

Abstract
Androgen receptor (AR) contributes to the progression of glioblastoma (GBM), and antiandrogen agents have the potential to be used for the treatment of GBM. However, AR mutation commonly happens in GBM, which makes the antiandrogen agents less effective. Heat shock 27 kDa protein (HSP27) is a well-documented chaperone protein to stabilize ARs. Inhibition of HSP27 results in AR degradation regardless of the mutation status of ARs, which makes HSP27 a good target to abolish ARs in GBM. Compound I is a HSP27 inhibitor that significantly induces AR degradation in GBM cells via the proteasomal pathway, and it selectively inhibits AR-overexpressed GBM cell growth with IC50 values around 5 nM. The compound also significantly inhibits in vivo GBM xenograft at 20 mg/kg and does not cause toxicity to mice up to 80 mg/kg. These results suggest that targeting HSP27 to induce AR degradation in GBM is a promising and novel treatment.
Graphical Abstract
1. INTRODUCTION
Glioblastoma (GBM) is the most common and malignant tumor of the brain. The prognosis of GBM patients remains poor, even with the progress in the development of new surgical techniques and the standard chemotherapy of temozolomide (TMZ) combined with radiotherapy.1–3 TMZ is an alkylating agent that is considered the most efficient chemo drug in GBM therapy because of its good capability to pass the blood–brain barrier (BBB). However, de novo and acquired resistance to TMZ treatment is very common in GBM patients, resulting in a poor outcome.3 Identification of new molecular targets to develop more effective drugs for GBM treatment is very urgent. On the other hand, the incidence of GBM in men is higher than that in women, which is also associated with men experiencing poorer outcome.4,5 Moreover, there are well-reported cellular, molecular, and imaging alterations that underline these sex differences.5,6 These findings suggest that involvement in sex hormones could play a role in GBM development.7 To elucidate this sex disparity, studies found that an androgen receptor (AR) is overexpressed in GBM and androgens contribute to the GBM tumor progression.7–11 The overexpression of ARs and the role of androgens in GBM are consistent with the gender-related discrepancy of this disease. Therefore, targeting ARs becomes a novel approach in GBM and an AR inhibitor Seviteronel has been investigated in clinical trials for AR-overexpressed GBM patients.12 Unfortunately, AR mutation happens in 30% of the AR-overexpressed GBM patients,8 which very likely limits the effectiveness of AR-targeting agents in the treatment of GBM. It is critical to find an alternative approach to diminish AR activity in GBM.
In fact, the AR is correlated to a small chaperone protein heat shock protein 27 kDa (HSP27) that exists in multiple oligomeric states within the cells. Overexpression of HSP27 increases the stability of its client proteins and protects cancer cells from various treatment-induced cell death.13 The critical connection between HSP27 and ARs is that the AR is a well-documented HSP27 client protein.13,14 HSP27 is responsible for AR stability and seems to be a good direct target for HSP27-AR pathway in GBM. HSP27 functions through an ATP-independent mechanism and therefore could not be inhibited with geldanamycin derivatives that target the ATP-binding site of chaperone proteins.15–17 Strategies to inhibit HSP27 at the mRNA level are alternative approaches to suppress the protein activity.13,18 Gene silencing strategies using short interfering RNA (siRNA) and antisense oligonucleotides (ASO) to disrupt HSP27 have been investigated, and the AR level was consequently reduced.13,14,19 Unfortunately, GBM is different from other cancer types because of the BBB protection. ASO and siRNA HSP27 inhibitors are not applicable in GBM because of their poor BBB penetration of these biological agents. In addition, it is impossible to study the transient dynamic characteristics between HSP27 and ARs with HSP27 gene silencing techniques because they do not directly interfere with ARs and HSP27 at the protein level. Small-molecule HSP27 inhibitors without the administration difficulties and high potential to pass the BBB would demonstrate promise for great clinical outcomes in GBM treatment. They can also be used to investigate the transient changes in HSP27-AR interaction, following rapid HSP27 inhibition.
In our previous study, we developed dual HSP27 and tubulin inhibitors based on nimesulide [cyclooxygenase 2 (COX-2) inhibitor] as a lead compound.20 The derivatives showed inhibitory activity to both HSP27 chaperone function and tubulin polymerization.21 Some of the compounds showed great potency against cancer cell growth with IC50 values at the low nanomoles.21,22 Because of the HSP27 targeting effect, we hypothesize that the small molecules could affect the stability and function of ARs, which could make these compounds potential candidates to target AR-overexpressed GBM.23,24 In the present study, we systematically investigated a promising lead compound for the in vitro and in vivo anti-GBM activity. The targeting effect of the compound to AR stability via HSP27 inhibition was confirmed. The compound showed selectivity to AR-overexpressed GBM cells and also induced AR degradation in both in vitro and in vivo GBM models. Through a toxicity study, we identified the structure moieties that are critical to the low toxicity profile and provided a new direction for future structural modification. Our results indicate that the HSP27 inhibitor has great potential to be a group of novel agents for the treatment of AR-overexpressed GBM.
2. RESULTS AND DISCUSSION
2.1. ARs and Mutated ARs are Detected in GBM Cell Lines.
It has been demonstrated that the AR is highly expressed in some of the GBM patients and contribute to higher disease accidents in men than in women.7,8,25 Because of the high mutation of ARs in GBM, AR antagonists could not be very effective for the treatment of GBM in this regard.8,9 Abolishing the AR protein in GBM cells has more advantages because both wild-type and mutated ARs could be eliminated by this strategy.8–12 Therefore, we initiated the novel approach to induce AR degradation via inhibition of HSP27 in GBM cells because the AR is a well-documented client protein of HSP27 and its stability is dependent on HSP27.
Multiple GBM cell lines are used in our study for the potential drug candidate mechanism and efficacy evaluation. To examine the AR and HSP27 status in these GBM cells, we used western blotting assay to check the protein levels in the four cell lines including T98G, U251, A172, and U87 (Figure 1). Highest AR expression in T98G cells is observed, and U87 shows weaker expression, whereas U251 and A172 do not show clear AR expression compared to other two cell lines. To further explore the mutated AR (AR-V7, N-terminus-truncated ARs with a molecular weight of 87 kDa) in these cells, we used an antibody specific for mutated ARs and found that only T98G cells express the mutated version of ARs. The other three cell lines do not show clear protein bands. HSP27 is observed in three cell lines with the higher levels in T98G and A172 cells, whereas U251 cells do not express detectable HSP27. It seems that when ARs and HSP27 are coexpressed in the cells, higher HSP27 could contribute to higher ARs, as indicated by T98G cells. Both T98G and U87 cells show detectable HSP27 and ARs, and they could be good models for the investigation if targeting HSP27 could induce AR degradation.
Figure 1.

AR, mutated AR (AR-V7), and HSP27 expressions in four GBM cell lines. The proteins were analyzed by western blotting with specific antibodies, and the results are the representative images and quantification. Data are expressed as mean ± SD (n = 3).
2.2. Four Drug Candidates Showed Promising Selectivity and Activity, Inhibiting the Growth of AR-Overexpressed GBM Cells.
To identify the most promising compounds targeting GBM, we investigated the in vitro activity of the four nimesulide analogues developed from our previous studies (Figure 2).21,22 These compounds were discovered as dual HSP27 and tubulin inhibitors, which showed inhibitory activity to the in vitro chaperone activity of HSP27.21,22 The HSP27 inhibition of the compound triggers our interest to investigate if the compound could target the AR function of the GBM cells. First, we examined if the compound could inhibit the growth of the GBM cells with cell proliferation assay. The IC50 values of the compounds against the proliferation of the four GBM cell lines were determined with multiple doses, and the results are shown in Table 1. All the IC50 values are less than 20 nM, indicating the great potency of the compounds in the in vitro cell proliferation assay. Together, all four compounds showed relatively better potency to T98G cells that express higher level of AR expression, suggesting that the selectivity of the compounds is correlated to the AR expression in the cells. Overall, compounds with the ethyl sulfonamide moiety are slightly less active than methyl sulfonamide compounds, suggesting that the bulky group in this moiety harms the activity. It is possible that the bulky group decreases the solubility of the compound, which might reduce the biological activity.
Figure 2.

Chemical structures of compounds I–IV.
Table 1.
Growth Inhibitory Effects of the Four Compounds in GBM Cells
| IC50 (nM) |
||||
|---|---|---|---|---|
| compounds | T98G | U251 | A172 | U87 |
| I | 2.01 ± 0.64 | 4.00 ± 1.20 | 6.24 ± 1.06 | 4.78 ± 1.62 |
| II | 4.87 ± 2.04 | 6.39 ± 1.94 | 11.96 ± 2.77 | 6.74 ± 2.73 |
| III | 1.85 ± 0.81 | 1.57 ± 0.33 | 3.13 ± 0.78 | 3.05 ± 1.16 |
| IV | 5.71 ± 1.99 | 6.97 ± 1.08 | 8.21 ± 2.26 | 7.83 ± 3.81 |
2.3. N-Methyl Group is the Key Moiety of the Compounds Causing In Vivo Toxicity.
To identify the most promising drug candidate from the four compounds for further investigation, we used in vivo toxicity as a criterion. Mice were exposed to the four compounds at 80 mg/kg per day to evaluate the toxicity. For compounds III and IV, mice started to show dehydration and reduced food consumption after 5 and 7 days, respectively, and were euthanized accordingly. For compounds I and II, mice did not show any syndromes of toxicity such as acute pain or distress and weight loss even at day 10. All the rest of the mice were euthanized at day 10. To quantitively determine the in vivo toxicity, the weight of the mice was analyzed. Compounds III and IV (N-methyl group) significantly (p < 0.05) decreased the body weight of mice by the fifth and seventh day of exposure, while compounds I and II (N-hydrogen group) had no change in weight compared to the control group (Figure 3). Also, the hematology results of all the mice are listed in Table S1 (Supporting Information). There is no significant change in the profile of the blood even for compounds III and IV that showed toxicity to the mice. The results reveal that compounds I and II without the N-methyl group are less toxic to the animals compared to the compounds with the N-methyl group, suggesting that the methyl group seems to play a critical role in this toxicity. Considering the in vitro selectivity and activity and in vivo toxicity, compound I has more potential to be a better drug candidate. Therefore, we focused on the molecular mechanism investigation of compound I in AR-overexpressed GBM cells.
Figure 3.

Effect of compounds I–IV on body weight with the IP at a dose of 80 mg/kg. CD-1 mice were exposed to both compounds I and II and III and IV for 10, 5, and 7 days, respectively. Their body weight was measured at the beginning and the end of the experiment. Data are expressed as mean ± SD (n = 4). *p < 0.05, **p < 0.01 compared to the DMSO group with the unpaired t-test.
2.4. Compound I Inhibits the Chaperone Activity of HSP27.
This group of compounds were previously identified to bind to both HSP27 and tubulin.26 Therefore, we determined if compound I affects the chaperone activity of HSP27. Our hypothesis is that the compound has the potential to downregulate ARs via HSP27 inhibition. It is well documented that HSP27 plays a vital role in the prevention of cell apoptosis and effectively prevents the aggregation or degradation of its client proteins.27 The cellular protective functions of HSP27 in the apoptotic pathway are regulated by its chaperone activity, and this activity contributes to the protection of cells from stress stimuli.28 To examine the in vitro chaperone activity, insulin is often used as a model substrate protein to mimic the protein aggregation and HSP27 serves as the chaperone to prevent aggregation.29,30 In this study, dithiothreitol (DTT) could denature insulin, which induces insulin B chain to aggregate. We used alpha crystallin, that is, the chaperone function domain of HSP27 to perform the chaperone assay.30 In the presence of the chaperone protein, the aggregation of insulin can be suppressed because of the formation of stable complexes between the chaperone and the unfolded B chain.31 The aggregated insulin could be examined via the absorbance at 400 nm. The capability of compound I to modulate the in vitro chaperone activity of HSP27 was evaluated by monitoring the DTT-induced insulin aggregation in the presence of alpha crystallin with or without compound I. As shown in Figure 4, alpha crystallin exhibits significant potency against DTT-induced insulin aggregation. Compound I does not interfere with DTT-induced insulin aggregation directly. When compound I and α-crystallin are combined together, the chaperone function of α-crystallin is reduced. Therefore, more insulin aggregation is observed and the curve is shifted up compared to the only α-crystallin with DTT-induced insulin aggregation. The results demonstrate the inhibitory activity of compound I to the in vitro chaperone function of HSP27.
Figure 4.

Inhibition of compound I to HSP27 chaperone function. α-crystallin lost the activity to prevent DTT-induced insulin aggregation in the presence of compound I. The kinetics of the DTT-induced insulin aggregation was monitored in the absence of a chaperone protein or in the presence of a chaperone protein without or with compound I. The mixture of insulin and DTT with or without other components in the assay buffer was incubated for 45 min at 37 °C, and the absorbance at 400 nm was measured. The compound at this concentration or below did not interfere with DTT and insulin interaction. The results are representative of three independent experiments, each curve was measured in triplicate, and the mean was used to generate the curve. The representative one of the three experiments is presented. The statistical analysis was performed for the end reading of the curve with the unpaired t-test, p < 0.05, with compound I versus without compound I.
To investigate molecular interaction of compound I with HSP27, we performed a docking study with the published crystal structure of HSP27 (6DV5.pdb), similar to previous studies recently published (Figure 5).32 The crystal structure of HSP27 is a multimeric system, of which a biologically relevant phosphorylation site is located between two monomers, with S78 and S82 important for HSP27 phosphorylation.32 We found that compound I is able to bind to this site and block S78 by a hydrogen bond between the serine residue and the nitrogen from the sulfonamide moiety. This interaction likely contributes to the biological profile of compound I, preventing phosphorylation of HSP27 and thereby preventing HSP27 interaction with the misfolded proteins in the cellular environment. The docking result is consistent with the chaperone inhibition study of compound I.
Figure 5.

Binding of compound I with HSP27. (A) Multimer HSP27 protein crystal in the ribbon structure (6DV5.pdb). (B) Binding pocket between two monomers with compounds shown in space fill; (C) compound I binding to the HSP27 phosphorylation site, showing that SER78 is blocked from phosphorylation; and (D) 2D ligand interaction diagram of compound I docked to the HSP27 pocket.
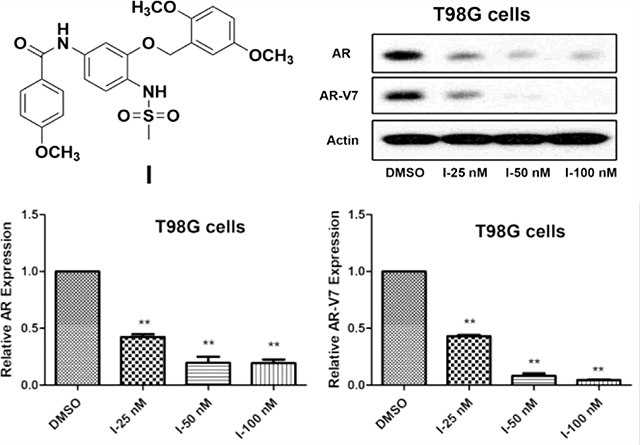
2.5. Compound I Dose-Dependently Decreases AR and Mutated AR Protein Levels in GBM Cells.
The AR is a well-studied client protein of HSP27 and inhibits HSP27 with siRNA-induced AR degradation, which has been reported in prostate cancer.14 Herein, we initiate the HSP27 inhibition strategy with a small molecule because of the potential BBB crossing activity, which is critical for GBM treatment. The results exhibit that compound I could downregulate AR protein levels in GBM cells dose-dependently (Figure 6). Compound I at 25, 50, and 100 nM all significantly induces degradation of ARs compared to the control in two GBM cell lines including T98G and U87. Furthermore, the mutated AR in T98G cells could be downregulated by compound I dose-dependently as well. The results demonstrate the great superiority of the strategy to targeting ARs compared to the AR antagonists because both wild-type and mutated ARs could be abolished by the compound. The AR downregulation activity of the compound is also consistent with the selectivity of the compound to AR-overexpressed cells in the cell proliferation study (Table 1). Regardless of the status of ARs in GBM cells, wild-type or mutated, a small-molecule HSP27 inhibitor could downregulate the AR protein level and inhibit the cell proliferation. So far, we demonstrate that compound I inhibits HSP27 chaperone function and downregulates ARs in GBM cells. However, whether the two activities are correlated to each other in GBM just like in prostate cancer still needs to be confirmed.
Figure 6.

Compound I abolishing ARs/mutated ARs in GBM. AR and AR-V7 expressions were analyzed by western blotting after the compound treatment. The experiment was repeated three times independently, and the representative image and quantification are shown. Data are expressed as mean ± SD (n = 3). *p < 0.05, **p < 0.01 compared to the DMSO treatment group by the unpaired t-test.
2.6. MG132 Reverses the Effect of Compound I on ARs in Both U87 and T98G Cells.
The AR is stabilized by HSP27, and without the protection of HSP27, ARs will be degraded by proteasomes, which has been reported in prostate cancer.14,19 We would like to determine if it is the same case in GBM cells that when HSP27 is inhibited by compound I, more AR could be degraded by proteasomes. This is based on the assumption that compound I affects HSP27 activity and thus accelerates AR degradation via the ubiquitin–proteasome pathway.33 MG132 is a proteasome inhibitor that can block proteasome-mediated protein degradation.34 We use MG132 as a tool to investigate whether compound I downregulates ARs via the proteasome pathway in T98G and U87 cells. The expression level of ARs in these cells was determined by western blotting assay with the treatment of compound I in the absence or presence of MG132, and the results are shown in Figure 7. Compound I could significantly decrease AR expression at 50 and 100 nM in U87 cells (p < 0.01, treatment vs control), whereas in the presence of MG132, the AR downregulation effect is significantly attenuated (p < 0.05, with MG132 vs without MG132). MG132 does not affect ARs when used alone. The same results are observed in T98G cells with wild-type of ARs and mutated ARs as well. Interestingly, it seems that MG132 does not fully rescue AR protein levels in the combination, particularly in U87 cells. It is possible that the compound could downregulate ARs with other mechanisms besides the proteasome pathway. In this case, even if the proteasome is blocked, ARs might be decreased by compound I with some other unknown pathways. Regardless, the MG132 rescue effect further connects HSP27 with ARs in GBM, and inhibiting HSP27 will lead to AR degradation via the proteasome pathway.
Figure 7.

Downregulation of ARs by compound I through the proteasome pathway. AR or mutated AR (AR-V7) expression was analyzed by western blotting after treatment by compound I with MG132 or without it. The experiment was repeated three times independently, and the representative image and quantification are shown. Data are expressed as mean ± SD (n = 3). **p < 0.01 compared to DMSO treatment by the unpaired t-test. #p < 0.05, ##p < 0.01 combination compared to only compound I treatment by the unpaired t-test.
2.7. Compound I Also Suppresses AR Transcription in GBM Cells.
We demonstrate that inhibiting HSP27 could downregulate ARs through the proteasome pathway to accelerate the protein degradation directly. The results from the combination of compound I and MG132 suggest that ARs might be regulated in multiple mechanisms by compound I because MG132 could not fully reverse the effect of compound I. Therefore, we examined the mRNA of ARs and the AR downstream genes PSA and NKX3.1 as well (Figure 8). The results listed here indicate that AR downstream genes PSA and NKX3.1 are significantly suppressed by compound I at 25 nM, which is consistent with the low AR protein level. The lower level of ARs could not effectively activate the androgen response element (ARE) and initiate the transcription. Interestingly, ARs are also suppressed via transcriptional regulation by compound I, and AR mRNA is downregulated significantly at 25 nM. The lower level of AR mRNA after the treatment of compound I is a new phenomenon, which has not been reported with HSP27 inhibition. It is possible that the downregulation of AR transcription is not the targeting effect of HSP27 inhibition. Lower AR mRNA could be due to the suppression of an AR promoter or degradation of mRNA via the post-transcriptional mechanism.35 For the AR promoter, there are several pathways such as nuclear factor κB (NF-κB) and cyclic adenosine monophosphate (cAMP) response element-binding protein involved in the regulation of the promoter.36,37 The AR itself actually also affects the AR mRNA. Post-transcriptional reregulation of the AR mRNA stability is far less investigated.35 At the current stage, it is difficult to elucidate the detailed mechanisms of the lower mRNA of ARs regulated by compound I. It is common that the small molecule has multiple molecular targets. In this case, compound I might affect other pathways to decrease AR transcription, which needs further investigation in the future.
Figure 8.

Transcriptional regulation of AR-related gene expression. The cells were treated with compound I for 12 h, and the RNA was extracted. AR, PSA, and NKX3.1 gene expressions were examined with real time PCR (polymerase chain reaction). The results of T98G cells (A) and U87 cells (B) are shown with the quantification results. Data are expressed as mean ± SD (n = 3). *p < 0.05, **p < 0.01 compared to the DMSO treatment group by the unpaired t-test.
2.8. Dihydrotestosterone Dissociates ARs from HSP27 and Decreases Compound I-Induced AR Degradation.
The AR initiates its biological function via testosterone binding and then translocates into the nucleus to activate the ARE. When the AR is in the nucleus, the HSP27-AR complex is broken, and AR degradation could only resume when the AR is out of the nucleus. To examine if HSP27-AR cooperation exists as we hypothesized in GBM, we used dihydrotestosterone (DHT) to force AR to translocate into the nucleus, which is a well-used method to initiate AR transcription activity. By this approach, the HSP27-AR complex will be temporarily broken because HSP27 will not enter the nucleus to continue stabilizing the AR. The immunofluorescence staining method could be used to examine the interaction of HSP27 and ARs. We observe that in T98G cells, the AR is distributed in both the nucleus and cytosol in the control group (Figure 9), which is due to the androgen residual in the cell culture medium. Part of the AR could be shuttled into the nucleus by the low level of androgens. HSP27 is distributed around the nucleus in the cytosol. When compound I is applied, the AR level is significantly lower compared to the control group. This is consistent with the results of the western blotting assay, and compound I induces AR degradation via HSP27 inhibition. When DHT is applied, all the ARs are concentrated in the nucleus and there is no distribution of ARs in the cytosol anymore. The results suggest that the AR in GBM is sensitive to the ligand DHT and responds to androgen stimulation very well. The high level of ARs condensed in the nucleus disrupts the HSP27-AR complex because HSP27 does not distribute in the nucleus. When DHT and compound I are combined, it is observed that the AR level in the nucleus is similar to only DHT treatment, suggesting that the HSP27-AR complex disruption eliminates the AR degradation induced by compound I. The results further demonstrate that compound I induces AR degradation through the HSP27-AR axis. The same phenomenon could be observed in U87 cells as well.
Figure 9.

Expression and location of ARs and HSP27 after T98G and U87 cells treated with compound 1 or DHT or their combination, and then the cells were analyzed by the immunofluorescence assay.
2.9. Compound I Inhibits Tubulin Polymerization via Interfering with the Colchicine Binding Domain.
This group of compounds were initially found to be binding to both HSP27 and tubulin.26 We observed that compound I inhibits HSP27 and induces AR degradation, and it shows selectivity to AR-overexpressed GBM cells (Table 1). However, compound I also inhibits the proliferation of GBM cells without AR expression, suggesting that the compound has general activity against GBM cells. This broad cell growth inhibition activity is unlikely to be correlated to AR degradation. A more general target of compound I might be responsible for the general cell growth inhibition. Herein, we examine the tubulin polymerization inhibition activity of compound I (Figure 10). The results exhibit that compound I dose-dependently inhibits tubulin polymerization in the in vitro enzyme assay. The result is consistent with our previous studies that the compound I is a dual inhibitor to HSP27 and tubulin,21,22 although it is more selective to AR-overexpressed T98G cells because of the AR downregulation activity via HSP27 inhibition. In terms of potency for the inhibition of tubulin polymerization, compound I at 1 μM is much more active than the positive control nocodazole, suggesting that compound I is also a potent tubulin inhibitor as a cancer therapeutic agent.
Figure 10.

Tubulin polymerization in the presence of different concentrations of compound I and positive control nocodazole. DMSO is used as a negative control. The results are representative of three independent experiments, and the mean value was used to generate the curve. The statistical analysis was performed with the end reading of the curve with the unpaired t-test, **p < 0.01 with compound I versus control.
A docking study reveals that compound I is able to occupy the binding pocket of the colchicine binding domain of tubulin. As can be seen from Figure 11, compound I binds to the colchicine binding site in tubulin (1SA0.pdb).38 Major hydrogen bond interactions can be seen between the sulfonamide moiety and ALA316 and LYS352. Also, a hydrogen bond is seen between nitrogen and SER178. The binding model supports the ability of compound I to destabilize normal tubulin function and the ability of compound I to act as a possible chemotherapeutic agent.
Figure 11.

Interaction of compound I with tubulin at the colchicine binding pocket (1SA0.pdb). (A) Dimer of tubulin shown as blue and pink, with compound I shown as space fill; (B) binding pocket of compound I with the solvent accessible surface shown; and (C) 2D ligand interaction plot with key amino acids contributing to the hydrogen bonding interactions.
2.10. Compound I Does Not Inhibit COX-2.
Compound I is a promising drug candidate for potential treatment of AR-overexpressed GBM. From the drug design point of view, we removed the initial N-methyl group of compound I, which actually was introduced to eliminate the COX-2 inhibition function of the very original lead compound nimesulide.39 The ionization of the sulfonamide moiety of nimesulide is critical for COX-2 inhibition. With a pka value of 5.93, the majority of nimesulide is ionized to form the negative charge, which is a key factor for COX-2 binding. Introducing the N-methyl group blocks ionization and eliminates COX-2 inhibition, which has been demonstrated in our previous study.39 However, the current investigation reveals that removing the methyl group could reduce in vivo toxicity and also improve solubility. There is a concern that whether removing this methyl group could bring COX-2 inhibition back. To clarify this potential problem, we measured the COX-2 inhibition of the compound using the prostaglandin E2 production assay (Figure 12). U87 cells were treated with compound I and nimesulide for 12 h. The cell culture medium was collected to determine the prostaglandin E2 level. The results show that prostaglandin E2 levels are significantly decreased compared to dimethyl sulfoxide (DMSO) (33.03 ± 6.03 pg/mL) with nimesulide (7.66 ± 1.18 pg/mL), as indicated in Figure 12, while there is no significant difference for compound I to control (35.33 ± 7.95 pg/mL). The results suggest that removing of the methyl group does not affect the COX-2 inhibition of compound I. It is speculated that the nitro group of nimesulide is critical to the COX-2 inhibition as well. In compound I, the nitro moiety is reduced to amino and formed a new benzamide moiety, and this change leads to the change in the pka value of compound I to 8.65. Under physiological pH conditions, compound I will not form negative charge anymore, which could eliminate the COX-2 inhibition as well. Therefore, the methyl group of the sulfonamide moiety is not critical for COX-2 inhibition anymore.
Figure 12.

Compound I does not change prostaglandin E2 production. U87 cells were treated with nimesulide (10 μM) and compound I (10 nM) for 12 h, the medium was collected, and the PGE2 level was examined with ELISA. Data are expressed as mean ± SD (n = 3). **p < 0.01 compared to the DMSO treatment group.
2.11. Compound I Significantly Reduces the Growth of Human GBM and the AR Level in the Xenograft Tumor Model.
As a potential drug candidate, it is critical to determine the in vivo activity of compound I with the GBM model. In order to investigate the compound, the xenograft tumor mouse model was established by subcutaneously injecting U87 cells into the left and right flank of nude mice. Unfortunately, we did not successfully form a xenograft model with T98G cells with the same procedure. After U87 tumor reached 200 mm3, compound I was administrated into mice via intraperitoneal injection (IP) and oral every other day for 2 weeks and tumor volume and mice weight were measured. As shown in Figure 13A, mice body weight is not affected by the compound I treatment either with IP or oral administration, which is consistent with the toxicity study (Figure 3). The tumor size of compound I with both IP and oral administration is significantly (p < 0.05) decreased compared with the DMSO group (Figure 13B), and the tumor weight of compound I with IP injection is significantly decreased compared with the DMSO group (Figure 13C). Unfortunately, because of the variability of the tumor with oral administration, the tumor weight does not reach statistical significance compared to the control. To confirm that the tumor inhibition effect is correlated with the AR protein, the AR expression level in tumor was examined via western blotting (Figure 13D). AR expression in both IP and oral administration groups for the compound I treatment is significantly (p < 0.01) decreased compared with the DMSO group. The results demonstrate the great in vivo activity of compound I in the animals and provide strong evidence that compound I is a promising new drug candidate to treat AR-overexpressed GBM.
Figure 13.

In vivo efficacy of compound I in human GBM. A total of 5 × 106 U87 cells were inoculated subcutaneously into nude mice, which were randomly assigned to DMSO and compound I-treated groups. Nude mice were treated with compound I at 20 mg/kg with IP injection and oral administration. Body weight (A, n = 4); tumor size (B, n = 8, data are expressed as mean ± SD. Oral ##p < 0.01, IP **p < 0.01 compared to the DMSO treatment group); and tumor weight (C, n = 8, data are expressed as mean ± SD. *p < 0.05 compared to the DMSO treatment group) were measured and recorded; AR expression in tumor was analyzed by western blotting, as shown by representative images and by quantification (D, n = 4, data are expressed as mean ± SD. *p < 0.05 compared to the DMSO treatment group).
3. CONCLUSIONS
GBM is the most aggressive and malignant primary human brain cancer with a high mortality rate.40 It is reported that a higher incidence rate happens in men compared to women (3:2),4,5,7 indicating that there is a sex disparity of the disease, which may be correlated with different sex hormone pathways.41 Further studies reveal that AR overexpression and mutation of ARs are frequently observed in human GBM, and suppressing AR expression could induce GBM cell death in vitro and in vivo.40,42 ARs play an important role in GBM progression.43 It is well known that HSP27 is a chaperone protein that could stabilize ARs.44 Targeting HSP27 to induce AR degradation becomes a novel approach for the treatment of AR-overexpressed GBM. Specifically inducing a protein degradation has been investigated via the proteolysis-targeting chimera (PROTAC) strategy.45 PROTAC achieves protein degradation through “hijacking” the cell’s ubiquitin–proteasome system. The PROTAC molecule consists of a ligand of the protein of interest and a covalently linked ligand of an E3 ubiquitin ligase. The ligand has to directly interact with the target protein.45 This strategy has been applied to induce oncoprotein degradation successfully in cells and even in animals. However, the PROTAC strategy-generated probe is always bulky and less water soluble, which limits its potential as a drug candidate. We aim to develop a small-molecule HSP27 inhibitor as potential drug candidates to abolish ARs in GBM, which makes this approach superior than the PROTAC approach because of the drug-like character of our compounds. The candidate does not directly bind to ARs. However, the compound binds to HSP27 and interferes with the HSP27-AR complex, which results in AR degradation. The identified candidate compound I shows great potency and selectivity to inhibit AR--overexpressed GBM cells. It inhibits HSP27 chaperone function and induces AR degradation, which is correlated to the selectivity to AR-overexpressed cells. The compound also inhibits tubulin polymerization, therefore showing general activity to inhibit cell proliferation. Compared to a similar analogue with an extra N-methyl group, compound I shows lower toxicity to mice. The removal of the N-methyl group does not bring the COX-2 inhibition back to the compound, suggesting that after the reduction of the nitro group in the very lead compound nimesulide, the COX-2 inhibition is eliminated in this scaffold. In addition to HSP27 and tubulin inhibition, compound I also suppressed AR transcription with an unknown mechanism because the mRNA of ARs is also decreased after the treatment. Lower AR mRNA could be due to the suppression of the AR promoter or degradation of mRNA via the post-transcriptional mechanism. At the current stage, it is difficult to elucidate the detailed mechanisms of the lower mRNA of ARs regulated by compound I. Taken together, it seems that the compound suppresses ARs in GBM with multiple mechanisms. The in vivo study reveals that compound I inhibits the U87 xenograft and abolishes the AR in the tumor samples as well. All the in vitro and in vivo activities demonstrate that compound I is a promising drug candidate for AR-overexpressed GBM.
4. EXPERIMENTAL SECTION
4.1. Reagents.
Compounds I–IV were prepared by our own lab. Prostaglandin E2 ELISA kit (Cayman Chemical Company, 514010) and thiazolyl blue tetrazolium bromide, 98% (Alfa Aesar, P31B064) were used. Tubulin (>99% pure) was isolated from bovine brain (Cytoskeleton, TL238). GTP (Cytoskeleton, BST06), tubulin glycerol buffer (Cytoskeleton, BST05–001), and tubulin general buffer (Cytoskeleton, BST01–010) were used. Insulin (Sigma-Aldrich, 91077C), α-crystallin (Sigma-Aldrich, C4163), DTT (Amresco, EC# 222–468-7), MG132 (Sigma-Aldrich, C2211), Dulbecco’s modified Eagle’s medium (DMEM) (Cleveland Clinic media laboratory, 11–500p), RPMI 1640 (Cleveland Clinic, 10–500p), fetal bovine serum (FBS) (Atlanta Biologicals, S11150), Pen/Strep solution (Cleveland Clinic, 725–100p), anti-β-actin antibody (Cell Signaling Technology, 4967S), RiboZol reagent (VWR, 97064–952), DNase I (Promega, M6101), ImProm-II reverse transcription system (Promega, A3800), 4% paraformaldehyde (VWR, J61899-AK), bovine serum albumin (Millipore Sigma, 2905-OP), DAPI (VWR, 95059–474), anti-HSP27 antibody (Cell Signaling Technology, 2402S), anti-AR antibody (Cell Signaling Technology, 5153S), anti-AR-V7 antibody (Cell Signaling Technology, 68492S), anti-rabbit IgG (Cell Signaling Technology, 7074S), anti-rabbit Alexa Fluor 488 secondary antibody (Thermo Scientific, A-21206), anti-mouse Alexa Fluor 594 secondary antibody (Thermo Scientific, SA5–10168), non-fat dry milk (Rockland, B51–0500), and A chemiluminescent substrate (Thermo Scientific, 34577) were used. All the other chemicals are of analytical grade.
4.2. Cell Culture.
T98G, A172, U87, and U251 cells were obtained from ATCC. The cells were maintained in RPMI 1640 or DMEM supplemented with 10% FBS, 100 U/mL penicillin, and 100 mg/mL streptomycin in a humidified incubator with 5% CO2 at 37 °C.
4.3. Compounds I–IV.
The synthesis of the four compounds has been published in our previous studies.21,22 All the final compounds exhibited purities above 97%. The chromatographic separation was performed on a C18 column (2.0 mm × 150 mm, 5 μm) obtained from Phenomenex (Torrance, CA). Two mobile phases (H2O/CH3OH and H2O/CH3CN) were employed for isocratic elution with a flow rate of 0.2 mL/min. The injection volume was 20 μL, and the UV detector was set up at 256 and 290 nm.
4.4. Cell Viability Analysis.
MTT assay was used to evaluate the effect of dual HSP27 and tubulin inhibitors on the growth of T98G, A172, U87, and U251 cells in eight replications. 3000 cells per well were seeded with RPMI 1640 or DMEM in 96-well flat-bottomed plates for 24 h and were then exposed to various concentrations of test compounds dissolved into DMSO (highest final concentration 0.1%) in the medium for 48 h. Controls received DMSO at a same concentration as that in highest does drug-treated cells. Cells were incubated in 100 μL of 1 mg/mL MTT reagent diluted in fresh media at 37 °C for 2 h. Supernatants were removed from the wells, and the precipitated MTT dye was dissolved in 200 μL/well DMSO. The absorbance at 570 nm was determined using a SpectraMax Plus 384 spectrophotometer (Molecular Devices).
4.5. Experimental Animals.
Male CD-1 mice and nude mice were purchased from Taconic lab. Mice were housed in Plexiglas cages, kept on a 12/12 h light–dark cycle, and received food and water ad libitum in a temperature- and humidity-controlled environment. All the experimental procedures involving animals were performed in accordance with the guide for the Care and Use of The Cleveland State University (CSU) Institutional Animal Care and Use Committee (IACUC).
4.6. Maximum Tolerated Dose Study.
Twenty CD-1 mice were randomly divided into five groups. Mice were injected with the vehicle (DMSO) or compounds I–IV [80 mg/kg in phosphate-buffered saline (PBS)] by IP three times weekly. Mice would be euthanized once the toxicity (e.g., dehydration, lethargy, disorientation, hunched posture, and ruffled coat) was observed. Body weights were determined at the start dosing day and the end dosing day. Non-fasted blood samples were collected from the heart immediately for hematology analysis after euthanizing. Hematology analysis was performed with a hematology analyzer—Element HT5 (Heska Corporation, USA).
4.7. Western Blotting.
Cells were cultured in six-well culture plates, incubated with DMSO or inhibitors, and then lysed with RIPA (Thermo Scientific, Prod# 89900) supplemented with a protease inhibitor cocktail (Thermo Scientific, Prod# 1861278). After incubating the cells on ice for 10 min, lysates were collected into a 1.5 mL centrifuge tube; then, the supernatant would be collected after being centrifuged at 10,000g for 10 min. Protein concentrations were determined by the Bradford protein assay kit (Bio-Rad). Fifty micrograms of the total protein lysate for each sample was boiled with 1× loading buffer for 10 min. Samples were then separated on a 10% SDS-polyacrylamide gel and transferred to a polyvinylidene fluoride membrane (Bio-Rad). The membrane was blocked for 2 h with 5% non-fat milk in 1× TBS-T (150 mM NaCl, 10 mM Tris, pH7.4, 0.1% Tween 20) at room temperature and then incubated with the primary antibody at 4 °C overnight. After the membrane was incubated with the primary antibody and washed three times with 1× TBS-T for 10 min each time, it was incubated with the secondary antibody for 60 min at room temperature. The membrane was washed three times again for 10 min each time with 1× TBS-T. Eventually, the membranes were incubated with the SuperSignal West Pico chemiluminescent substrate (Pierce) according to the protocol of the manufacturer.
4.8. Tubulin Polymerization Assay.
A mixture of 100 μL of microtubule-associated protein-rich tubulin (2 mg/mL, bovine brain, Cytoskeleton) in buffer containing 80 mM PIPES (pH 6.9), 2 mM MgCl2, 0.5 mM EGTA, and 5% glycerol was mixed with DMSO (as control) or various concentrations of compound I in DMSO and incubated at 37 °C. Then, 1 μL of 100 mM GTP was added to the mixture to initiate the tubulin polymerization, and the absorbance at 340 nm was monitored every single minute continuously for 45 min using the Molecular Devices SpectraMax Microplate reader.
4.9. HSP27 Chaperone Activity Assay.
24 μL of 1 mg/mL insulin stock solution was added to the single well of 384 well plates, 3 μL of 5 mg/mL α-crystallin (a segment of HSP27 responsible for the chaperone function of HSP27), and 71 μL of PBS with 10 μM compound I dissolved inside were added as well. The mixture was thoroughly mixed and incubated at 37 °C for 5 min, and then, 2 μL of 1 M DTT in water was added to initiate the insulin aggregation. The mixture of insulin in the absence or presence of α-crystallin with 0.1% DMSO was used as the control. The absorbance at 400 nm was monitored every 3 min continuously for 2 h using the Molecular Devices SpectraMax Microplate reader.
4.10. Quantitative Real-Time Polymerase Chain Reaction Analysis.
T98G and U87 cells were seeded at a density of 2 × 105 cells per well in a six-well plate, left in an incubator overnight to adhere, and were treated with compound I at 25 nM for 12 h. Total RNA was isolated using the RiboZol reagent according to the manufacturer’s instructions. The RNA yield and purity were determined spectrophotometrically at 260–280 nm, and the integrity of RNA was verified by electrophoresis by denaturing agarose gels stained with ethidium bromide. To remove DNA contamination from the RNA samples, 10 μg of the total RNA was incubated with 10 units of RNase-free DNase I at 37 °C for 30 min, followed by phenol/chloroform extraction and ethanol precipitation. Then, 1 μg of DNase I-treated RNA was reverse-transcribed into cDNA using the ImProm-II reverse transcription system according to the protocols. The iTaq Universal Sybr Green Super mix was obtained from Bio-Rad and used for setting up real-time PCR (RT-PCR) reactions. Also, the specific primers for RT-PCR were designed using the Primer Express software (v3.0; Applied Biosystems). The relative levels of each gene mRNA transcripts to 18S were determined. The primer sequences for AR, PSA, NKX3.1, and 18S were as follows: AR: GGCCAGGAAAGCGACTTCA (forward); CCCATTTCGCTTTTGACACA (reverse), P S A : TGTGCTTCAAGGTATCACGTCAT (forward); CTTGATCCACTTCCGGTAATGC (reverse), NKX3.1: CTTGGAGAAGCACTCCTCTTG (forward); CGCAGTACAGGTATGGGT AGTA (reverse), and 18S: TCGGAACTGAGGCCATGATT (forward); CTTTCGCTCTGGTCCGTCTT (reverse). The comparative cycle of the threshold fluorescence method was applied, and the relative transcript amount of the target gene was normalized to that of 18S using the 2−ΔΔCt method. Three replicates were performed per cDNA sample.
4.11. Immunofluorescence Assay.
T98G and U87 Cells were seeded in six-well plates. Cover slips were placed into the wells, and the cells could attach naturally. After 24 h, cells were treated with compound I at 100 nM, DHT at 10 nM, or combination for 12 h. Then, cells were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.1% Triton X-100 for 10 min, and blocked with 1% bovine serum albumin for 30 min. Several washing steps with TBST–BSA (5%) occurred in between fixation, permeabilization, and blocking. Then, incubation with the AR and HSP27 primary antibodies for 1 h, followed by washing with TBST–BSA (5%) and then fluorescein-labeled secondary antibodies for 1 h were performed. DAPI was incubated for 10 min to stain the nucleus. Images were visualized and analyzed using the GE Launches DeltaVision Ultra Microscopy System.
4.12. Computational Study.
Docking studies were done using the molecular modeling program MOE version 2019.0102 (Chemical Computing Group). Each protein target, HSP27 (6DV5.pdb) and tubulin (1SA0.pdb), was prepared for docking using the QuickPrep function, which adds hydrogen and optimizes the hydrogen orientation for pH 7.4; additionally, any missing loops or crystallographic steric classes were fixed. The binding pocket of HSP27 was identified from the literature,32 and for tubulin, the colchicine binding site was chosen as per our previous work.21 For the docking study, an induced-fit docking model was used because compound binding to proteins may induce changes in the amino acid orientation to allow for optimal binding between the ligand and protein. For each protein, 10 binding poses were generated and visually inspected for optimal binding interaction.
4.13. COX-2 Inhibition Assay.
From our previous research, all four inhibitors in this paper are derivatives of the COX-2 inhibitor nimesulide,20 and their COX-2 inhibition activity still remains to be determined. Prostaglandin E2 production is an indicator of COX-2 activity because COX-2 could catalyze the conversion of the substrate arachidonic acid to prostaglandins.46 In this study, compound I (10 nM) was treated with U87 cells for 12 h, and nimesulide (10 μM) was the positive control. After the treatment, the cell media were taken to determine prostaglandin E2 content via the prostaglandin E2 ELISA kit (Cayman Chemical, USA).
4.14. In Vivo Xenograft Study.
The animal protocol was approved by Cleveland State University (CSU) Institutional Animal Care and Use Committee (IACUC). NCRNU male nude mice were purchased from Taconic labs, NY. U87 cells were resuspended in sterile PBS (100 μL) and injected (5 × 106 cells/injection) subcutaneously at the left and right flank of a male nude mouse (5–6 weeks, n = 4/group, two tumors per mouse and eight tumors per group). Tumors and body weight were monitored with Vernier calipers three times weekly, and the tumor volume was calculated by the following formula: V = 2/3d1 × d22, where d1 is the larger diameter and d2 is the smaller diameter. When the tumor volume reached approximately 50 mm3, mice were injected with the vehicle (DMSO) or compound I (20 mg/kg in PBS) by IP or oral administration three times weekly for 14 days, and the tumor size was monitored and measured at the same time. In the end, mice were euthanized by exposure to excess CO2 and the tumors were removed and weighted. Also, every two tumors from every single mouse were mixed and homogenized with RIPA buffer (protein inhibitors and phenylmethylsulfonyl fluoride were added) to prepare the tumor lysates after the tumor was weighted.
4.15. Statistical Analysis.
Statistical and graphical information was determined using the GraphPad Prism software (GraphPad Software Incorporated) and Microsoft Excel (Microsoft Corporation). Gray values from western blotting were determined via Quantity One Software (Bio-Rad). The determination of IC50 values was performed using the nonlinear regression analysis. Statistically significant differences were calculated with the two-tailed unpaired Student’s t-test, and p values were reported at 95% confidence intervals.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by GRHD, Faculty Research Development (FRD) program, summer undergraduate research program of Cleveland State University, and National Institutes of Health instrumental grant (1S10OD025252-01).
ABBREVIATIONS
- AR
androgen receptor
- ASO
antisense oligonucleotides
- BBB
blood–brain barrier
- FBS
fetal bovine serum
- cAMP
cyclic adenosine monophosphate
- COX-2
cyclooxygenase 2
- GBM
glioblastoma
- HSP27
heat shock protein 27 kDa
- IP
intraperitoneal injection
- NF-κB
nuclear factor-κB
- PROTAC
proteolysis targeting chimera
- siRNA
short interfering RNA
- TMZ
temozolomide
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01537.
Compound purity (PDF)
Mice blood profile in the toxicity determination (PDF)
Compound molecular formula strings (CSV)
Contributor Information
Yaxin Li, Department of Chemistry, Center for Gene Regulation in Health and Disease, College of Sciences and Health Professions, Cleveland State University, Cleveland, Ohio 44115, United States.
Cody M. Orahoske, Department of Chemistry, Center for Gene Regulation in Health and Disease, College of Sciences and Health Professions, Cleveland State University, Cleveland, Ohio 44115, United States
Werner J. Geldenhuys, Department of Pharmaceutical Sciences, School of Pharmacy, West Virginia University, Morgantown, West Virginia 26506, United States.
Asmita Bhattarai, Department of Biological, Geological, and Environmental Sciences, Center for Gene Regulation in Health and Disease, College of Sciences and Health Professions, Cleveland State University, Cleveland, Ohio 44115, United States.
Abboud Sabbagh, Department of Chemistry, Center for Gene Regulation in Health and Disease, College of Sciences and Health Professions, Cleveland State University, Cleveland, Ohio 44115, United States.
Viharika Bobba, Department of Chemistry, Center for Gene Regulation in Health and Disease, College of Sciences and Health Professions, Cleveland State University, Cleveland, Ohio 44115, United States.
Fatma M. Salem, Department of Chemistry, Center for Gene Regulation in Health and Disease, College of Sciences and Health Professions, Cleveland State University, Cleveland, Ohio 44115, United States
Wenjing Zhang, Department of Chemistry, Center for Gene Regulation in Health and Disease, College of Sciences and Health Professions, Cleveland State University, Cleveland, Ohio 44115, United States.
Girish C. Shukla, Department of Biological, Geological, and Environmental Sciences, Center for Gene Regulation in Health and Disease, College of Sciences and Health Professions, Cleveland State University, Cleveland, Ohio 44115, United States
Justin D. Lathia, Department of Biological, Geological, and Environmental Sciences, Center for Gene Regulation in Health and Disease, College of Sciences and Health Professions, Cleveland State University, Cleveland, Ohio 44115, United States; Department of Molecular Medicine, Lerner Research Institute, Cleveland Clinic, and Case Comprehensive Cancer Center, Cleveland, Ohio 44195, United States
Bingcheng Wang, Rammelkamp Center for Research and Department of Medicine, MetroHealth Campus, Case Comprehensive Cancer Center, Case Western Reserve University School of Medicine, Cleveland, Ohio 44109, United States.
Bin Su, Department of Chemistry, Center for Gene Regulation in Health and Disease, College of Sciences and Health Professions, Cleveland State University, Cleveland, Ohio 44115, United States.
REFERENCES
- (1).Stupp R; Taillibert S; Kanner AA; Kesari S; Steinberg DM; Toms SA; Taylor LP; Lieberman F; Silvani A; Fink KL; Barnett GH; Zhu J-J; Henson JW; Engelhard HH; Chen TC; Tran DD; Sroubek J; Tran ND; Hottinger AF; Landolfi J; Desai R; Caroli M; Kew Y; Honnorat J; Idbaih A; Kirson ED; Weinberg U; Palti Y; Hegi ME; Ram Z Maintenance Therapy With Tumor-Treating Fields Plus Temozolomide vs Temozolomide Alone for Glioblastoma. JAMA 2015, 314, 2535. [DOI] [PubMed] [Google Scholar]
- (2).Stupp R; Taillibert S; Kanner A; Read W; Steinberg DM; Lhermitte B; Toms S; Idbaih A; Ahluwalia MS; Fink K; Di Meco F; Lieberman F; Zhu J-J; Stragliotto G; Tran DD; Brem S; Hottinger AF; Kirson ED; Lavy-Shahaf G; Weinberg U; Kim C-Y; Paek S-H; Nicholas G; Bruna J; Hirte H; Weller M; Palti Y; Hegi ME; Ram Z Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma. JAMA 2017, 318, 2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Lee SY Temozolomide Resistance in Glioblastoma Multiforme. Genes Dis. 2016, 3, 198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Ostrom QT; Cote DJ; Ascha M; Kruchko C; Barnholtz-Sloan JS Adult Glioma Incidence and Survival by Race or Ethnicity in the United States From 2000 to 2014. JAMA Oncol. 2018, 4, 1254–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ostrom QT; Rubin JB; Lathia JD; Berens ME; Barnholtz-Sloan JS Females Have the Survival Advantage in Glioblastoma. Neuro. Oncol. 2018, 20, 576–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Bayik D; Zhou Y; Park C; Hong C; Vail D; Silver DJ; Lauko A; Roversi G; Watson DC; Lo A; Alban TJ; McGraw M; Sorensen M; Grabowski MM; Otvos B; Vogelbaum MA; Horbinski C; Kristensen BW; Khalil AM; Hwang TH; Ahluwalia MS; Cheng F; Lathia JD Myeloid-Derived Suppressor Cell Subsets Drive Glioblastoma Growth in a Sex-Specific Manner. Cancer Discovery 2020, 10, 1210–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Yang W; Warrington NM; Taylor SJ; Whitmire P; Carrasco E; Singleton KW; Wu N; Lathia JD; Berens ME; Kim AH; Barnholtz-Sloan JS; Swanson KR; Luo J; Rubin JB Sex Differences in GBM Revealed by Analysis of Patient Imaging, Transcriptome, and Survival Data. Sci. Transl. Med. 2019, 11, No. eaao5253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zalcman N; Canello T; Ovadia H; Charbit H; Zelikovitch B; Mordechai A; Fellig Y; Rabani S; Shahar T; Lossos A; Lavon I Androgen Receptor: A Potential Therapeutic Target for Glioblastoma. Oncotarget 2018, 9, 19980–19993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bao D; Cheng C; Lan X; Xing R; Chen Z; Zhao H; Sun J; Wang Y; Niu C; Zhang B; Fang S Regulation of p53wt glioma cell proliferation by androgen receptor-mediated inhibition of small VCP/p97-interacting protein expression. Oncotarget 2017, 8, 23142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zalsman N; Canello T; Ovadia H; Charbit H; Zelikovitch B; Mordechai A; Fellig Y; Rabani S; Shahar T; Lossos A; Lavon I CSIG-24. ANDROGEN RECEPTOR IS A POTENTIAL THERAPEUTIC TARGET IN GLIOBLASTOMA. Neuro. Oncol. 2017, 19, vi54–vi55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lavon I; Zalsman N; Canello T; Charbit H; Zelikovitch B; Mordechai A; Fellig Y; Rabani S; Shahar T; Lossos A CSIG-13. ANDROGEN RECEPTOR IS INVOLVED IN GLIOBLASTOMA AND PRESENTS A POTENTIAL THERAPEUTIC TARGET. Neuro. Oncol. 2016, 18, vi43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Abstracts from the 23rd Annual Scientific Meeting and Education Day of the Society for Neuro-Oncology November 15–18, 2018 New Orleans, Louisiana; Neuro-Oncology, 2018; Vol. 20 (suppl_6). [Google Scholar]
- (13).Gibert B; Eckel B; Fasquelle L; Moulin M; Bouhallier F; Gonin V; Mellier G; Simon S; Kretz-Remy C; Arrigo A-P; Diaz-Latoud C Knock Down of Heat Shock Protein 27 (HspB1) Induces Degradation of Several Putative Client Proteins. PLoS One 2012, 7, No. e29719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zoubeidi A; Zardan A; Beraldi E; Fazli L; Sowery R; Rennie P; Nelson C; Gleave M Cooperative Interactions between Androgen Receptor (AR) and Heat-Shock Protein 27 Facilitate AR Transcriptional Activity. Cancer Res. 2007, 67, 10455–10465. [DOI] [PubMed] [Google Scholar]
- (15).Lelj-Garolla B; Mauk AG Roles of the N- and C-Terminal Sequences in Hsp27 Self-Association and Chaperone Activity. Protein Sci. 2012, 21, 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).McDonald ET; Bortolus M; Koteiche HA; Mchaourab HS Sequence, Structure, and Dynamic Determinants of Hsp27 (HspB1) Equilibrium Dissociation Are Encoded by the N-Terminal Domain. Biochemistry 2012, 51, 1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Neckers L; Workman P Hsp90 Molecular Chaperone Inhibitors: Are We There Yet? Clin. Cancer Res. 2012, 18, 64–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Jakubowicz-Gil J; Langner E; Bądziul D; Wertel I; Rzeski W Silencing of Hsp27 and Hsp72 in Glioma Cells as a Tool for Programmed Cell Death Induction upon Temozolomide and Quercetin Treatment. Toxicol. Appl. Pharmacol. 2013, 273, 580–589. [DOI] [PubMed] [Google Scholar]
- (19).Andrieu C; Taieb D; Baylot V; Ettinger S; Soubeyran P; De-Thonel A; Nelson C; Garrido C; So A; Fazli L; Bladou F; Gleave M; Iovanna JL; Rocchi P Heat Shock Protein 27 Confers Resistance to Androgen Ablation and Chemotherapy in Prostate Cancer Cells through EIF4E. Oncogene 2010, 29, 1883–1896. [DOI] [PubMed] [Google Scholar]
- (20).Yi X; Zhong B; Smith KM; Geldenhuys WJ; Feng Y; Pink JJ; Dowlati A; Xu Y; Zhou A; Su B Identification of a Class of Novel Tubulin Inhibitors. J. Med. Chem. 2012, 55, 3425–3435. [DOI] [PubMed] [Google Scholar]
- (21).Zhong B; Chennamaneni S; Lama R; Yi X; Geldenhuys WJ; Pink JJ; Dowlati A; Xu Y; Zhou A; Su B Synthesis and Anticancer Mechanism Investigation of Dual Hsp27 and Tubulin Inhibitors. J. Med. Chem. 2013, 56, 5306–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zhong B; Lama R; Kulman DG; Li B; Su B Lead Optimization of Dual Tubulin and Hsp27 Inhibitors. Eur. J. Med. Chem. 2014, 80, 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zhong B; Chennamaneni S; Lama R; Yi X; Geldenhuys WJ; Pink JJ; Dowlati A; Xu Y; Zhou A; Su B Synthesis and Anticancer Mechanism Investigation of Dual Hsp27 and Tubulin Inhibitors. J. Med. Chem. 2013, 56, 5306–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhong B; Lama R; Kulman DG; Li B; Su B Lead Optimization of Dual Tubulin and Hsp27 Inhibitors. Eur. J. Med. Chem. 2014, 80, 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bayik D; Zhou Y; Park C; Hong C; Vail D; Silver DJ; Lauko A; Roversi G; Watson DC; Lo A; Alban TJ; McGraw M; Sorensen M; Grabowski MM; Otvos B; Vogelbaum MA; Horbinski C; Kristensen BW; Khalil AM; Hwang TH; Ahluwalia MS; Cheng F; Lathia JD Myeloid-Derived Suppressor Cell Subsets Drive Glioblastoma Growth in a Sex-Specific Manner. Cancer Discov. 2020, 10, 1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Yi X; Zhong B; Smith KM; Geldenhuys WJ; Feng Y; Pink JJ; Dowlati A; Xu Y; Zhou A; Su B Identification of a Class of Novel Tubulin Inhibitors. J. Med. Chem. 2012, 55, 3425–3435. [DOI] [PubMed] [Google Scholar]
- (27).Concannon CG; Gorman AM; Samali A On the Role of Hsp27 in Regulating Apoptosis. Apoptosis 2003, 8, 61. [DOI] [PubMed] [Google Scholar]
- (28).Rogalla T; Ehrnsperger M; Preville X; Kotlyarov A; Lutsch G; Ducasse C; Paul C; Wieske M; Arrigo A-P; Buchner J; Gaestel M Regulation of Hsp27 Oligomerization, Chaperone Function, and Protective Activity against Oxidative Stress/Tumor Necrosis Factor α by Phosphorylation. J. Biol. Chem. 1999, 274, 18947–18956. [DOI] [PubMed] [Google Scholar]
- (29).Faiella L; Piaz FD; Bisio A; Tosco A; De Tommasi N A Chemical Proteomics Approach Reveals Hsp27 as a Target for Proapoptotic Clerodane Diterpenes. Mol. BioSyst. 2012, 8, 2637. [DOI] [PubMed] [Google Scholar]
- (30).Idippily ND; Zheng Q; Gan C; Quamine A; Ashcraft MM; Zhong B; Su B Copalic Acid Analogs Down-Regulate Androgen Receptor and Inhibit Small Chaperone Protein. Bioorg. Med. Chem. Lett. 2017, 27, 2292. [DOI] [PubMed] [Google Scholar]
- (31).Lelj-Garolla B; Mauk AG Self-Association and Chaperone Activity of Hsp27 Are Thermally Activated. J. Biol. Chem. 2006, 281, 8169–8174. [DOI] [PubMed] [Google Scholar]
- (32).Nappi L; Aguda AH; Nakouzi NA; Lelj-Garolla B; Beraldi E; Lallous N; Thi M; Moore S; Fazli L; Battsogt D; Stief S; Ban F; Nguyen NT; Saxena N; Dueva E; Zhang F; Yamazaki T; Zoubeidi A; Cherkasov A; Brayer GD; Gleave M Ivermectin Inhibits HSP27 and Potentiates Efficacy of Oncogene Targeting in Tumor Models. J. Clin. Invest. 2019, 130, 699–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Stope MB; Schubert T; Staar D; Rönnau C; Streitbörger A; Kroeger N; Kubisch C; Zimmermann U; Walther R; Burchardt M Effect of the Heat Shock Protein HSP27 on Androgen Receptor Expression and Function in Prostate Cancer Cells. World J. Urol. 2012, 30, 327–331. [DOI] [PubMed] [Google Scholar]
- (34).Yoshida T; Shiraishi T; Nakata S; Horinaka M; Wakada M; Mizutani Y; Miki T; Sakai T Proteasome Inhibitor MG132 Induces Death Receptor 5 through CCAAT/Enhancer-Binding Protein Homologous Protein. Cancer Res. 2005, 65, 5662–5667. [DOI] [PubMed] [Google Scholar]
- (35).Burnstein KL Regulation of Androgen Receptor Levels: Implications for Prostate Cancer Progression and Therapy. J. Cell. Biochem. 2005, 95, 657–669. [DOI] [PubMed] [Google Scholar]
- (36).Zhang L; Charron M; Wright WW; Chatterjee B; Song CS; Roy AK; Brown TR Nuclear Factor-κB Activates Transcription of the Androgen Receptor Gene in Sertoli Cells Isolated from Testes of Adult Rats. Endocrinology 2004, 145, 781–789. [DOI] [PubMed] [Google Scholar]
- (37).Supakar PC; Jung MH; Song CS; Chatterjee B; Roy AK Nuclear Factor κ B Functions as a Negative Regulator for the Rat Androgen Receptor Gene and NF- κ B Activity Increases during the Age-dependent Desensitization of the Liver. J. Biol. Chem. 1995, 270, 837–842. [DOI] [PubMed] [Google Scholar]
- (38).Ravelli RBG; Gigant B; Curmi PA; Jourdain I; Lachkar S; Sobel A; Knossow M Insight into Tubulin Regulation from a Complex with Colchicine and a Stathmin-like Domain. Nature 2004, 428, 198–202. [DOI] [PubMed] [Google Scholar]
- (39).Su B; Diaz-Cruz ES; Landini S; Brueggemeier RW Novel Sulfonanilide Analogues Suppress Aromatase Expression and Activity in Breast Cancer Cells Independent of COX-2 Inhibition. J. Med. Chem. 2006, 49, 1413–1419. [DOI] [PubMed] [Google Scholar]
- (40).Zalcman N; Canello T; Ovadia H; Charbit H; Zelikovitch B; Mordechai A; Fellig Y; Rabani S; Shahar T; Lossos A; Lavon I Androgen Receptor: A Potential Therapeutic Target for Glioblastoma. Oncotarget 2018, 9. DOI: DOI: 10.18632/oncotarget.25007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Hess KR; Broglio KR; Bondy ML Adult glioma incidence trends in the United States, 1977–2000. Cancer 2004, 101, 2293–2299. [DOI] [PubMed] [Google Scholar]
- (42).Yu X; Jiang Y; Wei W; Cong P; Ding Y; Xiang L; Wu K Androgen Receptor Signaling Regulates Growth of Glioblastoma Multiforme in Men. Tumor Biol. 2015, 36, 967–972. [DOI] [PubMed] [Google Scholar]
- (43).Rodríguez-Lozano DC; Piña-Medina AG; Hansberg-Pastor V; Bello-Alvarez C; Camacho-Arroyo I Testosterone Promotes Glioblastoma Cell Proliferation, Migration, and Invasion Through Androgen Receptor Activation. Front. Endocrinol. 2019, 10, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Li J; Fu X; Cao S; Li J; Xing S; Li D; Dong Y; Cardin D; Park H-W; Mauvais-Jarvis F; Zhang H Membrane-Associated Androgen Receptor (AR) Potentiates Its Transcriptional Activities by Activating Heat Shock Protein 27 (HSP27). J. Biol. Chem. 2018, 293, 12719–12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Winter GE; Buckley DL; Paulk J; Roberts JM; Souza A; Dhe-Paganon S; Bradner JE Phthalimide Conjugation as a Strategy for in Vivo Target Protein Degradation. Science 2015, 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Yaksh TL; Dirig DM; Conway CM; Svensson C; Luo ZD; Isakson PC The Acute Antihyperalgesic Action of Nonsteroidal, Anti-Inflammatory Drugs and Release of Spinal Prostaglandin E2Is Mediated by the Inhibition of Constitutive Spinal Cyclooxygenase-2 (COX-2) but not COX-1. J. Neurosci. 2001, 21, 5847–5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.