthesis strategies that utilizes a convergent approach to produce proteins from multiple segments. The workhorse methodology in protein semisynthesis is native chemical ligation (NCL), a reaction in which a peptide with a C-terminal thioester (α-thioester) condenses with a second peptide containing an N-terminal cysteine to form a native amide bond (Figure 2c) (36). The α-thioester can be generated during SPPS, and proteins with an N-terminal Cys can be produced via expression of a fusion protein and subsequent treatment with a site-selective protease (20). The α-thioester can also be generated recombinantly by fusion to inteins, a class of proteins that perform a splicing reaction that catalyzes their own excision from flanking polypeptides sequences (20). The protein splicing reaction can be interrupted with exogenous thiols, trapping an α-thioester intermediate which can subsequently be used for protein ligation, an approach termed expressed protein ligation (EPL) (37). Alternative strategies for generating amide bonds have also been developed through transpeptidation with sortase and subtiligase enzymes (20, 38, 39). Thus, protein semisynthesis offers an efficient marriage between the chemical control of peptide synthesis and the robustness of recombinant protein production. These methods have been used for the production of hundreds of semisynthetic protein substrates (20) and, in particular, have been crucial in providing synthetic access to an array of PTMs and chemical probes on histone proteins. A notable set of applications in histone semisynthesis is the production of ubiquitylated histone substrates, which we discuss in detail as a case study below (section 3.1).
With mature methodologies for producing modified histones in place, there has been an increasing demand for strategies to perform efficient and high-throughput biochemical screening of these substrates once incorporated into nucleosomes. The combination of protein chemistry techniques with DNA sequencing technology has led to the development of barcoded nucleosome libraries, which enable single-pot biochemical experiments of large numbers of nucleosome substrates, each containing a distinct histone PTM signature (Figure 2d) (40, 41). Such library experiments have been used to assess the activity of histone PTM reader and writer proteins, as well as ATP-dependent chromatin remodelers (40–43).
2.2. Tools based on nucleic acid synthesis
Early work to understand the fundamental structural and dynamic properties of nucleosomes made frequent use of labeled oligonucleotides as biophysical probes. Chemical crosslinking of nucleosomal DNA to histone proteins provided a course model of the arrangement of protein-DNA interactions in the complex (44). Radiolabeled DNA probes were used in competition experiments to determine the relative free energies of nucleosome formation associated with nucleosome positioning sequences (45). Förster resonance energy transfer (FRET) systems in which fluorescent labels were incorporated into DNA and adjacent histone residues were used to quantify DNA breathing in the nucleosome structure and demonstrated that transcription factors could bind nucleosomal DNA by stabilizing a transiently unwrapped conformation (46). Similar approaches were used to determine the effects of histone PTMs on DNA unwrapping and nucleosome disassembly (47, 48), as well as the conformational dynamics associated with compaction of synthetic chromatin arrays (49, 50). DNA probes have also been utilized in super resolution microscopy approaches for the direct visualization of chromatin states in single cells. Incorporation of the synthetic nucleotide 5-ethynyl-2’-deoxyuridine (EdU) into genomic DNA and subsequent click-chemistry tagging with a fluorescent dye provided significantly improved resolution of subchromosomal regions when combined with stochastic optical reconstruction microscopy (STORM) analytical techniques (51). Super-resolution techniques that use fluorescence in-situ hybridization (FISH) probes allow the visualization of chromatin conformation within targeted genomic regions, and have been used to visualize the organization of active and polycomb-repressed chromatin domains (52), as well as to characterize TAD-like domain structures in single cells at nanometer- and kilobase-scale resolution (53). These applications and others discussed here greatly benefit from the relative ease with which chemical probes can be synthetically or enzymatically incorporated into DNA oligos (Figure 3a). Indeed, a broad set of base-modified DNA sensors have been developed for the site-selective detection of protein binding, viscosity, and redox potential (54), although the applications of these probes in a chromatin context has been limited.
Figure 3.
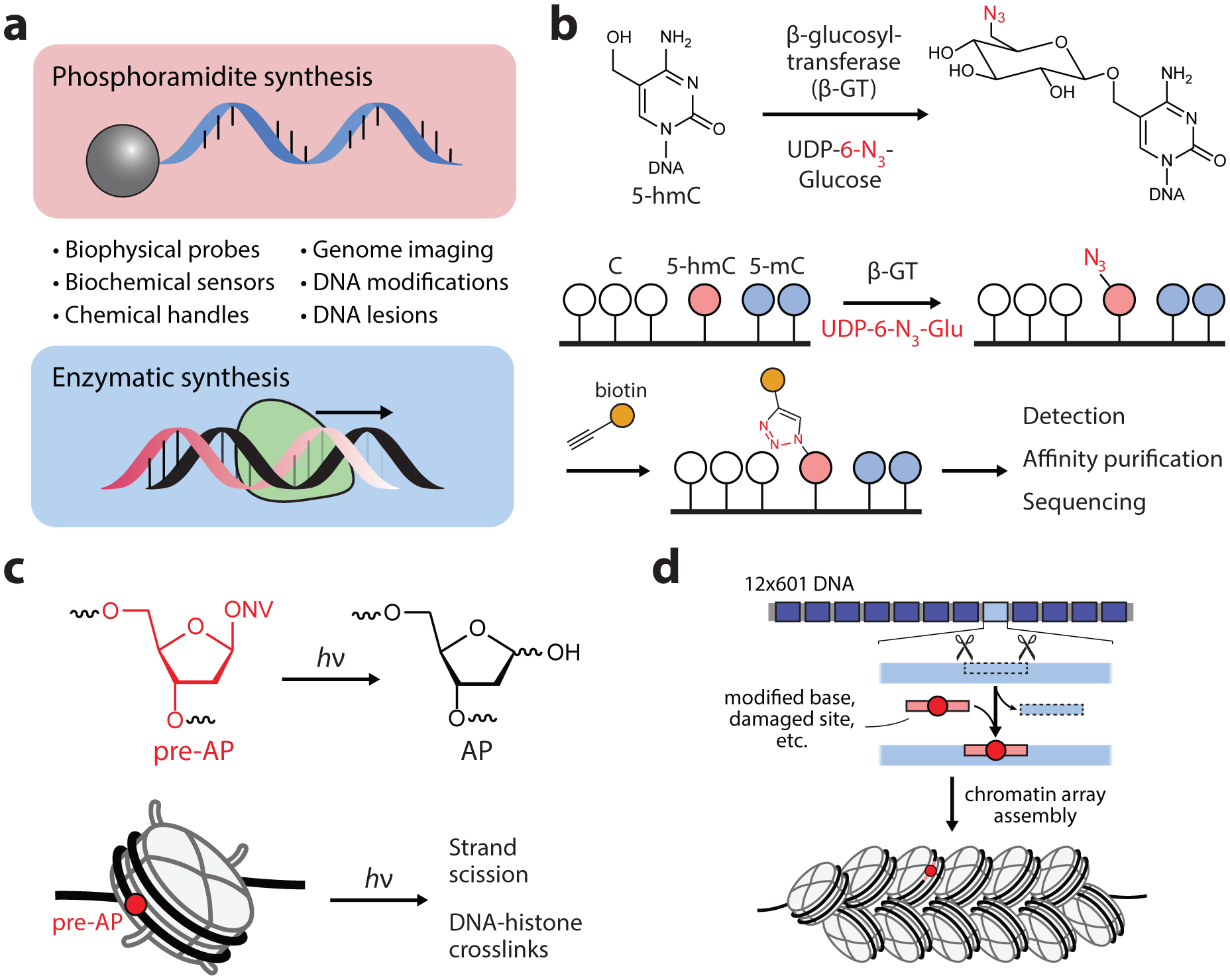
Tools based on nucleic acid synthesis. (a) Overview of oligonucleotide probes that can be generated by chemical and enzymatic DNA synthesis approaches. (b) Genomic profiling of 5-hmC by selective tagging with biorthogonal sugars. (c) Incorporation of photolabile precursors for the generation of apurinic/apyrimidinic (AP) sites in situ. ONV, o-nitroveratrole. (d) DNA nicking and gap-filling strategy for the incorporation of modified oligos into chromatin array DNA substrates.
Chemical approaches for modifying DNA has served a particularly significant role in studying the variety of chemical modifications that have been discovered on the eukaryotic genome. The most prominent and well-studied of these is 5-methylcytosine (5mC), a generally repressive mark that, in humans, is found on 60–80% of CpG dinucleotides (55). Methylated DNA probes were used in electrophoretic mobility shift assays and southwestern blotting experiments that identified the first 5mC reader proteins (56, 57). After the discovery of 5mC eraser proteins in 2009 (58), which function via oxidation of 5mC to 5-hydroxymethylcytosine (5hmC), the Carell lab introduced LC-MS methodologies for quantifying the abundance of 5mC and its oxidized derivatives. By enzymatically digesting isolated DNA into individual nucleosides and spiking in isotopically labeled synthetic standards, 5hmC was found to be present in all investigated mouse tissues at significant quantities (59). This approach was also used to confirm the presence of further oxidized forms of 5hmC, 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC), and continues to be the gold standard for quantification of modified DNA bases (60, 61). Nucleic acid tools for studying oxidized forms of 5mC have been crucial for providing evidence that these marks themselves serve as epigenetic regulators. These developments are discussed in a case study below (section 3.2).
Since the discovery of 5mC and its oxidized derivatives, a variety of techniques have been developed for the genome-wide mapping of these marks via deep sequencing. Bisulfite sequencing, for example, utilizes a chemical derivatization strategy to distinguish 5mC from cytosine (62), and has subsequently been integrated into experimental workflows for mapping other oxidized 5mC marks. These and other DNA sequencing methods for mapping base modifications have been reviewed extensively (63); here, we provide a few key examples of enzymatic and chemical tagging methods that have been used for the selective enrichment and analysis of cytosine modifications. Song et al. introduced an approach for the selective chemical labeling of 5hmC using a viral β-glucosyltransferase enzyme (β-GT) (64). The method, sometimes called hmC-seal, utilizes β-GT to catalyze the transfer of an azide-functionalized glucose moiety to the hydroxyl of 5hmC (Figure 3b). Subsequently click-chemistry-based biotinylation and affinity enrichment allowed the genome-wide profiling of 5hmC marks in mammalian genomes. In work that provided the first genome-wide mapping of 5fC, Raiber et al. utilized a chemical tagging approach to selectively label the aldehyde moiety of 5fC with a biotin-hydroxylamine probe (65). Methods that utilize synthetic oligonucleotides for the site-selective detection of cytosine modifications without the need for DNA sequencing have also been established (63).
Generally, the nucleosome complex has been found to protect DNA from exogenous sources of damage (66). Work utilizing synthetic oligos to introduce DNA lesions into nucleosomes with site-specificity, however, has revealed that histones can react with DNA damage intermediates with deleterious effects. Incorporation of photolabile precursors of apurinic/apyrimidinic (AP) lesions allowed for the generation of abasic DNA damage in situ, which was found to cause DNA-histone crosslinks and to drive strand scission at rates ~60-fold more rapidly in nucleosomes compared to that of naked DNA (Figure 3c) (67). Similar enhancements of strand breakage in nucleosomes were observed for other common abasic lesions (68–70). The effects of DNA damage from the highly efficacious chemotherapeutic agent cisplatin has also been investigated in the context of nucleosomes. Site-specific incorporation of single cisplatin intrastrand and interstrand crosslinks into nucleosomal DNA was found to inhibit ATP-independent DNA sliding and to stall transcription elongation (71, 72). Nucleosomal DNA has also been shown to be vulnerable to long-range damage via charge transport through the DNA base pair stack. Conjugation of photooxidants to the ends of nucleosomal DNA revealed significant long-range oxidation of guanine bases (73), and the efficiency of charge transfer was sensitive to environmental conditions that affect nucleosome structure (74).
Introduction of DNA modifications into nucleosomal DNA has primarily been achieved through a combination of chemical synthesis and cloning techniques. These approaches have also been used to generate multi-nucleosome arrays with site specific modifications, including oxidized bases (75), fluorescent probes (49), and methylated bases (76). In an effort to streamline the synthesis of site-modified DNA in chromatin array substrates, the Sczepanski lab introduced a “plug-and-play” strategy in which targeted sites are excised by nicking enzymes and subsequently gap-filled with labeled primers (Figure 3d) (77). Making use of this strategy, they determined that the extent of chromatin compaction significant affects the activity of base excision repair in synthetic chromatin arrays (77, 78).
2.3. Tools based on small molecule probes
Dysregulation of chromatin regulatory networks is a prominent feature in carcinogenesis (6), and efforts to develop chemical probes that modulate the activity of associated epigenetic machinery have been highly fruitful. Inhibitors are now available that target a broad spectrum of chromatin effector proteins, including DNA methyltransferases and writers, erasers, and readers of histone methylation and acetylation, several of which are currently used in clinical treatments or are undergoing pre-clinical testing (79). The biochemical control afforded by these molecules has been critical for mechanistic investigations of epigenetic signaling pathways. Indeed, this is an extremely active area of chemical biology that has benefited greatly from extensive and open collaboration between academic labs and the pharmaceutical/biotech sector (https://www.thesgc.org/). Here, we discuss approaches that leverage the activity of small molecule probes to develop discovery tools for chromatin biology.
The development of activity-based small molecule probes provides a powerful approach for assessing the biochemical context of chromatin effector enzymes (Figure 4a). As an example, Liu et al. developed an approach for characterizing the interactome of the H3K9-specific methyltransferase G9a (80). The inhibitor UNC0638, which specifically binds the active form of G9a, was immobilized on beads, allowing the affinity purification of active G9a complexes. Combining this approach with SILAC-based quantitative proteomics identified G9a-associated factors that were stimulated by the inflammation response in macrophages. A similar approach has been extended to the prominent HDAC inhibitor SAHA (suberoylanilide hydroxamic acid), which was developed as an oral therapy for cutaneous lymphoma. SAHA was modified to include a benzophenone photo-crosslinker and an alkyne handle (SAHA-BPyne), allowing the affinity purification of drug targets (81). In extracts and in live cells, SAHA-BPyne labeled endogenous HDACs 1, 2, 3, and 6, and was able to isolate HDAC-associated proteins (81). Immobilized SAHA and givinostat, another hydroxamate-based HDAC inhibitor, have also been used to evaluate the selectivity of additional HDAC inhibitors using a drug competition assay combined with chemoproteomic profiling (82). Similar activity-based approaches were used to determine the substrate of a series of pimelic diphenylamide HDAC inhibitors, identifying HDAC3 as their preferred target in cells (83). For inhibitors with broad substrate scope, conjugation to a histone peptide has been used to impart site-selective inhibition, and has been successfully applied to both acyltransferase and HDAC inhibitors (84, 85). The compound JQ1, a potent inhibitor of the two bromodomains in the BRD4 transcriptional regulator, has been utilized as a chemical probe in several systems for both studying and engineering chromatin, and is discussed in detail as a case study below (section 3.3).
Figure 4.
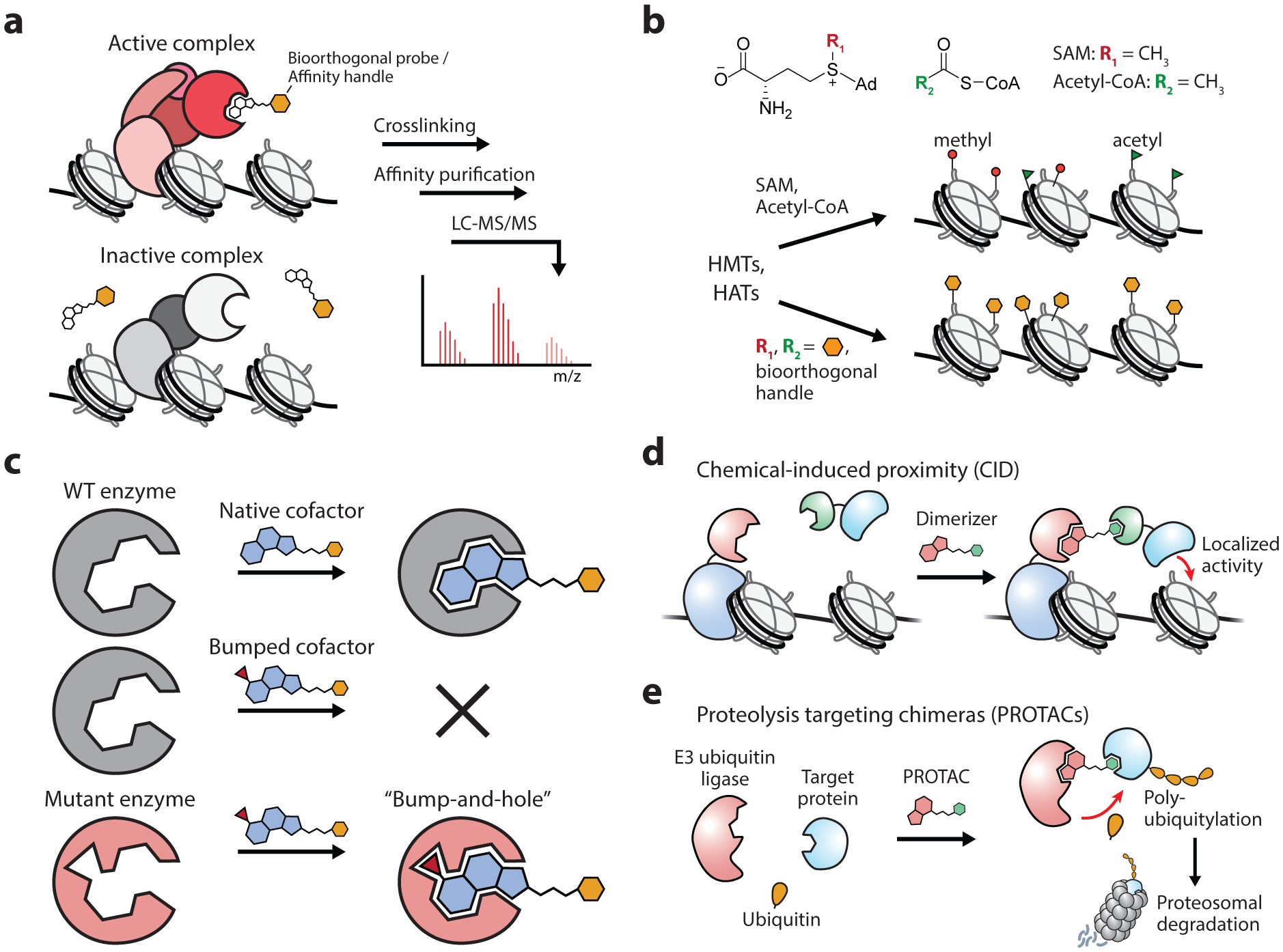
Tools based on small molecule probes. (a) Small molecule probes for activity-profiling of chromatin effectors. (b) Orthogonal probes for profiling epigenetic writers such as histone methyltransferases (HMTs) and histone acetyltransferases (HATs) which use SAM and acetyl-CoA cofactors, respectively. (c) Schematic of the “bump-and-hole” strategy, which can be used to broaden the substrate scope of enzymes for cofactors and inhibitors, as well as to enhance the specificity of chemical probes with their target proteins. (d) Use of heterobifunctional probes to chemically induce the proximity of target proteins and complexes. (e) Bifunctional probes that recruit E3 ligases induce the ubiquitin-mediated proteolytic degradation of target proteins.
Small molecule probes have also been developed to identify the substrates of epigenetic writers. These approaches are centered on a strategy that utilizes analogs of the co-substrates involved in group-transfer reactions, such as S-adenosyl-L-methionine (SAM) and acetyl-CoA. Corresponding methylation and acetylation reactions with these probes mark their targets with a chemical reporter, allowing the identification of new substrate sites (Figure 4b). Early work with SAM analogs determined that chemical replacement of the sulfonium methyl group was tolerated by DNA methyltransferases, allowing chemical modification of target substrates (86). Several SAM analogs have since been developed for use with protein methyltransferases, the most versatile of which tag substrates with azide or alkyne moieties which can subsequently be labeled with fluorescent probes for visualization or affinity tags for purification via click chemistry (87). SAM analogs, however, are tolerated by only a subset of methyltransferases, limiting their enzyme and substrate scope (88). In a clever application of the “bump and hole” engineering strategy, the Luo laboratory mutated individual methyltransferases that allowed them to utilize bulky SAM analogs that are not accepted by native enzymes (89, 90). This approach, dubbed bioorthogonal profiling of protein methylation (BPPM), allows identified substrate sites to be attributed to specific methyltransferase enzymes and has been utilized to characterize the substrate scope of histone lysine and arginine methyltransferases. Although these applications were initially confined to in vitro applications with cellular lysates due to the poor membrane permeability of SAM and its analogs, additional efforts have extended BPPM for use in cells (91). By engineering the human SAM synthetase to process cell-permeable methionine analogs, the corresponding SAM analogs could be synthesized inside living cells. Coupled with previously established BPPM methods, this allowed for genome-wide profiling of chromatin-modifying activity of histone methyltransferases G9a and GLP1. Acetyl-CoA analogs bearing biorthogonal functional groups have been analogously used to label the targets of chromatin acetylation (92). Additionally, “bump and hole” approaches have been applied to characterize the substrates of both histone acetyltransferases and kinase enzymes that act on chromatin (Figure 4c) (93, 94).
The development of chemical probes that induce the physical proximity of two biomolecules has had a transformative effect in biological discovery, and the chromatin field is no exception. These chemical inducers of proximity (CIPs) typically contain bivalent structural elements that bind two separate targets, allowing the precise and rapid temporal control of protein interactions in live cells (Figure 4d). Since their inception in 1993 (95), a suite of CIP systems have been developed and used to study a broad set of biological processes (96). In the chromatin field, CIPs have been used to investigate the formation and spreading of repressive chromatin domains by heterochromatin protein 1 (HP1) (97, 98), as well as the eviction of the polycomb repressive complexes 1 and 2 (PRC1 and PRC2) by the ATP-dependent chromatin remodeler, BAF (Brahma/Brg associated factor, aka the mammalian SWI/SNF) (99, 100). The combination of CIPs with gene-targeting dCas9 systems has allowed for the further control of epigenome editing and chromatin architecture with locus-specificity (101, 102). Photocaged CIPs can perform light-induced protein dimerization and have demonstrated the precise spatiotemporal control of protein associations at centromeres, kinetochores, and centrosomes in living cells (103).
A particularly promising subset of CIP systems are those that induce the degradation of target proteins. Recent advances in the development of proteolysis targeting chimeras (PROTACs) offer a modular and tunable approach toward targeted protein degradation (104). PROTAC molecules are heterobifunctional compounds consisting of a ubiquitin E3 ligase ligand coupled via a flexible linker to a moiety that binds a target protein of interest (Figure 4e). Ligand binding brings these proteins into close proximity, resulting in the ectopic ubiquitylation of the target protein and its subsequent degradation by the proteasome. Whereas early PROTACs utilized peptide ligands with poor membrane permeability (105), the development of all-small-molecule PROTACs has enabled their use in cells and as potential drug platforms (106–108). These molecules exhibit a catalytic mode of action, offer exquisite temporal control over protein abundance, and show promise in targeting high-value drug targets, including transcription factors and epigenetic regulators (104). Indeed, PROTACs have been designed that target the three main classes of epigenetic effectors: writers, readers, and erasers (109).
2.4. Tools based on analytical chemistry measurements
A now central tenet in chromatin biology is the “histone code” hypothesis (5), which proposes that the combinatorial nature of histone PTMs performs a fundamental signaling role that serves as an extension to the regulatory control imparted by the genetic code. Efforts to understand this role of histone PTMs necessarily require approaches for their robust identification and quantification. Although analytical techniques based on antibodies are routine for PTM detection and affinity purification, antibodies are poorly suited for identification of new PTM targets, and even high-quality antibodies are frequently affected by epitope occlusion and PTM cross-reactivity (110). As a result, the absolute gold standard for PTM identification and quantitation is mass spectrometry (MS) due its high specificity and mass accuracy (Figure 5a). Indeed, MS has been central to the site-specific identification of scores of histone PTMs (111). Moreover, middle-down MS approaches that examine large peptides in the 5–6 kDa range, typically the histone tails, allow for the combinatorial analysis of multiple, coexisting histone PTMs, a common regulatory feature of chromatin (5). MS-based methods have also been developed to profile the abundance of cellular metabolites (112), which have been shown to play direct roles in the determination of epigenetic cellular states (113).
Figure 5.
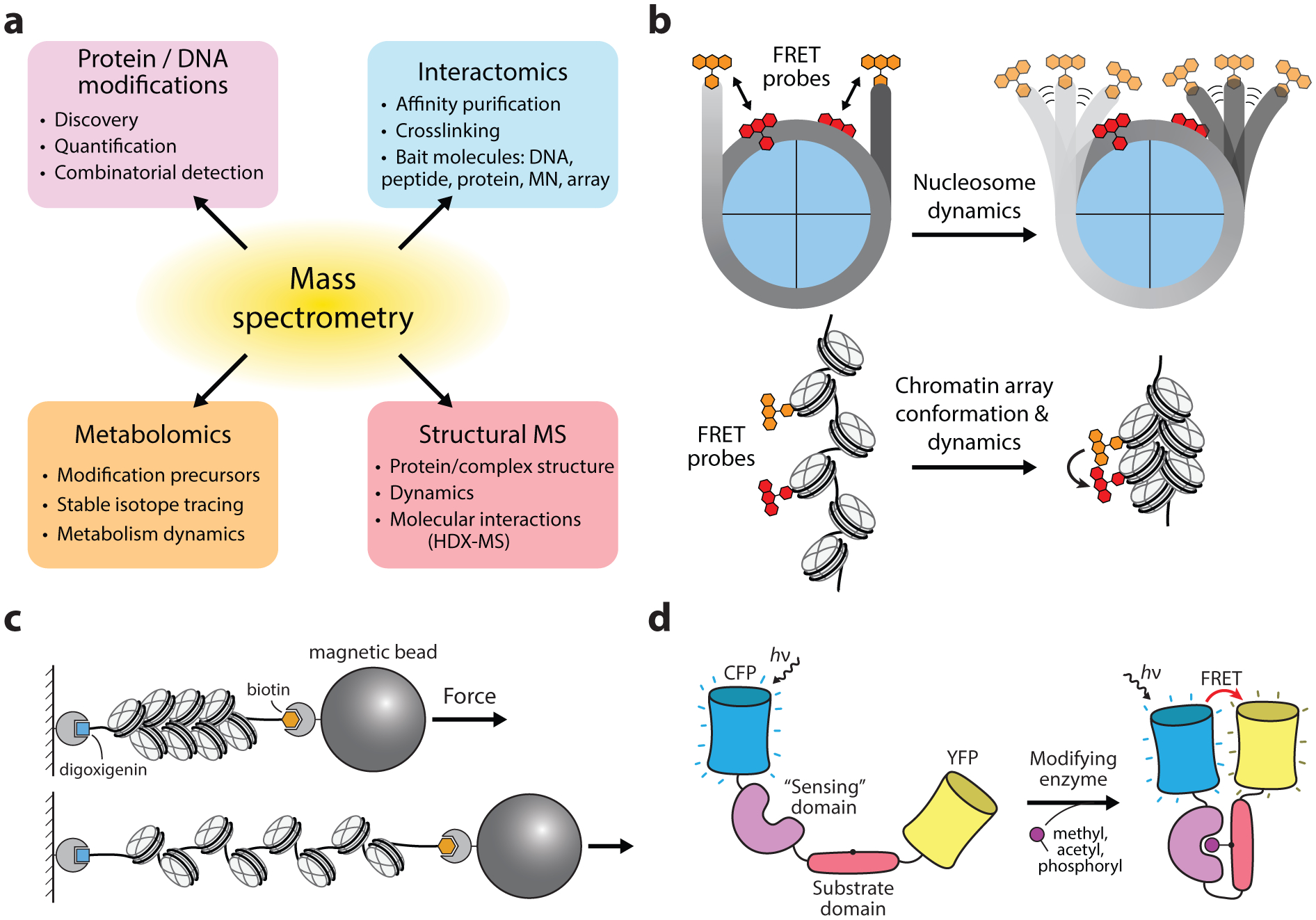
Tools based on analytical chemistry measurements. (a) The broad analytical scope of mass spectrometry in characterizing epigenetic modifications, proteins, and processes. (b) The use of optical probes for characterizing the dynamics and structural arrangements of nucleosomes and chromatin arrays. (c) Use of force spectroscopy for characterizing molecular interactions in nucleosomes and synthetic chromatin arrays. (d) Schematic of genetically-encoded chemical actuators designed to sense specific chemical modifications such as histone PTMs.
In addition to detecting histone PTMs, MS-based proteomics approaches have been used for characterizing the interactome of histone modifications. Early applications utilized modified histone peptides which were incubated with SILAC-labeled nuclear lysates to affinity purify and identify histone PTM readers (114). To better represent the chromatin context of histone PTMs, similar approaches have been utilized with modified nucleosome substrates (115). In order to capture weak or transient interactions, integration of crosslinking moieties into bait peptides has been used to covalently capture PTM-binding proteins (116). Additional strategies have been developed to analyze the interactome of native histone tails in a cellular context by the incorporation of photocrosslinking ncAAs into histones via amber suppression (33).
In recent years, development of hydrogen/deuterium exchange mass spectrometry (HDX-MS) has offered a powerful approach for analyzing the conformations of large protein complexes (117). By measuring the exchange of solvent deuterium with the amide hydrogens in the protein backbone, the solvent accessibility of proteins can be directly measured, providing information about protein structure, interactions, and dynamics. These approaches have been particularly useful for studying large, chromatin-associated complexes, and HDX-MS has been used for characterizing the dynamic and structural features of histone-chaperone complexes (118), nucleosome and sub-nucleosome assemblies (119), and chromatin sequestered into phase-separated liquid droplets (120).
Determining the structural and dynamic properties of nucleosomes, arrays, and chromatin-effector complexes is a fundamental prerequisite for understanding the roles of chromatin in controlling genetic processes (17). Early efforts focused on characterization of the nucleosome core particle, which largely benefited from the site-directed installment of fluorescent probes on both histones and DNA, permitting the analysis of specific structural elements via FRET (Figure 5b). Such applications have revealed the fundamental energies and time scales associated with the dynamic processes of nucleosome assembly/disassembly, histone eviction, and DNA accessibility (46, 121). Single-molecule force spectroscopy approaches have further scrutinized these processes and have elucidated the energies associated with the step-by-step disruption of histone-DNA interactions, providing a nucleosome unwrapping free-energy landscape with near-base resolution (122).
Biochemical methods for measuring chromatin compaction such as Mg2+ precipitation, ultracentrifugation, and restriction digest accessibility assays have been valuable for characterizing extended chromatin fibers, however, these approaches ultimately provide indirect and relatively coarse measurements of chromatin condensation. Analogous to experiments with mononucleosome substrates, fluorescence and force spectroscopy approaches have been extended for the analysis of the structure and dynamics of extended chromatin fibers. By homogeneously labeling histones with a fluorescent probe in chromatin arrays, chromatin compaction reduces the distance between fluorophores, promoting homo-FRET. The resulting excitation of fluorophores with polarized light can then be detected by corresponding decreases in emission steady-state anisotropy (SSA) (123). Single-molecule FRET approaches have further been developed for assessing the dynamics of chromatin arrays both immobilized on surfaces and in solution (50). Chromatin array compaction has also been characterized with force spectroscopy, which has been used to analyze the effects of chromatin effectors proteins on array stability and compaction (124) (Figure 5c). With regard to chromatin fiber compaction, a particularly fascinating set of investigations of heterochromatin protein 1 (HP1), a prominent factor in the formation of heterochromatin, has enlightened the complex and dynamic features of condensed chromatin and is discussed in detail in the corresponding case study in this review (section 3.4).
Genetically-encoded chemical actuators have allowed for the detection and visualization of chromatin-modifying processes in real-time (Figure 5d). To study the role of aurora B kinase in cell division, Fuller et al. designed a cyan and yellow fluorescent protein FRET sensor that is activated upon phosphorylation by aurora B (125). By targeting the sensor to either chromosome or centromere domains, phosphorylation dynamics were quantified throughout cell division, helping to define the role of aurora B kinase activity in establishing the cell-division plane. Similar sensors have been designed to detect the methylation and acetylation of histones proteins in live cells (126, 127). Recently, genetic systems have been developed for the photo-inducible control of phase separation of proteins containing intrinsically disordered domains (128). Given the rapid progression in understanding the role that phase separation plays in a variety of biological processes (129), these tools are particularly promising considering the experimental challenges associated with studying these non-equilibrium processes.
Advances in X-ray crystallography, NMR, and the rapidly maturing cryo-EM, have been critical for investigating chromatin structures as well as large chromatin effector complexes. While these approaches are reviewed extensively elsewhere (21, 130), we wish to highlight several labeling methodologies that have permitted NMR analysis of large chromatin substrates. Segmental isotopic labeling allows for subsections of large protein complexes to be the focus of NMR analysis and is readily accessible by the recombinant production of 15N and 13C-labeled substrates and subsequent protein assembly with inteins or alternative transpeptidase enzymes (21). The site-specific incorporation of magnetic resonance probes has further aided the analysis of large protein complexes (131, 132). Additionally, developments in methyl-TROSY NMR spectroscopy has enabled the analysis of large macromolecular assemblies, and has been used to characterize nucleosome stability and heterochromatin formation (120, 133).
2.5. Tools for epigenome engineering
Synthetic methods that can precisely define the chemical composition of nucleosomes and chromatin arrays has been essential in establishing causal relationships between chromatin biochemistry in vitro. Toward the goal of performing experiments on ever-more native chromatin substrates, researchers have developed tools for controlling epigenetic machinery in the cellular environment (134). A widely successful approach involves the repurposing of genome-editing tools such as zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and CRISPR-associated (Cas) systems for recruitment, rather than DNA-editing, allowing locus-specific control of epigenetic machinery (Figure 6a). By genetically fusing DNA-binding proteins to epigenetic effectors, these systems have successfully targeted the installation and removal of a broad set of epigenetic modifications, including DNA methylation (135, 136), histone methylation (137, 138), and histone acetylation (139). Importantly, these studies demonstrated the capability of epigenetic marks to directly activate and repress gene expression in cells. Small molecules have also been developed for altering transcriptional regulation at specific DNA sequences. Polyamides composed of N-methylpyrrole and N-methylimidazole repeats can be rationally designed to bind sequences via interactions with the DNA minor groove (140), which can be used to directly block transcription factor binding to DNA (141) or to recruit transcriptional machinery through a conjugated activator moiety (142). Locus-specific targeting of chromatin effectors has also been combined with protein inducers, allowing these systems to be triggered with small molecules or with light (143). Gene activation has further been demonstrated by the recruitment of synthetic cargo containing epigenetic inhibitors via Cas9 fusions (144) as well as bifunctional polyamides (145).
Figure 6.
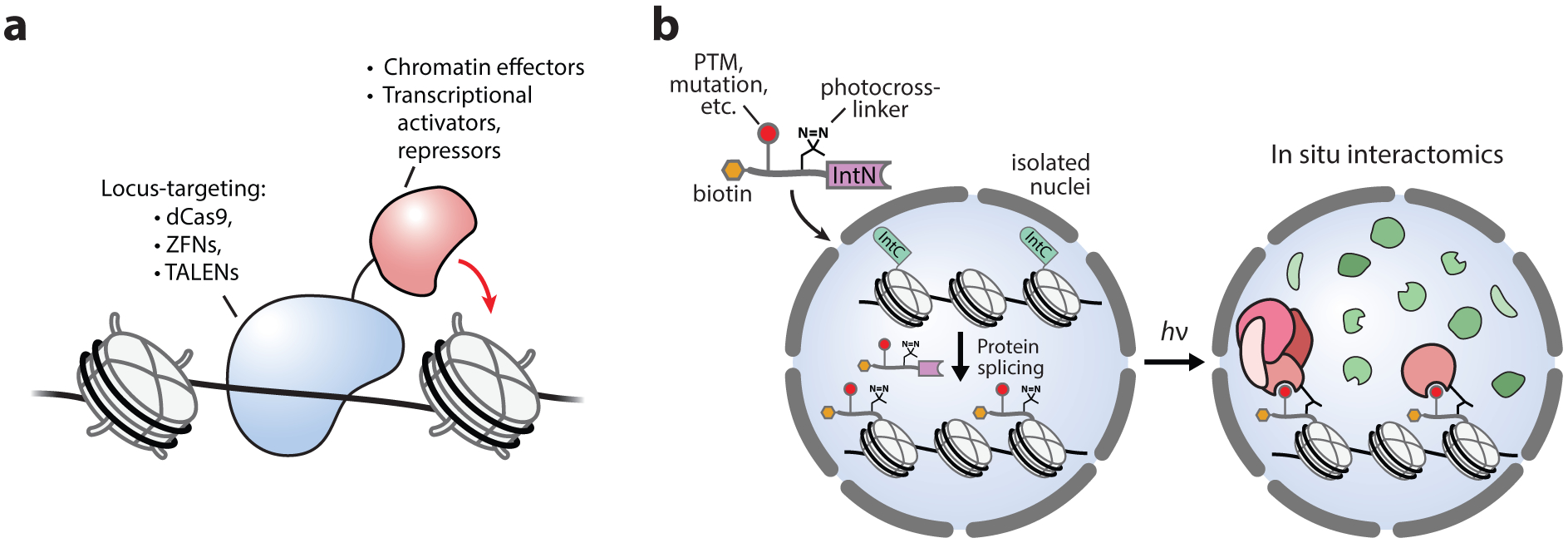
Tools for epigenome engineering. (a) Locus-specific targeting of transcriptional and epigenetic effectors. (b) Approaches for performing protein semisynthesis in nucleo (depicted) or in live cells, providing precise chemical control of chromatin composition in the native chromatin environment.
Our laboratory has recently developed tools that utilize highly efficient split-inteins to perform protein semisynthesis in live cells, allowing the traceless ligation of affinity probes, fluorophores, and PTMs onto native chromatin (146, 147). In isolated nuclei, this approach generated semisynthetic ubiquitylated H2B in situ, which was able to stimulate the methylation of H3K79 through an established cross-talk mechanism (146, 148). An extension of this approach incorporated histone PTMs together with an adjacent photoactivatable crosslinker, allowing the histone PTM interactome to be captured and profiled using MS-based proteomics (Figure 6b) (149). Overall, tools for epigenome engineering offer exciting potential for the direct manipulation of chromatin biochemistry and signaling in native cellular environments and will undoubtedly play an essential role in continued efforts to decipher the underlying mechanisms of the epigenetic regulatory landscape.
3. CASE STUDIES
3.1. Protein semisynthesis of ubiquitylated H2B
In chromatin the primary targets of ubiquitylation are histones H2A and H2B, which are monoubiquitylated near their C-termini and are associated with the regulation of gene expression (4). The size and chemical complexity of ubiquitin, itself a 76 amino acid protein, raised interesting questions regarding the mark’s biochemical and structural effects on chromatin. In vitro characterization of this mark, however, posed a unique challenge for chemists because ubiquitin is too large to be introduced as a single unit during peptide synthesis. This has since motivated the development of methods to synthesize and characterize the effects of ubiquitylated histones. In particular, studies on the primary site of H2B ubiquitylation at Lys120 have led to the development of a set of chemical tools, rooted in peptide and protein chemistry, that have elucidated the biochemical and physiological function of this mark in chromatin.
In work that signified a pivotal development toward the synthesis of ubiquitylated proteins, Chatterjee et al. (150) utilized EPL with a ligation auxiliary to generate a native ubiquitin linkage on Lys120 of a synthetic H2B peptide (Figure 7a). A key feature of this strategy was the use of a recombinantly generated ubiquitin(1–75)-α-thioester that omitted the C-terminal glycine at position 76. The missing residue was then reconstituted en route, first by reacting bromoacetic acid with the ε-amine of an orthogonally protected Lys120, forming an isopeptide bond on the synthetic histone peptide. The branch was then coupled to a photolabile ligation auxiliary and subsequently ligated to the ubiquitin thioester via EPL. Subsequent photolysis of the auxiliary yielded the synthetic H2B peptide with a native ubiquitin linkage at Lys120. In ensuing work, the addition of a second ligation step was used to generate full-length ubiquitylated H2B protein (Figure 7b) (151). To facilitate this, the native Ala117 in the H2B peptide corresponding to residues 117–125 was replaced with Cys and protected with an S-(o-nitrobenzyl) group. Following auxiliary-mediated ubiquitin ligation, photolysis both removed the ligation auxiliary and liberated the N-terminal Cys, which was subsequently ligated to recombinant H2B(1–116)-α-thioester. A final desulfurization step converted the single Cys back to Ala, yielding the full-length, native, H2BK120ub protein. Ubiquitylated H2B was then assembled with canonical histones and DNA to form nucleosomal substrates, and subsequent biochemical experiments determined that the H2BK120ub mark directly stimulated the methyltransferase activities of Dot1 (151) and Set1 (152) on H3K79 and H3K4, respectively.
Figure 7.
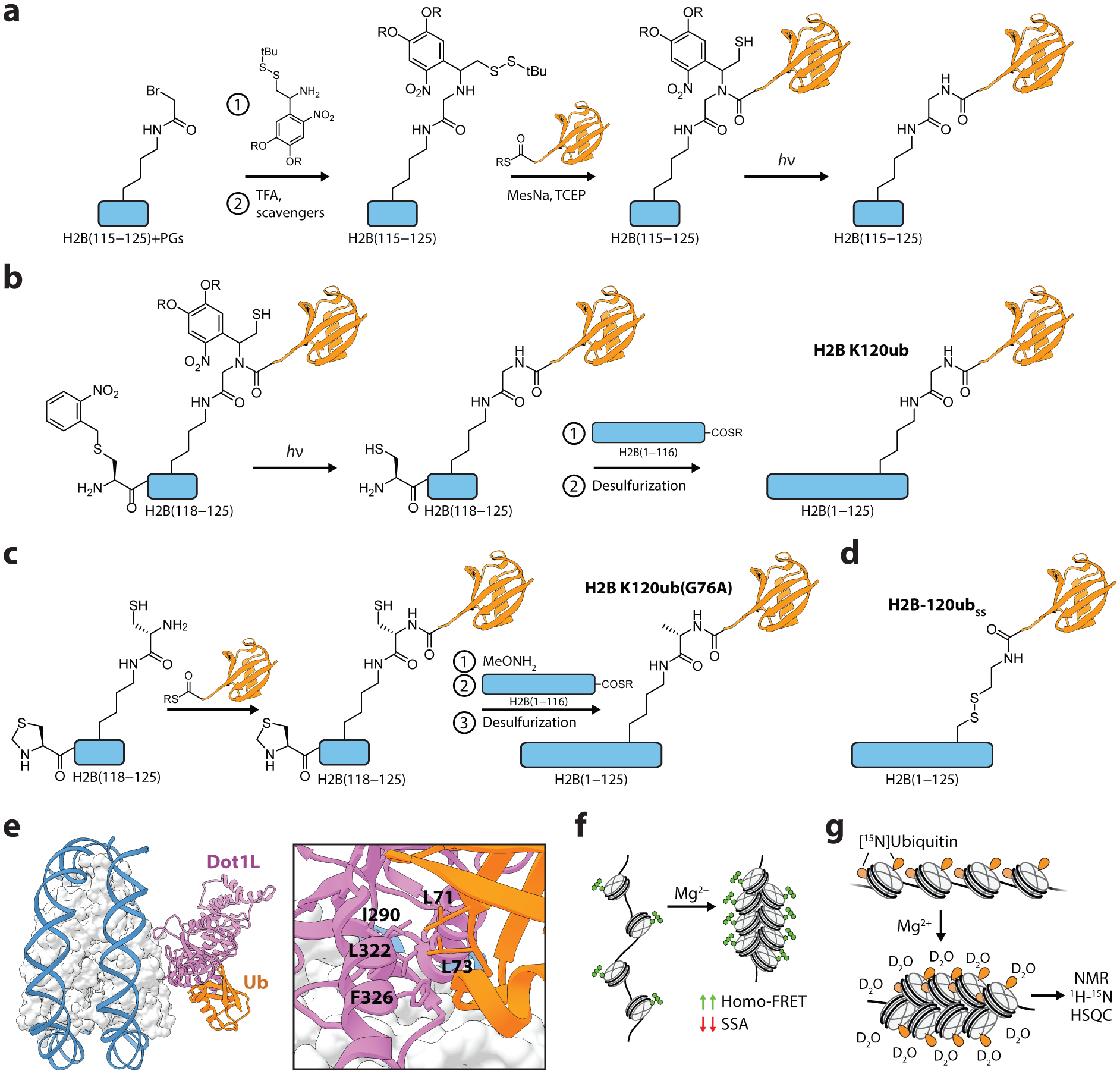
Protein semisynthesis and characterization of ubiquitylated H2B. (a) Synthesis of H2BK120ub peptides using an aminothiol ligation auxiliary. (b) Semisynthesis of the full-length, native H2BK120ub protein via two-step traceless ligation route. (c) Streamlined synthesis of full-length H2BK120ub with a G76A ubiquitin modification. (d) Structure of the disulfide-linked ubiquitin analog, H2B-120ubSS. (e) Structure of Dot1L engaging a nucleosome with the H2BK120ub modification. Residues mediating the Dot1L-ubiquitin interaction are shown. PDB, 6nn6. (f) Quantification of chromatin array compaction with homo-FRET, measured by changes in steady-state anisotropy (SSA). (g) Probing ubiquitin interactions in compacted chromatin arrays with H/D exchange and NMR spectroscopy. PGs, protecting groups. MesNa, sodium 2-mercaptoethanesulfonate. TCEP, tris(2-carboxyethyl)phosphine.
In order to perform structure-activity relationship studies of the H2BK120ub mark and Dot1 stimulation, McGinty et al. (153) developed a streamlined synthetic route to the semisynthesis of an H2BK120ub analog with a single G76A point mutation at the C-terminus of ubiquitin (Figure 7c). By coupling a Cys residue to the ε-amine of Lys120 on the synthetic H2B peptide, the need for a ligation auxiliary was bypassed, and ubiquitin(1–75)-α-thioester was ligated directly to the branched cysteine. After the second ligation step to form full-length H2B, desulfurization yielded H2BK120ub(G76A) with a single non-native methyl group at the C-terminus of ubiquitin which, importantly, was indistinguishable from the native linkage in a variety of biochemical assays. Using this approach, the authors synthesized a set of modified nucleosomes with mutations in either ubiquitin or the histones and tested their effects on human Dot1 activity. Surprisingly, this revealed that mutations in the canonical hydrophobic patch on ubiquitin centered on isoleucine-44 did not significantly affect Dot1 methyltransferase activity, suggesting an orthogonal site of interaction.
To further probe the positional requirements for H2BK120ub stimulation of Dot1, a disulfide-directed methodology was developed to facilitate the installment of ubiquitin at several positions throughout the nucleosome (154). Ubiquitin with a C-terminal sulfhydryl group was synthesized by the treatment of a recombinant ubiquitin-GyrA intein fusion with cysteamine. Cys residues were then introduced into histones H2A and H2B by mutagenesis and subsequently activated with 2,2’-dithiobis(5-nitropyridine), enabling the formation of the disulfide-linked H2B-120ubSS (Figure 7d). Compared to native ubiquitylation, the disulfide chemistry increases the linkage between ubiquitin and H2B by one atom, an alteration that did not alter the stimulation of Dot1 activity on nucleosome substrates. This development allowed the installment of ubiquitin at several positions throughout the nucleosome. Disulfide-mediated ubiquitylation at H2B-125 and H2A-22 exhibited levels of Dot1-stimulation similar to that of the H2B-120 position, demonstrating significant plasticity in Dot1 regulation. The disulfide ubiquitylation chemistry was later utilized to determine which ubiquitin residues were important for stimulation of methyltransferase activities (148). A panel of H2B-120ubSS proteins was synthesized to perform an alanine scan of the ubiquitin surface in the context of nucleosome substrates, which identified a ‘leucine patch’ (Leu71 and Leu73) as essential for the stimulation of Dot1 and Set1 methyltransferase activities. This work, along with subsequent biochemical studies employing a version of H2BK120ub containing a photocrosslinker within the ‘leucine patch’ (155), led to a model in which ubiquitin helps to restrict the position of Dot1 on the nucleosome surface, leading to an increase in H3K79 methylation. Recent structural studies support this corralling model (156, 157), revealing that the ‘leucine patch’ on ubiquitin directly binds to an epitope on Dot1 such that the enzyme is appropriately oriented for methylation of the substrate lysine (Figure 7e).
In addition to characterizing mechanisms of PTM crosstalk, protein semisynthesis has also enabled a set of biophysical investigations on the direct effects of H2B-K120ub on chromatin compaction. Sedimentation velocity experiments on chromatin arrays containing twelve nucleosomes (12-mers) determined that the H2B-120ubSS mark reduced chromatin compaction (123). The precise nature of these effects was investigated in experiments that measured array compaction via Förster resonance energy transfer measurements between like chromophores (homo-FRET) (Figure 7f). A fluorescent probe was incorporated into nucleosome arrays by labeling an H2AN110C mutant with fluorescein maleimide. Upon treatment with Mg2+, which induces compaction of arrays, changes in fluorescence anisotropy provided a direct read-out of chromatin array compaction. These experiments revealed that H2B-120ubSS prevented the later stages of chromatin compaction and could act synergistically with hyperacetylated H4 tails to impair inter-fiber interactions (123). To investigate how ubiquitin disrupted these interactions, Debelouchina et al. utilized a combination of hydrogen-deuterium (H/D) exchange and nuclear magnetic resonance (NMR) spectroscopy to map the residues on the ubiquitin surface that perturb chromatin array compaction (158). To perform NMR experiments on such complex substrates, ubiquitin was uniformly 15N-labeled during recombinant protein expression and seamlessly integrated into the synthetic route for the H2B-120ubSS protein. H/D exchange was performed on the segmentally labeled 12-mer chromatin arrays during Mg2+-induced compaction (Figure 7g). Following a quenching step, 2D-NMR heteronuclear single quantum correlation (HSQC) experiments were used to show that two acidic residues on ubiquitin, Glu16 and Glu18, are protected from H/D exchange, implicating this negatively charged epitope in the disruption of array compaction. Subsequent biochemical experiments indicated that this ‘glutamate patch’ disfavored both local and higher-order chromatin structures through electrostatic interactions with both histones and other ubiquitin moieties.
3.2. Oxidized 5-methylcytosine modifications as epigenetic regulators
In 1980 the roles of 5-methylcytosine (5mC) in gene repression were beginning to be established (159). Early investigations on the mechanism of 5mC benefitted from the use of the first epigenetic drug, 5-azacytidine, which was shown to reduce DNA methylation and, correspondingly, reactivate silenced genes (160). At promoter elements, 5mC-mediated gene repression was found to occur both indirectly by recruiting 5mC reader proteins that act as transcription repressors (161), and directly by preventing the binding of transcriptional activators (162). By 1989, proteins that install and read 5mC had been documented, but how these marks were removed remained enigmatic for two decades until the discovery of TET (ten-eleven translocation) proteins and their ability to oxidize 5mC, producing 5-hydroxymethylcytosine (5hmC) (58, 163). These enzymes were subsequently demonstrated to further oxidize 5hmC to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) (60, 61), which can be excised by thymine DNA glycosylase (TDG) and converted back into cytosine via base excision repair (BER) (Figure 8a) (164).
Figure 8.
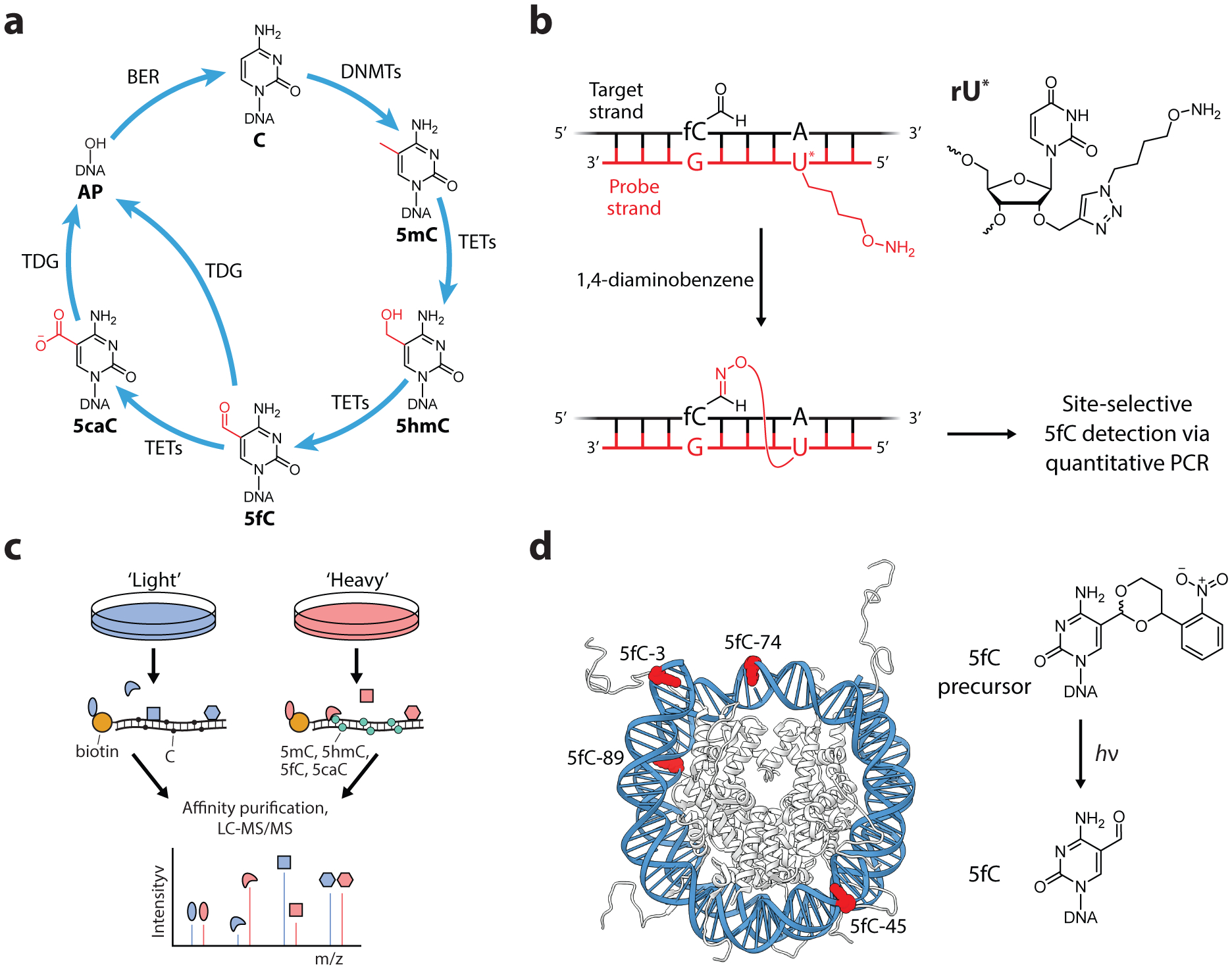
Oxidized 5-methylcytosine modifications as epigenetic regulators. (a) DNA modifications at the C5 position of cytosine. Successive steps of oxidation of 5mC serve as an active demethylation pathway, however, oxidized 5mC derivatives have been shown to serve distinct epigenetic regulatory functions in eukaryotic cells. (b) An oligo probe for the detection of 5fC at specific sites within eukaryotic DNA. Positioning a hydroxylamine-modified uracil four base pairs to the 5’-direction relative to 5fC exhibited highly efficient and selective reactions. Diaminobenzene is used as a catalyst to increase the rate of reaction. (c) Affinity purification of cytosine reader proteins using modified oligonucleotides as bait. Interacting proteins were identified by quantitative proteomics using the stable isotope labelling with amino acids in cell culture (SILAC) approach. (d) Generation of 5fC modifications by incorporation of photolabile 5fC precursors into nucleosome DNA. Irradiation produced the 5fC marks in situ, demonstrating that DNA-histone crosslinks can form within the folded nucleosome structure. PDB, 1kx5.
The development of tools for studying oxidized 5-methylcytosine derivatives in cells and in vitro has provided evidence that these marks, in addition to being intermediates for DNA demethylation, themselves function as epigenetic regulators. In key experiments that addressed the permanence of 5hmC and 5fC marks, the Balasubramanian lab fed mice a diet with deuterated methionine, a metabolic precursor to SAM, which allowed the dynamics of methylated DNA to be measured via LC-MS analysis. The slow turnover of 5hmC and 5fC, especially in brain tissues where the abundance of these marks are highest, suggested that 5hmC and 5fC modifications were largely permanent and not merely transient species (165, 166). The permanence of 5fC was also measured at single genome locations by the development of site-specific oligo probes (Figure 8b). DNA complementary to a target 5fC site was synthesized with a modified uracil base containing a hydroxylamine tether, which reacted with 5fC in close proximity (167). Using this labeling approach combined with a quantitative PCR readout, the authors determined that specific 5fC sites in mouse embryonic stem cells exhibited semi-permanence, indicating that the global trends of 5fC turnover are also reflected at individual sites.
The previously discussed hmC-seal method (Figure 3b) determined that 5hmC marks occupied gene bodies and gene proximal regions, and was enriched in highly expressed genes, suggesting a role in the activation or maintenance of gene expression (64). Interestingly, significant 5hmC depletion has been observed in many types of human cancer, raising the possibility that 5hmC could serve as a marker for cancer diagnostics, however, the low abundance of this mark makes detection in circulating cell-free DNA challenging (168, 169). Taking advantage of the high sensitivity of hmC-seal due to the selective tagging and enrichment of 5hmC sites, this approach has been adapted to perform genome-wide profiling of 5hmC in circulating cell-free DNA (170, 171). The results suggest that 5hmC signatures from blood samples could potentially be used to both identify cancer types and track tumor progression.
A recurrent role of epigenetic marks is the recruitment of effector proteins to localize protein activity to specific sites. Two studies searched for such “reader” proteins using biotinylated oligo probes containing C, 5mC, or oxidized 5mC marks to fish out proteins from cell lysates via affinity purification and MS-based proteomics (Figure 8c) (172, 173). 5hmC, 5fC, and 5caC were found to interact with transcriptional regulators as well as proteins associated with DNA damage response and repair, suggesting that these marks play roles in both active DNA demethylation as well as gene regulation. Surprisingly, readers for the individual cytosine modifications exhibited limited overlap, indicating that these marks have distinct biochemical interaction profiles.
The effects of oxidized 5mC marks on nucleosome structure have also been investigated using a combination of biochemical and biophysical assays. Incorporation of FRET labels into nucleosomal DNA and histone proteins revealed that the 5hmC mark significantly destabilized DNA interactions with the H2A/H2B dimer (174). Given the importance of nucleosome disassembly in transcription, these findings are consistent with the association of 5hmC with more actively transcribed genes (64). To determine the effects of 5fC on nucleosome structure, Ngo et al. utilized a fluorescence-force spectroscopy approach (175). Nucleosomes were anchored to a surface with a biotin-neutravidin interaction on one end of the DNA and subsequently pulled by an optical trap via a DNA-tethered bead on the opposite end. Nucleosome disassembly was monitored by DNA-incorporated FRET probes, which revealed that as little as two copies of 5fC resulted in a significant enhancement of nucleosome mechanical stability (175), a finding consistent with the enrichment of 5fC at poised enhancers and transcription start sites of low-expressed genes (176).
The reactivity of the formyl group of 5fC toward nucleophilic amines, hydrazides, and aminooxy derivatives, which has been exploited for the development of 5fC-specific probes, raised the possibility that 5fC may form DNA-protein crosslinks in the context of nucleosomes. The incorporation of photolabile 5fC precursors into nucleosome DNA allowed for the site-specific generation of single 5fC bases in situ (Figure 8d) (177). DNA-histone crosslinks in these nucleosomes were detected and found to be reversible, approaching equilibrium values ranging from 3–21% depending on the positioning of 5fC. Crosslink formation could be significantly reduced by mutating lysines in histone tails to arginine, suggesting the formation of a Schiff base with lysine ε-amines. The existence of these crosslinks were later confirmed in mouse embryonic stem cells and found to affect transcriptional elongation (178). Indeed, it is possible that 5fC-histone crosslinks are a contributing factor to the permanence of 5fC in cells. Additionally, recent work from the Sczepanski lab has also demonstrated that chromatin compaction can inhibit 5fC removal by TDG, preventing base excision repair. Using a “plug-and-play” approach (described above) to site-specifically incorporate 5fC into 12-mer chromatin arrays, 5fC excision by TDG was found to be inhibited in compacted chromatin arrays, demonstrating that active DNA demethylation is sensitive to local chromatin structure (78).
3.3. Tools for Manipulating the BRD4 Transcription Factor
The discovery that a conserved protein fold called the bromodomain (BD) recognizes and binds to acetyl-lysine residues was a critical step in understanding the role of lysine acetylation in transcriptional activation (179). While the physiochemical properties of lysine acetylation, namely charge neutralization and steric hindrance, were understood to have direct effects on nucleosome and chromatin structure, the frequency of bromodomains in epigenetic effector proteins, including histone PTM writers, chromatin remodelers, and transcription factors, established the integral role of acetyl-lysine-mediated recruitment of proteins in genetic regulation (180). The bromodomain and extra-terminal domain (BET) family of proteins (BRD2, BRD3, BRD4, and testis-specific BRDT) each contain two highly conserved bromodomains (BD1 and BD2) and have emerged as master regulators of transcription (181). In particular, BRD4 serves a prominent role in modulating the expression of essential oncogenes, making it a promising therapeutic target in multiple cancer types (182). In 2010, two independent groups identified potent BET protein inhibitors containing a thienotriazolodiazepine (JQ1) and benzotriazolodiazepine (I-BET) scaffold (Figure 9a) (183, 184). These and subsequent BET inhibitors have had substantial effects on the development of new epigenetic therapeutics, several of which are currently in clinical trials, and have also provided critical tools for probing the mechanisms of transcriptional regulation by BET proteins.
Figure 9.
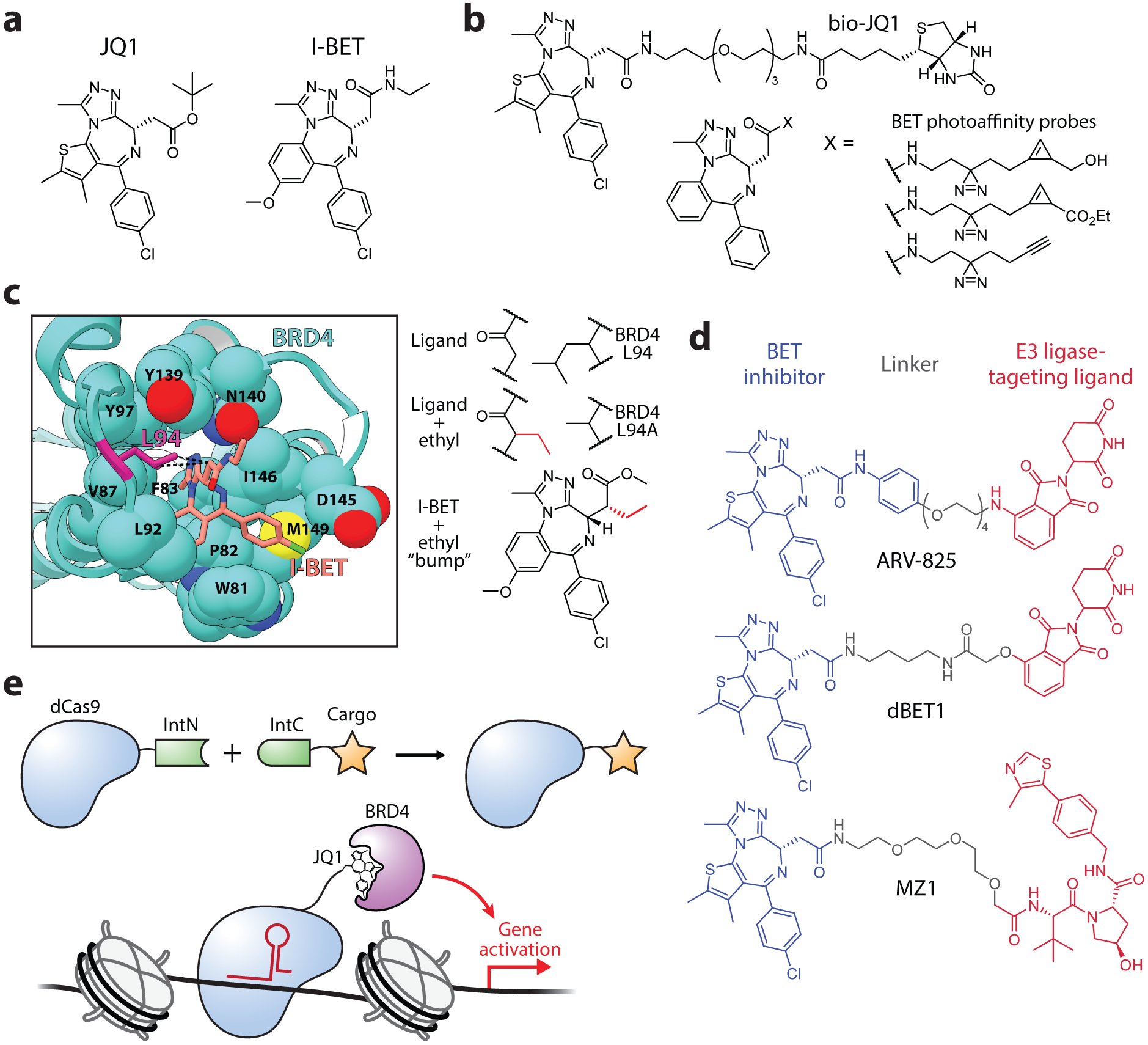
Tools for manipulating the BRD4 transcription factor. (a) Chemical structure of the BRD4 inhibitors JQ1 and I-BET. (b) Bioorthogonal probes for characterizing the function of JQ1/I-BET inhibitors in cells. (c) A bump-and-hole approach for studying BET inhibition. Crystal structure depicting the hydrophobic contact between BRD4 L94 and I-BET (left). Schematic depiction of the complementary modifications introduced into I-BET and BRD4 (right). PDB, 3p5o. (d) First-generation PROTACs for the targeted degradation of BRD4. (e) Use of protein trans-splicing for the synthesis of modified dCas9 proteins. A dCas9-JQ1 fusion drove the locus-specific recruitment of BRD4, resulting in selective gene activation.
Shortly after the development of the JQ1 and I-BET inhibitors, several probe derivatives were developed to further understand the effects of BET inhibition in cells. Anders et al. synthesized a JQ1 affinity probe with a biotinylated moiety at C-6 of the diazepine (bio-JQ1), a substitution found to have only modest effects on inhibitor activity (Figure 9b) (185). By treating cells with bio-JQ1 and performing formaldehyde crosslinking, DNA-protein complexes interacting with bio-JQ1 were affinity purified and identified by deep sequencing, revealing the genome-wide distribution of the inhibitor. By comparing these results to ChIP-seq signals from BET proteins, bio-JQ1 occupancy was found to be most highly correlated to that of BRD4, consistent with the in vitro affinities of JQ1 for BET proteins. Li et al. developed a versatile set of probes based on a JQ1 analog that contained a photocrosslinker (diazirine) together with a clickable label (cyclopropane or alkyne) (Figure 9b) (186). Cell treatment with the cyclopropane probe and subsequent labeling with a cell-permeable, tetrazine-containing fluorophore allowed for live-cell imaging of the inhibitor which, after fixing the cells, was determined to co-localize with BRD4 via immunofluorescence. Additionally, the photocrosslinking moiety was used to capture the probe’s interactome with and without competition with JQ1, identifying potential off-target proteins of the JQ1 inhibitor.
Although BD inhibitors such as JQ1 and I-BET have been effective tools for studying BET proteins, they exhibit poor selectivity for individual sub-family members. Thus, probing the functions of individual BET proteins or specific BDs within those proteins has been challenging with standard small-molecule inhibitors. The development of a bump-and-hole approach by the Ciulli group for BET protein inhibition allowed, for the first time, selective inhibition of individual BDs within the BET family (187). By analyzing the crystal structures of JQ1/I-BET bound to BET bromodomains, a conserved leucine residue (L94 in BRD4) was identified that forms a hydrophobic contact with the inhibitors. Mutating this leucine to alanine created a hole in the BD, which was found to be efficiently complemented by an ethyl bump introduced to a side-chain methylene on I-BET (Figure 9c). Importantly, because the leucine was a conserved residue, the same bump-and-hole strategy could be applied to the BD1s and BD2s in the entire BET family, offering an average selectivity of 160-fold over wild-type. The system was then used in cells to assess the relative functions of the two bromodomains in BRD4, and determined that inhibition of BD1 alone is sufficient to displace BRD4 from chromatin. Further optimization of this system developed improved bump-and-hole pairs which found that the second bromodomain of BRD4, although less involved in chromatin binding, was essential for transcription of NF-κB genes, highlighting the importance of BD interactions with non-histone proteins in genetic regulation (188).
In 2015, three groups published bifunctional compounds for the chemically-induced degradation of BRD4. The PROTACs dBET1 (107), ARV-825 (108), and MZ1 (189) all utilize a thienotriazolodiazepine scaffold as the targeting ligand and show similar potency in the submicromolar range, but differ in their linker design, recruited E3 ligase, and BET protein selectivity (Figure 9d). The Bradner group developed dBET1, which consists of JQ1 linked to thalidomide, a phthalimide immunomodulatory drug that binds to and recruits cereblon (CRBN), a component of an E3 ligase complex (107). dBET1 treatment efficiently degraded BRD4 within hours in human leukemia cells and, consistent with the pan-BET selectivity of JQ1, also degraded BRD2 and BRD3. dBET1 exhibited improved antitumor efficacy in a human leukemia xenograft compared to JQ1 alone, establishing an exciting precedent for the improved therapeutic benefits of PROTACs. The Crews group took a similar approach in the design of ARV-825, which utilized OTX015 (a JQ1 derivative with improved pharmacokinetic properties) coupled to pomalidomide, another CRBN-targeting ligand (108). ARV-825 degraded BRD4 with high potency, exhibiting a DC50 (50% of maximal degradation) below 1 nM, and exhibited increased apoptosis and suppressed proliferation relative to JQ1/OTX015 alone in lymphoma cells. For the development of MZ1, the Ciulli group utilized JQ1 coupled to ligands that targeted the von Hippel-Lindau (VHL) E3 ligase complex (189). Surprisingly, MZ1 exhibited selectivity for BRD4 over BRD2/BRD3, providing evidence that it may be possible to leverage the PROTAC design for improved drug-targeting specificity. Since these initial works, BRD4-targeting PROTACs have been further optimized, leading to compounds with very high potencies down to the picomolar range (190, 191). Furthermore, the recent development of photocaged and photoswitchable PROTACs for BRD4 degradation offer exciting potential for enhanced spatiotemporal control in both research and therapeutic contexts (192, 193).
BRD4 recruitment by JQ1 has also proven useful in efforts toward locus-specific epigenome manipulation. By fusing a nuclease-deficient Cas9 (dCas9) to the N-terminal fragment of a split intein (IntN), cargo fused to the corresponding IntC fragment could be tracelessly ligated to dCas9, offering a highly modular approach for targeting synthetic entities to the genome (144). This approach was used to couple JQ1 to dCas9 which, when delivered to cells, showed gene-specific activation based on the recruitment of endogenous BRD4 (Figure 9e). Demonstrating the versatility of this approach, a dCas9 coupled to a branched cargo peptide containing four JQ1 moieties was synthesized, which showed greatly enhanced locus-specific recruitment of a transiently transfected BRD4 construct.
3.4. Heterochromatin and HP1
Heterochromatin is a compacted chromatin state that is primarily found at genomic regions associated with transcriptional repression, such as the centromeres, telomeres, and inactive X-chromosome. Critical to the formation of heterochromatin is the presence of the epigenetic mark H3K9me2/3 and its interaction with heterochromatin protein 1 (HP1), a 25 kDa protein that stabilizes chromatin strand interactions and promotes chromatin compaction (Figure 10a) (194). In mammalian cells HP1 has three isoforms: α and β, which are found predominantly in heterochromatin, and γ, which is associated with both heterochromatin and euchromatin. All of these subtypes have three domains that mediate chromatin interactions, a chromodomain (CD) that binds H3K9me2/3, a hinge region that binds DNA and RNA, and a chromoshadow domain (CSD) that mediates HP1 dimerization. Thus, the binding of HP1 to chromatin exhibits a potential for multivalency that contributes to a complex and dynamic chromatin state. Accordingly, methods in protein chemistry and, in particular, analytical chemistry experiments have been central in efforts to understand the role of HP1 in heterochromatin formation as well as the molecular dynamics of the resulting macromolecular complex.
Figure 10.
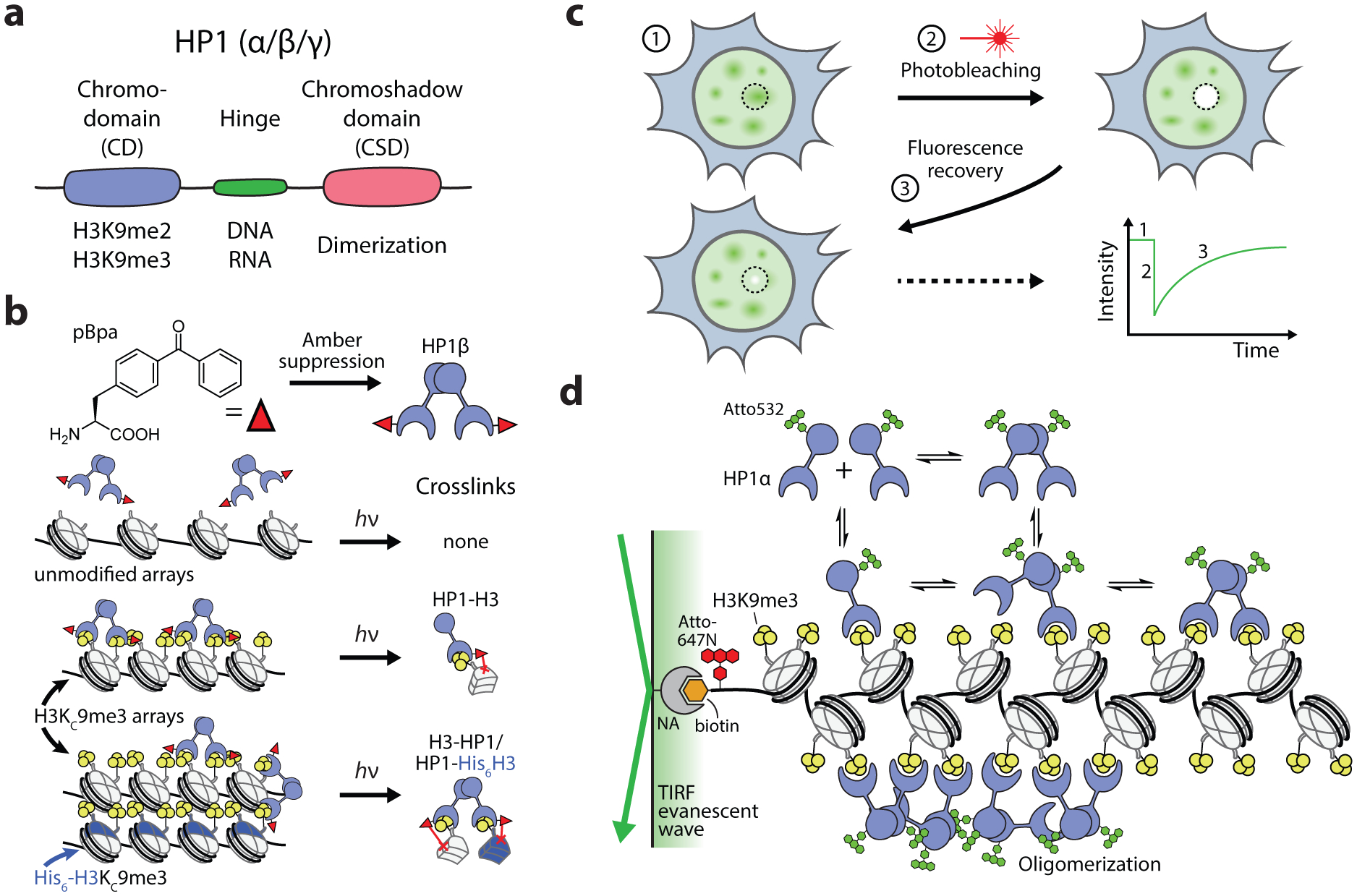
Biochemical and biophysical analyses of HP1-mediated heterochromatin formation. (a) Domain structure and interactions of HP1 proteins. (b) Site-selective incorporation of pBpa into HP1β and subsequent photocrosslinking with chromatin arrays demonstrated that HP1β mediates both intra- and inter-array interactions. H3KC9me3 histones were produced using the Cys-labeling approach depicted in Figure 2a. (c) Use of FRAP for quantifying the dynamics of fluorescently labeled proteins in live cells. (d) Single-molecule TIRF approach for measuring the interaction dynamics of HP1α proteins with methylated (H3K9me3) chromatin arrays. Real-time interaction kinetics were determined by observing the co-localization of surface-immobilized arrays (Atto647N-labeled) with diffusing HP1 protein (Atto532-labeled). NA, neutravidin.
Early work probing the role of the HP1-H3K9me3 interaction in heterochromatin benefited from the use of a cysteine mutagenesis and alkylation approach to generate a trimethyl-lysine mimic on position 9 of histone H3 (H3KC9me3). Assembling chromatin substrates with H3KC9me3, Canzio et al. demonstrated that, upon recognition of the methyl mark, the yeast HP1 homolog Swi6 exhibited oligomerization, establishing that HP1 binds chromatin in a cooperative fashion that bridges interactions between neighboring nucleosomes (195). Using similar substrates, the roles of the CD, hinge, and CSD domains of the human subtype HP1β was scrutinized in detailed biophysical experiments using TROSY-based NMR spectroscopy (196). CD binding of H3KC9me3 was shown to be a highly dynamic interaction, and the hinge region exhibited weak, non-specific interactions with DNA. To determine whether HP1 can bridge long-range interactions between different chromatin fibers, Hiragami-Hamada and coworkers incorporated the photo-crosslinkable ncAA p-benzoyl-L-phenylalanine (pBpa) into the CD of human HP1β via amber suppression (197). Synthetic chromatin arrays with tagged (His6) and untagged H3 were used to distinguish between intra- and inter-array interactions. Consistent with previous results, crosslinks were observed in the presence of H3K9me3 but not in unmodified arrays and, furthermore, crosslinks were detected that identified single HP1 dimers linked to both the tagged and untagged H3, demonstrating that HP1-H3K9me3 binding also mediates inter-fiber chromatin interactions (Figure 10b). HP1 interactions and dynamics in the context of cellular heterochromatin have been assessed by fluorescence recovery after photobleaching (FRAP) approaches (Figure 10c), which determined that HP1 exhibited short residence times in the range of milliseconds to seconds, consistent with the highly dynamic interactions found in vitro (198, 199). Enhanced association of HP1 with heterochromatin was found to be dependent on the presence of the histone methyltransferase that installs H3K9me3, Suv39h1/2, further supporting the role of the HP1-H3K9me3 interaction in heterochromatin formation in cells (198).
An interesting result of these studies is that, while HP1 is efficiently recruited to heterochromatin domains, the resulting network exhibits highly dynamic features. The role of multivalent binding domains in HP1-chromatin dynamics was investigated by Kilic et al., who developed an approach to measure the interaction dynamics of HP1α using single-molecule total internal reflection (smTIRF) microscopy (Figure 10d) (200). In this setup, chromatin arrays and HP1α molecules were labeled with distinct fluorescent probes and chromatin arrays were immobilized onto a coverslip via a biotin-neutravidin anchor, allowing the direct kinetic analysis of individual HP1α-chromatin interactions. A key strategic development to uncover the effects of HP1α multivalency was the synthesis of both a covalently-linked HP1α dimer as well as a peptide known to induce HP1α dimerization, allowing for the direct assessment of multivalent contributions to HP1α binding. Subsequent kinetic analyses revealed that multivalent interactions accelerated HP1α association and prolonged binding residence time. Dissociated HP1α was additionally found to rapidly rebind at neighboring mononucleosome sites. In order to understand how HP1 proteins control chromatin compaction, FRET probes were introduced into the 12×601 DNA template for chromatin assembly (50). These reporters were placed with base-pair precision to sense interactions of nucleosome stacking, inter-nucleosome interactions near the dyad, and the dynamics of the linking DNA region between nucleosomes. Single-molecule FRET experiments revealed a detailed set of exchange kinetics of HP1-mediated chromatin compaction, finding that HP1α transiently stabilizes interacting nucleosomes in the chromatin fiber through H3K9me3 interactions. Thus, multivalent interactions of HP1α promote recruitment to chromatin while maintaining a highly dynamic state.
The complexity of HP1-heterochromatin interactions was further extended by recent reports that mammalian HP1α and its Drosophila homolog HP1a can promote liquid-liquid phase separation in vitro (201, 202). Indeed, multivalent protein interactions has previously been proposed to drive phase separation as a way of compartmentalizing various biological processes (129). Larson et al. utilize a DNA curtain assay to visualize the real-time compaction of individual chromatin strands in a flow-cell using TIRF microscopy, revealing that mutations to a basic patch in the HP1α hinge region slow rates of compaction, presumably due to disruption of DNA binding capability (201). Heterochromatin domains in cells labeled with fluorescently-tagged HP1 manifest as defined foci, and mutations that prevent HP1 dimerization prevented foci formation (201, 202). Importantly, at the early stages of formation in Drosophila embryos, these foci exhibited several characteristics that are consistent with liquid-liquid phase separation, including spheroid shape, the ability to fuse, and reduced diffusion near heterochromatin boundaries (202). Further analysis with FRAP revealed that heterochromatin foci mature over time and exhibit a partially immobile fraction, indicating that the liquid condensate properties of heterochromatin may be altered and diminished by developmental biological processes (202). Indeed, acetylation of chromatin substrates was found to decrease droplet formation in HeLa cell nuclei (203). Unexpectedly, HDX-MS analysis of phase-separated condensates formed by the yeast HP1 protein Swi6 revealed increased accessibility of histone residues that are normally buried within the nucleosome structure (120), raising the potential for an additional layer of multivalent interactions in phase-separated heterochromatin.
SUMMARY
The progress made in the field of epigenetics in recent years has been remarkable. Increasingly sensitive genomics and proteomics tools allow us to dig ever deeper into the ‘fine structure’ of chromatin, revealing correlations between chromatin states and the regulation of DNA transactions. The chemical biology approaches summarized herein are united by the common goal of uncovering the biochemical processes driving such correlations. Indeed, one could argue that the back and forth that now exists between ‘omics’ and chemical biology approaches creates something of a virtuous cycle which drives the development of ever more sophisticated tools for characterizing and manipulating chromatin. These advances are unearthing new paradigms in epigenetics – for example, the existence of biochemical crosstalk between different chromatin marks (see case studies 1 and 2) or the role of phase separation in heterochromatin (see case study 4) – whilst also providing molecular tools that validate epigenetic targets in disease (see case study 3). Looking to the future, we imagine that continuing efforts will be made toward locus specific engineering of native cellular chromatin, ideally with the same level of molecular precision that is already possible in the test-tube. If fully realized, this capability will allow biochemical mechanisms established using reconstituted in vitro systems to be directly validated in a cellular context. Thus, as the current group of chemical biology tools for studying epigenetics come of age, we can already envision the exciting opportunities that will be afforded by the next generation.
ACKNOWLEDGEMENTS
We thank the National Institutes of Health (NIH, R37 GM086868 and P01 CA196539) for financial support. J.D.B. was supported by a postdoctoral NIH fellowship (GM123659). We also thank members of the Muir lab for helpful discussions.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. 1997. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 389:251–60 [DOI] [PubMed] [Google Scholar]
- 2.Clapier CR, Iwasa J, Cairns BR, Peterson CL. 2017. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol 18:407–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Woodcock CL, Ghosh RP. 2010. Chromatin higher-order structure and dynamics. Cold Spring Harb. Perspect. Biol 2:a000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang H, Sabari BR, Garcia BA, David Allis C, Zhao Y. 2014. SnapShot: Histone modifications. Cell. 159:458–458.e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jenuwein T, Allis CD. 2001. Translating the histone code. Science. 293:1074–80 [DOI] [PubMed] [Google Scholar]
- 6.Dawson MA, Kouzarides T. 2012. Cancer epigenetics: From mechanism to therapy. Cell. 150:12–27 [DOI] [PubMed] [Google Scholar]
- 7.Budhavarapu VN, Chavez M, Tyler JK. 2013. How is epigenetic information maintained through DNA replication? Epigenetics and Chromatin. 6:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allfrey VG, Littau VC, Mirsky AE. 1963. On the role of of histones in regulation ribonucleic acid synthesis in the cell nucleus. Proc. Natl. Acad. Sci. U.S.A 49:414–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang R-C, Bonner J. 1962. Histone, a suppressor of chromosomal RNA synthesis. Proc. Nat. Acad. Sci. U.S.A 48:1216–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allfrey VG, Faulkner R, Mirsky AE. 1964. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. U.S.A 51:786–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krieger DE, Vidali G, Erickson BW, Allfrey VG, Merrifield RB. 1979. The synthesis of diacetylated histone H4-(1–37) for studies on the mechanism of histone deacetylation. Bioorg. Chem 8:409–27 [Google Scholar]
- 12.Krieger DE, Levine R, Merrifield RB, Vidali G, Allfrey VG. 1974. Chemical Studies of Histone Acetylation. J. Biol. Chem 249:332–34 [PubMed] [Google Scholar]
- 13.Taunton J, Collins JL, Schreiber SL. 1996. Synthesis of natural and modified trapoxins, useful reagents for exploring histone deacetylase function. J. Am. Chem. Soc 118:10412–22 [Google Scholar]
- 14.Taunton J, Hassig CA, Schreiber SL. 1996. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 272:408–11 [DOI] [PubMed] [Google Scholar]
- 15.Müller MM, Muir TW. 2015. Histones: At the crossroads of peptide and protein chemistry. Chem. Rev 115:2296–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischle W, Schwarzer D. 2016. Probing Chromatin-modifying Enzymes with Chemical Tools. ACS Chem. Biol 11:689–705 [DOI] [PubMed] [Google Scholar]
- 17.Boichenko I, Fierz B. 2019. Chemical and biophysical methods to explore dynamic mechanisms of chromatin silencing. Curr. Opin. Chem. Biol 51:1–10 [DOI] [PubMed] [Google Scholar]
- 18.Booth MJ, Raiber E-A, Balasubramanian S. 2015. Chemical methods for decoding cytosine modifications in DNA. Chem. Rev 115:2240–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reuter JA, Spacek DV., Snyder MP. 2015. High-Throughput Sequencing Technologies. Mol. Cell 58:586–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thompson RE, Muir TW. 2020. Chemoenzymatic Semisynthesis of Proteins. Chem. Rev 120:3051–3126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Debelouchina GT, Muir TW. 2017. A molecular engineering toolbox for the structural biologist. Q. Rev. Biophys 50:e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simon MD, Chu F, Racki LR, de la Cruz CC, Burlingame AL, et al. 2007. The Site-Specific Installation of Methyl-Lysine Analogs into Recombinant Histones. Cell. 128:1003–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dadová J, Galan SR, Davis BG. 2018. Synthesis of modified proteins via functionalization of dehydroalanine. Curr. Opin. Chem. Biol 46:71–81 [DOI] [PubMed] [Google Scholar]
- 24.Lechner CC, Agashe ND, Fierz B. 2016. Traceless Synthesis of Asymmetrically Modified Bivalent Nucleosomes. Angew. Chemie Int. Ed 55:2903–6 [DOI] [PubMed] [Google Scholar]
- 25.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, et al. 2006. A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell. 125:315–26 [DOI] [PubMed] [Google Scholar]
- 26.Lang K, Chin JW. 2014. Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins. Chem. Rev 114:4764–4806 [DOI] [PubMed] [Google Scholar]
- 27.Nguyen DP, Alai MMG, Kapadnis PB, Neumann H, Chin JW. 2009. Genetically Encoding Nε-Methyl-L-lysine in Recombinant Histones. J. Am. Chem. Soc 131:14194–95 [DOI] [PubMed] [Google Scholar]
- 28.Neumann H, Peak-Chew SY, Chin JW. 2008. Genetically encoding N(epsilon)-acetyllysine in recombinant proteins. Nat. Chem. Biol 4:232–34 [DOI] [PubMed] [Google Scholar]
- 29.Lee S, Oh S, Yang A, Kim J, Söll D, et al. 2013. A Facile Strategy for Selective Incorporation of Phosphoserine into Histones. Angew. Chemie Int. Ed 52:5771–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gattner MJ, Vrabel M, Carell T. 2013. Synthesis of ε-N-propionyl-, ε-N-butyryl-, and ε-N-crotonyl-lysine containing histone H3 using the pyrrolysine system. Chem. Commun 49:379–81 [DOI] [PubMed] [Google Scholar]
- 31.Elsässer SJ, Ernst RJ, Walker OS, Chin JW. 2016. Genetic code expansion in stable cell lines enables encoded chromatin modification. Nat. Methods 13:158–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilkins BJ, Rall NA, Ostwal Y, Kruitwagen T, Hiragami-Hamada K, et al. 2014. A cascade of histone modifications induces chromatin condensation in mitosis. Science. 343:77–80 [DOI] [PubMed] [Google Scholar]
- 33.Kleiner RE, Hang LE, Molloy KR, Chait BT, Kapoor TM. 2018. A Chemical Proteomics Approach to Reveal Direct Protein-Protein Interactions in Living Cells. Cell Chem. Biol 25:110–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang T, Li X-M, Bao X, Fung YME, Li XD. 2016. Photo-lysine captures proteins that bind lysine post-translational modifications. Nat. Chem. Biol 12:70–72 [DOI] [PubMed] [Google Scholar]
- 35.Xie X, Li X-M, Qin F, Lin J, Zhang G, et al. 2017. Genetically Encoded Photoaffinity Histone Marks. J. Am. Chem. Soc 139:6522–25 [DOI] [PubMed] [Google Scholar]
- 36.Dawson PE, Muir TW, Clark-Lewis I, Kent SBH. 1994. Synthesis of proteins by native chemical ligation. Science. 266:776–79 [DOI] [PubMed] [Google Scholar]
- 37.Muir TW, Sondhi D, Cole PA. 1998. Expressed protein ligation: A general method for protein engineering. Proc. Natl. Acad. Sci. U.S.A 95:6705–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang TK, Jackson DY, Burnier JP, Wells JA. 1994. Subtiligase: A tool for semisynthesis of proteins. Proc. Natl. Acad. Sci. U.S.A 91:12544–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mao H, Hart SA, Schink A, Pollok BA. 2004. Sortase-Mediated Protein Ligation: A New Method for Protein Engineering. J. Am. Chem. Soc 126:2670–71 [DOI] [PubMed] [Google Scholar]
- 40.Nguyen UTT, Bittova L, Müller MM, Fierz B, David Y, et al. 2014. Accelerated chromatin biochemistry using DNA-barcoded nucleosome libraries. Nat. Methods 11:834–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dann GP, Liszczak GP, Bagert JD, Müller MM, Nguyen UTT, et al. 2017. ISWI chromatin remodellers sense nucleosome modifications to determine substrate preference. Nature. 548:607–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liszczak G, Diehl KL, Dann GP, Muir TW. 2018. Acetylation blocks DNA damage-induced chromatin ADP-ribosylation. Nat. Chem. Biol 14:837–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wojcik F, Dann GP, Beh LY, Debelouchina GT, Hofmann R, Muir TW. 2018. Functional crosstalk between histone H2B ubiquitylation and H2A modifications and variants. Nat. Commun 9:1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mirzabekov AD, Shick VV., Belyavsky AV., Karpov VL, Bavykin SG. 1978. The structure of nucleosomes: The arrangement of histones in the DNA grooves and along the DNA chain. Cold Spring Harb. Symp. Quant. Biol 42:149–55 [DOI] [PubMed] [Google Scholar]
- 45.Lowary PT, Widom J. 1998. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol 276:19–42 [DOI] [PubMed] [Google Scholar]
- 46.Li G, Widom J. 2004. Nucleosomes facilitate their own invasion. Nat. Struct. Mol. Biol 11:763–69 [DOI] [PubMed] [Google Scholar]
- 47.Simon M, North JA, Shimko JC, Forties RA, Ferdinand MB, et al. 2011. Histone fold modifications control nucleosome unwrapping and disassembly. Proc. Natl. Acad. Sci. U.S.A 108:12711–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neumann H, Hancock SM, Buning R, Routh A, Chapman L, et al. 2009. A Method for Genetically Installing Site-Specific Acetylation in Recombinant Histones Defines the Effects of H3 K56 Acetylation. Mol. Cell 36:153–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Poirier MG, Oh E, Tims HS, Widom J. 2009. Dynamics and function of compact nucleosome arrays. Nat. Struct. Mol. Biol 16:938–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kilic S, Felekyan S, Doroshenko O, Boichenko I, Dimura M, et al. 2018. Single-molecule FRET reveals multiscale chromatin dynamics modulated by HP1α. Nat. Commun 9:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fang K, Chen X, Li X, Shen Y, Sun J, et al. 2018. Super-resolution Imaging of Individual Human Subchromosomal Regions in Situ Reveals Nanoscopic Building Blocks of Higher-Order Structure. ACS Nano. 12:4909–18 [DOI] [PubMed] [Google Scholar]
- 52.Boettiger AN, Bintu B, Moffitt JR, Wang S, Beliveau BJ, et al. 2016. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature. 529:418–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bintu B, Mateo LJ, Su J-H, Sinnott-Armstrong NA, Parker M, et al. 2018. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science. 362:eaau1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hocek M 2019. Enzymatic Synthesis of Base-Functionalized Nucleic Acids for Sensing, Cross-linking, and Modulation of Protein-DNA Binding and Transcription. Acc. Chem. Res 52:1730–37 [DOI] [PubMed] [Google Scholar]
- 55.Smith ZD, Meissner A. 2013. DNA methylation: roles in mammalian development. Nat. Rev. Genet 14:204–20 [DOI] [PubMed] [Google Scholar]
- 56.Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, et al. 1992. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to Methylated DNA. Cell. 69:905–14 [DOI] [PubMed] [Google Scholar]
- 57.Meehan RR, Lewis JD, McKay S, Kleiner EL, Bird AP. 1989. Identification of a mammalian protein that binds specifically to DNA containing methylated CpGs. Cell. 58:499–507 [DOI] [PubMed] [Google Scholar]
- 58.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, et al. 2009. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 324:930–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Globisch D, Münzel M, Müller M, Michalakis S, Wagner M, et al. 2010. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One. 5:e15367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ito S, Shen L, Dai Q, Wu SC, Collins LB, et al. 2011. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 333:1300–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pfaffeneder T, Hackner B, Truß M, Münzel M, Müller M, et al. 2011. The discovery of 5-formylcytosine in embryonic stem cell DNA. Angew. Chemie Int. Ed 50:7008–12 [DOI] [PubMed] [Google Scholar]
- 62.Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, et al. 1992. A genomic sequencing protocol that yields a positive display of 5- methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. U.S.A 89:1827–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Berney M, McGouran JF. 2018. Methods for detection of cytosine and thymine modifications in DNA. Nat. Rev. Chem 2:332–48 [Google Scholar]
- 64.Song C-X, Szulwach KE, Fu Y, Dai Q, Yi C, et al. 2011. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol 29:68–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Raiber E-A, Beraldi D, Ficz G, Burgess HE, Branco MR, et al. 2012. Genome-wide distribution of 5-formylcytosine in embryonic stem cells is associated with transcription and depends on thymine DNA glycosylase. Genome Biol. 13:R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ren M, Bai J, Xi Z, Zhou C. 2019. DNA damage in nucleosomes. Sci. China Chem 62:561–70 [Google Scholar]
- 67.Sczepanski JT, Wong RS, McKnight JN, Bowman GD, Greenberg MM. 2010. Rapid DNA-protein cross-linking and strand scission by an abasic site in a nucleosome core particle. Proc. Natl. Acad. Sci. U.S.A 107:22475–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou C, Sczepanski JT, Greenberg MM. 2013. Histone modification via rapid cleavage of C4′-oxidized abasic sites in nucleosome core particles. J. Am. Chem. Soc 135:5274–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou C, Greenberg MM. 2012. Histone-catalyzed cleavage of nucleosomal DNA containing 2-deoxyribonolactone. J. Am. Chem. Soc 134:8090–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weng L, Greenberg MM. 2015. Rapid Histone-Catalyzed DNA Lesion Excision and Accompanying Protein Modification in Nucleosomes and Nucleosome Core Particles. J. Am. Chem. Soc 137:11022–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Todd RC, Lippard SJ. 2010. Consequences of cisplatin binding on nucleosome structure and dynamics. Chem. Biol 17:1334–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhu G, Song L, Lippard SJ. 2013. Visualizing inhibition of nucleosome mobility and transcription by cisplatin-DNA interstrand crosslinks in live mammalian cells. Cancer Res. 73:4451–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Núñez ME, Noyes KT, Barton JK. 2002. Oxidative charge transport through DNA in nucleosome core particles. Chem. Biol 9:403–15 [DOI] [PubMed] [Google Scholar]
- 74.Davis WB, Bjorklund CC, Deline M. 2012. Probing the effects of DNA-protein interactions on DNA hole transport: The N-terminal histone tails modulate the distribution of oxidative damage and chemical lesions in the nucleosome core particle. Biochemistry. 51:3129–42 [DOI] [PubMed] [Google Scholar]
- 75.Menoni H, Shukla MS, Gerson V, Dimitrov S, Angelov D. 2012. Base excision repair of 8-oxoG in dinucleosomes. Nucleic Acids Res. 40:692–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Beh LY, Debelouchina GT, Clay DM, Thompson RE, Lindblad KA, et al. 2019. Identification of a DNA N6-Adenine Methyltransferase Complex and Its Impact on Chromatin Organization. Cell. 177:1781–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Banerjee DR, Deckard CE, Elinski MB, Buzbee ML, Wang WW, et al. 2018. Plug-and-Play Approach for Preparing Chromatin Containing Site-Specific DNA Modifications: The Influence of Chromatin Structure on Base Excision Repair. J. Am. Chem. Soc 140:8260–67 [DOI] [PubMed] [Google Scholar]
- 78.Deckard CE, Banerjee DR, Sczepanski JT. 2019. Chromatin Structure and the Pioneering Transcription Factor FOXA1 Regulate TDG-Mediated Removal of 5-Formylcytosine from DNA. J. Am. Chem. Soc 141:14110–14 [DOI] [PubMed] [Google Scholar]
- 79.Shortt J, Ott CJ, Johnstone RW, Bradner JE. 2017. A chemical probe toolbox for dissecting the cancer epigenome. Nat. Rev. Cancer 17:160–83 [DOI] [PubMed] [Google Scholar]
- 80.Liu C, Yu Y, Liu F, Wei X, Wrobel JA, et al. 2014. A chromatin activity-based chemoproteomic approach reveals a transcriptional repressome for gene-specific silencing. Nat. Commun 5:5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Salisbury CM, Cravatt BF. 2007. Activity-based probes for proteomic profiling of histone deacetylase complexes. Proc. Natl. Acad. Sci 104:1171–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, et al. 2011. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol 29:255–68 [DOI] [PubMed] [Google Scholar]
- 83.Xu C, Soragni E, Chou CJ, Herman D, Plasterer HL, et al. 2009. Chemical Probes Identify a Role for Histone Deacetylase 3 in Friedreich’s Ataxia Gene Silencing. Chem. Biol 16:980–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lau OD, Kundu TK, Soccio RE, Ait-Si-Ali S, Khalil EM, et al. 2000. HATs off: Selective synthetic inhibitors of the histone acetyltransferases p300 and PCAF. Mol. Cell 5:589–95 [DOI] [PubMed] [Google Scholar]
- 85.Dose A, Sindlinger J, Bierlmeier J, Bakirbas A, Schulze-Osthoff K, et al. 2016. Interrogating substrate selectivity and composition of endogenous histone deacetylase complexes with chemical probes. Angew. Chemie Int. Ed 55:1192–95 [DOI] [PubMed] [Google Scholar]
- 86.Dalhoff C, Lukinavičius G, Klimašauskas S, Weinhold E. 2006. Direct transfer of extended groups from synthetic cofactors by DNA methyltransferases. Nat. Chem. Biol 2:31–32 [DOI] [PubMed] [Google Scholar]
- 87.Bothwell IR, Islam K, Chen Y, Zheng W, Blum G, et al. 2012. Se-adenosyl-L-selenomethionine cofactor analogue as a reporter of protein methylation. J. Am. Chem. Soc 134:14905–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang R, Ibáñez G, Islam K, Zheng W, Blum G, et al. 2011. Formulating a fluorogenic assay to evaluate S-adenosyl-L-methionine analogues as protein methyltransferase cofactors. Mol. Biosyst 7:2970–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang R, Zheng W, Yu H, Deng H, Luo M. 2011. Labeling substrates of protein arginine methyltransferase with engineered enzymes and matched S-adenosyl-l-methionine analogues. J. Am. Chem. Soc 133:7648–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Islam K, Zheng W, Yu H, Deng H, Luo M. 2011. Expanding cofactor repertoire of protein lysine methyltransferase for substrate labeling. ACS Chem. Biol 6:679–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang R, Islam K, Liu Y, Zheng W, Tang H, et al. 2013. Profiling genome-wide chromatin methylation with engineered posttranslation apparatus within living cells. J. Am. Chem. Soc 135:1048–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yang Y-Y, Hang HC. 2011. Chemical Approaches for the Detection and Synthesis of Acetylated Proteins. ChemBioChem. 12:314–22 [DOI] [PubMed] [Google Scholar]
- 93.Yang C, Mi J, Feng Y, Ngo L, Gao T, et al. 2013. Labeling lysine acetyltransferase substrates with engineered enzymes and functionalized cofactor surrogates. J. Am. Chem. Soc 135:7791–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hengeveld RCC, Hertz NT, Vromans MJM, Zhang C, Burlingame AL, et al. 2012. Development of a chemical genetic approach for human aurora B kinase identifies novel substrates of the chromosomal passenger complex. Mol. Cell. Proteomics 11:47–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Spencer DM, Wandless TJ, Schreiber SL, Crabtree GR. 1993. Controlling Signal Transduction with Synthetic Ligands. Science. 262:1019–24 [DOI] [PubMed] [Google Scholar]
- 96.Stanton BZ, Chory EJ, Crabtree GR. 2018. Chemically induced proximity in biology and medicine. Science. 359:eaao5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hathaway NA, Bell O, Hodges C, Miller EL, Neel DS, Crabtree GR. 2012. Dynamics and memory of heterochromatin in living cells. Cell. 149:1447–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Erickson BK, Rose CM, Braun CR, Erickson AR, Knott J, et al. 2017. A Strategy to Combine Sample Multiplexing with Targeted Proteomics Assays for High-Throughput Protein Signature Characterization. Mol. Cell 65:361–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kadoch C, Williams RT, Calarco JP, Miller EL, Weber CM, et al. 2017. Dynamics of BAF-Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat. Genet 49:213–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stanton BZ, Hodges C, Calarco JP, Braun SMG, Ku WL, et al. 2017. Smarca4 ATPase mutations disrupt direct eviction of PRC1 from chromatin. Nat. Genet 49:282–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Morgan SL, Mariano NC, Bermudez A, Arruda NL, Wu F, et al. 2017. Manipulation of nuclear architecture through CRISPR-mediated chromosomal looping. Nat. Commun 8:15993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Braun SMG, Kirkland JG, Chory EJ, Husmann D, Calarco JP, Crabtree GR. 2017. Rapid and reversible epigenome editing by endogenous chromatin regulators. Nat. Commun 8:560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ballister ER, Aonbangkhen C, Mayo AM, Lampson MA, Chenoweth DM. 2014. Localized light-induced protein dimerization in living cells using a photocaged dimerizer. Nat. Commun 5:5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Burslem GM, Crews CM. 2020. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell. 181:102–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. 2001. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U.S.A 98:8554–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schneekloth AR, Pucheault M, Tae HS, Crews CM. 2008. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorganic Med. Chem. Lett 18:5904–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, et al. 2015. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 348:1376–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lu J, Qian Y, Altieri M, Dong H, Wang J, et al. 2015. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol 22:755–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vogelmann A, Robaa D, Sippl W, Jung M. 2020. Proteolysis targeting chimeras (PROTACs) for epigenetics research. Curr. Opin. Chem. Biol 57:8–16 [DOI] [PubMed] [Google Scholar]
- 110.Rothbart SB, Dickson BM, Raab JR, Grzybowski AT, Krajewski K, et al. 2015. An Interactive Database for the Assessment of Histone Antibody Specificity. Mol. Cell 59:502–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhao Y, Garcia BA. 2015. Comprehensive catalog of currently documented histone modifications. Cold Spring Harb. Perspect. Biol 7:a025064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Simithy J, Sidoli S, Garcia BA. 2018. Integrating Proteomics and Targeted Metabolomics to Understand Global Changes in Histone Modifications. Proteomics. 18:1700309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fan J, Krautkramer KA, Feldman JL, Denu JM. 2015. Metabolic regulation of histone post-translational modifications. ACS Chem. Biol 10:95–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, et al. 2010. Quantitative Interaction Proteomics and Genome-wide Profiling of Epigenetic Histone Marks and Their Readers. Cell. 142:967–80 [DOI] [PubMed] [Google Scholar]
- 115.Bartke T, Vermeulen M, Xhemalce B, Robson SC, Mann M, Kouzarides T. 2010. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell. 143:470–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Li X, Foley EA, Molloy KR, Li Y, Chait BT, Kapoor TM. 2012. Quantitative Chemical Proteomics Approach To Identify Post- translational Modification-Mediated Protein − Protein Interactions. J. Am. Chem. Soc 134:16–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hodge EA, Benhaim MA, Lee KK. 2020. Bridging protein structure, dynamics, and function using hydrogen/deuterium-exchange mass spectrometry. Protein Sci. 29:843–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Denizio JE, Elsässer SJ, Black BE. 2014. DAXX co-folds with H3.3/H4 using high local stability conferred by the H3.3 variant recognition residues. Nucleic Acids Res. 42:4318–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Karch KR, Coradin M, Zandarashvili L, Kan Z-Y, Gerace M, et al. 2018. Hydrogen-Deuterium Exchange Coupled to Top- and Middle-Down Mass Spectrometry Reveals Histone Tail Dynamics before and after Nucleosome Assembly. Structure. 26:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sanulli S, Trnka MJ, Dharmarajan V, Tibble RW, Pascal BD, et al. 2019. HP1 reshapes nucleosome core to promote phase separation of heterochromatin. Nature. 575:390–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Böhm V, Hieb AR, Andrews AJ, Gansen A, Rocker A, et al. 2011. Nucleosome accessibility governed by the dimer/tetramer interface. Nucleic Acids Res. 39:3093–3102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hall MA, Shundrovsky A, Bai L, Fulbright RM, Lis JT, Wang MD. 2009. High-resolution dynamic mapping of histone-DNA interactions in a nucleosome. Nat. Struct. Mol. Biol 16:124–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fierz B, Chatterjee C, McGinty RK, Bar-Dagan M, Raleigh DP, Muir TW. 2011. Histone H2B ubiquitylation disrupts local and higher-order chromatin compaction. Nat. Chem. Biol 7:113–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li W, Chen P, Yu J, Dong L, Liang D, et al. 2016. FACT Remodels the Tetranucleosomal Unit of Chromatin Fibers for Gene Transcription. Mol. Cell 64:120–33 [DOI] [PubMed] [Google Scholar]
- 125.Fuller BG, Lampson MA, Foley EA, Rosasco-Nitcher S, Le KV., et al. 2008. Midzone activation of aurora B in anaphase produces an intracellular phosphorylation gradient. Nature. 453:1132–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sasaki K, Ito T, Nishino N, Khochbin S, Yoshida M. 2009. Real-time imaging of histone H4 hyperacetylation in living cells. Proc. Natl. Acad. Sci. U.S.A 106:16257–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lin C-W, Jao CY, Ting AY. 2004. Genetically Encoded Fluorescent Reporters of Histone Methylation in Living Cells. J. Am. Chem. Soc 126:5982–83 [DOI] [PubMed] [Google Scholar]
- 128.Bracha D, Walls MT, Wei M-T, Zhu L, Kurian M, et al. 2018. Mapping Local and Global Liquid Phase Behavior in Living Cells Using Photo-Oligomerizable Seeds. Cell. 175:1467–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Li P, Banjade S, Cheng H-C, Kim S, Chen B, et al. 2012. Phase transitions in the assembly of multivalent signalling proteins. Nature. 483:336–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jang S, Song J-J. 2019. The big picture of chromatin biology by cryo-EM. Curr. Opin. Struct. Biol 58:76–87 [DOI] [PubMed] [Google Scholar]
- 131.Zhou B-R, Feng H, Kato H, Dai L, Yang Y, et al. 2013. Structural insights into the histone H1-nucleosome complex. Proc. Natl. Acad. Sci. U.S.A 110:19390–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhang W, Tyl M, Ward R, Sobott F, Maman J, et al. 2013. Structural plasticity of histones H3–H4 facilitates their allosteric exchange between RbAp48 and ASF1. Nat. Struct. Mol. Biol 20:29–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rosenzweig R, Kay LE. 2014. Bringing Dynamic Molecular Machines into Focus by Methyl-TROSY NMR. Annu. Rev. Biochem 83:291–315 [DOI] [PubMed] [Google Scholar]
- 134.David Y, Muir TW. 2017. Emerging Chemistry Strategies for Engineering Native Chromatin. J. Am. Chem. Soc 139:9090–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Li F, Papworth M, Minczuk M, Rohde C, Zhang Y, et al. 2007. Chimeric DNA methyltransferases target DNA methylation to specific DNA sequences and repress expression of target genes. Nucleic Acids Res. 35:100–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Maeder ML, Angstman JF, Richardson ME, Linder SJ, Cascio VM, et al. 2013. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat. Biotechnol 31:1137–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Snowden AW, Gregory PD, Case CC, Pabo CO. 2002. Gene-specific targeting of H3K9 methylation is sufficient for initiating repression in vivo. Curr. Biol 12:2159–66 [DOI] [PubMed] [Google Scholar]
- 138.Kearns NA, Pham H, Tabak B, Genga RM, Silverstein NJ, et al. 2015. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat. Methods 12:401–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, et al. 2015. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol 33:510–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dervan PB, Edelson BS. 2003. Recognition of the DNA minor groove by pyrrole-imidazole polyamides. Curr. Opin. Struct. Biol 13:284–99 [DOI] [PubMed] [Google Scholar]
- 141.Gottesfeld JM, Neely L, Trauger JW, Baird EE, Dervan PB. 1997. Regulation of gene expression by small molecules. Nature. 387:202–5 [DOI] [PubMed] [Google Scholar]
- 142.Mapp AK, Ansari AZ, Ptashne M, Dervan PB. 2000. Activation of gene expression by small molecule transcription factors. Proc. Natl. Acad. Sci. U.S.A 97:3930–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zhao W, Wang Y, Liang F-S. 2020. Chemical and light inducible epigenome editing. Int. J. Mol. Sci 21:998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Liszczak GP, Brown ZZ, Kim SH, Oslund RC, David Y, Muir TW. 2017. Genomic targeting of epigenetic probes using a chemically tailored Cas9 system. Proc. Natl. Acad. Sci 114:681–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Erwin GS, Grieshop MP, Ali A, Qi J, Lawlor M, et al. 2017. Synthetic transcription elongation factors license transcription across repressive chromatin. Science. 358:1617–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.David Y, Vila-Perelló M, Verma S, Muir TW. 2015. Chemical tagging and customizing of cellular chromatin states using ultrafast trans-splicing inteins. Nat. Chem 7:394–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Burton AJ, Haugbro M, Parisi E, Muir TW. 2020. Live-cell protein engineering with an ultra-short split intein. Proc. Natl. Acad. Sci 117:12041–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Holt MT, David Y, Pollock S, Tang Z, Jeon J, et al. 2015. Identification of a functional hotspot on ubiquitin required for stimulation of methyltransferase activity on chromatin. Proc. Natl. Acad. Sci. U.S.A 112:10365–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Burton AJ, Haugbro M, Gates LA, Bagert JD, Allis CD, Muir TW. 2020. In situ chromatin interactomics using a chemical bait and trap approach. Nat. Chem 12:520–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chatterjee C, McGinty RK, Pellois J-P, Muir TW. 2007. Auxiliary-mediated site-specific peptide ubiquitylation. Angew. Chemie Int. Ed 46:2814–18 [DOI] [PubMed] [Google Scholar]
- 151.McGinty RK, Kim J, Chatterjee C, Roeder RG, Muir TW. 2008. Chemically ubiquitylated histone H2B stimulates hDot1L-mediated intranucleosomal methylation. Nature. 453:812–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kim J, Guermah M, McGinty RK, Lee J-S, Tang Z, et al. 2009. RAD6-Mediated Transcription-Coupled H2B Ubiquitylation Directly Stimulates H3K4 Methylation in Human Cells. Cell. 137:459–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.McGinty RK, Köhn M, Chatterjee C, Chiang KP, Pratt MR, Muir TW. 2009. Structure-activity analysis of semisynthetic nucleosomes: Mechanistic insights into the stimulation of Dot1L by ubiquitylated histone H2B. ACS Chem. Biol 4:958–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chatterjee C, McGinty RK, Fierz B, Muir TW. 2010. Disulfide-directed histone ubiquitylation reveals plasticity in hDot1L activation. Nat. Chem. Biol 6:267–69 [DOI] [PubMed] [Google Scholar]
- 155.Zhou L, Holt MT, Ohashi N, Zhao A, Müller MM, et al. 2016. Evidence that ubiquitylated H2B corrals hDot1L on the nucleosomal surface to induce H3K79 methylation. Nat. Commun 7:10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Anderson CJ, Baird MR, Hsu A, Barbour EH, Koyama Y, et al. 2019. Structural Basis for Recognition of Ubiquitylated Nucleosome by Dot1L Methyltransferase. Cell Rep. 26:1681–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Worden EJ, Hoffmann NA, Hicks CW, Wolberger C. 2019. Mechanism of Cross-talk between H2B Ubiquitination and H3 Methylation by Dot1L. Cell. 176:1490–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Debelouchina GT, Gerecht K, Muir TW. 2017. Ubiquitin utilizes an acidic surface patch to alter chromatin structure. Nat. Chem. Biol 13:105–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Razin A, Riggs AD. 1980. DNA methylation and gene function. Science. 210:604–10 [DOI] [PubMed] [Google Scholar]
- 160.Mohandas T, Sparkes RS, Shapiro LJ. 1981. Reactivation of an Inactive Human X Chromosome: Evidence for X Inactivation by DNA Methylation. Science. 211:393–96 [DOI] [PubMed] [Google Scholar]
- 161.Boyes J, Bird A. 1991. DNA methylation inhibits transcription indirectly via a methyl-CpG binding protein. Cell. 64:1123–34 [DOI] [PubMed] [Google Scholar]
- 162.Watt F, Molloy PL. 1988. Cytosine methylation prevents binding to DNA of a HeLa cell transcription factor required for optimal expression of the adenovirus major late promoter. Genes Dev. 2:1136–43 [DOI] [PubMed] [Google Scholar]
- 163.Ito S, D’Alessio AC, Taranova OV., Hong K, Sowers LC, Zhang Y. 2010. Role of tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 466:1129–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kohli RM, Zhang Y. 2013. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 502:472–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Bachman M, Uribe-Lewis S, Yang X, Burgess HE, Iurlaro M, et al. 2015. 5-Formylcytosine can be a stable DNA modification in mammals. Nat. Chem. Biol 11:555–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Bachman M, Uribe-Lewis S, Yang X, Williams M, Murrell A, Balasubramanian S. 2014. 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat. Chem 6:1049–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Su M, Kirchner A, Stazzoni S, Müller M, Wagner M, et al. 2016. 5-Formylcytosine Could Be a Semipermanent Base in Specific Genome Sites. Angew. Chemie Int. Ed 55:11797–800 [DOI] [PubMed] [Google Scholar]
- 168.Jin S-G, Jiang Y, Qiu R, Rauch TA, Wang Y, et al. 2011. 5-hydroxymethylcytosine is strongly depleted in human cancers but its levels do not correlate with IDH1 mutations. Cancer Res. 71:7360–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Vasanthakumar A, Godley LA. 2015. 5-hydroxymethylcytosine in cancer: significance in diagnosis and therapy. Cancer Genet. 208:167–77 [DOI] [PubMed] [Google Scholar]
- 170.Song C-X, Yin S, Ma L, Wheeler A, Chen Y, et al. 2017. 5-Hydroxymethylcytosine signatures in cell-free DNA provide information about tumor types and stages. Cell Res. 27:1231–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Li W, Zhang X, Lu X, You L, Song Y, et al. 2017. 5-Hydroxymethylcytosine signatures in circulating cell-free DNA as diagnostic biomarkers for human cancers. Cell Res. 27:1243–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PWTC, et al. 2013. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 152:1146–59 [DOI] [PubMed] [Google Scholar]
- 173.Iurlaro M, Ficz G, Oxley D, Raiber E-A, Bachman M, et al. 2013. A screen for hydroxymethylcytosine and formylcytosine binding proteins suggests functions in transcription and chromatin regulation. Genome Biol. 14:R119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Mendonca A, Chang EH, Liu W, Yuan C. 2014. Hydroxymethylation of DNA influences nucleosomal conformation and stability in vitro. Biochim. Biophys. Acta - Gene Regul. Mech 1839:1323–29 [DOI] [PubMed] [Google Scholar]
- 175.Ngo TTM, Yoo J, Dai Q, Zhang Q, He C, et al. 2016. Effects of cytosine modifications on DNA flexibility and nucleosome mechanical stability. Nat. Commun 7:10813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Song C-X, Szulwach KE, Dai Q, Fu Y, Mao SQ, et al. 2013. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell. 153:678–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Li F, Zhang Y, Bai J, Greenberg MM, Xi Z, Zhou C. 2017. 5-Formylcytosine Yields DNA-Protein Cross-Links in Nucleosome Core Particles. J. Am. Chem. Soc 139:10617–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Raiber E-A, Portella G, Cuesta SM, Hardisty R, Murat P, et al. 2018. 5-Formylcytosine organizes nucleosomes and forms Schiff base interactions with histones in mouse embryonic stem cells. Nat. Chem 10:1258–66 [DOI] [PubMed] [Google Scholar]
- 179.Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou M-M. 1999. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 399:491–96 [DOI] [PubMed] [Google Scholar]
- 180.Josling GA, Selvarajah SA, Petter M, Duffy MF. 2012. The role of bromodomain proteins in regulating gene expression. Genes (Basel). 3:320–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Shi J, Vakoc CR. 2014. The Mechanisms behind the Therapeutic Activity of BET Bromodomain Inhibition. Mol. Cell 54:728–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Cochran AG, Conery AR, Sims RJ. 2019. Bromodomains: a new target class for drug development. Nat. Rev. Drug Discov 18:609–28 [DOI] [PubMed] [Google Scholar]
- 183.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, et al. 2010. Selective inhibition of BET bromodomains. Nature. 468:1067–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, et al. 2010. Suppression of inflammation by a synthetic histone mimic. Nature. 468:1119–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Anders L, Guenther MG, Qi J, Fan ZP, Marineau JJ, et al. 2014. Genome-wide localization of small molecules. Nat. Biotechnol 32:92–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Li Z, Wang D, Li L, Pan S, Na Z, et al. 2014. “Minimalist” cyclopropene-containing photo-cross-linkers suitable for live-cell imaging and affinity-based protein labeling. J. Am. Chem. Soc 136:9990–98 [DOI] [PubMed] [Google Scholar]
- 187.Baud MGJ, Lin-Shiao E, Cardote T, Tallant C, Pschibul A, et al. 2014. A bump-and-hole approach to engineer controlled selectivity of BET bromodomain chemical probes. Science. 346:638–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Runcie AC, Zengerle M, Chan K-H, Testa A, van Beurden L, et al. 2018. Optimization of a “bump-and-hole” approach to allele-selective BET bromodomain inhibition. Chem. Sci 9:2452–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zengerle M, Chan K-H, Ciulli A. 2015. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol 10:1770–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Qin C, Hu Y, Zhou B, Fernandez-Salas E, Yang C-Y, et al. 2018. Discovery of QCA570 as an Exceptionally Potent and Efficacious Proteolysis Targeting Chimera (PROTAC) Degrader of the Bromodomain and Extra-Terminal (BET) Proteins Capable of Inducing Complete and Durable Tumor Regression. J. Med. Chem 61:6685–6704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zhou B, Hu J, Xu F, Chen Z, Bai L, et al. 2018. Discovery of a Small-Molecule Degrader of Bromodomain and Extra-Terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression. J. Med. Chem 61:462–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Reynders M, Matsuura BS, Bérouti M, Simoneschi D, Marzio A, et al. 2020. PHOTACs enable optical control of protein degradation. Sci. Adv 6:eaay5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Naro Y, Darrah K, Deiters A. 2020. Optical Control of Small Molecule-Induced Protein Degradation. J. Am. Chem. Soc 142:2193–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Maison C, Almouzni G. 2004. HP1 and the dynamics of heterochromatin maintenance. Nat. Rev. Mol. Cell Biol 5:296–304 [DOI] [PubMed] [Google Scholar]
- 195.Canzio D, Chang EY, Shankar S, Kuchenbecker KM, Simon MD, et al. 2011. Chromodomain-mediated oligomerization of HP1 suggests a nucleosome-bridging mechanism for heterochromatin assembly. Mol. Cell 41:67–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Munari F, Soeroes S, Zenn HM, Schomburg A, Kost N, et al. 2012. Methylation of lysine 9 in histone H3 directs alternative modes of highly dynamic interaction of heterochromatin protein hHP1β with the nucleosome. J. Biol. Chem 287:33756–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Hiragami-Hamada K, Soeroes S, Nikolov M, Wilkins B, Kreuz S, et al. 2016. Dynamic and flexible H3K9me3 bridging via HP1β dimerization establishes a plastic state of condensed chromatin. Nat. Commun 7:11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Müller KP, Erdel F, Caudron-Herger M, Marth C, Fodor BD, et al. 2009. Multiscale analysis of dynamics and interactions of heterochromatin protein 1 by fluorescence fluctuation microscopy. Biophys. J 97:2876–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Cheutin T, McNairn AJ, Jenuwein T, Gilbert DM, Singh PB, Misteli T. 2003. Maintenance of stable heterochromatin domains by dynamic HP1 binding. Science. 299:721–25 [DOI] [PubMed] [Google Scholar]
- 200.Kilic S, Bachmann AL, Bryan LC, Fierz B. 2015. Multivalency governs HP1α association dynamics with the silent chromatin state. Nat. Commun 6:7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, et al. 2017. Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature. 547:236–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Strom AR, Emelyanov AV., Mir M, Fyodorov DV., Darzacq X, Karpen GH. 2017. Phase separation drives heterochromatin domain formation. Nature. 547:241–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Gibson BA, Doolittle LK, Schneider MWG, Jensen LE, Gamarra N, et al. 2019. Organization of Chromatin by Intrinsic and Regulated Phase Separation. Cell. 179:470–84 [DOI] [PMC free article] [PubMed] [Google Scholar]